

Abstract
Recently, we identified 1,189 CpG sites whose DNA methylation level in blood associated with Crohn’s disease. Here, we examined associations between DNA methylation and genetic variants to identify methylation quantitative trait loci across disease states in (1) 402 blood samples from 164 newly diagnosed pediatric Crohn’s disease patients taken at 2 time points (diagnosis and follow-up), and 74 non-inflammatory bowel disease controls, (2) 780 blood samples from a non-Crohn’s disease adult population, and (3) 40 ileal biopsies (17 Crohn’s disease cases and 23 non-inflammatory bowel disease controls) from group (1). Genome-wide DNAm profiling and genotyping were performed using the Illumina MethylationEPIC and Illumina Multi-Ethnic arrays. SNP-CpG associations were identified via linear models adjusted for age, sex, disease status, disease subtype, estimated cell proportions, and genotype-based principal components. In total, we observed 535,448 SNP-CpG associations between 287,881 SNPs and 12,843 CpG sites (P < 8.21 × 10−14). Associations were highly consistent across different ages, races, disease states, and tissue types, suggesting that the majority of these methylation quantitative trait loci participate in common gene regulation. However, genes near CpGs associated with inflammatory bowel disease SNPs were enriched for 18 KEGG pathways relevant to inflammatory bowel disease-linked immune function and inflammatory responses. We observed suggestive evidence for a small number of tissue-specific associations and disease-specific associations in ileum, though larger studies will be needed to confirm these results. Our study concludes that the vast majority of blood-derived methylation quantitative trait loci are common across individuals, though a subset may be involved in processes related to Crohn’s disease. Independent cohort studies will be required to validate these findings.
Keywords: methylation quantitative trait loci (mQTL), inflammatory bowel disease (IBD), Crohn’s disease (CD), DNA methylation (DNAm)
Introduction
A majority of inflammatory bowel disease (IBD) trait-associated genomic variants identified in genome-wide association studies (GWAS) reside in the genome’s noncoding regions (Farh et al. 2015). In the past decade, studies have focused on demonstrating the potential of these variants to influence specific traits or diseases through transcriptomic regulation (Rockman and Kruglyak 2006; Farber and Lusis 2008; Ward and Kellis 2012). Previous expression quantitative trait loci (eQTL) studies in IBD proposed that the genetic influences on gene expression are tissue-specific (Venkateswaran et al. 2018), disease-specific (Mo et al. 2018), and inflammation-specific (Thalayasingam et al. 2018; Venkateswaran et al. 2018). Meanwhile, recent advances in high-throughput array technologies have enabled genome-wide DNA methylation (DNAm) profiling for >850,000 CpGs. DNAm is one of the primary epigenetic mechanisms involved in gene expression regulation. Investigation of DNAm has the potential to advance our understanding of complex disease pathogenesis (Handy et al. 2011; Moore et al. 2013). In an epigenome-wide association study (EWAS), we recently identified 1189 CpGs showing differential DNAm in Crohn’s disease (CD) patients at diagnosis, compared to non-IBD controls (Somineni et al. 2019). A Mendelian randomization (MR) approach revealed that most of the DNAm changes observed in CD patients are consequences of the disease rather than causal factors influencing disease risk (Somineni et al. 2019). Since DNAm can be influenced by both environmental and genetic factors, in this study, we will focus on identification of methylation quantitative trait loci (mQTLs), or SNPs that associate with DNAm at specific CpG sites. We will compare mQTLs found within blood across disease state and progression, as well as age and race. Finally, we will compare patterns found in blood to those found in ileal (disease) tissue. In general, we are looking for mQTL patterns that distinguish CD cases from CD controls, and mQTLs that are context-specific.
Identifying SNP-CpG associations and comparing them between disease groups will provide epigenetic annotation of IBD variants, facilitating their functional characterization. The comparative mQTL study has the potential to reveal differences in interactions between genetic and epigenetic mechanisms in IBD patients vs controls. Further, the identification of mQTLs can aid MR approaches through identification of potential instruments to distinguish the casual vs consequential role of DNAm in complex diseases (Somineni et al. 2019), and can shed light on causal relationships between gene expression and DNAm in complex diseases (Hannon et al. 2018; Wu et al. 2018).
Investigating whether SNP-CpG associations are stable or dynamic over the course of the diseases can help inform whether the association might be a useful biomarker for disease, a useful marker for treatment, a potential cause of disease, or without obvious utility. A recent longitudinal analysis at 5 different stages showed that blood mQTLs are generally stable across the life span (Gaunt et al. 2016). However, among the 1,189 differentially methylated CpG sites identified in our previous study from blood derived DNA, we observed that during the CD disease course (at 1–3 years of follow-up), DNAm patterns in cases reverted back to levels observed in healthy controls. (Somineni et al. 2019). We also noted high correlations between DNAm at these 1,189 sites and levels of the inflammatory marker C-reactive protein (CRP) (Somineni et al. 2019). Investigation of mQTLs in CD patients pre- and post-treatment will reveal whether the genetic influence on DNAm at these sites varies over the disease course.
CD commonly affects the terminal ileum. Thus, in contrast to DNAm patterns in blood, methylation patterns in the ileum might be considered patterns seen at the “site of disease.” Consequently, ileal DNAm is more likely to be causative, compared to patterns observed in other tissues. Thus, we examine ileum-specific mQTL associations in CD patients.
Here, we used blood samples obtained from a total of 238 pediatric subjects; 164 patients with CD and 74 healthy controls sampled from the Risk Stratification and Identification of Immunogenetics and Microbial Markers of Rapid Disease Progression in Children with Crohn’s Disease (RISK) study (Kugathasan et al. 2017). We replicated our analysis in an independent cohort of 780 blood samples obtained from prospectively recruited adults in the Grady Trauma Project (GTP). Enrichment analysis showed that the previously identified IBD GWAS SNPs are significantly enriched for mQTLs in the CD cohort, but not in the adult cohort. To assess whether the mQTLs are specific to disease, disease course, or tissue type, we performed separate mQTL analyses in 164 CD cases at diagnosis, in cases at follow-up (1–3 years from the day of diagnosis), in 74 healthy controls and in 40 ileal biopsies (obtained from a subset of subjects included in the blood mQTL analysis; 23 controls and 17 patients with CD at diagnosis).
Methods
Study population and design of discovery cohort
The discovery cohort was a subset of pediatric subjects recruited under the Risk Stratification and Identification of Immunogenetic and Microbial Markers of Rapid Disease Progression in Children with Crohn’s Disease (RISK) study (Kugathasan et al. 2017) for whom DNAm data had been collected. A detailed description of the dataset is provided in our previous study (Somineni et al. 2019). In total, 164 newly diagnosed, pediatric patients with Crohn’s disease (cases) at 2 time-points (diagnosis and 3-year follow-up) and 74 non-IBD controls had DNAm measured for this study. Patients with no bowel pathology upon endoscopy, negative gut inflammation, and continued presentation as asymptomatic for IBD during follow-up were considered as non-IBD controls.
Blood genotype data for CD cohort
Peripheral blood DNA genotypes were obtained for 238 subjects using Infinium Multi-Ethnic Global-8 Kit (Illumina, San Diego, CA, USA) and processed with GenomeStudio software. Detailed quality control (QC) steps for both samples and variants are provided in our earlier study (Somineni et al. 2019). Briefly, sample QC was performed by checking that (1) genotype call rates >95%, (2) inferred gender is consistent with clinical records, and (3) subjects are unrelated according to a pairwise identity-by-descent test. Similarly, SNP QC was performed by identifying and removing SNPs with low call rate (<95%), (1) Hardy-Weinberg disequilibrium (P < 1.0 × 10−3), or (2) minor allele frequency (MAF) < 5%, as well as (3) nonautosomal variants and (4) variants mapping to multiple locations. This resulted in a discovery data set consisting of 636,006 high-quality variants. These variants for all 238 subjects were then used to impute genotypes from the 1000 Genomes Project Phase3 autosomal reference panel, using IMPUTE2 software (Howie et al. 2012). Imputed SNPs with MAF < 1% and imputation quality scores <80% were removed. We also removed imputed variants with no dbSNP annotation, INDELs/CNV, and SNPs with missing genotype information in >5% of the samples. Last, SNPs with <5 individuals in each group [homologous reference (AA), heterozygous (AB), and homologous variant (BB)] were excluded. In total, 3,109,863 high-quality SNPs for all 238 subjects were retained across the entire genome.
Blood and Ileal DNA methylation data for CD cohort
DNAm data was profiled for 402 samples (164 CD cases at diagnosis and 3-years follow-up and 74 healthy controls at diagnosis) as described in our previous study (Somineni et al. 2019). In total, our discovery dataset consists of 77.7% Caucasian, 16.8% African American, and 5.5% other ancestries (Supplementary Table 1). In this study, we have added DNAm data for 40 ileal biopsies, performed in 17 CD cases and 23 non-IBD controls from the discovery cohort. Genome-wide DNA methylation for these samples was profiled at single-base resolution using MethylationEPIC BeadChips. QC for the blood DNA methylation is described in detail in Somineni et al. (2019). We applied the same QC protocol on the ileal biopsies and retained all 40 ileal biopsies. Data were normalized using the beta-mixture quantile dilation (BMIQ) method (Teschendorff et al. 2013). Estimated cell counts for CD4+ T cells, CD8+ T cells, NK cells, B cells, monocytes, and granulocytes were calculated using Houseman’s approach (Houseman et al. 2012). After exclusion of CpGs with SNPs in their probe sequences, DNA methylation of 609,192 CpG sites in 402 samples from 238 subjects was used in this study.
Blood genotypes for GTP cohort
Study design and genotyping of the replication cohort was previously described in recent studies (Almli et al. 2018; Nievergelt et al. 2019). Briefly, GTP individuals were prospectively recruited from waiting rooms at Grady Memorial Hospital in Atlanta, GA between 2005 and 2008 for a study of stressful life events and symptoms of post-traumatic stress disorder (PTSD). Seven hundred and ninety-one subjects from GTP were genotyped using Illumina HumanOmni1-Quad, HumanOmniExpress or Multi-Ethnic Global arrays. Genotypes were called using Illumina’s GenomeStudio software. QC analyses on the genetic data from each chip were done separately, and untyped variants were imputed with IMPUTE2 software (Howie et al. 2012) using 1000 Genomes Project Phase3 as a reference panel. Individuals with > 5% missing data were removed. SNPs with >95% call rate, MAF > 1% and Hardy–Weinberg equilibrium (HWE) P > 10−5 were included. As a result, 5,971,966 genomic variants for 780 samples were retained. We used PLINK software (Purcell et al. 2007) to perform genotype QC in both discovery and replication cohorts.
Blood DNAm for GTP cohort
DNA was extracted from whole blood and interrogated using the MethylationEPIC BeadChip according to manufacturer’s instructions. Raw methylation beta values from the Infinium MethylationEPIC BeadChip were determined via the Illumina GenomeStudio program. Samples with probe detection call rates <90% and those with an average intensity value of either <50% of the experiment-wide sample mean or <2,000 arbitrary units (AU) were removed using the R package CpGassoc (Barfield et al. 2012). Data points with detection P-values > 0.01 were set to missing. CpG sites that cross-hybridize between autosomes and sex chromosomes were removed. BMIQ was used to normalize the distribution of types I and II probes (Teschendorff et al. 2013). In total, methylation of 608,245 CpG sites was examined in 780 blood samples of the replication cohort.
mQTL study
All SNPs passing QC were tested for association with ∼600K CpG sites using a linear mixed model implemented in matrixeQTL (Shabalin 2012). The model was adjusted for age, gender, estimated cell proportions, and 3 genotype-based principal components (PCs) to adjust for population sub-structure, and treated CpG-specific methylation as the outcome and SNP allele count as an explanatory variable. First, we tested each CpG site against all SNPs for both cis- and trans-SNP-CpG associations in the discovery cohort of 238 samples at diagnosis (164 CD cases and 74 controls), with adjustment for disease status in addition to other covariates. A stringent Bonferroni threshold of P < 8.21 × 10−14 was used to identify mQTLs in the CD (discovery) cohort. To replicate our findings, we further tested all significant associations in the GTP cohort using the same model as in the discovery cohort, adjusting for age, gender, PTSD status, first 3 PCs from genotype data, and estimated cell proportions from DNA methylation data. To identify whether these associations are specifically observed in the disease- and longitudinal datasets, we tested and compared them in the 74 healthy controls, 164 CD cases at diagnosis and at 3-year follow-up, separately. We also tested the significant blood mQTLs in a cohort of ileal samples (n = 40, which is a subset of individuals from the discovery cohort) to assess tissue-specific associations. To detect all the possible mQTLs for known IBD SNPs, we selected the mQTLs for those SNPs from the discovery cohort results, and calculated FDR on the actual test P-values using the Benjamini-Hochberg method, implemented in the p.adjust function in R. For these follow-up analyses in ileum, we restricted analysis to SNPs that had at least 2 individuals in each genotype category (AA/AB/BB) to ensure sufficient genetic variation. A statistical significance threshold of P < 0.05 was used for replication analyses of the disease-, longitudinal-, and tissue- associated mQTLs. To formally test for disease-specific differences in CpG-SNP associations in ileum, we performed interaction tests by refitting the original model with the addition of a term for SNP × disease status. We used the clump command in PLINK (Purcell et al. 2007) to identify the number of independent associations for each genomic variant with more than 1 significant SNP-CpG association with default parameters (distance threshold = 250 kb and r2 threshold = 0.50).
Annotation of SNPs and CpG sites
We used the annovar package (Wang et al. 2010) to annotate SNPs, and the MethylationEPIC annotation file to annotate CpG sites.
KEGG pathway enrichment analysis
We used missMethyl (Phipson et al. 2016), a R/Bioconductor package, to identify KEGG database pathways that are more likely to occur in the associated CpGs than would be expected by chance. Genes with more CpG probes on the MethylationEPIC array are more likely to have differentially methylated CpGs, which could introduce potential bias when performing pathway enrichment analysis. The gometh function implemented in missMethyl considers the varying number of CpGs per gene by computing prior probability for each gene based on the gene length and the number of CpGs probed per gene on the array.
Results
Blood mQTL discovery
To identify blood mQTLs, we tested the association between 3,109,863 SNPs and 609,192 CpG sites from 238 blood samples taken from 164 pediatric patients with CD at diagnosis and 74 matched non-IBD controls (Supplementary Table 1). We identified 535,448 associated SNP-CpG pairs involving 303,986 (9.8%) unique SNPs and 13,823 (2.3%) unique CpGs, at a Bonferroni adjusted genome-wide significance of P < 8.21 × 10−14 (Fig. 1a). On average, each CpG site was associated with ∼38 SNPs [interquartile range (IQR): 4–42], which likely reflects linkage disequilibrium (LD) between proximal SNPs, while each mQTL variant associated with 1.73 CpGs (IQR: 1–2). Using the clump command in PLINK (Purcell et al. 2007), we identified 16,313 LD pruned proximal SNPs that were associated with 13,202 CpG sites, which made a total of 24,523 associations.
Fig. 1.
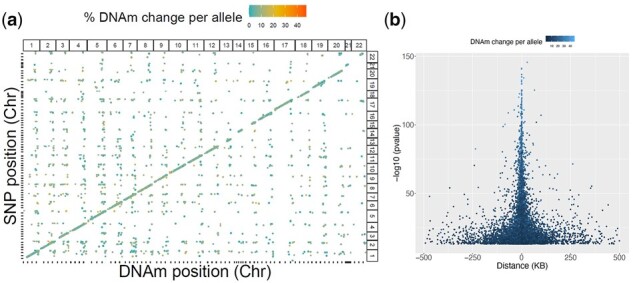
Genetic associations with DNAm genome-wide in discovery cohort. a) In total 535,448 SNP-CpG pairs are plotted, including both cis- and trans-associations. The x-axis indicates genomic variant position, whereas the y-axis indicates CpG site position. The color of each point represents absolute differences in DNAm per allele relative to the reference allele. Point size represents the significance of the association. A clear positive diagonal line indicates that most of them are cis-associations. b) In total, 14,979 cis-associations are plotted by position of CpG relative to SNP. The strongest associations are seen for proximal pairs of SNPs and CpG sites.
Ninety-two percent (n = 22566) of the SNP-CpG pairs involved associations between the LD-pruned genetic variants (n = 14,979) and CpG sites (n = 12,260) within 1 MB (±500 kb; Fig. 1b) and were considered cis-acting. The remaining 8% appeared to be trans-acting (n = 1,966). Overall, the full set of cis-acting mQTLs (regardless of LD) tended to have larger effect size (change in DNAm per allele) with a mean absolute value of 0.087 [IQR: 0.053–0.108], compared to trans-acting mQTLs with a mean absolute value of 0.069 [IQR: 0.028–0.094] (Mann–Whitney–Wilcoxon P < 2.2 × 10−16 (Supplementary Fig. 1). A list of all cis- and trans- SNP-CpG pairs identified from 238 blood samples is provided in Supplementary Table 2. Enrichment analysis of cis-mQTL associated SNPs (n = 287,881) showed that they are more likely than other SNPs on the array to occur 1 kb upstream of transcription start site (TSS), 1 kb downstream of transcription end site (TES), in exons, the 3′ or 5′ untranslated regions, or within 2 bp of a splicing junction (P < 2 × 10−10; Supplementary Table 3). Similarly, cis-mQTL associated CpGs (n = 12,843) are more likely than other CpGs on the array to occur in CpG islands and shores (P < 2.2 × 10−16; Supplementary Tables 4 and 5).
Blood mQTLs are highly replicable in an independent dataset regardless of age and race
To evaluate whether our blood mQTLs are unique to IBD, or more common to the human condition, we tested all cis associations in an independent cohort of 780 blood samples (GTP cohort) collected previously (Almli et al. 2018). More details about demographic information of this dataset are available in Almli et al. (2018) as well as in our methods section. Notably, while our discovery dataset consists of pediatric patients with predominantly Caucasian ancestry (78%; Supplementary Table 1), the GTP dataset is composed of prospectively recruited adult individuals with predominantly African American ancestry (93%), with a mean age of 42.26 (SD: 12.31) years (Supplementary Table 1). After QC, 5,971,966 genomic variants and 608,245 CpG sites were retained for 780 blood samples. To test for the replicability of blood mQTLs from the discovery cohort, we focused only on the SNPs and CpG sites identified in the discovery cohort. In total, 253,000 out of 287,881 SNPs (87.9%) and 12,808 out of 12,843 CpG sites (99.7%) were available to be tested in the replication cohort, representing a total of 425,288 CpG-SNP relationships. Applying a standardized genome-wide significance threshold of P < 5 × 10−8, we replicated ∼84% of the CD dataset associations (358,804 SNP-CpG pairs out of 425,288 tested). This includes 87% (n = 220,468) of SNPs, and 85% (n = 10,905) of CpG sites. At a less stringent nominal significance threshold of P < 0.05, we detected 388,609 associations, of which 91.3% (n = 388,217) were considered replications, where the effect sizes of both CD and GTP cohort associations are directionally consistent. Test statistics for these associations were highly correlated in the discovery vs replication cohorts (r = 0.93; Fig. 2), indicating that most blood SNP-CpG associations are consistent regardless of age, race, and disease status. Summary statistics for the 388,609 SNP-CpG associations are provided in Supplementary Table 6. Among the few mQTLs showing associations in the opposite direction (n = 393; 358 unique SNPs and 119 CpG sites) between the CD and GTP cohorts (i.e. an SNP associated with increased methylation in 1 cohort, and decreased methylation of the same CpG in another cohort), we did not observe significant enrichment for any KEGG pathways, including inflammation or immune-related pathways (data not shown). Similarly, none of the previously identified IBD-associated SNPs (Liu et al. 2015; de Lange et al. 2017) or CD-associated CpGs (Somineni et al. 2019) were involved in these 393 mQTL associations. These results are consistent with our previous (Smith et al. 2014) findings that blood mQTLs are stable across ancestries (African American vs Caucasian), and developmental stages (neonate vs adult). Collectively, our results indicate that the vast majority of genetic influence on changes in blood DNAm are highly consistent regardless of disease, race and age groups, and thus unlikely to be casually related to CD in any substantial way.
Fig. 2.

Replication of mQTL associations in replication cohort. Effect sizes for 425,288 SNP-CpG pairs are compared between discovery and replication cohorts. Associations significant in both discovery cohort (P < 8.21 × 10−14) and replication cohort (P < 5 × 10−8) are marked in maroon. Gray dots indicate associations significant only in discovery cohort.
CD-specific mQTLs in blood samples
To assess whether mQTLs vary with respect to disease status, we retested each of the 498,261 significant SNP-CpG pairs separately within controls (n = 74), within CD cases at baseline (n = 164), and within CD cases during the course of the disease (n = 164; Supplementary Table 7). We observed a strong positive correlation (r = 0.98) between effect sizes estimated from controls vs cases both at baseline and at disease course (Fig. 3, a and c). Collectively, our data establish that while methylation levels may vary with CD at specific sites (CD-associated CpG sites) (Somineni et al. 2019), the influence of SNPs on CpG methylation levels at CpG sites remains consistent in the presence or absence of disease.
Fig. 3.

Disease-, disease course-, or tissue-specific associations in discovery cohort. For 498,261 cis-SNP-CpG pairs significant in the discovery cohort of 238 blood samples, Figure 3 shows effect sizes estimated in the following subgroups: a) 74 healthy controls vs 164 CD cases at diagnosis, (b) 164 CD cases at diagnosis v. their 3-year follow-up, (c) 74 healthy controls at baseline vs 164 CD cases at follow-up, and (d) all 238 blood samples vs 40 ileal biopsies. Maroon color indicates the associations that are significant (P < 5 × 10−8) in both subgroups. Gray indicates the associations that are significant in 1 subgroup but not the other.
Longitudinal profiling of mQTLs
In our previous report, we demonstrated that with treatment, DNAm changes associated with CD at diagnosis revert to the levels seen in healthy controls (Somineni et al. 2019). To examine the longitudinal trajectory of SNP-CpG associations and their dynamics during the course of the disease, we took advantage of our longitudinal DNAm data generated from same individuals from the discovery cohort (164 CD cases at diagnosis vs follow-up). We found that mQTL effect sizes were similar between diagnosis and at 3-year follow-up in CD patients, showing a strong positive correlation in magnitude and direction of effect (r = 0.99; Fig. 3b). The 2 outlier clusters in Fig. 3b represent 2 CpGs (cg22715629 and cg00924943) that are associated with 136 SNPs in LD. All discovery SNP-CpG associations (n = 497,689) were nominally significant at baseline (P < 0.05), and 99.5% (n = 497,443) were nominally significant during the course of the disease (P < 0.05). Therefore, our results suggest that these genetic associations are independent of disease status or treatment.
Tissue-specific associations between blood and ileum
To further evaluate how the mQTLs identified in our blood-based discovery dataset act in tissues more directly relevant to disease, we estimated SNP-CpG effect sizes using DNAm data from ileal biopsies (n = 40) and compared them to effects observed in blood. With the standard replication threshold of P < 0.05, we replicated 50.4% (n = 238,400) blood SNP-CpG associations in ileal biopsies. Overall, we noted an extremely strong correlation in effect sizes between ileal and blood samples, with 91.4% (n = 473,382) of mQTLs showing directional consistency (r = 0.75; Fig. 3d). A list of all ileal biopsy mQTL results are provided in Supplementary Table 8. We further investigated CpGs (n = 2,188) from SNP-CpG associations (n = 24,879) with directionally inconsistent effects between blood and biopsy. Although some of the directionally inconsistent effects may reflect the smaller sample size used in the ileal analysis, we did a KEGG pathway analysis to investigate enrichment for potential tissue-specific biological functions, and observed that these sites were more likely to localize to genes involved in metabolic pathways (P = 3.34 × 10−8; Supplementary Table 9). Using summary statistics from 2 different studies, Qi et al. (2018) recently showed that mQTLs are highly correlated among the blood and brain tissues. Similarly, using blood and 4 regions of postmortem brain samples, we previously showed a strong mQTL correlation among the tissues, especially within the 4 brain regions (Smith et al. 2014). Our current results show a similar pattern for blood vs ileum taken from the same patients, where the mQTL effects are highly correlated between blood and ileal biopsies.
Enrichment of previously identified IBD susceptible SNPs in blood mQTLs
To date, large-scale GWAS meta-analysis have identified 241 susceptibility loci for IBD (Liu et al. 2015; de Lange et al. 2017). A majority of these (>90%) localize to noncoding regions, presenting a key challenge to identify the functional variants and the relevant genes they act upon. Of these 241 previously identified IBD SNPs, 104 were genotyped and 64 were successfully imputed in our discovery dataset. We found that 37 (22.2%) of these 168 SNPs associated with 69 CpG sites in our discovery set of mQTL associations, which is greater than the 9.8% expected by chance (OR = 2.6, Fisher’s exact test P = 3e-6; Supplementary Fig. 2a; Supplementary Table 10). All but one of the associations were cis-mQTL associations: 90% (n = 62) of these CpGs were located within 100 kb from the associated GWAS variant. Of these 62 CpGs, 13% are located within 1.5 kb of a transcriptional start site (TSS200 or TSS1500), and 47.8% are between the start and stop codon. These results provide an additional 31 genes potentially associated with the previous-identified IBD SNPs, where the associated CpGs may be located within the gene body or near the TSS or promotor regions of the gene. Four of the established IBD-associated SNPs (rs13407913, rs798502, rs59043219, and rs1819333) showed association with >5 CpGs located within the gene bodies of ADCY3, GNA12, IRF6, and RNASET2/RPS6KA2, respectively. The 37 IBD-associated SNPs comprised 0.013% of the total number of mQTL SNPs observed in our discovery cohort (Supplementary Table 10). For comparison, in our GTP cohort, which did not include IBD cases, we observed associations between 87 IBD-associated SNPs and 252 nearby CpGs. Due to the larger sample size, a greater number of mQTL associations were observed in total in this larger cohort, but the 87 IBD-associated SNPs comprised a smaller percentage of the total (0.006%; Supplementary Table 10). Summary statistics for 69 and 253 SNP-CpG associations from the IBD susceptibility SNPs identified in both discovery and replication cohorts are provided in Table 1.
Table 1.
Summary of blood mQTLs identified in discovery and replication cohorts.
| Cohorts | Number of mQTL associations in total | IBD genomic variants from de Lange et al. (2017; n = 241) | CD-associated CpGs from Somineni et al. (2019; n = 1,189) | |
|---|---|---|---|---|
| Discovery cohort (n = 238) | Cis-associations | |||
| SNP-CpG associations | 498,261 | 69 | 565 | |
| Unique SNPs | 287,881 | 37 | 565 | |
| Unique CpGs | 12,843 | 69 | 22 | |
| Trans-associations | ||||
| SNP-CpG associations | 37,187 | 1 | 0 | |
| Unique SNPs | 27,664 | 1 | 0 | |
| Unique CpGs | 1,118 | 1 | 0 | |
| Replication cohort (n = 780) | Cis-associations | |||
| SNP-CpG associations | 3,140,805 | 253 | 9,587 | |
| Unique SNPs | 1,429,468 | 87 | 8,848 | |
| Unique CpGs | 62,456 | 252 | 261 | |
| Trans-associations | ||||
| SNP-CpG associations | 175,588 | 4 | 184 | |
| Unique SNPs | 106,900 | 4 | 85 | |
| Unique CpGs | 7901 | 4 | 11 | |
We further searched for previously identified IBD SNPs in a wider set of mQTL results identified using a less stringent significance threshold (FDR < 0.05) in the discovery cohort. At this threshold, 64.2% (n = 108) of IBD SNPs were associated with 453 CpGs within 1 MB, comprising 454 cis-mQTL associations (Supplementary Table 10, Fig. 4a). Of these CpGs, 84.5% (n = 384) were located within 100 kb of the associated GWAS variant. Further disease-specific analysis on the mQTLs showed that similar to SNPs across the genome, associations between IBD-associated SNP and DNAm in blood are extremely consistent between CD cases (n = 164) vs controls (n = 74) (r = 0.94; Fig. 4b).
Fig. 4.

Comparing CpG-specific associations with IBD SNPs in blood vs ileum. a) Frequency of CpG sites that are associated with previously identified IBD SNPs in discovery cohort at FDR < 0.05. In total, 454 associations are considered. b) Effect sizes of 454 SNP-CpG associations compared in CD cases vs controls in blood. x-axis shows the effect sizes in CD cases (n = 164) and y-axis shows the effect sizes for the same associations in controls (n = 74) (c) Effect sizes of 370 SNP-CpG associations found in blood are compared to effect sizes observed in ileum. The nominally significant associations in ileum (P < 0.05) are marked in maroon. (D) Effect sizes of 269 SNP-CpG associations compared across the disease groups in ileum. The x-xis shows mQTL effect sizes in CD cases (n = 17) and y-axis shows effect sizes for the same associations in controls (n = 23). Maroon points indicate nominally significant (P < 0.05) interactions between SNP and disease group, suggesting differences in strength and/or direction of SNP-CpG association for cases vs controls.
Pathway enrichment of CpG sites that are under the influence of IBD-associated SNPs
To investigate the utility of DNAm as a tool to identify functional variants and underlying regulatory mechanisms that may contribute to our fundamental understanding of CD susceptibility and pathogenesis, we examined whether CpG sites associated with IBD-associated SNPs localize near genes associated with specific pathways. We first evaluated all 69 CpGs whose blood DNAm associated with IBD SNPs in the discovery analysis (P < 8.21 × 10−14) to identify the novel pathways that are relevant to CD. Our pathway enrichment analysis identified 18 KEGG pathways that were more likely to occur in the IBD SNP–associated CpGs than would be expected by chance (FDR < 0.05; Supplementary Table 11). Among these, 5 were relevant to immune function and other pathways, including human cytomegalovirus infection, insulin resistance, vascular smooth muscle contraction, hepatitis B, and chemokine signaling. Pathway enrichment analysis on an expanded set of CpG sites (n = 454) that associated with IBD SNPs according to a looser significance threshold (FDR < 0.05) showed enrichment for 7 KEGG pathways (FDR < 0.05), including a natural killer cell mediated toxicity pathway. We also observed nominally significant (P < 0.05) enrichment for immune and inflammatory related pathways such as MAPK signaling pathway, cytokine-cytokine receptor interaction, IBD, JAK-STAT signaling pathway, and Th17 cell differentiation (Supplementary Table 12).
Influence of previously identified IBD SNPs on DNAm in ileum mQTLs
We next investigated the 37 SNPs that were both IBD-associated (de Lange et al. 2017) and associated with CpGs in our discovery cohort (P < 8.21 × 10−14) to identify whether they were also mQTLs in ileum (Supplementary Table 13). Seventeen of 37 SNPs were nominally significant mQTLs (P < 0.05) in ileum as well, and effect sizes showed a strong positive correlation between ileum and blood (r = 0.789, P < 7.87 × 10−16; Supplementary Fig. 2c).
We also followed up on the 108 FDR-significant IBD-associated SNPs in ileum. We tested 370 of the 454 blood mQTL pairs for association in ileum. These mQTL pairs included 85 of the 108 IBD-associated SNPs; the remaining 23 SNPs were not tested due to insufficient genetic variation in the smaller number of ileum samples. At a nominal threshold of P < 0.05, we detected associations for 51 of 85 IBD-associated SNPs. In total, 91 (24.6%) of 370 blood mQTLs in ileum were nominally significant, and the effect sizes were consistent in direction for 89 of these, indicating replication (Fig. 4c; Supplementary Table 14). The 2 IBD SNPs showing association in the opposite direction in ileum (rs35320439, and rs516246) were independently associated with CpGs cg07689252 and cg26360664 (Fig. 4c). Neither of these CpGs were associated with CD in [10]. The strong positive correlation and directionally consistent effects (r = 0.68; Fig. 4c) suggest that the IBD-associated SNPs influence the DNAm levels similarly across tissues for most CpGs. Performing the ileal mQTL analysis separately in CD cases (n = 17) and controls (n = 23) showed little correlation between effect sizes observed for cases vs controls (r = 0.18, Fig. 4d). In an interaction analysis of all ileal samples, the interaction between case-control status and SNP genotype was nominally significant (P < 0.05) for 17 SNP-CpG pairs involving 14 of the SNPs (Fig. 4d; Supplementary Table 14). These results suggest there may be disease-specific SNP-CpG associations in disease-relevant tissues such as ileum for CD. However, given our smaller sample size (n = 40) and the nominal significance level, further confirmation is needed in larger studies of disease-relevant tissues such as ileum.
CD-associated differentially methylated CpGs in blood mQTLs
Recently, we identified 1,189 CD-associated CpG sites whose methylation level in blood distinguished CD cases from non-IBD controls (Somineni et al. 2019). After exclusion of 212 CpGs with one or more SNPs in their probe sequence, we tested 977 of our previously identified CpG sites for associations with SNPs within 1MB. In the discovery cohort, 22 (1.8%) of these 977 CpGs associated with 565 unique SNPs for a total of 565 SNP-CpG associations (P < 2.81 × 10−14; Supplementary Fig. 2b; Supplementary Table 15). Although we did not observe an enrichment of CD-associated CpG sites compared to the replication cohort (Table 1), 1 CD-associated CpG site cg20406979 had a strong association (P = 7.60 × 10−25) with SNP rs1819333, previously reported to associate with CD (Somineni et al. 2019). This SNP and CpG site were located 314 nucleotides apart, 3 kb upstream of the TSS for the gene RNASET2.
We further investigated the 977 CD-associated differentially methylated CpGs using an FDR-based significance threshold (FDR < 0.05) and observed a total of 7,819 cis-associations. 29% (n = 285) of 977 CD-associated CpGs were associated with 7,120 nearby genomic variants (Supplementary Table 15), with 59.9% of these CpGs associating with more than 5 SNPs (Fig. 5a). Further, separate analysis on CD cases (n = 164) and controls (n = 74) indicates that the mQTLs associated with CD-associated DNAm sites in the blood are extremely consistent across the disease types (r = 0.91; Fig. 5b).
Fig. 5.

Testing the effects of CD-associated CpG sites in blood and ileum. a) Frequency of SNPs that are associated with CD-associated CpG sites in discovery cohort at FDR < 0.05 are plotted. In total, 7,819 associations with 289 CD-associated CpG sites are considered. b) Effect sizes of 7,819 SNP-CpG associations compared in cases vs controls in blood. The x-axis shows the effect sizes in CD cases (n = 164) and y-axis shows the effect sizes for the same associations in controls (n = 74) (c) Effect sizes of 6,221 SNP-CpG associations from blood are compared to effect sizes observed in ileum. Associations nominally significant in ileum (P < 0.05) are marked in maroon. d) Effect sizes of 4,142 SNP-CpG associations compared in cases vs controls in ileum. x-axis shows effect sizes in CD cases (n = 17) and y-axis shows effect sizes for the same associations in controls (n = 23). Maroon points indicate nominally significant (P < 0.05) interactions between SNP and disease group, suggesting differences in strength and/or direction of SNP-CpG association for cases vs controls.
Next, we investigated whether the CD-associated CpGs associated with 108 previously identified IBD risk SNPs within 1MB. We observed 2 IBD SNPs (rs1819333, rs3853824) associated with 4 CD-associated CpGs (cg20406979, cg23216724, cg15706657, and cg15815084) for a total of 4 cis associations. Of those, 3 CpGs cg20406979, cg23216724, and cg15706657 had stronger association with 1 IBD-associated SNP rs1819333 (P = 7.60 × 10−25, P = 1.2 × 10−11, and P = 9.3 × 10−07, respectively; Supplementary Table 15).
Influence of SNPs on CD-associated CpGs in Ileum mQTLs
Next, we investigated 22 of the 1,189 CpGs previously associated with CD in blood (Somineni et al. 2019) that were also blood mQTLs (P < 8.21 × 10−14) to see if they are also mQTLs in ileum. In blood, these CpGs were involved in 565 significant SNP-CpG associations. We replicated 173 (30.6%) of these associations in ileal biopsies using a nominal threshold of P < 0.05, and observed effect sizes are consistent in magnitude and direction in ileum vs blood (r = 0.82; P < 2.2 × 10−16; Supplementary Fig. 2d; Supplementary Table 16).
We performed similar comparisons for the 285 CD-associated CpGs that were significant at FDR < 0.05 in blood, which were involved in 7,819 significant SNP-CpG associations in blood. We followed up on 6,227 of these SNP-CpG pairs in ileum; the remainder were not tested due to insufficient genetic variation among the 40 ileal samples. At a nominal threshold of P < 0.05, we detected 47 of 285 significant CD-associated CpGs and 1,002 (16.1%) out of 6,227 blood SNP-CpG pairs in ileum (P < 0.05). Among these 1002, 65.4% (n = 655) showed directionally consistent associations (r = 0.15; P < 2.2 × 10−16; Fig. 5c). The 34.6% (n = 347) of CpG-SNP associations that showed association in the opposite direction in blood vs ileum (Supplementary Table 16) comprised 12 CD-associated CpGs that were associated with 341 SNPs. Boxplots for the top SNP associated with the 12 CD-associated CpGs are compared between blood and ileum (Supplementary Table 16). None of the IBD SNPs showed the opposite mQTL effects between blood and ileum. Overall, only 1 IBD SNP (rs1819333) was nominally associated with a CD-associated CpG (cg15706657) in ileum (P = 0.040), and the association was directionally consistent with blood results in our discovery cohort. Our analysis stratified by CD suggested possibly distinct effects for cases (n = 17) vs controls (n = 23), with nominally significant (P < 0.05) interactions between SNP and disease status for 177 mQTLs (177 SNPs and 32 CD-associated CpGs) (Fig. 5d, Supplementary Table 17). However, given the nominal significance levels, this is merely suggestive, and a much larger sample would be required to test this formally.
Discussion
The primary focus of this study was to identify and characterize mQTLs in blood from 238 pediatric samples of largely (78%) European ancestry. Our findings establish that blood mQTLs are robust and reproducible, supporting their generalizability across age groups, ancestries, disease status, and DNA sample source, and offer a valuable source for future studies examining blood mQTLs. Recent studies have identified different sets of differential DNAm patterns in various tissue types (Zhang et al. 2013; Lokk et al. 2014), and previous epigenomic and transcriptomic studies in IBD suggest that disease-relevant tissue samples are needed to develop precise biomarkers for the disease subtypes (Sonawane et al. 2017; Barbeira et al. 2018; Venkateswaran et al. 2018; Yang et al. 2018). Our findings show that among genome-wide-significant mQTLs, genetic influences on DNAm are generally consistent across blood and ileum, even among IBD SNPs and CD-associated CpG sites. However, we observed lower cross-tissue reproducibility using a relaxed FDR-based significance criterion. Using a threshold of FDR < 0.05 in blood and nominal association [P < 0.05] in ileum, we identified 2 IBD SNPs (Fig. 4c) and 12 CD-associated CpGs (Fig. 5c; Supplementary Fig. 3) with associations in the opposite direction between blood and ileum. This could suggest that a small portion of mQTLs act in a tissue-specific manner, but could also be due to the low power and relaxed significance criteria in the ileal analysis. Studies with large numbers of samples from both blood and ileal biopsies from the same cohort are needed to formally test for tissue-specific associations for the previously identified IBD SNPs or CD-associated CpGs.
Blood mQTLs were also consistent in comparisons of CD patients pre- vs post-treatment and in CD patients compared to healthy matched controls or to an external epidemiologic sample of predominantly African American adults. This trend was observed even when we focused on previously identified IBD SNPs and CD-associated CpGs, though was not as clear when studied in the ileal biopsies, again likely due to the small numbers.
Our study has several limitations. We did not explore associations involving rare variants in this study given limited power to identify such associations. In particular, because of the limited number of ileal samples used in this study, power was limited to identify tissue-specific mQTLs or to detect functionally important associations involving previously identified IBD variants in the disease-relevant ileum.
Data availability
The blood DNA methylation data for all the 238 subjects (402 samples) included in this study have been deposited in the Gene Expression Omnibus (GEO) and are accessible through GEO series accession GSE112611. DNA methylation data for the 40 ileal biopsies are also deposited in GEO and can be retrieved through the accession number GSE135905. All the supplementary files related to this manuscript are placed in GSA figshare: https://doi.org/10.25387/g3.17430605.
Author Contributions
SV, KNC, DJC, AKS, and SK conceived of and designed the study. JP, DTO, and SK, processed samples for methylation profiling. VS, HKS, and VK performed the QC on the data. VS performed the analysis with input from DJC and KNC. AKS, SV, HKS, LAD, JSH, DJC, KNC, GG, AKS, and SK interpreted the results and wrote the manuscript. All authors reviewed and approved the manuscript prior to submission.
Ethics approval and consent to participate
Protocols including signed consent of all participants and/or consent of parents or legal guardians in the case of minors were approved by the IRBs of Emory University for CD RISK study supported by a research initiative grant from the Crohn’s and Colitis Foundation. The GTP study was approved by the Institutional Review Board of Emory University School of Medicine and the Grady Health Systems Research Oversight Committee. All participants provided written informed consent.
Funding
This work was supported by a research initiative grant from the Crohn’s and Colitis Foundation, New York, NY to the individual study institutions participating in the RISK study. This research was also supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health, under grant numbers RO1-DK098231 and RO1-DK087694 to SK.
Conflicts of interest
None declared.
Acknowledgments
The authors are grateful to Anne Dodd, Jason Matthews, and all other RISK investigators for their support and helpful comments.
Literature cited
- Almli LM, Lori A, Meyers JL, Shin J, Fani N, Maihofer AX, Nievergelt CM, Smith AK, Mercer KB, Kerley K, et al. Problematic alcohol use associates with sodium channel and clathrin linker 1 (SCLT1) in trauma-exposed populations. Addict Biol. 2018;23(5):1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, Torstenson ES, Shah KP, Garcia T, Edwards TL, et al. ; GTEx Consortium. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018;9(1):1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barfield RT, Kilaru V, Smith AK, Conneely KN.. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics. 2012;28(9):1280–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez-Achury J, Ji SG, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49(2):256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber CR, Lusis AJ.. Integrating global gene expression analysis and genetics. Adv Genet. 2008;60:571–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunt TR, Shihab HA, Hemani G, Min JL, Woodward G, Lyttleton O, Zheng J, Duggirala A, McArdle WL, Ho K, et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016;17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy DE, Castro R, Loscalzo J.. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123(19):2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon E, Gorrie-Stone TJ, Smart MC, Burrage J, Hughes A, Bao Y, Kumari M, Schalkwyk LC, Mill J.. Leveraging DNA-methylation quantitative-trait loci to characterize the relationship between methylomic variation, gene expression, and complex traits. Am J Hum Genet. 2018;103(5):654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT.. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR.. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugathasan S, Denson LA, Walters TD, Kim MO, Marigorta UM, Schirmer M, Mondal K, Liu C, Griffiths A, Noe JD, et al. Prediction of complicated disease course for children newly diagnosed with Crohn's disease: a multicentre inception cohort study. Lancet. 2017;389(10080):1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. ; International IBD Genetics Consortium. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokk K, Modhukur V, Rajashekar B, Martens K, Magi R, Kolde R, Koltsina M, Nilsson TK, Vilo J, Salumets A, et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014;15(4):r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Marigorta UM, Arafat D, Chan LHK, Ponder L, Jang SR, Prince J, Kugathasan S, Prahalad S, Gibson G.. Disease-specific regulation of gene expression in a comparative analysis of juvenile idiopathic arthritis and inflammatory bowel disease. Genome Med. 2018;10(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LD, Le T, Fan G.. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nievergelt CM, Maihofer AX, Klengel T, Atkinson EG, Chen CY, Choi KW, Coleman JRI, Dalvie S, Duncan LE, Gelernter J, et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat Commun. 2019;10(1):4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B, Maksimovic J, Oshlack A.. Missmethyl: an R package for analyzing data from illumina's humanmethylation450 platform. Bioinformatics. 2016;32(2):286–288. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. Plink: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi T, Wu Y, Zeng J, Zhang FT, Xue AL, Jiang LD, Zhu ZH, Kemper K, Yengo L, Zheng ZL, et al. ; eQTLGen Consortium. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun. 2018;9(1):2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L.. Genetics of global gene expression. Nat Rev Genet. 2006;7(11):862–872. [DOI] [PubMed] [Google Scholar]
- Shabalin AA. Matrix eqtl: ultra fast eqtl analysis via large matrix operations. Bioinformatics. 2012;28(10):1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AK, Kilaru V, Kocak M, Almli LM, Mercer KB, Ressler KJ, Tylavsky FA, Conneely KN.. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics. 2014;15:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somineni HK, Venkateswaran S, Kilaru V, Marigorta UM, Mo A, Okou DT, Kellermayer R, Mondal K, Cobb D, Walters TD, et al. Blood-derived DNA methylation signatures of Crohn's disease and severity of intestinal inflammation. Gastroenterology. 2019;156(8):2254–2265.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane AR, Platig J, Fagny M, Chen CY, Paulson JN, Lopes-Ramos CM, DeMeo DL, Quackenbush J, Glass K, Kuijjer ML.. Understanding tissue-specific gene regulation. Cell Rep. 2017;21(4):1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S.. A beta-mixture quantile normalization method for correcting probe design bias in illumina infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalayasingam N, Nair N, Skelton AJ, Massey J, Anderson AE, Clark AD, Diboll J, Lendrem DW, Reynard LN, Cordell HJ, et al. Cd4+ and b lymphocyte expression quantitative traits at rheumatoid arthritis risk loci in patients with untreated early arthritis implications for causal gene identification. Arthritis Rheumatol. 2018;70(3):361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateswaran S, Marigorta UM, Denson LA, Hyams JS, Gibson G, Kugathasan S.. Bowel location rather than disease subtype dominates transcriptomic heterogeneity in pediatric IBD. Cell Mol Gastroenterol Hepatol. 2018;6(4):474–476.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H.. Annovar: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M.. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 2012;30(11):1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, Lloyd-Jones LR, Marioni RE, Martin NG, Montgomery GW, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun. 2018;9(1):918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RY, Quan J, Sodaie R, Aguet F, Segre AV, Allen JA, Lanz TA, Reinhart V, Crawford M, Hasson S.. A systematic survey of human tissue-specific gene expression and splicing reveals new opportunities for therapeutic target identification and evaluation. bioRxiv. 2018;311563. [Google Scholar]
- Zhang B, Zhou Y, Lin N, Lowdon RF, Hong CB, Nagarajan RP, Cheng JB, Li DF, Stevens M, Lee HJ, et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the m&m algorithm. Genome Res. 2013;23(9):1522–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The blood DNA methylation data for all the 238 subjects (402 samples) included in this study have been deposited in the Gene Expression Omnibus (GEO) and are accessible through GEO series accession GSE112611. DNA methylation data for the 40 ileal biopsies are also deposited in GEO and can be retrieved through the accession number GSE135905. All the supplementary files related to this manuscript are placed in GSA figshare: https://doi.org/10.25387/g3.17430605.