

Abstract
In situ hybridization is a powerful technique to identify specific RNA or DNA sequences within individual cells in tissue sections, providing important insights into physiological processes and disease pathogenesis. In situ hybridization (ISH) has been used for many years to assess the location of cells infected by viruses, but recently a next-generation ISH approach was developed with a unique probe design strategy that allows simultaneous signal amplification and background suppression to achieve single-molecule visualization while preserving tissue morphology. This next-generation ISH is based on an approach like branched PCR, but performed in situ and is more facile, sensitive, and reproducible than classical ISH methods or in situ PCR approaches in routinely detecting RNA or DNA in formalin-fixed paraffin embedded (FFPE) tissues. For the last several years our laboratory has been applying this ISH platform for the detection of human immunodeficiency (HIV) and simian immunodeficiency (SIV) viral RNA (vRNA) and/or viral DNA (vDNA) positive cells within a multitude of FFPE tissues. With this detailed technical manuscript, we would like to share our knowledge and advice with all individuals interested in using next-generation ISH in their research.
Introduction
ISH is the experimental approach used for targeting and visualizing complementary DNA, RNA, or modified nucleic acid strands (i.e., probes) to specific DNA or RNA sequences within a cell or section of tissue. ISH allows for the specific localization and visualization of specific nucleic sequences in tissues, important to understand the expression level, organization, distribution, and interactions between the target and its cellular environment, which is valuable information not obtainable with the use of other popular techniques, such as qPCR. Until recently, ISH was commonly performed with either a labeled complementary DNA or a complementary RNA (riboprobe).
These probes were either directly conjugated with radio-, fluorescent-, or antigen-labeled bases (e.g., 35 S, FITC, and digoxigenin) then localized and quantified in the tissue using either autoradiography, fluorescence microscopy, or immunohistochemistry detection approaches, respectively. While these in situ technologies continue to be valuable approaches, there is ample room for improvement to develop approaches that are less labor intensive, simpler, faster, sensitive, and specific.
An alternative commercial next-generation ISH approach (e.g., RNAscope assay), first described in 2012, for the detection of host messenger RNA (mRNA) is based on branched PCR. The mRNA detection is performed in FFPE cells and tissues, with a sensitivity approaching single-RNA molecule visualization in individual cells1. The specificity of this approach is achieved under the unique condition that two double-Z target probes bind contiguously to their respective complementary RNA (or DNA) sequences for a signal preamplifier to sequentially bind1. This allows the initiation of a signal amplification cascade via subsequent hybridization steps similar to branched DNA (bDNA)1, 2. In addition, this approach is remarkably rapid and easy, with results obtained in just 1 day (<8 h), a significant advantage compared to up to 4 weeks with alternative techniques, including Radio-ISH1, 2. This next-generation ISH has opened new perspectives and opportunities for HIV/SIV research. The major obstacles to a HIV cure are the cellular and tissue reservoirs that are established during the early stages of the disease3, 4. The overall goal of this technique is to identify, localize, and ultimately understand the principal tissue compartments that act as a viral reservoir and are persistent within an infected host. This will in turn help in the development of effective cure strategies against HIV.
In this manuscript, we explain our duplex next-generation RNA/DNA multiplex ISH protocol (e.g., RNAscope/DNAscope) in detail and explain how we modified the existing RNA ISH protocol to optimize the next-generation ISH to our samples and specific targets. This protocol allows visualization, localization, and quantification of HIV/SIV viral RNA and viral DNA within 5 μm tissue sections. Simultaneous visualization of both vRNA and vDNA is performed by combining two custom probe sets: one sense, targeting the vDNA coding strand (C1 SIVmac239 Gag-Pol-Sense probe [416141-C1]), and one anti-sense, targeting vRNA transcripts (C2 SIVmac239 Vif-Env-Nef-Tar-Anti-Sense probe [416131-C2]) covering different regions of the viral genome (Table 1), using two different visualization channels, C1 and C2. In this protocol, channels C1 and C2 allow us to visualize signals in different colors (i.e., AP in red and HRP in brown) and detect the probes with different approaches. Excluding the tissue fixation processing and cutting, this assay takes 2 days. Presented here is the duplex vRNA and vDNA in situ hybridization protocol that can be performed on cell pellets or tissue sections.
Table 1:
List of probes to target vRNA and vDNA of HIV-1 and SIV.
| Single Plex RNA/DNAscope Probe Sets | |||
|---|---|---|---|
| Name | ACD Catalog # | Number of ZZ | Description |
| SIVmac239 (Anti-Sense) | 312811 | 83 | Anti-sense probe targeting within 1251-9420bp of D01065.1 (gag, pol, vif, vpx, vpr, tat, env, and nef) |
| SIVmac239 (Sense) | 314071 | 83 | Sense probe targeting reverse strand within 1251-9420bp of D01065.1 (gag, pol, vif, vpx, vpr, tat, env, and nef) |
| V-HIV1-Clade A (Anti-Sense) | 416101 | 80 | Anti-sense probe targeting within 879-7629bp of HIV-1 Clade A Consensus (gag, pol, vif, vpr, tat, rev, vpu, env, and nef) |
| V-HIV1-Clade A (Sense) | 426341 | 80 | Sense probe targeting reverse strand within 879-7629bp of HIV-1 Clade A Consensus (gag, pol, vif, vpr, tat, rev, vpu, env, and nef) |
| V-HIV1-Clade B (Anti-Sense) | 416111 | 78 | Anti-sense probe targeting within 854-8291bp of AF324493.2, HIV-1 Clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env, and nef) |
| V-HIV1-Clade B (Sense) | 425531 | 78 | Sense probe targeting reverse strand within 854-8291bp of AF324493.2, HIV-1 Clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env, and nef) |
| V-HIV1-Clade D (Anti-Sense) | 416121 | 76 | Anti-sense probe targeting within 894-7697bp of HIV-1 Clade D Consensus (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| V-HIV1-Clade D (Sense) | 426351 | 76 | Sense probe targeting reverse strand within 894-7697bp of HIV-1 Clade D Consensus (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| Multi-Plex RNA/DNAscope Probe Sets | |||
| Name | ACD Catalog # | Number of ZZ | Description |
| V-SIVmac239-gag-pol-Sense-C1 | 416141-C1 | 40 | Sense probe targeting reverse strand within 1251-4093bp of D01065.1 (gag and pol) |
| V-SIVmac239-vif-env-nef-tar-C2 (Anti-sense) | 416131-C2 | 47 | Anti-sense probe targeting within 5381-10257bp of D01065.1(vif, vpx, vpr, tat, env, nef, and the TAR element) |
| V-HIV1-Clade_B-gag-pol-sense-C1 | 444051-C1 | 40 | Sense probe targeting reverse strand within 854-3940bp of AF324493.2, HIV-1 Clade B NL4-3 (gag and pol) |
| V-HIV1-Clade_B-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Anti-sense) | 444061-C2 | 40 | Anti-sense probe targeting within 5042-9673bp of AF324493.2,, HIV-1 Clade B NL4-3 (vif, vpr, tat, env, nef, and the TAR element) |
| V-HIV1-Clade_C-gag-pol-sense-C1 | 444021-C1 | 48 | Sense probe targeting reverse strand within 888-5032bp of HIV-1 Clade C concensus sequence (gag and pol) |
| V-HIV1-Clade_C-vif-vpr-rev-vpu-env-nef-tar-C2 (Anti-sense) | 444041-C2 | 49 | Anti-sense probe targeting within 5078-9698bp of HIV-1 Clade C concensus sequence (vif, vpr, tat, env, nef, and the TAR element) |
| V-HIV1-Clade_AE-gag-pol-sense-C1 | 444011-C1 | 55 | Sense probe targeting reverse strand within 890-4812bp of AF259954.1, HIV-1 Clade AE (gag and pol) |
| V-HIV1-Clade_AE-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Anti-sense) | 444031-C2 | 57 | Anti-sense probe targeting within 5052-9694bp of AF259954.1, HIV-1 Clade AE (vif, vpr, tat, env, nef, and the TAR element) |
Protocol
1. Section and slide preparations
-
Trim the paraffin blocks and use a microtome to cut 5 +/−1 µm sections. Mount the sections or cell pellets onto charged microscope slides in a 40–45 °C RNase free water bath. Air dry slides overnight at 37 °C or RT.
NOTE: The slides can be stored for up to 3 months at room temperature (RT) and 6 months at 4 °C.
-
Deparaffinize FFPE slides.
Bake slides in a dry oven for 1 h at 60 °C.
In a fume hood, fill two slide staining dishes with ~200 mL of fresh xylene, and two additional staining dishes with ~200 mL of fresh 100% ethanol. Cover the containers with lids.
Place the slides in a rack and submerge in the first xylene-containing dish. Incubate for 5–10 min at RT with agitation.
Place the slides into the second xylene-containing dish and incubate for 5–10 min at RT with agitation.
Immediately place the slides in the dish containing 100% ethanol. Incubate the slides for 5–10 min at RT with agitation.
Immediately place the slides in the second dish containing 100% ethanol and incubate for 5–10 min at RT with agitation.
Remove the rack from the ethanol, gently tap the side of the rack to remove excess ethanol, and rinse in RNase free water for 5–15 min.
2. Oven preparation
Turn on the hybridization oven and set the temperature to 40 °C.
Place a cloth or sturdy absorbent paper towel in a tray and wet completely with double-distilled water to allow for humidity control.
Insert the covered tray into the oven and close the oven door. Warm the tray for at least 30 min at 40 °C before use. Keep the tray in the oven when not in use.
3. Heat-induced epitope retrieval
Prepare 0.5x citrate based ISH hybridization target retrieval buffer (10 nmol/L, pH = 6, see Table of Materials). Bring it to a boil in a beaker on the heating plate.
Perform heat-induced epitope retrieval by placing the slides into boiling target retrieval buffer for 30 min.
Remove the slides from the target retrieval buffer and immediately wash in double-distilled water. Dehydrate in 100% ethanol for 5 min before air drying.
Once the slides have air-dried, apply hydrophobic barrier pen to encircle the tissue section on the slide. Be sure to allow the hydrophobic barrier to air dry completely.
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| ACD HybEZII Hybridization system (110V) with ACD EZ-Batch Slides system | ACD | 321710 | Hybridization oven |
| CAT Hematoxylin | Biocare medical | CATHE-GAL | colorstain |
| Clear-Mount | ELECTRON MICROSCOPY SCIENCES | 17985-15 | mounting reagent for red chromogen |
| Immpact DAB Peroxidase Kit | Vector | SK-4105 | Used to reveal HRP - DAB (Brown) to replace the DAB coming in the ACD kit |
| lithium carbonate | Fisher chemical | L119-500 | bluing solution |
| paraformaldehyde | ELECTRON MICROSCOPY SCIENCES | 15714-S | for tissue fixation (4%) |
| PBS | life technology | 14190-136 | |
| Permount Mounting Medium | ThermoFisher Scientific | SP15-100 | mounting regaent for brown chromogen |
| Prolong Gold | ThermoFisher Scientific | P36930 | mounting regaent for fluorescence |
| ribonucleases A | ThermoFisher Scientific | 12091039 | for RNAse treatment in DNAscope protocol |
| ribonucleases T1 | Roche | R1003 | for RNAse treatment in DNAscope protocol |
| RNAscope 2.5, 2-plex detection reagent | ACD | 322430 | Brown and red kit chromogen detection |
| RNAscope Target Retrieval Reagents | ACD | 322000 | retrieval buffer |
| SuperFrost Plus Glass Slides | ThermoFisher Scientific | 12-550-17 | |
| TBS | BOSTON BIOPRODUCTS | BM-301-4L | for washes |
| TSA Plus Fluorescence palette kit (Cy3, Cy5, TMR, Fluorescein) | Perkin elmer | NEL760001KT | HRP Fluorescence detection |
| Tween 20 | SIGMA | P1379-1L | for washes |
| XYLENE 20LT | ThermoFisher Scientific | AC422680200 |
4. Protease pretreatment
-
Place the dried slides on a locking slide rack, then prepare the protease pretreatment reagents (protease digestion solution, 2.5 µg/mL) by diluting with sterile, cold, PBS in a 1:5 ratio. Mix well.
NOTE: Three different protease reagents with different concentrations are provided in the commercially available kit. Protease III (standard), Protease IV (strong), and Protease Plus (mild). Empirically test the protease digestion time and dilution prior to implementation in a study because optimal conditions will vary based on tissue type, fixation, and thickness (see Discussion).
Dispense the diluted protease solution on slides to completely cover the tissue sections. Immediately incubate the slides for 20 min at 40 °C in an oven (prepared in step 1.4), ensuring that the slides are sealed in the humid hybridization tray. Do not let the tissue sections dry out for the remainder of the protocol.
Immediately rinse 3x by submerging the locking slide rack into a wash tray filled with double-distilled water.
Perform the endogenous peroxidase block by dropping the peroxidase solution onto each tissue section to completely cover it. Incubate the slides for 10 min at RT. Once done, rinse the sections 3x in double-distilled water.
5. Probe hybridization and signal amplification
NOTE: To prevent evaporation, make sure the humidity-controlled tray seals properly so that the tissues do not dry out during the incubation steps. Place the humidity chamber back in the oven during the washing step to ensure it remains at 40 °C.
-
Mix the C2 probe and C1 probe in a 1:50 ratio by pipetting 1 volume of C2 probe to 50 volumes of C1 probe into a tube as suggested by the manufacturer. Invert the tube several times. Prewarm the target probe mixture in 40 °C oven for ~10 min to dissolve any precipitation prior to use.
NOTE: The mixed target probes can be stored at 4 °C for up to 6 months.
Remove the slides from the water. Rinse, and tap or flick the slides to remove excess water from the tissue sections. Immediately dispense the probe on the slides, ensuring each tissue section is completely covered without any air bubbles. Incubate the probe mix in the humidity chamber overnight at 40 °C.
The next day, remove the slides from the oven and place them in the wash tray containing 0.5x wash buffer for 5 min at RT. Repeat the wash step one more time.
Remove the slides from the wash buffer. Rinse, and tap or flick the slides to remove excess wash buffer from the tissue sections.
Dispense commercially AMP 1 reagent ready-to-use (2 nmol/L) in hybridization buffer B (20% formamide, 5x SSC, 0.3% lithiumdodecyl sulfate, 10% dextran sulfate, blocking reagents) on the slides, ensuring complete coverage of tissue section without any air bubbles. Incubate for 30 min at 40 °C in the humidity chamber. Repeat steps 1.6.3–1.6.4, performing the wash for 2 min each.
Dispense commercially available AMP 2. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 15 min at 40 °C. Repeat steps 5.3–5.4, performing the wash for 2 min each.
Dispense commercially available AMP 3. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 30 min at 40 °C. Repeat steps 5.3–5.4, performing the wash for 2 min each.
Dispense commercially available AMP 4. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 15 min at 40 °C. Repeat steps 5.3–5.4, performing the wash for 2 min each.
Dispense commercially available AMP 5. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 30 min at 40 °C. Repeat step 5.3–5.4, performing the wash for 2 min each.
Dispense commercially available AMP 6. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 15 min at 40 °C. Repeat step 5.3–5.4, performing the wash for 2 min each.
Before detection, rinse the slides one time in 1x TBS-Tween 20 (0.05% v/v). Remove the slides from the wash buffer, rinse, and tap or flick the slides to remove excess wash buffer from the tissue sections. Immediately place in the wash tray filled with 1x TBS-Tween buffer.
6. Channel 1 (C1) target signal detection
NOTE: This is performed using the Red Alkaline Phosphatase and Fast Red Chromogen Amplification 6 from 2-plex detection kits (see Table of Materials) containing alkaline phosphatase labels, and chromogenic detection. It uses fast red as a substrate to generate a red signal.
-
Prepare fast red (FR) working solution using a 1:60 dilution of Fast RED-B to Fast RED-A. Mix well. To reduce precipitate and obtain a cleaner signal, filter the chromogen solution through a 0.45 µm MCE membrane using a syringe.
NOTE: Use the Fast RED-B solution within 5 min. Do not expose to direct sunlight or UV light.
Remove the slides from the TBS-Tween, rinse, and tap or flick the slides to remove excess buffer from the tissue sections.
Dispense mixed, filtered FR solution onto each tissue section, being sure that each section is completely covered. Incubate at RT for 6–8 min. Observe under microscope.
Rinse the slides in 0.5x wash buffer 2x. Remove the slides from the wash buffer, rinse, and tap or flick the slides to remove excess wash buffer from the tissue sections.
Dispense commercially available AMP 7. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 10 min at 40 °C. Repeat step 5.3–5.4, performing the wash for 2 min each.
Dispense commercially available AMP 8. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 15 min at 40 °C. Repeat step 5.3–5.4, performing the wash for 2 min each.
Dispense commercially available AMP 9. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 30 min at 40 °C. Repeat step 5.3–5.4, performing the wash for 2 min each.
Dispense commercially available AMP 10. Ensure that the tissue section is completely covered without any air bubbles. Incubate the slides in the humidity chamber for 30 min at 40 °C. Repeat step 5.3–5.4, performing the wash for 2 min each.
7. Channel 2 (C2) target signal detection
NOTE: This is performed using commercially available Brown HRP and DAB Chromogen Kits (see Table of Materials). The amplification 10 from the 2-plex detection contains horseradish peroxidase labels, and chromogenic detection is performed using DAB to generate a brown signal.
For optimal detection of the DAB signal, use commercially available kits and follow the manufacturer’s instructions (see Table of Materials). Observe under a microscope. CAUTION: DAB is toxic. Follow appropriate precautions and safety guidelines when handling and disposing of this chemical.
8. Counterstaining and mounting
-
Counterstain the slides with hematoxylin.
Counterstain the slides for 30 s with 50% fresh filtered hematoxylin while agitating the slides. Slides will appear purple. Immediately rinse in running water while agitating the slides up and down until the water is clear. The tissue sections will remain purple.
To obtain a better contrast, place counterstained slides in distilled water saturated with lithium carbonate for 1 min. Rinse in running water thoroughly at least 3x while agitating the slides. Use double-distilled water for the final rinse.
Because FR is sensitive to organic solvents, slides stained with FR will need to be covered with water-based mounting medium and dried overnight at RT.
-
Mount the slides.
Make sure the tissue sections covered with water-based mounting medium are dry.
Dip slides in xylene before cover slipping using mounting reagent. Be sure to prevent or remove any air bubbles between the coverslip and tissue section and let dry for 16 hours at RT.
9. Quantitative image analysis protocol for RNAscope using CellProfiler5
Briefly, ensure that the software will separate hematoxylin and FR stains into separate images. Identify and measure the objects of interest: nuclei, virions, positive cells, and aggregate FR positive staining. Store measurements in a CSV file and save the analyzed image.
Select the option to “Unmix colors”, which separates stains, splitting the original region of interest (ROI) into separate hematoxylin and FR images.
If hemosiderin, tattoo, or similar features interfere with the analysis, add a second “Unmix” step with hematoxylin, FR, and DAB. Use the second FR image to find the most intense FR pixels. This second image will be used as a mask for true FR staining.
Optionally, smooth the stained images before thresholding them. This is an empirical decision and not always needed. Threshold the individual stained images using “IdentifyPrimaryObjects” to select positive pixels.
-
Identify the three different types of objects (virion, virion aggregates, productive cell).
Ensure that the nuclei are 4–100 pixels (px) diameter objects stained with hematoxylin. Declump and fill holes after thresholding. “IntenseFastRed” objects are 4–100 px diameter and include virions, positive cells, and aggregate positive staining, such as seen on follicular dendritic cells (FDCs) in B cell follicles (BCF). This image is used to filter out false positives (e.g., hemosiderin).
Ensure that FR small positives are 2–12 px diameter objects. This measurement includes virions and vDNA+ cells. Discard any objects outside this range. Declump and fill holes after thresholding.
-
Ensure that FR large positives: 9–100 px diameter objects. This measurement includes vRNA+ positive cells and aggregate positive staining. Declump after thresholding. The size of small and large FR positive objects can overlap. They will be separated in a later step.
NOTE: The software (e.g., Cellprofiler) uses pixels for object sizes, and the ones used here are derived from slides scanned at 0.2510 µm/pixel (40x).
-
Define and extract results.
After any objects are identified, define and extract results for number of virions, productive infected cells, and nuclei.
Identify virions by masking FastRedSmallPositives with the IntenseFastRed object set to remove false positives (e.g., hemosiderin).
Next, identify the positive cells and aggregate FR positive staining. Remove false positives and virions from FastRedLargePositives by masking it with IntenseFastRed and with the virion object set.
Extract positive cells from the refined FastRedLargePositives by masking again with Nuclei. Split objects that are no longer touching, and filter the results by object area, removing small objects (≤6 px). This removes the specks created by masking the nuclei overlap. The result is the positive cells.
Finally, define aggregate FR positives. This step uses IdentifyTertiaryObjects, finding objects contained in a larger, parent object. In this case, the refined FastRedLargePositives object set is the parent and positive cells are subtracted.
Count the number of virions and positive cells.
Measure the aggregate positive area and convert it from pixels to mm2. Optionally, record the area occupied by virions and positive cells in mm2 if the analysis requires using a standard cell and virion sizes instead of direct counts.
-
Overlay the positive objects on the original image and save the result.
NOTE: The ISH data is reported by the number of virions per 106 nuclei (cells) and the number of productively infected vRNA+ cells per 106 nuclei (cells) for a better understanding and to facilitate comparison with qPCR data, but results could also be reported by area of tissue in mm2.
Representative Results
In a previous manuscript2, 6, 7, 8, 9, 10, 11, we reported that the next-generation ISH platforms detecting either vRNA or vDNA can be combined, using sense probes (vDNA) targeting the 5’ gag-pol portion of the SIV/HIV genome and antisense probes (vRNA) targeting genes in the 3’ half of the genome (vif, vpx, vpr, tat, env, and nef) as well as the TAR element in the 5’ genome (Table 1). This approach distinguishes transcriptionally active cells (vRNA +, vDNA+) from transcriptionally inactive (putatively latently) infected cells or cells harboring transcriptionally incompetent proviruses (vRNA-, vDNA+) in the same tissue section2 (Figure 1).
Figure 1: Viral RNA and vDNA detection in the same tissue section.
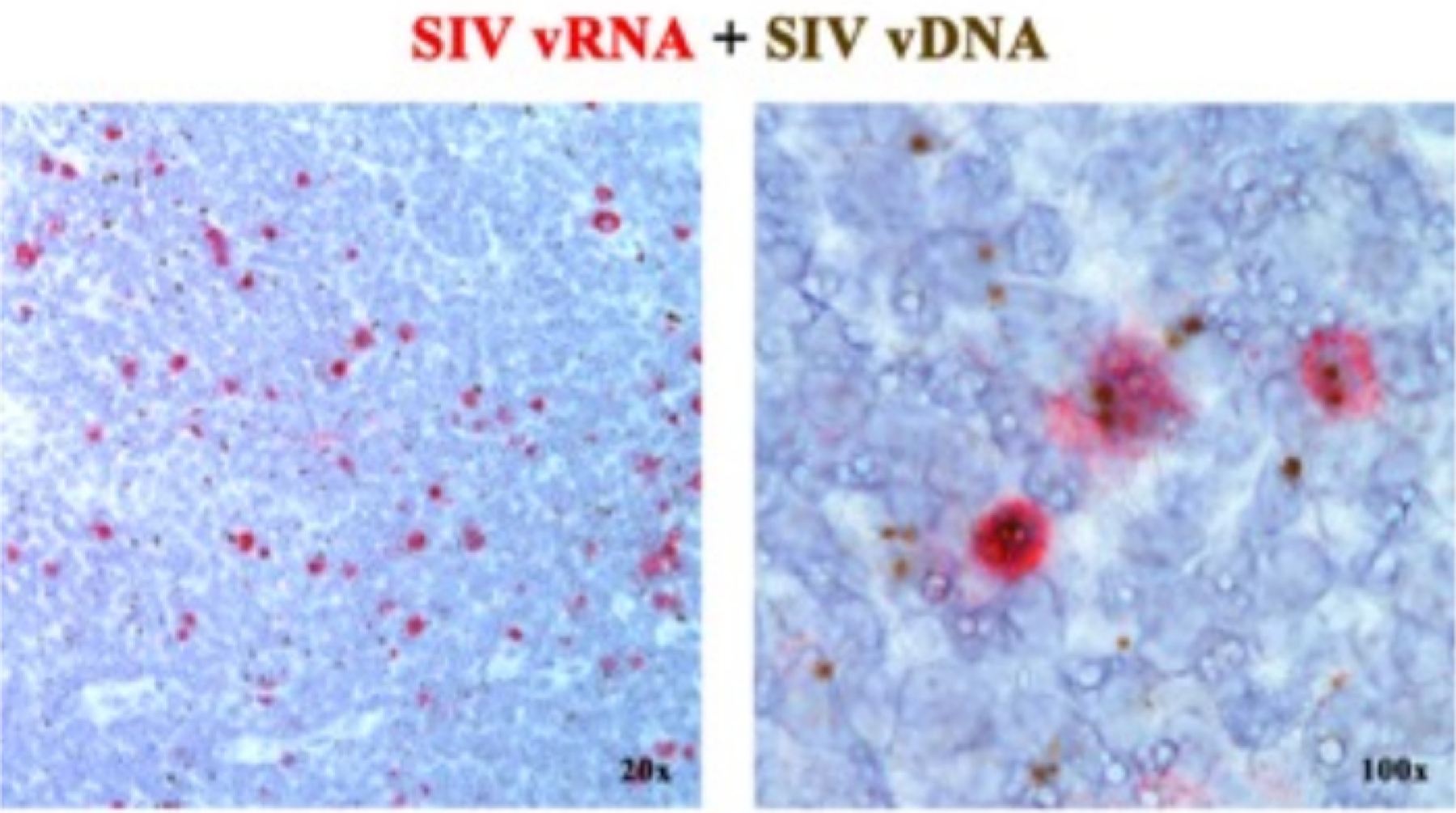
Combination of both RNA hybridization (red) and DNA hybridization (brown) assay in an acutely SIV-infected RM lymph node demonstrating the ability to detect vRNA and vDNA in the same tissue section and providing a powerful approach to identify transcriptionally silent vDNA+ vRNA-cells in situ. This figure has been modified from Deleage C. et al.2.
Discussion
In situ hybridization is a meticulous assay that requires rigor and basic knowledge of nucleic acid chemistry, cell biology, and histology to be able to adapt each critical step to localize a target in a well conserved environment. In this discussion we would like to highlight the critical steps where troubleshooting is crucial to obtain accurate and interpretable results.
The fixation and processing of the tissues are critical and should be addressed upfront to make sure the assay can yield the best results. Neutral buffered PFA (4% freshly prepared) fixative is optimal for the duplex assay. However, the assay can also be performed on frozen tissues (OCT) with the appropriate post cryosectioning fixation conditions.
Pretreatment of the tissue sections is a crucial step. There are two pretreatment steps in this assay: the first is a heat-induced epitope retrieval (HIER). This step is important for the reversal of methylene bridge cross-links and restoration of protein structures, which is needed in fixed tissues. The efficiency of this treatment depends on time, temperature, type of retrieval buffer, and pH. The second pretreatment is a protease-induced epitope retrieval (PIER). This step cleaves peptides, exposing the antigen or nucleotides, and uses enzymes including proteinase K, trypsin, and pepsin. This is an extremely sensitive step that could potentially damage both tissue morphology and the target of interest. The concentration of the enzyme, as well as the time and temperature of incubation are critical in this process. Overdigestion leads to poor nuclei demarcation and difficulty in quantification steps. It is critical to find a balance between optimal access to the RNA/DNA target and pretreatment conditions that do not damage the tissue or target of interest. Each tissue type has a different level of sensitivity to each of these pretreatments and each parameter (enzyme concentration, time, temperature) should be empirically tested.
The stringency of the wash buffer is based on three major parameters: temperature, concentration of salts and detergent, and time. The wash buffer is a saline sodium citrate buffer (SSC), and the salt concentration within the buffer controls the stringency during the wash steps. In their protocol, ACD advises to use the wash buffer at a final concentration of 0.1x SSC, 0.03% lithium dodecyl sulfate. While working on DNAscope and multiplex optimization, we determined that using the wash buffer at a final concentration of 0.05x SSC gave us better results to visualize the DNA signal and considerably helped reduce nonspecific off target hybridization resulting from the overnight incubation of the sense probe.
The choice of detection approach, chromogen (red or brown) versus fluorescence needs to be thought through based on tissue type and goal before starting the assay. The red chromogenic approach will give a nice contrast, because red is not naturally found in tissues. Brown chromogen will give similar results to red chromogen. However, it is important to keep in mind that some blood degradation products present in the tissue have similar color, and tattoo ink will be difficult to separate from the brown signal while quantifying. A fluorescence detection approach will allow a clear distinction of different cellular markers and the multiplexing will offer a perfect assay to phenotype the cells harboring vRNA and/or vDNA.
Multiple controls are necessary to ensure the specificity of the probes and the quality of the assay. Each newly designed probe must be tested on known positive and negative control tissues or cell pellets. We often generate plasmids containing our targeted sequence and perform transfection into cell lines to generate positive controls. For each run we add a known negative tissue (HIV or SIV negative), a no probe control containing only the probe diluent, and an RNase treated control to ensure the quality and specificity of the assay.
The quantification is an extremely important step and should be performed using the appropriate tools and algorithm based on the question asked. In this manuscript we presented an image analysis software (e.g., Cellprofiler), that we chose after evaluation of multiple options. We estimated that this software was the best software for our needs, but there are numerous image analysis software programs that could be used.
Acknowledgments
This project has been funded in whole with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and by the Oregon National Primate Research Center NIH grant award P51OD011092 (J.D.E). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The duplex was developed with the help of Advanced Cell Diagnostics.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/60318.
Disclosures
The authors have nothing to disclose.
References
- 1.Wang F et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnosis 14 (1), 22–29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deleage C et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathogens and Immunity 1 (1), 68–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sengupta S, Siliciano RF Targeting the Latent Reservoir for HIV-1. Immunity 48 (5), 872–895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R HIV reservoirs: what, where and how to target them. Nature Reviews Microbiology 14 (1), 55–60 (2016). [DOI] [PubMed] [Google Scholar]
- 5.McQuin C et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biology 16 (7), e2005970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deleage C, Chan CN, Busman-Sahay K, Estes JD Next-generation in situ hybridization approaches to define and quantify HIV and SIV reservoirs in tissue microenvironments. Retrovirology 15 (1), 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deleage C, Turkbey B, Estes JD Imaging lymphoid tissues in nonhuman primates to understand SIV pathogenesis and persistence. Current Opinion in Virology 19, 77–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Estes JD et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nature Medicine 23 (11), 1271–1276 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mavigner M et al. Simian Immunodeficiency Virus Persistence in Cellular and Anatomic Reservoirs in Antiretroviral Therapy-Suppressed Infant Rhesus Macaques. Journal of Virology 92 (18) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson CW et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathogen 14 (4), e1006956 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deleage C et al. Impact of early cART in the gut during acute HIV infection. Journal of Clinical Investigation Insight 1 (10), e87065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]