

Abstract
The present study investigated the effect of graphene oxide in cellulose acetate-based composite nanofibers on the transdermal delivery of naproxen. The composite nanofibers were successfully produced via the electrospinning process by directly mixing cellulose acetate, graphene oxide, and naproxen solution with varied compositions. The formation of the nanofibers was confirmed by electron microscopy and other characterization techniques to prove the existence of graphene oxide and naproxen itself. Surprisingly, graphene oxide encourages the production of nanofibers with smaller average diameter, higher conductivity, higher mechanical strength, and higher naproxen release from the cellulose acetate nanofibers. Once combined with naproxen, the composite nanofiber exhibited antibacterial activity with an inhibitory zone of 9.15 mm. The cytotoxicity evaluation also showed that the addition of naproxen increased the death of HeLa cells with a CC50 of up to 29.33 μg mL−1. The kinetic model of naproxen release follows the Korsmeyer–Peppas and Higuchi models with acceleration at neutral pH. These results are promising for further applications for wound healing purposes.
The present study investigated the effect of graphene oxide in cellulose acetate-based composite nanofibers on the transdermal delivery of naproxen.
1. Introduction
Skin is the largest human organ with a surface area of 1–2 m2.1,2 The skin has a vital function protecting human organs from external damage, including physical, biological, and environmental. Physical damage such as an injury can be a severe problem without proper treatment.3 Open injuries can be an opportunity for bacteria to enter4 and increase the risk of infections, such as tetanus, chronic wounds, bone infections,5 and even death. Wound dressing technology provide promising aspect on tissue treatment well-known as wound healing term.6 The fast development of multifunctional and ideal wound dressings has become an excellent solution for this issue. Ideal wound dressings have varied functions, such as moisturizing the wound area, air exchange, preventing wound infection, and proliferating fast cell healing.7 Nanofibers have attracted the attention of researchers around the world in recent years due to their numerous advantages that make them ideal candidates for wound dressing, such as small pore size and a very high surface-to-volume ratio.8
In the biomedical field, three major groups of nanofibers have been widely studied, namely tissue scaffolding, drug delivery systems, and modern wound dressings.9,10 The wound management method that uses the principle of moisture balance is known as modern wound dressing, which is stated to be more effective in wound healing technology.11 The characteristics, composition and formulation of the materials in wound dressings influence the effectiveness of wound healing.12 Wound management systems containing drugs can act as drug delivery systems and affect the healing process precisely. Development of transdermal delivery systems took a big step forward by applying nanofibers to control drug movement and systemic delivery. Several designs of drug-containing nanofibers have been reported, including the influence of nanofiber materials on drug action and release.8,13–15
Considering the development of eco-friendly nanofibers, some researchers have designed novel nanofiber from natural polymers, synthetic polymers, and hybrid blends.8 Cellulose acetate (CA) is a cellulose derivative, a semi-synthetic polymer produced by the catalyst-based reaction of cellulose with acetic anhydride. Up to now, CA-based nanofibers have received attention because their fabrication and disposal do not cause environmental damage.16 CA is also well-used in various applications because of its excellent mechanical properties and compatibility, such as in laminates, fiber adhesives, and films.8,12,14,17 CA nanofibers also have high potential for use in bone regeneration,18 gas sensors,19 supercapacitors,20 filtration,21 and drug delivery.22 The preparation of CA nanofibers using the coaxial electrospinning method has several advantages, such as the controlled and continuous release of drugs over a period and administration of multiple drugs.22
In recent years, many researchers have modified nanofiber membranes to enhance their effectivity and efficiency for some desired processes, such as increasing the drug delivery capacity by adding materials, biomolecules, drugs, nanoparticles, or natural-based material compounds. The addition of graphene to the polymer matrix has been reported to improve the mechanical properties of nanofibers, specifically the tensile strength and Young's modulus.23 As a carbon-based nanomaterial, graphene has good electrochemical stability, high surface area and high adsorption capacity, and can be easily modified for use in various applications, such as transistors, polymers, composites, energy storage, and hydrogen production. Moreover, graphene has been also developed for photocatalytic applications.24 In particular, graphene can be transformed to graphene oxide (GO) by modifying the graphene with oxygen-based functional groups, such as hydroxyl, carboxylic acids, and epoxides; typically, the oxygen to carbon ratio is about one to three.25 The addition of various functional groups on GO allows the formation of better bonds with other organic molecules.26 The characteristic high surface area of GO allows a high drug loading capacity.27 These properties make GO very attractive as a drug carrier. GO can be used for biomedical applications, improving the mechanical properties and stability of drugs in the polymer matrix.28 In this research, nanofibers will be made from CA as a semi-synthetic biopolymer. The nanofiber biopolymer will be combined with graphene and naproxen as an anti-inflammatory agent.
Naproxen (NA) is a nonsteroidal anti-inflammatory drug (NSAID) that is often used for the treatment of rheumatism, osteoarthritis, and joint pain disorders.17 NA also has analgesic and antipyretic properties;17 when encapsulated in nanofibers, it tends to be released through the mechanism of desorption and diffusion.14
Several studies have been carried out combining graphene with CA. Aboamera et al. succeeded in making ultrafine CA nanofibers combined with GO by electrospinning method, which improved the mechanical properties of the nanofibers with the tensile strength and Young's modulus of the CA–GO composites increasing (by up to 75%) compared to those of the pure CA nanofibers.23 Nevertheless, the potential improvements achieved by incorporating a commercial drug like NA with this CA-based nanofiber have not been intensively studied. Moreover, no studies on the kinetic aspects of drug release from CA–GO nanofibers have been reported yet. Thus, it is crucial to promote multifunctional CA-based nanofibers for treating various diseases. Based on the data above, it is known that the addition of GO can improve the mechanical properties and drug release profile of CA nanofibers. In this research, the effects of incorporating GO and naproxen into CA nanofibers were analyzed. Several characterization techniques were used to prove that GO and CA–GO nanofibers were obtained, along with analysis of drug release profiles in the form of naproxen release kinetics, which became an important aspect of the present study. It is expected that the addition of graphene and naproxen to CA nanofibers will improve the drug-carrying ability in drug delivery applications.
2. Experimental
2.1. Material and methods
2.1.1. Chemicals
Cellulose powder, sodium hydroxide (98.5%, NaOH), cellulose acetate (CA, Mw 30 000 g mol−1), acetone (99.5%), sodium naproxen (NA, 98%), buffer solutions (pH 4, 7 and 9), phosphate-buffered saline (PBS, 100%) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, 97%) were purchased from Sigma-Aldrich (USA). All chemicals were directly used without specific purification.
2.1.2. Methods
2.1.2.1. Synthesis of GO
GO nanosheets were prepared by pyrolyzing cellulose as the carbon source according to a previously reported protocol.29 In detail, about 1 gram of cellulose was placed in a small glass vial and heated at a rate of 30 °C min−1 to a temperature of 300 °C for 3 h under nitrogen flow. After cooling down, the black precipitate obtained was marked as GO and stored for further investigation.
2.1.2.2. Electrospinning process for preparing CA–GO nanofibers
Before the electrospinning process, the CA–GO polymer solution, to be used as the dope solution, was firstly prepared by dissolving CA onto acetone with vigorous stirring to obtain a homogeneous solution. Varying amounts of GO (0.1, 0.5, and 1 g) were dissolved in acetone and then added into the CA polymer solution followed by ultrasonic treatment for 1 h. The obtained mixture was then set as the dope solution for the electrospinning process. The flow rate of the CA–GO solution was firstly optimized by varying from 0.01, 0.05, 0.1, to 0.5 μL h−1 with a running time of 3 h. The needle tip and the target were set to 12 cm. Besides using the CA–GO solution to produce CA–GO nanofibers, CA–GO–NA, bare CA, and CA–NA nanofibers were obtained by preparing dope solutions using the above protocol, where NA was firstly dissolved in acetone with varied amounts of GO (10–800 μg mL−1) before mixed with the CA solution. The electrospinning process was performed by placing 10 mL of the dope solution in a syringe set with a syringe pump and a metallic needle (20 G). The electrospinning was equipped with an aluminium foil-covered cylinder collector and operated on a 23 kV power supply, as illustrated in Scheme 1a. All the electrospinning settings and preparation of the dope solutions followed previous reports.10,30 The composite nanofiber with 0.5 mg of GO was denoted as CA–GO0.5–NA.
Scheme 1. (a) Schematic illustration of the preparation of CA–GO–NA nanofibers. Naproxen was embedded into the nanofibers. (b) Mechanism for the synthesis of graphene oxide from cellulose.

2.1.2.3. Cytotoxicity analysis
The cytotoxicity of the produced nanofibers was evaluated through in vitro MTT assay upon HeLa cancer cells following a reported protocol.29–31 In the cell culturing step, HeLa cells were cultured on DMEM medium and placed in an incubator at a temperature of 37 °C with 5% CO2. Once the cells are harvested, nanofiber samples with a dimension of 0.5 × 0.5 cm2 are placed in the wells of 12-well plates with 200 μL of medium. The HeLa cell-containing medium was then added with a density of about 5.4 × 104 cells per mL and further incubated for 24 h. After removing the medium and washing with PBS, 300 μL of MTT solution was added to each well. The cells were incubated for 5 h to produce dark formazan in each well. This formazan was collected and re-dissolved in DMSO and the absorbance was measured at 560 nm. The intensity of the absorbance correlates with the number of living cells. By comparing with untreated HeLa cells (as a control), the viability percentage was determined, with a percentage of over 80% indicating non-toxic.
2.1.2.4. Antibacterial assay
In the bacterial culture, Gram-positive Staphylococcus aureus (S. aureus) was used as the pathogenic bacteria, where its strain was obtained from ATCC 29213, and all of the materials and reagents were sterilized at 120 °C for 20 min. About 10 mL of nutrient broth (NB) media was used for bacterial culture for 24 h on a shaker incubator at 37 °C and then 100 μL was re-suspended in 9.9 mL of NB media and incubated at 37 °C for 4 h. This sub-cultured bacterium was used in experiments with a bacterial inoculation concentration of 106 CFU mL−1. The antimicrobial assay was performed using the disc diffusion method and observing the diameter of the inhibition zone. Nanofibers with dimensions of 0.6 × 0.6 cm were used for this assay and placed on a Petri dish with agar media as the medium for bacterial growth. The bacterium was then inoculated in the Petri dishes and incubated for 48 hours before observation.
2.1.2.5. Characterization
The characterization of the obtained GO was carried out using an Xplora Raman spectrometer (Horiba, Japan) using a diode laser with a wavelength of 523 nm and 1200 lines per mm grating. The morphology and diameter of the nanofibers were investigated using an EVO MA-10 scanning electron microscope (SEM) (Zeiss, Germany). Detailed fiber diameters were determined from the SEM images after processing with Image J software. Functional group analyses of the nanofiber composites was done using an IRTracer-100 Fourier transform infrared (FTIR) spectrophotometer (Shimadzu, Japan) combined with an attenuated total reflection (ATR) unit in the range from 400 to 4000 cm−1. A Max-2BX X-ray diffractometer (XRD) (Rigaku D, Tokyo Japan) was used to determine the crystal structure of GO with a Cu Kα radiation source in the 2θ range of 5–65°. Thermal stability evaluation of the obtained nanofiber composite was achieved using thermal gravimetric analysis (TGA; TGA 4000, PerkinElmer, Massachusetts, United States) from 40 to 995 °C at a rate 10 °C min−1. The conductivity of the nanofibers was determined using a four-point probe (Tech RTS-9, China). The mechanical strength and properties of the obtained nanofibers were determined using a uniaxial tension prober (AUTOGRAPH AG-B, Shimadzu, Kyoto, Japan). The nanofiber specimens (n = 3) were adjusted to dimensions of 40 × 10 mm and stretched with a cell load of 50 N (20 mm initial length, speed of 10 mm min−1). The thickness of the nanofibers (n = 3) was assessed using digital micrometer SF Gold Electronics Co., Ltd China (with 0–12.7 mm range and 0.001 mm resolution).
2.1.2.6. Data analyses
Some of the research data, like mechanical tests, cytotoxicity tests, antibacterial tests, drug homogeneity data in nanofibers, cytotoxicity CC50, and drug release data, were analyzed statistically using Origin software (version 8.0724, OriginLab Inc., Northampton, MA, USA) and some NA release data were fitted on some models using Solver in Microsoft Excel 2019 (professional plus version). Some data were also obtained in triplicate, and means were compared using a paired-sample t-test.
3. Theory
3.1. Release kinetics of NA
The nanofibers' release kinetics were evaluated by inserting fiber samples with dimensions of 2 × 2 cm dimension on a dialysis membrane with a molecular weight cut-off of 40 kDa. This setup was then immersed in 50 mL of DI water and mixed with a magnetic stirrer at 200 rpm. 1 mL samples of DI water were taken from the outer side of the membrane at set time intervals and the total volume was kept at 50 mL by replacing with fresh DI water. The concentration of released NA was determined by converting NA's absorbance at 330.5 nm and referring it to standard solutions. The corrected concentration of NA was determined using the following equation:
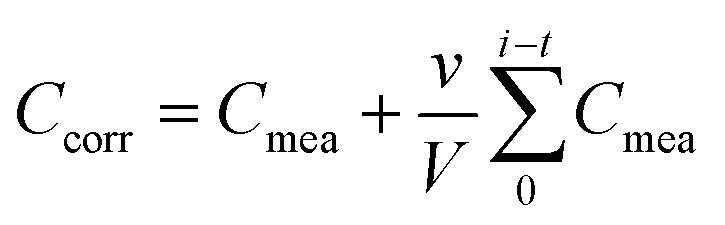 |
1 |
where Ccorr (ppm) is the corrected concentration at time t (s), Cmea is the measured concentration at time t (ppm), v is the volume of the DI water taken (mL), and V is the total volume of DI water (mL). Furthermore, the release mechanism was examined based on four kinetic models, namely the zero-order, first-order, Higuchi, and Korsmeyer–Peppas models, shown in eqn (2)–(5), respectively.
| Ft = K0t | 2 |
where Ft refers to the fraction of NA released at time t and K0 is the zero-order release constant (s−1).
| ln(1 − Ft) = −K1t | 3 |
where Ft refers to the fraction of NA released at time t and K1 is the first-order release constant (s−1).
| FH = K2t1/2 | 4 |
where FH refers to the fraction of NA released at time t and K2 is the Higuchi release constant (s−0.5).1
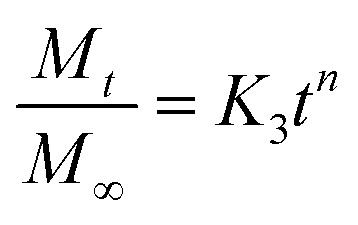 |
5 |
where M∞ and Mt refer to the concentration of naproxen released at time ∞ and t, respectively, n is the release exponent, and K3 is the Korsmeyer–Peppas release constant. The value of n refers to the Fickian diffusion model, where n ≤ 0.5 is non-Fickian, 0.5 < n < 1 is anomalous phenomena, and n ≥ 1 indicates lack of time dependence in the release kinetics; i.e., zero-order kinetics.
4. Result and discussion
4.1. Result
4.1.1. Synthesis and characterization of GO
GO nanomaterial was prepared by the pyrolysis method with cellulose as the raw material. This pyrolysis drives cellulose to dehydration at 100 °C, and hereafter, carbonization occurs at 240–340 °C. This carbonization will allow cellulose to re-arrange its structure, producing GO structure in some regions (as shown in Scheme 1b). The GO regions in this study carried a particular advantage in straightening physical interactions with the nanofibers and supporting the NA that is loaded on the nanofibers. Confirmation of the production of GO as well as its purity and crystallinity was obtained by XRD. The XRD pattern in Fig. 1a shows a noisy pattern indicating the low crystallinity of the sample. Moreover, the obtained GO was interrupted with the pattern for cellulose (JCPDS 50-2241), which indicates that the cellulose structure was still maintained. The obtained graphene-like structure in this study was confirmed by the existence of a particular signal matching with the pattern for C60 buckminsterfullerene (JCPDS 40-0558). The formation of GO was then strongly confirmed by Raman spectra (Fig. 1b). The peaks at 1379 cm−1 and 1588 cm−1, which refer to the D and G bands, respectively, reveal that GO was obtained from the synthesis process.32–35 When compared with Raman spectra of bare cellulose, it could be understood that the obtained GO was utterly different from cellulose by the disappearance of the peaks at 1095 cm−1 and around 456 cm−1, which are typical peaks for the C–O–C symmetric vibration and C–C–C ring deformation of cellulose, respectively.36
Fig. 1. (a) XRD pattern of cellulose-based GO compared with database patterns for carbon (JCPDS 44-0558) and cellulose (JCPDS 50-2241). (b) Raman spectra of the obtained GO (red line) and bare cellulose (green line). SEM images of (c) CA, (d) CA–GO, and (e) CA–GO–NA nanofibers.

4.2. Preparation and characterization of composite nanofibers
The nanofibers were prepared via the electrospinning method. Basically, the high voltage would create a strong electric field to pull the dope solution from the needle to the target during the spinning process (as shown in Scheme 1a). The formation of the Taylor cone indicates that the electrospinning process is running well. The fullness and stability of the Taylor cone affect the form and morphology of the produced nanofibers. Hence, we firstly improved the Taylor cone by optimizing the flow rate of the dope solution (CA–GO0.5–NA) in the electrospinning process by varying it from 0.01 to 0.05, 0.1, and 0.5 μL h−1. The photographs from the needle tip during the running process (Fig. S1, ESI†) show that the optimum Taylor cone was obtained with a flow rate of 0.05 μL h−1. The properties of the raw materials in the dope solution and the optimum flow rate contributed to the formation of the Taylor cone. At a low flow rate (0.01 μL h−1), the Taylor cone did not perform well and could not be tracked, while at the high flow rate (0.1 and 0.5 μL h−1), the Taylor cone exhibited tailing due to the flow rate of the dope solution being higher than the voltage attraction and evaporation rate of the solvent in the dope solution. Thus, 0.05 μL h−1 was set as the optimum flow rate to obtain the composite nanofibers.
For further characterization, SEM-EDX was carried out for morphological observation of the composite nanofiber (Fig. 1c–e). The SEM images show that composite fibers were successfully obtained from the electrospinning process. Moreover, it can be clearly seen in Fig. 1d and e that the addition of GO and NA to the dope solution resulted in smaller nanofiber diameter than those of the CA nanofibers. To prove the above, we used Image J software to modify the SEM figures and obtain the average diameter for each type of nanofiber (as shown in Table 1). We further improved SEM findings by the electrical conductivity approach. Interestingly, electrical conductivity of material on dope solution influences its speed to reach the target and diameter of obtained nanofiber. The electrical conductivity data, in Table 1, shows that the CA solution is approximately 0.8 μS cm−1 and this value further slightly increased after the addition of GO and NA to 1.0 and 1.5 μS cm−1, respectively. These data proved that the conductivity of the nanofibers influenced the nanofiber diameter. The improvement on these results has been made by accessing conductivity data upon the composite nanofibers with varied GO (as shown in Table S1, ESI†), where the value was increased by adding GO concentration.
Average diameter size and conductivity data.
| Nanofiber sample | Diametera (nm) | Conductivity (μS cm−1) |
|---|---|---|
| CA | 802 ± 25 | 0.8 |
| CA–GO0.5 | 652 ± 20 | 1.0 |
| CA–GO0.5–NA | 447 ± 15 | 1.5 |
Diameter data are presented as mean ± SD (n = 50).
Next, on SEM investigation, some additional data was found by exploring the EDX of the SEM images (Fig. S2, ESI†). The data inform qualitatively elements composed on the nanofibers (on area of the SEM image), which is confirming that carbon and oxygen elements are predominately contained on CA, and CA–GO nanofiber; whereas EDX data of CA–GO–NA performs sodium element from sodium naproxen. This firstly indicates that GO and NA were successfully incorporated in the CA nanofibers (forming CA–GO–NA). Moreover, these data can also reveal qualitatively for the addition of GO and NA onto CA nanofiber. It is well-known that CA, GO, and NA are composed mainly of carbon with oxygen as the second most abundant element. The addition of GO to CA would increase the carbon to oxygen ratio from 1.58 to 1.99. However, this role is not acceptable for the addition of NA due to the existence of sodium affecting the carbon to oxygen ratio (about 1.17). Furthermore, FTIR analysis was carried out next to investigate the existence of CA, GO, and NA (shown in Fig. 2). The FTIR spectrum for pure SA shows a particular band at 1760 cm−1, which indicates stretching of the C O group, together with a band at 902 cm−1 assigned to the acetyl C–O stretching. Next, the GO exhibits a unique vibration at 3608 cm−1 and a shoulder peak at 1624 cm−1 referring to O–H starching and C C aromatic functional groups, respectively, while the naproxen spectrum contains vibrations for O–H at 3145 cm−1; O–CH3 at 3003 cm−1; –CH3 at 2962 cm−1; and C O at 1726 cm−1 and 1681 cm−1 associated with the non-hydrogen stretching of –C O and the hydrogen stretching of –C O, respectively. Because most of the naproxen molecules are not involved in hydrogen bonding, the intensity of the vibration band at 1726 cm−1 is more dominant. For the combined CA–GO nanofiber, the FTIR spectrum shows the presence of a low C C vibration on 1500–1681 cm−1 coming from the GO. This band is a hexagonal carbon bond with an sp2 hybridized state as the basis of all the structures of graphene and GO. A C O double bond vibration was also observed for a carbonyl group. The other contribution of GO in the spectrum for the CA–GO nanofiber is an O–H vibration that changes from slightly narrow to broad form (marked as a on Fig. 2a). A narrow band O–H vibration in the spectrum for GO indicates delimitation of the hydrogen bonding effect on the hydroxyl group. In contrast, the broad band in the spectrum for CA–GO (marked with b on Fig. 2a) indicates that hydrogen bonding is occurring and is responsible for the CA–GO attraction. For the CA–GO–NA nanofiber, the FTIR spectral analysis becomes difficult due to the three components having similar organic functional groups overlapping each other. However, a crowded band from NA was observed at 900–600 cm−1 (marked with c and d on Fig. 2a) referring to C–H bending vibrations in its aromatic structure.
Fig. 2. (a) FTIR spectra and (b) TGA curves, as derivative weight%, of the composite nanofiber compared with the bare materials.

Furthermore, TGA analysis was carried out to obtain more accurate data proving the composition of the obtained nanofibers. The TGA data are shown in Fig. S3 (ESI†), where the main degradation stage occurs from 300 to 450 °C. To obtain detailed degradation stage data for each nanofiber, the TGA results are based on derivative weight (shown in Fig. 2b). In general, the CA nanofiber shows thermal degradation that is similar to that of pure CA. The CA–GO nanofiber composites show rapid decomposition above 400 °C, which indicates that the nanofibers have high thermal stability. The first decrease occurs at 104.95 °C for GO confirming water evaporation, and it also happens for all of graphene-containing nanofibers. The second signal for GO occurs at 469.81–570.47 °C due to decomposition of cellulose as its raw material. Based on these data, the GO shows good thermal conductivity, which can produce central degradation and decrease the thermal stability of the composite.37 Moreover, CA–GO–NA nanofibers showed slightly lower thermal stability than pure CA or CA–GO nanofibers due to the addition of NA. This data was supported by Karimi et al., who reported that naproxen began to degrade at around 150 °C and completely degraded at around 280 °C. Another report confirmed a decomposition temperature of 200 °C associated with the free destruction of the naproxen complex.13 Furthermore, in the present study, the NA decomposes in the range of 300–400 °C. The degradation stage at lower temperature for the CA–GO–NA nanofiber well-supported the FTIR data indicating an interaction of hydrogen bonds between cellulose acetate and its mixture.
Mechanical analyses were further addressed to the obtained composite nanofibers to prove their physical performance (Table 2). From these analyses, it was confirmed that the addition of GO enhances the strength of the composite nanofibers. In detail, increasing the GO content might increase the stress and decrease the fiber's strain ability, thus increasing the Young's modulus of the nanofibers.
Physical and mechanical properties of the prepared nanofibers.
| Sample name | Thicknessa (μm) | Stress (10−5 MPa) | Strain (%) | Young's modulus (10−7 MPa) |
|---|---|---|---|---|
| CA | 225 ± 23 | 2.46 ± 0.022 | 53.95 ± 0.52 | 4.56 |
| CA–GO0.1 | 245 ± 18 | 2.75 ± 0.015 | 40.22 ± 0.24 | 6.84 |
| CA–GO0.5 | 297 ± 22 | 2.77 ± 0.011 | 30.31 ± 0.84 | 9.14 |
| CA–GO1 | 347 ± 22 | 4.94 ± 0.027 | 19.05 ± 0.24 | 25.93 |
All data are presented as mean ± SD (n = 3).
4.3. Cytotoxicity assessment
The cytotoxicity evaluation was performed by placing a particular nanofiber shape on a cellular plate and adding HeLa-containing medium to assess the cell viability via MTT assay. HeLa cells were incubated with various nanofibers, including bare CA, CA–GO, CA–NA, and CA–GO–NA (Fig. 3a). The cytotoxicity results furnished that the viability of HeLa cells with CA and CA–GO nanofibers is above 80%, even with an incubation time of over 7 h. In contrast, the presence of NA on the CA–NA and CA–GO–NA nanofibers decreased the cell viability from the first hour.
Fig. 3. (a) Cell viability data of HeLa cell after adjusted times incubation with CA (grey); CA–GO0.5 (red); CA–NA (cyan); and CA–GO0.5–NA (magenta) nanofibers. (b) Cell viability data of HeLa cell after 5 h incubated with varied concentrations of NA on CA–NA (cyan) and CA–GO0.5–NA (magenta) nanofibers. All data are represented as means ± SD (n = 3).

Next, CA–NA and CA–GO0.5–NA composites with varying NA concentrations were used to evaluate the performance of the healing process (Fig. 3b). The data revealed that the toxicity of the NA-containing nanofibers was concentration-dependent, with increasing NA concentration resulting in lower cell viability. Compared with CA–NA, the presence of GO on CA–GO0.5–NA also increases the cell viability. For further toxicity exploration, using CA–NA and CA–GO0.5–NA nanofibers in varying concentrations, we investigated the cytotoxicity concentrations of the nanofibers to give 50% viability (CC50) (Fig. 4a and b), which is shown in the supporting data in Fig. 3b. The CC50 of CA–NA and CA–GO0.5–NA were 115.53 and 29.33 μg mL−1, respectively. This data revealed that the GO plays an important role in connecting the CA nanofibers with NA.
Fig. 4. HeLa cell viability plot after 5 h incubation with varying concentrations of NA on CA–NA (a) and (b) CA–GO–NA nanofibers (c). Inhibition diameter of S. aureus upon the nanofibers.

4.4. Antibacterial evaluation
The antibacterial activity of the CA, CA–GO0.5, CA–NA and CA–GO0.5–NA nanofibers was assess using a pathogenic bacterium, namely S. aureus, a Gram-positive bacterium. S. aureus was chosen for the present study due to its sensitivity to nonpolar material.38 Thus, it brings an advantage to the next potential application of nanofibers as a wound healing product for cancer treatment. The antibacterial activity was determined by measuring the inhibition area of each nanofiber after bacteria exposure (Fig. S4, ESI†) and is plotted in Fig. 4c.
4.5. In vitro NAP release and its kinetic evaluation
The dissolution tests were carried out next to ensure the facile mechanism of NA release from the nanofiber composites; the NA release pattern was further fitted with four kinetic models, namely zero-order, first-order, Higuchi, and Korsmeyer–Peppas models. From the data in Fig. 5a, it can be seen that the burst release of NA from the nanofiber occurs in the initial stage (first 40 min), followed by sustained release. Moreover, the figure also shows a comparison of the suggested kinetic model and its experimental results. It can be observed that the Higuchi model gives the best fit with experimental data (by having the closest R2 to 1 and the highest χ2 value) for the CA–NA nanofiber, whereas the addition of GO on CA–GO0.5–NA turned the kinetic release model close to the Korsmeyer–Peppas model. Those statements are supported by detailed data in Table 3, where the R2 and χ2 values match the Korsmeyer–Peppas model with Fickian diffusion (n < 0.5).
Fig. 5. (a) Dissolution rate of NA from CA–NA ( ), CA–GO0.1–NA (
), CA–GO0.1–NA ( ), CA–GO0.5–NA (
), CA–GO0.5–NA ( ) and CA–GO1–NA (
) and CA–GO1–NA ( ) nanofibers. Fitted curves following the kinetic models for each sample: zero order (green line); first order (blue line); Higuchi (brown line); and Korsmeyer–Peppas (grey line). (b) Dissolution rate of NA from CA–GO0.5–NA at pH 4 (
) nanofibers. Fitted curves following the kinetic models for each sample: zero order (green line); first order (blue line); Higuchi (brown line); and Korsmeyer–Peppas (grey line). (b) Dissolution rate of NA from CA–GO0.5–NA at pH 4 ( ), 7 (
), 7 ( ), and 9 (
), and 9 ( ). Fitted curves following kinetic models for each sample: zero order (green line); first order (blue line); Higuchi (brown line); and Korsmeyer–Peppas (grey line).
). Fitted curves following kinetic models for each sample: zero order (green line); first order (blue line); Higuchi (brown line); and Korsmeyer–Peppas (grey line).

Release kinetics of NA from the nanofibers with varying amounts of GO.
| Sample name | Kinetic model | |||
|---|---|---|---|---|
| Zero order | First order | Higuchi | Korsmeyer–Peppas | |
| CA–NA | k = 0.0000179 | k = 0.000018 | k = 0.041342 | k = 0.081857 |
| R 2 = 0.915354 | R 2 = 0.915538 | R 2 = 0.939039 | R 2 = 0.928745 | |
| χ 2 = 25.231426 | χ 2 = 25.061711 | χ 2 = 0.2017329 | n = 0.382855 | |
| χ 2 = 0.7491128 | ||||
| CA–GO0.1–NA | k = 0.106539 | k = 0.0000374 | k = 0.082659 | k = 0.106539 |
| R 2 = 0.834155 | R 2 = 0.834155 | R 2 = 0.848590 | R 2 = 0.920029 | |
| χ 2 = 110.55573 | χ 2 = 109.18690 | χ 2 = 4.5618367 | n = 0.456744 | |
| χ 2 = 3.2978708 | ||||
| CA–GO0.5–NA | k = 0.000087 | k = 0.000089 | k = 0.191593 | k = 0.088019 |
| R 2 = 0.940850 | R 2 = 0.944407 | R 2 = 0.981685 | R 2 = 0.986237 | |
| χ 2 = 7.6669838 | χ 2 = 7.2589723 | χ 2 = 1.2965448 | n = 0.227448 | |
| χ 2 = 0.4603844 | ||||
| CA–GO1–NA | k = 0.000047 | k = 0.000048 | k = 0.112240 | k = 0.309269 |
| R 2 = 0.880740 | R 2 = 0.883455 | R 2 = 0.964627 | R 2 = 0.974845 | |
| χ 2 = 178.67849 | χ 2 = 175.42864 | χ 2 = 4.4288032 | n = 0.325378 | |
| χ 2 = 0.3676479 | ||||
Lastly, the focus of the study was further arrayed on investigating the effect of the pH medium on releasing NA. As shown in Fig. 5b and Table 4, the nanofibers release the NA at all pH values and follow the Korsmeyer–Peppas kinetic model with Fickian diffusion (n < 0.5) at pH 4 7. In contrast, the nanofibers also exhibited non-Fickian diffusion (n > 0.5) at pH 9. In particular the release rate (variation in dissolution percentage with time) at pH 7 is higher than those at pH 4 and 9.
Kinetic release of NA from nanofibers with varied pH.
| Sample name | Kinetic model | |||
|---|---|---|---|---|
| Zero order | First order | Higuchi | Korsmeyer–Peppas | |
| CA–GO0.5–NA, pH 4 | k = 0.000022 | k = 0.000022 | k = 0.060864 | k = 0.088019 |
| R 2 = 0.485708 | R 2 = 0.487025 | R 2 = 0.639793 | R 2 = 0.745309 | |
| χ 2 = 590.47390 | χ 2 = 584.97213 | χ 2 = 22.953652 | n = 0.084771 | |
| χ 2 = 0.1495669 | ||||
| CA–GO0.5–NA, pH 7 | k = 0.000087 | k = 0.000089 | k = 0.191593 | k = 0.6404710 |
| R 2 = 0.940850 | R 2 = 0.944407 | R 2 = 0.981685 | R 2 = 0.986237 | |
| χ 2 = 7.6669838 | χ 2 = 7.2589723 | χ 2 = 1.2965448 | n = 0.227448 | |
| χ 2 = 0.4603844 | ||||
| CA–GO0.5–NA, pH 9 | k = 0.000065 | k = 0.000067 | k = 0.147093 | k = 0.108501 |
| R 2 = 0.921403 | R 2 = 0.924577 | R 2 = 0.992016 | R 2 = 0.992433 | |
| χ 2 = 13.205178 | χ 2 = 12.758705 | χ 2 = 0.4538900 | n = 0.551476 | |
| χ 2 = 0.3057432 | ||||
5. Discussion
In the present study, we designed CA-based nanofibers and modified them by adding GO and NA to enhance the potential for application in NA delivery. The use of supporting CA was considered to improve the mechanism of releasing NA. However, before any application, it is imperative to investigate the character of the nanofiber composites and their composition. XRD first proved the success of the synthesis process in obtaining GO. The XRD pattern of GO showed that it was composed of cellulose (JCPDS 50-2241) as the raw material of GO and a carbon-based fullerene material (JCPDS 40-0558). The fullerene-like structure was attributed to cellulose self-conjugating during the pyrolysis process (as shown on Scheme 1b), which results from reactions with the functional group on the cellulose chain, instead of an inter-conjugating reaction. This finding was supported by a previous report claiming the formation of GO from the fullerene XRD database, indicating the possibility of GO formation in the present study.39 Raman data supported the findings from the XRD data by showing both a G band and a D band that ensure GO formation. The G band signal is produced from symmetrical in-plane vibration of sp2 ordered carbons atoms, while the edging vibration of sp3-ordered carbon atoms is responsible for the D band signal, and is significantly different from the cellulose pattern. The present study showed that the higher G band peak drives the ID/IG ratio lower to about 0.85, indicating that the graphene-like structure was predominant in the obtained GO. Formation of GO with a low ID/IG ratio is standard and has been reported in previous studies.40–42 The composite nanofiber in the present study was also prepared through a high voltage electrospinning process. The formation of a stable Taylor cone, which is suggested for producing good nanofibers, was controlled by the applied voltage and flow rate. Moreover, adding GO and NA to the CA nanofiber affected the nanofiber properties. Table 1 shows that the diameters of the nanofibers composed of CA, GO, and NA are lower than those of the bare CA nanofibers. On this condition, we assume that GO and NA's electrostatic charge has a positive influence by accelerating the pulling process of the dope solution during the electrospinning process, leading to a smaller fiber diameter. Both GO and NA have negative electrostatic charges, and CA–GO–NA was predicted to have the highest negative charge; this reason was improved by Table S1 (ESI†). This also explains why CA–GO–NA has a relatively low nanofiber diameter.43–46 Further investigation to support the above statement was carried out with conductivity analysis of each nanofiber, where the higher conductivity drives the electrospinning process to obtain smaller nanofibers. We confirmed that GO and NA had been incorporated into the CA nanofiber by EDX, FTIR, and TGA data, where each analysis confirmed the successful union of GO and NA with the CA nanofibers. In particular, using the FTIR and TGA data, it could be proved that both GO and NA are incorporated into the nanofiber via physical interaction. This interaction is favorable for a drug delivery system due to minimizing the energy of the nanofibers for the release of NA and encouraging responsive nanofibers in biological conditions.15 Investigation of the mechanical properties of the composite nanofiber become an important part, not only for confirming its value for bioapplications, but also for confirming the role of the GO in the nanofibers. Table 2 confirms that GO promotes the composite nanofiber to higher stress, thus, resulting in a high Young's modulus. Even though GO is incorporated into the composite through physical interaction, it is very practical for strengthening the nanofibers and is not easily released from the fibers.
The promise for application in the clinical field made cytotoxic evaluation an important aspect that must be observed for the nanofibers. Data in Fig. 3a and b prove that both CA and GO are non-toxic and also reveal that the nanofibers have good biocompatibility. At the same time, the existence of NA drives the nanofibers to exhibit toxicity. Fig. 3a shows that the addition of 25 μg mL−1 NA to the nanofiber decreased the HeLa viability in their first 7 h of incubation (below 40%). Next, we tried to deeply evaluate the toxicity of NA with and without GO via MTT assay and found that GO promotes the toxic effect of the nanofiber, even if the concentration of NA was raised to 800 μg mL−1. This statement was proved by the determination of the CC50, where the CC50 of CA–NA was higher than that of CA–GO0.5–NA. This finding also shows that GO acts on binding NA with the CA nanofiber and influences its release from the nanofiber. Further assessment of the antibacterial activity of the obtained nanofibers proved that the addition of NA increased the antibacterial activity against S. aureus. As a drug-carrying nanofiber, the release of NA and its mechanism were crucial parts to investigate. Therefore, we fitted the NA release with some kinetic models. Starting with the GO effect, the NA release follows the Korsmeyer–Peppas model closely; the R2 value is the highest with the lowest Chi-square (χ2) for this model (Table 3). The Korsmeyer–Peppas model in the present study indicates that the NA release is influenced by the polymeric system of the CA nanofibers, in which NA needs more energy to overcome the polymeric obstacle during the release process.47 Moreover, compared with the fibres without GO or a low GO concentration, the high-GO nanofiber (CA–GO1–NA) releases the NA relatively faster. Furthermore, the n value for all Korsmeyer–Peppas models for all of the nanofibers indicates Fickian diffusion (n < 0.5) and refers to the release of NA owing to a concentration effect. In detail, without GO, the NA release from CA–NA tends to follow the Higuchi model (based on the R2 and χ2 values; Table 3), in which removing GO would minimize the polymeric effect, component distribution and relaxational component of the fibers.48 This data also indicates that the addition of GO drives the release of the NA to follow the Korsmeyer–Peppas model. The release of NA at different pHs also follows the Korsmeyer–Peppas model with Fickian diffusion (Fig. 5b and Table 4).The release rate of NA on neutral pH also dominates compared it with acid or base conditions. We assumed that this data was caused by the electrostatic effect presented by both NA and the CA–GO nanofiber. CA and GO give a stronger attraction that prohibits the diffusion and release of NA in acid or basic conditions. Therefore, neutral conditions became favorable for facile release. These data inform us that the addition of GO to the nanofiber composite tends to change the release behavior from Higuchi to the Korsmeyer–Peppas model. The well-fitted data with Korsmeyer–Peppas model indicates that the release of NA was influenced by CA as the polymeric material and associated GO. The release performance of the CA–NA nanofiber fits with the Higuchi model, indicating that the system follows a standard drug release model. However, the existence of GO made the release of NA more complicated due to the additional interaction between the components of the nanofiber.
6. Conclusions
The composite nanofibers were successfully synthesized via the electrospinning of a dope solution containing a mixture of CA, GO, and NA. Starting with the good characterization of the nanofiber and its component, other observations like mechanical strength, cytotoxicity, and antibacterial analyses support each other to prove the good potency of the obtained nanofiber to be applied in bio-applications, as well as the non-toxic feature of the nanofiber in the absence of NA. GO acted as a reliable component to enhance the mechanical strength of the fiber, drives the production of a lower diameter of the fiber and keeps the NA well present on the nanofiber. A kinetic study of the CA nanofiber proved that the release of NA in the presence of GO follows the Korsmeyer–Peppas model, while it matches the Higuchi kinetic model for the nanofiber without GO. The GO on the CA–GO–NA nanofiber promotes the facile release of NA under neutral pH conditions. The present study can be a good reference for further investigating controlled administration of naproxen using emerging innovative systemic delivery systems.
Author contributions
Wulan Purnamasari: investigation. Titah Aldila Budiastanti: writing-original draft. Aminatun Aminatun: data curation. Ulfa Rahmah: project administration. Sri Sumarsih: investigation. Jia-Yaw Chang: conceptualization. Mochamad Zakki Fahmi: conceptualization, writing – review and editing, supervision.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors thank Universitas Airlangga and Southeast Asia Taiwan University (SATU) Joint Research Scheme for financial support [grant number: 1312/UN3.15/PT/2021]. Part of this research also supported by the LPDP program Republic of Indonesia.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09293f
Notes and references
- Kolarsick P. A. Kolarsick M. A. Goodwin C. Anatomy and physiology of the skin. Journal of the Dermatology Nurses' Association. 2011;3:203–213. doi: 10.1097/JDN.0b013e3182274a98. [DOI] [Google Scholar]
- Rezvani Ghomi E. Khalili S. Nouri Khorasani S. Esmaeely Neisiany R. Ramakrishna S. Wound dressings: current advances and future directions. J. Appl. Polym. Sci. 2019;136:47738. doi: 10.1002/app.47738. [DOI] [Google Scholar]
- Singer A. J. Clark R. A. Cutaneous wound healing. N. Engl. J. Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Martin C. Low W. L. Amin M. C. I. M. Radecka I. Raj P. Kenward K. Current trends in the development of wound dressings, biomaterials and devices. Pharm. Pat. Anal. 2013;2:341–359. doi: 10.4155/ppa.13.18. [DOI] [PubMed] [Google Scholar]
- Serra R. Grande R. Butrico L. Rossi A. Settimio U. F. Caroleo B. Amato B. Gallelli L. de Franciscis S. Chronic wound infections: the role of Pseudomonas aeruginosa and Staphylococcus aureus. Expert Rev. Anti-Infect. Ther. 2015;13:605–613. doi: 10.1586/14787210.2015.1023291. [DOI] [PubMed] [Google Scholar]
- Hart A., Wound management, in Operational and Medical Management of Explosive and Blast Incidents, Springer, 2020, pp. 597–609 [Google Scholar]
- Jeckson T. A. Neo Y. P. Sisinthy S. P. Gorain B. Delivery of therapeutics from layer-by-layer electrospun nanofiber matrix for wound healing: an update. J. Pharm. Sci. 2021;110:635–653. doi: 10.1016/j.xphs.2020.10.003. [DOI] [PubMed] [Google Scholar]
- Hu X. Liu S. Zhou G. Huang Y. Xie Z. Jing X. Electrospinning of polymeric nanofibers for drug delivery applications. J. Controlled Release. 2014;185:12–21. doi: 10.1016/j.jconrel.2014.04.018. [DOI] [PubMed] [Google Scholar]
- Rošic R. Kocbek P. Pelipenko J. Kristl J. Baumgartner S. Nanofibers and their biomedical use. Acta Pharm. 2013;63:295–304. doi: 10.2478/acph-2013-0024. [DOI] [PubMed] [Google Scholar]
- Dzikri M. Armedya T. Khairunisa S. Sakti S. Raharjo Y. Purnamasari W. Nasronudin N. Fahmi M. Design of cellulose acetate-collagen nanofiber and its in vitro assessment as wound dressing candidate. Digest Journal of Nanomaterials and Biostructures. 2019;14:203–212. [Google Scholar]
- Vachhrajani V. and Khakhkhar P., Moisturising Dressings, in Science of Wound Healing and Dressing Materials, Springer, 2020, pp. 69–71 [Google Scholar]
- Armedya T. P. Dzikri M. F. Sakti S. C. W. Abdulloh A. Raharjo Y. Wafiroh S. Fahmi M. Z. Kinetical release study of copper ferrite nanoparticle incorporated on PCL/collagen nanofiber for naproxen delivery. Bionanoscience. 2019;9(2):274–284. doi: 10.1007/s12668-019-00618-y. [DOI] [Google Scholar]
- Karimi S. Ghaee A. Barzin J. Preparation and characterization of a piezoelectric poly(vinylidene fluoride)/nanohydroxyapatite scaffold capable of naproxen delivery. Eur. Polym. J. 2019;112:442–451. doi: 10.1016/j.eurpolymj.2019.01.027. [DOI] [Google Scholar]
- Wang Y. Qiao W. Wang B. Zhang Y. Shao P. Yin T. Electrospun composite nanofibers containing nanoparticles for the programmable release of dual drugs. Polym. J. 2011;43:478–483. doi: 10.1038/pj.2011.11. [DOI] [Google Scholar]
- Ma C. Shi Y. Pena D. A. Peng L. Yu G. Thermally responsive hydrogel blends: a general drug carrier model for controlled drug release. Angew. Chem. 2015;127:7484–7488. doi: 10.1002/ange.201501705. [DOI] [PubMed] [Google Scholar]
- Konwarh R. Karak N. Misra M. Electrospun cellulose acetate nanofibers: the present status and gamut of biotechnological applications. Biotechnol. Adv. 2013;31:421–437. doi: 10.1016/j.biotechadv.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Akduman C. Özgüney I. Kumbasar E. P. A. Preparation and characterization of naproxen-loaded electrospun thermoplastic polyurethane nanofibers as a drug delivery system. Mater. Sci. Eng., C. 2016;64:383–390. doi: 10.1016/j.msec.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K. Prabakaran M. Ke M. Gang X. Chung I. M. Um I. C. Gopiraman M. Kim I. S. Highly dispersed nanoscale hydroxyapatite on cellulose nanofibers for bone regeneration. Mater. Lett. 2016;168:56–61. doi: 10.1016/j.matlet.2016.01.010. [DOI] [Google Scholar]
- Petropoulou A. Kralj S. Karagiorgis X. Savva I. Loizides E. Panagi M. Krasia-Christoforou T. Riziotis C. Multifunctional Gas and pH Fluorescent Sensors Based on Cellulose Acetate Electrospun Fibers Decorated with Rhodamine B-Functionalised Core-Shell Ferrous Nanoparticles. Sci. Rep. 2020;10:1–14. doi: 10.1038/s41598-019-56847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Y. Yang Y. Hu Z. Guo B. Wang X. Yang X. Zhang Q. Wu H. High-performance symmetric supercapacitors based on carbon nanosheets framework with graphene hydrogel architecture derived from cellulose acetate. J. Power Sources. 2017;337:45–53. doi: 10.1016/j.jpowsour.2016.10.112. [DOI] [Google Scholar]
- Kaiser A. Stark W. J. Grass R. N. Rapid production of a porous cellulose acetate membrane for water filtration using readily available chemicals. J. Chem. Educ. 2017;94:483–487. doi: 10.1021/acs.jchemed.6b00776. [DOI] [Google Scholar]
- Khoshnevisan K. Maleki H. Samadian H. Shahsavari S. Sarrafzadeh M. H. Larijani B. Dorkoosh F. A. Haghpanah V. Khorramizadeh M. R. Cellulose acetate electrospun nanofibers for drug delivery systems: applications and recent advances. Carbohydr. Polym. 2018;198:131–141. doi: 10.1016/j.carbpol.2018.06.072. [DOI] [PubMed] [Google Scholar]
- Aboamera N. M. Mohamed A. Salama A. Osman T. Khattab A. An effective removal of organic dyes using surface functionalized cellulose acetate/graphene oxide composite nanofibers. Cellulose. 2018;25:4155–4166. doi: 10.1007/s10570-018-1870-8. [DOI] [Google Scholar]
- Pang J. Mendes R. G. Bachmatiuk A. Zhao L. Ta H. Q. Gemming T. Liu H. Liu Z. Rummeli M. H. Applications of 2D MXenes in energy conversion and storage systems. Chem. Soc. Rev. 2019;48:72–133. doi: 10.1039/C8CS00324F. [DOI] [PubMed] [Google Scholar]
- Compton O. C. Nguyen S. T. Graphene oxide, highly reduced graphene oxide, and graphene: versatile building blocks for carbon-based materials. Small. 2010;6:711–723. doi: 10.1002/smll.200901934. [DOI] [PubMed] [Google Scholar]
- Pan S. Aksay I. A. Factors controlling the size of graphene oxide sheets produced via the graphite oxide route. ACS Nano. 2011;5:4073–4083. doi: 10.1021/nn200666r. [DOI] [PubMed] [Google Scholar]
- Mahmoud A. E. D. Stolle A. Stelter M. Sustainable synthesis of high-surface-area graphite oxide via dry ball milling. ACS Sustainable Chem. Eng. 2018;6:6358–6369. doi: 10.1021/acssuschemeng.8b00147. [DOI] [Google Scholar]
- Rasoulzadeh M. Namazi H. Carboxymethyl cellulose/graphene oxide bio-nanocomposite hydrogel beads as anticancer drug carrier agent. Carbohydr. Polym. 2017;168:320–326. doi: 10.1016/j.carbpol.2017.03.014. [DOI] [PubMed] [Google Scholar]
- Fahmi M. Wathoniyyah M. Khasanah M. Rahardjo Y. Wafiroh S. Incorporation of graphene oxide in polyethersulfone mixed matrix membranes to enhance hemodialysis membrane performance. RSC Adv. 2018;8:931–937. doi: 10.1039/C7RA11247E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahmi M. Z. Prasetya R. A. Dzikri M. F. Sakti S. C. W. Yuliarto B. MnFe2O4 nanoparticles/cellulose acetate composite nanofiber for controllable release of naproxen. Mater. Chem. Phys. 2020:123055. doi: 10.1016/j.matchemphys.2020.123055. [DOI] [Google Scholar]
- Fahmi M. Z. Chang J. Y. A facile strategy to enable nanoparticles for simultaneous phase transfer, folate receptor targeting, and cisplatin delivery. RSC Adv. 2014;4(100):56713–56721. doi: 10.1039/C4RA11582A. [DOI] [Google Scholar]
- Lin D. Tseng C.-Y. Lim Q. F. Tan M. J. Kong K. V. A rapid and highly sensitive strain-effect graphene-based bio-sensor for the detection of stroke and cancer bio-markers. J. Mater. Chem. B. 2018;6:2536–2540. doi: 10.1039/C8TB00404H. [DOI] [PubMed] [Google Scholar]
- Zinin P. Velikovskii D. Y. Sharma S. Misra A. Filonenko V. Anokhin A. Kutuza I. Bhat S. Riedel R. Near-Ir Raman Spectroscopy of Graphitic B–C–N Materials. Glass Ceram. 2020:1–7. [Google Scholar]
- Gui Y. Sun H. Yan H. Wang H. Zhang Y. Song X. M. Jia R. Effects of substrates on Raman spectroscopy in chemical vapor deposition grown graphene transferred with poly(methyl methacrylate) Solid State Commun. 2017;264:31–34. doi: 10.1016/j.ssc.2017.04.010. [DOI] [Google Scholar]
- Wibrianto A. Putri D. F. Sakti S. C. W. Lee H. V. Fahmi M. Z. Naproxen release aspect from boron-doped carbon nanodots as a bifunctional agent in cancer therapy. RSC Adv. 2021;11(59):37375–37382. doi: 10.1039/D1RA06148H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenzel K. Fischer S. NIR FT Raman spectroscopy–a rapid analytical tool for detecting the transformation of cellulose polymorphs. Cellulose. 2001;8:49–57. doi: 10.1023/A:1016616920539. [DOI] [Google Scholar]
- Chen L. Xu C. Liu J. Fang X. Zhang Z. Optical absorption property and photo-thermal conversion performance of graphene oxide/water nanofluids with excellent dispersion stability. Sol. Energy. 2017;148:17–24. doi: 10.1016/j.solener.2017.03.073. [DOI] [Google Scholar]
- Jonquieres R. Bierne H. Fiedler F. Gounon P. Cossart P. Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: a novel mechanism of protein association at the surface of Gram-positive bacteria. Mol. Microbiol. 1999;34:902–914. doi: 10.1046/j.1365-2958.1999.01652.x. [DOI] [PubMed] [Google Scholar]
- Imran H. Manikandan P. N. Dharuman V. Graphene oxide supported liposomes for efficient label free electrochemical DNA biosensing. Sens. Actuators, B. 2018;260:841–851. doi: 10.1016/j.snb.2018.01.103. [DOI] [Google Scholar]
- Jeong S. Y. Kim S. H. Han J. T. Jeong H. J. Yang S. Lee G.-W. High-performance transparent conductive films using rheologically derived reduced graphene oxide. ACS Nano. 2011;5:870–878. doi: 10.1021/nn102017f. [DOI] [PubMed] [Google Scholar]
- Ma J. Liu R. Wang X. Liu Q. Chen Y. Valle R. P. Zuo Y. Y. Xia T. Liu S. Crucial role of lateral size for graphene oxide in activating macrophages and stimulating pro-inflammatory responses in cells and animals. ACS Nano. 2015;9:10498–10515. doi: 10.1021/acsnano.5b04751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minitha C. R. and Rajendrakumar R. T., Synthesis and characterization of reduced graphene oxide, in Advanced Materials Research, Trans Tech Publ, 2013, pp. 56–60 [Google Scholar]
- Ren W. Fang Y. Wang E. A binary functional substrate for enrichment and ultrasensitive SERS spectroscopic detection of folic acid using graphene oxide/Ag nanoparticle hybrids. ACS Nano. 2011;5:6425–6433. doi: 10.1021/nn201606r. [DOI] [PubMed] [Google Scholar]
- Madadrang C. J. Kim H. Y. Gao G. Wang N. Zhu J. Feng H. Gorring M. Kasner M. L. Hou S. Adsorption behavior of EDTA-graphene oxide for Pb(II) removal. ACS Appl. Mater. Interfaces. 2012;4:1186–1193. doi: 10.1021/am201645g. [DOI] [PubMed] [Google Scholar]
- Bešter-Rogač M. Nonsteroidal Anti-Inflammatory Drugs Ion Mobility: A Conductometric Study of Salicylate, Naproxen, Diclofenac and Ibuprofen Dilute Aqueous Solutions. Acta Chim. Slov. 2009;56:70–77. [Google Scholar]
- Moghimipour E. Salimi A. Eftekhari S. Design and characterization of microemulsion systems for naproxen. Adv. Pharm. Bull. 2013;3:63. doi: 10.5681/apb.2013.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer R. W. Lustig S. R. Peppas N. A. Solute and penetrant diffusion in swellable polymers. I. Mathematical modeling. J. Polym. Sci., Part B: Polym. Phys. 1986;24:395–408. doi: 10.1002/polb.1986.090240214. [DOI] [Google Scholar]
- Siepmann J. Peppas N. A. Higuchi equation: derivation, applications, use and misuse. Int. J. Pharm. 2011;418:6–12. doi: 10.1016/j.ijpharm.2011.03.051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.