

Abstract
Skin wound repair is essential for organismal survival and failure of which leads to non‐healing wounds, a leading health issue worldwide. However, mechanistic understanding of chronic wounds remains a major challenge due to lack of appropriate genetic mouse models. αSMA+ myofibroblasts, a unique class of dermal fibroblasts, are associated with cutaneous wound healing but their precise function remains unknown. We demonstrate that genetic depletion of αSMA+ myofibroblasts leads to pleiotropic wound healing defects, including lack of reepithelialization and granulation, dampened angiogenesis, and heightened hypoxia, hallmarks of chronic non‐healing wounds. Other wound‐associated FAP+ and FSP1+ fibroblasts do not exhibit such dominant functions. While type I collagen (COL1) expressing cells play a role in the repair process, COL1 produced by αSMA+ myofibroblasts is surprisingly dispensable for wound repair. In contrast, we show that β1 integrin from αSMA+ myofibroblasts, but not TGFβRII, is essential for wound healing, facilitating contractility, reepithelization, and vascularization. Collectively, our study provides evidence for the functions of myofibroblasts in β1 integrin‐mediated wound repair with potential implications for treating chronic non‐healing wounds.
Keywords: extracellular matrix, myofibroblasts, wound healing
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Skin; Stem Cells & Regenerative Medicine
Cutaneous chronic wound healing in the mouse requires contribution of aSMA+ dermal myofibroblasts.

Introduction
Traditionally rendered as generic extracellular matrix (ECM) producing cells that are integral for tissue architecture, fibroblasts are increasingly recognized to exhibit lineage specificity within and across tissue types and function as active drivers of physiological and pathological responses (Sorrell & Caplan, 2009; Wynn & Ramalingam, 2012; Ohlund et al, 2014; Kalluri, 2016; Lynch & Watt, 2018). The profound phenotypic heterogeneity of fibroblasts is, in part, reflected by the extensive list of marker genes used to define them (Kalluri & Zeisberg, 2006; Kalluri, 2016; LeBleu & Kalluri, 2018; Lynch & Watt, 2018). A wide variety of proteins expressed at higher levels by fibroblasts have been used to describe distinct populations, including the intermediate filament vimentin (Franke et al, 1978), the cell surface glycoprotein Thy‐1 (also known as CD90) (Rettig et al, 1988), the actin filament alpha‐smooth muscle actin (αSMA) (Darby et al, 1990), the cell surface peptidase fibroblast activation protein (FAP) (Rettig et al, 1988; Mathew et al, 1995), the intracellular protein fibroblast‐specific protein 1 (FSP1, also known as S100A4; Strutz et al, 1995), the cell surface proteins discoidin domain‐containing receptor 2 (DDR2) (Olaso et al, 2002), platelet‐derived growth factor receptor alpha (PDGFRα) (Collins et al, 2011), and dipeptidyl aminopeptidase 4 (DPP4, also known as CD26) (Driskell et al, 2013; Rinkevich et al, 2015; Philippeos et al, 2018; Shook et al, 2018; Tabib et al, 2018), among many others.
Skin represents a genetically accessible model to address whether dermal fibroblasts are essential for cutaneous wound healing. Skin fibroblasts are thought to function by promoting ECM remodeling, lending contractile properties to the scar tissue, secreting signaling molecules and growth factors, and communicating with other cell types in the wound microenvironment to regulate effective wound closure (Grinnell, 1994; Martin, 1997; Werner et al, 2007; Gurtner et al, 2008; Sorrell & Caplan, 2009; Driskell & Watt, 2015; Leavitt et al, 2016). Multiple fibroblast lineages have been implicated in cutaneous wound repair. It has been shown that during skin wound repair, the lower reticular dermal fibroblasts, originating from the embryonic delta‐like homologue 1 (Dlk1) lineage and responsible for bulk fibrillar ECM synthesis, are recruited during early stages (Driskell et al, 2013). In contrast, upper papillary dermal fibroblasts are recruited during late reepithelialization phase, which originate from the embryonic B lymphocyte‐induced maturation protein 1 (Blimp1) lineage and control hair follicle growth and arrector pili muscle formation (Driskell et al, 2013). Meanwhile, the embryonic engrailed 1 (Eng1) fibroblast lineage marked by DPP4/CD26 was shown to deposit ECM in acute and fibrotic conditions, including cutaneous wound healing, and represent a functional difference between scarring versus non‐scarring repair in cutaneous and oral injury, respectively (Rinkevich et al, 2015; Jiang et al, 2018). Genetic blockade or pharmacological inhibition of this lineage delayed wound closure and ameliorated cutaneous scarring (Rinkevich et al, 2015). Furthermore, multiple fibroblast populations were recently described in skin wounding, including adipocyte progenitors (marked by SCA1+CD34+CD29+) that are activated by a subset of macrophages (Shook et al, 2018). Despite the extensive marker genes identified for fibroblasts and their well‐recognized heterogeneity (Kalluri & Zeisberg, 2006; Lynch & Watt, 2018), the role of activated fibroblasts, often referred to as myofibroblasts, with a defining feature of having bundles of actin filament protein αSMA (Powell et al, 1999; Tomasek et al, 2002; Hinz et al, 2007; Gurtner et al, 2008; Li & Wang, 2011; Darby et al, 2016), remains largely unknown.
Here, we tackle the functional role of fibroblasts using skin, a genetically accessible model that relies critically on fibroblasts for mechanical support and wound repair (Grinnell, 1994; Martin, 1997; Sorrell & Caplan, 2004; Gurtner et al, 2008; Driskell & Watt, 2015). We specifically focus on myofibroblasts defined by expression of αSMA, which is postulated to regulate dermal contractility, ECM remodeling, and intercellular communications during skin wound repair (Tomasek et al, 2005; Hinz, 2007; Gurtner et al, 2008; Darby et al, 2016). The downstream mediators executing each of the functions of αSMA+ myofibroblasts have not been carefully dissected. Acta2 (the gene encoding αSMA) null mice do not exhibit wound repair defects (Tomasek et al, 2013; Ibrahim et al, 2015), suggesting that proteins other than αSMA are likely mediators of the wound healing properties of myofibroblasts.
Results
To directly address the functional role of αSMA+ myofibroblasts in wound healing, we engineered transgenic mouse lines carrying the herpes simplex virus thymidine kinase (HSV‐TK) transgene, a previously established conditional fibroblast depletion system (Iwano et al, 2001; LeBleu et al, 2013a), driven by the promoter of Acta2 (encoding αSMA). Daily treatment with ganciclovir (GCV) initiated upon wounding efficiently depleted only proliferating wound‐induced αSMA+ myofibroblasts (Fig 1A and B, hereafter referred to as αSMA‐TK) and completely blocked wound closure with continued GCV administration (Fig 1C and D). αSMA functions in cell contractility (Tomasek et al, 2002; Hinz, 2007; Darby et al, 2016), and depletion of αSMA+ myofibroblasts could alter wound contraction which is known to influence murine wound repair (Galiano et al, 2004). However, closure is similar between wildtype (WT) and αSMA‐TK wounds at early stages of wound repair (prior to day 6), where wound contraction typically occurs, suggesting that the delayed closure is likely not a result of alterations in wound contraction. Non‐proliferating myofibroblasts or αSMA+ cells are spared in this model system, hence significantly decreasing systemic toxicity. In addition to being expressed by myofibroblasts, αSMA has also been reported to be expressed by pericytes/vascular smooth muscle cells (Kalluri & Zeisberg, 2006; Plikus et al, 2017) and arrector pili muscle cells (Rahmani et al, 2014). The abundance of αSMA+ cells associated with CD31+ endothelial cells, presumably pericytes/vascular smooth muscle cells, was unchanged within the wounds of αSMA‐TK mice (Fig EV1A and B). In addition, αSMA associated with the hair follicles of surrounding unwounded skin was similar between WT and αSMA‐TK mice (Fig EV1C and D). Similarly, αSMA depletion was not observed in other organs, including liver, kidney, and lung (Fig EV1E and F), likely a reflection of the non‐proliferative status of αSMA+ cells under normal physiological conditions. Treatment with GCV starting 3 days prior to wounding followed by cessation of GCV treatment at day 21 post‐wounding resulted in αSMA+ myofibroblasts restoration within the wounds of αSMA‐TK mice (Fig EV2A) and complete wound closure by day 39 post‐wounding (Fig 1E), further validating the on‐target specificity of our system. Importantly, the GCV treated WT mice did not exhibit any wound closure defects or systemic toxicity.
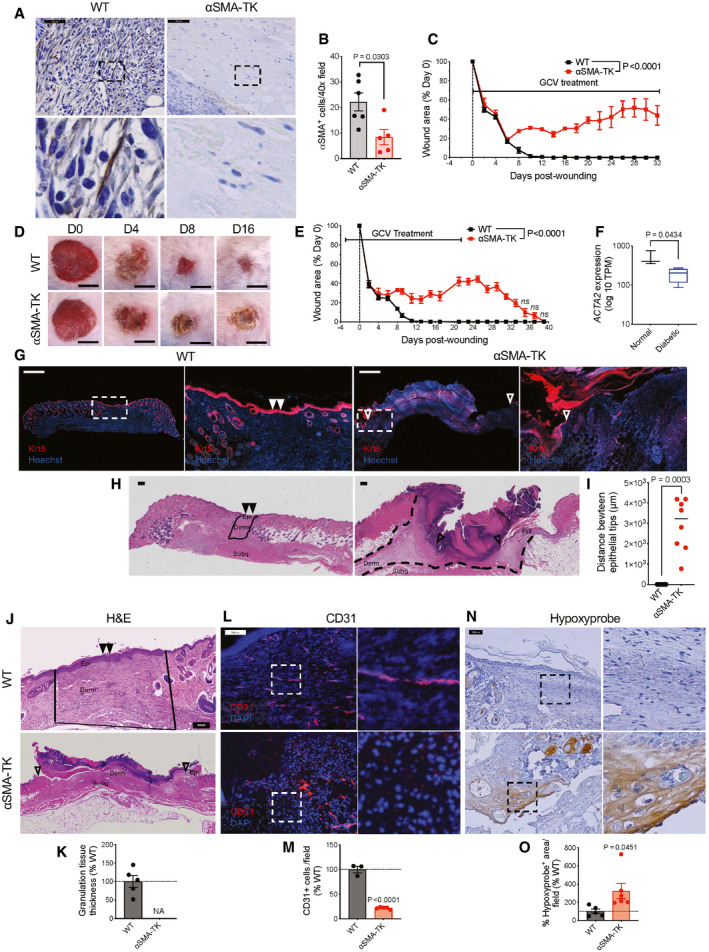
Figure 1. αSMA+ myofibroblasts are required for proper wound closure.
-
A–EImpaired wound closure in αSMA‐TK mice. (A) IHC for αSMA+ cells in wound tissue sections at day 6 post‐wounding. Representative images are shown. Scale bar, 50 µm. Black boxes denote zoomed regions displayed in bottom panels. (B) Quantification of αSMA+ cells at day 6 post‐wounding. WT, N = 6; TK, N = 5 biological replicates. Two‐tailed non‐parametric Mann–Whitney test performed. (C) WT and αSMA‐TK mice were treated with GCV for 32 days. Wounds were measured at indicated timepoints and wound area is presented as percentage of day 0 value. WT, N = 5; TK, N = 4 biological replicates per group, data are from one experiment. Two‐way ANOVA performed comparing WT to TK mice. (D) Representative gross images of wounds. Scale bar, 5 mm. (E) αSMA‐TK mice and WT littermates were placed on GCV regimen 3 days prior to wounding (day −3). On day 21, GCV treatment was stopped and the mice monitored until day 39. WT, N = 7; TK, N = 9 biological replicates. Data are from two independent experiments. Two‐way ANOVA and Sidak’s multiple comparison test were performed comparing WT to TK mice.
-
FExpression levels of ACTA2 in normal and diabetic human wound tissue. RNA‐seq reads are derived from Davis et al (2020c), Data ref: Davis et al (2020a). Normal, N = 3; diabetic, N = 4 biological replicates. Data are presented as a box and whisker plot of min to max values. Unpaired t‐test performed.
-
G–IImpaired re‐epithelialization of the wounds in αSMA‐TK mice at day 17 post‐wounding. (G) Representative images of keratin 5 (Krt5, red) and Hoechst (blue) stained wound sections at day 17 post‐wounding. White boxes denote zoomed regions displayed in right panels, triangles (open: incomplete closure, filled: closed wounds) denote the epithelial tongues. Scale bar, 1,000 µm. (H) Representative H&E images of wound sections. Wound bed is marked with black lines, triangles (open: incomplete closure, filled: closed wounds) denote the epithelial tongues. Epi, epidermis; derm, dermis; subq, subcutaneous regions denoted. Scale bar, 200 µm. (I) Measurements of the distances between epithelial tips in the wounded skins of WT and αSMA‐TK mice at day 17 post‐wounding. WT, N = 7; TK, N = 8 biological replicates. Black line denotes median value. Mann–Whitney test performed.
-
J–OImpaired granulation tissue formation and angiogenesis in αSMA‐TK mice. (J) Representative H&E images of wound tissue sections at day 17 post‐wounding. Granulation areas are marked by black lines, triangles (open: incomplete closure, filled: closed wounds) denote the epithelial tongues. Epi, epidermis; derm, dermis regions denoted. Scale bar: 200 μm. (K) Granulation tissue thickness measured on digital images and normalized to the average values in the WT control (set to 100%). NA (not assessed) reflects the lack of granulation in αSMA‐TK mice. WT, N = 5; TK, N = 4 biological replicates. (L) Immunofluorescent staining for CD31 (red) and nuclei (blue) on wound tissue sections at day 17 post‐wounding. White boxes denote zoomed regions depicted in right panels. Scale bar, 100 μm. (M) Quantification of CD31+ cells per visual field normalized to WT mice. WT, N = 3; TK, N = 5 biological replicates. (N) IHC for hypoxyprobe (brown) on wound tissue sections at day 17 post‐wounding. Black boxes denote zoomed regions depicted in right panels. Scale bar, 100 μm. (O) Quantification of hypoxyprobe area per visual field normalized to WT mice. WT, N = 5; TK, N = 6 biological replicates.
Data information: Data are reported as mean ± s.e.m. for (B, C, E, K, M, and O). Data are presented as a box and whisker plot of min to max values for (F). Individual data points (circles) and the median (black line) are reported for (I). Exact P‐values are reported. One sample t‐test performed comparing values to WT average for (K, M, and O).
Source data are available online for this figure.
Figure EV1. Depletion of αSMA+ cells is specific to myofibroblasts.
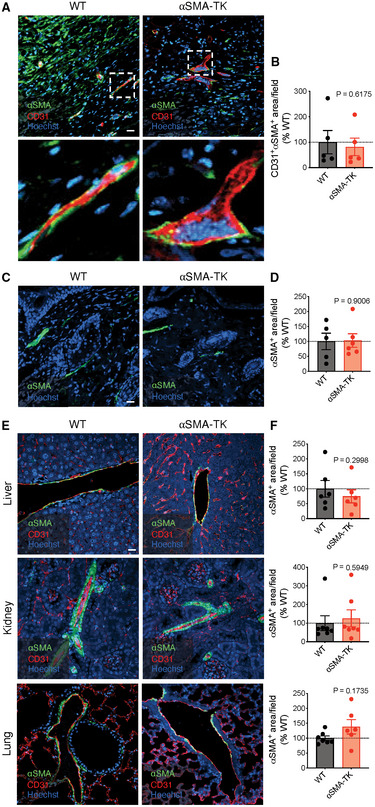
- Representative images of wound tissue at day 17 post‐wounding stained for αSMA (green), CD31 (red), and Hoechst (blue). Scale bar, 20 μm. White boxes denote zoomed regions displayed in bottom panels.
- Quantification of CD31+αSMA+ cells in wound tissue at day 17 post‐wounding. WT, N = 5; TK, N = 5 biological replicates.
- Representative images of hair follicles in normal skin surrounding wound tissue at day 17 post‐wounding stained for αSMA (green) and Hoechst (blue). Scale bar, 20 μm.
- Quantification of αSMA+ cells in normal skin surrounding wound tissue at day 17 post‐wounding. WT, N = 5; TK, N = 6 biological replicates.
- Representative images of liver, kidney, and lung stained for αSMA (green), CD31 (red), and Hoechst (blue). Scale bar, 20 μm.
- Quantification of αSMA+ cells in liver, kidney, and lung at day 17 post‐wounding. Liver: WT, N = 6; TK, N = 6 biological replicates. Kidney: WT, N = 7; TK, N = 7 biological replicates. Lung: WT, N = 7; TK, N = 6 biological replicates.
Data information: Data are reported as mean ± s.e.m. One sample t‐test performed comparing values to WT average for (B, D, F). Exact P‐values are reported.
Source data are available online for this figure.
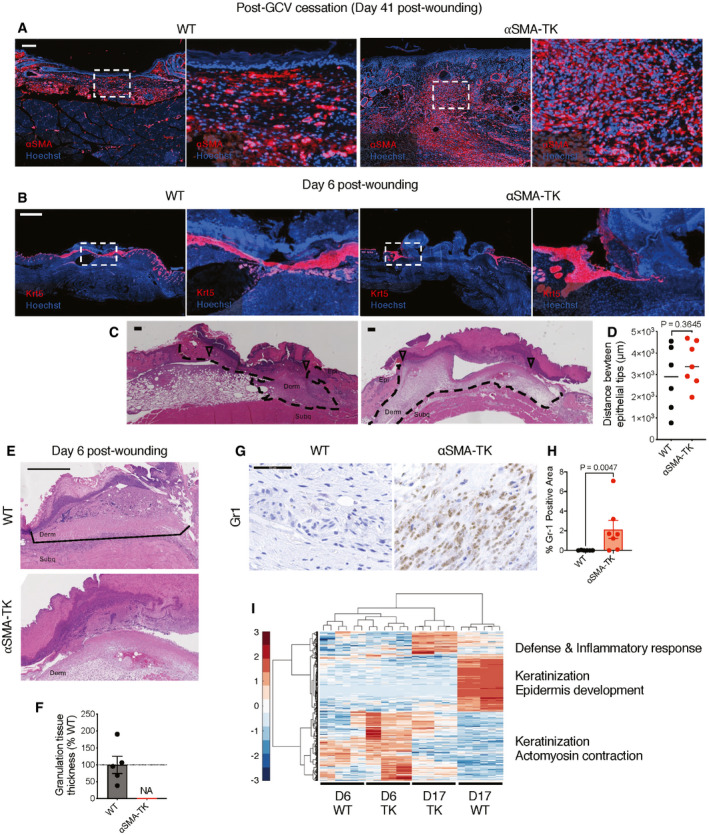
Figure EV2. Phenotypes associated with depletion of αSMA+ myofibroblasts.
-
ARepresentative images of wound tissue at day 41 post‐wounding stained for αSMA (red) and Hoechst (blue) from mice treated with GCV initiated at 3 days prior to wounding and continued until 21 days post‐wounding followed by withdrawal of GCV treatment (related to Fig 1E). White boxes denote zoomed regions displayed in right panels. Scale bar, 100 µm.
-
B, CImpaired re‐epithelialization of the wounds in αSMA‐TK mice at day 6 post‐wounding. (B) Representative images of keratin 5 (Krt5, red) and Hoechst (blue) stained wound sections. White boxes denote zoomed regions displayed in right panels. Scale bar, 1,000 µm. (C) Representative H&E images of wound sections. Wound area is marked with black dashed lines, open triangles denote the epithelial tongues. Epi, epidermis; derm, dermis; subq, subcutaneous regions denoted. Scale bar, 200 µm.
-
DMeasurements of the distances between epithelial tips in the wounded skins of WT and αSMA‐TK mice at day 6 post‐wounding. WT, N = 6; TK, N = 7 biological replicates. Black lines denote median values. Unpaired Student’s t‐test performed.
-
E, FImpaired granulation tissue formation in αSMA‐TK mice at day 6 post‐wounding. (E) Representative H&E images of wound tissue sections at day 6 post‐wounding. Granulation areas are marked by black lines. Derm, dermis and subq, subcutaneous regions denoted. Scale bar, 500 μm. (F) Granulation tissue thickness measured on digital images and normalized to the average values in the WT control (set to 100%). NA (not assessed) reflects the lack of granulation in αSMA‐TK mice. WT, N = 5; TK, N = 7 biological replicates.
-
GRepresentative IHC images of day 17 wound tissue stained with Gr1 antibody. Scale bar, 50 μm.
-
HQuantitative analysis of Gr1+ area in day 17 wound tissue. WT, N = 6; TK, N = 7 biological replicates. Two‐tailed Mann–Whitney test performed comparing WT to TK.
-
IUnsupervised clustering of gene expression of day 6 wounds, when the highest numbers of αSMA+ fibroblasts are observed, and day 17, when the wound closure is achieved in WT control mice. The main gene categories altered in αSMA‐TK mice are marked on the right. For day 6 wounds, N = 3 biological replicates per group were included, and each sample was analyzed in technical duplicates. Day 17 analysis was performed with N = 2 biological replicates per group with each sample analyzed in technical triplicates.
Data information: Data are reported as mean ± s.e.m. for (F and H). Individual data points (circles) and the median (black line) are reported for (D). Exact P‐values are reported.
Source data are available online for this figure.
Our results provided direct genetic modeling support for the non‐redundant function of αSMA+ myofibroblasts in skin wound healing, and with relevance to non‐healing wounds. In this regard, a significant decline of αSMA expression is observed in chronic wounds associated with diabetic patients when compared to acute wounds from non‐diabetic donors (Fig 1F; Davis et al, 2020c). Analyzing further the wound closure defects in αSMA‐TK mice, we found permanently stalled epithelial migration fronts as determined by Keratin 5 (Krt5) immunofluorescence (Figs 1G and EV2B) and histology (Figs 1H and I, and EV2C and D). Such open wounds were accompanied by a lack of granulation tissue formation in the wound bed (Figs 1J and K, and EV2E and F), compromised vascularization (Fig 1L and M), increased hypoxic response (Fig 1N and O), and increased infiltration of Gr1+ cells (Fig EV2G and H), all of which are cardinal features of non‐healing wounds (Singer & Clark, 1999). Consistently, transcriptional changes measured by microarray corroborated the altered keratinization and defense responses to infection in αSMA‐TK wounds (Fig EV2I). Global gene expression profiles of the day 17 αSMA‐TK wounds resemble those of day 6 open wounds in the control mice, based on hierarchical clustering (Fig EV2I). Thus, genetic depletion of αSMA+ myofibroblasts renders skin wounds into a state with multiple features of chronic non‐healing wounds, thereby providing a robust model to dissect downstream events regulated by myofibroblasts.
Next, we mapped the dynamic accumulation of αSMA+ myofibroblasts during wound repair and noted its induction around 6–9 days post‐wounding (Fig 2A). The wound‐induced αSMA+ myofibroblasts could originate either from local expansion and migration of skin‐resident cells (Reynolds et al, 1993; Higashiyama et al, 2011; Driskell et al, 2013; Shook et al, 2018, 2020), or de novo recruitment and differentiation of bone marrow (BM)‐derived mesenchymal cells (Opalenik & Davidson, 2005; Guerrero‐Juarez et al, 2019). To further define such mechanistic possibilities, we employed a cell tracking reporter mouse line expressing the RFP transgene under the αSMA promoter (αSMA‐RFP; LeBleu et al, 2013b), and confirmed αSMA‐RFP induction around 6–14 days post‐wounding (Figs 2B and EV3A), similar to antibody‐labeled αSMA+ myofibroblasts (Fig 2A). Next, we performed BM reconstitution experiments in which sub‐lethally irradiated female WT mice were transplanted with BM from αSMA‐RFP+ male mice (Figs 2C and EV3B). Less than 1% αSMA‐RFP+ cells repopulated the skin wounds 6 days post‐wounding (Figs 2D and E, and EV3C), suggesting a minor contribution of BM‐derived cells to the accumulation of myofibroblasts. In inverse studies where sub‐lethally irradiated αSMA‐RFP mice were transplanted with WT BM, or control αSMA‐RFP mice transplanted with αSMA‐RFP BM (Fig 2C), we consistently observed 23–30% RFP+ cells in the wound bed (Figs 2D and E, and EV3C). These results together supported a primarily skin‐resident origin of αSMA+ myofibroblasts in cutaneous wound repair.
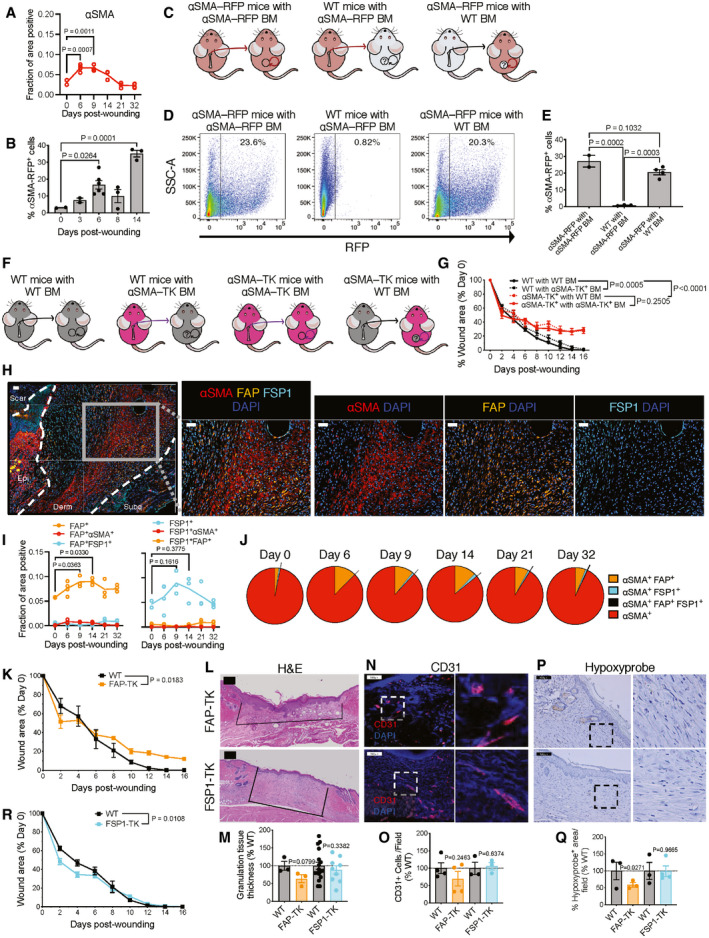
Figure 2. Skin‐resident αSMA+ myofibroblasts are critical for wound repair.
-
AAnalysis of multiplex immunostaining of full‐thickness skin wounds performed at indicated time points. Data are presented as the fractional area of each image positive for αSMA and negative for CD31. Day 0, N = 2; Day 6, N = 4; Day 9, N = 3; Day 14, N = 3; Day 21, N = 3; Day 32, N = 4 biological replicates. One‐way ANOVA with Dunnett’s multiple comparison test performed.
-
BFACS quantification of αSMA‐RFP+ cells in wounds. Day 0, N = 2; day 3, N = 2; day 6, N = 6; day 8, N = 3; day 14, N = 3 biological replicates. One‐way ANOVA with Dunnett’s multiple comparison test performed comparing each timepoint to day 0.
-
CSchematic of bone marrow transplants between WT and αSMA‐RFP mice.
-
DRepresentative FACS plots of digested skin from bone marrow (BM) transplanted αSMA‐RFP mice 17 days post‐wounding.
-
EFACS quantification of αSMA‐RFP+ cells in wounds of BM transplanted mice. αSMA‐RFP with αSMA‐RFP BM, N = 2; WT with αSMA‐RFP BM, N = 3; αSMA‐RFP with WT BM, N = 4 biological replicates. One‐way ANOVA with Tukey’s multiple comparison test performed comparing WT with αSMA‐RFP BM and αSMA‐RFP with WT BM to αSMA‐RFP with αSMA‐RFP BM.
-
FSchematic of bone marrow transplants between WT and αSMA‐TK mice.
-
GWound area measurements in WT and ⍺SMA‐TK mice transplanted with WT or αSMA‐TK BM. WT with WT BM and WT with αSMA‐TK BM, N = 5; αSMA‐TK with WT BM and αSMA‐TK with αSMA‐TK BM, N = 4 biological replicates. Two‐way ANOVA performed with the indicated comparisons.
-
H–JMultispectral imaging of changes in fibroblast subsets during wound repair. (H) Representative images (20× magnification) of orthogonal wound tissue sections at day 6 post‐wounding displaying αSMA, FAP, and FSP1 overlaid with DAPI in the wound bed. Subcutaneous (subq), dermis (derm), epidermis (epi), and scar regions are denoted. Individual αSMA, FAP, and FSP1 channel images overlaid with DAPI. Scale bar, 50 µm. (I) Analysis of multiplex immunostaining of full‐thickness skin wounds performed at indicated time points. Data are presented as the fractional area of each image positive for the indicated markers and negative for CD31. Day 0, N = 2; Day 6, N = 4; Day 9, N = 3; Day 14, N = 3; Day 21, N = 3; Day 32, N = 4 biological replicates. One‐way ANOVA with Dunnett’s multiple comparison test performed. (J) Percent overlap of αSMA with FAP and FSP1 throughout wound healing. αSMA+ denotes cells positive for αSMA but not FAP or FSP1. Day 0, N = 2; Day 6, N = 4; Day 9, N = 3; Day 14, N = 3; Day 21, N = 3; Day 32, N = 4 biological replicates.
-
KWound closure in FAP‐TK and wild‐type littermate mice. N = 4 biological replicates per group, data are from one experiment. Two‐way ANOVA performed comparing WT to TK mice.
-
LRepresentative H&E images of wound tissue sections at day 17 post‐wounding. Granulation areas are marked by black lines. Scale bar, 200 μm.
-
MGranulation tissue thickness measured on digital images and normalized to the average values in the WT control. WT, N = 3; FAP‐TK, N = 3; WT, N = 17, FSP1‐TK, N = 9 biological replicates.
-
NImmunofluorescent staining for CD31 (red) and nuclei on wound tissue sections at day 17 post‐wounding. White boxes denote zoomed regions depicted in right panels. Scale bar, 100 μm.
-
OQuantification of CD31+ cells per visual field normalized to WT mice. WT, N = 4; FAP‐TK, N = 4; WT, N = 3; FSP1‐TK, N = 3 biological replicates.
-
PIHC for hypoxyprobe (brown) on wound tissue sections at day 17 post‐wounding. Black boxes denote zoomed regions depicted in right panels. Scale bar, 100 μm.
-
QQuantification of hypoxyprobe area per visual field normalized to WT mice. WT, N = 3; FAP‐TK, N = 3; WT, N = 3; FSP1‐TK, N = 4 biological replicates.
-
RWound closure in FSP1‐TK and WT littermate mice. N = 10 biological replicates per group, data are from two independent experiments. Two‐way ANOVA performed comparing WT to TK mice.
Data information: Data are reported as individual values for (A and I). Data are reported as mean ± s.e.m. for (B, E, G, K, M, O, Q, and R). One sample t‐test performed comparing values to WT average for (M, O and Q). Exact P‐values are reported.
Source data are available online for this figure.
Figure EV3. Depletion of specific fibroblast populations using viral thymidine kinase transgene.
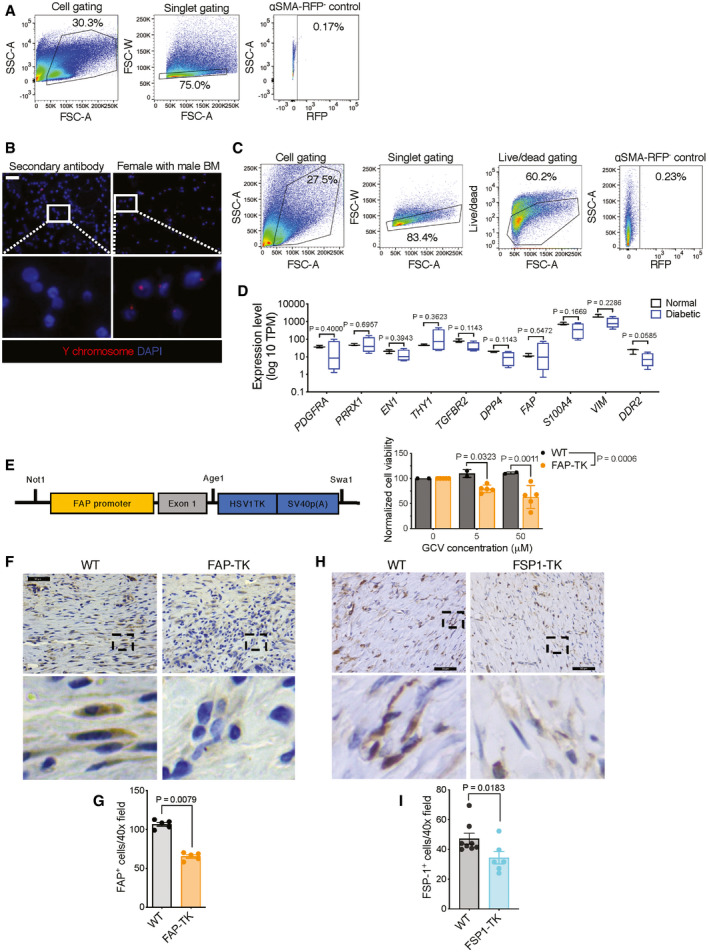
- FACS gating strategy for Fig 2B. αSMA‐RFP‐ control: skin tissue from an αSMA‐RFP‐ (WT) mouse.
- Representative Y‐chromosome (red) fluorescent in situ hybridization (FISH) images of bone marrow cells after bone marrow transplantation. White boxes denote zoomed regions depicted in bottom panels. Negative control: secondary antibody alone stained tissue. Scale bar, 50 μm.
- FACS gating strategy for Fig 2D and E. αSMA‐RFP‐ control: skin tissue from an αSMA‐RFP‐ (WT) mouse.
- Expression level of putative fibroblast marker genes in normal and diabetic human wound tissue. RNA‐seq reads are derived from Davis et al (2020c), Data ref: Davis et al (2020a). Normal, N = 3; diabetic, N = 4 biological replicates. Data are presented as a box and whisker plot of min to max values. PDGFRA, TGFBR2, DPP4, VIM: Mann–Whitney test performed; PRRX1, THY1, FAP: Welch’s t‐test performed; EN1, S100A4, DDR2: unpaired t‐test performed.
- Construct for newly generated FAP‐TK transgenic mice used in the study (left panel). The transgenic cassette contains the promoter and exon 1 regions (extended promoter) of FAP followed by HSV thymidine kinase and SV40 polyadenylation site. Dose‐dependent ablation of cultured fibroblasts from the ears of FAP‐TK mice and their wild‐type littermates (right panel). Data are normalized to the viability at 0 μM. WT, N = 2; TK, N = 5 biological replicates. Two‐way ANOVA with Sidak’s multiple comparison test performed comparing WT to TK.
- Representative IHC images of FAP in day 17 wound tissue. Black boxes denote zoomed regions depicted in bottom panels. Scale bar, 50 μm.
- Quantification of FAP+ cells in day 17 wound tissue. N = 5 biological replicates per group. Two‐tailed non‐parametric Mann–Whitney test performed comparing WT to TK mice.
- Representative IHC images of FSP1 in day 17 wound tissue. Black boxes denote zoomed regions depicted in bottom panels. Scale bar, 50 µm.
- Quantification of FSP1+ cells in day 17 wound tissue. WT, N = 8; TK+, N = 6 biological replicates. Two‐tailed non‐parametric Mann–Whitney test performed comparing WT to TK mice.
Data information: Data are reported as mean ± s.e.m for (E, G, and I). Data are presented as a box and whisker plot of min to max values for (D). Exact P‐values are reported.
Source data are available online for this figure.
In order to further determine if the rare population of αSMA+ myofibroblasts recruited from the BM could functionally contribute to wound healing, we repeated our BM transplant experiments, this time reconstituting WT mice with BM from αSMA‐TK+ mice. In addition, we performed the reverse experiment of transplanting WT BM into αSMA‐TK+ mice (Fig 2F). We observed a marginal delay healing of wounds in the WT mice transplanted with αSMA‐TK+ BM (Fig 2G). Of importance, wounds in WT mice healed faster than αSMA‐TK mice, regardless of the BM transfer regime (Fig 2G), suggesting that the major stratifier of these two groups is local cell types rather than systemic influences. Based on these data, it is likely the tissue‐resident rather than BM‐derived αSMA+ myofibroblasts that primarily contribute to wound repair. However, it is possible that αSMA+ myofibroblasts recruited from other non‐BM tissues can functionally contribute to cutaneous wound healing, despite the fact that αSMA+ cells in other tissues are largely not targeted by the αSMA‐TK system (Fig EV1E and F).
Given the well‐recognized heterogeneity of skin‐resident fibroblasts (Lynch & Watt, 2018), we next sought to compare the function of αSMA+ myofibroblasts in wound healing to other types of dermal fibroblasts. Surveying for a variety of known fibroblast markers, we noticed that many fibroblast biomarker genes exhibited a trend toward decreased expression in diabetic wounds (Fig EV3D), albeit the differences in gene expression were smaller than the decreased expression observed with ACTA2 (encoding αSMA, Fig 1F). We further evaluated two subsets of fibroblasts, marked by the cell surface peptidase fibroblast activation protein (FAP; Rettig et al, 1988; Mathew et al, 1995) and intracellular protein fibroblast specific protein 1 (FSP1, encoded by S100a4; Strutz et al, 1995), respectively. Both FAP and FSP1 are putative fibroblast biomarkers that are known to be involved in tissue fibrosis (Strutz et al, 1995; Iwano et al, 2001; Jacob et al, 2012) but are under‐characterized in skin wound repair.
We first analyzed the expression dynamics of FAP+ and FSP1+ fibroblasts in relation to αSMA+ myofibroblasts by co‐staining wounds for these proteins (Fig 2H). FAP+ and FSP1+ fibroblasts emerged around 9 days post‐wounding (Fig 2H and I), with moderate overlap with αSMA+ myofibroblasts (Fig 2H–J). We employed a similar TK strategy to ablate wound‐recruited FAP+ (Fig EV3E–G) or FSP1+ fibroblasts (Fig EV3H and I), as previously utilized to ablate αSMA+ myofibroblasts. Depletion of FAP+ fibroblasts only mildly delayed wound closure, and granulation (Fig 2K–Q), while suppression of FSP1+ fibroblasts did not appear to impact overall wound repair, except for a minor acceleration of wound closure at early phase (Fig 2L–R). These results highlight skin resident and locally derived αSMA+ myofibroblasts as key functional orchestrators of cutaneous wound repair, with critical functions that cannot be recapitulated or compensated by other types of fibroblasts.
Next, to mechanistically identify downstream mediators of αSMA+ myofibroblasts dependent wound repair, we analyzed single‐cell RNA sequencing data derived from murine skin wounds (Haensel et al, 2020b; Appendix Fig S1A). Focusing here on fibroblasts, we identified four subtypes, three of which expressed Acta2 (αSMA, Fig 3A and B). A number of chemokines and cytokines (Cxcl1, Ccl2, and Cxcl5) were enriched in αSMA cluster 1 (αSMA‐C1) (Fig 3C). αSMA cluster 2 (αSMA‐C2) displayed enrichment of genes associated with actin remodeling and contraction, including Tagln, Myl9, and Mylk (Fig 3D). On the other hand, αSMA cluster 3 (αSMA C3) expressed Cd82, Gpx3, and Gapdh (Fig 3E).
Figure 3. Transcriptionally distinct αSMA+ myofibroblasts subpopulations emerge during wound repair.

- UMAP plot defining cell clusters. Expression of defining markers for each cluster is presented in Appendix Fig S1A.
- UMAP plot of Acta2 (encoding αSMA) expression in the fibroblast cluster (left panel). UMAP plot of fibroblast subclusters (right panel).
- Enrichment plot of genes in αSMA C1 (left panel). Violin plots for genes enriched in αSMA C1 (right panels).
- Enrichment plot of genes in αSMA C2 (left panel). Violin plots for genes enriched in αSMA C2 (right panels).
Data information: Data are reported as median (open circle) with interquartile range indicated with bar for violin plots (C, D, and E, right panels).
We next evaluated the role of αSMA+ myofibroblast‐derived ECM, as production of collagen is considered a major function of myofibroblasts (Darby et al, 2016). In the non‐healing wounds of αSMA‐TK mice, Col1 expression and fibers were significantly decreased (Figs 4A–D and EV4A), accompanied by a notable decrease in wound tissue stiffness and tensile strength, as measured by atomic force microscopy (Fig EV4B and C). These results suggest that Col1 may mediate the function of αSMA+ myofibroblasts in cutaneous wound repair. We specifically deleted Col1 in αSMA+ myofibroblasts (αSMA‐Cre‐ERT2; Col1a1loxP/loxP , Col1α1cKO) through daily administration of tamoxifen initiated at wounding, which led to a significant reduction of Col1 abundance and collagen fibers in the wounds (Fig 4E–H). However, we observed insignificant changes in closure of wounds (Fig 4I), re‐epithelialization (Fig 4J and K), or vascularization in Col1α1cKO mice (Fig 4L and M). These data suggest that Col1 production does not specifically contribute to wound repair function of αSMA+ myofibroblasts. scRNA seq analysis of wound fibroblasts revealed that while a subset of αSMA+ myofibroblasts express Col1a1, expression of Col1a1 is also high in the αSMA‐negative cluster (Fig 4N), suggesting that other wound‐associated fibroblasts may contribute to Col1 production.
Figure 4. Collagen I produced by αSMA+ myofibroblasts is dispensable for wound closure.
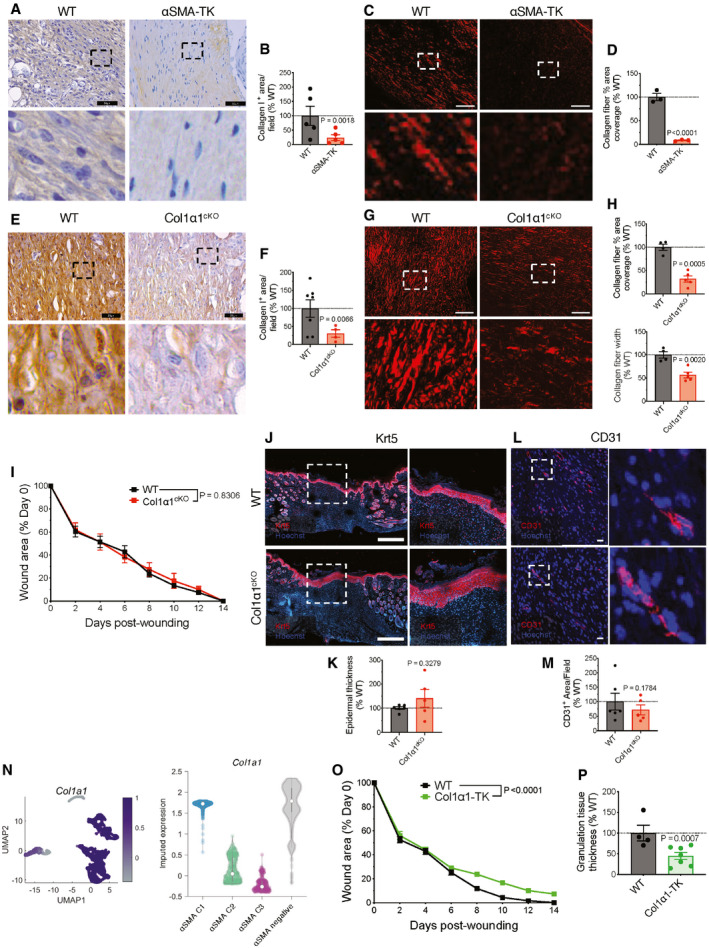
-
AIHC for collagen I on wound tissue sections at day 17 post‐wounding (brown stain). Black boxes denote zoomed regions depicted in bottom panels. Scale bar: 50 μm.
-
BQuantification of collagen I‐positive area normalized to the average area in WT controls. WT, N = 5; TK, N = 5 biological replicates.
-
CPolarized light images of picrosirius red stained wound tissues at day 17 post‐wounding. Red: collagen fibers. White boxes denote zoomed regions depicted in bottom panels. Scale bar, 50 µm.
-
DQuantification of collagen fiber (picrosirius red) percent area normalized to the average area in WT controls. WT, N = 3; TK, N = 3 biological replicates.
-
E–HReduced collagen deposition in Col1α1cKO wounds. (E) IHC for collagen I on wound tissue sections at day 14 post‐wounding (brown stain). Black boxes denote zoomed regions depicted in bottom panels. Scale bar: 25 μm. (F) Quantification of collagen I‐positive area normalized to the average area in WT controls. WT, N = 7; Col1α1cKO, N = 4 biological replicates. (G) Polarized light images of picrosirius red stained wound tissues at day 14 post‐wounding. Red: collagen fibers. White boxes denote zoomed regions depicted in bottom panels. Scale bar, 50 µm. (H) Quantification of collagen fiber percent area and fiber width normalized to the average area and width, respectively, in WT controls. WT, N = 4; Col1α1cKO, N = 5 biological replicates.
-
IWound area measurements in WT and Col1α1cKO mice. WT, N = 7; Col1α1cKO, N = 5 biological replicates. Data are from two independent experiments. Two‐way ANOVA performed comparing WT and Col1α1cKO mice.
-
J–MReepithelization and vascularization in WT and Col1α1cKO mice. (J) Representative images of keratin 5 (Krt5, red) stained wounds at day 14 post‐wounding. White boxes denote zoomed regions depicted in right panels. Scale bar, 500 µm. (K) Quantification of epidermal thickness. WT, N = 6; Col1α1cKO, N = 5 biological replicates. (L) Immunofluorescent staining for CD31 (red) and nuclei (blue) on wound tissue sections at day 14 post‐wounding. White boxes denote zoomed regions depicted in right panels. Scale bar, 20 μm. (M) Quantification of CD31+ area per visual field normalized to WT mice. WT, N = 6; Col1α1cKO, N = 5 biological replicates.
- N
-
OWound area measurements in Col1α1‐TK and WT littermate mice. WT, N = 13; Col1α1‐TK, N = 12 biological replicates, data are from two independent experiments. Two‐way ANOVA performed comparing WT to TK mice.
-
PGranulation tissue thickness measured on digital images and normalized to the average values in the WT control. WT, N = 4; TK, N = 7 biological replicates.
Data information: Data are reported as mean ± s.e.m. for (B, D, F, H, I, K, M, O, and P). Data are reported as median (open circle) with interquartile range indicated with bar for violin plots (N, right panel). One sample t‐test performed comparing values to WT average for (B, D, F, H, K, M, and P). Exact P‐values are reported.
Source data are available online for this figure.
Figure EV4. Col1 expression during wound healing.
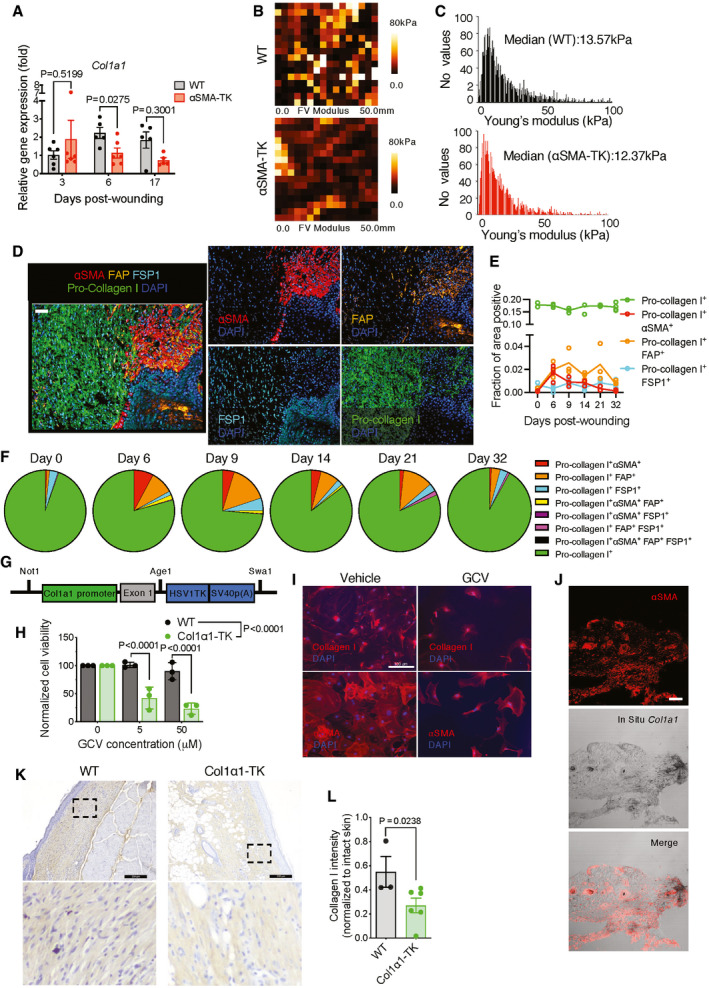
-
AFold change in mRNA expression of Col1a1 in wound tissue. Expression is normalized to day 0 (D0, unwounded skin) values. Day 0 WT, Day 3 WT, Day 3 αSMA‐TK, and Day 6 αSMA‐TK, N = 6 mice; Day 6 WT, Day 17 WT, and Day 17 αSMA‐TK, N = 5 biological replicates. Unpaired t‐test comparing WT to αSMA‐TK for each time point performed based on ΔCT values.
-
B, CAssessment of the mechanical properties of wound sections from WT and αSMA‐TK mice at day 6 post‐wounding. N = 3 biological replicates per group, four positions per wound, 257 measurements per position. (B) Representative elasticity maps. The color map indicates the modulus of elasticity in kPa evaluated using force curves generated for each point in the field. This measurement provides AFM heights relative to the lowest contact point in the field. (C) Distribution of the Young’s modulus throughout the samples and the median values indicated on each histogram.
-
DRepresentative image (20× magnification) at day 6 post‐wounding displaying αSMA (red), FAP (orange), FSP1 (teal), and pro‐collagen I (green) overlaid with DAPI (blue, left panel). Individual αSMA, FAP, FSP1, and pro‐collagen I channel images overlaid with DAPI (right panel). Scale bar: 50 µm.
-
EAnalysis of multiplex immunostaining of full‐thickness skin wounds performed at indicated time points. Data are presented as the fractional area of each image positive for a given marker and negative for CD31. Day 0, N = 2; Day 6, N = 4; Day 9, N = 3; Day 14, N = 3; Day 21, N = 3; Day 32, N = 4 biological replicates.
-
FPercent overlap of pro‐collagen I with other markers throughout wound healing. Pro‐collagen I+ denotes cells positive for pro‐collagen I but not αSMA, FAP, or FSP1.
-
GConstruct for newly generated Col1α1‐TK transgenic mice used in the study. The transgenic cassette contains the promoter and exon 1 regions (extended promoter) of Col1a1 followed by HSV thymidine kinase and SV40 polyadenylation site.
-
HDose‐dependent ablation of cultured fibroblasts from the ears of Col1α1‐TK mice and their wild‐type littermates. WT, N = 3; Col1α1‐TK+, N = 3 biological replicates. Two‐way ANOVA with Sidak’s multiple comparison test performed comparing WT to TK.
-
IImmunofluorescent images of ear fibroblasts from Col1α1‐TK mice stained for collagen I (red, top panels)) and αSMA (red, bottom panels) after GCV or vehicle (PBS) treatment for 5 days. Scale bar, 100 μm.
-
JRepresentative images of αSMA staining (red) and Col1a1 ISH (black) in day 17 WT wound tissue. Scale bar, 100 μm.
-
KRepresentative IHC images of collagen I in day 17 wound tissue. Black boxes denote zoomed regions depicted in bottom panels. Scale bar, 200 μm.
-
LQuantification of collagen I intensity in day 17 wound tissue. Data are normalized to collagen I intensity of unwounded skin. WT, N = 3; Col1α1‐TK+, N = 6 biological replicates. Two‐tailed non‐parametric Mann–Whitney test performed comparing WT to TK.
Data information: Data are reported as individual values for (E). Data are reported as mean ± s.e.m. for (A, H, and L). Exact P‐values are reported.
Source data are available online for this figure.
In this regard, we noted that during wound healing, unlike the dynamic induction and eventual decrease of αSMA+ myofibroblasts, Col1 level remained consistently high throughout the course of wound repair (Fig EV4D and E). Additionally, Col1+ fibroblasts showed only partial overlaps with αSMA+ myofibroblasts and other fibroblast markers we examined (Fig EV4E and F). This suggests that besides αSMA+ myofibroblasts (Tomasek et al, 2002), many other fibroblasts may contribute to collagen I production during wound repair (vide supra). In order to directly evaluate the functional role of Col1, we engineered new transgenic TK mice driven by the Col1a1 promoter (Col1α1‐TK, Fig EV4G–J). Genetic depletion of all wound‐induced proliferative Col1+ cells (Fig EV4K and L) significantly suppressed wound closure (Fig 4O) and inhibited granulation tissue formation (Fig 4P).
While seeking additional mediators, we noted previous reports of extensive overlaps between αSMA+ myofibroblasts and CD29+ adipocytes (Shook et al, 2018, 2020). Interestingly, in both murine (Fig 5A) and human wounds (Fig 5B), β1 integrin (also known as CD29, encoded by Itgb1) was strongly associated with αSMA. β1 integrin serves as a collagen receptor typically in conjunction with the α1 integrin subunit to facilitate signal transduction (Kalluri & Zeisberg, 2006; Liu et al, 2010). The non‐healing wounds of αSMA‐TK mice displayed a significant reduction of αSMA+β1 integrin+ cells (Fig 5C and D). We, therefore, evaluated whether β1 integrin might serve as a potential effector of αSMA+ myofibroblasts in wound repair. To this end, we deleted β1 integrin in αSMA+ myofibroblasts (αSMA‐Cre; Itgb1loxP/loxP , Itgb1cKO). The Itgb1cKO mice exhibited a significant delay in wound closure specifically during the early phases of wound closure (P = 0.0424, Fig 5E), and correspondingly exhibited reduced reepithelization (Fig 5F–G) and vascularization (Fig 5H and I). We did not observe a difference in overall collagen fiber abundance in the wounds of Itgb1cKO mice when compared to control mice (Fig 5J and K). However, there was a reduction in the collagen fiber width in the wounds of Itgb1cKO mice (Fig 5L), suggesting that β1 integrin in αSMA+ myofibroblasts may act to facilitate collagen crosslinking independent of collagen deposition. β1 integrin has previously been implicated in cell proliferation and survival (Varner & Cheresh, 1996; Schwartz, 2001); thus, we evaluated if deletion of β1 integrin altered αSMA+ myofibroblast abundance. Immunostaining revealed that Itgb1cKO wounds have similar amounts of αSMA+ myofibroblasts compared to WT wounds (Fig EV5A and B). While TGFβ signaling is known to induce the myofibroblast phenotype (MacLellan et al, 1994; Hautmann et al, 1997) and TGFβ receptor II (TGFβRII, encoded by Tgfbr2) is expressed by αSMA+ myofibroblasts (Fig EV5C), deleting TGFβRII from αSMA+ myofibroblasts (αSMA‐Cre; Tgfbr2loxP/loxP , Tgfbr2cKO) did not have a detectable impact on matrix deposition or wound closure in vivo (Fig EV5D–F), in contrast to the altered wound repair that was observed upon deletion of Tgfbr2 in Col1+ fibroblasts (Denton et al, 2009).
Figure 5. β1 integrin facilitates αSMA+ myofibroblast‐mediated wound repair.

- A
- B
-
CIF for β1 integrin (red) and αSMA (green) on wound tissue sections at day 17 post‐wounding. White boxes denote zoomed regions depicted in right panels. Scale bar: 20 μm.
-
DQuantification of the number of αSMA+ β1 integrin+ cells. WT, N = 3; TK, N = 3 biological replicates. Unpaired t‐test performed comparing WT to TK.
-
EWound area measurements in WT and Itgb1cKO mice. WT, N = 5; Itgb1cKO, N = 6 biological replicates. Two‐way ANOVA performed comparing WT and Itgb1cKO mice.
-
F–KReepithelization, granulation tissue formation, and vascularization in WT and Itgb1cKO mice. (F) Representative images of keratin 5 (Krt5, red) stained wounds at day 14 post‐wounding. White boxes denote zoomed regions depicted in right panels. Scale bar, 500 µm. (G) Quantification of epidermal thickness. WT, N = 4; Itgb1cKO, N = 6 biological replicates. (H) Immunofluorescent staining for CD31 (red) and nuclei (blue) on wound tissue sections at day 14 post‐wounding. White boxes denote zoomed regions depicted in right panels. Scale bar, 20 μm. (I) Quantification of CD31+ area per visual field normalized to WT mice. WT, N = 6; Itgb1cKO, N = 6 biological replicates. (J) Polarized light images of picrosirius red stained wound tissues at day 14 post‐wounding. Red: collagen fibers. White boxes denote zoomed regions depicted in right panels. Scale bar, 50 µm. (K) Quantification of collagen fiber percent area normalized to the average area in WT controls. WT, N = 6; Itgb1cKO, N = 6 biological replicates.
-
LQuantification of collagen fiber width normalized to the average width in WT controls. WT, N = 6; Itgb1cKO, N = 6 biological replicates.
Data information: Data are reported as mean ± s.e.m. for (D, E, G, I, K, and L). Data are reported as median (open circle) with interquartile range indicated with bar for violin plots (A). One sample t‐test performed comparing values to WT average for (G, I, K, and L). Exact P‐values are reported.
Source data are available online for this figure.
Figure EV5. TGFβRII signaling in αSMA+ fibroblasts is not required for cutaneous wound repair.
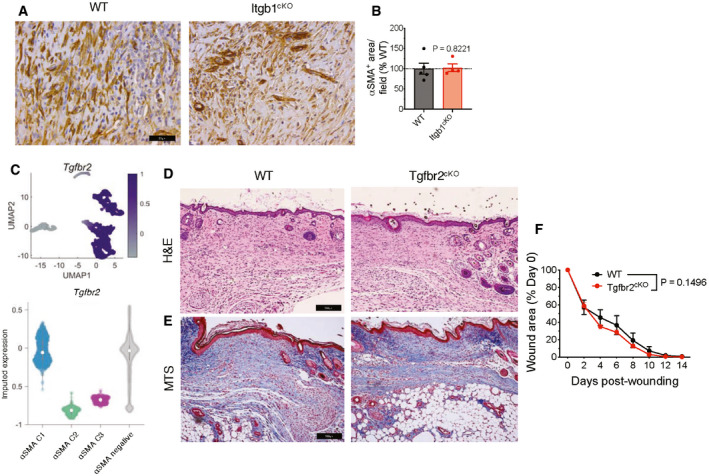
-
AIHC for αSMA in day 17 wound tissue. Representative images are shown. Scale bar, 25 µm.
-
BQuantification of αSMA+ area in day 17 wound tissue per visual field normalized to WT mice. WT, N = 5; Itgb1cKO, N = 4 biological replicates. One sample t‐test performed comparing values to WT average.
- C
-
D, ERepresentative H&E (D) and Masson’s trichrome (E) images of wound sections of WT and Tgfbr2cKO mice at day 17 post‐wounding. Scale bar, 100 μm.
-
FWound closure in WT and Tgfbr2cKO mice. N = 9 biological replicates per group, data are from two independent experiments. Two‐way ANOVA performed comparing WT to Tgfbr2cKO mice.
Data information: Data are reported as mean ± s.e.m. for (B and F). Data are reported as median (open circle) with interquartile range indicated with bar for violin plots in (C). Exact P‐values are reported.
Source data are available online for this figure.
These results further underscore the functional heterogeneity of fibroblasts and highlight the specific role of β1 integrin in mediating αSMA+ myofibroblast‐regulated skin wound repair.
Discussion
αSMA+ myofibroblasts are known to exhibit diverse origins and functions. Both the Delta like homologue 1 (Dlk1)+ reticular dermal fibroblasts involved in bulk ECM synthesis (Driskell et al, 2013) and Engrailed‐1 (En1)‐lineage+ fibroblasts involved in fibrosis (Rinkevich et al, 2015) express αSMA. Acta2, Col1a1, Fap, and S100a4 are expressed at similar levels in En1‐lineage positive and negative fibroblasts (Rinkevich et al, 2015), indicating that these myofibroblast subtypes are likely not solely derived from the En1‐lineage. In addition, αSMA+ myofibroblasts can arise from CD29 (β1 integrin)+ adipocyte precursors (Shook et al, 2018) and mature adipocytes (Shook et al, 2020) that communicate with wound macrophages. Under specific wounding contexts, αSMA+ myofibroblasts are also derived from a rare myeloid lineage (Guerrero‐Juarez et al, 2019) and involved in wound‐induced adipocyte formation (Plikus et al, 2017). While we found wound‐induced αSMA+ myofibroblasts are predominantly locally derived, rather than bone marrow derived, it is conceivable that some may have a tissue‐resident myeloid lineage origin. Among the known fibroblast‐associated genes (Appendix Fig S2A and B) (Driskell & Watt, 2015; Rinkevich et al, 2015; Kalluri, 2016; Shook et al, 2018; Guerrero‐Juarez et al, 2019; Ahangar et al, 2020; Griffin et al, 2020; Leavitt et al, 2020; Mascharak et al, 2021), many are significantly decreased in αSMA‐TK wounds, suggesting additional potential mediators of αSMA+ myofibroblast‐orchestrated wound repair.
Our work assigns functional attributes to the skin‐resident and wound‐induced αSMA+ myofibroblasts that cannot be compensated by any other fibroblasts in the skin. The sustained depletion of αSMA+ myofibroblasts leads to non‐healing wounds, recapitulating many aspects of human chronic wounds, including those found in diabetic patients (Fig 1), including hindered granulation tissue formation and reepithelization and increased hypoxia. In contrast to αSMA‐null animals where compensation from other actin isoforms might mask the phenotype (Tomasek et al, 2013; Ibrahim et al, 2015), here we demonstrated using genetic models the essential non‐redundant function of αSMA+ myofibroblasts. It is important to note that deletion of Acta2 assesses the function of the αSMA protein, while our studies use αSMA as marker for a class of fibroblasts (named myofibroblasts due to their expression of αSMA) to assess the function of this fibroblast subset with respect to wound healing. It has been assumed largely based on in vitro culture experiments that αSMA+ myofibroblast production of collagen I is critical for wound repair (Tomasek et al, 2002). Several studies have connected fibroblast lineages with production of ECM (Driskell et al, 2013; Rinkevich et al, 2015; Shook et al, 2018, 2020; Leavitt et al, 2020; Mascharak et al, 2021); however, these findings were at best correlative and a functional dissection of the role of Col1 in wound repair had not been performed. While the effectors of αSMA+ myofibroblasts likely govern multiple aspects of skin wound repair, our genetic models substantiate that Col1 produced by αSMA+ myofibroblasts is dispensable for wound healing. Expression of Col1a1 is detected in non‐Acta2 cells, suggesting that other fibroblast populations or other cell types may produce Col1 in wounds, as was demonstrated in this study using the newly generated Col1α1‐TK mice. Alternatively, previous studies have demonstrated that fascia fibroblasts mobilize fascia into wounds (Jiang et al, 2020), indicating that de novo production of collagen may not be required for proper wound repair. We previously reported that mice with deletion of Col1 (αSMA‐Cre; Col1a1loxP/loxP ) (Chen et al, 2021) and TGFβRII (αSMA‐Cre; Tgfbr2loxP/loxP ) (LeBleu et al, 2013a) in αSMA+ myofibroblasts exhibit normal tissue architecture and lifespan, indicating that the deletion of such genes largely does not result in overt abnormalities. Moreover, deletion of Col1 in αSMA+ myofibroblasts did not alter vascular integrity or basement membranes (Chen et al, 2021) supporting that perivascular or vascular smooth muscle cells are largely unaffected by αSMA‐Cre‐mediated deletion of genes expressed by myofibroblasts. Previous studies have demonstrated that αSMA‐Cre‐ERT2 labels myofibroblasts in wound tissue (Plikus et al, 2017), suggesting that off‐target effects of gene deletion are likely minimal. Nonetheless, potential off‐target deletion of genes (i.e., in non‐myofibroblasts) remains a limitation of Cre‐based systems and a potential confounder associated with the use of such model systems.
Besides the functional role of αSMA+ cells, our genetic models also addressed the function of two other important myofibroblast populations in skin wound repair. Recent studies indicated that FAP does not co‐localize with αSMA in the fibrotic stroma of tumors (Ozdemir et al, 2014; Kilvaer et al, 2018) and FSP1 and αSMA do not overlap in the heart after myocardial infarction (Saraswati et al, 2019), suggesting that these markers label discrete cell types. Depletion of αSMA+ (LeBleu et al, 2013a) or FSP1+ (Iwano et al, 2001) fibroblasts was associated with attenuation of kidney fibrosis, where bone‐marrow‐derived αSMA+ myofibroblasts were more prevalent in fibrotic kidneys and were dependent on Tgfbr2 for differentiation into myofibroblasts (LeBleu et al, 2013a). In contrast, during cutaneous wound repair, skin‐resident αSMA+ myofibroblasts were the main contributors, and Tgfbr2 in αSMA+ myofibroblasts appeared functionally dispensable, although it is possible that other TGFβ receptors might compensate for Tgfbr2 loss. Our current observations alongside this previous knowledge illustrate the molecular and functional divergence of myofibroblasts in a tissue and disease‐dependent manner.
αSMA+ myofibroblasts, mediated by β1 integrin, facilitate granulation tissue formation, and promote reepithelization and angiogenesis. GSEA of Acta2 + Itgb1 + myofibroblasts revealed an enrichment of pathways related to extracellular matrix organization and inflammatory response (Appendix Fig S2C and D). Enriched genes related to extracellular matrix organization include Fbn1 (encoding fibronectin), Col5a1, Col5a2, and Col5a3 (encoding the α1, α2, and α3 chains of type VI collagen, respectively), Col6a1 and Col6a2 (encoding the α1 and α2 chains of type VI collagen, respectively), Lama4 and Lamb1 (encoding the α4 and β1 chains of laminin, respectively), Lum (encoding lumican), Mmp2, and Lox (encoding lysyl oxidase). Basement membrane proteins of the epidermis, including type IV and VII collagens, laminin, and fibronectin, regulate the proliferation, migration, and differentiation of epidermal stem cells through direct signaling as well as growth factor presentation (Chermnykh et al, 2018). Inflammation‐related genes that were enriched in Acta2 + Itgb1 + myofibroblasts included Ccl2, Cxcl10, and Il1b. CCL2 produced by mesenchymal cells induces expression of antimicrobial peptides by keratinocytes (Marx et al, 2021). Deletion of the receptor for CXCL10, CXCR3, inhibits reepithelization and regeneration of basement membrane in wounds (Yates et al, 2009). IL‐1β induces keratinocyte proliferation (Ristow, 1987; Takei et al, 1998) and secretion of several soluble factors involved in T‐cell activation (Sanmiguel et al, 2009). Together, these data suggest that αSMA+ myofibroblasts may influence keratinocytes through remodeling of the extracellular matrix and secretion of chemokines and cytokines. Alternatively, reduced reepithelization could be a reflection of the delayed wound closure in Itgb1cKO mice (i.e., reepithelization is a correlate of wound repair). Further mechanistic dissection of the interactions between αSMA+ myofibroblasts and keratinocytes will clarify their precise role in regulating reepithelization. Altogether, our work provides a novel genetic model for the study of chronic non‐healing wounds, providing an appealing platform for identifying therapeutic targets to accelerate skin wound repair.
Materials and Methods
Mice
All animal experiments were conducted in agreement with the National Institutes of Health guidelines. All protocols were approved by the Animal Care and Use Committees of Beth Israel Deaconess Medical Center, Boston, MA, and the University of Texas MD Anderson Cancer Center, Houston, TX. Mice were housed in individually ventilated cages on a 12‐h light/dark cycle at 21–23°C and 40–60% humidity. Several of the GEMMs in this study were described previously, including αSMA‐TK, αSMA‐RFP, and αSMA‐Cre mice (LeBleu et al, 2013a, 2013b), FSP1/S100A4‐TK mice (Iwano et al, 2001), αSMA‐Cre‐ERT2 mice (Wendling et al, 2009), Col1a1loxP/loxP (Chen et al, 2021), Itgb1loxP/loxP (Raghavan et al, 2000), and Tgfbr2loxP/loxP (Chytil et al, 2002). Nomenclature of conditional knockout mice is as follows: Col1α1cKO, αSMA‐Cre‐ERT2; Col1a1loxP/loxP. Itgb1cKO, αSMA‐Cre; Itgb1loxP/loxP . Tgfbr2cKO, αSMA‐Cre; Tgfbr2loxP/loxP . Newly generated transgenics include FAP‐TK and Col1a1‐TK (see Figs EV3E and EV4G). Specifically, extended Fap and Col1a1 promoters were cloned into the Not1‐Swa1 sites of pORF‐HSV1‐TK vector (Invivogen), the constructs were gel‐purified and injected into fertilized oocytes. Fertilized oocytes were subsequently implanted to pregnant dams to generate chimeras (performed at the Genetically Engineered Mouse Facility, UT MDACC). Male and female adult mice were used between ages of 8 and 12 weeks. For detailed information on mouse strains and genotyping procedures, see Appendix Table S1 and Appendix Table S2. The αSMA‐TK depletion system prevents the accumulation of actively proliferating αSMA‐expressing cells, and depletion of αSMA+ cells is not observed in normal/non‐pathological tissues where αSMA+ cells are largely non‐proliferative. Moreover, overt abnormalities indicative of off‐target effects are not observed in αSMA‐TK mice (LeBleu et al, 2013a; McAndrews et al, 2021). In order to account for variability across mice, animal numbers were based on power calculations in order to derive statistically meaningful results and effective depletion of cells or deletion of genes was confirmed for the experimental models used. Sample size was chosen based on an effect size of 3.922 to detect statistically meaningful differences between the two groups with a power of 0.95 and α error probability of 0.05.
Full‐thickness wound model
The full‐thickness wound model has been described previously (Shinzawa et al, 2007; Driskell et al, 2013). Briefly, mice were shaved, epilated, and two full‐thickness wounds were created bilaterally with a sterile 8‐mm punch biopsy on the flanks. In experiments utilizing the TK transgenic animals, specific fibroblast populations were ablated by daily intraperitoneal administration of ganciclovir (GCV) at a dose of 50 mg kg−1. GCV (Invivogen) was prepared according to the manufacturer’s instructions. αSMA‐Cre‐ERT2; Col1a1loxP/loxP mice were administered tamoxifen (Sigma Aldrich, 100 mg kg−1 in corn oil) i.p. daily from day 0 to 14. The time frame of treatment is detailed for each individual experiment. Wound closure was measured using microcalipers at the indicated time points, the wound areas were calculated as Area = π/4 × length × width and expressed as percent of initial wound area (day 0). The wound closures for the two wounds were averaged per mouse. Mice that were euthanized prior to the study endpoint were excluded from analysis. Skin fragments encompassing the wounded area were excised and utilized as described below. Groups are compared to respective wild‐type controls to account for potential variability arising from mice backgrounds or genetics. Mice were not randomized, as animals were allocated to groups based on their genotype; thus, randomization was not possible. Investigators were not blinded to group allocation, as knowledge of animal genotype was required for assignment to experimental groups.
Immunohistochemical multiplex analysis
The process for multiplexed staining and multispectral imaging has been described previously (Carstens et al, 2017). Briefly, 5‐μm‐thick sections of paraffin‐embedded tissues were deparaffinized and fixed with formaldehyde:methanol (1:10) for 20 min at room temperature, prior to antigen retrieval with heated citric acid buffer (pH 6.0) 15 min, 98°C (EZ Retriever Microwave, BioGenex). For FAP staining, antigen retrieval was performed with TE buffer (pH 9.0). Each section was subjected to sequential rounds of staining, each including a blocking step with 1% BSA, primary antibody step (for antibodies see Appendix Table S3), secondary horseradish peroxidase‐conjugated polymer, and a specific fluorophore (Appendix Table S3 for tyramide signal amplification). Each staining round was followed by additional antigen retrieval in heated citrate buffer (pH 6.0) for 15 min to remove bound antibodies while leaving covalently bound fluorophores. In the next step, sections were counterstained with DAPI or Hoechst 33342 and mounted with fluoroshield histology mounting medium (Sigma‐Aldrich) or Vectashield mounting medium (Vector Labs). Stained slides were scanned using the Vectra Multispectral Imaging System version 3 (Perkin Elmer) to generate digital images at 200× magnification. The dermis region was imaged for subsequent analysis. Spectral unmixing was then performed using the inForm 2.3 software (Perkin‐Elmer), using single stains as a reference. For quantitative analysis of the changes in cell populations during wound healing, a custom‐written MATLAB algorithm was used (Mathworks). The wound tissue area was identified by integration of all fluorescent channels for further analysis. Following smoothing with a median filter, a standard deviation threshold within the wound area was used to segment positively stained regions. Areas positive for CD31 (pericytes) were excluded from analysis. Data are presented as fraction of wound area positive for the indicated marker(s).
Histology
For hematoxylin and eosin (H&E) or Sirius red staining, tissues were formalin fixed and paraffin‐embedded. H&E staining was performed with ST Infinity H&E staining system (Leica) and Sirius red staining performed with 0.1% picrosirius red (Direct Red 80, Sigma Aldrich) according to manufacturer’s instructions. H&E‐stained slides were imaged with a Leica DM 1000 LED microscope or scanned with a Leica Aperio AT2, Keyence BZ‐X700, or Zeiss Axio Scan.Z1. Picrosirius red stained slides were imaged using a Zeiss Axio Observer.Z1 equipped with a light polarizer. Collagen fiber quantification was performed using a previously described Matlab algorithm (McGrail et al, 2012).
Immunolabeling of tissues
For immunohistochemistry (IHC) and immunofluorescence (IF), tissues were formalin fixed and paraffin‐embedded. Five‐micrometer sections were rehydrated and antigen retrieval was performed in 10 mM citrate buffer (pH 6.0) or TE buffer (pH 9.0) for a period of 15 min to 1 h. After 1 h blocking with appropriate blocking buffer (Appendix Table S3) at room temperature, the sections were incubated overnight at 4°C with primary antibody and then with secondary antibody for 1 h at room temperature. For IHC, ABC reagent (Vector Laboratories, West Grove, PA) was applied and the sections were developed using diaminobenzidine reagent (DAB) according to the manufacturer’s instructions.
For frozen specimens, 8‐μm‐thick cryosections of OCT‐embedded tissues were fixed in 4% buffered paraformaldehyde (PFA) for 20 min and permeabilized in 0.3% Triton‐X for 30 min. After blocking with appropriate blocking buffer (30 min at room temperature), the sections were incubated overnight at 4°C with primary antibodies (Appendix Table S3), followed by secondary antibodies for 1 h at room temperature, and 30‐min incubation with nuclear stain (DAPI).
Stained slides were imaged with a Leica DM 1000 LED microscope (IHC), Zeiss LSM800 confocal microscope (IF), Zeiss Axio Observer.Z1 (IF), Zeiss Axioskop2 Plus (IF), or Zeiss Axio Scan.Z1 (IF). The counts of marker‐positive cells detectable by IHC/IF were determined on digital images using the count tool of Adobe Photoshop suite. Mean positive area and signal intensity were also measured on digital images using ImageJ. Alternatively, images were thresholded based on negative control images (tissue stained with secondary antibody) followed by manual counting of positive cells using the cell counter function in ImageJ. Epidermal thickness was evaluated by measuring the Krt5+ distance within the wound bed. Unless otherwise noted, all analyses of marker‐positive cells (number, signal intensity) were limited to the wound granulation tissue. For details on the antibodies used in the study, see Appendix Table S3.
Bulk gene expression profiling
For microarray analyses, wounds were excised and total RNA was extracted with Trizol reagent (Invitrogen) following manufacturer’s instructions. Isolated RNA (500 ng) was labeled and hybridized according to the manufacturer’s protocols (Illumina, Inc, San Diego, CA) with the Illumina MouseWG‐6 v2.0 expression BeadChips. The BeadChips were scanned with Illumina BeadArray Reader (Illumina, Inc). The data were extracted and normalized with Bead Studio 3.7 (Illumina, Inc.) after background subtraction. Normalized gene‐level expression data were generated using GenomeStudio (Illumina) and then exported for further analysis using a custom algorithm in MATLAB (Mathworks). For unsupervised hierarchical clustering, data were log2‐transformed and highly variable genes (standard deviation > 2) selected for clustering. All clustering was performed using Ward’s minimum variance method. Gene set enrichment analysis (GSEA) was performed as described previously (Subramanian et al, 2005), using 103 iterations on a list of differentially expressed genes (FDR < 0.1) ranked by average log2‐transformed fold change. Bulk RNA sequencing data of normal and diabetic human wounds (transcripts per million) were acquired from GSE154556 (Davis et al, 2020c).
Bone marrow transplant experiments
Unfractionated bone marrow (BM) was harvested from the tibia and femurs of WT, ⍺SMA‐TK, and ⍺SMA‐RFP male donor mice. Single‐cell suspensions were generated and viable cells counted by trypan blue exclusion. Recipient female mice received sublethal dose of γ‐irradiation (8 Gy/mouse from 60Co source) and were injected on the following day with 2 × 106 BM cells in 100‐μl PBS via the retro‐orbital plexus. Mice were allowed 3 weeks for BM repopulation before use in wounding studies. The degree of BM chimerism was assessed by fluorescent in situ hybridization (FISH) of bone marrow smears for the presence of donor Y‐chromosome as described previously (LeBleu et al, 2013a). Negative control was secondary antibody alone labeled tissues.
Ex vivo fibroblast culture
Fibroblasts were isolated from the ears of transgenic mice and treated with GCV as described previously (O'Connell et al, 2011; LeBleu et al, 2013a). Briefly, adult WT and TK+ mice were euthanized, ear tissues were harvested, minced, and digested overnight at 37°C with type collagenase IV (400 U/ml, Gibco) in RPMI (Corning) at 37°C, 1% penicillin–streptomycin (P/S, Corning) and antimycotics (A/M, Corning), then transferred into RPMI supplemented with 10% FBS, 1% P/S, and A/M. Cells were expanded ex vivo for no more than 2 weeks, then seeded into 96‐well plates at 1 × 104 cells per well, treated with control medium or GCV at 5 and 50 μM for 5 days, and cell viability was measured by trypan blue exclusion.
scRNA‐seq analysis
Single‐cell RNA sequencing data of murine wounds and intact skin were acquired from Haensel et al (2020b) (Data ref: Haensel et al (2020a)). Single‐cell RNA sequencing data of normal and diabetic human wounds were acquired from Davis et al (2020c) (Data ref: Davis et al (2020b)). Genes not expressed in 95% of cells were excluded. Cells that had below 500 reads or contained over 20% of reads mapping to mitochondrial genes were excluded from further analysis. Following normalization of counts for library size, we used Markov affinity‐based graph imputation of cells (MAGIC) to denoise the expression matrix (van Dijk et al, 2018). Dimensionality reduction was performed by uniform manifold approximation and projection (UMAP), and clusters identified using Euclidean distance as a similarity metric (Becht et al, 2018). All analyses were performed in Matlab R2019a. Gene set enrichment analysis (GSEA) was performed based on preranked gene expression of Acta2+Itgb1+ fibroblasts compared to other fibroblasts in GSEA 4.1.0.
Quantitative real‐time PCR
RNA was isolated from snap‐frozen tissues and extracted with Trizol reagent (Invitrogen) according to the manufacturer’s instruction. cDNA was synthesized using High‐Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific). Real‐time quantitative PCR was carried out using SYBR Green Real‐Time PCR Master Mix on QuantStudio™ 7 Flex Real‐Time PCR System (ThermoFisher Scientific). For primer details, see Appendix Table S2.
Atomic force microscopy
Atomic force microscopy was conducted at the University of Texas Health Science Center Core Facility using a BioScope IITM Controller (Bruker Corporation; Santa Barbara, CA). The image acquisition and analyses were performed with Research NanoScope software (Bruker Corporation, versions 7.30 and 1.40, respectively) integrated into a Nikon TE2000‐E inverted optical microscope (Nikon, Lewisville, TX) to facilitate bright‐field imaging. To investigate the nano‐mechanical properties of the tissue surface, 40‐µm‐thick cryosections of OCT‐imbedded skin wound samples from ⍺SMA‐TK and wild‐type mice were attached poly‐L‐lysine‐coated slides (Sigma P8920) and hydrated in water. The elastic (Young’s) modulus was measured on hydrated tissue sections using DNP‐S triangular cantilevers (resonant frequency of 2–24 kHz, spring constant of 0.06 N/m, radius of curvature equal to 10 nM, Bruker Corporation). Wound areas of the tissue sections were selected using optical microscopy. Force volume images were captured on 50 µm2 scan areas with 16 × 16 force measurements per line. Force curves were probed using a ramp size of 2 µm and a scan rate of 1.0 Hz, with a force load of 15 nN. The probe spring constant was determined prior to each experiment, by thermal tuning. The elastic modulus was calculated by fitting to a standard Sneddon model for a conical indenter and a Poisson’s ratio of 0.5. At least three areas were analyzed for every sample and a minimum of 256 force measurements captured per area. Topographical AFM imaging was performed on dry tissue sections using RTESP cantilevers (fo = 237–289 kHz, k = 20–80 N/m, Bruker Corporation). The structure of the tissue was determined using tapping mode operated in air to a scan rate of 0.7 Hz. Images of the extracellular matrix were captured to a scan range of 10, 5, and 2.5 µm2.
In situ hybridization
Col1a1 in situ hybridization was performed as previously described (Keskin et al, 2015; Zheng et al, 2015). Ten‐micrometer frozen wound sections were hybridized with Col1a1 probe overnight at 65°C. The PCR primers used for generating digoxigenin‐labeled riboprobes via in vitro transcription (Promega and Roche) are as follows: Anti‐sense: Forward: 5’‐AGCACGTCTGGTTTGGAGAG‐3’, Reverse: 5’‐TAATACGACTCACTATAGGGAGA‐3’. Sense: Forward: 5’‐AATTAACCCTCACTAAAGGGAGAGCACGTCTGGTTTGGAGAG‐3’, Reverse: 5’‐TTGGTCACGTTCAGTTGGTCA‐3’. Sections were co‐labeled with αSMA‐Cy3 antibody (Sigma, Appendix Table S3) and imaged with a Zeiss LSM700 confocal microscope.
Statistical analysis
All quantitative data represent the results obtained in biological replicates denoted in the figure legends and expressed as mean value ± standard error (s.e.m.) unless otherwise noted. All statistical analyses were performed using GraphPad Prism software, Version 9. Normal distribution was assessed using Kolmogorov–Smirnov test and non‐normal datasets were compared using non‐parametric statistical tests. The tests used to determine statistical significance are indicated in figure legends, with exact P‐values included.
Author contributions
Kathleen M McAndrews: Conceptualization; Data curation; Formal analysis; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Toru Miyake: Conceptualization; Data curation; Formal analysis; Investigation; Visualization; Methodology. Ehsan A Ehsanipour: Data curation; Formal analysis; Investigation; Visualization; Methodology. Patience J Kelly: Investigation. Lisa M Becker: Investigation. Daniel J McGrail: Software; Formal analysis; Visualization. Hikaru Sugimoto: Investigation. Valerie S LeBleu: Supervision. Yejing Ge: Conceptualization; Formal analysis; Supervision; Writing—original draft; Writing—review and editing. Raghu Kalluri: Conceptualization; Supervision; Funding acquisition; Writing—original draft; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
KMM, EAE, TM, PJK, LMB, and HS performed the experiments. KMM, EAE, and TM designed the experiments, generated the data, and generated the figures. TM designed and generated the FAP‐TK and Col1α1‐TK mice. KMM, EAE, and TM, analyzed the data. DJM analyzed the microarray and scRNA‐seq data and generated the script for multispectral imaging analysis. KMM, YG, and RK wrote the manuscript. RK and VSL participated in discussions, provided intellectual input, and supervised experimental discussion. TM and RK conceived the project. RK conceptually designed the strategy for this study.
Disclosure and Competing Interest Statement
KMM received speaker honorarium from Stellanova Therapeutics. VSL is a Scientific Advisory Board member and stockholder of Stellanova Therapeutics.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank P. Correa de Sampaio, J.L. Carstens, and P.P. Phillips for technical assistance with the optimization of the multiplexed panel and scanning of resultant slides, F. Kugeratski, M. Tang, L.M. Snowden, and L. Gibson for assistance in mouse maintenance and experimental procedures, C. Kwak for assistance with flow cytometry analysis, and O.V. Volpert for assistance with manuscript writing. FSP1‐TK mice were kindly provided by Eric Neilson (Northwestern University), Tgfbr2loxP/loxP mice were kindly provided by Harold Moses (Vanderbilt University), and αSMA‐Cre‐ERT2 mice were kindly provided by Pierre Chambon (Université de Strasbourg). This work was primarily supported by the Cancer Prevention and Research Institute of Texas (R.K.). Y.G. and R.K. are CPRIT Scholars of Cancer Research. Y.G. is supported by NIAMS 1K01AR072132 and CPRIT FP00006955. K.M.M. is supported by the Ergon Foundation postdoctoral fellowship. D.J.M. is supported by NCI grant K99CA240689. The UT MDACC Flow Cytometry & Cellular Imaging Facility, UT MDACC Atomic Force Microscopy Core Facility, UT MDACC Advanced Technology Genomics Core, and UT MDACC Genetically Engineered Mouse Facility are supported by NIH P30CA016672.
The EMBO Journal (2022) 41: e109470.
Contributor Information
Yejing Ge, Email: yge1@mdanderson.org.
Raghu Kalluri, Email: rkalluri@mdanderson.org.
Data availability
Source data for each figure are included and all custom‐written algorithms are available upon request. The datasets produced in this study are available in the following databases: Gene expression (microarray) data: Gene Expression Omnibus GSE178902 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178902).
References
- Ahangar P, Mills SJ, Cowin AJ (2020) Mesenchymal stem cell secretome as an emerging cell‐free alternative for improving wound repair. Int J Mol Sci 21: 7038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, Newell EW (2018) Dimensionality reduction for visualizing single‐cell data using UMAP. Nat Biotechnol 37: 38–44. [DOI] [PubMed] [Google Scholar]
- Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, Allison JP, LeBleu VS, Kalluri R (2017) Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun 8: 15095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, LeBleu VS, Kalluri R (2021) Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39: 548–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chermnykh E, Kalabusheva E, Vorotelyak E (2018) Extracellular matrix as a regulator of epidermal stem cell fate. Int J Mol Sci 19: 1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chytil A, Magnuson MA, Wright CV, Moses HL (2002) Conditional inactivation of the TGF‐beta type II receptor using Cre:Lox. Genesis 32: 73–75 [DOI] [PubMed] [Google Scholar]
- Collins CA, Kretzschmar K, Watt FM (2011) Reprogramming adult dermis to a neonatal state through epidermal activation of beta‐catenin. Development 138: 5189–5199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby I, Skalli O, Gabbiani G (1990) Alpha‐smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest 63: 21–29 [PubMed] [Google Scholar]
- Darby IA, Zakuan N, Billet F, Desmouliere A (2016) The myofibroblast, a key cell in normal and pathological tissue repair. Cell Mol Life Sci 73: 1145–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Gallagher KA, Tsoi LC (2020a) Gene Expression Omnibus GSE154556 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154556). [DATASET]
- Davis FM, Gallagher KA, Tsoi LC (2020b) Gene Expression Omnibus GSE154557 (https://www‐ncbi‐nlm‐nih‐gov.ezproxy.u‐pec.fr/geo/query/acc.cgi?acc=GSE154557). [DATASET]
- Davis FM, Tsoi LC, Wasikowski R, denDekker A, Joshi A, Wilke C, Deng H, Wolf S, Obi A, Huang S et al (2020c) Epigenetic regulation of the PGE2 pathway modulates macrophage phenotype in normal and pathologic wound repair. JCI Insight 5: e138443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton CP, Khan K, Hoyles RK, Shiwen X, Leoni P, Chen Y, Eastwood M, Abraham DJ (2009) Inducible lineage‐specific deletion of TbetaRII in fibroblasts defines a pivotal regulatory role during adult skin wound healing. J Invest Dermatol 129: 194–204 [DOI] [PubMed] [Google Scholar]
- van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D et al (2018) Recovering gene interactions from single‐cell data using data diffusion. Cell 174: 716–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, Ferron SR, Herault Y, Pavlovic G, Ferguson‐Smith AC et al (2013) Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504: 277–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Watt FM (2015) Understanding fibroblast heterogeneity in the skin. Trends Cell Biol 25: 92–99 [DOI] [PubMed] [Google Scholar]
- Franke WW, Schmid E, Osborn M, Weber K (1978) Different intermediate‐sized filaments distinguished by immunofluorescence microscopy. Proc Natl Acad Sci USA 75: 5034–5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiano RD, Jt M, Dobryansky M, Levine JP, Gurtner GC (2004) Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen 12: 485–492 [DOI] [PubMed] [Google Scholar]
- Griffin MF, desJardins‐Park HE, Mascharak S, Borrelli MR, Longaker MT (2020) Understanding the impact of fibroblast heterogeneity on skin fibrosis. Dis Model Mech 13: dmm044164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell F (1994) Fibroblasts, myofibroblasts, and wound contraction. J Cell Biol 124: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero‐Juarez CF, Dedhia PH, Jin S, Ruiz‐Vega R, Ma D, Liu Y, Yamaga K, Shestova O, Gay DL, Yang Z et al (2019) Single‐cell analysis reveals fibroblast heterogeneity and myeloid‐derived adipocyte progenitors in murine skin wounds. Nat Commun 10: 650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT (2008) Wound repair and regeneration. Nature 453: 314–321 [DOI] [PubMed] [Google Scholar]
- Haensel D, Dai X (2020a) Gene Expression Omnibus GSE142471 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142471). [DATASET]
- Haensel D, Jin S, Sun P, Cinco R, Dragan M, Nguyen Q, Cang Z, Gong Y, Vu R, MacLean AL et al (2020b) Defining epidermal basal cell states during skin homeostasis and wound healing using single‐cell transcriptomics. Cell Rep 30: 3932–3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautmann MB, Madsen CS, Owens GK (1997) A transforming growth factor beta (TGFbeta) control element drives TGFbeta‐induced stimulation of smooth muscle alpha‐actin gene expression in concert with two CArG elements. J Biol Chem 272: 10948–10956 [DOI] [PubMed] [Google Scholar]
- Higashiyama R, Nakao S, Shibusawa Y, Ishikawa O, Moro T, Mikami K, Fukumitsu H, Ueda Y, Minakawa K, Tabata Y et al (2011) Differential contribution of dermal resident and bone marrow‐derived cells to collagen production during wound healing and fibrogenesis in mice. J Invest Dermatol 131: 529–536 [DOI] [PubMed] [Google Scholar]
- Hinz B (2007) Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 127: 526–537 [DOI] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton‐Piallat ML, Gabbiani G (2007) The myofibroblast: one function, multiple origins. Am J Pathol 170: 1807–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Chen L, Bond JE, Medina MA, Ren L, Kokosis G, Selim AM, Levinson H (2015) Myofibroblasts contribute to but are not necessary for wound contraction. Lab Invest 95: 1429–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano M, Fischer A, Okada H, Plieth D, Xue C, Danoff TM, Neilson EG (2001) Conditional abatement of tissue fibrosis using nucleoside analogs to selectively corrupt DNA replication in transgenic fibroblasts. Mol Ther 3: 149–159 [DOI] [PubMed] [Google Scholar]
- Jacob M, Chang L, Pure E (2012) Fibroblast activation protein in remodeling tissues. Curr Mol Med 12: 1220–1243 [DOI] [PubMed] [Google Scholar]
- Jiang D, Christ S, Correa‐Gallegos D, Ramesh P, Kalgudde Gopal S, Wannemacher J, Mayr CH, Lupperger V, Yu Q, Ye H et al (2020) Injury triggers fascia fibroblast collective cell migration to drive scar formation through N‐cadherin. Nat Commun 11: 5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Correa‐Gallegos D, Christ S, Stefanska A, Liu J, Ramesh P, Rajendran V, De Santis MM, Wagner DE, Rinkevich Y (2018) Two succeeding fibroblastic lineages drive dermal development and the transition from regeneration to scarring. Nat Cell Biol 20: 422–431 [DOI] [PubMed] [Google Scholar]
- Kalluri R (2016) The biology and function of fibroblasts in cancer. Nat Rev Cancer 16: 582–598 [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6: 392–401 [DOI] [PubMed] [Google Scholar]
- Keskin D, Kim J, Cooke VG, Wu CC, Sugimoto H, Gu C, De Palma M, Kalluri R, LeBleu VS (2015) Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin‐2. Cell Rep 10: 1066–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilvaer TK, Rakaee M, Hellevik T, Ostman A, Strell C, Bremnes RM, Busund LT, Donnem T, Martinez‐Zubiaurre I (2018) Tissue analyses reveal a potential immune‐adjuvant function of FAP‐1 positive fibroblasts in non‐small cell lung cancer. PLoS One 13: e0192157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt T, Hu MS, Marshall CD, Barnes LA, Lorenz HP, Longaker MT (2016) Scarless wound healing: finding the right cells and signals. Cell Tissue Res 365: 483–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt T, Hu MS, Borrelli MR, Januszyk M, Garcia JT, Ransom RC, Mascharak S, desJardins‐Park HE, Litzenburger UM, Walmsley GG et al (2020) Prrx1 Fibroblasts Represent a Pro‐fibrotic Lineage in the Mouse Ventral Dermis. Cell Rep 33: 108356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Kalluri R (2018) A peek into cancer‐associated fibroblasts: origins, functions and translational impact. Dis Model Mech 11: dmm029447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R (2013a) Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Teng Y, O'Connell JT, Charytan D, Muller GA, Muller CA, Sugimoto H, Kalluri R (2013b) Identification of human epididymis protein‐4 as a fibroblast‐derived mediator of fibrosis. Nat Med 19: 227–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Wang JH (2011) Fibroblasts and myofibroblasts in wound healing: force generation and measurement. J Tissue Viability 20: 108–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Xu SW, Blumbach K, Eastwood M, Denton CP, Eckes B, Krieg T, Abraham DJ, Leask A (2010) Expression of integrin beta1 by fibroblasts is required for tissue repair in vivo . J Cell Sci 123: 3674–3682 [DOI] [PubMed] [Google Scholar]
- Lynch MD, Watt FM (2018) Fibroblast heterogeneity: implications for human disease. J Clin Investig 128: 26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan WR, Lee TC, Schwartz RJ, Schneider MD (1994) Transforming growth factor‐beta response elements of the skeletal alpha‐actin gene. Combinatorial action of serum response factor, YY1, and the SV40 enhancer‐binding protein, TEF‐1. J Biol Chem 269: 16754–16760 [PubMed] [Google Scholar]
- Martin P (1997) Wound healing‐‐aiming for perfect skin regeneration. Science 276: 75–81 [DOI] [PubMed] [Google Scholar]
- Marx C, Gardner S, Harman RM, Wagner B, Van de Walle GR (2021) Mesenchymal stromal cell‐secreted CCL2 promotes antibacterial defense mechanisms through increased antimicrobial peptide expression in keratinocytes. Stem Cells Transl Med 10: 1666–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascharak S, des Jardins‐Park HE, Davitt MF, Griffin M, Borrelli MR, Moore AL, Chen K, Duoto B, Chinta M, Foster DS et al (2021) Preventing Engrailed‐1 activation in fibroblasts yields wound regeneration without scarring. Science 372: eaba2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew S, Scanlan MJ, Mohan Raj BK, Murty VV, Garin‐Chesa P, Old LJ, Rettig WJ, Chaganti RS (1995) The gene for fibroblast activation protein alpha (FAP), a putative cell surface‐bound serine protease expressed in cancer stroma and wound healing, maps to chromosome band 2q23. Genomics 25: 335–337 [DOI] [PubMed] [Google Scholar]
- McAndrews KM, Vazquez‐Arreguin K, Kwak C, Sugimoto H, Zheng X, Li B, Kirtley ML, LeBleu VS, Kalluri R (2021) alphaSMA(+) fibroblasts suppress Lgr5(+) cancer stem cells and restrain colorectal cancer progression. Oncogene 40: 4440–4452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrail DJ, Ghosh D, Quach ND, Dawson MR (2012) Differential mechanical response of mesenchymal stem cells and fibroblasts to tumor‐secreted soluble factors. PLoS One 7: e33248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, Dewar R, Rocha RM, Brentani RR, Resnick MB et al (2011) VEGF‐A and Tenascin‐C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci 108: 16002–16007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlund D, Elyada E, Tuveson D (2014) Fibroblast heterogeneity in the cancer wound. J Exp Med 211: 1503–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaso E, Labrador JP, Wang L, Ikeda K, Eng FJ, Klein R, Lovett DH, Lin HC, Friedman SL (2002) Discoidin domain receptor 2 regulates fibroblast proliferation and migration through the extracellular matrix in association with transcriptional activation of matrix metalloproteinase‐2. J Biol Chem 277: 3606–3613 [DOI] [PubMed] [Google Scholar]
- Opalenik SR, Davidson JM (2005) Fibroblast differentiation of bone marrow‐derived cells during wound repair. FASEB J 19: 1561–1563 [DOI] [PubMed] [Google Scholar]
- Özdemir B, Pentcheva‐Hoang T, Carstens J, Zheng X, Wu C‐C, Simpson T, Laklai H, Sugimoto H, Kahlert C, Novitskiy S et al (2014) Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25: 719–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippeos C, Telerman SB, Oulès B, Pisco AO, Shaw TJ, Elgueta R, Lombardi G, Driskell RR, Soldin M, Lynch MD et al (2018) Spatial and single‐cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J Invest Dermatol 138: 811–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plikus MV, Guerrero‐Juarez CF, Ito M, Li YR, Dedhia PH, Zheng Y, Shao M, Gay DL, Ramos R, Hsi T‐C et al (2017) Regeneration of fat cells from myofibroblasts during wound healing. Science 355: 748–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB (1999) Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol 277: C1–C9 [DOI] [PubMed] [Google Scholar]
- Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E (2000) Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol 150: 1149–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani W, Abbasi S, Hagner A, Raharjo E, Kumar R, Hotta A, Magness S, Metzger D, Biernaskie J (2014) Hair follicle dermal stem cells regenerate the dermal sheath, repopulate the dermal papilla, and modulate hair type. Dev Cell 31: 543–558 [DOI] [PubMed] [Google Scholar]
- Rettig WJ, Garin‐Chesa P, Beresford HR, Oettgen HF, Melamed MR, Old LJ (1988) Cell‐surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proc Natl Acad Sci USA 85: 3110–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AJ, Chaponnier C, Jahoda CA, Gabbiani G (1993) A quantitative study of the differential expression of alpha‐smooth muscle actin in cell populations of follicular and non‐follicular origin. J Invest Dermatol 101: 577–583 [DOI] [PubMed] [Google Scholar]
- Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP et al (2015) Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348: aaa2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow HJ (1987) A major factor contributing to epidermal proliferation in inflammatory skin diseases appears to be interleukin 1 or a related protein. Proc Natl Acad Sci USA 84: 1940–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmiguel JC, Olaru F, Li J, Mohr E, Jensen LE (2009) Interleukin‐1 regulates keratinocyte expression of T cell targeting chemokines through interleukin‐1 receptor associated kinase‐1 (IRAK1) dependent and independent pathways. Cell Signal 21: 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraswati S, Marrow SMW, Watch LA, Young PP (2019) Identification of a pro‐angiogenic functional role for FSP1‐positive fibroblast subtype in wound healing. Nat Commun 10: 3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA (2001) Integrin signaling revisited. Trends Cell Biol 11: 466–470 [DOI] [PubMed] [Google Scholar]
- Shinzawa H, Takeda A, Sone Y, Murashita K, Uchinuma E (2007) Wound healing process of a full‐thickness skin wound model in rats. Int Surg 92: 63–72 [PubMed] [Google Scholar]
- Shook BA, Wasko RR, Rivera‐Gonzalez GC, Salazar‐Gatzimas E, López‐Giráldez F, Dash BC, Muñoz‐Rojas AR, Aultman KD, Zwick RK, Lei V et al (2018) Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 362: eaar2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook BA, Wasko RR, Mano O, Rutenberg‐Schoenberg M, Rudolph MC, Zirak B, Rivera‐Gonzalez GC, López‐Giráldez F, Zarini S, Rezza A et al (2020) Dermal adipocyte lipolysis and myofibroblast conversion are required for efficient skin repair. Cell Stem Cell 26: 880–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer AJ, Clark RA (1999) Cutaneous wound healing. N Engl J Med 341: 738–746 [DOI] [PubMed] [Google Scholar]
- Sorrell JM, Caplan AI (2004) Fibroblast heterogeneity: more than skin deep. J Cell Sci 117: 667–675 [DOI] [PubMed] [Google Scholar]
- Sorrell JM, Caplan AI (2009) Fibroblasts‐a diverse population at the center of it all. Int Rev Cell Mol Biol 276: 161–214 [DOI] [PubMed] [Google Scholar]
- Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG (1995) Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130: 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabib T, Morse C, Wang T, Chen W, Lafyatis R (2018) SFRP2/DPP4 and FMO1/LSP1 define major fibroblast populations in human skin. J Invest Dermatol 138: 802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei T, Kito H, Du W, Mills I, Sumpio BE (1998) Induction of interleukin (IL)‐1 alpha and beta gene expression in human keratinocytes exposed to repetitive strain: their role in strain‐induced keratinocyte proliferation and morphological change. J Cell Biochem 69: 95–103 [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002) Myofibroblasts and mechano‐regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363 [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Haaksma CJ, Schwartz RJ, Howard EW (2013) Whole animal knockout of smooth muscle alpha‐actin does not alter excisional wound healing or the fibroblast‐to‐myofibroblast transition. Wound Repair Regen 21: 166–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, McRae J, Owens GK, Haaksma CJ (2005) Regulation of alpha‐smooth muscle actin expression in granulation tissue myofibroblasts is dependent on the intronic CArG element and the transforming growth factor‐beta1 control element. Am J Pathol 166: 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varner JA, Cheresh DA (1996) Integrins and cancer. Curr Opin Cell Biol 8: 724–730 [DOI] [PubMed] [Google Scholar]
- Wendling O, Bornert JM, Chambon P, Metzger D (2009) Efficient temporally‐controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis 47: 14–18 [DOI] [PubMed] [Google Scholar]
- Werner S, Krieg T, Smola H (2007) Keratinocyte‐fibroblast interactions in wound healing. J Invest Dermatol 127: 998–1008 [DOI] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates CC, Whaley D, Hooda S, Hebda PA, Bodnar RJ, Wells A (2009) Delayed reepithelialization and basement membrane regeneration after wounding in mice lacking CXCR3. Wound Repair Regen 17: 34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R (2015) Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527: 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]