

Abstract
Background
Heart failure (HF) is one of the leading causes of death worldwide and is associated with cardiac metabolic perturbations. Human Type 2 Berardinelli‐Seip Congenital Lipodystrophy (BSCL2) disease is caused by mutations in the BSCL2 gene. Global lipodystrophic Bscl2−/− mice exhibit hypertrophic cardiomyopathy with reduced cardiac steatosis. Whether BSCL2 plays a direct role in regulating cardiac substrate metabolism and/or contractile function remains unknown.
Methods
We generated mice with cardiomyocyte‐specific deletion of Bscl2 (Bscl2cKO ) and studied their cardiac substrate utilisation, bioenergetics, lipidomics and contractile function under baseline or after either a treatment regimen using fatty acid oxidation (FAO) inhibitor trimetazidine (TMZ) or a prevention regimen with high‐fat diet (HFD) feeding. Mice with partial ATGL deletion and cardiac‐specific deletion of Bscl2 were also generated followed by cardiac phenotyping.
Results
Different from hypertrophic cardiomyopathy in Bscl2−/− mice, mice with cardiac‐specific deletion of Bscl2 developed systolic dysfunction with dilation. Myocardial BSCL2 deletion led to elevated ATGL expression and FAO along with reduced cardiac lipid contents. Cardiac dysfunction in Bscl2cKO mice was independent of mitochondrial dysfunction and oxidative stress, but associated with decreased metabolic reserve and ATP levels. Importantly, cardiac dysfunction in Bscl2cKO mice could be partially reversed by FAO inhibitor TMZ, or prevented by genetic abolishment of one ATGL allele or HFD feeding. Lipidomic analysis further identified markedly reduced glycerolipids, glycerophospholipids, NEFA and acylcarnitines in Bscl2cKO hearts, which were partially normalised by TMZ or HFD.
Conclusions
We identified a new form of cardiac dysfunction with excessive lipid utilisation which ultimately causes cardiac substrate depletion and bioenergetics failure. Our findings also uncover a crucial role of BSCL2 in controlling cardiac lipid catabolism and contractile function and provide novel insights into metabolically treating energy‐starved HF using FAO inhibitor or HFD.
Keywords: BSCL2/Seipin, heart failure, lipid metabolism, lipidomics
Cardiac deletion of BSCL2 causes ATGL overexpression, excessive fatty acid oxidation (FAO), drastic reduction of cardiac lipidome and ultimately energetic and contractile dysfunction in mice. Inhibiting FAO by partial ATGL deletion or trimetazidine and increasing lipid supply via high fat diet (HFD) feeding alleviate cardiac dysfunction caused by BSCL2 deficiency. BSCL2 is a novel protein indispensable for cardiac lipid metabolism and function.
1. INTRODUCTION
Heart failure (HF) is one of the leading causes of morbidity and mortality worldwide. In healthy individuals, the heart exhibits striking metabolic flexibility, being capable of utilising carbohydrate, lipid, amino acids and/or ketone bodies to meet energetic demands, cellular constituent turnover and metabolic signalling; these processes are critical for maintenance of mechanical work. Oxidation of fatty acids (FAs) predominates, accounting for 60–70% of myocardial oxygen consumption. 1 In patients with hypertrophied and failing hearts, derangements of substrate utilisation include an increased reliance on glycolysis concomitant with an overall reduced oxidative metabolism. 2 The severely failing heart usually demonstrates a lower concentration of ATP, supporting the concept that energy starvation contributes significantly to the pathogenesis of HF. 3 Indeed, there is a striking correlation between cardiac energetic status and survival in HF patients. 4 Thus, targeting metabolic processes in the heart may represent a promising way to develop new therapeutic approaches for HF.
Cardiac lipid metabolism is precisely controlled to maintain a balance between FA uptake, triglyceride (TG) synthesis, TG hydrolysis and FA oxidation (FAO). 5 Imbalances in these processes are commonly seen in obese and diabetic patients and animal models, which are associated with cardiac lipotoxicity (i.e., accumulation of toxic lipid intermediates) and contractile dysfunction. 5 Recent preclinical and clinical evidence argues for an important role of adipose triglyceride lipase (ATGL)‐mediated cardiac lipolysis in promoting mitochondrial FAO and ATP production thus contractile function. 6 Constitutive Atgl −/− mice develop severe cardiac steatosis and HF, associated with a high mortality. 7 Conversely, cardiomyocyte‐specific overexpression of ATGL maintains normal cardiac function in lean mice and reduces cardiac TG content and improves cardiac function during diabetes and obesity.8, 9 The precise mechanisms linking cardiac lipid catabolism with contractile dysfunction remain to be further explored.
Type 2 Berardinelli‐Seip Congenital Lipodystrophy (BSCL2) is caused by mutations in a gene called BSCL2 (also called Seipin), a highly conserved endoplasmic reticulum (ER) protein expressed in most tissues, with the highest levels in testis, neuronal and adipose tissue. 10 , 11 In addition to metabolic abnormalities including insulin resistance and type 2 diabetes, 12 , 13 patients with BSCL2 develop progressive hypertrophic cardiomyopathy, highlighted by concentric left ventricular hypertrophy and cardiac failure. 14 , 15 , 16 , 17 Global Bscl2‐deficient (Bscl2 −/− ) mice recapitulate human BSCL2 disease, exhibiting congenital lipodystrophy and severe insulin resistance. 18 , 19 , 20 Various molecular functions of BSCL2 have been proposed, ranging from regulating lipid droplet (LD) biogenesis 21 , 22 to mitochondrial metabolism. 23 , 24 BSCL2 forms oligomers of 10–12 subunits and plays crucial roles in lipid transfer and/or LD formation. 25 , 26 BSCL2 has been shown to interact with 1‐acylglycerol‐3‐phosphate O‐acyltransferase 2 (AGPAT2), 27 LIPIN1, 28 glycerol‐3‐phosphate acyltransferase 3 (GPAT3), 29 and Promethin. 30 , 31 We and others demonstrate that BSCL2 plays a key role in regulating cyclic AMP/protein kinase A (cAMP/PKA)‐mediated TG lipolysis essential for white adipocyte differentiation. 18 , 20 Deletion of BSCL2 in mature white and brown adipose tissue also triggers cAMP/PKA‐mediated lipolysis and FAO resulting in adipose tissue loss. 32 , 33 , 34 Recently, we reported that Bscl2 −/− mice develop hypertrophic cardiomyopathy with reduced cardiac steatosis. 35 Despite the relatively low expression of BSCL2 in murine hearts, whether BSCL2 plays a cell‐autonomous role in modulating cardiac lipid metabolism and function has not been fully addressed.
The present study highlights a new form of metabolic cardiomyopathy caused by deletion of BSCL2 in hearts. Cardiomyocyte‐specific deletion of BSCL2 enhanced ATGL expression and FAO, resulting in markedly reduced cardiac lipid reserve, associated with compromised ATP production and contractile dysfunction. Inhibition of FAO or supplying FAs by high‐fat diet (HFD) feeding partially alleviated cardiac energetic stress and augmented contractile function. These findings improve our understanding of how perturbations in lipid utilisation/storage contribute towards HF development and provide preclinical insights into metabolic treatment of HF.
2. MATERIALS AND METHODS
2.1. Mice
Cre+;Bscl2f/f mice (i.e., Bscl2cKO ) were generated by breeding Bscl2f/f mice 18 with transgenic mice expressing Cre recombinase under the cardiac‐specific alpha myosin‐heavy chain 6 (Myh6) promoter (JAX Cat#:011038). 36 All mice were under C57BL/6J background. The Myh6‐Cre mice (i.e., Cre+;Bscl2w/w ) were included as controls when assessing cardiac and mitochondrial functions. Cre−;Bscl2f/f mice were used as controls (Ctrl) for the majority of the studies. Atgl+/–;Bscl2cKO mice were generated by breeding heterozygous Atgl+/– mice (B6;129P2‐Pnpla2tm1Rze/J, Jackson stock #: 019003) with Bscl2cKO mice. Littermates generated from Atgl+/–;Bscl2f/f and Atgl+/–;Bscl2cKO mating [i.e., Atgl+/+;Bscl2f/f ;Myh6‐Cre− (Ctrl), Atgl+/+;Bscl2f/f;Myh6‐Cre+ (BcKO ) and Atgl+/–;Bscl2f/f;Myh6‐Cre+ (AhBcKO )] were used for all experiments. Most experiments were performed in ad libitum male mice with at least two independent cohorts. Some experiments were also repeated in female mice. To inhibit mitochondrial β‐oxidation, 6‐month‐old Ctrl and Bscl2cKO mice were i.p. injected with PBS or trimetazidine (TMZ) at 15 mg/kg/day for 6 weeks, a dose that does not induce whole‐body insulin resistance. 37 Ctrl and Bscl2cKO mice were fed a 60% HFD (Research Diets; D12492) starting at 3 months of age for a duration of 3 months. Most experiments were performed in ad libitum male mice and repeated in female mice. Body compositions were measured using a Bruker small animal NMR system (Bruker minispec LF90II). Transthoracic 2D and M‐mode echocardiography was performed with a VisualSonics Vevo 2100 equipped with a 30 MHz probe (VisualSonics). Mice were initially anaesthetised with a 3% isoflurane, followed by maintenance at 1–2%. Cardiac gravimetric and histological measurements were performed as described elsewhere. 35 All mice were housed in the central animal facility with room temperature controlled at 21°C, and an artificial 12 h:12 h light:dark cycle (lights on at 06:00 am). Mice were directly sacrificed by cervical dislocation and hearts were rapidly excised. In ex vivo perfused working heart experiments, hearts were rapidly excised following anaesthesia via intraperitoneal ketamine/xylazine (80/10 mg/kg) injection.
2.2. Myocardial substrate utilisation and contractile function in isolated working mouse hearts
All hearts from 3‐month‐old male Ctrl and Bscl2cKO mice were prepared and perfused in the working mode (non‐recirculating manner) for 30 min with a preload of 12.5 mmHg and an afterload of 50 mmHg as described. 38 Standard Krebs–Henseleit buffer was supplemented with 8 mM glucose, 0.4 mM oleate conjugated to 3% BSA (fraction V, FA‐free; dialysed), 10 μU/ml insulin (basal/fasting concentration), 0.05 mM l‐carnitine and 0.13 mM glycerol. Metabolic fluxes were assessed through the use of distinct radiolabelled tracers: (1) [U‐14C]‐glucose (0.12 mCi/L from MP Biomedicals; glucose oxidation) and (2) [9, 10–3H]‐oleate (0.067 mCi/L from Sigma–Aldrich; β‐oxidation). Measures of cardiac metabolism (e.g., oleate and glucose oxidation and oxygen consumption) and function (e.g., cardiac power) were determined. At the end of the perfusion period, hearts were snap‐frozen and stored at −80°C prior to analysis. Data were presented as steady‐state values (i.e., the mean of the last two time points during a distinct perfusion condition for each individual heart).
2.3. Lipidomic analysis by high‐resolution/accurate mass spectrometry and tandem mass spectrometry
Total lipids from frozen ventricles were extracted and resuspended in isopropanol:methanol:chloroform (4:2:1, v:v:v) containing 20 mM ammonium formate followed by untargeted lipidomic analysis. Relative quantification of abundances between samples was performed by normalising target lipid ion peak areas to the PC (14:0/14:0) internal standard followed by normalisation to tissue weights as previously described. 35
2.4. FA and glucose oxidation assays
FAO and glucose oxidation reaction assays with LV homogenates were prepared and carried out as detailed previously. 39
2.5. Mitochondrial isolation and measurement of mitochondrial respiration
Fresh ventricles were isolated and minced for mitochondrial isolation and measurement of mitochondrial respiration by XFe24 Extracellular Flux Analyzer (Seahorse Bioscience) as previously described. 34 See Supplemental Materials and Methods for details.
2.6. Tissue ATP measurement
ATP content was determined by using ATP Bioluminescent Assay Kit (FL‐AA; Sigma–Aldrich, Saint Louis, MO, USA) according to the manufacturer's procedure. Briefly, frozen heart tissues were homogenised in cold 10% trichloroacetic acid buffer, centrifuged at 5000×g for 10 min at 4°C followed by neutralisation with 50 mM Tris–acetate (pH 7.8). The ATP content was then determined by a microplate reader with luminescence luminometer (FLUOstar Omega; BMG Labtech). Data were normalised to tissue weight.
2.7. RNA isolation and quantitative real‐time PCR
Total RNA was extracted with Trizol Reagent (Thermo Fisher) and reverse‐transcribed using MLV‐V reverse transcriptase using random primers (Invitrogen). Real‐time quantitative RT‐PCR was performed on the Strategene MX3005 system. Data were normalised to two housekeeping genes (Actb and 36B4) based on Genorm algorithm (medgen.ugent.be/genorm/) and expressed as fold changes. All tissue gene expression studies were performed in non‐fasted mice. qRT‐PCR primers were listed in Table S1.
2.8. Immunoblotting
Protein extraction, determination of protein concentrations, Western blotting and quantification were performed as previously described. 35 Specific antibodies were listed in Supplemental Materials.
2.9. Statistical analysis
Quantitative data were presented as means ± SEM. Statistical comparisons were made by using unpaired t‐test, one‐way ANOVA or two‐way ANOVA followed by Tukey's post‐hoc tests or multiple t‐tests after correction using the Holm–Sidak method using the built‐in statistics of GraphPad Prism 9 software. The number of independent biological replicates is indicated as n in the figure legends. ANOVA F‐test was used to check homogeneity of variance, and Shapiro–Wilk test was used to check normality before unpaired t‐test using GraphPad Prism 9 software. A p value of less than .05 was considered statistically significant.
Additional methodological details are included in the Supplemental Materials.
3. RESULTS
3.1. Myocardial deletion of BSCL2 induces systolic heart dysfunction with dilation
Previously, we demonstrated lipodystrophic Bscl2 −/− mice develop mild hypertrophic cadiomyopathy. 35 In order to interrogate the specific role of cardiac BSCL2, we generated a mouse model with a cardiomyocyte‐specific deletion of BSCL2 (Bscl2cKO ) via Myh6‐Cre. Gene expression analysis confirmed an approximate 75% reduction of Bscl2 specifically in cardiac muscle but not in liver, skeletal muscle and epididymal white adipose tissue (eWAT) of Bscl2cKO mice (Figure S1(A)). Cardiac‐specific deletion of Bscl2 also resulted in no change in body fat and lean masses (Figure S1(B)). We were not able to confirm the cardiac deletion of BSCL2 at protein level as no antibody is sensitive enough to detect the low‐abundance endogenous BSCL2 protein in murine hearts.
We next determined the impact of cardiomyocyte‐specific deletion of Bscl2 on cardiac function, in comparison with two distinct control groups [Ctrl and Cre+;Bscl2w/w ]. No changes were observed in total body weights (Table S2), ventricle mass in proportion to body weight (Figure 1(A)) or tibia length (Figure S1(C)) at either 3 or 6 months of age between experimental groups. Three‐month‐old mice also did not display noticeable changes in left ventricular functions between the three experimental groups (Figures 1(B)–1(H)). Six‐month‐old Cre+,Bscl2w/w mice demonstrated no differences in left ventricle posterior wall thickness at systole (LVPWs) or diastole (LVPWd) (Figures 1(B) and 1(C)), left ventricle anterior wall thickness at systole (LVAWs) (Figure 1(D)) or diastole (LVAWd) (Table S2) and left ventricle internal diameter at systole (LVIDs) or diastole (LVIDd) (Figures 1(E) and 1(F)). However, they exhibited a minor reduction in ejection fraction and fractional shortening compared with Ctrl mice (Figures 1(G) and 1(H)), in agreement with a previous report. 40 In contrast, 6‐month‐old Bscl2cKO mice exhibited decreased wall thickness, especially at systole (Figures 1(B)–1(D)) and increased dilation at both systoles and diastoles (Figures 1(E), 1(F), and 1(I)), along with marked reductions in contractile function compared with Ctrl and Cre+,Bscl2w/w mice (Figures 1(G) and 1(H)). To further pinpoint a cell‐autonomous role of BSCL2 in mediating cardiac function, we also compared echocardiography in 5‐month‐old mice, an age when the cardiac function of Myh6‐Cre+ (Cre+,Bscl2w/w ) mice is not compromised due to prolonged Cre expression. Not surprisingly, while the wall thicknesses of 5‐month‐old Bscl2cKO mice were marginally reduced, these mice still demonstrated significantly reduced contractile function accompanied with increased dilation compared to both Ctrl and Cre+, Bscl2w/w mice (Figures 1(B)–1(H)). In addition, we observed a greater induction of brain natriuretic peptide (Nppb) and growth differentiation factor 15 (Gdf15), and reduction of adult Myh6 gene expression, in 6‐month‐old Bscl2cKO hearts versus Ctrl hearts (Figure 1(J)). The BSCL2 deletion‐induced cardiac dysfunction was not accompanied by abnormal cardiomyocyte morphology and excessive myocardial fibrosis (assessed by trichrome staining) in hearts of Bscl2cKO mice (Figures S1(D) and S1(E)). Together, these data suggest that cardiomyocyte‐specific BSCL2 deletion leads to systolic heart dysfunction independent of the long‐term expression of transgene Myh6‐Cre.
FIGURE 1.

Mice with cardiac‐specific deletion of BSCL2 develop systolic heart dysfunction with dilation. (A) Ventricle weight (VW) normalised to body weight (BW) in 3‐month‐old (3 M), 5‐month‐old (5 M) and 6‐month‐old (6 M) male Cre‐;Bscl2f/f (Ctrl), Cre+;Bscl2w/w , and Cre+;Bscl2f/f (cKO) mice. 3 M old: Ctrl, n = 6; Cre+;Bscl2w/w , n = 5; cKO, n = 6. 5 M old: Ctrl, n = 6; Cre+;Bscl2w/w , n = 6; cKO, n = 6. 6 M old: Ctrl, n = 6; Cre+;Bscl2w/w , n = 6; cKO, n = 9. (B and C) left ventricle post wall thickness at end systole (LVPWs, mm) and at end diastole (LVPWd, mm), respectively; (D) left ventricle anterior wall thickness at end systole (LVAWs, mm); (E and F) left ventricle internal diameter at end systole (LVIDs, mm) or end diastole (LVIDd, mm); (G) ejection fraction (%) and (H) fractional shortening (%) in 3 M, 5 M and 6 M old male mice. 3 M old: Ctrl, n = 8; Cre+;Bscl2w/w , n = 8; cKO, n = 12. 5 M old: Ctrl, n = 6; Cre+;Bscl2w/w , n = 8; cKO, n = 10. 6 M old: Ctrl, n = 11; Cre+;Bscl2w/w , n = 8; cKO, n = 9. (I) Representative echocardiography, and (J) qRT‐PCR analysis of cardiac stress genes in ventricles of 6‐month‐old male mice. n = 6 per group. *, p < .05; **, p < .005. Two‐way ANOVA followed by Tukey's post‐hoc tests
3.2. Myocardial‐specific deletion of BSCL2 causes elevated TG turnover and FAO preceding functional decline
It remains to be determined whether loss of BSCL2 in cardiomyocytes directly reduces cardiac TG contents as we have observed in lipodystrophic Bscl2 −/− hearts. 35 Indeed, we found reduced ventricular TG content (by 57%) (Figure 2(A)), increased protein expression of ATGL (≈2.5‐fold), but not HSL (Figure 2(B)), in 3‐month‐old Bscl2cKO mice compared with Ctrl mice. Changes in ATGL protein expression were not attributed to alterations in the mRNA level of Pnpla2, the gene encoding ATGL (Figure S2(A)). Isolated adult Bscl2cKO cardiomyocytes displayed similar upregulation of ATGL, but not HSL, compared with Ctrl cells (Figures 2(C) and 2(D)). Ctrl and Bscl2cKO cardiomyocytes responded similarly to the stimulation of isoproterenol in terms of PKA‐mediated phospholamban (PL) phosphorylation (Figure S2(B)). Analysis of whole heart lysates also revealed comparable basal PKA‐mediated substrate phosphorylation between Ctrl and Bscl2cKO mice (Figure S2(C)). These data suggest that cardiac‐specific deletion of BSCL2 did not affect cAMP/PKA signalling but induced posttranscriptional ATGL upregulation in hearts, different from what we previously observed in BSCL2‐deleted adipose tissue. 32 , 33 , 34
FIGURE 2.
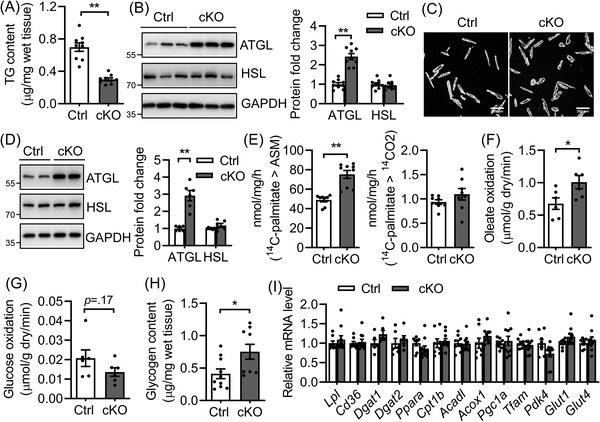
Cardiac‐specific deletion of BSCL2 induces cardiac triglyceride turnover and excessive fatty acid oxidation. (A) Ventricular TG content as normalised to wet tissue weight. n = 9 per group. (B) Representative Western blotting and quantification of lipolytic proteins in heart homogenates. n = 9 per group. (C and D) Isolated adult cardiomyocytes, representative Western blotting in duplicates and quantification from three independent experiments. (E) Acid soluble metabolites (ASM) and CO2 production after incubating heart crude mitochondrial fraction with 14 C‐palmitate. Ctrl, n = 8; cKO, n = 9. (F) Oleate oxidation rate; (G) glucose oxidation rate in ex vivo perfused working hearts. n = 6 per group. (H) Glycogen content. Ctrl, n = 10; cKO, n = 9. (I) qRT‐PCR analysis of genes involved in fatty acid metabolism, mitochondrial biogenesis and glucose metabolism. Ctrl, n = 8; cKO, n = 10. All experiments used 3‐month‐old male Cre−;Bscl2f/f (Ctrl) and Bscl2cKO (cKO) mice. *, p < .05; **, p < .005, unpaired t‐tests (parametric)
Higher ATGL‐mediated lipolysis is coupled with cardiac FAO. 6 When incubating heart homogenates with 14C‐palmitate, 3‐month‐old Bscl2cKO mice demonstrated elevated release of 14C‐labelled acid soluble metabolites despite a lack of change in 14CO2 production (Figure 2(E)). Ex vivo perfusions in working hearts from 3‐month‐old male Ctrl and Bscl2cKO mice identified no differences in heart rates, cardiac oxygen consumption, cardiac efficiency as well as cardiac power between two genotypes (Figures S2(D) and S2(G)), suggesting maintained cardiac function at this age. However, the Bscl2cKO hearts demonstrated a 33% increase in oleate oxidation (Figure 2(F)), concomitant with a tendency of lower glucose oxidation (Figure 2(G)) and an 83% increase of baseline glycogen content (Figure 2(H)). In spite of enhanced FAO and glycogen accumulation, Bscl2cKO hearts demonstrated no changes in the expression of genes involved in FA uptake (e.g., Lpl, Cd36), TG synthesis (e.g., Dgat1, Dgat2), mitochondrial and peroxisomal β‐oxidation (e.g., Pparα, Cpt1β, Acadl, Acox1), mitochondrial biogenesis (e.g., Pgc1α, Tfam) and glucose metabolism (e.g., Pdk4, Glut1, Glut4) (Figure 2(I)). Lack of changes in mitochondrial biogenesis was further confirmed by the similar ratios of mitochondrial DNA to nuclear DNA (Figure S2(H)) and protein levels of each of the electron transport chain (ETC) complexes between heart lysates of 3‐month‐old Ctrl and Bscl2cKO mice (Figure S2(I)). Together, these data clearly suggest that cardiomyocyte‐specific BSCL2 deficiency results in higher rates of cardiac TG turnover and FAO independent of transcriptional changes of mitochondrial and extra‐mitochondrial metabolic genes.
3.3. Chronic derangements in myocardial FAO leads to massive lipid remodelling and reduced endogenous substrates in Bscl2cKO hearts
To identify mechanisms underlying the development of HF, we performed untargeted lipidomic analyses of ventricles from 6‐month‐old Ctrl and Bscl2cKO mice. Total normalised lipid ion abundances identified in Bscl2cKO hearts were reduced by about 55% (Figure 3(A)). Heatmap analysis revealed massive reductions in the absolute levels of five broadly classified lipid classes defined by the Lipid MAPS consortium; that is, glycerophospholipids, fatty acyls [mainly non‐esterified fatty acids (NEFA)], sphingolipids, sterol lipids and glycerolipids in Bscl2cKO mice (Figure 3(B)). When comparing the % distributions of these five lipid classes, the proportions of glycerophospholipids, sphingolipids and sterol lipids were significantly higher in Bscl2cKO hearts compared with Ctrl hearts (Figure 3(C)). The proportions of NEFA were relatively comparable between two genotypes, whereas the proportion of glycerolipids was markedly reduced by 58% in Bscl2cKO hearts (Figure 3(C)). Analysis of the absolute levels of glycerolipids revealed 78%, 50% and 75% reductions in TG, diacylglycerol (DAG) and monoacylglycerol (MAG), respectively, in Bscl2cKO hearts (Figure 3(D)). The absolute levels of NEFA and total acylcarnitines (ACs) were significantly lower in Bscl2cKO hearts (Figure 3(E)). Specifically, the abundances of the major long‐chain ACs (AC16:0, AC16:1, AC18:0, AC18:1, AC18:2) were all reduced by approximately 70% (Figure 3(F)). These data suggest myocardial BSCL2 deletion results in a dramatic remodelling of lipid compositions highlighted by reduced levels of energy providing substrates indicative of impairment of cardiac metabolic reserve. Furthermore, only Bscl2cKO hearts displayed reduced cardiac TG contents (Figure S3(A)) and increased ATGL expression (Figure S3(B)), and no differences were observed between Ctrl and Cre+;Bscl2w/w hearts. The expression of PPARα and its target proteins (CD36, CPT1β) were also similar between three genotypes (Figure S3(B)). Collectively, these data emphasise a BSCL2‐specific regulation of lipid remodelling in Bscl2cKO hearts independent of transcriptional activation of PPARα.
FIGURE 3.

Bscl2cKO mice develop massive cardiac lipid remodelling and exhibit reduced metabolic reserve. (A) The total lipid ion abundance normalised to tissue weight. (B) Heatmap of lipid species including glycerophospholipids [i.e., phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), phosphatidylglycerol (PG), cardiolipin (CL), phosphatidic acid (PA) and acylcarnitines (AC)], non‐esterified free fatty acids (NEFA), sphingolipids, sterols and glycerolipids based on Z‐scores calculated from the summed ion abundances normalised to tissue weight. (C) Pie chart representing the proportional (%) distribution of summed ion abundances of glycerolipid, glycerophospholipid, sphingolipid, NEFA and sterol lipid classes. (D–F) Comparison of the total normalised ion abundances for (D) glycerolipids including TG, DAG and MAG, (E) NEFA and ACs, and (F) specific AC and hydroxyl acylcarnitines (OHAC) species. Global lipidomic analysis of ventricles by shotgun mass spectrometry was performed in non‐fasting 6‐month‐old male Cre−;Bscl2f/f (Ctrl) and Bscl2cKO (cKO) mice. n = 4 per group with each pooled from three animals. *, p < .05; **, p < .005, unpaired t‐tests (nonparametric)
3.4. Cardiomyopathy in aged Bscl2cKO mice is associated with energy deficiency independent of overt mitochondrial dysfunction and oxidative stress
We next examined whether the massive lipid remodelling exerts maladaptive effects on mitochondrial function, leading to the progressive development of HF when Bscl2cKO mice age. Transmission electron microscopy images revealed a complete lack of LDs in 6‐month‐old Bscl2cKO hearts, in support of cardiac TG reduction (Figure 4(A)). Sarcomere arrangement, mitochondrial morphology and sizes, as well as mitochondria distribution were generally preserved in both Ctrl and Bscl2cKO hearts (Figure 4(A)). Respirational analysis of isolated mitochondria from hearts of Ctrl and Bscl2cKO mice identified similar basal (in the presence of exogenous succinate and rotenone) and maximal oxygen consumption rates, as well as coupled (ADP‐driven, Complex V) and uncoupled respiration (Figures 4(B)–4(C)). Interestingly, the activity of Complex I (CI) was significantly increased, whereas the activities of CII and CIII trended higher in Bscl2cKO mitochondria compared with those from Ctrl or Cre+;Bscl2w/w mice (Figure 4(D)). Again, the expression of marker proteins per mitochondrial mass for each mitochondrial ETC complex remained comparable (Figure 4(E)), which was also the case in the whole hearts of 6‐month‐old Bscl2cKO mice (Figure S4(A)). There were no differences in the expressions of PGC1α and mitochondrial stress and uncoupling proteins (Prohibitin, UCP2 and UCP3) in Bscl2cKO hearts compared to both Ctrl and Cre+;Bscl2w/w hearts (Figure S4(A)). Six‐month‐old Bscl2cKO hearts still possessed a tendency of higher capacity to oxidise exogenous palmitate relative to Ctrl hearts (Figure 4(F)); while its glucose oxidation rate trended higher (Figure 4(G)), resulting in a tendency toward lower cardiac glycogen content in these mice (Figure 4(H)). Oxidation of 2′, 7′‐dichlorofluorescein diacetate was not augmented in heart extracts of 6‐month‐old Bscl2cKO mice (Figure S4(B)). The level of lipid peroxides malondialdehyde was even reduced in Bscl2cKO hearts compared with Ctrl hearts (Figure S4(C)). Consistent with lack of oxidative stress, there were no differences in the expression of SOD1, SOD2 and Catalase in Bscl2cKO hearts relative to both Ctrl and Cre+;Bscl2w/w hearts (Figure S4(A)). Furthermore, genes regulating glutathione metabolism (e.g., Gclc, Gsr, Gss) were largely unaltered although GPX3 expression showed a trend toward increase in both Cre+;Bscl2w/w and Bscl2cKO hearts compared with Ctrl hearts (Figure S4(D)). These data suggest that mitochondrial dysfunction and oxidative stress are unlikely to play roles in maladaptive cardiac remodelling and progression of HF in Bscl2cKO mice.
FIGURE 4.
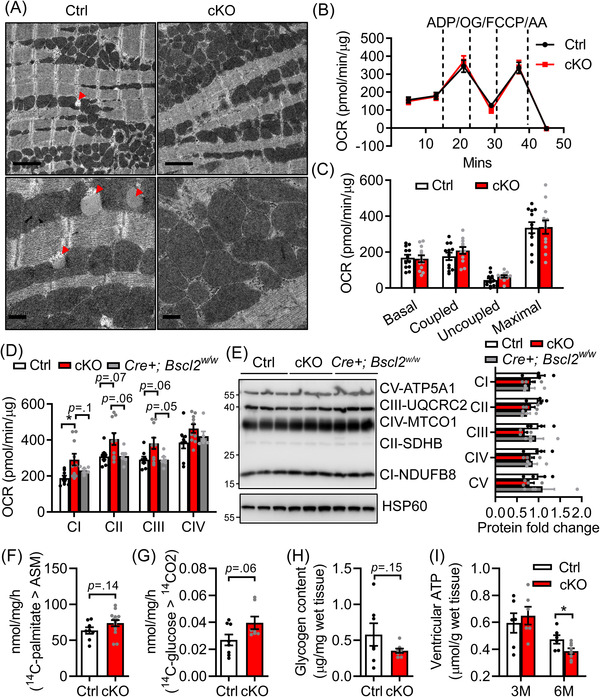
Cardiac dysfunction in Bscl2cKO mice is associated with bioenergetics failure independent of mitochondrial respiratory dysfunction. (A) Representative transmission electron micrographs of ventricles from 6‐month‐old Cre−;Bscl2f/f (Ctrl) and Bscl2cKO (cKO) mice. Red arrowheads: lipid droplets (LDs). Upper panels: scale bar = 2 μm; lower panels: scale bar = 0.5 μm. n = 3 per group. (B) Succinate/rotenone‐driven mitochondrial oxygen consumption rates (OCR) were measured in isolated mitochondria from hearts of 6‐month‐old Ctrl and cKO mice by Seahorse XF24 analyser with sequential addition of ADP, oligomycin (OG), FCCP and antimycin A (AA). Basal, coupled, uncoupled and maximal mitochondrial respiration were shown in (C). n = 4 in triplicates per group. Two‐way ANOVA followed by Tukey's post‐hoc tests. (D) Electron flow assays measuring complex I–IV activities (n = 3 in triplicates) and (E) representative Western blotting and quantification in isolated mitochondria from ventricles of 6‐month‐old Ctrl, Bscl2cKO , Cre+;Bscl2w/w mice. n = 4 per group. Mitochondrial HSP60 was used as loading control. (F) Acid‐soluble metabolites (ASM) after incubating heart crude mitochondrial fraction with 14 C‐palmitate. Ctrl, n = 4 in duplicates; cKO, n = 6 in duplicates. (G) Glucose oxidation rate after incubating heart crude mitochondrial fraction with 14 C‐glucose. n = 6 per group. (H) Ventricular glycogen contents, n = 7 per group. Panels F and G were performed in 6‐month‐old Ctrl and cKO mice. Unpaired t‐tests (parametric). (I) ventricular ATP contents in non‐fasting 3 and 6‐month‐old male Ctrl and Bscl2cKO mice. 3 M: Ctrl, n = 6; cKO, n = 7, 6 M: Ctrl, n = 6; cKO, n = 8. *, p < .05, multiple unpaired t‐tests with Holm–Sidak method
While the ventricular ATP content was maintained in 3‐month‐old Bscl2cKO mice, it was significantly lower when Bscl2cKO mice reached 6‐month‐old (Figure 4(I)), in line with the reduced cardiac metabolic reserve (i.e., lipid and glycogen stores). Furthermore, there were no differences in the plasma concentrations of glucose and lipid substrates (TG, cholesterol, NEFA and glycerol) between 6‐month‐old Ctrl and Bscl2cKO mice (Table S3). These data suggest that deprivation of myocellular endogenous substrates is likely responsible for the progressive metabolic transition leading to cardiac energetic and contractile failure in Bscl2cKO mice.
3.5. Inhibition of cardiac TG lipolysis or FAO partially rescues cardiac dysfunction in Bscl2cKO mice
Since cardiac dysfunction in Bscl2cKO mice is associated with higher ATGL expression and FAO, we first generated mice with partial loss of ATGL in Bscl2cKO mice (AhBcKO ) (Figure S5(A)). ATGL haploinsufficiency indeed abolished cardiac ATGL upregulation in BcKO mice, with only 50% myocardial ATGL present compared with Ctrl hearts (Figure S5(B)). AhBcKO mice showed no alterations in body weights, and VW to TL ratios when compared with Ctrl and BcKO mice (Figure S5(C) and Table S4). Partial ATGL loss neither significantly restored the wall thicknesses nor reduced dilation of BcKO mice (Figure S5(D) and Table S4). However, the cardiac contractile function was markedly improved in AhBcKO mice when compared with BcKO mice, although still impaired relative to Ctrl mice (Figures S5(E) and S5(F), respectively). Partial deletion of ATGL led to a minimal elevation of ventricular TG accumulation (Figure S5(G)). These data suggest cardiac ATGL upregulation indeed contributes to the development of cardiac dysfunction in Bscl2cKO mice.
To further dissect whether elevated FAO is involved in the pathogenesis of HF in Bscl2cKO mice, we treated mice with TMZ, a specific 3‐Ketoacyl‐CoA thiolase (3‐KAT) inhibitor that inhibits FAO, 37 for up to 6 weeks starting at 6‐month‐old when Bscl2cKO mice already developed cardiac dysfunction (Figure 5(A)). TMZ did not alter body weight (Figure S6(A)) nor liver weight (Figure S6(B)), but tended to increase white fat mass in both genotypes (Figure S6(C)). TMZ elevated circulating cholesterol levels in both Ctrl and Bscl2cKO mice, but there were no differences in plasma glucose, NEFA, glycerol or TG levels in the experimental groups (Table S5). Ctrl and Bscl2cKO mice also demonstrated similar ventricle to body weight (Figure S6(D)) and tibia length (Figure 5(B)) ratios after TMZ treatment. TMZ produced no changes in cardiac dilation and contractile function of Ctrl mice. However, it was able to significantly improve LVPWs and cardiac contractile function (including increasing fractional shortening by 20.6%) without notably normalising dilatation in Bscl2cKO hearts (Figures 5(C)–5(E) and Table S5). Attenuation of cardiac dysfunction by TMZ was also evident in female Bscl2cKO mice (Figure S6(E)–S6(H)).
FIGURE 5.
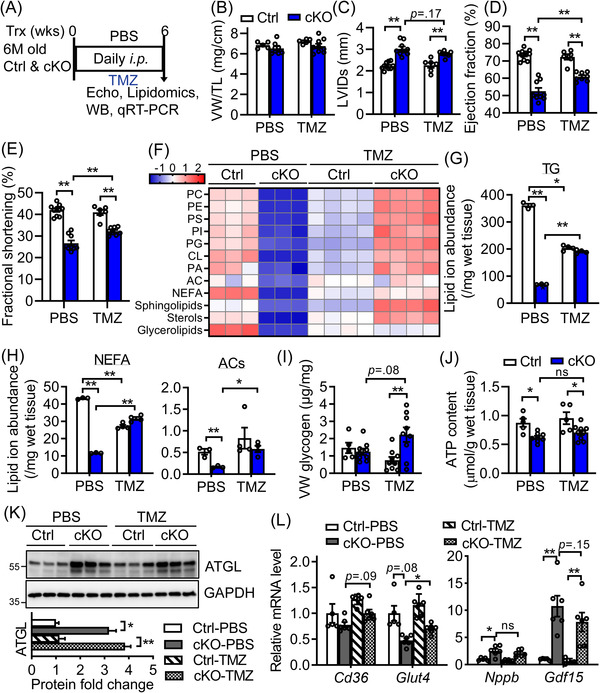
Inhibition of fatty acid oxidation partially improves metabolic reserve and cardiac function in Bscl2cKO mice. (A) 6‐month‐old male Cre−;Bscl2f/f (Ctrl) and Bscl2cKO (cKO) mice received daily i.p. injection with PBS or trimetazidine (TMZ) at 15 mg/kg body weights (BW) for a total of 6 weeks. (B) Ratio of ventricle weight (VW) to tibia length (TL). Ctrl, n = 5; cKO, n = 9. (C) Left ventricular internal diameter in end systole (LVIDs); (D) ejection fraction and (E) fractional shortening. PBS‐Ctrl, n = 10; PBS‐cKO, n = 9. n = 9 per TMZ‐treated group. (F) Heatmap of major lipid species based on Z‐score calculated from the summed ion abundances normalised to milligram of wet tissue; (G and H) comparison of the total normalised ion abundances for (G) glycerolipids as well as (H) NEFA and ACs. n = 3 per PBS‐treated group. n = 4 per TMZ‐treated group. Each sample was pooled from three animals. (I) Ventricular glycogen. PBS‐Ctrl, n = 5; PBS‐cKO, n = 9. n = 9 per TMZ‐treated group. (J) Ventricular ATP contents normalised to gram of wet tissue. PBS‐Ctrl, n = 5; PBS‐cKO, n = 8. TMZ‐Ctrl, n = 5, TMZ‐cKO, n = 9. (K) Representative Western blotting and quantification of total cell extracts from ventricles of 6‐month‐old PBS and TMZ‐treated Ctrl and Bscl2cKO mice. n = 6 per group. Data were normalised to GAPDH with PBS‐treated Ctrl set to 1. Two independent experiments. (L) qRT‐PCR analysis of lipid and glucose transport genes and cardiac stress genes in ventricles of 6‐month‐old PBS and TMZ‐treated male mice. n = 6 per group. *, p < .05; **, p < .005. ns: not significant. Two‐way ANOVA followed by Tukey's post‐hoc tests
We performed another independent set of untargeted lipidomics to examine the effect of TMZ on cardiac lipidome. As indicated in Figure 5(F), PBS‐treated Bscl2cKO hearts recapitulated all changes in cardiac lipid contents as we previously observed in 6‐month‐old Bscl2cKO hearts (Figure 3). TMZ unexpectedly reduced the absolute abundances of cardiac lipids in all categories except ACs in Ctrl hearts. In contrast, TMZ dramatically enhanced the accumulation of these lipid classes in Bscl2cKO hearts relative to PBS‐treated Bscl2cKO hearts. Especially, the absolute levels of phospholipids, sphingolipids, and sterols were even greater in TMZ‐treated Bscl2cKO than PBS‐treated Ctrl hearts (Figure 5(F)). Specifically, TMZ markedly increased TG, NEFAs and ACs in Bscl2cKO hearts (Figures 5(G) and 5(H)). While it slightly lowered glycogen in Ctrl hearts, it caused more glycogen accumulation in Bscl2cKO hearts (Figure 5(I)). Despite a significant upregulation of metabolic reserve, there was only a very minimal but non‐significant improvement of ventricular ATP content in TMZ‐treated Bscl2cKO hearts (Figure 5(J)). As expected, TMZ itself did not abolish ATGL upregulation in Bscl2cKO hearts, since it acts on FAO downstream of ATGL (Figure 5(K)). Downregulation of lipid and glycogen contents in TMZ‐treated Ctrl heart could not be explained by similar expression levels of Cd36 and Glut4, two key genes governing cardiac lipid and glucose entries. However, we did observe a slight upregulation of these two genes in TMZ‐treated compared to PBS‐treated Bscl2cKO hearts, in line with their elevated lipid and glycogen contents (Figure 5(L)). Furthermore, there was also a trend for TMZ to attenuate the gene expression of cardiac stress markers in Bscl2cKO hearts (Figure 5(L)). Together, our data suggest that inhibiting FAO in Bscl2cKO hearts is able to partially restore cardiac function, potentially through modifying cardiac metabolism.
3.6. Preventative treatment of HFD significantly attenuates cardiac bioenergetics deficiency and dysfunction in Bscl2cKO mice
To gain further insight into the importance of metabolic reserve in energy‐starved Bscl2cKO hearts, we also fed 3‐month‐old Ctrl and Bscl2cKO male mice a normal chow diet (NCD) or a 60% HFD for a period of 3 months (Figure 6(A)). By 6 months old, Ctrl and Bscl2cKO mice exhibited similar obese phenotype with comparable weight gains after HFD feeding (Table S3). They exhibited similarly higher levels of plasma glucose, cholesterol, NEFA and glycerol relative to their NCD‐fed counterparts, and there were no differences in serum TG concentrations in all four groups (Table S3). HFD increased comparable cardiac hypertrophy as evidenced by ventricle weight to tibia length ratios and a tendency of increase in wall thickness in both genotypes (Figure 6(B) and Table S3). There were no significant changes in dilation and contractile function in Ctrl mice after 3 months of HFD feeding (Figures 6(C) and 6(D) and Table S3). However, HFD was able to increase wall thickness (LVPWs) and improve cardiac contractile function in Bscl2cKO mice to the levels similar to HFD‐fed Ctrl mice despite exerting no effect on dilation (Figures 6(C) and 6(D) and Table S3). Interestingly, in Ctrl hearts, we found HFD reduced the amounts of phospholipids (i.e., PC, PS, PI, PG, CL and PA) and sphingolipids, but preferentially increased the levels of sterols and glycerolipids when compared with NCD (Figure 6(E)). The amounts of almost all lipid classes were increased in Bscl2cKO hearts after HFD feeding, albeit still lower than HFD‐fed Ctrl hearts (Figure 6(E)). Specifically, TG levels were greatly upregulated in both Ctrl and Bscl2cKO hearts after HFD feeding, whereas the levels of NEFA and ACs in Bscl2cKO hearts were increased to a lesser extent by HFD when compared with NCD (Figures 6(F) and 6(G)). Nevertheless, while Bscl2cKO hearts contained less ATP under NCD, the ventricular ATP contents in HFD‐fed Bscl2cKO mice was partially recovered to the levels of HFD‐fed Ctrl hearts, suggesting improved cardiac energetics (Figure 6(H)). Immunoblot analysis revealed similar upregulation of ATGL in Bscl2cKO relative to Ctrl hearts regardless of diets (Figure 6(I)). Collectively, HFD could restore cardiac function by improving cardiac energetics via increasing energy supply in Bscl2cKO mice.
FIGURE 6.

High‐fat diet feeding increases metabolic reserve and prevents cardiac dysfunction in Bscl2cKO mice. (A) 3‐month‐old Bscl2f/f (Ctrl) and Bscl2cKO (cKO) mice were fed with normal chow diet (NCD) or high‐fat diet (HFD, 60% fat calories) for a total of 3 months (M). (B) Ratio of ventricle weight (VW) to tibial length (TL). NCD‐Ctrl, n = 6; NCD‐cKO, n = 7; HFD‐Ctrl, n = 7; HFD‐cKO, n = 11. (C) Left ventricular internal diameter in end systole (LVIDs); and (D) fractional shortening. NCD‐Ctrl, n = 9; NCD‐cKO, n = 12; HFD‐Ctrl, n = 12; HFD‐cKO, n = 16. (E) Heatmap of major lipid species based on Z‐score calculated from summed ion abundances normalised to milligram of wet tissue; (F and G) comparison of the total normalised ion abundances for (F) glycerolipids and NEFA as well as (G) ACs. n = 3 per NCD group. n = 4 per HFD group. Each sample was pooled from three animals. (H) Myocardial ATP content as normalised to gram of wet tissue. NCD‐Ctrl, n = 8; NCD‐cKO, n = 8; HFD‐Ctrl, n = 7; HFD‐cKO, n = 11. (I) Representative Western blotting of total heart extracts from ventricles of Ctrl and Bscl2cKO mice. n = 3 per group. Two independent experiments. *, p < .05; **, p < .005. Two‐way ANOVA followed by Tukey's post‐hoc tests
4. DISCUSSION
In this study, we show that genetic deletion of BSCL2 in cardiomyocytes leads to dramatic cardiac lipid remodelling and systolic heart dysfunction with dilation in mice. Mechanistically, cardiac BSCL2 ablation causes ATGL overexpression, excessive FAO and overt cardiac lipid remodelling. Increased lipid catabolism ultimately exhausts intramyocellular lipid and glycogen reserve and is likely responsible for the energetic and contractile dysfunction in Bscl2cKO mice. Importantly, inhibiting TG turnover by partial genetic deletion of ATGL or suppressing FAO by promoting substrate switch or HFD feeding via increasing lipid supply can partially mitigate cardiac dysfunction in Bscl2cKO mice (Figure 7). Our results thus identify a novel and indispensable role of BSCL2 in regulating a preferential oxidation of FAs from endogenous cardiac TG lipolysis which governs cardiac energetics and function.
FIGURE 7.

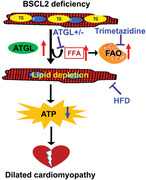
Schematic diagram of the indispensable role of BSCL2 in regulating cardiac lipid metabolism and function. Cardiac deletion of BSCL2 causes ATGL overexpression and excessive fatty acid oxidation (FAO) which exhaust intramyocellular triglyceride (TG) and induce drastic depletion of cardiac lipidome, ultimately resulting in energetic and contractile failure in mice. Partial deletion of ATGL, inhibiting FAO by trimetazidine or increasing lipid supply via high‐fat diet (HFD) feeding replenishes cardiac lipidome and alleviates cardiac dysfunction caused by loss of BSCL2
BSCL2 deletion enhances cAMP/PKA triggered ATGL‐mediated lipolysis and FAO in adipose tissue. 32 , 33 , 34 Similar to global Bscl2 −/− hearts, 35 Bscl2cKO hearts exhibited higher ATGL expression associated with reduced TG contents and accelerated FAO, highlighting the cell‐autonomous role of cardiac BSCL2 in lipid catabolism. Interestingly, lipodystrophic Bscl2−/− mice develop cardiac hypertrophy due to increased basal IGF1 receptor (IGF1R)‐mediated PI3K/AKT signalling secondary to hyperinsulinemia. 35 These mice develop mild cardiomyopathy, which can be attenuated by partially restoring fat mass and/or improving whole‐body and cardiac insulin resistance. 35 , 41 This suggests that insulin resistance largely accounts for the pathophysiology of metabolic cardiomyopathy in lipodystrophic Bscl2 −/− mice, resembling diabetic hearts. Indeed, in patients with congenital generalised lipodystrophy, leptin treatment, which is known to improve metabolic complications including insulin resistance, has been recently shown to attenuate cardiac hypertrophy and increase contractile function. 42 On the other hand, Bscl2cKO mice exhibit neither left ventricular hypertrophy nor functional changes at 3 months old despite an increase in FAO. By 6 months old, these mice tend to have thinner left ventricular wall and develop energy deficit‐induced systolic dysfunction but not end‐stage HF. Such changes occur independent of profound structural remodelling (e.g., hypertrophy and fibrosis) (Figure 1) and insulin resistance in Bscl2cKO hearts (data not shown). Notably, a previous report identified no differences in heart weights of 1‐year‐old mice with cardiac deletion of BSCL2 driven by the same Myh6‐Cre. 43 However, cardiac function was not directly assessed and it remains unknown whether BSCL2 directly controls cardiac function in their model.
Alleviation of cardiac dysfunction by partial ATGL loss in Bscl2cKO mice provides solid evidence that an uncontrolled ATGL‐mediated lipid catabolism plays a part in impairing cardiac performance (Figure S5). ATGL‐mediated fat catabolism has been directly linked to cardiac PGC‐1/PPARα expression and FAO rates. 6 While Bscl2cKO hearts clearly exhibited excessive ATGL‐coupled FAO preceding impaired cardiac function, none of PGC‐1α and PPARα and their downstream target genes were altered (Figure 2). However, lean mice with myocardial ATGL overexpression8, 9 or acetyl‐CoA carboxylase 2 (ACC2) deletion 44 maintain normal cardiac energetics and performance despite higher TG turnover or FAO rates. Thus, other changes in cellular metabolism independent of ATGL‐mediated intramyocardial lipid catabolism may exist to contribute to the cardiac energy deficit and progressive deterioration of cardiac function in Bscl2cKO mice at baseline, which warrants further investigation.
ATGL is known to be ubiquitinated (Ref. 45 and data not shown) and we have previously demonstrated enhanced ATGL stability in Bscl2 −/− cardiomyocytes and mouse embryonic fibroblasts (MEFs). 35 However, we were not able to pull down endogenous ATGL using current available antibodies, which prevents us from examining ATGL ubiquitination in BSCL2‐deleted hearts. Therefore, the molecular events for the posttranscriptional regulation of ATGL in the absence of BSCL2 remain to be identified.
Reliance on FAO in obesity and/or diabetes is correlated with lower cardiac efficiency, impaired mitochondrial respiratory function and increased ROS production. 46 Alteration of the cardiac lipidome may also mediate functional impairment through dampening mitochondrial function. 47 However, none of these abnormalities occurred in our Bscl2cKO mice despite massive reduction of cardiac lipid contents. In fact, mitochondrial function still trends higher even in BSCL2‐deleted hearts with reduced ATP levels. Such finding is seemingly counterintuitive, as it is hard to associate higher myocardial mitochondrial function with reduced ATP level. It is worth to mention that mitochondrial hyperactivation in Bscl2cKO hearts was derived from in vitro or ex vivo assays performed in the presence of excessive exogenous substrates, whereas this may not be the case in vivo. Higher cardiac FAO is normally associated with increased exogenous lipid import as observed in diabetic hearts or hearts with cardiac‐restricted overexpression of PPARα. 48 This was also not true with our Bscl2cKO mice which displayed similar expression of lipid uptake genes and comparable circulating lipid metabolites. Therefore, imbalance of lipid consumption and supply within Bscl2cKO myocardium leads to downregulation of the vast amounts of lipid substrates (Figure 3), contributing to the ATP deficit in Bscl2cKO hearts. In addition, AC levels tightly reflect the FAO rates, and AC profiling has been used to identify FAO dysregulation. 49 Notably, Bscl2cKO hearts demonstrated no alterations in CPT1 expression, suggesting intact carnitine shuttle (Figure S3(B)). The almost unanimous reduction of long‐chain ACs in Bscl2cKO hearts highlights the presence of increased FAO which may eventually deplete mitochondrial ACs thus reducing substrates entering TCA cycles and causing energy deficit. While it is plausible that BSCL2 deletion is still causing constant hydrolysis of the remaining TGs and channelling FAs to mitochondria for combustion in a compartmentalised domain in vivo, the energy status of the whole myocardium is ultimately compromised due to the limited availability of substrates. One limitation of our study is that the amount of myocardial ATP was not directly measured by NMR and probably does not reflect the whole myocardium ATP reserve. Nevertheless, our Bscl2cKO mice constitute as the first animal model that demonstrates excessive myocardial lipid catabolism associated with deterioration of metabolic reserve and cardiac dysfunction in the absence of elevated lipid uptake.
Treatment with TMZ results in an insignificant increase of skeletal muscle TG content and higher plasma AC in HFD‐induced obese mice. 37 However, thus far, no study has examined the effects of TMZ treatment on myocardial lipid and glycogen contents. Surprisingly, we identified TMZ drastically remodels cardiac lipidome by downregulating the lipid contents of almost all lipid classes in normal mouse hearts. Whether this is primary to the cardiac effect of TMZ remains unknown. TMZ improves cardiac function in ischemia/reperfusion injury predominantly by shifting energy production from NEFA to glucose oxidation in the heart. 50 , 51 , 52 , 53 The mechanisms underlying the cardioprotective role of TMZ in our Bscl2cKO mice could not be simply explained by alleviation of energy deficit, as TMZ failed to significantly improve intracellular ATP levels in Bscl2cKO hearts. Conversely, TMZ promotes drastic lipid and glycogen accumulation in metabolic‐stressed Bscl2cKO hearts. The prominence of glycogen in TMZ‐treated Bscl2cKO hearts may reflect enhanced glucose utilisation as glycogen content has also been shown to commensurate with augmented carbohydrate metabolism such as in GLUT1‐overexpressing hearts. 54 Interestingly, aside from glucose utilisation, TMZ has been recently shown to induce cardiac β‐hydroxybutyrate flux to attenuate isopropanol‐induced rat HF. 55 Whether TMZ indeed induces substrate switch to glucose and ketone bodies in Bscl2cKO hearts needs to be further dissected by ex vivo heart perfusion experiments. Meanwhile, it is not clear whether TMZ treatment benefits the chronically dysfunctional myocardium by metabolically acting on heart itself or other organs such as liver, skeletal muscle and adipose tissue to ultimately moderate cardiac substrate utilisation. In addition, whether TMZ inhibits cardiac FAO in vivo still remains controversial, 56 and we also cannot exclude the non‐metabolic effects of TMZ in preventing left ventricle remodelling independent of its inhibitory activity on FAO. 57 , 58 Nevertheless, our study underscores that TMZ has potential in ameliorating cardiac function and slows HF progression in a non‐ischemic model of HF.
HFD alone has been shown to be cardioprotective especially in alleviating energy‐compromised HF. 59 In the present study, we find that decreased cardiac function in Bscl2cKO mice can be attenuated by HFD feeding. Lipidomics study further confirmed improved cardiac metabolic substrates mainly in the form of TG in HFD‐fed Bscl2cKO mice. This was in line with the notion that HFD feeding provides more coronary circulation of substrates to match up the rate of enhanced lipid utilisation thus attenuating the myocardial ATP deficit in the energy‐deprived failing Bscl2cKO hearts. In spite of an improvement in cardiac contractile function, we were not able to observe a significant reduction of cardiac stress markers in HFD‐treated Bscl2cKO mice (not shown). Nonetheless, results from our HFD feeding studies support the cardio‐protective effect of HFD on the energy‐deprived HF.
5. CONCLUSIONS
Our study highlights an important link between BSCL2 and myocardial energy metabolism and function and advances our understanding of the relationship between TG dynamics, FAO rates and the pathogenesis of HF. It may also provide insights into the therapeutic approaches in the treatment of cardiac disorders related to dysregulated lipid metabolism in general.
CONFLICT OF INTEREST
None declared.
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting information
ACKNOWLEDGEMENTS
We thank the Electron Microscopy and Histology Core at Augusta University for technical assistance and electron microscope imaging.
This work was supported by National Heart, Lung and Blood Institute (R01HL132182 to W. C.), National Institute of General Medical Sciences (R01GM124108 to J. L.), National Heart, Lung and Blood Institute (R01HL158515 to J. L.) and the American Heart Association Career Development Award (18CDA34080244 to H. Z.).
Zhou H, Li J, Su H, et al. BSCL2/Seipin deficiency in hearts causes cardiac energy deficit and dysfunction via inducing excessive lipid catabolism. Clin Transl Med. 2022;12:e736. 10.1002/ctm2.736
REFERENCES
- 1. van der Vusse GJ, Glatz JF, Stam HC, Reneman RS. Fatty acid homeostasis in the normoxic and ischemic heart. Physiol Rev. 1992;72:881–940. [DOI] [PubMed] [Google Scholar]
- 2. Kolwicz SC Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011;90:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–145. [DOI] [PubMed] [Google Scholar]
- 4. Neubauer S, Horn M, Cramer M, et al. Myocardial phosphocreatine‐to‐ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. [DOI] [PubMed] [Google Scholar]
- 5. Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haemmerle G, Moustafa T, Woelkart G, et al. ATGL‐mediated fat catabolism regulates cardiac mitochondrial function via PPAR‐alpha and PGC‐1. Nat Med. 2011;17:1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. [DOI] [PubMed] [Google Scholar]
- 8. Pulinilkunnil T, Kienesberger PC, Nagendran J, et al. Cardiac‐specific adipose triglyceride lipase overexpression protects from cardiac steatosis and dilated cardiomyopathy following diet‐induced obesity. Int J Obes (Lond). 2014;38:205–215. [DOI] [PubMed] [Google Scholar]
- 9. Pulinilkunnil T, Kienesberger PC, Nagendran J, et al. Myocardial adipose triglyceride lipase overexpression protects diabetic mice from the development of lipotoxic cardiomyopathy. Diabetes. 2013;62:1464–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Windpassinger C, Auer‐Grumbach M, Irobi J, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet. 2004;36:271–276. [DOI] [PubMed] [Google Scholar]
- 11. Magre J, Delepine M, Khallouf E, et al. Identification of the gene altered in Berardinelli‐Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28:365–370. [DOI] [PubMed] [Google Scholar]
- 12. Berardinelli W. An undiagnosed endocrinometabolic syndrome: report of 2 cases. J Clin Endocrinol Metab. 1954;14:193–204. [DOI] [PubMed] [Google Scholar]
- 13. Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatr Suppl. 1996;413:2–28. [DOI] [PubMed] [Google Scholar]
- 14. Bjornstad PG, Foerster A, Ihlen H. Cardiac findings in generalized lipodystrophy. Acta Paediatr Suppl. 1996;413:39–43. [DOI] [PubMed] [Google Scholar]
- 15. Bhayana S, Siu VM, Joubert GI, et al. Cardiomyopathy in congenital complete lipodystrophy. Clin Genet. 2002;61:283–287. [DOI] [PubMed] [Google Scholar]
- 16. Friguls B, Coroleu W, del Alcazar R, et al. Severe cardiac phenotype of Berardinelli‐Seip congenital lipodystrophy in an infant with homozygous E189X BSCL2 mutation. Eur J Med Genet. 2009;52:14–16. [DOI] [PubMed] [Google Scholar]
- 17. Lupsa BC, Sachdev V, Lungu AO, Rosing DR, Gorden P. Cardiomyopathy in congenital and acquired generalized lipodystrophy: a clinical assessment. Medicine (Baltimore). 2010;89:245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen W, Chang B, Saha P, et al. Berardinelli‐seip congenital lipodystrophy 2/seipin is a cell‐autonomous regulator of lipolysis essential for adipocyte differentiation. Mol Cell Biol. 2012;32:1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cui X, Wang Y, Tang Y, et al. Seipin ablation in mice results in severe generalized lipodystrophy. Hum Mol Genet. 2011;20:3022–3030. [DOI] [PubMed] [Google Scholar]
- 20. Prieur X, Dollet L, Takahashi M, et al. Thiazolidinediones partially reverse the metabolic disturbances observed in Bscl2/seipin‐deficient mice. Diabetologia. 2013;56:1813–1825. [DOI] [PubMed] [Google Scholar]
- 21. Fei W, Shui G, Gaeta B, et al. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J Cell Biol. 2008;180:473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Szymanski KM, Binns D, Bartz R, et al. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci USA. 2007;104:20890–20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bi J, Wang W, Liu Z, et al. Seipin promotes adipose tissue fat storage through the ER Ca(2)(+)‐ATPase SERCA. Cell Metab. 2014;19:861–871. [DOI] [PubMed] [Google Scholar]
- 24. Ding L, Yang X, Tian H, et al. Seipin regulates lipid homeostasis by ensuring calcium‐dependent mitochondrial metabolism. EMBO J. 2018;37:e97572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yan R, Qian H, Lukmantara I, et al. Human SEIPIN binds anionic phospholipids. Dev Cell. 2018;47:248–256 e244. [DOI] [PubMed] [Google Scholar]
- 26. Sui X, Arlt H, Brock KP, et al. Cryo‐electron microscopy structure of the lipid droplet‐formation protein seipin. J Cell Biol. 2018;217:4080–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Talukder MM, Sim MF, O'Rahilly S, Edwardson JM, Rochford JJ. Seipin oligomers can interact directly with AGPAT2 and lipin 1, physically scaffolding critical regulators of adipogenesis. Mol Metab. 2015;4:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sim MF, Dennis RJ, Aubry EM, et al. The human lipodystrophy protein seipin is an ER membrane adaptor for the adipogenic PA phosphatase lipin 1. Mol Metab. 2012;2:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pagac M, Cooper DE, Qi Y, et al. SEIPIN regulates lipid droplet expansion and adipocyte development by modulating the activity of glycerol‐3‐phosphate acyltransferase. Cell Rep. 2016;17:1546–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Castro IG, Eisenberg‐Bord M, Persiani E, et al. Promethin is a conserved seipin partner protein. Cells. 2019;8:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chung J, Wu X, Lambert TJ, et al. LDAF1 and seipin form a lipid droplet assembly complex. Dev Cell. 2019;51:551–563 e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou H, Black SM, Benson TW, Weintraub NL, Chen W. Berardinelli‐Seip congenital lipodystrophy 2/Seipin is not required for brown adipogenesis but regulates brown adipose tissue development and function. Mol Cell Biol. 2016;36:2027–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou H, Lei X, Benson T, et al. Berardinelli‐Seip congenital lipodystrophy 2 regulates adipocyte lipolysis, browning, and energy balance in adult animals. J Lipid Res. 2015;56:1912–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou H, Xu C, Lee H, Yoon Y, Chen W. Berardinelli‐Seip congenital lipodystrophy 2/SEIPIN determines brown adipose tissue maintenance and thermogenic programing. Mol Metab. 2020;36:100971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou H, Lei X, Yan Y, et al. Targeting ATGL to rescue BSCL2 lipodystrophy and its associated cardiomyopathy. JCI Insight. 2019;4:e129781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agah R, Frenkel PA, French BA, et al. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac‐restricted, site‐specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ussher JR, Keung W, Fillmore N, et al. Treatment with the 3‐ketoacyl‐CoA thiolase inhibitor trimetazidine does not exacerbate whole‐body insulin resistance in obese mice. J Pharmacol Exp Ther. 2014;349:487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsai JY, Kienesberger PC, Pulinilkunnil T, et al. Direct regulation of myocardial triglyceride metabolism by the cardiomyocyte circadian clock. J Biol Chem. 2010;285:2918–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu W, Zhou H, Xuan H, et al. Novel metabolic disorders in skeletal muscle of Lipodystrophic Bscl2/Seipin deficient mice. Mol Cell Endocrinol. 2019;482:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pugach EK, Richmond PA, Azofeifa JG, Dowell RD, Leinwand LA. Prolonged Cre expression driven by the alpha‐myosin heavy chain promoter can be cardiotoxic. J Mol Cell Cardiol. 2015;86:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joubert M, Jagu B, Montaigne D, et al. The sodium‐glucose cotransporter 2 inhibitor dapagliflozin prevents cardiomyopathy in a diabetic lipodystrophic mouse model. Diabetes. 2017;66:1030–1040. [DOI] [PubMed] [Google Scholar]
- 42. Nguyen ML, Sachdev V, Burklow TR, et al. Leptin attenuates cardiac hypertrophy in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2021;106:e4327–e4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bai B, Yang W, Fu Y, et al. Seipin knockout mice develop heart failure with preserved ejection fraction. JACC Basic Transl Sci. 2019;4:924–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kolwicz SC Jr, Olson DP, Marney LC, et al. Cardiac‐specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure‐overload hypertrophy. Circ Res. 2012;111:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghosh M, Niyogi S, Bhattacharyya M, et al. Ubiquitin ligase COP1 controls hepatic fat metabolism by targeting ATGL for degradation. Diabetes. 2016;65:3561–3572. [DOI] [PubMed] [Google Scholar]
- 46. Boudina S, Sena S, Theobald H, et al. Mitochondrial energetics in the heart in obesity‐related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. [DOI] [PubMed] [Google Scholar]
- 47. Kiebish MA, Yang K, Liu X, et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J Lipid Res. 2013;54:1312–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Finck BN, Lehman JJ, Leone TC, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mihalik SJ, Goodpaster BH, Kelley DE, et al. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring). 2010;18:1695–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dyck JR, Cheng JF, Stanley WC, et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. 2004;94:e78–84. [DOI] [PubMed] [Google Scholar]
- 51. Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long‐chain 3‐ketoacyl coenzyme A thiolase. Circ Res. 2000;86:580–588. [DOI] [PubMed] [Google Scholar]
- 52. Liu Z, Chen JM, Huang H, et al. The protective effect of trimetazidine on myocardial ischemia/reperfusion injury through activating AMPK and ERK signaling pathway. Metabolism. 2016;65:122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tuunanen H, Engblom E, Naum A, et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118:1250–1258. [DOI] [PubMed] [Google Scholar]
- 54. Luptak I, Balschi JA, Xing Y, et al. Decreased contractile and metabolic reserve in peroxisome proliferator‐activated receptor‐alpha‐null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005;112:2339–2346. [DOI] [PubMed] [Google Scholar]
- 55. Li H, Ma Z, Zhai Y, et al. Trimetazidine ameliorates myocardial metabolic remodeling in isoproterenol‐induced rats through regulating ketone body metabolism via activating AMPK and PPAR alpha. Front Pharmacol. 2020;11:1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cavar M, Ljubkovic M, Bulat C, et al. Trimetazidine does not alter metabolic substrate oxidation in cardiac mitochondria of target patient population. Br J Pharmacol. 2016;173:1529–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Di Napoli P, DiGiovanni P, Gaeta MA, D'Apolito G, Barsotti A. Beneficial effects of trimetazidine treatment on exercise tolerance and B‐type natriuretic peptide and troponin T plasma levels in patients with stable ischemic cardiomyopathy. Am Heart J. 2007;154:602 e601–605. [DOI] [PubMed] [Google Scholar]
- 58. Liu F, Yin L, Zhang L, et al. Trimetazidine improves right ventricular function by increasing miR‐21 expression. Int J Mol Med. 2012;30:849–855. [DOI] [PubMed] [Google Scholar]
- 59. Stanley WC, Dabkowski ER, Ribeiro RF, O'Connell KA. Dietary fat and heart failure: moving from lipotoxicity to lipoprotection. Circ Res. 2012;110:764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information