

Abstract
DNA interstrand crosslinks (ICLs) are cytotoxic lesions that threaten genome integrity. The Fanconi anemia (FA) pathway orchestrates ICL repair during DNA replication, with ubiquitylated FANCI‐FANCD2 (ID2) marking the activation step that triggers incisions on DNA to unhook the ICL. Restoration of intact DNA requires the coordinated actions of polymerase ζ (Polζ)‐mediated translesion synthesis (TLS) and homologous recombination (HR). While the proteins mediating FA pathway activation have been well characterized, the effectors regulating repair pathway choice to promote error‐free ICL resolution remain poorly defined. Here, we uncover an indispensable role of SCAI in ensuring error‐free ICL repair upon activation of the FA pathway. We show that SCAI forms a complex with Polζ and localizes to ICLs during DNA replication. SCAI‐deficient cells are exquisitely sensitive to ICL‐inducing drugs and display major hallmarks of FA gene inactivation. In the absence of SCAI, HR‐mediated ICL repair is defective, and breaks are instead re‐ligated by polymerase θ‐dependent microhomology‐mediated end‐joining, generating deletions spanning the ICL site and radial chromosomes. Our work establishes SCAI as an integral FA pathway component, acting at the interface between TLS and HR to promote error‐free ICL repair.
Keywords: DNA interstrand crosslinks (ICLs), DNA repair, DNA replication, genome stability, translesion DNA synthesis (TLS)
Subject Categories: DNA Replication, Recombination & Repair
This work establishes SCAI as a critical pathway choice regulator ensuring error‐free interstrand crosslink (ICL) repair via the Fanconi anemia (FA) pathway.

Introduction
DNA repair pathways counteract a wide spectrum of DNA lesions to protect the genome from detrimental mutations, breaks, and chromosome rearrangements. While some DNA lesions are directly removed by dedicated enzymes (e.g., oxidized DNA bases are readily restored by base excision repair), other and more deleterious lesions such as DNA interstrand crosslinks (ICLs) require precise coordination of multiple enzymatic activities to re‐establish intact DNA. ICLs are highly cytotoxic lesions, whose repair is tightly coupled to DNA replication (Akkari et al, 2000; Räschle et al, 2008). A large number of proteins participate in ICL repair, defects in at least 22 of which are causative of Fanconi anemia (FA), a rare, inherited disease characterized by developmental birth defects, bone marrow failure, and cancer predisposition (Rageul & Kim, 2020). These factors can be functionally subdivided according to where they operate in the multistep FA ICL repair pathway (Wang, 2007; Ceccaldi et al, 2016; Rageul & Kim, 2020). First, the FA core complex (comprising FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM, FAAP20, and FAAP100) associates with damaged chromatin and monoubiquitylates the heterodimer FANCI‐FANCD2 (ID2) via UBE2T (FANCT) (Garcia‐Higuera et al, 2001; Smogorzewska et al, 2007; Hira et al, 2015; Rickman et al, 2015). Monoubiquitylated ID2 marks the activation of the FA pathway and associates with the ICL to promote repair of the crosslink via downstream effector proteins (Knipscheer et al, 2009; Douwel et al, 2014). These include DNA nucleases that nick one DNA strand on both sides of the ICL to unhook it (SLX4 (FANCP)‐XPF (FANCQ)‐ERCC1 complex), a translesion DNA synthesis (TLS) polymerase that bypasses the ICL adduct (REV1‐Polζ), and homologous recombination (HR) proteins that repair the two‐ended DNA double‐strand break (DSB) (Rageul & Kim, 2020). This final DSB repair step is distinct from other HR processes, as it requires prior restoration of the repair template by TLS. Thus, HR and TLS must be tightly coordinated to ensure faithful ICL repair, but the proteins regulating these processes remain largely unknown. Here, we demonstrate that the protein SCAI (suppressor of cancer cell invasion) has a critical role in ICL repair via the FA pathway, operating at the interface between the TLS and HR steps to prevent erroneous repair of DSB intermediates via Polθ‐dependent microhomology‐mediated end joining (MMEJ), thereby ensuring faithful ICL resolution.
Results
Loss of SCAI manifests with principal hallmarks of FA pathway deficiency
To identify novel factors involved in ICL repair, we carried out a genome‐scale CRISPR‐Cas9 dropout screen for genes required for survival in the presence of a low dose of the ICL‐inducing drug mitomycin C (MMC), corresponding to 20% lethality (LD20). To this end, human RPE‐1 cells with targeted knockout (KO) of p53 were transfected with a lentiviral sgRNA library and propagated in the presence or absence of MMC for 12 days (Fig 1A). Validating our screening approach, many FA and FA‐associated genes as well as HR factors scored among the top hits (Figs 1B and EV1A; Dataset EV1). In fact, KO of 20 of the 22 known FA genes manifested with hypersensitivity to MMC in this screen (Fig 1B). Unexpectedly, the screen also revealed that sgRNAs targeting SCAI were selectively depleted from cells exposed to MMC (Fig 1B; Dataset EV1). SCAI was originally identified as a transcriptional regulator and suppressor of cell migration (Brandt et al, 2009) but was later found to play a role in DSB repair via interaction with 53BP1 (Hansen et al, 2016; Isobe et al, 2017). However, unlike SCAI, sgRNAs targeting 53BP1 did not show significant dropout in MMC‐treated cells (Dataset EV1). Our screen thus suggested a previously unrecognized function of SCAI during ICL repair, which we set out to further explore. In accordance with our screen, SCAI KO cells were exquisitely sensitive to MMC and cisplatin, another ICL inducer, and this could be fully rescued by stable re‐expression of SCAI in these cells (Figs 1C–E and EV1B and C). As observed for many established FA genes, SCAI KO cells displayed strong accumulation in G2 phase accompanied by an increased level of DNA damage foci following MMC treatment (Figs 1F–H and EV1D–H). Likewise, metaphase spreads from SCAI KO cells exhibited a marked increase in chromosome breaks/gaps and radial chromosome formation upon MMC treatment, a characteristic feature of FA patient cells (Fig 1I–K; García‐de‐Teresa et al, 2020). Again, all of these phenotypes were fully complemented by re‐expression of ectopic SCAI (Fig 1F–K). We conclude that loss of SCAI hypersensitizes cells to ICL‐inducing agents and phenocopies major hallmarks of defective ICL repair resulting from FA gene inactivation.
Figure 1. SCAI‐deficient cells display major hallmarks of FA gene dysfunction.
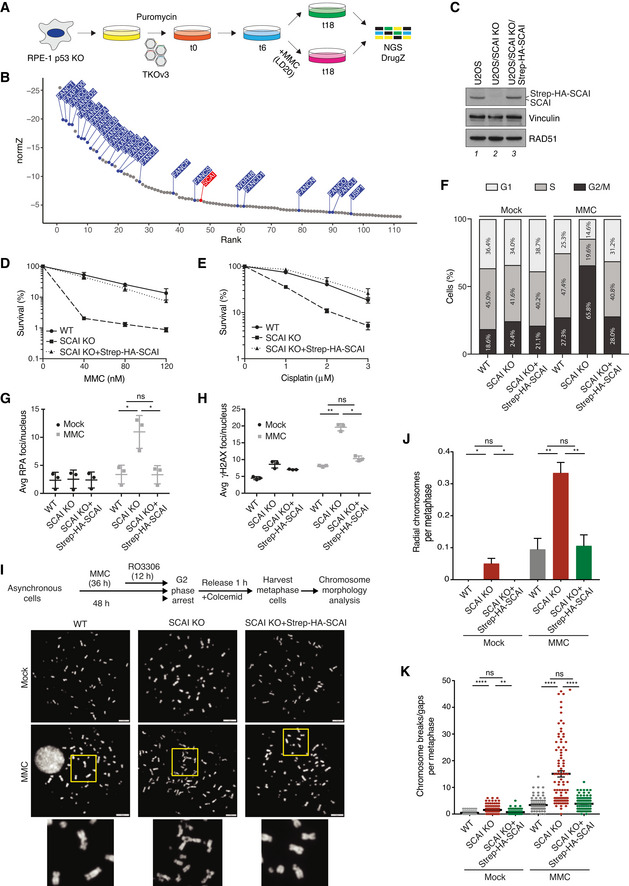
- Schematic outline of genome‐scale CRISPR‐Cas9 screen for genes whose KO sensitizes human RPE‐1 cells to MMC. LD20, 20% lethal dose; NGS, next‐generation sequencing.
- DrugZ analysis of sgRNA depletion in CRISPR screen in (A) following low‐dose MMC treatment (n = 2 technical replicates). FA genes are highlighted in blue; SCAI is highlighted in red.
- Immunoblot analysis of U2OS WT, U2OS/SCAI KO, and U2OS/SCAI KO cells stably reconstituted with Strep‐HA‐SCAI (U2OS/SCAI KO/Strep‐HA‐SCAI).
- Clonogenic survival of U2OS WT, U2OS/SCAI KO, and U2OS/SCAI KO/Strep‐HA‐SCAI cells subjected to indicated doses of MMC for 24 h (mean ± SEM; n = 3 independent experiments).
- As in (D), except that cells were treated with Cisplatin for 24 h (mean ± SEM; n = 3 independent experiments).
- U2OS WT, U2OS/SCAI KO, and U2OS/SCAI KO/Strep‐HA‐SCAI cells were treated or not with MMC (9 nM) for 48 h, fixed, and co‐stained with PCNA antibody and DAPI. Cell cycle distribution was analyzed by quantitative image‐based cytometry (QIBC) (≥ 2,000 cells analyzed per condition). Data from a representative experiment are shown.
- U2OS WT, U2OS/SCAI KO, and U2OS/SCAI KO/Strep‐HA‐SCAI cells were treated or not with MMC (90 nM) for 1 h, fixed 24 h later, and co‐stained with RPA2 antibody and DAPI. RPA2 foci were quantified by QIBC (≥ 3,000 cells analyzed per condition; mean ± SD; n = 3 independent experiments; *P < 0.05; ns, not significant, two‐tailed paired t‐test).
- As in (G), except that cells were co‐stained with γH2AX antibody and DAPI (≥ 3,000 cells analyzed per condition; mean ± SD; n = 3 independent experiments; *P < 0.05; **P < 0.01; ns, not significant, two‐tailed paired t‐test).
- Experimental workflow for metaphase chromosome morphology analysis (top) and representative images of metaphase spreads from indicated cell lines treated or not with MMC (bottom). DNA was stained with DAPI. Scale bars, 10 µm.
- Quantification of radial chromosomes in (I) (mean ± SD; 180 cells analyzed per condition; n = 3 independent experiments; *P < 0.05; **P < 0.01, ns, not significant, two‐tailed t‐test).
- Quantification of chromosomal breaks/gaps in (I) (mean ± SEM; 99 metaphase cells analyzed for each condition pooled from three independent experiments; **P < 0.01; ****P < 0.0001, ns, not significant, Mann–Whitney U test).
Figure EV1. SCAI‐deficient cells display major hallmarks of FA gene dysfunction (related to Figs 1 and 2).
- GO term analysis of significant hits (NormZ < −3) in CRISPR‐Cas9 screen in Fig 1A, using Reactome pathways from PANTHER16.0.
- Clonogenic survival of U2OS WT and U2OS/SCAI KO cells subjected to indicated doses of MMC for 24 h (mean ± SEM; n = 3 independent experiments).
- As in (B), except that cells were treated with indicated doses of cisplatin for 24 h (mean ± SEM; n = 3 independent experiments).
- Scatter plot showing cell cycle distribution of cells in Fig 1F. Light grey, G1 phase; dark grey, S phase; red: G2/M phase. Proportion of cells in G2/M phase is indicated.
- U2OS WT, U2OS/SCAI KO, and U2OS/SCAI KO/Strep‐HA‐SCAI cells were treated or not with MMC (90 nM) for 1 h, fixed 24 h later, and co‐stained with 53BP1 antibody and DAPI. 53BP1 foci were quantified by QIBC (≥ 3,000 cells analyzed per condition; mean ± SD; n = 3 independent experiments; *P < 0.05; two‐tailed paired t‐test).
- As in (E), except that cells were stained with RAD51 antibody (≥ 3,000 cells analyzed per condition; mean ± SD; n = 3 independent experiments; *P < 0.05; ns, not significant, two‐tailed paired t‐test).
- Representative images of the experiments in Fig 1G and H. Scale bar, 10 µm.
- Representative images of the experiments in (E) and (F). Scale bar, 10 µm.
- Immunoblot analysis of FANCD2 siRNA knockdown efficiency in U2OS cells.
- Clonogenic survival of U2OS and U2OS/SCAI KO cells transfected with non‐targeting control (CTRL) or FANCD2 siRNAs and subjected to indicated doses of MMC for 24 h (mean ± SEM; n = 3 independent experiments).
- Immunoblot analysis of U2OS WT and U2OS/SCAI KO cells harvested at the indicated times after exposure to MMC (0.5 µM).
Preventing FA pathway activation alleviates ICL hypersensitivity of SCAI‐deficient cells
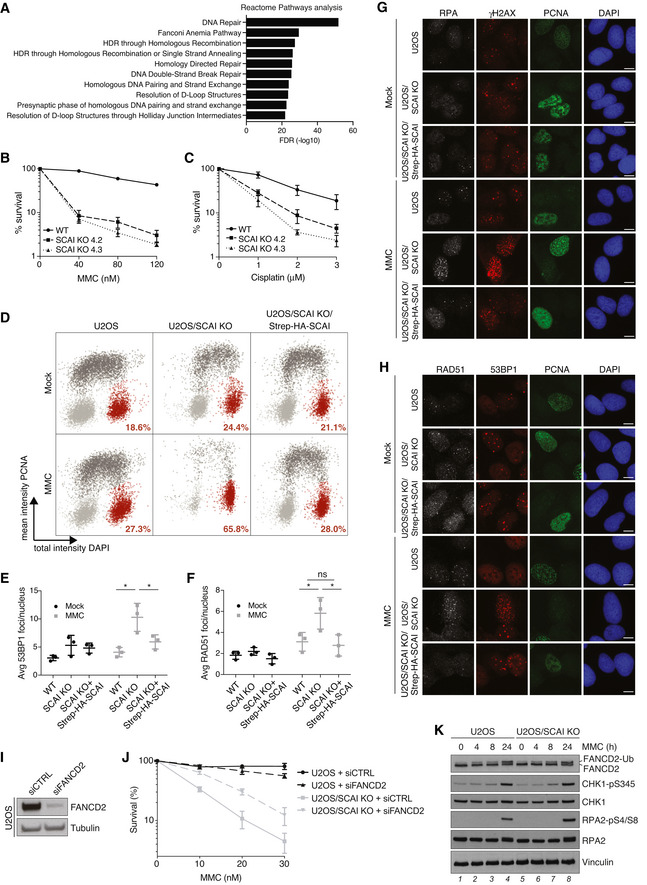
To better define the emerging role of SCAI in ICL repair, we carried out a complementary genome‐scale CRISPR‐Cas9 screen for genes whose loss suppresses the hypersensitivity of SCAI‐deficient cells to MMC. To this aim, SCAI KO cells transduced with a lentiviral sgRNA library were propagated in the presence of a near‐lethal dose (LD80) of MMC (Fig 2A). Strikingly, gene ontology (GO) term analysis revealed that FA pathway components were the most enriched class of genes whose inactivation conferred significantly increased resistance to MMC in SCAI KO cells (Fig 2B and C). These comprised FA core complex factors and associated proteins (FANCA, FANCB, FANCC, FANCF, FANCL, FAAP20, and FAAP100), the ID2 complex, and proteins regulating its deubiquitylation (USP1 and WDR48), all of which ordinarily have critical roles in activating the FA pathway to protect against ICL toxicity (Fig 2B and C; Dataset EV2; Ceccaldi et al, 2016; Rageul & Kim, 2020). By contrast, effectors of ICL repair via this pathway functioning downstream of ID2 ubiquitylation did not score as hits in this screen (Dataset EV2). Genes encoding factors involved in chromatin remodeling via histone acetylation and regulators of cell proliferation and survival via p53 also conferred resistance to MMC in SCAI KO cells but were far less prominently represented than FA genes (Fig 2C; Dataset EV2). Verifying these screen results, we found that FANCA depletion by siRNA largely restored survival of SCAI KO cells exposed to MMC and suppressed the accumulation of DSBs (Fig 2D–G). Knockdown of FANCD2 similarly alleviated MMC hypersensitivity of SCAI KO cells (Fig EV1I and J). Notably, SCAI KO conferred much stronger MMC hypersensitivity than FANCA or FANCD2 knockdown by siRNAs in a U2OS background (Figs 2D and E, and EV1I and J). To exclude the possibility that this was a consequence of residual FA pathway activation due to incomplete knockdown efficiency of the siRNAs, we knocked out FANCA in U2OS and U2OS SCAI KO cells (Fig 2H). This confirmed that SCAI KO cells are indeed more sensitive to MMC than FANCA KO cells, and that loss of FANCA strongly alleviates the hypersensitivity of SCAI KO cells to MMC (Fig 2I and J). Importantly, whereas FANCD2 monoubiquitylation upon MMC treatment was abolished by FANCA KO as expected, loss of SCAI had no impact (Figs 2K and EV1K). Collectively, these data suggest that SCAI has an essential role in ensuring proper repair of ICLs once their processing via the FA pathway is initiated, but not for ID2 ubiquitylation and FA pathway activation per se.
Figure 2. Preventing FA pathway activation alleviates ICL hypersensitivity of SCAI‐deficient cells.
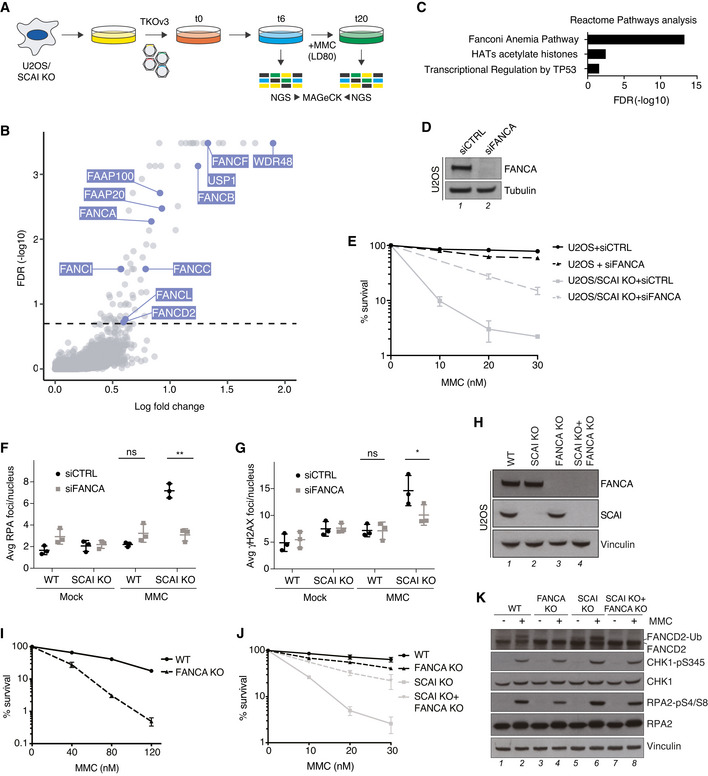
- Schematic outline of genome‐scale CRISPR‐Cas9 screen for genes whose KO suppresses MMC hypersensitivity of SCAI KO cells. LD80, 80% lethal dose; NGS, next‐generation sequencing.
- MaGECK analysis of sgRNA enrichment in CRISPR screen in (A) following MMC treatment (n = 3 technical replicates; false discovery rate (FDR) of 0.2 indicated by dotted line). FA genes are highlighted in blue.
- GO term analysis of significantly enriched genes (FDR < 0.2) in CRISPR‐Cas9 screen in (A), using Reactome pathways from PANTHER16.0.
- Immunoblot analysis of FANCA siRNA knockdown efficiency in U2OS cells.
- Clonogenic survival of U2OS and U2OS/SCAI KO cells transfected with non‐targeting control (CTRL) or FANCA siRNAs and subjected to indicated doses of MMC for 24 h (mean ± SEM; n = 3 independent experiments).
- U2OS and U2OS/SCAI KO cells transfected with indicated siRNAs for 48 h were treated or not with MMC (90 nM) for 1 h, fixed 24 h later, and co‐stained with RPA2 antibody and DAPI. RPA2 foci were quantified by QIBC (≥ 3,000 cells analyzed per condition; mean ± SD; n = 3 independent experiments; *P < 0.05; ns, not significant, two‐tailed paired t‐test).
- As in (F), except that cells were stained with γH2AX antibody (≥ 3,000 cells analyzed per condition; mean ± SD; n = 3 independent experiments; **P < 0.01; ns, not significant, two‐tailed paired t‐test).
- Immunoblot analysis of the indicated U2OS cell lines.
- Clonogenic survival of U2OS and U2OS/FANCA KO cells subjected to indicated doses of MMC for 24 h (mean ± SEM; n = 3 independent experiments).
- Clonogenic survival of U2OS, U2OS/SCAI KO, U2OS/FANCA KO, and U2OS/FANCA+SCAI double KO (DKO) cells subjected to indicated doses of MMC for 24 h (mean ± SEM; n = 3 independent experiments).
- Immunoblot analysis of U2OS, U2OS/SCAI KO, U2OS/FANCA KO, and U2OS/FANCA SCAI DKO cell lines exposed or not to MMC as indicated.
SCAI promotes replication‐coupled ICL repair downstream of ID2 ubiquitylation
To elucidate how SCAI functions in ICL repair we used Xenopus egg extracts, which efficiently recapitulate the replication‐coupled repair of a plasmid containing a site‐specific cisplatin ICL (pICLpt) (Räschle et al, 2008). In this system, two replisomes quickly converge on the lesion (Fig 3A, i). Upon collision, CMG is first ubiquitylated by TRAIP and unloaded by p97 (Fig 3A, ii) (Fullbright et al, 2016; Wu et al, 2019). One of the leading strands is then extended to within one nucleotide (nt) of the crosslink (−1 position) (Räschle et al, 2008), at which point one of the forks undergoes reversal to produce a substrate suitable for DNA incisions (Fig 3A, iii) (Amunugama et al, 2018). Ubiquitylated ID2 localizes to the ICL and promotes incisions by the SLX4‐XPF‐ERCC1 complex (Fig 3A, iv), generating a DSB in one of the daughter molecules while leaving a gap containing an adduct in the other (Knipscheer et al, 2009; Douwel et al, 2014). The adducted molecule is restored by TLS in a two‐step process that involves insertion across the adducted base by an unknown polymerase, and extension past the adduct by REV1‐Polζ (Fig 3A, v) (Budzowska et al, 2015). Finally, the DSB is repaired by HR utilizing the intact daughter molecule as a template (Fig 3A, vi–vii; Long et al, 2011).
Figure 3. SCAI promotes replication‐coupled ICL repair.
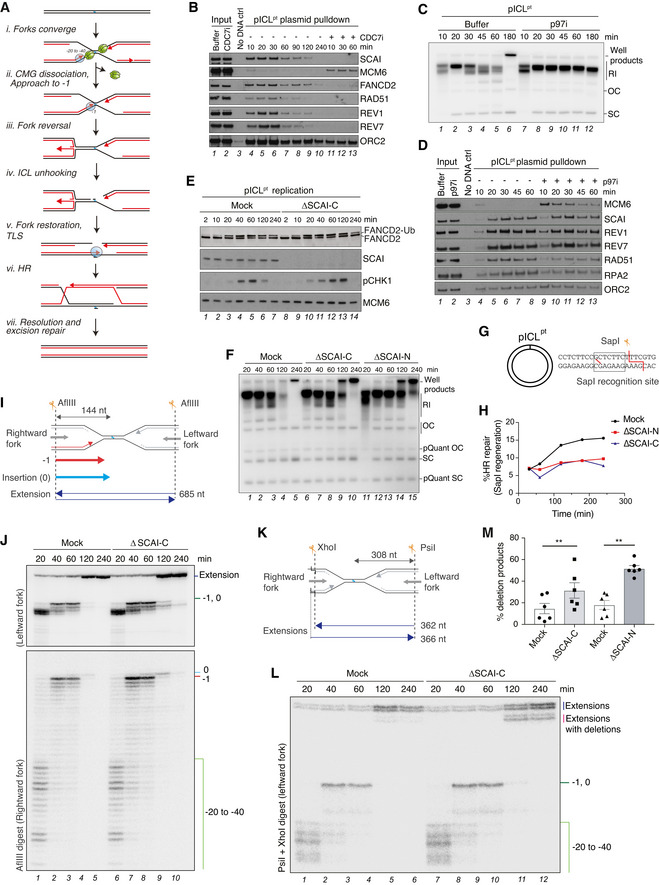
- Current model of ICLpt repair in Xenopus egg extracts (Amunugama et al, 2018).
- pICLpt was isolated via plasmid pull down at the indicated times after incubation in Xenopus egg extract in the presence or absence of CDC7i (100 µM) and associated proteins were analyzed by immunoblotting.
- pICLpt was replicated in egg extracts in the presence of [α‐32P]dATP for the indicated times, and reactions were analyzed by native agarose gel electrophoresis. The p97 inhibitor NMS‐873 (p97i; 200 μM) was supplemented to the reaction where indicated. Note that in the absence of p97 activity, CMGs are no longer unloaded from the plasmid, leading to accumulation of replication intermediate products (RI) (Fullbright et al, 2016). OC, open circular; SC, supercoiled.
- Samples from (C) were recovered via plasmid pull down at the indicated time points as in (B) and analyzed by immunoblotting.
- pICLpt was replicated in mock‐ or SCAI‐C‐depleted egg extracts, and samples collected at the indicated time points were analyzed by immunoblotting.
- pICLpt was replicated in mock‐ or SCAI‐depleted egg extract in the presence of [α‐32P]dATP for the indicated times, and reactions were analyzed by native agarose gel electrophoresis. ΔSCAI‐C and ΔSCAI‐N denote SCAI immunodepletion with an antibody raised against the C or N terminus of SCAI, respectively; RI, replication intermediates; OC, open circular; SC, supercoiled.
- Schematic of pICLpt illustrating the SapI site, which is regenerated upon replication‐coupled repair (Knipscheer et al, 2009).
- Quantification of SapI regeneration in mock or SCAI‐depleted extracts. Note that plasmids containing a SapI site but no ICL account for ~ 5–7% of each pICLpt preparation. Primary data are shown in Fig EV2F and G. A representative of two independent experiments is shown.
- Schematic of intermediates and extension products generated by AflIII digest of pICLpt.
- pICLpt was replicated in mock‐ or SCAI‐depleted egg extract in the presence of [α‐32P]dATP for the indicated times, and reactions were digested with AflIII and analyzed on a denaturing polyacrylamide gel. Stalling points relative to the ICL site are indicated.
- Schematic of intermediates and extension products generated by PsiI+XhoI double digest of pICLpt.
- Samples in (J) were digested with PsiI+XhoI and analyzed on a denaturing polyacrylamide gel. Stalling points relative to the ICL site are indicated.
- Quantification of extensions with deletions in (L) at 240 min (mean ± SEM; n = 6 independent experiments; **P < 0.01; two‐tailed paired t‐test).
Consistent with the role of SCAI in promoting ICL repair in human cells, SCAI was efficiently recruited to the ICL‐containing plasmid in a replication‐dependent manner (Figs 3B and EV2A). SCAI recruitment to the ICL occurred early in the reaction and was unaffected by inhibition of p97, which blocks CMG unloading and downstream unhooking of the ICL (Fig 3C and D, compare lanes 4–8 to 9–13) (Fullbright et al, 2016; Wu et al, 2019). To monitor the impact of SCAI loss on ICL repair, we replicated pICLpt in mock‐ or SCAI‐depleted egg extracts. As observed in human cells, FANCD2 monoubiquitylation occurred normally in the absence of SCAI (Fig 3E). Consistently, the appearance of incisions on parental DNA was also largely unaffected by SCAI immunodepletion with either of two independent antibodies (Fig EV2B–G; note that upon SCAI‐N immunodepletion incised strands are stabilized (Fig EV2F, lanes 6–10), likely because this antibody but not the SCAI‐C antibody also co‐depletes CtIP (Dataset EV4)). Notably, however, SCAI immunodepletion led to an accumulation of replication‐dependent well products during pICLpt replication (Fig 3F, compare lanes 4–5, 9–10, and 14–15), which were previously suggested to arise from HR events (Long et al, 2011; Semlow et al, 2016). Despite the increase in well product formation, regeneration of the SapI site, which is mediated by error‐free repair, was completely abolished in the absence of SCAI, indicating a failure to complete HR (Figs 3G and H, and EV2E–G; Long et al, 2011). To probe the impact of SCAI depletion on pICLpt replication, we analyzed replication intermediates on a denaturing acrylamide gel following AflIII digestion (Fig 3I). Nascent strands in SCAI‐depleted extracts approached the crosslink with similar kinetics as in the mock‐depleted reaction (−1 position), indicating that fork convergence and CMG unloading occurred normally in the absence of SCAI (Fig 3J; compare lanes 1–2 and 6–7; Fig 3A, i–ii). However, SCAI‐depleted extracts exhibited a modest delay in performing TLS across the adduct, as evidenced by the prolonged presence of the nascent strand product at the 0 position (Fig 3J; compare lanes 4–5 and 9–10; Fig EV2H). Strikingly, despite the mild delay in TLS and lack of HR, we detected an increase in extension product formation when SCAI was absent (Fig 3J; compare lanes 4–5 and 9–10; Fig EV2H). Resolution of extension products by a different restriction digest (Fig 3K) revealed a defined deletion product that accumulated in SCAI‐depleted egg extracts (Figs 3L and M, and EV2I; note that mock‐depleted extracts display variable deletion product formation, as previously reported (Budzowska et al, 2015), and this was consistently increased upon SCAI immunodepletion), suggesting that some of the DNA ends undergo repair by an alternative, HR‐independent mechanism. We conclude that SCAI depletion in egg extracts corrupts ICL repair downstream of ID2 ubiquitylation, leading to a TLS delay, loss of HR‐mediated repair, and appearance of extension products containing deletions.
Figure EV2. SCAI promotes replication‐coupled ICL repair (related to Fig 3).
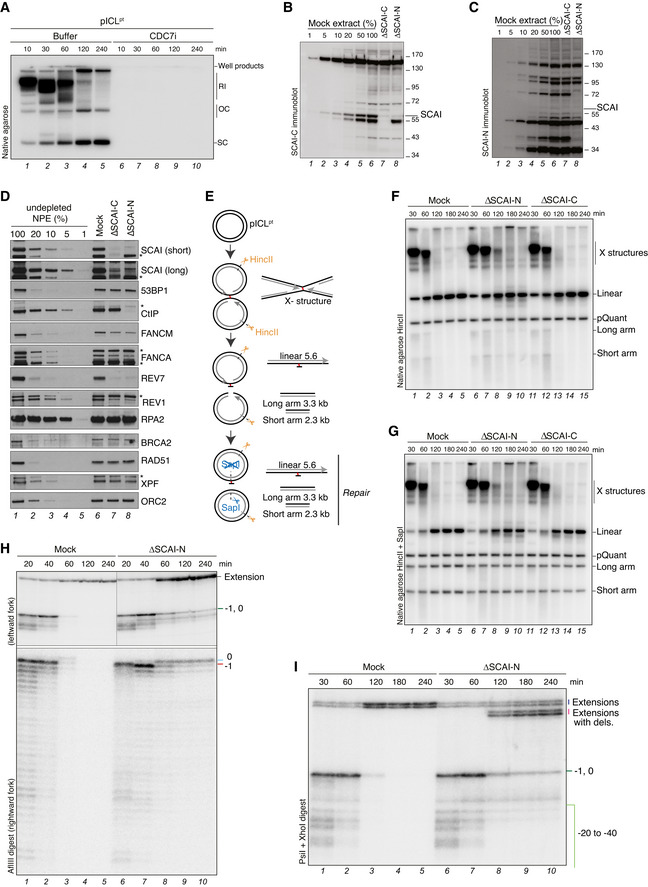
-
ASamples in Fig 3B were replicated in the presence of [α‐32P]dATP and analyzed by native agarose gel electrophoresis. RI, replication intermediates; OC, open circular; SC, supercoiled.
-
BSCAI‐C immunoblot analysis of Xenopus egg extracts (NPE) subjected to immunodepletion with SCAI‐C, SCAI‐N, or an IgG control antibody.
-
CSCAI‐N immunoblot analysis of samples from (B).
-
DImmunoblot analysis of Xenopus egg extracts subjected to immunodepletion with antibodies targeting the C‐terminal (SCAI‐C) or N‐terminal (SCAI‐N) region of SCAI or an IgG control. Asterisks denote antibody cross‐reactivity. Note that SCAI‐N but not SCAI‐C immunodepletion also co‐depletes CtIP.
-
ESchematic of the products generated by HincII or HincII‐SapI digest of pICLpt.
-
F, GpICLpt was replicated in mock‐ or SCAI‐depleted extracts and samples were digested with HincII (F) or HincII and SapI (G) and analyzed by agarose gel electrophoresis. DSB repair by HR restores the SapI site (Long et al, 2011). pQuant is used as a recovery control (Knipscheer et al, 2012) and the quantification of this experiment is shown in Fig 3H.
-
HpICLpt was replicated in mock‐ or SCAI‐N‐depleted egg extracts in the presence of [α‐32P]dATP. Samples were digested with AflIII and analyzed on a denaturing polyacrylamide gel. Stalling points relative to the ICL site are indicated. See Fig 3I for schematic of intermediates and extension products generated by AflIII digest of pICLpt.
-
ISamples from (H) were digested with PsiI and XhoI and analyzed on a denaturing polyacrylamide gel. Stalling points relative to the ICL site are indicated. See Fig 3I for schematic of intermediates and extension products generated by PsiI and XhoI double digest of pICLpt.
SCAI interacts with Polζ but prevents erroneous ICL repair independently of Polζ
To explore potential interactions between SCAI and ICL repair factors, we performed label‐free mass spectrometry analysis of SCAI immunoprecipitates (IPs) from egg extracts to identify its interacting partners. Consistent with a role of SCAI in ICL repair, combined analysis of SCAI IPs with either antibody showed predominant enrichment of many FA proteins, as well as transcription and chromatin remodeling factors (Fig 4A; Dataset EV3). Strikingly, all five subunits of the polymerase ζ (Polζ) complex (REV1, REV3, REV7, POLD2, and POLD3) were strongly enriched in SCAI IPs (Fig 4A). Correspondingly, SCAI was enriched in REV1, REV7, and REV3 IPs (Fig 4B). SCAI immunodepletion with either antibody co‐depleted REV3 and to a lesser extent REV1 and REV7 (Figs 4C and EV2D; Dataset EV4). This suggested that SCAI is tightly associated with REV1‐Polζ in egg extracts, and that the TLS delay observed in the absence of SCAI is likely caused by co‐depletion of REV1‐Polζ. In contrast, although SCAI IPs also contained many FA pathway activators, depletion of SCAI did not functionally co‐deplete the FA core complex as evidenced by intact ID2 monoubiquitylation during replication‐coupled ICL repair (Figs 3E and EV2D; Dataset EV4). In accordance with an interaction between SCAI and the Polζ complex, these proteins also accumulated with similar kinetics on plasmids containing a site‐specific DNA–protein crosslink (DPC), whose repair occurs in the absence of DSB formation and ID2 but requires Polζ for bypass of the DNA–peptide adduct (Fig EV3A and (Duxin et al, 2014; Gallina et al, 2021)). In this setting, SCAI depletion specifically delayed Polζ‐mediated bypass of the peptide adduct, as evidenced by the specific persistence of −1, 0, and +1 nascent strand products compared to the control reaction (Fig EV3B–D). SCAI‐Polζ interaction was also readily observed in human cells and was largely mediated by REV3 (Figs 4D and EV3E and F). Inspection of the REV3 sequence revealed a conserved region within its PCD domain that shares notable homology with AHDC1, another SCAI interactor (Fig 4E) (Hansen et al, 2016). Indeed, for both REV3 and AHDC1, this region was required for their interaction with SCAI in human cells (Figs 4F and G, and EV3G). Importantly, a Xenopus REV3 peptide spanning this sequence efficiently bound purified recombinant SCAI, demonstrating that the SCAI‐REV3 interaction is direct (Fig EV3H and I). Finally, in line with a potential role of SCAI in regulating REV1‐Polζ via direct interaction independently of DSB repair, we found that SCAI is also enriched at UV‐C‐damaged chromatin unlike 53BP1 and that SCAI KO cells show moderate sensitivity to UV‐C radiation (Fig EV4A–I; Dataset EV5). We conclude that SCAI is tightly associated with REV1‐Polζ via a conserved region in REV3.
Figure 4. SCAI interacts with Polζ but protects against erroneous ICL repair independently of Polζ.
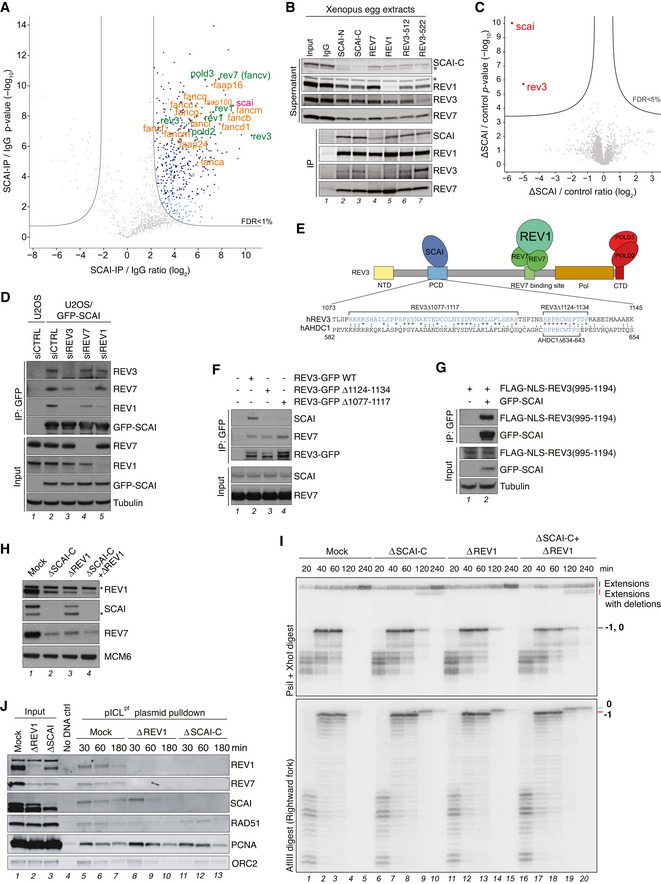
- Analysis of SCAI‐interacting proteins in Xenopus egg extracts. Volcano plot shows enrichment of proteins in both SCAI‐C and SCAI‐N immunoprecipitates (IPs) compared to an IgG control, plotted against the P‐value (n = 4 independent experiments; permutation‐based FDR < 0.01, s0 = 2).
- SCAI, REV7, REV1, and REV3 immunoprecipitates (IP) in NPE were immunoblotted with the indicated antibodies. Asterisks denote antibody cross‐reactivity.
- Whole proteome MS analysis of mock‐ versus SCAI‐depleted egg extracts. The volcano plot shows the difference in abundance of proteins between the mock reaction and SCAI‐depleted samples (with either N or C antibodies) (X‐axis), plotted against the P‐value resulting from two‐tailed Student’s t‐test (Y‐axis). Proteins significantly down‐regulated (FDR < 5%) in SCAI‐depleted extracts are highlighted in red (n = 4 biochemical replicates; FDR < 5% corresponds to a permutation‐based FDR‐adjusted q‐value of < 0.05).
- GFP IPs from U2OS or U2OS/GFP‐SCAI cells transfected with indicated siRNAs were immunoblotted with indicated antibodies. Note that a small fraction of REV7 is still immunoprecipitated with GFP‐SCAI in REV3 knockdown cells. This could be due to incomplete knockdown, and/or additional interactions of SCAI with the REV1‐Polζ complex that are independent of REV3.
- Schematic of SCAI interaction with the REV1‐Polζ complex. The SCAI‐interacting region of human REV3 and its alignment with a homologous region in human AHDC is indicated. NTD, N‐terminal domain; PCD, positively charged domain; Pol, polymerase domain; CTD, C‐terminal domain.
- GFP IPs from U2OS cells transfected with indicated REV3‐GFP expression constructs were immunoblotted with indicated antibodies.
- As in (F), except that U2OS or U2OS/GFP‐SCAI cells were transfected with FLAG‐NLS‐REV3(995–1194) expression plasmid.
- Mock‐, SCAI‐, REV1‐, or SCAI/REV1‐depleted egg extracts were analyzed by immunoblotting with indicated antibodies. Asterisks denote antibody cross‐reactivity.
- pICLpt was replicated in egg extracts immunodepleted with the indicated antibodies and processed as in Fig 3J and L.
- pICLpt was replicated in egg extracts immunodepleted with indicated antibodies, isolated at various times via plasmid pull down, and associated proteins were analyzed by immunoblotting.
Figure EV3. SCAI interacts with Polζ (related to Fig 4).
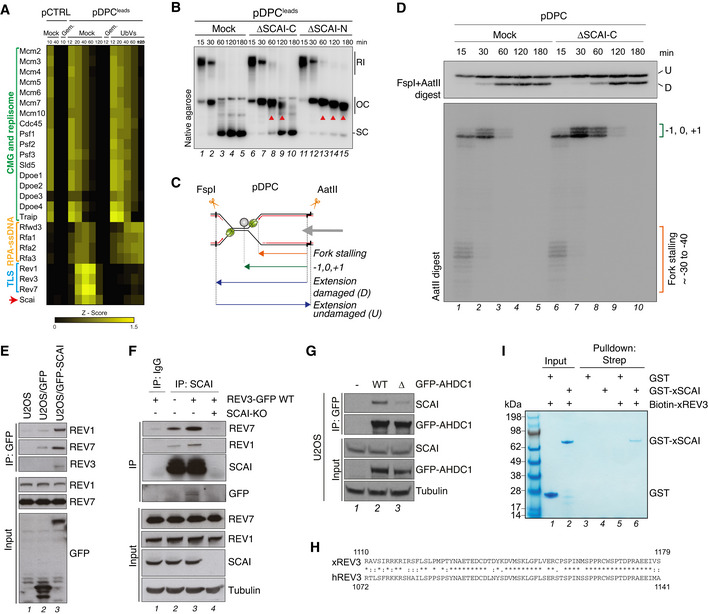
- Heat map depicting the mean of the Z‐scored Log2 label‐free quantification LFQ intensity from four biochemical replicates of pCTRL and pDPCLeads replication in Xenopus egg extracts (data originally published in Larsen et al (2019)). Where indicated, Geminin was added to block DNA replication, and ubiquitin‐vinyl sulfone (Ub‐VS) was added to deplete the pool of free ubiquitin from the extracts.
- pDPCLeads was replicated in mock‐, SCAI‐C‐, and SCAI‐N‐depleted egg extracts in the presence of [α‐32P]dATP. Reactions were analyzed by native agarose gel electrophoresis. Red arrowheads indicate accumulation of open circular (OC) molecules caused by a TLS defect.
- Nascent leading strand and extension products generated upon FspI+AatII digest of pDPC. Double digestion generates shorter damaged and longer undamaged extension products. The CMG helicase is depicted in green and the crosslinked M.HpaII protein in grey.
- pDPC was replicated in mock‐ or SCAI‐depleted Xenopus egg extract in the presence of [α‐32P]dATP. Samples were digested with FspI+AatII and analyzed on a denaturing polyacrylamide gel. Stalling points relative to the DPC site are indicated. U, undamaged; D, damaged extension product.
- U2OS cells or derivative lines stably expressing GFP or GFP‐SCAI were subjected to GFP IP followed by immunoblotting with indicated antibodies.
- U2OS WT or SCAI KO cells that were transfected with REV3‐GFP where indicated were subjected to IP with IgG or SCAI antibody followed by immunoblotting.
- GFP IPs from U2OS cells transfected with indicated GFP‐AHDC1 expression constructs were analyzed by immunoblotting.
- Alignment of the SCAI‐interacting regions of human and Xenopus REV3 proteins. Asterisk indicates positions with a conserved amino acid; colon indicates conservation between amino acids with strongly similar properties; period indicates conservation between amino acids with weakly similar properties.
- Recombinant GST or GST‐xSCAI proteins incubated or not with Biotin‐xREV3 peptide (aa 1110–1179) were subjected to streptavidin pull down and analyzed by Coomassie staining.
Figure EV4. SCAI is recruited to UV‐induced DNA lesions (related to Fig 4).
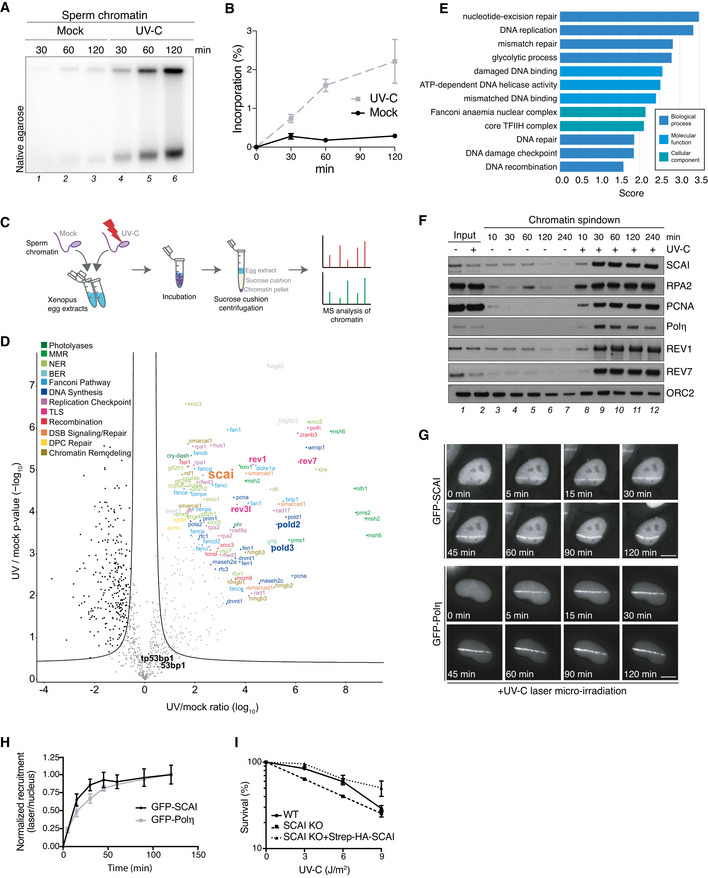
- Sperm chromatin was left untreated or exposed to UV‐C (2,000 J/m2), incubated in HSS/NPE mix in the presence of [α‐32P]dATP and nucleotide incorporation into chromatin was analyzed at the indicated time points via native agarose gel electrophoresis.
- Quantification of data in (A) (mean ± SEM; n = 3 independent experiments).
- Schematic outline of CHROMASS workflow to analyze protein recruitment to UV‐damaged chromatin. Sperm chromatin left untreated or irradiated with UV‐C (2,000 J/m2) was incubated in Xenopus egg extract for 30 min, isolated by sucrose cushion centrifugation, and analyzed by label‐free mass spectrometry (MS).
- Protein recruitment to UV‐damaged chromatin compared to an undamaged control. Volcano plot shows enrichment of individual proteins (UV/mock ratio) plotted against the P‐value (n = 4 independent experiments; FDR < 0.05, s0 = 0.5).
- Term enrichment analysis showing GO terms corresponding to proteins significantly recruited to UV‐C‐damaged chromatin (Dataset EV5). All displayed terms were significant with P < 0.02, as determined through Fisher exact testing with Benjamini–Hochberg correction.
- Sperm chromatin left untreated or irradiated with UV‐C (2,000 J/m2) was isolated at the indicated time points and analyzed by immunoblotting.
- Representative images of GFP‐SCAI and GFP‐Polη recruitment to UV‐C laser micro‐irradiation in U2OS cells at indicated time points. Scale bar, 10 µm.
- U2OS cells stably expressing GFP‐SCAI or transiently transfected with GFP‐Polη were subjected to UV‐C laser micro‐irradiation. At indicated time points, GFP fluorescence intensities at damage sites over the nuclear background were quantified. Recruitment was normalized to the time point of maximal recruitment (mean ± SEM; n = 3 independent experiments; at least 30 cells analyzed per condition).
- Clonogenic survival of U2OS cells subjected to indicated doses of UV‐C (mean ± SEM; n = 3 independent experiments).
Because immunodepletion of SCAI co‐depletes REV1‐Polζ (Fig EV2D), we next addressed whether erroneous ICL repair and the appearance of deletions were caused by REV1‐Polζ co‐depletion and/or a TLS defect. Importantly, unlike SCAI immunodepletion, the absence of REV1‐Polζ did not increase deletion product formation, although a TLS delay was apparent (Fig 4H and I, compare lanes 4–5 and 14–15). While REV1 depletion partially co‐depleted SCAI (Fig 4H), the residual SCAI pool still underwent enrichment at the ICL plasmid in the absence of REV1‐Polζ (Fig 4J, compare lanes 5–7 and 8–10), preventing the appearance of extension products containing deletions (Fig 4I, compare lanes 14–15 and 19–20). Thus, although SCAI associates with REV1‐Polζ and may thereby also contribute to TLS, its function in suppressing deletions during ICL repair is not shared by REV1‐Polζ. This suggests both REV1‐Polζ‐dependent and ‐independent functions of SCAI in ICL repair.
SCAI prevents microhomology‐mediated end joining by Polθ during ICL repair
We next addressed the mechanism of error‐prone DSB repair triggered by the absence of SCAI.
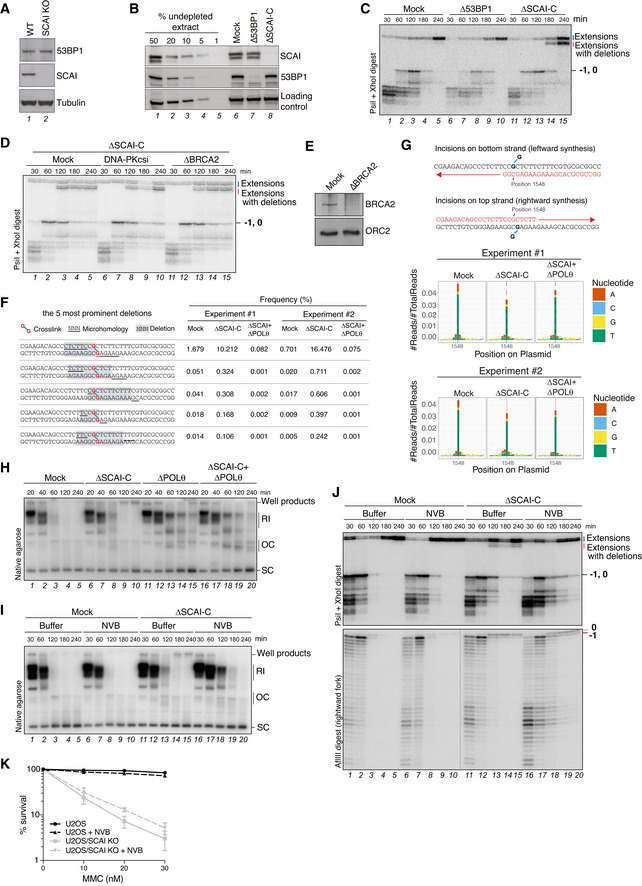
Although 53BP1 was previously shown to interact with and recruit SCAI to DSBs (Hansen et al, 2016; Isobe et al, 2017), 53BP1 levels were neither affected by SCAI KO in human cells nor SCAI immunodepletion in egg extracts, and 53BP1 immunodepletion from egg extracts did not induce deletion products upon repair of the ICL plasmid (Fig EV5A–C), indicating that the role of SCAI in ensuring error‐free ICL repair is not shared by 53BP1. Moreover, immunodepletion of the HR factor BRCA2 or inhibition of DNA‐PKcs, which is essential for classical non‐homologous end joining, did not suppress the formation of extension products with deletions in SCAI‐depleted egg extracts, suggesting the involvement of an alternative mechanism of end ligation (Fig EV5D and E). Analysis of repair products by next‐generation sequencing revealed predominant accumulation of an 8‐nt deletion product generated in the absence of SCAI, accompanied by other deletions of varying lengths (Figs 5A and EV5F). These deletions occurred around the ICL and contained sequences with microhomology at the ligation site, indicative of repair via microhomology‐mediated end joining (MMEJ) (Fig EV5F). The preferential appearance of the 8‐nt deletion can be explained by the presence of a 6‐nt tandem repeat flanking the ICL site (Fig EV5F). In contrast, when we monitored repair products without deletions, mutagenesis across the ICL was only modestly affected by the absence of SCAI (Fig EV5G). Importantly, immunodepletion of DNA Polθ, the main mediator of MMEJ (Chan et al, 2010; Roerink et al, 2014; Ceccaldi et al, 2015; Mateos‐Gomez et al, 2015), abrogated deletion products generated in the absence of SCAI (Fig 5A–C, compare lanes 8–10 and 18–20; Fig EV5F and H). Inhibition of Polθ by Novobiocin treatment (preprint: Zhou et al, 2020) had a similar effect (Fig EV5I and J). Consistently, depletion of Polθ significantly reduced both the appearance of radial chromosomes and chromosome gaps and breaks in MMC‐treated SCAI KO cells (Fig 5D–G). Moreover, Polθ knockdown or inhibition by Novobiocin improved long‐term survival of SCAI KO cells after MMC treatment, albeit only to a modest extent, likely due to a failure to restore HR‐mediated repair in the absence of SCAI and/or defective REV1‐Polζ‐mediated TLS (Figs 5H and EV5K). Together, these findings indicate that SCAI has a critical function in suppressing illegitimate processing of DNA ends during ICL repair via the FA pathway, providing a molecular explanation for the exquisite sensitivity of SCAI‐deficient cells to ICL‐inducing drugs that can be alleviated by blocking FA pathway activation.
Figure EV5. SCAI prevents Polθ‐mediated MMEJ during ICL repair (related to Fig 5).
- Whole cell extracts of U2OS and U2OS/SCAI KO cells were analyzed by immunoblotting with indicated antibodies.
- Mock‐, 53BP1‐, or SCAI‐C‐depleted egg extracts were analyzed by immunoblotting with indicated antibodies.
- pICLpt was replicated in extracts immunodepleted in mock‐, 53BP1‐, or SCAI‐C‐depleted egg extracts digested with PsiI+XhoI and resolved on a denaturing polyacrylamide gel.
- pICLpt was replicated in SCAI‐C‐depleted extracts that were also depleted of BRCA2, or supplemented with DNA‐PKcs inhibitor (NU7441; 100 µM) where indicated. Reaction samples were digested with PsiI+XhoI and analyzed on a denaturing polyacrylamide gel.
- Mock‐ or BRCA2‐depleted egg extracts were analyzed by immunoblotting with indicated antibodies.
- Most frequent deletion products in sequencing data in Fig 5A. In red, crosslinked guanines; highlighted in grey, deletions; underlines, microhomology regions.
- Schematic representation of DNA synthesis occurring across the adducted base following DNA incisions on either the top or bottom strand. The denoted nucleotide position 1,548 corresponds to the insertion 0 position when incisions occur in the bottom strand or to the −1 position when incisions occur in the top strand. Sequencing products of correct length from Fig 5A were analyzed for their nucleotide misincorporation distribution (bottom graphs). Misincorporation is based on the bottom strand read.
- Samples in Fig 5C were analyzed by native agarose gel electrophoresis.
- pICLpt was replicated in mock‐ or SCAI‐depleted extracts treated or not with Novobiocin (NVB; 150 µM) in the presence of [α‐32P]dATP, and reactions were analyzed by native agarose gel electrophoresis. RI, replication intermediates; OC, open circular; SC, supercoiled.
- Samples in (I) were digested with PsiI+XhoI or AflIII and analyzed on a denaturing polyacrylamide gel.
- Clonogenic survival of U2OS and U2OS/SCAI KO cells treated or not with Novobiocin (NVB; 50 µM) and subjected to various doses of MMC for 24 h (mean ± SEM; n = 3 independent experiments).
Figure 5. SCAI prevents Polθ‐mediated MMEJ during ICL repair.
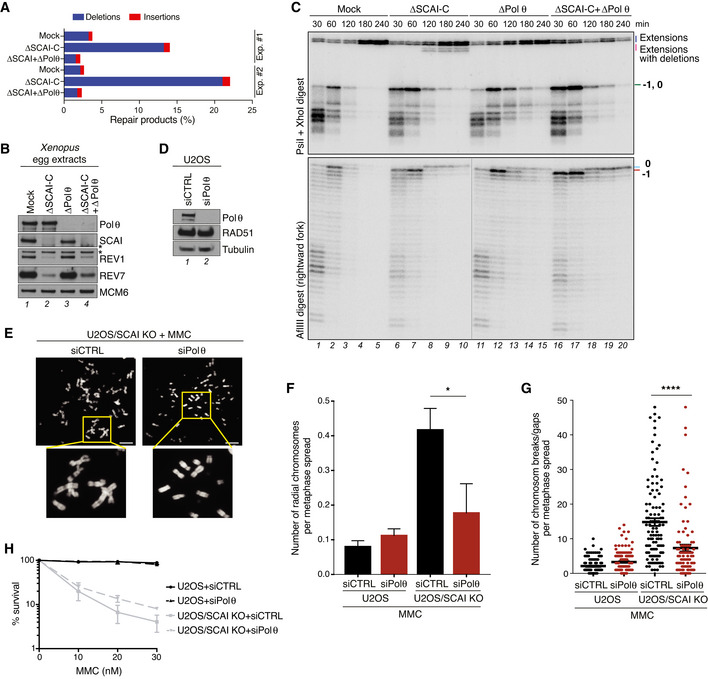
- pICLpt was replicated in egg extracts immunodepleted with indicated antibodies. Replication samples were amplified by PCR and analyzed by next‐generation sequencing for deletions and insertions. Results from two independent experiments are shown.
- Immunoblot analysis of extracts immunodepleted with indicated antibodies. Asterisks denote antibody cross‐reactivity.
- pICLpt was replicated in extracts immunodepleted as in (B), digested with PsiI+XhoI or AflIII and resolved on a denaturing polyacrylamide gel.
- Immunoblot analysis of Polθ siRNA knockdown efficiency in U2OS cells.
- Representative images of metaphase spreads from U2OS/SCAI KO cells transfected with indicated siRNAs and processed as in Fig 1I. Scale bars, 10 µm.
- Quantification of radial chromosomes in (E) (mean ± SD; 1,197, 241, 155, and 90 metaphase cells analyzed per condition (left to right) pooled from three independent experiments; *P < 0.05, two‐tailed t‐test).
- Quantification of chromosomal breaks/gaps in (E) (mean ± SEM; 150, 150, 114, and 90 metaphase cells analyzed per condition (left to right) pooled from three independent experiments; ****P < 0.0001, Mann–Whitney U test).
- Clonogenic survival of U2OS and U2OS/SCAI KO cells transfected with non‐targeting control (CTRL) or Polθ siRNAs and subjected to indicated doses of MMC for 24 h (mean ± SEM; n = 3 independent experiments).
Discussion
While much is known about the mechanisms underlying FA pathway activation, it remains largely unclear how individual steps in the processing of ICL repair intermediates are coordinated. Our findings reveal that SCAI exerts a unique and essential function during ICL resolution via the FA pathway by promoting error‐free repair (Fig 6). Accordingly, SCAI KO cells display extreme sensitivity to ICL‐inducing agents and other major hallmarks of FA pathway dysfunction (Fig 1). In the absence of SCAI, HR‐mediated resolution of ICL repair intermediates is impaired (Fig 3) and DSBs are instead preferentially processed by Polθ‐mediated MMEJ, leading to deletions and chromosomal aberrations (Fig 5). Underscoring a crucial role of SCAI in suppressing faulty repair, initiating ICL resolution via the otherwise protective FA pathway becomes highly toxic when SCAI is absent (Fig 2). Taken together, our data firmly establish SCAI as a novel and integral effector of faithful ICL repair via the FA pathway.
Figure 6. Model of SCAI function in ICL repair via the FA pathway.
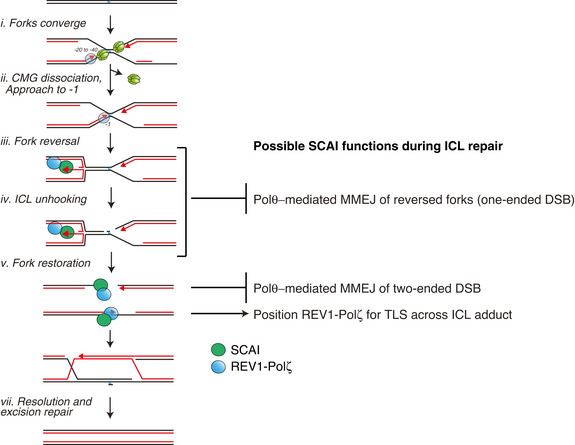
See main text for details.
A protective function of SCAI during ICL repair
A recent study published during the revision of our manuscript independently confirmed our finding that SCAI and REV3 interact and ensure efficient ICL repair (Adeyemi et al, 2021). In their study, Adeyemi et al (2021) showed that SCAI and REV3 form a complex (that they named Protexin) that protects stalled forks arising during ICL repair and upon other forms of genotoxic stress, thereby promoting downstream repair by HR. Our identification of a direct SCAI‐binding motif in REV3 corroborates and extends these findings by providing a molecular explanation for how the SCAI‐REV3 complex is formed (Fig 4E). Curiously, data from Adeyemi et al (2021) suggest that SCAI‐REV3 function during ICL repair is independent of other Polζ components (i.e., REV1 and REV7). In contrast, our proteomic analysis clearly identified all REV1‐Polζ components in SCAI immunoprecipitates and our data suggest that SCAI forms a constitutive complex with REV1‐Polζ (Fig 4). We note in this context that SCAI interacts with REV3 via a motif that does not overlap with its REV7‐ and POLD2‐interacting regions (Fig 4E). Hence, SCAI would not be expected to compete with other Polζ components for binding to REV3, and whether SCAI can form a complex with REV3 that excludes other Polζ components, which are known to greatly stimulate REV3 catalytic activity (Nelson et al, 1996), remains to be established.
Functionally, Adeyemi et al (2021) provided evidence that the SCAI‐REV3 complex protects stalled forks from hyper‐resection by EXO1. Importantly, upon fork convergence at an ICL, one of the two forks undergoes reversal to generate a substrate suitable for incisions by XPF‐ERCC1 (Fig 6, iii‐iv) (Amunugama et al, 2018). This fork reversal generates a one‐ended DSB, which could be the main structure protected by SCAI (Fig 6iii). Adeyemi et al (2021) further showed that the protective function of SCAI is dependent on FANCM translocase activity, which is presumed to generate the reversed fork during ICL repair (Gari et al, 2008; Zellweger et al, 2015; Amunugama et al, 2018). Thus, it is tempting to speculate that a primary function of SCAI in ICL repair is to protect the reversed fork from Polθ‐mediated MMEJ, which we clearly show here (Fig 5), and/or end resection. The accumulation of replication‐dependent well products observed in SCAI‐depleted extracts would be consistent with slow‐migrating branched structures generated by the fusions of reversed forks on plasmids (Fig 3F). However, we note that our assays monitoring repair products on denaturing gels (e.g., Fig 3L) or via DNA sequencing analysis (Figs 5A and EV5F) cannot discriminate between ligation products arising from reversed forks (Fig 6iii) or two‐ended DSBs (Fig 6v), and more work will thus be needed to elucidate the exact nature of these fusions/deletion products. Nevertheless, our data show that SCAI‐Polζ recruitment during replication‐coupled ICL repair occurs early in the process, prior to ICL unhooking and two‐ended DSB generation (Fig 3D). Thus, we favor the idea that the rapid recruitment of SCAI to ICLs might position it at reversed forks to prevent illegitimate repair of the one‐ended DSB. Although MMEJ is ordinarily thought to be a salvage pathway for DSB repair of HR‐deficient cells (Ceccaldi et al, 2015; Mateos‐Gomez et al, 2015), a single reversed fork undergoing fusion to another DSB in cells would likely be detrimental as it would disrupt DNA replication and trigger chromosome rearrangements. Consistently, we found that Polθ deletion suppressed radial chromosome formation in SCAI KO cells and moderately alleviated their sensitivity to MMC, suggesting a toxic function of Polθ in ICL repair that becomes evident in the absence of SCAI (Fig 5).
SCAI and repair pathway choice during ICL resolution
During classical two‐ended DSB repair, SCAI was suggested to regulate pathway choice and facilitate HR by binding to 53BP1 at DSBs, thereby counteracting RIF1 function in promoting repair by NHEJ (Isobe et al, 2017). In contrast, during ICL repair, SCAI appears to have an important role in regulating the choice between HR and MMEJ. This difference in pathway regulation could reflect the nature of DSB intermediates generated during ICL repair. Indeed, the one‐ended DSB generated upon fork reversal contains a long 3′ ssDNA overhang that could span between ~ 30 and a few hundred nucleotides in length, depending on where the last Okazaki fragment was synthesized (Fig 6iii). Following ICL unhooking, one end of the two‐ended DSB also contains a similar 3′ overhang (Fig 6v). In either case, the DSB is already pre‐resected, and this 3′ ssDNA tail is likely inhibitory to Ku70/80 association and downstream repair by NHEJ (Daley & Wilson, 2005; Krasner et al, 2015), favoring repair by Polθ‐mediated MMEJ (Wyatt et al, 2016). Thus, DSB repair pathway utilization during ICL repair via the FA pathway might be inherently channeled toward HR and MMEJ, and in analogy to its proposed role at two‐ended DSBs we found that SCAI is a critical regulator of the choice between these pathways. However, more work is needed to clarify whether this function of SCAI also involves regulation of resection via the RIF1‐53BP1 axis.
SCAI and REV1‐Polζ function
In addition to its crucial function in protecting against erroneous repair of ICLs, our data suggest that SCAI may also impact TLS as part of the REV1‐Polζ complex. Consistently, a recent study on the function of Polζ at heterochromatin regions also reported an interaction between SCAI and REV3 (Yamin et al, 2021). In egg extracts, SCAI immunodepletion co‐depletes REV3, suggesting tight REV3‐SCAI association, and SCAI is likely a novel constitutive component of REV1‐Polζ (note that the converse is not true as some SCAI is not associated with REV1‐Polζ). This interaction between SCAI and REV1‐Polζ, which we show is largely mediated by direct SCAI binding to a conserved peptide motif within the REV3 PCD domain, may at least partly explain how SCAI is recruited to DPCs or UV‐damaged chromatin in the absence of DSBs (Figs EV3A–D and EV4). Consistent with the possibility that SCAI positively regulates REV1‐Polζ function, we found that SCAI KO cells are sensitive to UV treatment albeit less so than to MMC (Fig EV4I). We speculate that this mild sensitivity to UV damage reflects REV1‐Polζ defects in performing TLS across UV lesions (i.e., 6–4 photoproducts). In contrast, the extreme hypersensitivity of SCAI KO cells to MMC is likely due to multiple functions of SCAI during ICL repair (i.e., protecting DSBs from MMEJ as described above and stimulating REV1‐Polζ function). During ICL repair, the rapid recruitment of SCAI to ICLs might position it at reversed forks to prevent illegitimate repair, while simultaneously placing REV1‐Polζ close to the 3′ end to ensure downstream TLS across the unhooked ICL (Fig 6). This would be an elegant mechanism for coupling fork protection with TLS‐mediated bypass of the lesion. Unfortunately, we have not been able to produce recombinant REV1‐Polζ, precluding us from assessing REV1‐Polζ function in the presence or absence of SCAI in egg extracts since REV1‐Polζ is co‐depleted upon SCAI immunodepletion.
SCAI and FA
We found that loss of SCAI manifests with principal cellular hallmarks of FA gene dysfunctionality (Fig 1). Consistently, our previous studies revealed that Scai KO mice display subfertility and meiotic defects but overtly normal development (Hansen et al, 2016), similar to many FA mouse models (Bakker et al, 2013). Accordingly, it is possible that loss‐of‐function SCAI mutations may exist in patients exhibiting FA‐like symptoms but no alterations of known complementation groups. This will be an interesting prospect for future research.
Materials and Methods
Cell culture and colony survival assays
Human U2OS cells were obtained from ATCC, cultured in DMEM containing 10% FBS, and regularly tested negative for mycoplasma infection. The cell line was not authenticated. U2OS cell lines stably expressing GFP‐SCAI were described previously (Hansen et al, 2016). To generate U2OS SCAI knockout (KO) cell lines, parental U2OS cells were co‐transfected with sgRNA plasmid and pX459/Cas9‐(pSpCas9(BB)‐2A‐Puro) and selected briefly with puromycin during clonal selection. Clones were screened for SCAI expression by immunoblotting. U2OS/SCAI KO#4.2 and U2OS/SCAI KO#4.3 cell lines were derived from independent clones. U2OS/SCAI KO#4.2 cells stably reconstituted with Strep‐HA‐tagged full‐length SCAI were generated by transfection of pcDNA4/TO/Strep‐HA‐SCAI and selection of positive clones in medium containing zeocin (Invitrogen).
For colony formation assays, 300–3,000 cells were seeded in duplicates in 6 cm dishes and allowed to adhere for a minimum of 16 h. If cells were treated with siRNAs, transfections were performed 48 h prior to reseeding to 6 cm dishes. Cells were then treated with indicated doses of UV‐C or indicated concentrations of cisplatin or MMC for 24 h, washed and released into drug‐free medium for 7–8 days. For experiments with Novobiocin, cells were treated with Novobiocin (50 µM) 16 h post‐seeding, and MMC was added after an additional 8 h. Twenty‐four hours later, cells were washed and incubated in Novobiocin‐containing medium for 7–8 days. Plates were then washed twice in PBS, left to dry and stained with cell staining solution (0.5% w/v Crystal Violet, 25% v/v methanol). Finally, the plates were washed extensively in deionized water. Colonies were counted with GelCount Colony Counter (Oxford Optronix) and surviving fraction calculated as: colonies/(seeded colonies x plating efficiency).
Plasmids and siRNAs (human)
Full‐length REV3‐GFP cDNA (kind gift from Christine Canman, University of Michigan) was inserted into pcDNA5/FRT/hCMV (for high expression) (kind gift from Jakob Nilsson, University of Copenhagen, Denmark) using XhoI and PmeI restriction enzymes, and SCAI‐binding‐deficient versions were generated by Q5 mutagenesis (New England Biolabs), deleting either aa 1124–1134 or aa 1077–1117. To generate pcDNA4/TO‐Flag‐3xNLS‐REV3 (995–1194) expression construct, REV3 (995–1194) was PCR amplified from REV3 cDNA and inserted into pcDNA4/TO‐FLAG‐3xNLS using KpnI and XhoI restriction enzymes. Plasmid encoding pEGFP‐Polη was a kind gift from Alan Lehmann and was described previously (Kannouche et al, 2001). pEGFP‐C1‐AHDC1 WT was generated by inserting AHDC1 cDNA into pEGFP‐C1 vector, and the SCAI‐binding‐deficient version was generated by Q5 mutagenesis (New England Biolabs), deleting aa 634‐643 (SPPRCWTPS). Plasmid for generation of SCAI knockout cells by CRISPR/Cas9 was described previously (Hansen et al, 2016). The sgRNA sequence used was as follows: sgRNA‐SCAI#4.1: 5′‐GGCTTGAAGCGCTGGCAAATAGG‐3′ and sgRNA‐FANCA: 5′‐TTCTGGAAAGCAGACAACCA‐3′.
siRNAs used in this study included: Non‐targeting control (siCTRL): 5′‐GGGAUACCUAGACGUUCUA‐3′; siPolθ#2: 5′‐GGAUAAAGCUUAAUACAGATT‐3′; siPolθ#4: 5′‐UUACUGACUCCAAAAGCGGTATT‐3′ (used only for metaphase spread experiments); siREV3: 5′‐GCAAUUUUGAACCUUAUGGTT‐3′; siREV7: 5′‐GAUGCAGCUUUACGUGGAATT‐3′; siREV1: 5′‐CUGCCAGGUCCAAGCAAUAUATT‐3′; siFANCA: 5′‐GGAGGUCACGGUUGAUGUATT‐3′.
Immunochemical methods (human)
For immunoblotting and immunoprecipitation, which were essentially done as described by Poulsen et al (2012), cells were lysed in EBC buffer (50 mM Tris, pH 7.5; 150 mM NaCl; 1 mM EDTA; 0.5% NP40; 1 mM DTT) supplemented with protease and phosphatase inhibitors. All immunoprecipitations were benzonase‐treated and sonicated to minimize chromatin‐mediated interactions. Cleared lysates were then incubated with GFP‐Trap Agarose (Chromotek) for 2 h on an end‐over‐end rotator at 4°C and washed in EBC buffer. All immunoblotting experiments were performed at least twice, and representative data are shown.
Antibodies to human proteins used in this study included: RPA2 (NA19L, Calbiochem (1:500)); RPA2‐pS4/S8 (A300‐245A, Bethyl (1:1,000)); SCAI (custom‐made sheep antibody raised against full‐length human SCAI (1:1,000) (Hansen et al, 2016); 12892S, Cell Signaling (1:500)); Vinculin (V9131, Sigma (1:10,000)); RAD51 (PC130, Ab‐1, Millipore (1:500)); REV3 (GTX17515, GeneTex (1:250)); REV7 (ab180579, Abcam (1:250)); REV1 (sc‐393022, Santa Cruz (1:250)); GFP (11814460001, Roche Diagnostics GmbH (1:500)); Tubulin alpha (T9026, Sigma‐Aldrich (1:5000)); FLAG (F‐1804, Sigma (1:500)); FANCA (A301‐980A, Bethyl (1:1,000)); FANCD2 (NB100‐182, Novusbio (1:1,000)); CHK1 pS345 (2348, Cell Signaling (1:500)); CHK1 (sc‐8408 (Clone G4), Santa Cruz (1:1,000)); XPA (A301‐780A, Bethyl (1:1000)); and Polθ (custom‐made antibody raised against Xenopus Polθ, see below for details (1:1,000)).
Immunofluorescence and high‐content imaging analysis (human)
Cells were pre‐extracted in PBS containing 0.2% Triton X‐100 for 2 min on ice, before fixation with 4% formaldehyde for 15 min, blocked with 5% BSA‐PBS for 1 h, and incubated with primary antibodies diluted in blocking solution for 1 h at room temperature (RT). This was followed by staining with secondary antibodies (Alexa Fluor; Life Technologies) and 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI, 0.5 μg/ml, DNA staining) diluted in blocking solution for 1 h at RT. Coverslips were then mounted onto glass slides using Mowiol‐based mounting medium. CPD staining was performed according to manufacturer's guidelines (Cosmo Bio LTD).
Images were acquired with a Leica AF6000 wide‐field microscope (Leica Microsystems) equipped with HC Plan‐Apochromat 63×/1.4 oil immersion objective, using standard settings. Image acquisition and analysis were carried out with Leica Application Suite X software (Leica Microsystems). Raw images were exported as TIFF files, and if adjustments in image contrast and brightness were applied, identical settings were used on all images of a given experiment.
Quantitative image‐based cytometry (QIBC) was performed as described previously (Toledo et al, 2013). Upon staining as described above, images were acquired with a ScanR inverted high‐content screening microscope (Olympus) equipped with wide‐field optics, UPLSAPO dry objectives (20x, 0.75‐NA), fast excitation, and emission filter‐wheel devices for DAPI, FITC (fluorescein isothiocyanate), Cy3 and Cy5 wavelengths, an MT20 illumination system, and a digital monochrome Hamamatsu ORCA‐R2 CCD camera. Automated and unbiased image analysis was carried out with the ScanR analysis software. Data were exported and processed using Spotfire software (Tibco). Antibodies used for immunofluorescence in this study included: PCNA (#2037, Triolab Immunoconcepts (1:500)); γH2AX (05‐636 (Clone JBW301), Millipore (1:500)); RPA1 (#79398, Abcam (1:1,000)); RAD51 (70‐002, BioAcademia (1:300)); 53BP1 (M3802, Merck (1:1,000)); and CPDs (NM‐DND‐001, Cosmo Bio LTD (1:500)).
UV‐C laser micro‐irradiation (human)
UV‐C laser micro‐irradiation was performed essentially as described previously (Apelt et al, 2020). Briefly, U2OS cells stably expressing GFP‐SCAI or transiently transfected with GFP‐Polη were grown on 18‐mm quartz coverslips. Medium was replaced with CO2‐independent Leibovitz L‐15 medium (Thermo Fisher Scientific) and coverslips were transferred to a Chamlide CMB magnetic chamber. UV‐C laser tracks were generated with a diode‐pumped solid‐state 266‐nm Yttrium Aluminum Garnet laser (average power 5 mW, repetition rate up to 10 kHz, and pulse length 1 ns), which was integrated in a UGA‐42‐Caliburn/2L Spot Illumination system (Rapp OptoElectronic). Micro‐irradiation was combined with live‐cell imaging in an environmental chamber set to 37°C on an all‐quartz wide‐field fluorescence Zeiss Axio Observer 7 microscope, using a 100× (1.2‐NA) ultrafluar glycerol immersion objective. The laser system is coupled to the microscope via a TriggerBox, and a neutral density (ND‐1) filter blocks 90% of the laser light. An HXP 120‐V metal‐halide lamp was used for excitation. Images were acquired in Zeiss ZEN and quantified in ImageJ.
Genome‐scale CRISPR screening (human)
Viral particles of the LCV2::TKOv3 and pLCKO2::TKOv3 sgRNA libraries were produced as previously described (Hart et al, 2017). The TKOv3 library contains 71,090 sgRNA sequences targeting 18,053 human protein coding genes with a modal number of four sgRNAs per gene. To identify genes required for cell survival in the presence of MMC, RPE1‐hTERT FLAG‐Cas9 TP53 −/− cells (kind gift from Daniel Durocher, Lunenfeld‐Tanenbaum Institute, Toronto, Canada) were transduced with the pLCKO2::TKOv3 library at an MOI of 0.19 at a coverage of 355‐fold sgRNA representation, which was maintained throughout the screen at each cell passage point. One day after transduction, transduced cells were selected for 24 h with 25 µg/ml puromycin. Cells were trypsinized and reseeded in the same plates while maintaining puromycin selection for another 24 h. Three days after transduction, which was considered the initial time point (t0), cells were pooled and passaged while cell pellets of two replicates of 2.7 × 107 cells were frozen for downstream processing. Cells were passaged after another 3 days, and 9 days after transduction (t6) cells were split into technical duplicates that were either mock treated or propagated in the presence of a low dose of MMC (21 nM) equivalent to pre‐determined LD20 concentrations in uninfected RPE1‐hTERT FLAG‐Cas9 TP53‐/‐ cells. Cells were subcultured every 3 days (t9, t12, and t15) in medium with or without MMC until the final time point at t18, at which point cell pellets from 4 × 107 cells were frozen from each replicate.
To identify genes whose loss suppresses the hypersensitivity of SCAI KO cells to MMC, U2OS/SCAI KO (#4.2) cells were transduced with the LCV2::TKOv3 library at an MOI of 0.3 at a coverage of 450‐fold sgRNA representation. 24 h after transduction, transduced cells were selected for 48 h with 1 µg/ml puromycin. Three days after transduction, which was considered the initial time point (t0), the transduced cells were split into three technical replicates (coverage of sgRNA representation was maintained at > 450× per replicate). Cells were passaged at day 3 and 6 after transduction, and 9 days after transduction (t6), three replicates of 3.6 × 107 cells were frozen for downstream processing. Ten days after infection, a near‐lethal dose (LD80) of MMC (4.5 nM) was added to the cells. Twenty‐four hours after addition of MMC (t8), cells were washed once in PBS, supplied with fresh medium and passaged 2 days later (t10). This regimen of MMC exposure was carried out a total of three times (MMC treatment at t7, t11, and t15). Surviving cells were collected 23 days post‐infection (t20) for downstream processing.
Genomic DNA from cells collected at T0 and T18 (dropout screen) or T6 and T20 (suppressor screen) was isolated as previously described (Chen et al, 2015) and sgRNA sequences amplified by PCR using Q5 Mastermix Next Ultra II (New England Biolabs) with the following primers: pLCKO2_forward: 5′‐GAGGGCCTATTTCCCATGATTC‐3′ and pLCKO2_reverse: 5′‐CAAACCCAGGGCTGCCTTGGAA‐3′; or LCV2_forward: 5′‐GAGGGCCTATTTCCCATGATTC‐3′ and LCV2_reverse: 5′‐GTTGCGAAAAAGAACGTTCACGG‐3′. This was followed by a second PCR reaction containing i5 and i7 multiplexing barcodes and final gel‐purified products were sequenced on Illumina NextSeq500. Fastq files were generated using bcl2fastq v2.19.1 and reads were trimmed to 20 bp using cutadapt 1.18 (Martin, 2011) removing a variable number of bp at start and end depending on the size of the primer stagger. MAGeCK 0.5.8 (Li et al, 2014) was used to assign the trimmed reads to the guides in the TKOv3 library and create the count matrix. To identify genes required for cell survival in the presence of MMC, gene scores (normZ values) were estimated from the count matrix using the drugZ algorithm (Colic et al, 2019), applying a NormZ value of < −3 as a cut‐off for significant hits. To identify genes whose loss suppresses the MMC hypersensitivity of SCAI KO cells, sgRNAs enriched at t20 following MMC treatment as compared to t6 were identified using MAGeCK analysis, applying an FDR value of < 0.2 as cut‐off for significant hits. To assess data quality of the CRISPR screens, we generated precision–recall curves through the BAGEL.py “pr” function (Hart & Moffat, 2016) using the core essential (CEGv2.txt) and non‐essential (NEGv1.txt) gene lists from https://github.com/hart‐lab/bagel, comparing T0 to T18 for mock‐treated cells.
Metaphase chromosome spread analysis (human)
To obtain metaphase cells, asynchronous cells were first arrested at G2/M boundary by exposure to the CDK1 inhibitor RO‐3306 (7 μM; APExBIO) for 12 h, or in the last 12 h of MMC (30 nM) and/or siRNA treatment. Cells were then released into pre‐warmed medium containing 100 ng/ml KaryoMAX (Colcemid; Life Technologies) for 1 h to allow progression into metaphase. Metaphase cells were collected by mitotic shake‐off. To prepare metaphase chromosome spreads, mitotic cells were swollen with pre‐warmed KCl (75 mM) at 37°C for 20 min and collected by centrifugation at 300 g for 5 min at RT. Cells were fixed by adding ice‐cold methanol:acetic acid (3:1) in a drop‐wise manner while vortexing gently. Fixed cells were dropped onto slides with moist to obtain chromosome spreads. Slides were mounted using Vectashield mounting medium with DAPI (Vector Labs). Images were captured using an Olympus BX63 microscope. All images were analyzed manually.
Production of recombinant proteins in bacteria
Full‐length Xenopus SCAI was sub‐cloned from cDNA into pGEX vector for expression of recombinant N‐terminally GST‐tagged products. Briefly, Escherichia coli (DE3) cells harboring vectors expressing GST‐xSCAI were grown in TB media supplemented with 100 mg ampicillin at 37°C at an OD600 of 0.8–1.0. Then, the temperature was reduced to 20°C and protein expression was induced with 0.5 mM IPTG for 16 h. Cells were harvested at 4,600 g for 15 min. Bacterial pellets were resuspended in lysis buffer containing 50 mM HEPES, pH 7.5; 500 mM NaCl; 10% glycerol; 2 mM DTT; and 10 mM MgCl2. Resuspended cells were lysed by sonication and cleared by centrifugation at 100,000 g at 4°C for 30 min. The cleared lysate was incubated with Glutathione agarose beads (Thermo Fischer) and incubated at 4°C for 2 h. After extensive washing, the GST‐tagged constructs were eluted in lysis buffer supplemented with 30 mM reduced glutathione. Fractions containing GST‐xSCAI were concentrated in a 10 kDa MWCO and loaded onto a Superdex200 10/300 column equilibrated in 20 mM HEPES, pH 7.5; 2.5% glycerol; 300 mM NaCl; and 1 mM DTT. The corresponding peak fractions were collected and concentrated in a 10 kDa MWCO concentrator and then flash frozen in liquid N2 and stored at −80°C until further use.
In vitro pull‐down assays
Streptavidin agarose beads were incubated in pull‐down buffer consisting of 20 mM HEPES pH 7.5; 5% glycerol; 150 mM NaCl; 1 mM DTT; and 0.1% Triton‐X‐100. Biotin‐xREV3 peptide (aa 1110–1179) (Biotin‐RAVSIRRKRIRSFLSLPMPTYNAETEDCDTDYKDVMSKLGFLVERCPSPINMSPPRCWSPTDPRAEEIVS) (2 µg) was used as a bait and incubated with either GST or GST‐xSCAI (5 µg) as prey. The bait was loaded onto 20 µl of streptavidin agarose beads before adding the prey. The reaction volume was topped up to 40 µl with buffer and incubated at 4°C for 1 h. Beads were spun down at 500 g for 3 min. The supernatant was removed, and beads were washed three times with 1 ml pull‐down buffer. Supernatant was removed completely, samples were boiled in 15 µl Laemmli sample loading buffer and run on a 4–12% Bis‐Tris gel. Bands were visualized by Coomassie brilliant blue staining.
Xenopus egg extracts and DNA replication reactions
Preparation of Xenopus egg extracts was performed as described previously (Lebofsky et al, 2009). For plasmid DNA replication, plasmids were first licensed in high‐speed supernatant (HSS) for 20–30 min at RT at a final concentration of 7.5 ng/μl. Replication was then initiated by addition of two volumes of nucleoplasmic egg extract (NPE). To visualize DNA replication intermediates, replication reactions were supplemented with [α‐32P]dATP. For each time point, 1 µl reaction was added to 5 μl of stop buffer (5% SDS, 80 mM Tris, pH 8.0; 0.13% phosphoric acid; and 10% Ficoll) supplemented with 1 μl of proteinase K (20 mg/ml) (Roche). Samples were incubated at 37°C for 1 h, separated by 0.9% native agarose gel electrophoresis and visualized by phosphorimager (Typhoon FLA 7000). Radioactive signal was quantified using ImageJ (NIH, USA).
Preparation of DNA constructs (Xenopus)
pDPC and pDPCLeads were generated by crosslinking M.HpaII site specifically on a fluorinated cytosine as previously described (Larsen et al, 2019). The cisplatin ICL containing plasmid (pICLpt) was a kind gift from Johannes Walter and Puck Knipscheer (Enoiu et al, 2012).
Antibodies and immunodepletions (Xenopus)
Antibodies against REV1 (REV1‐N and REV1‐C (Budzowska et al, 2015)), REV7 (Räschle et al, 2008), PCNA (Kochaniak et al, 2009), FANCD2 (Knipscheer et al, 2009), MCM6 (Semlow et al, 2016), RPA (Walter & Newport, 2000), ORC2 (Fang & Newport, 1993), RAD51 (Long et al, 2011), XPF (Douwel et al, 2014), FANCM (Sparks et al, 2019), and Polη (Gallina et al, 2021) were described previously. The antibody against phosphorylated CHK1 (S345) (Cell Signaling, #2341) is commercially available. The following antibodies were raised against the indicated peptides: SCAI‐C (Ac‐CDVRNLYLESTMDEY‐OH, New England Peptide), SCAI‐N (H2N‐MSGANEDDIPQVERKC‐amide, New England Peptide), BRCA2 (Ac‐KPHIKEDQNEPESNSEYC‐amide, New England Peptide) (Ambjørn et al, 2021), RAD51 (CAEAMFAINADGVGDAKD, Proteogenix), CtIP (CPRRRQPYNAIFTSKIKEQKT, Bethyl laboratories), FANCA (Ac‐CSFKAPDDYDDLFFEPVF‐OH, New England Peptide), 53BP1 (Ac‐CKHNTHPKYKHNHVSD‐OH, New England Peptide), REV3‐512 (Ac‐CLADLSIPQLD‐OH, New England Peptide), and REV3‐522 (H2N‐GTADENSDNPC‐amid, New England Peptide). The Polθ antibody was raised against a protein fragment of Xenopus laevis Polθ spanning amino acids 1239–1533 (Abgent) according to Deng et al (2019). The fragment was tagged on the C terminus with His6 and purified from bacteria.
To immunodeplete Xenopus egg extracts, Protein A Sepharose Fast Flow (PAS) (GE Health Care) beads were bound to indicated antibodies overnight at 4°C. When serum and affinity‐purified antibodies were used together for double depletions, affinity‐purified antibody or IgG was incubated for 1 h at RT on an end‐over‐end rotator, before serum antibody or pre‐immune serum were added and beads were incubated overnight at 4°C. The beads were then washed twice with 500 μl PBS, once with ELB buffer (10 mM HEPES, pH 7.7; 50 mM KCl; 2.5 mM MgCl2; and 250 mM sucrose), twice with ELB buffer supplemented with 0.5 M NaCl, and twice with ELB buffer. One volume of NPE or pre‐cleared HSS was then depleted by mixing with 0.2 volumes of antibody‐bound beads and incubating at RT for 15 min on an end‐over‐end rotator, before being harvested. This was repeated for indicated number of rounds.
The following volume ratios and rounds of depletions were used: REV1, beads–antibody ratio 1:3, HSS: 1 round REV1‐N and 1 round REV1‐C, NPE: 2 rounds REV1‐N and 1 round REV1‐C; SCAI (both antibodies), beads–antibody ratio 1:5, HSS: 2 rounds, NPE: 3–4 rounds; 53BP1, beads–antibody ratio 1:5, HSS: 2 rounds, NPE: 4 rounds; Polθ, beads–antibody ratio 1:5, HSS: 2 rounds, NPE: 3–4 rounds; BRCA2, beads–antibody ratio 1:5, HSS: 2 rounds, NPE: 3–4 rounds. All affinity purified antibody stocks are at 1 mg/ml.
Nascent leading strand analysis (Xenopus)
For nascent leading strand analysis, 3–4 µl of replication reaction was added into 10 volumes of transparent stop buffer (50 mM Tris‐HCl, pH 7.5; 0.5% SDS; 25 mM EDTA). Samples were treated with 2 µl RNase (4 mg/ml) for 30 min at 37°C and 2 µl proteinase K (20 mg/ml) overnight at RT. Replication intermediates were purified by phenol/chloroform extraction and ethanol precipitation and resolved in 8 µl 10 mM Tris‐HCl, pH 8.0, as described previously (Räschle et al, 2008; Knipscheer et al, 2009). DNA was digested with the indicated restriction enzymes for 2 h at 37°C and supplemented with 0.5 volumes of denaturing PAGE Gel Loading Buffer II (Life Technologies). The digested DNA products were resolved on a 6% polyacrylamide sequencing gel.
Immunoprecipitation (Xenopus)
For immunoprecipitations, 5 µl of Protein A Sepharose Fast Flow (PAS) (GE Health Care) beads were incubated with 10 µg antibody overnight at 4°C. Beads were washed four times with ELB buffer, dried with an ultrafine tip, and diluted NPE was added (5 µl NPE; 20 µl ELB buffer; and 0.625 µl 10% NP‐40). Beads were incubated for 2 h on an end‐over‐end rotator at 4°C and washed 4× with ELB buffer supplemented with 0.25% NP‐40 and resuspended in 2× Laemmli sample buffer.
pICLpt repair assay (Xenopus)
Timing of dual incisions on pICLpt was monitored as described previously (Knipscheer et al, 2012). Briefly, replication intermediates were purified by phenol/chloroform extraction and ethanol precipitation, followed by digestion with the HincII restriction enzyme (New England Biolabs) for 3.5 h at 37°C, and separation of the repair intermediates on a 0.8% native agarose gel. To monitor repair via HR, the same replication intermediates were digested by HincII and SapI. Percent repair was calculated as described previously and normalized using pQuant plasmid (Knipscheer et al, 2012).
Plasmid pull down (Xenopus)
Plasmid pull downs were performed as described previously (Budzowska et al, 2015). Briefly, 6 μl streptavidin‐coupled magnetic beads (Dynabead M‐280, Invitrogen) per pull‐down reaction were equilibrated with wash buffer 1 (50 mM Tris‐HCl, pH 7.5; 150 mM NaCl; 1mM EDTA, pH 8.0; 0.02% Tween‐20) and then incubated with 12 pmol of biotinylated LacI at RT for 40 min. The beads were washed four times with pull‐down buffer 1 (10 mM HEPES pH 7.7; 50 mM KCl; 2.5 mM MgCl2; 250 mM sucrose, 0.25 mg/ml BSA; 0.02% Tween‐20) and stored on ice. At the indicated time points, 8 μl of reaction was added to the beads and samples were rotated for 30 min at 4°C. The beads were subsequently washed twice in wash buffer 2 (10 mM HEPES, pH 7.7; 50 mM KCl; 2.5 mM MgCl2; 0.25 mg/ml BSA; 0.03% Tween‐20) and resuspended in 2× Laemmli sample buffer.
Chromatin spin down (Xenopus)
Demembranated Xenopus sperm chromatin was prepared as described (Sparks & Walter, 2019) and stored at −80°C at a concentration of 100,000 sperm chromatin/μl (320 ng/μl). For analysis of UV‐damaged chromatin, sperm chromatin was diluted to 25,000 sperm chromatin/μl in ELB buffer (10 mM HEPES, pH 7.7; 50 mM KCl; 2.5 mM MgCl2; and 250 mM sucrose), deposited on parafilm, and irradiated with the indicated dose of UV‐C. To analyze replication‐independent repair in non‐licensing egg extracts, one volume of HSS and two volumes of NPE were premixed prior to the addition of sperm chromatin (final concentration of 16 ng/μl). At the indicated time points, 8 μl of replication reaction was stopped with 60 μl of ELB buffer supplemented with 0.2% Triton‐X. The mixture was carefully layered on top of a sucrose cushion (10 mM HEPES, pH 7.7; 50 mM KCl; 2.5 mM MgCl2; and 500 mM sucrose) and spun for 1 min at 6,800 g in a swing bucket centrifuge at 4°C. The chromatin pellet was carefully washed twice with 200 μl of ice‐cold ELB buffer and resuspended in 2× Laemmli buffer.
Sequencing of replication products (Xenopus)
Sequencing of replication products was performed as previously described (Budzowska et al, 2015). In brief, pICLpt was replicated in mock‐, SCAI‐, or SCAI‐/Polθ‐depleted Xenopus egg extract, and DNA samples (7.5 μl) were collected at the end of the replication reaction (240 min). Samples were diluted in transparent stop buffer (50 mM Tris‐HCl, pH 7.5; 0.5% SDS; 10 mM EDTA), supplemented with 4 μl RNase A (4 mg/ml) at 37°C for 30 min, followed by incubation with 4 μl proteinase K (20 mg/ml) overnight at RT. DNA was phenol/chloroform extracted, ethanol precipitated, and resuspended in 7 μl 10 mM Tris‐HCl, pH 8.0. Samples were amplified with KAPA HiFi DNA polymerase (Kapa Biosystems) for 12 PCR cycles (primers are F1: NNNN‐ATGAAGATCCCTCGACCTGC; R1: NNNN‐CCAATACGCAAACCGCCTC, where N represent random nucleotides to optimize sequencing.) The PCR samples were resolved on an 8% TBE polyacrylamide gel, stained with SYBR Gold, and PCR products of the appropriate size (around 174 nt) were excised. The gel slices were minced into smaller pieces, elution buffer (10 mM Tris, pH 8.0, 1 mM EDTA pH 8.0; 300 mM NaCl) was added to the gel pieces, and the mixture was agitated in Eppendorf thermomixer overnight at 30°C. The eluted DNA was recovered by centrifugation through a homemade nitex column. The recovered supernatant was precipitated with 100% ethanol in the presence of 0.3 M sodium acetate pH 5.3. DNA was resuspended in 12 µl of 10 mM Tris, pH 8.0, ligated to Illumina adaptors according to the manufacturer's instructions, and sequenced using a MiSeq sequencer (150 bp read length, paired ends) at the Danish National High‐Throughput Sequencing Center.
Sequencing data analysis
Sequencing data analysis was performed as described previously (Gallina et al, 2021). Sequencing data were demultiplexed and exported to paired FASTQ files. The paired FASTQ files were adaptor trimmed and quality controlled using AdapterRemoval (v. 2.2.2. –minlength 30,–mm 3–trimns–trimqualities–minquality 2–mm 3) (Schubert et al, 2016). Each read pair was collapsed into a single consensus sequence with a minimum overlapping length of 11 bases. In order to compute the base‐specific variation on the region of interest, quality‐controlled reads were then mapped to amplicon sequence using bwa mem alignment (Li & Durbin, 2009). BAM files were then filtered for high‐quality mapping reads using samtools (v. 1.9. ‐q30 –F4) (Li & Durbin, 2009) and sorted by mapping region with Picard (v. 2.9.1; http://broadinstitute.github.io/picard/). The position‐wise read coverage and base distribution could then be computed by processing the output of samtools mpileup (v. 1.9), using an in‐house python script for each sample. To adjust for sample differences in sequencing depth, the relative base frequency was used for the downstream analysis. Subsequent statistical analysis and plotting were conducted using Rstudio (v. 3.5.2—code available by request).
CHROMASS (Xenopus)
CHROMASS experiments were performed as previously described (Räschle et al, 2015). Briefly, isolated sperm chromatin was left untreated or exposed to UV‐C (2,000 J/m2) in quadruplicate. The sperm chromatin was then incubated at a final concentration of 16 ng/μl in non‐licensing egg extracts. Reactions were stopped after 30 min with 60 μl of ELB buffer supplemented with 0.2% Triton‐X, and chromatin spin down was performed as described above. The chromatin pellet was then resuspended in 50 μl denaturation buffer (8 M urea; 100 mM Tris‐HCl, pH 8.0), and transferred to a new low binding tube. Cysteines were reduced (1 mM DTT for 15 min at RT) and alkylated (5 mM chloroacetamide for 40 min at RT protected from light). Proteins were first digested with 0.5 μg LysC (2.5 h at RT) and then with 0.5 μg sequencing‐grade trypsin at 30°C overnight. Peptides were acidified by addition of 10% trifluoroacetic acid, reducing pH to < 4, followed by addition of 400 mM NaCl, and purified by StageTip (C18 material). For this, StageTips were first activated in 100% methanol, then equilibrated in 80% acetonitrile in 0.1% formic acid, and finally washed twice in 0.1% formic acid. Samples were loaded on the equilibrated StageTips and washed twice with 50 μl 0.1% formic acid. StageTip elution was performed with 80 μl of 25% acetonitrile in 0.1% formic acid, eluted samples were dried to completion in a SpeedVac at 60°C, dissolved in 10 μl 0.1% formic acid, and stored at −20°C until MS analysis.
SCAI immunoprecipitation for MS (Xenopus)
SCAI immunoprecipitations were performed essentially as described above using IgG, SCAI‐N, or SCAI‐C antibodies, additionally benzonase was added 1:100 to egg extracts and IPs were washed 3× with ELB buffer supplemented with 500 mM NaCl and 0.25% NP‐40 and 3x with ELB buffer supplemented with 500 nM NaCl. Beads were then dried using an ultrafine tip, after which 300 μl of ice‐cold 50 mM ammonium bicarbonate was added. Subsequently, 0.5 μg sequence‐grade trypsin was added, and samples were incubated shaking at 1,200 rpm at 4°C for 1 h. Samples were then allowed to warm to RT and incubated overnight while shaking at 1,200 rpm. Digested peptides were cleared by centrifugation through 0.45 μm spin filters, after which tris(2‐carboxyethyl) phosphine and chloroacetamide were added to final concentrations of 5 and 10 mM, respectively. Samples were incubated in the dark, shaking at 350 rpm, and 30°C for 30 min, prior to acidification by addition of trifluoroacetic acid to a final concentration of 0.5% (vol/vol). Peptides were purified via StageTip as described above.
SCAI immunodepletion proteome analysis
NPE proteome analyses were performed in quadruplicate, using mock‐depleted NPE or NPE depleted for SCAI using either the ΔSCAI‐C or ΔSCAI‐N antibody. Per sample, 4 μl of mock‐ or SCAI‐depleted NPE, containing ~ 100 μg of protein, was used for digestion. One μg sequencing‐grade Trypsin (Sigma) was added directly to the samples, after which they were incubated at RT for 10 min. Subsequently, samples were diluted 10‐fold with 50 mM TRIS pH 8.5, and another 1 μg of trypsin was added for digestion overnight at RT. Next, tris(2‐carboxyethyl)phosphine (TCEP) and chloroacetamide (CAA) were added to final concentrations of 10 mM, and samples were incubated at 30°C for 30 min. Samples were clarified of minor precipitation by passing them through 0.45 μm spin filters. Tryptic peptides were fractionated on‐StageTip at high pH essentially as described previously (Hendriks et al, 2018). Just prior to basifying and loading the samples on StageTips, another 1 μg of trypsin was added for 30 min at RT. Peptides were eluted from StageTips as eight fractions (F1–8) using 80 μl of 2, 4, 7, 10, 13, 16, 20, and 25% acetonitrile (ACN) in 50 mM ammonium hydroxide. All fractions were dried to completion in LoBind tubes, using a SpeedVac for 2 h at 60°C, after which the dried peptides were dissolved using 20 μl of 0.1% formic acid. Between 3 and 12 μl were used from each fraction, depending on peptide content, to equalize protein load for analysis by MS.
MS data acquisition (CHROMASS and SCAI‐IP)
MS samples were analyzed on an EASY‐nLC 1200 system (Thermo) coupled to a Q Exactive HF‐X Hybrid Quadrupole‐Orbitrap mass spectrometer (Thermo). Separation of peptides was performed using 15‐cm columns (75 μm internal diameter) packed in‐house with ReproSil‐Pur 120 C18‐AQ 1.9 μm beads (Dr. Maisch). Elution of peptides from the column was achieved using a gradient ranging from buffer A (0.1% formic acid) to buffer B (80% acetonitrile in 0.1% formic acid), at a flow rate of 250 nl/min. Gradient length was 80 min per sample, including ramp‐up and wash‐out. The analytical gradient was 57.5 min ranging from 8% to 28% buffer B for CHROMASS samples, or 57.0 min ranging from 7% to 28% buffer B for SCAI‐IP samples. The column was heated to 40°C using a column oven, and ionization was achieved using a NanoSpray Flex ion source (Thermo). Spray voltage was set at 2 kV, ion transfer tube temperature to 275°C, and RF funnel level to 40%. Measurements were performed with a full scan range 300–1,750 m/z, MS1 resolution of 60,000, MS1 AGC target of 3,000,000, and MS1 maximum injection time of 60 ms. Precursors with charges 2–6 were selected for fragmentation using an isolation width of 1.3 m/z and fragmented using higher‐energy collision disassociation (HCD) with a normalized collision energy of 25. Precursors were excluded from re‐sequencing by setting a dynamic exclusion of 60 s (CHROMASS) or 80 s (SCAI‐IP). MS2 AGC target was set to 200,000 and minimum MS2 AGC target to 20,000. For CHROMASS runs, MS2 maximum injection time was 90 ms, MS2 resolution was 45,000, and loop count was 9. For SCAI‐IP runs, MS2 maximum injection time was 60 ms, MS2 resolution was 30,000, and loop count was 14. CHROMASS samples were measured as two technical replicates using identical instrument settings.
MS data acquisition (NPE proteome)
NPE proteome samples were analyzed on an EASY‐nLC 1200 system (Thermo) coupled to an Orbitrap Exploris™ 480 mass spectrometer (Thermo). Separation of peptides was performed using 15‐cm columns (75 µm internal diameter) packed in‐house with ReproSil‐Pur 120 C18‐AQ 1.9 µm beads (Dr. Maisch). Elution of peptides from the column was achieved using a gradient ranging from buffer A (0.1% formic acid) to buffer B (80% acetonitrile in 0.1% formic acid), at a flow rate of 250 nl/min. Gradient length was 80 min per sample, including ramp‐up and wash‐out, with an analytical gradient of 60 min ranging in buffer B from 5 to 29% for F1, 5 to 33% for F2, 5 to 35% for F3, 6 to 37% for F4, 7 to 39% for F5, 8 to 41% for F6, 9 to 43% for F7, and 10 to 45% for F8. The column was heated to 40°C using a column oven, and ionization was achieved using a NanoSpray Flex™ NG ion source (Thermo). Spray voltage was set at 2 kV, ion transfer tube temperature to 275°C, and RF funnel level to 40%. Full scan range was set to 300–1,300 m/z, MS1 resolution to 120,000, MS1 AGC target to “200” (2,000,000 charges), and MS1 maximum injection time to “Auto.” Precursors with charges 2–6 were selected for fragmentation using an isolation width of 1.3 m/z, and fragmented using higher‐energy collision disassociation (HCD) with normalized collision energy of 25. Monoisotopic precursor selection (MIPS) was enabled in “Peptide” mode. Precursors were prevented from being repeatedly sequenced by setting expected peak width to 30 s, and setting dynamic exclusion duration to 60 s, with an exclusion mass tolerance of 15 ppm, exclusion of isotopes, and exclusion of alternate charge states for the same precursor. MS/MS resolution was set to 15,000, MS/MS AGC target to “200” (200,000 charges), MS/MS intensity threshold to 430,000, MS/MS maximum injection time to “Auto”, and TopN to 18.
MS data analysis
All MS RAW data were analyzed using the freely available MaxQuant software (Cox & Mann, 2008; Cox et al, 2011), v. 1.5.3.30. CHROMASS data and SCAI‐IP data were processed in separate computational runs. Default MaxQuant settings were used, with exceptions specified below. For generation of theoretical spectral libraries, the Xenopus laevis FASTA database was downloaded from UniProt on the February 10th 2018. In silico digestion of proteins to generate theoretical peptides was performed with trypsin, allowing up to three missed cleavages. Allowed variable modifications were oxidation of methionine (default) and protein N‐terminal acetylation (default) for all samples. Maximum variable modifications per peptide were reduced to 3. Label‐free quantification (LFQ) was enabled (Cox et al, 2014), with “Fast LFQ” disabled for all data, and “Skip normalization” enabled for SCAI‐IP data. Stringent MaxQuant 1% FDR data filtering at the PSM and protein levels was applied (default). Second peptide search was enabled. Matching between runs was enabled, with an alignment window of 20 min and a match time window of 1 min.
MS data annotation and quantification
The Xenopus laevis FASTA database downloaded from UniProt lacked comprehensive gene name annotation. Missing or uninformative gene names were, when possible, semi‐automatically curated, as described previously (Gallina et al, 2021). Quantification of the MaxQuant output files (“proteinGroups.txt”) was performed using Perseus software (Tyanova et al, 2016). For quantification purposes, all protein LFQ intensity values were log2 transformed, and filtered for presence in 4 of 4 replicates (n = 4/4) in at least one experimental condition. Missing values were imputed below the global experimental detection limit at a downshift of 1.8 and a randomized width of 0.3 (in log2 space; Perseus default). Statistical significance of differences was tested using two‐tailed Student's t‐testing, with permutation‐based FDR control applied at s0 values of 0.5 (CHROMASS; UV/mock), 1 (CHROMASS; mock/no‐DNA and UV/no‐DNA), or 2 (SCAI‐IP). In case of CHROMASS, proteins were filtered to be significantly enriched at FDR < 5% in either the mock/no‐DNA or UV/no‐DNA two‐sample t‐tests, to qualify for the UV/mock two‐sample t‐test. To assign proteins as highly significantly enriched in the SCAI‐IP, proteins were filtered for significance at FDR < 1% for both separate SCAI antibodies as well as both antibodies combined. Further filtering for SCAI interactors was performed by demanding significant enrichment when performing spectral counting at P < 5%. All P values and FDR‐adjusted q‐values are reported in Dataset EV3 (SCAI‐IP), Dataset EV4 (NPE proteome), and Dataset EV5 (CHROMASS).
Author contributions
Lisa Schubert: Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Project administration. Ivo Alexander Hendriks: Data curation; Investigation; Methodology. Emil P T Hertz: Data curation; Formal analysis; Investigation; Methodology. Wei Wu: Formal analysis; Investigation. Selene Sellés‐Baiget: Investigation. Saskia Hoffmann: Investigation. Keerthana Viswalingam: Investigation. Irene Gallina: Investigation. Satyakrishna Pentakota: Investigation. Bente Benedict: Investigation. Joachim Johansen: Formal analysis; Investigation. Katja Apelt: Investigation. Martijn Luijsterburg: Supervision. Simon Rasmussen: Supervision. Michael Lisby: Supervision. Ying Liu: Supervision. Michael Lund Nielsen: Supervision. Niels Mailand: Conceptualization; Supervision; Funding acquisition; Investigation; Writing—original draft; Project administration; Writing—review & editing. Julien Duxin: Conceptualization; Supervision; Funding acquisition; Investigation; Writing—original draft; Project administration; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
LS performed most of the experiments. Unless otherwise stated, experiments in human cells were performed under the supervision of NM, while experiments in Xenopus egg extracts were done under the supervision of JPD, IAH performed and analyzed MS experiments under the supervision of MLN; EPTH and KSV performed and analyzed genome‐scale CRISPR/Cas9 screens under the supervision of ML and NM; WW performed and analyzed the metaphase spread experiments under the supervision of YL; SP performed the experiment in Fig EV3I; IG performed the experiment in Fig EV5A; SSB performed the experiments in Figure EV2B,C; SH performed the experiments in Fig EV3E and F, as well as the experiments in Fig 2H–J with assistance of EPTH; and BB performed the experiment in Fig 4B. Experiment shown in Fig EV4G and H was performed in the laboratory of MSL with assistance of KA; JJ analyzed the sequencing data under the supervision of SR; LS, NM and JPD designed and analyzed experiments, and NM and JPD wrote the manuscript. All authors edited the manuscript.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Acknowledgements
We thank Daniel Durocher, Christine Canman, Jakob Nilsson, Alan Lehmann, Johannes Walter, and Puck Knipscheer for providing reagents, members of the Duxin and Mailand laboratories for helpful discussions, and Markus Räschle for preparation of Fig EV3A. This work was supported by grants from Novo Nordisk Foundation (grants no. NNF14CC0001, NNF16CC0020906, NNF19OC0055203, and NNF18OC0030752), Lundbeck Foundation (grant no. R223‐2016‐281), Independent Research Fund Denmark (grant no. 7016‐00055B), European Research Council (grants no. 616236‐DDRegulation and 715975‐DPC_REPAIR), European Commission's Horizon 2020 Research and Innovation Programme (Marie Skłodowska‐Curie Innovative Training Networks agreement no. 859853 (Antihelix)), and Danish National Research Foundation (grant no. DNRF115). We thank Magali Michaut and the DanStem Genomics Platform for technical support and use of instruments. Data processing and analysis were performed using the DeiC National Life Science Supercomputer at DTU (www.computerome.dk).
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
EMBO reports (2022) 23: e53639.
Contributor Information
Niels Mailand, Email: niels.mailand@cpr.ku.dk.
Julien P Duxin, Email: julien.duxin@cpr.ku.dk.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository. For CHROMASS and SCAI IP: PXD024280 (http://www.ebi.ac.uk/pride/archive/projects/PXD024280); For NPE proteome: PXD029409 (http://www.ebi.ac.uk/pride/archive/projects/PXD029409).
References
- Adeyemi RO, Willis NA, Elia AEH, Clairmont C, Li S, Wu X, D’Andrea AD, Scully R, Elledge SJ (2021) The Protexin complex counters resection on stalled forks to promote homologous recombination and crosslink repair. Mol Cell 81: 4440–4456.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M (2000) DNA replication is required To elicit cellular responses to psoralen‐induced DNA interstrand cross‐links. Mol Cell Biol 20: 8283–8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambjørn SM, Duxin JP, Hertz EPT, Nasa I, Duro J, Kruse T, Lopez‐Mendez B, Rymarczyk B, Cressey LE, van Overeem Hansen T et al (2021) A complex of BRCA2 and PP2A‐B56 is required for DNA repair by homologous recombination. Nat Commun 12: 5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunugama R, Willcox S, Wu RA, Abdullah UB, El‐Sagheer AH, Brown T, McHugh PJ, Griffith JD, Walter JC (2018) Replication fork reversal during DNA interstrand crosslink repair requires CMG unloading. Cell Rep 23: 3419–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelt K, Zoutendijk I, Gout DY, Wondergem AP, van den Heuvel D, Luijsterburg MS (2020) Human HMGN1 and HMGN2 are not required for transcription‐coupled DNA repair. Sci Rep 10: 4332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker ST, de Winter JP, te Riele H (2013) Learning from a paradox: recent insights into Fanconi anaemia through studying mouse models. Dis Model Mech 6: 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt DT, Baarlink C, Kitzing TM, Kremmer E, Ivaska J, Nollau P, Grosse R (2009) SCAI acts as a suppressor of cancer cell invasion through the transcriptional control of beta1‐integrin. Nat Cell Biol 11: 557–568 [DOI] [PubMed] [Google Scholar]
- Budzowska M, Graham TGW, Sobeck A, Waga S, Walter JC (2015) Regulation of the Rev1‐pol zeta complex during bypass of a DNA interstrand cross‐link. EMBO J 34: 1971–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ et al (2015) Homologous‐recombination‐deficient tumours are dependent on Polθ‐mediated repair. Nature 518: 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, D’Andrea AD (2016) The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 17: 337–349 [DOI] [PubMed] [Google Scholar]
- Chan SH, Yu AM, McVey M (2010) Dual roles for DNA polymerase theta in alternative end‐joining repair of double‐strand breaks in Drosophila . PLoS Genet 6: e1001005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sanjana N, Zheng K, Shalem O, Lee K, Shi XI, Scott D, Song J, Pan J, Weissleder R et al (2015) Genome‐wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160: 1246–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colic M, Wang G, Zimmermann M, Mascall K, McLaughlin M, Bertolet L, Lenoir WF, Moffat J, Angers S, Durocher D et al (2019) Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med 11: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Daley JM, Wilson TE (2005) Rejoining of DNA double‐strand breaks as a function of overhang length. Mol Cell Biol 25: 896–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wu RA, Sonneville R, Kochenova OV, Labib K, Pellman D, Walter JC (2019) Mitotic CDK promotes replisome disassembly, fork breakage, and complex DNA rearrangements. Mol Cell 73: 915–929.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douwel DK, Boonen RACM, Long DT, Szypowska AA, Räschle M, Walter JC, Knipscheer P (2014) XPF‐ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54: 460–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, Walter JC (2014) Repair of a DNA‐protein crosslink by replication‐coupled proteolysis. Cell 159: 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoiu M, Ho TV, Long DT, Walter JC, Schärer OD (2012) Construction of plasmids containing site‐specific DNA interstrand cross‐links for biochemical and cell biological studies. Methods Mol Biol 920: 203–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F, Newport JW (1993) Distinct roles of cdk2 and cdc2 in RP‐A phosphorylation during the cell cycle. J Cell Sci 106(Pt 3): 983–994 [DOI] [PubMed] [Google Scholar]
- Fullbright G, Rycenga HB, Gruber JD, Long DT (2016) p97 promotes a conserved mechanism of helicase unloading during DNA cross‐link repair. Mol Cell Biol 36: 2983–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina I, Hendriks IA, Hoffmann S, Larsen NB, Johansen J, Colding‐Christensen CS, Schubert L, Sellés‐Baiget S, Fábián Z, Kühbacher U et al (2021) The ubiquitin ligase RFWD3 is required for translesion DNA synthesis. Mol Cell 81: 442–458.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐de‐Teresa B, Rodríguez A, Frias S (2020) Chromosome instability in Fanconi anemia: from breaks to phenotypic consequences. Genes 11: 1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262 [DOI] [PubMed] [Google Scholar]
- Gari K, Décaillet C, Delannoy M, Wu L, Constantinou A (2008) Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci USA 105: 16107–16112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RK, Mund A, Poulsen SL, Sandoval M, Klement K, Tsouroula K, Tollenaere MAX, Räschle M, Soria R, Offermanns S et al (2016) SCAI promotes DNA double‐strand break repair in distinct chromosomal contexts. Nat Cell Biol 18: 1357–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Moffat J (2016) BAGEL: a computational framework for identifying essential genes from pooled library screens. BMC Bioinformatics 17: 164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Tong AHY, Chan K, Van Leeuwen J, Seetharaman A, Aregger M, Chandrashekhar M, Hustedt N, Seth S, Noonan A et al (2017) Evaluation and design of genome‐wide CRISPR/SpCas9 knockout screens G3 7: 2719–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks IA, Lyon D, Su D, Skotte NH, Daniel JA, Jensen LJ, Nielsen ML (2018) Site‐specific characterization of endogenous SUMOylation across species and organs. Nat Commun 10.1038/s41467-018-04957-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Shimamoto A, Tahara H et al (2015) Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet 96: 1001–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe S‐Y, Nagao K, Nozaki N, Kimura H, Obuse C (2017) Inhibition of RIF1 by SCAI allows BRCA1‐mediated repair. Cell Rep 20: 297–307 [DOI] [PubMed] [Google Scholar]
- Kannouche P, Broughton BC, Volker M, Hanaoka F, Mullenders LH, Lehmann AR (2001) Domain structure, localization, and function of DNA polymerase eta, defective in xeroderma pigmentosum variant cells. Genes Dev 15: 158–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC (2009) The Fanconi anemia pathway promotes replication‐dependent DNA interstrand cross‐link repair. Science 326: 1698–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Schärer OD, Walter JC (2012) Replication‐coupled DNA interstrand cross‐link repair in Xenopus egg extracts. Methods Mol Biol 920: 221–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochaniak AB, Habuchi S, Loparo JJ, Chang DJ, Cimprich KA, Walter JC, van Oijen AM (2009) Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J Biol Chem 284: 17700–17710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasner DS, Daley JM, Sung P, Niu H (2015) Interplay between Ku and replication protein A in the restriction of Exo1‐mediated DNA break end resection. J Biol Chem 290: 18806–18816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Räschle M, Walter JC, Duxin JP (2019) Replication‐coupled DNA‐protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Mol Cell 73: 574–588.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebofsky R, Takahashi T, Walter JC (2009) DNA replication in nucleus‐free Xenopus egg extracts. Methods Mol Biol 521: 229–252 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS (2014) MAGeCK enables robust identification of essential genes from genome‐scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Raschle M, Joukov V, Walter JC (2011) Mechanism of RAD51‐dependent DNA interstrand cross‐link repair. Science 333: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17: 3–12 [Google Scholar]
- Mateos‐Gomez PA, Gong F, Nair N, Miller KM, Lazzerini‐Denchi E, Sfeir A (2015) Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518: 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JR, Lawrence CW, Hinkle DC (1996) Thymine‐thymine dimer bypass by yeast DNA polymerase zeta. Science 272: 1646–1649 [DOI] [PubMed] [Google Scholar]
- Poulsen M, Lukas C, Lukas J, Bekker‐Jensen S, Mailand N (2012) Human RNF169 is a negative regulator of the ubiquitin‐dependent response to DNA double‐strand breaks. J Cell Biol 197: 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rageul J, Kim H (2020) Fanconi anemia and the underlying causes of genomic instability. Environ Mol Mutagen 61: 693–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC (2008) Mechanism of replication‐coupled DNA interstrand crosslink repair. Cell 134: 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räschle M, Smeenk G, Hansen RK, Temu T, Oka Y, Hein MY, Nagaraj N, Long DT, Walter JC, Hofmann K et al (2015) DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross‐links. Science 348: 1253671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman K, Lach F, Abhyankar A, Donovan F, Sanborn E, Kennedy J, Sougnez C, Gabriel S, Elemento O, Chandrasekharappa S et al (2015) Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA‐T Subtype of Fanconi anemia. Cell Rep 12: 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roerink SF, van Schendel R, Tijsterman M (2014) Polymerase theta‐mediated end joining of replication‐associated DNA breaks in C. elegans. Genome Res 24: 954–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Lindgreen S, Orlando L (2016) AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes 9: 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC (2016) Replication‐dependent unhooking of DNA interstrand cross‐links by the NEIL3 glycosylase. Cell 167: 498–511.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, Hurov KE, Luo JI, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD et al (2007) Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129: 289–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Chistol G, Gao AO, Räschle M, Larsen NB, Mann M, Duxin JP, Walter JC (2019) The CMG helicase bypasses DNA‐protein cross‐links to facilitate their repair. Cell 176: 167–181.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks J, Walter JC (2019) Extracts for analysis of DNA replication in a nucleus‐free system. Cold Spring Harb Protoc 2019: pdb.prot097154 [DOI] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask M‐B, Lukas C, Larsen DH, Povlsen LK, Bekker‐Jensen S, Mailand N, Bartek J, Lukas J (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Walter J, Newport J (2000) Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha. Mol Cell 5: 617–627 [DOI] [PubMed] [Google Scholar]
- Wang W (2007) Emergence of a DNA‐damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 8: 735–748 [DOI] [PubMed] [Google Scholar]
- Wu RA, Semlow DR, Kamimae‐Lanning AN, Kochenova OV, Chistol G, Hodskinson MR, Amunugama R, Sparks JL, Wang M, Deng L et al (2019) TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 567: 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Mieczkowski P, Wood RD, Gupta GP, Ramsden DA (2016) Essential roles for polymerase θ‐mediated end joining in the repair of chromosome breaks. Mol Cell 63: 662–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamin BB, Ahmed‐Seghir S, Tomida J, Despras E, Pouvelle C, Yurchenko A, Goulas J, Corre R, Delacour Q, Droin N et al (2021) DNA polymerase zeta contributes to heterochromatin replication to prevent genome instability. EMBO J 40: e104543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51‐mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, Farkkila A, Kochupurakkal B, Syed A, Shapiro GI et al (2020) Polymerase theta inhibition kills homologous recombination deficient tumors. bioRxiv 10.1101/2020.05.23.111658 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]