

Abstract
Preservation of nucleosomes during replication has been extensively studied, while the maintenance of nucleosomes during transcription has gotten less attention. The histone chaperone FACT has a role in transcription elongation, although whether it disassembles or assembles nucleosomes during this process is unclear. To elucidate the function of FACT in mammals, we deleted the Ssrp1 subunit of FACT in adult mice. FACT loss is lethal, possibly due to the loss of the earliest progenitors in bone marrow and intestine, while more differentiated cells are not affected. Using cells isolated from several tissues, we show that FACT loss reduces the viability of stem cells but not of cells differentiated in vitro. FACT depletion increases chromatin accessibility in a transcription‐dependent manner in adipose mesenchymal stem cells, indicating that nucleosomes are lost in these cells during transcription in the absence of FACT. We also observe activation of interferon (IFN) signaling and the accumulation of immunocytes in organs sensitive to FACT loss. Our data indicate that FACT maintains chromatin integrity during transcription in mammalian adult stem cells, suggesting that chromatin transcription in stem cells and differentiated cells is different.
Keywords: nucleosome, SPT16, SSRP1, stem cell, transcription
Subject Categories: Chromatin, Transcription & Genomics; Stem Cells & Regenerative Medicine
Loss of histone chaperone FACT in adult mice is lethal and causes stem cell death in bone marrow and intestine. FACT loss increases chromatin accessibility in MSC in a transcription‐dependent manner suggesting that nucleosome organization is different in undifferentiated and differentiated mammalian cells.

Introduction
Chromatin organization defines cell identity by limiting the access of transcription machinery to DNA in a region‐specific manner, making the existence of cell‐ and condition‐specific transcriptional programs possible. Although significant progress has been made in elucidating the structure and mechanism of action of factors involved in chromatin maintenance, studies have mainly used cell‐free conditions or simplified model organisms (e.g., yeast or human tumor cells). The role of these factors in modifying gene expression in a cell‐specific manner in multicellular organisms, including the spatio‐temporal regulation of their activities, is still far behind.
One group of such factors is the histone chaperones that have long been considered a component of the basic replication and transcription machinery responsible for the assembly and disassembly of nucleosomes (Hammond et al, 2017). However, studies done in multicellular organisms demonstrated that they are not ubiquitously expressed and that their depletion has different consequences for different cells (Voon et al, 2015; Dilg et al, 2016; Messiaen et al, 2016; Shi et al, 2016). These characteristics suggest that either their function is not unique or the control of chromatin integrity may not be required in all cells.
Histone chaperone FACT (FAcilitates Chromatin Transcription) is especially interesting in this regard. In recent years, the view of its function as an essential transcription elongation factor facilitating the access of the transcriptional machinery to nucleosomal DNA has been questioned by several mechanistic and structural studies (Formosa & Winston, 2020a), (Zhou et al, 2020). FACT does not bind to nucleosomes fully wrapped with DNA but binds to nucleosomes with partially unwrapped DNA (Tsunaka et al, 2016; Safina et al, 2017; Liu et al, 2020). Reduced FACT levels in several cell types increase chromatin accessibility at different genomic regions and are accompanied by elevated transcription (Mylonas & Tessarz, 2018; Sandlesh et al, 2020), suggesting that FACT may restrict the access of the transcriptional machinery to DNA. The phenotypes of FACT knockout in C. elegans or D. rerio do not support the proposed function of FACT as an essential transcription elongation factor (Koltowska et al, 2013; Kolundzic et al, 2018). Different laboratories have demonstrated that several normal mammalian cell types do not depend on FACT for growth or viability (Garcia et al, 2013; Kolundzic et al, 2018; Mylonas & Tessarz, 2018); however, most mouse and human tumor cells either cannot grow or die after FACT knockdown (Fleyshman et al, 2017; Sandlesh et al, 2020). It is still not clear what makes tumor cells dependent on FACT. This knowledge will be valuable for cancer research. The absence of FACT expression in most adult mammalian cells (Garcia et al, 2011) suggests that FACT may have cell‐specific functions or that some, but not all, cells require FACT due to a specific chromatin state. To understand FACT function in multicellular eukaryotes, it is critical to identify which mammalian cells depend on FACT and which processes are disturbed in these cells upon FACT loss.
FACT consists of two subunits, SPT16 (Suppressor of Ty 16) and SSRP1 (Structure‐Specific Recognition Protein 1). General knockout (KO) of the Ssrp1 gene in mice is lethal at the blastocyst stage (Cao et al, 2003) when expression of the FACT subunits is the highest in the life span of mice (Garcia et al, 2011). However, whether FACT is essential for the viability of adult mice when its expression is very low is not known. The stability of both FACT subunits depends on their binding to each other, and depletion of one leads to the disappearance of the other (Safina et al, 2013). Based on this knowledge, we generated an Ssrp1 conditional knockout (KO) mouse model to eliminate FACT and observe cell‐specific and age‐dependent consequences of FACT loss. Specifically, we crossed mice with LoxP sites inserted into the introns of the Ssrp1 gene and R26‐CreERT2 mice to achieve FACT depletion in adult animals using tamoxifen administration (Sandlesh et al, 2018). In this study, we determined the phenotypes of FACT loss in mice at different ages and identified the FACT‐dependent cells in FACT‐sensitive organs. We also propose the reasons for this selective FACT dependency.
Results
FACT loss in mice is lethal at different ages
For this study, we compared mice homozygous for the Ssrp1 mutant allele, Ssrp1fl / fl, with heterozygous and wild‐type animals (Sandlesh et al, 2018). Consistent with a previous report (Cao et al, 2003), we demonstrated that mice with one functional Ssrp1 allele lacked any phenotype. Therefore, we used these heterozygous mice as controls in many experiments. These mice were crossed with Rosa26 CreERT2 (CreERT2 +/+) mice to obtain mice homozygous for both alleles since the presence of one copy of CreERT2 results in poor or no excision of Ssrp1 (Sandlesh et al, 2018). Treatment of the mutant mice with tamoxifen for 5 days led to the reduction and disappearance of both FACT subunits in the spleen, the only organ in adult mice where SSRP1 and SPT16 can be detected by immunoblotting (Fig EV1A). Based on this finding, tamoxifen was administered for 5 days in subsequent experiments.
Figure EV1. Effect of tamoxifen administration on mice with wild type or floxed Ssrp1 alleles.
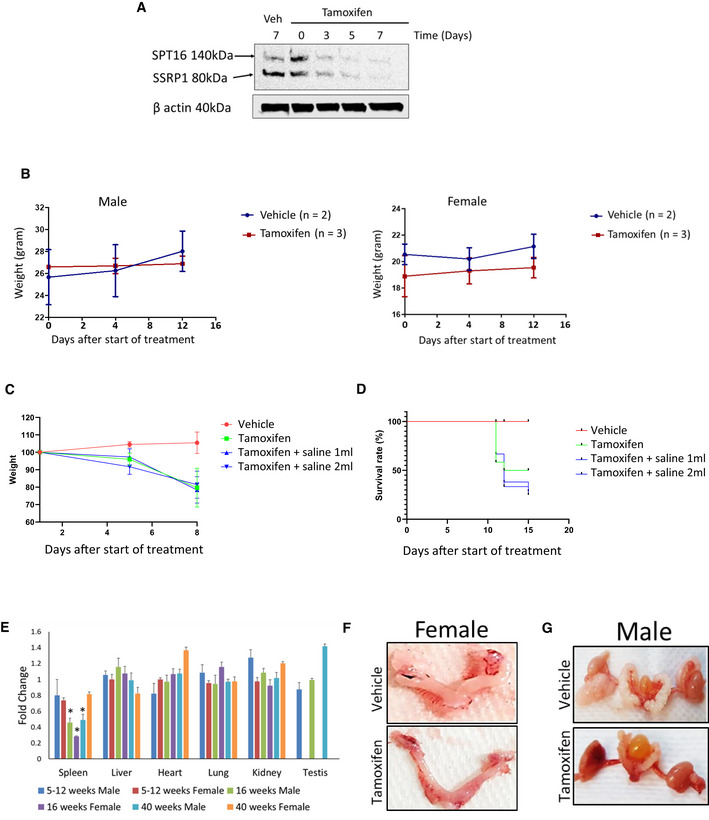
-
AKinetic of FACT protein subunit loss in spleen of Ssrp1fl / fl ; CreERT2 +/+ mice upon tamoxifen administration. Western blotting of total protein extracts of spleen collected from mice at indicated time points. Veh is vehicle.
-
BKinetic of weight changes of Ssrp1 +/+; CreERT2 +/+ mice upon tamoxifen administration. Mean ± SD for tamoxifen treated mice and mean ± range for vehicle treated mice.
-
CEffect of rehydration on weight of Ssrp1 +/+; CreERT2 +/+ mice treated with vehicle or tamoxifen. Kinetic of weight loss in control and tamoxifen treated animals either provided with subcutaneous saline injection (1 or 2 ml/day) or not. Mean ± SD, n = 3.
-
DEffect of rehydration on survival of Ssrp1 +/+; CreERT2 +/+ mice treated with vehicle or tamoxifen. Kaplan‐Mayer curves of control and tamoxifen treated groups either provided with subcutaneous saline injection (1 or 2 ml/day) or not. n = 3.
-
EChanges in organ weight in Ssrp1fl / fl ; CreERT2 +/+ mice treated with tamoxifen versus vehicle treated animals. Mean ± SD. *P‐value < 0.05. n = 5.
-
F, GRepresentative photographs of female (F) and male (G) reproductive organs excised from vehicle and tamoxifen treated mice.
One week after the end of tamoxifen administration, both female and male mice began losing weight and died or were euthanized between 12 and 50 days after the end of treatment (Fig 1A and B). Because Cre can cause DNA damage due to its nonspecific recombinase activity, we tested the effect of tamoxifen administration on mice with two CreERT2 alleles and wild‐type Ssrp1 and did not observe any toxicity (Fig EV1B), indicating that FACT loss caused the deaths of Ssrp1fl / fl mice. Older male mice (24–40 weeks at the start of tamoxifen administration) lived longer than younger males or female mice; however, they still died after FACT loss (Fig 1B). A comparison of the complete blood counts (CBC) between male and female Ssrp1fl / fl ; CreERT2 +/+ mice treated with tamoxifen or vehicle control showed a significantly reduced number of white blood cells (i.e., lymphocytes and granulocytes) but no changes in the numbers of red blood cells and platelets (Fig 1C). Serum biochemistry analysis did not find a significant deviation between tamoxifen‐treated Ssrp1fl / fl ; CreERT2 +/+ and Ssrp1fl /+; CreERT2 +/− mice except slight reduction of lipase in homozygous mice (Dataset EV1). Tamoxifen‐treated Ssrp1fl / fl ; CreERT2 +/+ mice showed signs of dehydration, and some animals developed atopic dermatitis‐like skin lesions (Fig 1D). Subcutaneous fluids (1–2 ml/day) did not rescue these mice (Fig EV1C and D). Thus, although FACT is only present in a limited number of cells in several organs of adult mice (Garcia et al, 2011), it is essential for the viability of adult mice. For subsequent experiments, we used mice between 10 and 15 weeks of age.
Figure 1. Effect of Ssrp1 KO in adult mice.
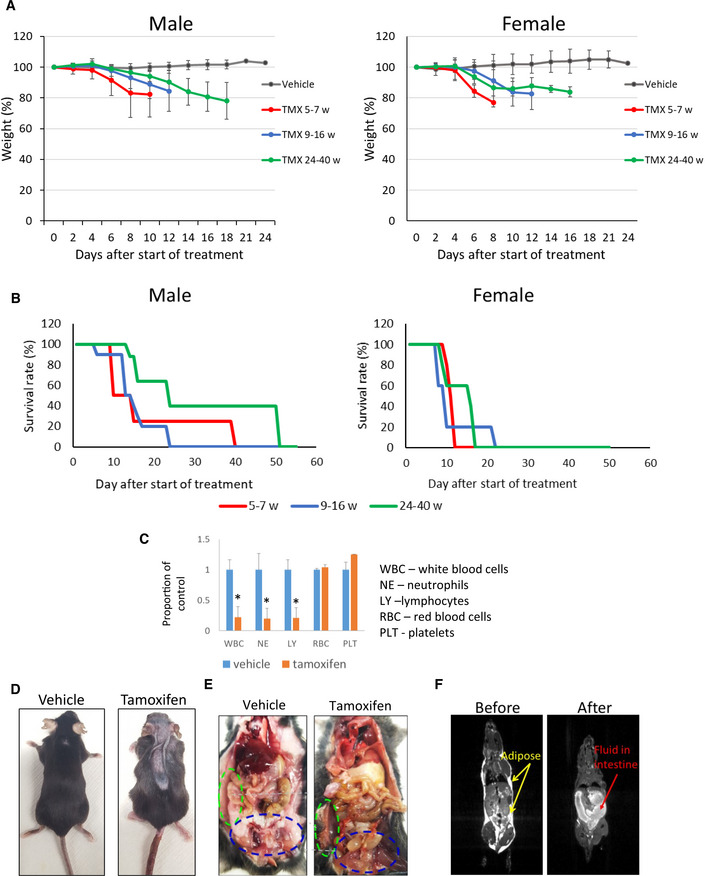
-
A, BNormalized weight (A) and survival (B) of Ssrp1fl / fl ; CreERT2 +/+ mice of different ages (weeks) after the start of tamoxifen (TMX) treatment. A. Mean weight (%) ± SD. P < 0.05 for all tamoxifen treated groups versus vehicle (ANOVA). B. Kaplan–Meier survival curves. Log‐rank test P = 0.032 for the oldest male age group versus all other male groups; P = 0.013 for all females versus all males; for all other comparisons P > 0.05. n = 6–10 mice.
-
CNormalized blood cell counts in Ssrp1fl / fl ; CreERT2 +/+ mice treated with vehicle or tamoxifen on day 7 after start of treatment. Mean ± SD, * indicates P < 0.05 (unpaired t‐test). n = 3 mice.
-
DPhotographs of vehicle‐ or tamoxifen‐treated Ssrp1fl / fl ; CreERT2 +/+ mice 7 days after start of treatment.
-
EGross pathology examination of Ssrp1fl / fl ; CreERT2 +/+ mouse euthanized due to 20% weight loss after treatment with tamoxifen. Vehicle‐treated mouse is shown for comparison. Green and blue ovals indicate subcutaneous and inguinal fat deposits, respectively.
-
FMRI image of Ssrp1fl / fl ; CreERT2 +/+ mouse before and after tamoxifen treatment.
Several tissues are sensitive to FACT depletion
Gross pathology examination revealed complete loss of adipose tissue (Fig 1E and F) and accumulation of liquid in the large intestine (Fig 1E) following the loss of FACT. The weights of the major parenchymal organs were not significantly changed, except the spleen, which was smaller in the tamoxifen‐treated groups (Fig EV1E). The ovaries were also significantly reduced in the tamoxifen‐treated mice (Fig EV1F); however, these changes have been observed in mice with wild‐type Ssrp1 and reported in the literature as a direct effect of tamoxifen treatment (Luo et al, 1997). In contrast, the testes, which also have high FACT expression (Garcia et al, 2011), were not reduced by tamoxifen treatment (Fig EV1E and G).
Upon FACT depletion, histological changes were found in organs that express FACT subunits in basal conditions (Garcia et al, 2011). Reduced cellularity was observed in the bone marrow and spleen, including atrophic changes in the red pulp (Fig 2A), areas where FACT is present in adult mice (Garcia et al, 2011). High FACT levels are also present in cells at the bottom of the crypts of the small and large intestines (Garcia et al, 2011). Ssrp1 KO resulted in the thinning of the colon villi and the emergence of a small number of degenerative crypts (Fig 2A and B). No histological changes were seen in the high FACT‐expressing testes (Fig EV1G). There was also no reduction in FACT expression in the testes after tamoxifen administration (Fig EV2A), suggesting that recombination did not occur in the testes, most probably due to the limited penetration of tamoxifen through the blood–testis barrier (Su et al, 2011). Among the FACT‐negative organs, tamoxifen administration causes histological changes only in the liver; there was a loss of glycogen granules in the hepatocytes. Some of the hepatocytes were enlarged or undergoing mitosis, which is not typical for the adult liver. Lymphoid cell infiltration was also seen in some areas (Fig 2C). The skin was excluded from the histological analysis in this study because the skin lesions first appeared in mice when they lost almost 20% of their weight, requiring euthanasia. Accurate assessment of the consequences of FACT loss in the skin will require mouse models with keratinocyte‐specific expression of CreERT2.
Figure 2. Pathological findings in Ssrp1fl / fl ; CreERT2 +/+ mice after tamoxifen treatment.
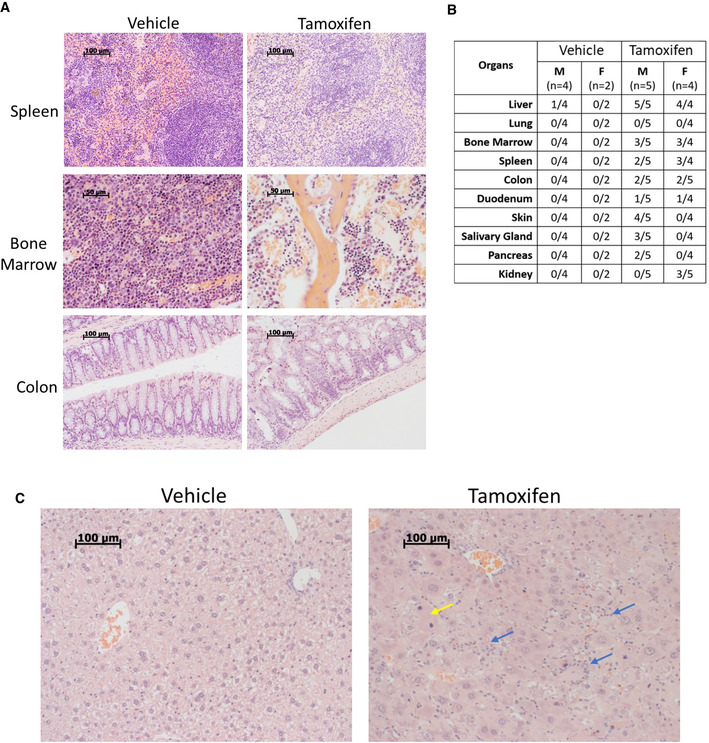
- H&E staining of mouse organ sections. Scale bars: spleen, colon 100 µm, bone marrow—50 µm.
- Incidence of pathological findings (number of mice with pathology/total number of mice).
- H&E staining of liver sections. Yellow arrow—a mitotic hepatocyte; blue arrows—lymphocyte infiltration. Scale bars—100 µm.
Figure EV2. Effect of FACT loss on DNA replication and apoptosis in mouse organs.
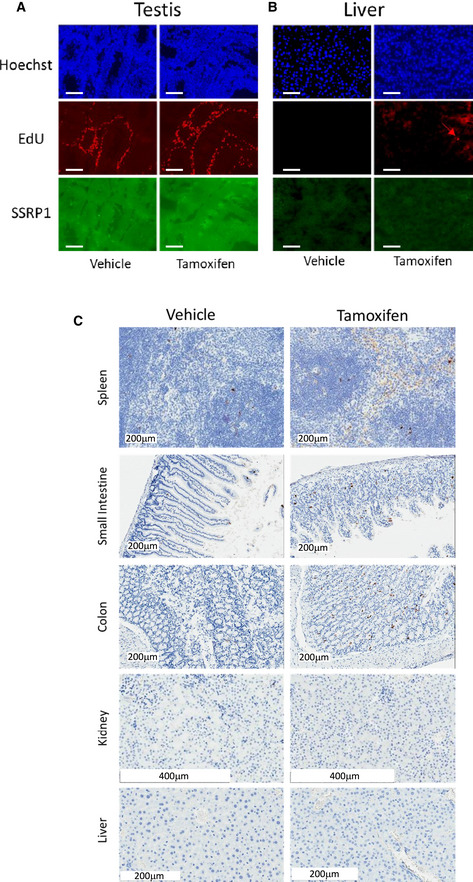
-
A, BEffect of tamoxifen administration to Ssrp1fl / fl ; CreERT2 +/+ mice on replication in testis (A) and liver (B). EdU was given to mice 3 days after stop of tamoxifen treatment for 1 h followed by organ isolation and staining for total DNA (Hoechst), EdU and SSRP1. Red arrow indicates EdU positive nucleus in liver. Scale bars – 100 µm.
-
CStaining of sections of several organs of in Ssrp1fl / fl ; CreERT2 +/+ mice for cleaved caspase 3, 3 days after stop of treatment with tamoxifen or vehicle.
Altogether, these findings may indicate the suppression of hematopoiesis and the suppression of the renewal capacity of the intestine, leading to atrophic changes that could explain the observed dehydration because the large intestine is the major organ of water absorption. Changes in the liver and reduction of lipase concentration in serum are likely secondary to weight loss. The loss of adipose tissue could explain the weight loss and might be either primary (death of adipocytes) or secondary (loss of fat from cells due to malnutrition and dehydration) to FACT loss.
FACT loss impairs proliferation and induces cell death in several organs
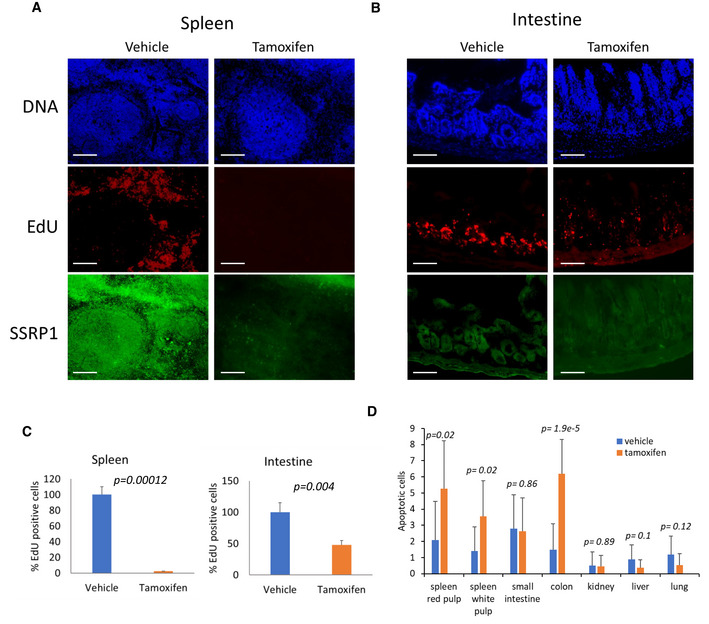
To better understand the consequences of FACT depletion, we compared the levels of proliferation and cell death in the major parenchymal organs between vehicle‐ and tamoxifen‐treated mice using EdU labeling as a marker of DNA replication and cleaved caspase‐3 staining as a marker of apoptosis. Three days after the end of tamoxifen treatment, the number of EdU‐positive cells was significantly reduced in the spleen and intestines of Ssrp1fl / fl ; CreERT2 +/+ mice (Fig 3A–C). Although EdU‐positive cells are normally absent from the liver, some EDU‐positive cells did appear following tamoxifen treatment, consistent with the appearance of mitotic cells in the H&E‐stained liver sections (Figs 2C and EV2B). Changes in EdU positive cells in the small and large intestines were moderate, with some sections with no changes in the number of EdU‐positive cells and some areas with a reduced frequency of replicating cells in the lower portion and the spread of replicating cells toward the upper portion of the crypts (Fig 3B and C). In the spleen, intestines, and colon, we observed a slight increase in the number of cleaved caspase‐3‐ positive cells; however, they were not concentrated in any specific cellular zone in these organs (Figs 3D and EV2C). Thus, FACT depletion reduced the number of cells in the hematological organs and intestines. This loss of cells was predominantly due to reduced cell proliferation. Interestingly, hepatocytes started proliferating in the liver upon Ssrp1 KO. Thus, a general impairment of DNA replication or mitosis, processes involving FACT activity (Tan et al, 2006; Zeng et al, 2010; Abe et al, 2011; Yang et al, 2016; Prendergast et al, 2020), cannot explain the toxicity of Ssrp1 KO.
Figure 3. FACT depletion in mice leads to reduced proliferation and cells death in several organs.
-
A, BEffect of tamoxifen on replication in the spleen (A) and intestine (B) of Ssrp1fl / fl ; CreERT2 +/+ mice. EdU was given to mice 3 days after the end of tamoxifen treatment (1 h before tissue collection). Organ sections were stained for total DNA (Hoechst), EdU, and SSRP1. Scale bars—100 µm.
-
CQuantitation of EdU staining. Mean number of EdU positive cells per field of view ± SD. n ≥ 5 fields of view per mouse. 3 mice were analyzed.
-
DQuantitation of cleaved caspase‐3 staining. Mean number of positive cells per fields of view ± SD, n ≥ 5 fields of view per mouse. Two male and female mice were analyzed.
Data information: P‐values for C and D were calculated using the unpaired t‐test.
FACT depletion is accompanied by the loss of long‐term hematopoietic stem cells from bone marrow and Lgr5 positive stem cells from the intestines
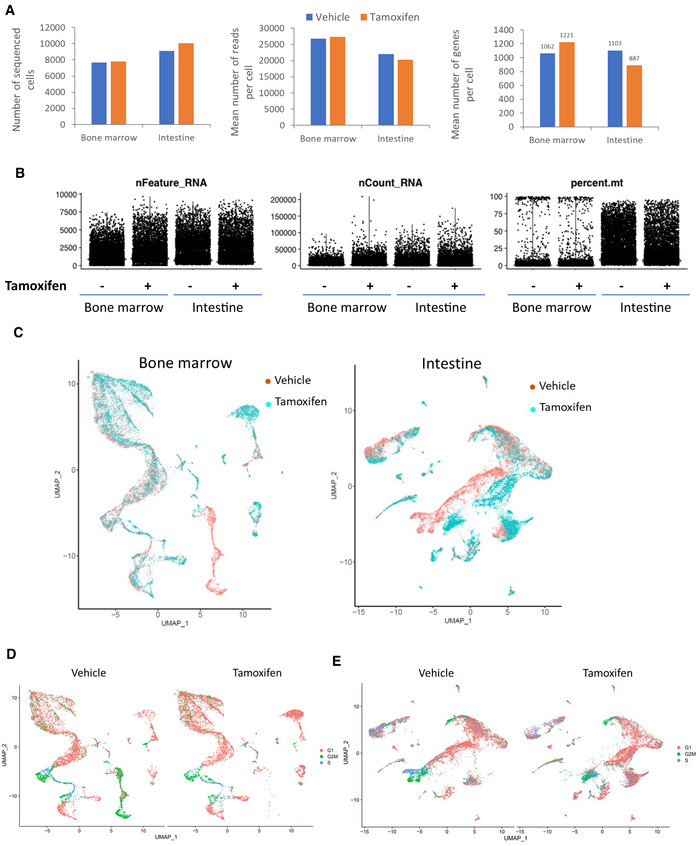
To identify cells sensitive to FACT loss in hematopoietic and intestinal tissues, we performed single‐cell RNA sequencing (scRNA‐seq). Quality control confirmed the integrity of the samples with viabilities of > 90% and > 75% for the cells obtained from the bone marrow and intestines, respectively. There were a comparable number of cells sequenced (about 8,000–10,000 per sample), the number of reads generated (about 20,000–27,000), and the number of genes quantified (approximately 1,000 per cell) (Fig EV3A and B).
Figure EV3. Analyses of scRNA sequencing of bone marrow and intestine of in Ssrp1fl / fl ; CreERT2 +/+ mice next day after stop of treatment with vehicle or tamoxifen.
-
AComparison of the numbers of sequenced cells, mean reads per cell and mean identified genes between samples.
-
BDot plots showing the number of features (nFeature_RNA, the number of genes detected in each cell), RNA molecules (nCount_RNA, the total number of molecules detected within a cell) and proportion of reads corresponding to mitochondrial RNA (percent.mt) in each sample.
-
COverlapped UMAP plots of bone marrow and intestinal samples color coded according to the treatment type.
-
D, EUMAP plots of bone marrow (D) and intestine (E) color‐coded based on the expression of markers of cell cycle.
The Uniform Manifold Approximation and Projection (UMAP) plots for vehicle‐ and tamoxifen‐treated samples showed significant overlap, suggesting that, in general, the tissue architecture was well preserved in both organs (Fig EV3C). There were no significant changes in the distribution of cells at the different phases of the cell cycle within the clusters between the vehicle‐ and tamoxifen‐treated samples and no preferential depletion of cells in any particular phase of the cell cycle (Fig EV3D and E), suggesting that FACT loss did not block any specific phase.
A comparison of the bone marrow UMAP plots revealed several clusters with visibly changed numbers of cells. Three clusters (14, 15, and 16, Appendix Figs S1–S4) almost completely disappeared after FACT depletion (Fig 4A). Using the Immunological Genome Project (ImmGen) database (Immunological Genome, 2020) as a reference, we identified the major types of bone marrow cells in our samples (Fig 4B). There were no dramatic differences in the proportions of the different cell types between samples from the vehicle‐ and tamoxifen‐treated mice (Fig 4C). In several instances, the clusters with increased or decreased numbers of cells belonged to the same cell type (e.g., the disappearing clusters 14–16 and the increasing cluster 9 were all identified as “stem cells”) (Fig 4D, Appendix Figs S1–S4). The same was true for the neutrophils and B cells. Several clusters consisting of one cell type (cluster 11—dendritic cells (DC), 20—basophils (Bph), 21—T cells, and 22—NK cells) were slightly increased in number (Fig 4C and D). Therefore, we examined the differences between the decreased and increased clusters of the same cell type.
Figure 4. Effect of FACT depletion on cell composition of bone marrow from Ssrp1fl / fl ; CreERT2 +/+ mice.
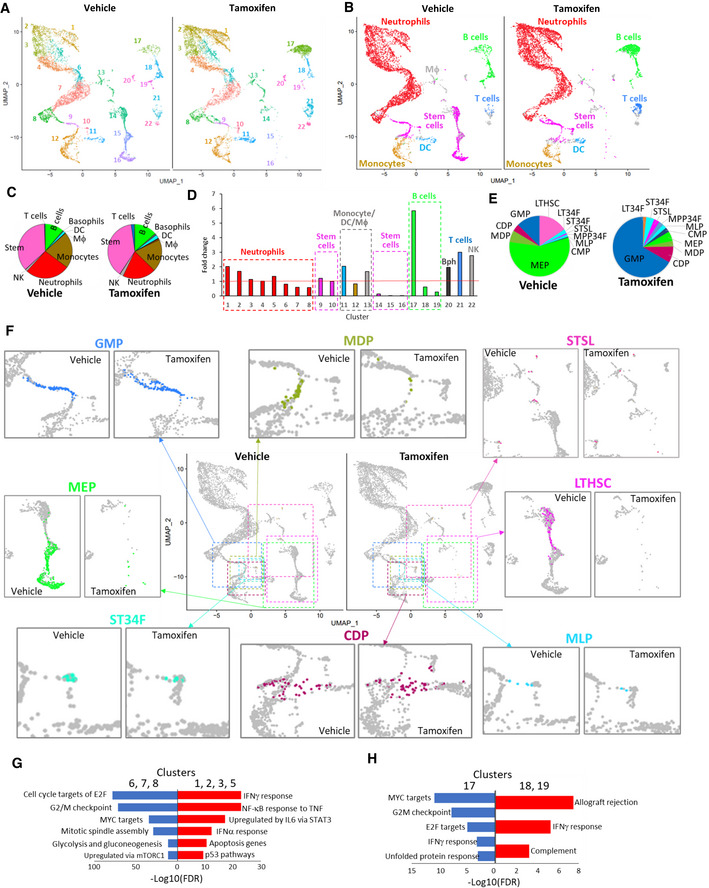
-
AUMAP plots with unbiasedly identified clusters shown in different colors.
-
BUMAP plots colored according to cell type identified by transcriptional signatures using the Immgen database as a reference.
-
CDistribution of bone marrow cell types in vehicle‐ and tamoxifen‐treated mice. The pie charts are colored accordingly to panel B. Only cell types with more than ten cells in any of the conditions are included.
-
DFold‐change in the proportion of cells in each cluster shown in panel A in samples from tamoxifen‐treated versus vehicle‐treated mice. Cell types are identified by color as in panel B.
-
EDistribution of different types of bone marrow stem cells from vehicle‐ and tamoxifen‐treated mice. Stem cell types were identified by transcriptional signatures using the Immgen stem cell database as a reference. Only cell types with more than five cells in any of the conditions identified are presented. LTHSC—long‐term hematopoietic stem cells (HSC), ST34F—CD34+Flk2‐ short‐term (ST) HSC, STSL—ST reconstituting stem cells, MPP34F—CD34+Flk2+ multipotent progenitors, MLP—multi‐lymphoid progenitors, CMP.DR—common myeloid progenitor, MEP—megakaryocyte‐erythroid progenitors, MDP—monocyte‐dendritic cell progenitors, CDP—common dendritic cell progenitors, GMP‐granulocyte–monocyte progenitor.
-
FChanges in the abundance and distribution of major bone marrow stem cell types. Center—UMAP plots showing enlarged regions as colored dashed rectangles. The color of a rectangle corresponds to the color of the stem cells in individual sections. Around central UMAPs—enlarged sections of the central UMAP plot showing individual stem cell subtypes. Colored dots—corresponding stem cells, gray dots—other cells.
-
G, HGSEA of gene markers of neutrophil (G) or B cell (H) in clusters enlarged upon FACT loss (red) or reduced upon FACT loss (blue).
Data information: Pooled data from two mice on day 1 after the end of treatment.
In the control samples, stem cells formed two separate groups: a central group isolated from other cells (clusters 14–16) and a group connecting the monocytes and granulocytes (clusters 9, 10) (Fig 4B). The central group of stem cells disappeared, while the cluster connecting the granulocytes and monocytes slightly increased (Fig 4B). We used the ImmGen datasets specific for bone marrow stem cells to determine the differences between these subpopulations. Using this approach, we identified 11 types of stem cells in our dataset (Fig 4E). The most dramatic change was observed for the long‐term hematopoietic stem cells (LTHSCs), which completely disappeared after Ssrp1 deletion (Fig 4E and F). CD34− LTHSCs gave rise to CD34+ LTHSCs (LT34Fs), which increased in number; however, the total cell count was very low and unreliable (one cell in the control and four cells after FACT loss). The LT34Fs were followed by short‐term HSCs (ST‐HSCs), consisting of ST34Fs (“CD34+Flk2− ST‐HSC”) and STSLs (“short‐term reconstituting stem cell”), whose numbers were slightly reduced or increased upon FACT loss, respectively (Fig 4E and F). Lineage‐committed progenitors changed in different directions. For example, the number of multipotent progenitors (MPP34F, CD34+Flk2+ multipotent progenitors) was increased, while the number of committed progenitors changed either up (MLP—multilymphocyte progenitors, CMP—common myeloid progenitors, CDP—common dendritic cell progenitors, GMP—granulocyte‐monocyte progenitor) or down (MEP—megakaryocyte–erythroid progenitors, MDP—monocyte dendritic cell progenitors) (Fig 4E and F). Thus, using gene expression signature as a method of cell identification, the stem cells most sensitive to FACT depletion in bone marrow were the earliest hematopoietic progenitors, LTHSCs. Changes in the number of other stem cells may be direct (in case their viability depends on FACT) or secondary (redistribution in response to loss of cells dependent on FACT).
To understand the differences in the neutrophils (increased in clusters 1–3, 5; decreased in clusters 6–8), we ran Gene Set Enrichment Analysis (GSEA, MSigDB) using gene markers specific for these clusters (Fig 4G and Dataset EV2). We found that FACT loss resulted in more neutrophils expressing genes of the Interferon (IFN) gamma response and induced by other inflammatory cytokines (e.g., TNF and IL‐6), while genes expressed in neutrophils that decreased in number were enriched in gene lists for proliferative response and elevated metabolic turnover (Fig 4F and Dataset EV2). Common genes between the two sets of clusters were enriched in several neutrophil datasets (Dataset EV2).
We ran the same analysis for B cells by comparing cluster 17 (increased) with clusters 18 and 19 (decreased). The total number of B cells increased almost two‐fold (6.5% versus 13.1%, Fig 4C), suggesting that they might migrate from outside the bone marrow. Results of this comparison paralleled the neutrophils (Fig 4H and Dataset EV2, Appendix Figs S1–S4). Upon FACT loss, there were more B cells with a transcriptional program similar to “Allograft rejection” and “IFN gamma response,” while in the control bone marrow, there were more B cells expressing genes enriched for “MYC targets,” “G2 M checkpoint,” and “ E2F targets.” Thus, these data suggest that FACT depletion in neutrophils and B cells resulted in the inhibition of the expression of genes involved in proliferation and metabolism and the induction of genes induced by IFNs and other inflammatory cytokines.
Intestine UMAP plots showed that three neighboring clusters were completely lost (Fig 5A, clusters 1–3), while several clusters increased (clusters 10–14) or emerged de novo (clusters 4, 8, 9, 16, which had no or a very few cells in control sample, Appendix Figs S5–S7). To recognize intestinal cells, we used data published by Gao et al (2018) and Haber et al (2017). All clusters were ascribed to certain intestinal cell types, except cluster 12, which was not identified (Fig 5B and Dataset EV3, Appendix Figs S5–S7). In most cases, comparison of our data with both published datasets resulted in assignment of cells to the same type. The transcription signature of the most prominently lost cluster (1) included “large intestine Lgr5‐positive stem cells” (Gao et al, 2018). Cells of this cluster also had high expression of Gkn3, a marker of small intestine stem cells (Haber et al, 2017) (Fig 5C and D).
Figure 5. Effect of FACT depletion on cell composition of the intestine from Ssrp1fl / fl ; CreERT2 +/+ mice.
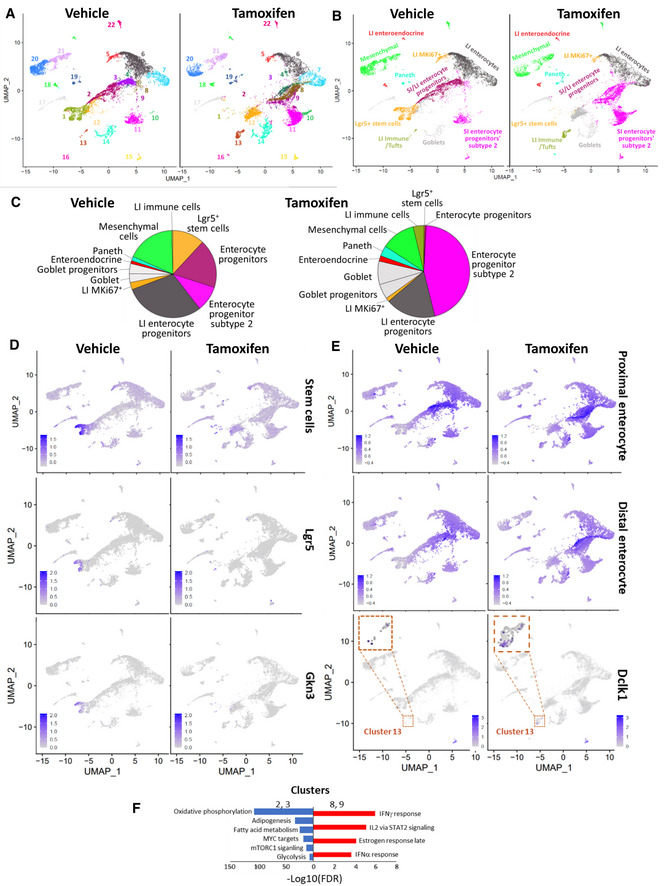
- UMAP plots with unbiasedly identified clusters shown in different colors.
- UMAP plots are colored according to cell type identified by transcriptional signatures using data from (Gao et al, 2018) as a reference. LI—large intestine, SI—small intestine.
- Distribution of intestinal cell types in samples from vehicle‐ and tamoxifen‐treated mice. Pie charts are colored accordingly to panel B.
- UMAP plots with color‐coded intestinal stem cells identified by transcriptional signatures of intestinal stem cells from (Haber et al, 2017) or by expression of Lgr5 or Gkn3 genes.
- UMAP plots showing the distribution of cells with transcriptional signatures of proximal and distal enterocytes from (Haber et al, 2017) and cells expressing the Dclk1 gene.
- GSEA of gene markers of clusters 2 and 3 (lost upon Ssrp1 KO, blue) versus clusters 8 and 9 (emerged upon Ssrp1 KO, red).
Data information: Pooled data from two mice on day 1 after the end of treatment.
No other cell types were completely lost upon FACT depletion. Lost clusters 2 and 3 were recognized as small and large intestine enterocyte progenitors using the Gao dataset (Fig 5B and C) or proximal enterocytes using the Haber dataset (Fig 5E). However, in the FACT‐depleted intestine, similar cells were found within de novo emerged clusters 4, 8, and the increased clusters 9–11 (Fig 5B, C and E). The close location of the reduced and increased clusters and their similar classification suggested that the enterocyte progenitors did not die upon FACT loss but had their transcriptional program shifted. Thus, we looked for differences in gene markers of clusters 2 and 3 (almost completely lost in the Ssrp1 KO sample) and clusters 8 and 9 (emerged in the Ssrp1 KO sample) (Fig 5F) using GSEA. This comparison showed that FACT depletion in enterocyte progenitors resulted in downregulation of genes involved in “oxidative phosphorylation,” “adipogenesis,” “ mTORC1 signaling,” and “glycolysis,” or being “MYC targets,” while the upregulated genes were part of categories “IFN gamma and alpha response,” “activated by IL2 via STAT2 signaling,” and “late estrogen response” (Fig 5F and Dataset EV3, Appendix Figs S5–S7). Surprisingly, these results were similar to what we observed with bone marrow neutrophils and B cells.
There were little changes in the number of cells in clusters 6 and 7 (Fig 5A), composed of distal enterocytes according to Haber or large intestinal enterocytes according to Gao. Quantitation revealed a slight increase in the number of Goblet cells and their progenitors and Paneth cells (Fig 5B and C). Cluster 13 had an increased number of cells upon FACT loss (Fig 5A). These cells had a gene expression signature for large intestine immune cells (FDR q‐value = 9.93 e‐65) and were positive for Dclk1, a marker of intestinal Tuft cells (Fig 5E). Changes in intestinal cell composition were similar to those seen in bone marrow, namely, the disappearance of the earliest intestinal stem cells and an increase in the number of more mature cell types. Cells in a state between stem and fully differentiated likely respond to FACT loss by reducing proliferation and inducing the IFN response.
Thus, scRNA‐seq data suggest that the cell types most sensitive to FACT loss are the undifferentiated stem cells. More mature differentiated cells tolerate FACT depletion; however, these findings need to be confirmed by additional methods.
Stem cells from several organs cannot expand in vitro in the absence of FACT
To confirm that stem cells are dependent on FACT for viability and growth, we compared the colony‐forming ability of stem cells from FACT‐sensitive organs. Cells isolated from bone marrow, small intestine, and colon were plated in stem cell medium and treated with 4‐hydroxytamoxifen (4‐OHT, active metabolite of tamoxifen) to induce Ssrp1 deletion in vitro. A similar experiment was performed with mesenchymal stem cells (MSCs) from adipose tissue to determine whether the loss of adipose tissue is a primary or secondary effect of Ssrp1 KO. While the growth of skin fibroblasts from the tails of the same mice was not affected by Ssrp1 deletion (Sandlesh et al, 2020), the growth of all stem cells was significantly reduced. No colonies formed from bone marrow isolated from Ssrp1fl / fl ; CreERT2 mice in the presence of 4‐OHT; however, 4‐OHT did not interfere with the growth of bone marrow cells from genotypes not resulting in bi‐allelic Ssrp1 deletions (Fig 6A and B). No organoids formed from the small intestine or colon stem cells from Ssrp1fl / fl ; CreERT2 mice upon 4‐OHT administration (Fig 6C and D). The adipose MSCs were growing in medium with 4‐OHT but at a slower rate than the vehicle‐treated cells, and their numbers were reduced due to increased apoptosis upon FACT depletion (Fig 6E and H).
Figure 6. FACT depletion affects the viability of stem cells but not differentiated cells.
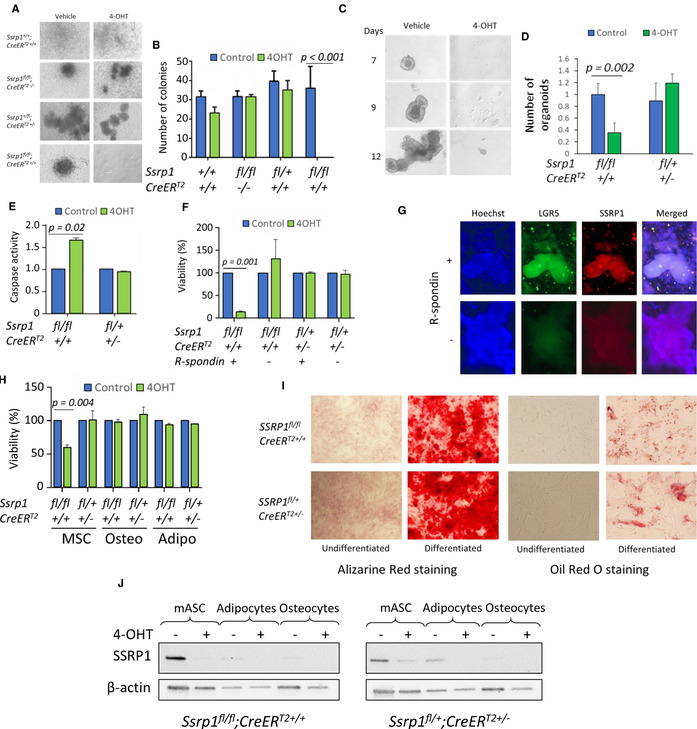
-
A, BEffect of 4‐OHT administration on colony formation by bone marrow cells of different genotypes. A. Photographs of colonies from bone marrow cells kept in culture for 7 days in the presence or absence of 4‐OHT. B. Quantitation of bone marrow colony assay. Mean colony number per plate ± SD, n = 3 plates per mouse. P values < 0.05 are shown (unpaired t‐test). 2 mice of each genotype were analyzed.
-
C, DGrowth of intestinal organoids in the presence or absence of 4‐OHT. C. Photographs of small intestine organoids on different days after plating. D. Number of organoids in the presence and absence of 4‐OHT. Mean fold change between number of organoids in control and 4‐OHT treated wells ± SD, n = 4 wells per mouse. P < 0.05 is shown (unpaired t‐test). Organoids were isolated from one mouse of each genotype in two independent experiments.
-
ECaspase activity in lysates of control and 4‐OHT treated MSCs of different genotypes. Mean fold change ± SD, n = 3 plates per mouse. P < 0.05 is shown (unpaired t‐test). MSCs were isolated from 2 to 3 mice of each genotype.
-
F, GEffect of 4‐OHT administration on intestinal organoids of different genotypes kept in the presence (undifferentiated) or absence (differentiated) of R‐spondin‐1. F. The viability of organoids assessed by the resorufin assay. Mean ± SD, n = 3 wells per mouse. P < 0.05 are shown (unpaired t‐test). Organoids were isolated from two mice of each genotype. G. IF images of intestinal organoids from Ssrp1fl / fl ; CreERT2 +/+ mice kept in the presence or absence of R‐Spondin‐1. The organoids were stained with antibodies to LGR5 and SSRP1 and counterstained with Hoechst (DNA).
-
H–JEffect of 4‐OHT administration on MSCs and their differentiated counterparts. H. Methylene blue viability assay for undifferentiated MSCs or MSCs differentiated into osteocytes or adipocytes for 21 days and then treated with vehicle or 4‐OHT. Mean ± SD, n = 3 plates per mouse. P < 0.05 is shown (unpaired t‐test). MSCs were isolated from two mice of each genotype. I. Staining of MSCs, osteocytes, and adipocytes with Alizarine Red (osteocytes) or Oil Red (adipocytes). J. Western blotting of protein extracts from MSCs, adipocytes, and osteocytes from mice of different genotypes treated with vehicle or 4‐OHT.
To determine if cells lose their dependence on FACT for viability upon differentiation, we isolated intestinal and adipose stem cells and grew them under conditions that either maintained their undifferentiated state or induced differentiation in vitro (Taha & Hedayati, 2010; Wittenburg et al, 2014; Lindeboom et al, 2018). For both types of cells, 4‐OHT administration was toxic only to undifferentiated cells. Intestinal organoids maintained in the absence of R‐spondin 1 differentiated into more mature enterocyte‐like cells (Lindeboom et al, 2018), lost expression of Lgr5 and became insensitive to FACT depletion (Fig 6F and G). In the case of the adipose MSCs, we obtained cultures of differentiated adipocytes and osteocytes using specific differentiation media (Huang et al, 2013) and demonstrated that neither adipocytes nor osteocytes died upon FACT depletion (Fig 6H and I). Thus, only undifferentiated stem cells are dependent on FACT for viability. Consistent with this data, FACT levels decrease upon differentiation (Fig 6G and J).
FACT loss in stem cells leads to significant changes in chromatin accessibility in a transcription‐dependent manner
To understand the cause of stem cell loss upon FACT depletion, we investigated the changes in transcription and chromatin structure in adipose MSCs, cells which viability and growth depend on FACT, but which do not die upon FACT loss. First, we compared general transcription in MSCs before and after FACT depletion by measuring the incorporation of modified ribonucleotide 5‐Ethynyl Uridine (EU) into newly synthesized RNA. We observed a slight but highly reproducible increase in EU incorporation in cells without FACT compared to those expressing FACT (Fig 7A and B), suggesting that general transcription was increased in the MSCs upon FACT loss. This phenomenon was not the result of 4‐OHT treatment since cells heterozygous for the Ssrp1fl allele showed almost identical EU incorporation before and after treatment (Fig EV4A and B).
Figure 7. Comparison of transcription between Ssrp1fl / fl ; CreERT2 +/+ MSCs treated with vehicle or 4‐OHT.
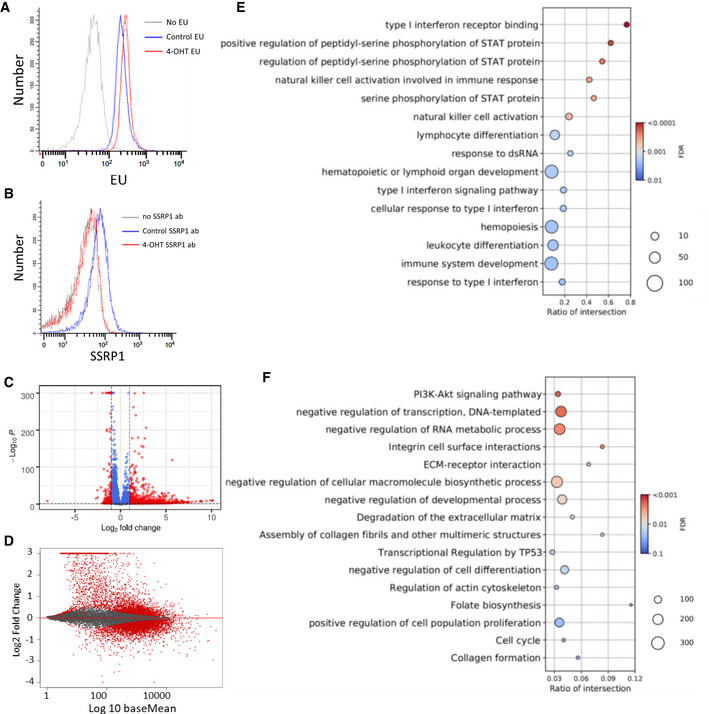
-
AEU incorporation measured by flow cytometry. Histograms of EU fluorescent signals from two biological replicates for each condition (except no EU).
-
BSSRP1 staining of the cells in A.
-
CVolcano plot of changes in transcript counts in 4‐OHT‐treated cells versus control. Red dots indicate transcripts with fold‐change ≥ 2 and FDR < 0.05. n = 2 biological replicates.
-
DMA plot showing the difference between the changes in transcript counts in 4‐OHT‐treated MSCs vs. control and the average expression of the same transcripts between conditions (baseMean). n = 2 cell cultures.
-
E, FDot plots with the results of GO enrichment for genes upregulated (C) and downregulated (D) upon FACT depletion.
Figure EV4. Comparison of changes in transcription in MSC of two genotypes treated with vehicle of 4‐OHT for 5 days and then split and analyzed 24 h after.
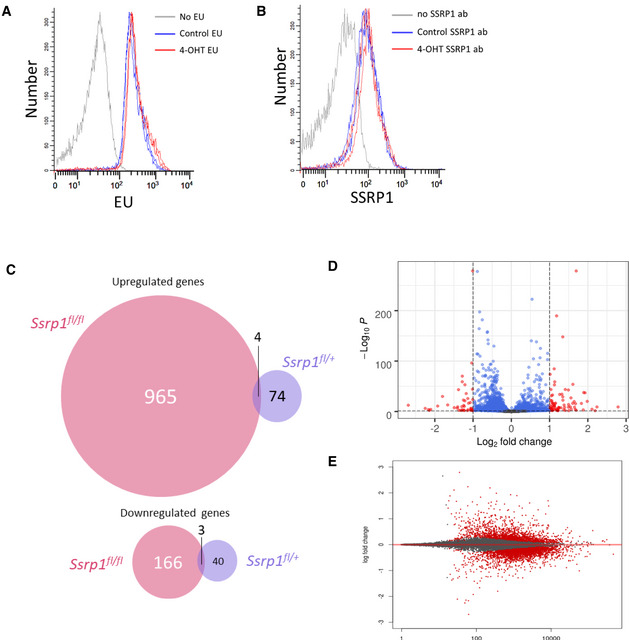
-
A, BEU incorporation assay (30 min) (A) and staining with SSRP1 antibody (B) of Ssrp1fl /+; CreERT2 +/+ MSC. Two replicates are shown.
-
C–EData of RNA‐seq experiment with two replicates of MSC. C. Venn diagrams showing number of up‐ and downregulated genes in homo‐ and heterozygous cells. D. Volcano plot of gene expression changes in heterozygous cells upon 4‐OHT treatment. E. MA plot of gene expression changes in heterozygous cells upon 4‐OHT treatment.
Next, we performed RNA‐seq on samples from MSCs before and after 4‐OHT treatment. There was a smaller number of genes for which expression changed in Ssrp1fl /+ heterozygous cells than in homozygous Ssrp1fl / fl cells. There was almost no overlap in the genes whose expression was changed between the homozygous and heterozygous cells in response to 4‐OHT (Fig EV4C), confirming that the gene expression changes in the Ssrp1fl / fl cells were in response to FACT loss and not 4‐OHT administration. In the homozygous cells, many more genes were upregulated upon FACT loss than downregulated (Fig 7C and D), whereas the changes in the gene expression of heterozygous cells were more random (Fig EV4C and D).
Gene ontology (GO) analysis of the upregulated genes showed that the most significantly enriched category was the IFN response (Fig 7E and Dataset EV4). The downregulated genes did not have a dominant category but belonged to the “PI3K‐AKT signaling pathway,” “negative regulation of transcription,” “extracellular matrix (ECM) remodeling,” “negative regulation of cell differentiation,” and “positive regulation of cell proliferation” categories (Fig 7F and Dataset EV4). Many of these categories reflected the observed phenotypic changes in these cells.
The chromatin state in the MSCs before and after 4‐OHT treatment was analyzed using the Assay for Transposase‐Accessible Chromatin with high‐throughput sequencing (ATAC‐seq, Appendix Figs S8 and S9) (Buenrostro et al, 2013). Because cells in different phases of the cell cycle have different degrees of chromatin accessibility, we limited the influence of this factor by maintaining cells for 48 h in serum‐depleted medium after 4‐OHT treatment. Profile plots were built for the distribution of ATAC‐seq reads at genomic regions flanking all known mouse genes, 3 kb before the transcriptional start site (TSS), 3 kb after the transcription end site (TES), and in between. Transposase inserts adaptors into genomic regions lacking nucleosomes (i.e., nucleosome‐free or linker DNA of accessible chromatin). These adapters can be used as primers for PCR amplification. Selection of the length of the amplified fragments allows the identification of nucleosome‐free regions (i.e., reads with the length shorter than nucleosomal DNA [< 120 bp]) or regions occupied by one nucleosome (i.e., reads with fragment lengths corresponding to mononucleosomes with linker DNA [> 120 and < 250 bp]) as described by Buenrostro et al (2013). The profile plots showed very good overlap between replicates of the vehicle‐ and 4‐OHT‐treated cells (Fig 8), suggesting high reproducibility of the effect. The plots built from the nucleosome‐free and mononucleosome fragments showed increased chromatin accessibility upon FACT depletion around the TSS and at gene bodies (Fig 8A and B). The increase in the gene bodies was more pronounced for the mononucleosome fragments than the nucleosome‐free fragments (Fig 8C and D).
Figure 8. FACT depletion in Ssrp1fl / fl ; CreERT2 +/+ MSCs leads to increase in chromatin accessibility in a transcription‐dependent manner.
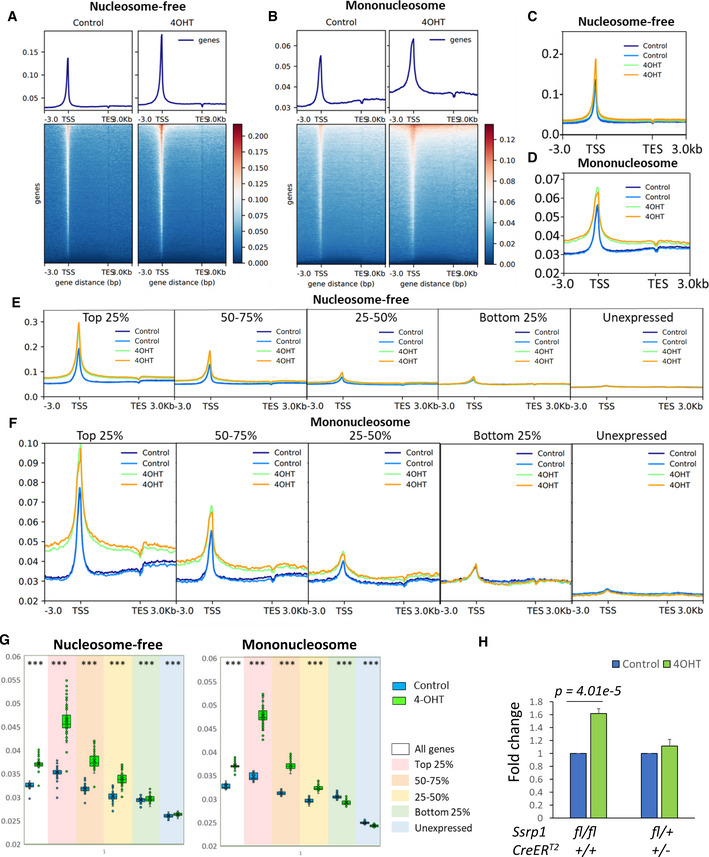
-
A, BProfiles and heatmaps for normalized ATAC‐seq read distribution averaged for all known mouse genes in control or 4‐OHT‐treated MSCs for reads below 120 bp (A, nucleosome‐free) and between 120 and 250 bp (B, mononucleosomes). One of two replicates is shown.
-
C, DATAC‐seq read density profile for two biological replicates of control and treated MSCs for all mouse genes of the nucleosome‐free (C) and mononucleosome (D) fragments.
-
E, FATAC‐seq read density profiles for nucleosome‐free (E) and mononucleosome (F) fragments for differentially expressed genes.
-
GBox plots of mean read density (y‐axis) at the gene body (bins 21–100 out of 100 between TSS – TES) for all genes and genes expressed at different levels. ***P < 1e‐10 (paired t‐test). n = 2 biological replicates. Central band is mean, box is interquartile range, whiskers are minimum and maximum.
-
HComparison of Hoechst 33342 binding to live control and 4‐OHT‐treated MSCs from different genotypes. Data are presented as the median fluorescence intensity ± SD, n = 4 biological replicates. P < 0.05 is shown (unpaired t‐test).
To test whether FACT‐mediated control of chromatin integrity depended on transcription, we used RNA‐seq data to classify the genes based on their expression levels in cells with FACT. First, we identified genes that were not expressed in any of the tested conditions, which consisted of 26,577 genes out of the 55,401 total mouse genes analyzed. The remaining 28,824 genes were divided into four quantiles based on their average expression levels in cells with FACT. The most dramatic change was seen with the top 25% of the expressed genes, for which chromatin accessibility was significantly increased around the TSS and at gene body (Fig 7D). The differences were gradually reduced from the 5′–3′ ends and behind the TES. The differences were also reduced with the reduction in transcription but remained significant for the three top quantiles of the transcribed genes independent of whether nucleosome‐free or mononucleosome fragments were analyzed (Fig 8E and F).
The differences in chromatin accessibility were not seen for the bottom 25% of the expressed and unexpressed genes when we analyzed the nucleosome‐free fragments, while the plots for the mononucleosome fragments for the same categories of genes at the gene bodies were even reverted (i.e., FACT loss caused a slight reduction in chromatin accessibility at the gene bodies with very low or no transcription). To quantify these effects, we compared the mean read density at the gene bodies (excluding the region immediately after the TSS; see the Materials and Methods for details) for the different categories of genes and fragment lengths (Fig 8G). This analysis confirmed that there were fewer mononucleosome fragments at poorly transcribed genes upon FACT loss.
Untranscribed genes constitute more than half of all the genes used for the analysis and may represent other non‐transcribed regions that constitute most of the genome in mammals. Thus, we used a Hoechst 33342 to measure the overall effect of FACT loss on chromatin accessibility. Hoechst 33342 is a cell‐permeable minor groove‐binding molecule whose fluorescence is many‐fold increased when bound to DNA. An access of small molecules to the minor groove of DNA is limited when DNA is wrapped around nucleosomes (Fitzgerald & Anderson, 1999; Suto et al, 2003). We incubated FACT‐positive and negative adipose MSC cells with Hoechst 33342 and then measured the fluorescence of individual cells using flow cytometry. To avoid potential differences in the amount of DNA per cell due to different cell cycle phases, we arrested all cells in G1 by serum withdrawal for 48 h. As a positive control, we used the small molecule curaxin CBL0137, which disrupts DNA/histone interactions in cells (Safina et al, 2017). As expected, curaxin treatment significantly increased the fluorescence of both homozygous and heterozygous cells (Fig EV5A). The same effect was observed after 4‐OHT treatment of homozygous but not heterozygous cells (Figs 8H and EV5B and C).
Figure EV5. Assessment of general chromatin accessibility by staining DNA in live cells with Hoechst 33342. Histograms of fluorescent intensity of MSC stained with 3 µM of Hoechst 33342 for 15 min.
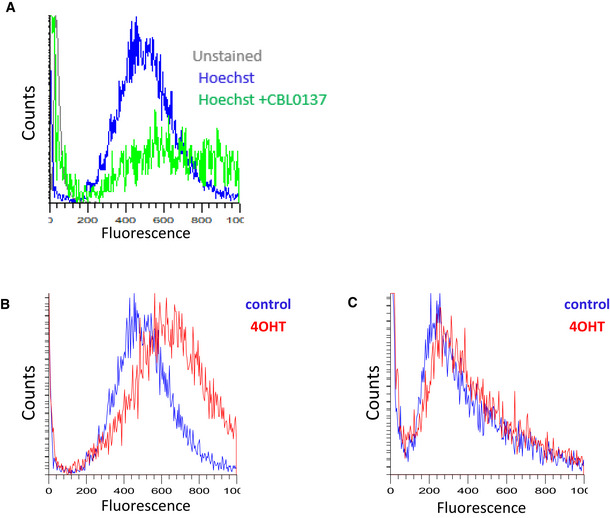
- After incubation with CBL0137 (1 µM) for 30 min. This plot also includes overlap of histogram of negative control (cells unstained with Hoechst).
- Ssrp1fl / fl ; CreERT2 +/+ cells untreated or treated with 4OHT.
- Ssrp1fl /+; CreERT2 +/− cells untreated or treated with 4OHT.
Thus, FACT loss resulted in increased chromatin accessibility, the degree of which was dependent on the level of transcription. The increase in both nucleosome‐free and mononucleosomal fragments was likely due to the opening or partial unwrapping and loss of nucleosomes.
Discussion
Transcription research has mainly focused on how RNA polymerase accesses nucleosomal DNA and what factors facilitate this process. FACT has been viewed as a factor facilitating transcription through chromatin (Orphanides et al, 1998); however, accumulating data questions FACT’s role in opening nucleosomes to RNA polymerase (reviewed in (Gurova et al, 2018; Formosa & Winston, 2020b)). These data may be summarized as follows: (i) there is no evidence that FACT removal from eukaryotic cells results in inhibition of transcription, while it has been shown that FACT depletion in several model systems results in no change or even an increase in the absolute transcription levels (Fleyshman et al, 2017; Sandlesh et al, 2020); (ii) there is no strong data that FACT can bind and destabilize nucleosomes with fully wrapped DNA; however, there is data indicating that FACT binds all nucleosomal components and nucleosomes not fully wrapped with DNA (reviewed in (Gurova et al, 2018)); (iii) decreased FACT levels in cells results in loss of histones from chromatin (Mason & Struhl, 2003; Carvalho et al, 2013; True et al, 2016) and increased accessibility of chromatin (Mylonas & Tessarz, 2018). These observations led to the emergence of a new model for the role of FACT in transcription: FACT binds the components of nucleosomes, histone dimers/tetramers, and possibly DNA, to preserve nucleosomes at their positions during RNA polymerase passage; without FACT, nucleosomes are prone to disassembly by transcription machinery or associated factors. This model can also explain the initially puzzling observation that while FACT enrichment at mammalian genes is highly proportional to the transcription level, the transcription of genes bound by FACT is not inhibited by FACT removal (Garcia et al, 2013; Nesher et al, 2018; Sandlesh et al, 2020). The greater the transcription levels (i.e., more frequent passage of RNA polymerase through a region), the more frequently nucleosomes are disturbed and the more opportunities for FACT to bind. As previously demonstrated, the re‐establishment of histone‐DNA contacts displaces FACT because of the higher affinity of histones for DNA than for FACT (Hsieh et al, 2013). Therefore, genes with lower transcription (i.e., less frequent passage of RNA polymerase) accumulate less FACT. At the same time, if FACT loss is accompanied by the loss of nucleosomes from the transcribed region, we might expect a facilitated passage of RNA polymerase through this region in the absence of nucleosomes, that is, elevated transcription.
In this study, we tested and confirmed this proposed model in a mammalian system. Although in this study we genetically excised only Ssrp1 gene, we have never observed in mammalian cells existence of SPT16 in the absence of SSRP1 and unbound to SSRP1. Thus, we discuss below the consequences of the loss of the whole complex rather than SSRP1 subunit, though theoretically SSRP1 may have independent activity in some cells or conditions.
We demonstrated that FACT is the major factor responsible for the maintenance of chromatin during transcription and is essential for the viability of a small fraction of cells in adult mice. However, these cells are vital to the viability of adult mice because Ssrp1 KO was lethal for mice at all tested ages (5–40 weeks). Together with previously published data that general Ssrp1 KO is lethal at 3.5 dpc (Cao et al, 2003), our new data revealed that Ssrp1 is an essential gene in mammals because it is critical for the survival of stem cells. Since we did not use aged mice in our experiments, consequences of FACT loss in geriatric animals requires further investigation.
The hematopoietic and intestinal tissues were the most sensitive to FACT loss. Both tissue types are dependent on the constant replenishing of cells from tissue‐specific stem cells. The earliest progenitors in both systems were completely lost upon Ssrp1 KO. The stem cells in other tissues may also be sensitive to FACT loss, as indicated by the depletion of adipose tissue and the presence of skin abnormalities. For testing the role of FACT in the viability of other tissues, tissue‐specific Ssrp1 KO is needed to prevent the death of animals from the bone marrow and intestinal failure.
FACT is not essential for many types of adult cells (Fleyshman et al, 2017; Sandlesh et al, 2020). Here, we established that number of many cell types, mostly differentiated but also some progenitors, increased upon FACT loss. This increase is likely due to FACT‐independent, compensatory redistribution of cell populations; however, time‐dependent monitoring of organ cell composition upon Ssrp1 KO is needed to establish this. In addition, the abundance of these cell types, even among stem and progenitor cells, suggests that multiple adult cells do not need FACT for survival. Importantly, there were many proliferating cells among the FACT‐independent cell types. We also did not see changes in the cell‐cycle distribution upon FACT loss, suggesting that stem cells disappear not due to problems with replication or mitosis, processes where FACT activity has been observed (Zeng et al, 2010; Abe et al, 2011). Thus, an important question is what makes these cells sensitive to FACT loss. To address this question, we showed that FACT loss resulted in a significant increase in chromatin accessibility. Previously, we observed that FACT removal from differentiated fibroblasts, which express low FACT levels and are not sensitive to Ssrp1 KO, does not lead to the loss of nucleosomes, while it does in oncogene‐transformed cells (Sandlesh et al, 2020). Therefore, we propose that there is a specific chromatin state in some stem cells and oncogene‐transformed cells, where nucleosomes are not as stable as in other cells what makes these nucleosomes as well as viability of cells‐dependent FACT.
There is a clear difference between the sensitivity of chromatin and FACT loss depending on transcription, that is, the higher the transcription rate, the greater the increase in chromatin accessibility upon FACT removal for most of transcribed genes (top 75% of genes). These changes were in line with the increase in global transcription in MSCs upon FACT depletion. However, there was a small but highly significant reduction in chromatin accessibility at untranscribed genes or those with low transcription rates upon FACT loss (at least if comparing mononucleosome fragments). This finding could potentially be explained by the role of FACT in preventing the stalking of folded nucleosomes through binding to the acidic patches (Li et al, 2016). In this case, the loss of FACT in the absence of transcription might lead to a tighter interaction between nucleosomes, leaving fewer opportunities for transposase to invade the linker DNA.
An interesting observation that may shed light on the physiological consequences of chromatin destabilization is the increase in the number of immune cells in the bone marrow and intestine. The increase in B cells in bone marrow may be due to a shift in differentiation, whereas the number of T cells in the bone marrow and intestine may only be increased due to migration. This accumulation may be related to the fact that the most upregulated pathway in response to Ssrp1 KO in all cell systems examined was the IFN response. We previously observed activation of the IFN response in mice treated with curaxin CBL0137, which disrupts histone/DNA binding (Safina et al, 2017). IFNs are well‐established factors that attract immune cells to the sites of viral invasion and inflammation (Stetson & Medzhitov, 2006). The mechanism of IFN activation upon chromatin destabilization is not fully established; however, in the case of curaxin, it resulted from dsRNA accumulating in cells due to the transcription of pericentromeric repeats (Leonova et al, 2018). The same mechanism may be involved in the effects of FACT depletion along with other mechanisms, such as the emergence of cryptic initiation from uncapped RNA sensed by the RIG/MAVS5 pathway (Unterholzner, 2013) or the binding of nuclear cGAS to DNA losing nucleosomes (Michalski et al, 2020).
In this study, we established that FACT performs a unique function in the earliest progenitor stem cells in several organs of adult mammals. We propose that in these cells, FACT is critical for the maintenance of chromatin during transcription. The loss of FACT results in the loss of nucleosomes at transcribed genes and elevated transcription. The most significantly upregulated pathway in response to the increased chromatin accessibility is IFN signaling, which is likely responsible for the accumulation of immune cells in the sensitive organs in response to FACT loss.
Materials and Methods
Reagents and Tools table
| Reagent | Source | Catalogue # | Application | |||
|---|---|---|---|---|---|---|
| Chemicals and kits | ||||||
| DMEM‐High Glucose with Pyruvate And l‐Glutamine | Corning | MT 10‐013‐CV | Cell culture | |||
| 0.05% Trypsin EDTA | Corning | MT 25‐052‐Cl | Cell culture | |||
| 10,000 U Penicillin‐Streptomycin | Corning | MT 30‐002‐CI | Cell culture | |||
| FBS, USDA, Regular, Heat Inactivated | Corning | MT 35‐011‐CV | Cell culture | |||
| α‐MEM | Stem Cell Technologies | 36450 | Cell culture | |||
| Advanced DMEM/F‐12 | Gibco | 12634010 | Intestinal organoid culture | |||
| GlutaMAX™ Supplement | Gibco | 35050061 | Intestinal organoid culture | |||
| lscove's Modified Dulbecco’s Medium (IMDM) with l‐Glutamine | Gibco | MT 10‐016‐CV | Bone marrow colony‐forming assay | |||
| MethoCult™ GF M3434 | Gibco | 03434 | Bone marrow colony‐forming assay | |||
| KnockOut DMEM | Gibco | 10829018 | mESC cell culture | |||
| Complete Protease Inhibitors—Cocktail, Tablets | Roche | 04693132001 | Preparation of protein extracts | |||
| Laemmli sample buffer | BioRad | 1610737 | Sample preparation for immunoblotting | |||
| Quick Start™ Bradford 1× dye reagent | BioRad | 500‐0204 | Measurement of protein concentrations | |||
| 4–20% gradient precast gels | BioRad | 4561094 | PAGE | |||
| PVDF membranes | BioRad | 1620177 | Immunoblotting | |||
| Western Lightning Plus‐ECL | PerkinElmer | NEL104001EA | Enhanced chemiluminescence for immunoblotting | |||
| Hoechst 33342 | Invitrogen | H3570 | DNA staining | |||
| 4‐Hydroxytamoxifen | Millipore Sigma Aldrich | H7904‐5MG | Induction of Ssrp1 deletion (in vitro) | |||
| Tamoxifen | Millipore Sigma Aldrich | T5648‐1G | Induction of Ssrp1 deletion (in vivo) | |||
| Caspase‐3 substrate (Ac‐DEVD‐AMC) | Enzo Life Sciences | ALX‐260‐031‐M001 | Apoptosis assay | |||
| Click‐iT™ EdU Alexa Fluor™ 488/594 imaging kit | Thermo Fischer Scientific | C10337 | Measurement of DNA replication | |||
| Click‐iT™ EU Alexa Fluor™ 488/594 imaging kit | Thermo Fischer Scientific | C10632 | Measurement i=of general transcription | |||
| Alizarine Red S | Millipore Sigma Aldrich | A5533‐25G | Staining of differentiated osteocytes | |||
| Oil Red O | Millipore Sigma Aldrich | O0625‐25G | Staining of differentiated adipocytes | |||
| Methylene blue | Millipore Sigma Aldrich | M4159 | Measuring cell viability | |||
| Resazurin sodium salt | Millipore Sigma Aldrich | R7017‐1G | Measuring the viability of intestinal organoids | |||
| Collagenase A | Millipore Sigma Aldrich | 10103578001 | Isolation of fibroblasts and mesenchymal stem cells | |||
| Dexamethasone | Millipore Sigma Aldrich | D4902 | Differentiation of mASCs | |||
| Isobutylmethylxanthine (IBMX) | Millipore Sigma Aldrich | I5879‐100MG | Differentiation of mASCs | |||
| Insulin | Gibco | 12585014 | Differentiation of mASCs | |||
| β‐glycerophosphate | Millipore Sigma Aldrich | 50020‐100G | Differentiation of mASCs | |||
| L‐ascorbic acid | Millipore Sigma Aldrich | A92902‐25G | Differentiation of mASC | |||
| Gentamycin | Gibco | 15710064 | Culturing of intestinal organoids | |||
| Murine recombinant EGF | PeproTech | 315‐09 | Culturing of intestinal organoids | |||
| Murine recombinant Noggin | PeproTech | 250‐38 | Culturing of intestinal organoids | |||
| Human recombinant R‐Spondin‐1 | PeproTech | 120‐38 | Culturing of intestinal organoids | |||
| Matrigel | BD Biosciences | 356237 | Culturing of intestinal organoids | |||
| Valproic acid sodium salt | Millipore Sigma Aldrich | P4543‐10G | Culturing of intestinal organoids | |||
| GSK3 inhibitor CHIR99021 | Reprocell | 04‐0004 | Culturing of intestinal organoids | |||
| Nextera kit | Illumina | FC‐121‐1030 | ATAC‐seq | |||
| NEBNext High‐Fidelity 2X PCR Master Mix | New England Biolabs | M0541 | ATAC‐seq | |||
| MinElute PCR Purification kit | Qiagen | 28004 | ATAC‐seq | |||
| mRNA HyperPrep kit | KAPA BIOSYSTEMS | KK8580 | RNA‐seq | |||
| Antibodies | ||||||
| SSRP1 mouse monoclonal 10D1 | BioLegend | 609702 |
Immunoblotting: 1:3,000 Immunohistochemistry: 1:200 |
|||
| SPT16 monoclonal mouse 8D2 | BioLegend | 607002 | Immunoblotting: 1:1,000 | |||
| beta‐actin mouse monoclonal (HRP conjugated) | Millipore Sigma Aldrich | A3854 | Immunoblotting 1:20,000 | |||
| Mouse Lgr5/GPR49 Antibody | R&D Systems | MAB8240‐SP | Immunohistochemistry: 1:200 | |||
| Cleaved caspase 3 | Thermo Fisher Scientific | 700182 | Immunohistochemistry: 1:200 | |||
| m‐IgGκ BP‐HRP | Santa Cruz Biotechnology | sc‐516102 | Immunoblotting: 1:4,000 | |||
| Secondary anti‐mouse Alexa Fluor® (488 and 594) | Life Technologies | A‐11062 and 11001 | Immunohistochemistry: 1:500 | |||
Methods and Protocols
The 10 mg/ml of tamoxifen solution were prepared by mixing 100 mg of tamoxifen powder in 100 µl of 99% proof ethanol and then by slowly dissolving this in 9.9 ml of sterile corn oil at 37°C overnight. A 5 mM of stock solution of 4‐hydroxytamoxifen (4‐OHT) was prepared by dissolving 4.8 mg of 4‐OHT powder in 2.5 ml of 99% proof ethanol.
Alizarin Red S was prepared by dissolving 2 g powder in 100 ml of distilled water. The pH of the solution was adjusted to 4.1–4.3 with 10% ammonium hydroxide. The solution was stored at 4°C away from light.
Oil red O stock solution was prepared by dissolving 0.7 g powder in 200 ml of isopropanol, stirring overnight, and filtering through a 0.2‐µm filter. The solution was stored at 4°C away from light. The working solution was prepared fresh by diluting six parts of oil red O stock into four parts of distilled water. The mixture was incubated at room temperature for 20 min and filtered through a 0.2‐µm filter.
2.5 mM of resazurin solution (36×) was prepared by dissolving 0.629 g of resazurin sodium salt (Milipore Sigma) in Dulbecco’s phosphate‐buffered saline (DPBS) and sterilizing through a 0.22‐µm filter. The solution was stored at −20°C protected from light. The working solution was prepared by diluting this stock to 6×. The working solution was stored at 4°C protected from light for up to 3 months. The 6× solution was used at a final concentration of 70 µM.
Animal experiments
The mouse model described in this study was deposited to MMRRC ID:46323.
Animal breeding and treatment
All animal breeding, procedures, and experiments were conducted in accordance with the Institute Animal Care and Use Committee (IACUC) at Roswell Park Comprehensive Cancer Center under an approved IACUC protocol. Mice were maintained in the Laboratory Animal Shared Resource with controlled air, light, and temperature conditions and fed ad libitum with 5% fat chow with free access to water.
The conditional FACT Knockout mouse model with the following genotypes was generated as previously described (Sandlesh et al, 2018): SSRP1fl / fl ; CreERT2 +/+ (KO mice), SSRP1 +/+; CreERT2 −/− (wild type), SSRP1 +/+; CreERT2 +/+, SSRP1fl / fl ; CreERT2 −/−, SSRP1fl / fl ; CreERT2 −/−, and SSRP1fl /+; CreERT2 +/− (controls).
Mice of different ages were treated with 100 µl of tamoxifen (1 mg) or vehicle by daily IP injection for 5 days. In some experiments, mice received daily subcutaneous injections of 0.9% saline (1 ml). Mouse weights and cage‐side observations were recorded daily. Animals were euthanized by CO2 asphyxiation and cervical dislocation upon losing 20% of their body weight. All cells, tissues, and organs were harvested under sterile conditions.
Mouse tissue collection and staining
Mouse tissues were harvested and preserved in 10% buffered formalin. Paraffin‐embedded tissue sections (5 μm) were stained with hematoxylin and eosin (H&E) and anti‐SSRP1 and Anti‐cleaved caspase‐3 antibodies in the Immunohistochemistry Laboratory of the Roswell Park Pathology Resource Network using standard techniques. All slides were scanned using the Aperio system (Aperio Technologies, Inc). Images were collected using Image Scope software (Aperio Technologies, Inc.).
EdU staining was performed 3 days after the end of tamoxifen treatment. Briefly, mice were injected with 50 mg/kg EdU (Invitrogen, Thermo Fisher Scientific) 1 h before euthanasia. Tissue sections were stained using the Click‐iT EdU Alexa 488 Fluor Imaging kit (Thermo Fisher Scientific) and counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) (Life Technologies) following the manufacturer’s instructions. The stained samples were examined using a Zeiss AxioImager A1 microscope equipped with an epifluorescent light source. Images were captured with an AxioCam MRc digital camera and processed with Zeiss Axio Imager Z1 microscope software (Carl Zeiss, Germany).
Magnetic resonance imaging (MRI)
Two mice of each gender and genotype were imaged. MRI was performed on day 0 before treatment and days 6, 8, and 10 after the start of treatment with tamoxifen or vehicle. Images were acquired on a 4.7 Tesla preclinical MRI scanner using the ParaVision 3.0.2 platform (Bruker Biospin, Billerica MA). A 72‐mm (ID) quadrature RF coil was used to capture the volumetric data for the entire mouse body. Two NMR tubes filled with 1 mM and 3 mM of CuSO4 in 1% agarose were included in each session for signal normalization. Following the scout scans, a three‐dimensional, fast spin echo scan was acquired using the following parameters: TE/TR = 6.2/100 ms; echo train length = 4; averages = 4; FOV = 113 × 55 × 35 mm; matrix = 256 × 128 × 128; zero‐filled to achieve an isotropic voxel size of 273 microns. Image signal intensities were normalized by a three‐point correction curve using two phantoms and background noise, and the whole body volume was segmented and calculated using the region‐growing tool in Analyze 7.0 software (AnalyzeDirect, Overland Park KS). Adipose tissue was segmented from lean tissue using a fixed intensity threshold derived from the average threshold of all datasets, as reported by the Otsu method (REF) and confirmed by visual inspection. The percent adipose tissue volume was calculated as a ratio of voxels segmented as adipose tissue divided by the total body voxels.
Complete blood counts (CBC) and serum biochemistry
Blood (50 µl) was collected from the saphenous veins of mice before treatment (baseline) and on days 7 and 12 after the start of tamoxifen treatment, as previously described (Parasuraman et al, 2010). For serum biochemistry, blood was collected from deeply anesthetized mice via cardiac puncture into serum separator tubes (Sarstedt). The samples were clotted at room temperature for at least 15 min and then centrifuged at 10,000 RCF for 10 min. The serum was analyzed using an IDEXX Catalyst DX Chemistry Analyzer (IDEXX Laboratories).
Cells
Mesenchymal stem cells (MSCs)
MSCs were harvested from the adipose tissue of 8–10‐week‐old mice. The tissue was minced and digested for 30 min via shaking in a solution of 2 mg/ml collagenase A in PBS at 37°C. The digested tissue was filtered through a 40‐µm strainer and centrifuged at 300 g for 5 min. The cells were resuspended in DMEM with 10% FBS and 1% penicillin–streptomycin and plated. To differentiate the MSCs into adipocytes and osteocytes, MSCs were plated on 60‐mm plates. When cells reached 80% confluence, differentiation into adipocytes was induced by incubating the cells in medium containing 250 nM of dexamethasone, 66 nM of insulin, and 0.5 mM of isobutylmethylxanthine (IBMX) for 21 days. Differentiation into osteocytes was induced by incubating in medium containing 0.1 µM of dexamethasone, 10 mM of β‐glycerophosphate, and 50 µM of L‐ascorbic acid for 14 days. The differentiation media were changed every 2–3 days. Differentiation into adipocytes and osteocytes was determined by staining with Oil Red O and Alizarin Red, respectively. After differentiation, the cells were treated with 2 µM of 4‐OHT for 5 days, and viability was assessed by resorufin staining.
Staining for adipocyte and osteocyte differentiation (Oil Red O and Alizarin Red S, respectively) was done by removing the medium, washing cells with PBS, and fixing with 10% neutral buffered formalin for 30 min with gentle shaking at room temperature. The cells were washed twice with distilled water, and then 60% isopropanol was added to the cells for 5 min. The cells were incubated with the appropriate stain for 20–45 min at room temperature, protected from light. The plates were then washed twice with distilled water. The cells were covered with water, and images were taken with a light microscope.
Intestinal stem cell isolation and organoid culture
Intestinal stem cells were collected as previously described (Sato et al, 2009). Briefly, approximately 15 mm of the intestines (small, immediately before cecum; large, immediately after cecum; cecum excluded) was harvested from mice and placed in cold PBS. The intestine was cut open longitudinally and flushed with cold PBS multiple times to remove all the contents. The tissue was cut into 5‐mm pieces and incubated at 4°C in PBS containing 2 mM of EDTA and 50 µg/ml of gentamycin with gentle shaking for 30 min. The tissue was then resuspended in cold PBS and vigorously shaken to dislodge the crypts. After allowing the suspension to settle by gravity for 5 min, the supernatant containing the villus fraction was discarded. The tissue was resuspended in cold PBS and shaken vigorously to enrich the supernatant with the crypt fraction. The crypt fraction was filtered through a 70‐µm strainer and centrifuged at 200 g for 3 min at 4°C. The pellet was resuspended in crypt culture medium (advanced DMEM:F12 containing 2% FBS, 50 µg/ml of gentamycin, 2 mM of l‐glutamine, 50 ng/ml of EGF, 500 ng/ml of R‐Spondin‐1, 100 ng/ml of Noggin, 1 mM of valproic acid sodium salt, and 3 µM of GSK3 inhibitor CHIR99021. The crypts were counted using a hemocytometer, and 500 crypts were mixed with 50 µl of Matrigel at 4°C and pipetted into the center of the wells of 24‐well plates prewarmed to 37°C. The domes were allowed to set for 10 min in a 37°C incubator, and then crypt culture medium (500 µl) was gently added to each well. Organoids formed over 7–10 days, with medium changed every 2–3 days. The crypts were passaged as previously described (Sato et al, 2009). After two passages, the organoids were treated with 2 µM of 4‐OHT for 5 days. On day 6, the organoids were split into new wells and allowed to grow for 5–7 days. The ability to form colonies was assessed by counting the number of organoids in the control‐ and 4‐OHT‐treated wells. The data were normalized to the number of colonies formed in the control wells.
To differentiate the organoids, the intestinal stem cells were cultured in medium without R‐Spondin‐1 for 2.5 days as previously described (Lindeboom et al, 2018). The undifferentiated organoids were maintained in medium containing R‐Spondin‐1. The differentiated and undifferentiated organoids were treated with 2 µM of 4‐OHT for 5 days, and viability was tested at the end of treatment using resorufin staining.
For immunofluorescence, the organoids were fixed in 4% paraformaldehyde (PFA) for 30 min, which partly dissolved the Matrigel. The organoids were collected by centrifugation at 900 g for 5 min at room temperature. The pellets were washed with PBS, and the organoids were permeabilized with PBS containing 0.1% Triton X‐100 and 3% bovine serum albumin at room temperature with rotating. The samples were then blocked in PBS containing 0.05% Triton X‐100 and 1% BSA for 1 h with rotating. The organoids were stained with primary antibodies (SSRP1 [1:200], Lgr5 [1:200]) at 4°C overnight with rotating. The organoids were washed with blocking solution and incubated for 1 h at room temperature with the following secondary antibodies: anti‐rat Alexa Fluor 488 (1:500) against Lgr5 and anti‐mouse Alexa Fluor 594 (1:500) against SSRP1. The organoids were washed with blocking solution and PBS and then counterstained with Hoechst (1:2,000 in PBS) for 30 min at room temperature with rotation, protected from light. After staining, the organoids were washed with PBS and then plated in 96‐well plates. Images were taken using a Zeiss Axio Imager Z1 microscope (Carl Zeiss, Germany).
Bone marrow colony‐forming assay
The femurs and tibias of mice were excised from the hindlimbs by removing the muscles and connective tissue and cleaned with 70% ethanol. The bones were crushed using a sterile mortar and pestle in PBS to generate a single cell suspension that was filtered through a 70‐µm cell strainer. This process was repeated until the bones appeared white, indicating all the marrow was removed. The cells were counted using a hemocytometer, and a suspension containing 4.5 × 105 nucleated cells/ml was prepared. Complete IMDM medium was prepared by adding 20 ml of IMDM to 80 ml of MethoCult M3231. 4‐OHT (2 µM) or DMSO (control) was added to 4.1 ml of complete IMDM by inverting the tube and vortexing for 5 s twice. The tube was placed in a refrigerator for 5 min to allow bubbles to escape. The 4.5 × 105 cells/ml bone marrow cell suspension (400 µl) was added to each tube of medium and the 4‐OHT and control mixtures. The tubes were mixed gently by inverting the tube two times and vortexing three times for 8 s each at maximum speed, followed by bubble release. The cell + medium + 4‐OHT or control mixture (1 ml) was plated in 35‐mm dishes using a primed 5‐ml syringe with a 19G needle. The final cell density was 40,000 per plate. The plates were incubated at 37°C, and colonies were allowed to form on methylcellulose in the presence or absence of 4‐OHT for 7 days.
Viability assays
For the methylene blue assay, 0.5% methylene blue in 50% methanol was added to plates after removing the medium. The plates were incubated for 15 min at room temperature with gentle shaking. The plates were washed with distilled water and air‐dried. The dye was eluted for 30 min at room temperature with 1% sodium dodecyl sulfate (SDS) in PBS with shaking. The absorbance was read at 595 nm.
The resorufin assay was used to measure the viability of the intestinal organoids. Undifferentiated and differentiated Intestinal organoids were treated with or without 2 µM of 4‐OHT for 5 days in 96‐well plates (100 µl). After treatment, 20 µl 6× resorufin solution was added to each well for a final concentration of 70 µM, and the plates were incubated at 37°C for 6 h. The absorbance was read at 570 nm.
EdU staining
Three days after the last treatment with vehicle or tamoxifen, 50 mg/kg EdU was administered intraperitoneally to the mice. After 2 h, the mice were euthanized, and the tissues were harvested and snap‐frozen. Tissue sections were fixed with 3.7% paraformaldehyde for 15 min at room temperature. For EdU treatment of cells, the cells were treated with 2 µM of 4‐OHT for 5 days and then split. After 24 h, the cells were incubated with 20 µM EdU for 1 h. The cells were then fixed with 3.7% PFA in PBS for 15 min at room temperature. Cells and tissue sections were permeabilized with 0.1% Triton X‐100 in PBS for 20 min at room temperature. The incorporated EdU was detected using the Click‐iT® Plus EdU Imaging Kit (Invitrogen), according to the manufacturer’s instructions.
EU staining
MSC cells were treated with vehicle or tamoxifen for 5 days and then split. After 24 h, the cells were treated with 0.2 mM of EU for 1 h. After the incubation, the cells were trypsinized, harvested, and washed with PBS in 1% BSA. The cells were fixed with 100 µl of Click‐iT fixative for 15 min at room temperature. The cells were stained with an SSRP1 antibody using a standard protocol and then for EU according to the manufacturer’s instructions. The cells were counterstained with Hoechst 33342. Cell staining was analyzed by flow cytometry.
Immunoblotting
Cells were washed with PBS and lysed on ice with RIPA buffer (150 mM of sodium chloride, 1.0% NP‐40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 50 mM of Tris, pH 8.0) containing protease inhibitor (Roche) for 30 min. The extracts were centrifuged at 15,000 g for 15 min to remove the debris. The protein concentrations of the supernatants were determined using Quick Start™ Bradford 1× dye reagent. Equal amounts of protein were boiled with Laemmli buffer at 100°C for 5 min. The samples were briefly centrifuged and then separated on precast 4–20% gradient gels. The proteins were transferred to PVDF membranes, which were blocked with 5% skim milk at room temperature for 1 h with gentle shaking. The blots were incubated with primary mouse SSRP1 or SPT16 antibodies at 4°C overnight. After the incubation, the blots were washed and incubated with HRP‐conjugated, anti‐mouse secondary antibodies for 1 h at room temperature. As a control, the blots were incubated with HRP‐conjugated mouse β‐actin antibody at room temperature for 1 h. The blots were washed for 1 min with Western Lightning Plus‐ECL, and images were acquired using the BioRad ChemiDoc™ Touch Imaging System.
Caspase‐3 assay
The caspase‐3 fluorogenic substrate Ac‐DEVD‐AMC was reconstituted in DMSO at a concentration of 1 mg/ml. Cells were lysed with 1X CCLR buffer, and the protein concentration was estimated using the Pierce BCA assay. The protein was used at a concentration of 2 µg/µl (50 µl). Ac‐DEVD‐AMC (20 µM in 20 mM HEPES, 10% glycerol, and 2 mM DTT) was added to each sample in a 96‐well black plate (BD Falcon). The plates were incubated at 37°C for 1 h, and then the fluorescence was measured.
DNA minor groove binding
Cells (500,000) were treated with 4‐OHT for 5 days and then serum‐starved (0.5% FBS) for 48 h. For the positive control, cells were treated with 1 µM of CBL0137 for 30 min. The cells were washed with PBS and stained with 3 µM Hoechst 33342 in 3% BSA‐PBS at 37°C for 15 min.
Flow cytometry
Flow cytometry was performed using LSRB II (Becton Dickinson). Histograms were constructed using WinList 3D software (Verity Software House).
ATAC‐seq
Two biological replicates were used for each condition. Cells were treated with 4‐OHT for 5 days and then split. After attachment, the cells were cultured in medium containing 0.5% FBS for 48 h. Cells (50,000) were prepared for ATAC‐seq as previously described (Buenrostro et al, 2013), with slight modifications. Briefly, the cells were washed with PBS, lysed, and incubated with the transposition reaction mixture at 37°C for 30 min. The DNA was purified using the Qiagen MinElute PCR Purification Kit and amplified using PCR primer 1 and barcoded primer 2 (1 cycle of 72°C for 5 min, 98°C for 30 s, 13 cycles of 98°C for 10 s, 63°C for 30 s, 72°C for 1 min). The primers were custom synthesized by Integrated DNA Technologies (IDT) using previously described sequences (Buenrostro et al, 2013). The PCR products were purified using AmpureXP beads (Beckman Coulter). The resulting libraries were evaluated on High‐Sensitivity D1000 ScreenTape using the TapeStation 4200 (Agilent Technologies). They were quantitated using the Kapa Biosystems qPCR quantitation kit for Illumina. The libraries were pooled, denatured, and diluted to 350 pM, with a 1% PhiX control library added. The resulting pool was loaded into a NovaSeq Reagent cartridge and sequenced using a NovaSeq6000 following the manufacturer’s recommended protocol (Illumina Inc.).
RNA‐seq
Two biological replicates were used for each condition. Cells were treated with 4‐OHT for 5 days, then split and allowed to grow for 24 h. The sequencing libraries were prepared from 500 ng total RNA using the mRNA HyperPrep kit (KAPA BIOSYSTEMS) following the manufacturer’s instructions. Briefly, PolyA RNA was purified using the mRNA Capture Beads. The purified RNA was fragmented and primed for cDNA synthesis. Fragmented RNA was reverse transcribed into first‐strand cDNA using random primers. Second‐strand cDNA synthesis converted the cDNA:RNA hybrids to double‐strand cDNA (dscDNA), with the 2nd strand marked by incorporating dUTP in place of dTTP. Pure beads were used to separate the dscDNA from the second‐strand reaction mix, resulting in blunt‐ended cDNA. A single “A” nucleotide was added to the 3′ ends of the blunt fragments. Multiple indexing adapters containing a single “T” nucleotide on the 3′ end were ligated to the ends of the dscDNA to prepare them for hybridization onto a flow cell. The adapter‐ligated libraries were amplified by PCR, purified using Pure Beads, and validated for the appropriate size using a 4200 TapeStation D1000 ScreenTape (Agilent Technologies, Inc.). The DNA libraries were quantitated using the KAPA Biosystems qPCR kit and pooled in an equimolar fashion. Each pool was denatured and diluted to 350 pM, with a 1% PhiX control library added. The resulting pools were loaded into NovaSeq Reagent cartridges for 100‐cycle single‐read analysis and sequenced with a NovaSeq6000 following the manufacturer’s recommended protocol (Illumina Inc.).
Sample preparation for single‐cell RNA‐sequencing
Bone marrow and intestinal cells from vehicle‐ and tamoxifen‐treated mice were used for single‐cell gene expression using 10× Genomics technology. For bone marrow samples, mouse femurs were cleaned with alcohol, washed with ice‐cold PBS containing antibiotics, and the metaphysis removed. The marrow was flushed with buffer (2 mM of EDTA + 0.5% BSA in PBS) using a 23G syringe. The cells were gently mixed, filtered through a 40‐µm cell strainer, and centrifuged at 300 g for 10 min. The pellets were resuspended in 0.04% BSA‐PBS and counted using trypan blue. A total of 1 × 105 cells were used for the analysis (1,000 cells/µl).
For the intestinal samples, 2 cm of intestine above and below cecum, excluding cecum, were excised and flushed with ice‐cold PBS containing gentamycin using a 20G needle. The intestine was placed in a dish with fresh PBS and cut open longitudinally. The walls were washed to remove any residual matter. The intestine was incubated in ice‐cold PBS containing 10 mM EDTA (10 ml) at 4°C with gentle rocking for 30 min. The tissue was into ice‐cold PBS and shaken with vigorous bursts to release the epithelial cells. The supernatant was centrifuged at 300 g for 5 min at 4°C. The pellet was washed once with ice‐cold PBS, resuspended in PBS containing 1 mg/ml collagenase, and incubated at 37°C for 5 min. The tube was then placed immediately on ice, followed by the addition of 5% (v/v) FBS. The cell suspension was centrifuged, washed with PBS, and filtered through a 40‐µm cell strainer. The cells were resuspended in 0.04% BSA‐PBS and counted using trypan blue. A total of 1 × 105 cells were used for the analysis (1,000 cells/µl).
10× Genomics
Single‐cell libraries were generated using the 10× Genomics platform. Cell suspensions were assessed by trypan blue using the Countess FL automated cell counter (ThermoFisher) to determine the concentration, viability, and absence of clumps and debris that could interfere with single‐cell capture. The cells were loaded into the Chromium Controller (10× Genomics), where they were partitioned into nanoliter‐scale Gel Beads‐in‐emulsion with a single barcode per cell. Reverse transcription was performed, and the resulting cDNA was amplified. The full‐length amplified cDNA was used to generate gene expression libraries by enzymatic fragmentation, end‐repair, a‐tailing, adapter ligation, and PCR to add Illumina‐compatible sequencing adapters. The resulting libraries were evaluated on D1000 ScreenTape using the TapeStation 4200 (Agilent Technologies) and quantitated using the Kapa Biosystems qPCR quantitation kit for Illumina. The libraries were then pooled, denatured, and diluted to 300 pM, with 1% PhiX control library added. The resulting pool was loaded into the appropriate NovaSeq Reagent cartridge and sequenced on a NovaSeq6000, following the manufacturer’s recommended protocol (Illumina Inc.).
Bioinformatics analysis
RNA‐seq fastq files were aligned to the mouse mm10 genome using STAR (Dobin et al, 2013) with GENCODE v25 gene annotation. Differential gene expression was assessed using DESeq2 (Love et al, 2014) with default parameters. Genes with a 1.5‐fold change between conditions and FDR < 0.05 were considered differentially expressed. GSEA was run using the MSigDB website (Broad Institute), and GO enrichment was run using g:Profiler (Raudvere et al, 2019) (Ensembl v104). The gene lists with FDR q‐value ≤ 1e‐4 were considered significant. The baseMean column from the DESeq2 differential expression result table was used to classify genes based on expression levels. A list of unexpressed genes was created from the genes with baseMean 0. The genes with baseMean > 0 were used for ranking based on expression levels. For this ranking, the baseMean values were divided by gene length and then ranked and divided into four quantiles.
ATAC‐seq fastq files were aligned to the same genome using bowtie2 (Langmead & Salzberg, 2012) with the following arguments: ‐‐very‐sensitive ‐‐no‐mixed ‐‐no‐unal ‐‐no‐discordant. Duplicates were filtered using the Picard MarkDuplicates function. The bigwig files with CPM‐normalized profiles were obtained using deepTools (Ramirez et al, 2014) and parameters maxFragmentlength and minFragmentlength to account for the fragment sizes. Pile‐ups were calculated using the computeMatrix function in the scale regions or reference‐point modes and visualized using the plotProfile and plotHeatmap functions from deepTools. The mean density reads at the gene bodies were calculated using the ComputeMatrix function from deepTools with the following parameters: scale regions ‐o results.tab.gz ‐‐regionBodyLength 5000 ‐a 0 ‐b 0 ‐‐binSize 50 ‐‐missingDataAsZero ‐‐skipZeros. The results.tab files for each expression category from two replicates were combined into one table. Bins 21–100 were used for the calculation of the mean density reads. These parameters were compared between conditions using the paired t‐test.
Single‐cell RNA‐seq analysis: The raw sequencing data were processed using Cellranger (version 5.0.0) software (10× Genomics). The filtered gene‐barcode matrices, which contained the barcodes with the Unique Molecular Identifier (UMI) counts that passed the cell detection algorithm, were used for further analysis. The Seurat single‐cell data analysis R package was used for read normalization, dimension reduction, and cell clustering (Satija et al, 2015). Cells with low RNA content (detected transcripts < 200), higher mitochondrial RNA content (> 20% of total reads), or very high RNA content (> 5,000 detected transcripts, likely doublets) were filtered out from the analysis. The mitochondrial RNA content filter was not applied for the intestine data as some of the cells had very high mtRNA content, as expected (Osorio & Cai, 2021). Additionally, the cell‐cycle scores for the S and G2/M phases were determined using the CellCycleScoring method based on the mouse orthologous genes for the cc.genes in the Seurat package. The lists of the cell cycle genes from Buettner et al (2015) were used to calculate the S and G2/M scores for each cell. The score was the average expression of the cell cycle genes subtracted by the aggregated expression of the control feature sets, which had the same number of genes and similar average gene expression value as the cell cycle genes.
The genes used for the S phase were Mcm4, Exo1, Slbp, Gmnn, Cdc45, Msh2, Mcm6, Rrm2, Pold3, Blm, Ubr7, Mcm5, Clspn, Hells, Nasp, Rpa2, Rad51ap1, Tyms, Rrm1, Rfc2, Prim1, Brip1, Usp1, Ung, Pola1, Mcm2, Fen1, Tipin, Pcna, Cdca7, Uhrf1, Casp8ap2, Cdc6, Dscc1, Wdr76, E2f8, Dtl, Ccne2, Atad2, Gins2, Chaf1b, and Pcna‐ps2. The G2/M genes were Nuf2, Psrc1, Ncapd2, Ccnb2, Smc4, Lbr, Tacc3, Cenpa, Kif23, Cdca2, Anp32e, G2e3, Cdca3, Anln, Cenpe, Gas2l3, Tubb4b, Cenpf, Dlgap5, Hjurp, Cks1brt, Gtse1, Bub1, Birc5, Ube2c, Rangap1, Hmmr, Ect2, Tpx2, Ckap5, Cbx5, Nek2, Ttk, Cdca8, Nusap1, Ctcf, Cdc20, Cks2, Mki67, Tmpo, Ckap2l, Aurkb, Kif2c, Cdk1, Kif20b, Top2a, Aurka, Ckap2, Hmgb2, Cdc25c, Ndc80, and Kif11.
The normalized and scaled UMI counts were calculated using the SCTransform method and regressed against the cell‐cycle scores. Dimension reductions, including principal component analysis (PCA) and UMAP, were carried out. Data clustering was identified using shared nearest neighbor (SNN)‐based clustering on the first 30 principal components. Marker genes for each cluster were identified using the default parameters of the FindMarkers method in the Seurat package. For the bone marrow data, cell annotation was done by the singleR (Aran et al, 2019) R package using the Immgen database as a reference. For the small intestine, marker genes from the published single‐cell data (Haber et al, 2017; Gao et al, 2018) were used to annotate the cell types.
Venn diagrams were built using https://bioinformatics.psb.ugent.be/webtools/Venn/.
GSEA was run using MSigDB https://www.gsea‐msigdb.org/gsea/msigdb/.
GO was run using https://biit.cs.ut.ee/gprofiler/gost.
Statistical analyses
All experiments, except scRNA sequencing, were run with at least two biological replicates and included additional technical replicates (independent well, slides, or fields of view). Animals were randomized between groups based on genotype, gender, and age. No blinding was done for experiments with mice, except pathology examination of H&E stained slides. Mice experiments including measurement of mouse viability and weight included groups of 5 and more mice of the same gender. Other animal experiments including collection of samples for different assays were done with at least 2 animals of the same gender per group. The exact number of animals is provided in figure legends. Pathology examination was done without knowledge of mouse genotype. Student’s t‐test was used to compare data between 2 groups. Details of the t‐test used (paired or unpaired, one or two tails distribution) are provided in figure legends. One‐way ANOVA with Tukey posthoc tests was used to compare data between multiple groups.
Author contributions
Imon Goswami: Investigation; Writing—original draft; Project administration. Poorva Sandlesh: Investigation; Methodology; Project administration; Writing—review & editing. Aimee Stablewski: Resources; Methodology. Ilya Toshkov: Investigation; Pathology evaluation. Alfiya Safina: Investigation. Mikhail Magnitov: Data curation; Formal analysis; Writing—review & editing. Jianmin Wang: Data curation; Formal analysis; Supervision; Investigation; Project administration; Writing—review & editing. Katerina Gurova: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Project administration; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
IG performed part of the animal experiments and most of the cell‐based experiments and drafted the manuscript. PS generated the mouse model, characterized the phenotypes of the mice upon FACT depletion, and edited the manuscript. AS generated the genetically‐modified mouse embryonic stem cells and performed the in vitro fertilization. IT is an animal pathologist who reviewed and evaluated all H&E slides. AFS performed the EU incorporation assays and assisted with other experiments. MM and JW assisted with the bioinformatic analyses, and MM edited the manuscript. KG developed the concept of the study, analyzed all the data, and drafted and edited the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Acknowledgements
We are grateful for the help and support of the administrative office of the Cell Stress Biology Department of Roswell Park Comprehensive Cancer Center, specifically Mary Morgan, Sarah Tyrpak and Bruce Specht. We thank the members of the shared resources facilities for help and advice with the design and processing of the experiments, specifically Dr. Prashant Singh, Director of the Genomics Shared Resource, Dr. Joseph Spernyak, Director of the Translational Imaging Shared Resource, and Dr. Leslie Curtin, veterinarian in the Laboratory Animal Shared Resource. We also thank Shawn Matott, a senior systems analyst, for the help with the High Power Computer resources. This study was partially funded by a National Institutes of Health (NIH) National Cancer Institute (NCI) grant to KG (R21CA198395), Roswell Park Alliance Foundation grant to KG, NCI NIH Core grant to RPCCC (P30CA16056), NIH Office of Research Infrastructure Program grant to the Center for Computational Research at the University at Buffalo (S10OD024973). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
EMBO reports (2022) 23: e53684.
Contributor Information
Jianmin Wang, Email: jianmin.wang@roswellpark.org.
Katerina Gurova, Email: katerina.gurova@roswellpark.org.
Data availability
The datasets supporting the conclusions in this article are available in the GEO Datasets repository. scRNA‐seq data are available as GSE189866, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE189866; ATAC‐seq and RNA‐seq data are available as GSE189663, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE189663.
References
- Abe T, Sugimura K, Hosono Y, Takami Y, Akita M, Yoshimura A, Tada S, Nakayama T, Murofushi H, Okumura K et al (2011) The histone chaperone facilitates chromatin transcription (FACT) protein maintains normal replication fork rates. J Biol Chem 286: 30504–30512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar SV, Aguilar O, Allan R, Amir EAD, Angeli V, Artyomov MN, Asinovski N, Astarita J, Austen KF, Bajpai G et al (2020) ImmGen at 15. Nat Immunol 21: 700–703 [DOI] [PubMed] [Google Scholar]
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR et al (2019) Reference‐based analysis of lung single‐cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20: 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods 10: 1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O (2015) Computational analysis of cell‐to‐cell heterogeneity in single‐cell RNA‐sequencing data reveals hidden subpopulations of cells. Nat Biotechnol 33: 155–160 [DOI] [PubMed] [Google Scholar]
- Cao S, Bendall H, Hicks GG, Nashabi A, Sakano H, Shinkai Y, Gariglio M, Oltz EM, Ruley HE (2003) The high‐mobility‐group box protein SSRP1/T160 is essential for cell viability in day 3.5 mouse embryos. Mol Cell Biol 23: 5301–5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo‐Fonseca M, de Almeida SF (2013) Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res 41: 2881–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilg D, Saleh RN, Phelps SE, Rose Y, Dupays L, Murphy C, Mohun T, Anderson RH, Scambler PJ, Chapgier AL (2016) HIRA is required for heart development and directly regulates Tnni2 and Tnnt3. PLoS One 11: e0161096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DJ, Anderson JN (1999) Selective nucleosome disruption by drugs that bind in the minor groove of DNA. J Biol Chem 274: 27128–27138 [DOI] [PubMed] [Google Scholar]
- Fleyshman D, Prendergast L, Safina A, Paszkiewicz G, Commane M, Morgan K, Attwood K, Gurova K (2017) Level of FACT defines the transcriptional landscape and aggressive phenotype of breast cancer cells. Oncotarget 8: 20525–20542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa T, Winston F (2020a) The role of FACT in managing chromatin: disruption, assembly, or repair? Nucleic Acids Res 48: 11929–11941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa T, Winston F (2020b) The role of FACT in managing chromatin: disruption, assembly, or repair? Nucleic Acids Res 48: 11929–11941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Yan L, Wang R, Li J, Yong J, Zhou X, Wei Y, Wu X, Wang X, Fan X et al (2018) Tracing the temporal‐spatial transcriptome landscapes of the human fetal digestive tract using single‐cell RNA‐sequencing. Nat Cell Biol 20: 721–734 [DOI] [PubMed] [Google Scholar]
- Garcia H, Fleyshman D, Kolesnikova K, Safina A, Commane M, Paszkiewicz G, Omelian A, Morrison C, Gurova K (2011) Expression of FACT in mammalian tissues suggests its role in maintaining of undifferentiated state of cells. Oncotarget 2: 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia H, Miecznikowski J, Safina A, Commane M, Ruusulehto A, Kilpinen S, Leach R, Attwood K, Li Y, Degan S et al (2013) Facilitates chromatin transcription complex is an "accelerator" of tumor transformation and potential marker and target of aggressive cancers. Cell Rep 4: 159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurova K, Chang HW, Valieva ME, Sandlesh P, Studitsky VM (2018) Structure and function of the histone chaperone FACT ‐ Resolving FACTual issues. Biochim Biophys Acta Gene Regul Mech 1861: 892–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y et al (2017) A single‐cell survey of the small intestinal epithelium. Nature 551: 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond CM, Stromme CB, Huang H, Patel DJ, Groth A (2017) Histone chaperone networks shaping chromatin function. Nat Rev Mol Cell Biol 18: 141–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh FK, Kulaeva OI, Patel SS, Dyer PN, Luger K, Reinberg D, Studitsky VM (2013) Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc Natl Acad Sci USA 110: 7654–7659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SJ, Fu RH, Shyu WC, Liu SP, Jong GP, Chiu YW, Wu HS, Tsou YA, Cheng CW, Lin SZ (2013) Adipose‐derived stem cells: isolation, characterization, and differentiation potential. Cell Transplant 22: 701–709 [DOI] [PubMed] [Google Scholar]
- Koltowska K, Apitz H, Stamataki D, Hirst EM, Verkade H, Salecker I, Ober EA (2013) Ssrp1a controls organogenesis by promoting cell cycle progression and RNA synthesis. Development 140: 1912–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolundzic E, Ofenbauer A, Bulut SI, Uyar B, Baytek G, Sommermeier A, Seelk S, He M, Hirsekorn A, Vucicevic D et al (2018) FACT Sets a barrier for cell fate reprogramming in Caenorhabditis elegans and human cells. Dev Cell 46: 611–626.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonova K, Safina A, Nesher E, Sandlesh P, Pratt R, Burkhart C, Lipchick B, Gitlin I, Frangou C, Koman I et al (2018) TRAIN (Transcription of Repeats Activates INterferon) in response to chromatin destabilization induced by small molecules in mammalian cells. Elife 7: e30842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Chen P, Yu J, Dong L, Liang D, Feng J, Yan J, Wang P‐Y, Li Q, Zhang Z et al (2016) FACT remodels the tetranucleosomal unit of chromatin fibers for gene transcription. Mol Cell 64: 120–133 [DOI] [PubMed] [Google Scholar]
- Lindeboom RGH, Voorthuijsen L, Oost KC, Rodríguez‐Colman MJ, Luna‐Velez MV, Furlan C, Baraille F, Jansen PWTC, Ribeiro A, Burgering BMT et al (2018) Integrative multi‐omics analysis of intestinal organoid differentiation. Mol Syst Biol 14: e8227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou K, Zhang N, Wei H, Tan YZ, Zhang Z, Carragher B, Potter CS, D'Arcy S, Luger K (2020) FACT caught in the act of manipulating the nucleosome. Nature 577: 426–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Martel C, Sourla A, Gauthier S, Merand Y, Belanger A, Labrie C, Labrie F (1997) Comparative effects of 28‐day treatment with the new anti‐estrogen EM‐800 and tamoxifen on estrogen‐sensitive parameters in intact mice. Int J Cancer 73: 381–391 [DOI] [PubMed] [Google Scholar]
- Mason PB, Struhl K (2003) The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo . Mol Cell Biol 23: 8323–8333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messiaen S, Guiard J, Aigueperse C, Fliniaux I, Tourpin S, Barroca V, Allemand I, Fouchet P, Livera G, Vernet M (2016) Loss of the histone chaperone ASF1B reduces female reproductive capacity in mice. Reproduction 151: 477–489 [DOI] [PubMed] [Google Scholar]
- Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, Hornung V, Hopfner KP (2020) Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587: 678–682 [DOI] [PubMed] [Google Scholar]
- Mylonas C, Tessarz P (2018) Transcriptional repression by FACT is linked to regulation of chromatin accessibility at the promoter of ES cells. Life Science Alliance 1: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesher E, Safina A, Aljahdali I, Portwood S, Wang ES, Koman I, Wang J, Gurova KV (2018) Role of chromatin damage and chromatin trapping of FACT in mediating the anticancer cytotoxicity of DNA‐binding small molecule drugs. Cancer Res 78: 1431–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D (1998) FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92: 105–116 [DOI] [PubMed] [Google Scholar]
- Osorio D, Cai JJ (2021) Systematic determination of the mitochondrial proportion in human and mice tissues for single‐cell RNA‐sequencing data quality control. Bioinformatics 37: 963–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasuraman S, Raveendran R, Kesavan R (2010) Blood sample collection in small laboratory animals. J Pharmacol Pharmacother 1: 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast L, Hong E, Safina A, Poe D, Katerina G (2020) Histone chaperone FACT is essential to overcome replication stress in mammalian cells. Oncogene 39: 5124–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T (2014) deepTools: a flexible platform for exploring deep‐sequencing data. Nucleic Acids Res 42: W187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J (2019) g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47: W191–W198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safina A, Cheney P, Pal M, Brodsky L, Ivanov A, Kirsanov K, Lesovaya E, Naberezhnov D, Nesher E, Koman I et al (2017) FACT is a sensor of DNA torsional stress in eukaryotic cells. Nucleic Acids Res 45: 1925–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safina A, Garcia H, Commane M, Guryanova O, Degan S, Kolesnikova K, Gurova KV (2013) Complex mutual regulation of facilitates chromatin transcription (FACT) subunits on both mRNA and protein levels in human cells. Cell Cycle 12: 2423–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandlesh P, Juang T, Safina A, Higgins MJ, Gurova KV (2018) Uncovering the fine print of the CreERT2‐LoxP system while generating a conditional knockout mouse model of Ssrp1 gene. PLoS One 13: e0199785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandlesh P, Safina A, Goswami I, Prendergust L, Rosario S, Gomez E, Wang J, Gurova K (2020) Prevention of chromatin destabilization by FACT is crucial for malignant transformation. iScience 23: 101177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, Regev A (2015) Spatial reconstruction of single‐cell gene expression data. Nat Biotechnol 33: 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ et al (2009) Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 459: 262–265 [DOI] [PubMed] [Google Scholar]
- Shi JX, Li JS, Hu R, Li XM, Wang H (2016) CAF1‐knockout mice are more susceptive to lipopolysaccharide‐induced acute lung injury. J Inflamm Res 9: 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R (2006) Type I interferons in host defense. Immunity 25: 373–381 [DOI] [PubMed] [Google Scholar]
- Su L, Mruk DD, Cheng CY (2011) Drug transporters, the blood‐testis barrier, and spermatogenesis. J Endocrinol 208: 207–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto RK, Edayathumangalam RS, White CL, Melander C, Gottesfeld JM, Dervan PB, Luger K (2003) Crystal structures of nucleosome core particles in complex with minor groove DNA‐binding ligands. J Mol Biol 326: 371–380 [DOI] [PubMed] [Google Scholar]
- Taha MF, Hedayati V (2010) Isolation, identification and multipotential differentiation of mouse adipose tissue‐derived stem cells. Tissue Cell 42: 211–216 [DOI] [PubMed] [Google Scholar]
- Tan BC, Chien CT, Hirose S, Lee SC (2006) Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J 25: 3975–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- True JD, Muldoon JJ, Carver MN, Poorey K, Shetty SJ, Bekiranov S, Auble DT (2016) The modifier of transcription 1 (Mot1) ATPase and Spt16 histone chaperone co‐regulate transcription through preinitiation complex assembly and nucleosome organization. J Biol Chem 291: 15307–15319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunaka Y, Fujiwara Y, Oyama T, Hirose S, Morikawa K (2016) Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev 30: 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L (2013) The interferon response to intracellular DNA: why so many receptors? Immunobiology 218: 1312–1321 [DOI] [PubMed] [Google Scholar]
- Voon HP, Hughes JR, Rode C, De La Rosa‐Velazquez IA, Jenuwein T, Feil R, Higgs DR, Gibbons RJ (2015) ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep 11: 405–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenburg G, Flade V, Garbe AI, Lauer G, Labudde D (2014) Scaffold preferences of mesenchymal stromal cells and adipose‐derived stem cells from green fluorescent protein transgenic mice influence the tissue engineering of bone. Br J Oral Maxillofac Surg 52: 409–414 [DOI] [PubMed] [Google Scholar]
- Yang J, Zhang XU, Feng J, Leng HE, Li S, Xiao J, Liu S, Xu Z, Xu J, Li DI et al (2016) The histone chaperone FACT contributes to DNA replication‐coupled nucleosome assembly. Cell Rep 14: 1128–1141 [DOI] [PubMed] [Google Scholar]
- Zeng SX, Li Y, Jin Y, Zhang QI, Keller DM, McQuaw CM, Barklis E, Stone S, Hoatlin M, Zhao Y et al (2010) Structure‐specific recognition protein 1 facilitates microtubule growth and bundling required for mitosis. Mol Cell Biol 30: 935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Liu Y, Luger K (2020) Histone chaperone FACT FAcilitates Chromatin Transcription: mechanistic and structural insights. Curr Opin Struct Biol 65: 26–32 [DOI] [PubMed] [Google Scholar]