

Abstract
Severe burns remain a leading cause of death and disability worldwide. Despite advances in patient care, the excessive and uncontrolled hypermetabolic stress response induced by this trauma inevitably affects every organ system causing substantial morbidity and mortality. Recent evidence suggests interleukin-6 (IL-6) is a major culprit underlying post-burn hypermetabolism. Indeed, genetic deletion of IL-6 alleviates various complications associated with poor clinical outcomes including the adverse remodeling of adipose tissue, cachexia and hepatic steatosis. Thus, pharmacological blockade of IL-6 may be a more favorable treatment option to fully restore metabolic function after injury. To test this, we investigated the safety and effectiveness of blocking IL-6 for post-burn hypermetabolism using a validated anti-IL-6 monoclonal antibody (mAb) in our experimental murine model. Here, we show daily anti-IL-6 mAb administration protects against burn-induced weight loss (P < .0001) without any adverse effect on mortality. At the organ level, post-burn treatment with the IL-6 blocker suppressed the thermogenic activation of adipose tissue (P < .01) and its associated wasting (P < .05). The reduction of browning-induced lipolysis (P < .0001) indirectly decreased hepatic lipotoxicity (P < .01) which improved liver dysfunction (P < .05). Importantly, the beneficial effects of this anti-IL-6 agent extended to the skin, reflected by the decrease in excessive collagen deposition (P < .001) and genes involved in pathologic fibrosis and scarring (P < .05). Together, our results indicate that post-burn IL-6 blockade leads to significant improvements in systemic hypermetabolism by inhibiting pathological alterations in key immunometabolic organs. These findings support the therapeutic potential of anti-IL-6 interventions to improve care, quality of life, and survival in burned patients.
Keywords: anti-IL-6 mAb, burn, effectiveness, hypermetabolism, interleukin-6, safety
1 |. INTRODUCTION
Hypermetabolic reprogramming triggered by thermal injury is characterized by a marked early inflammatory response and concomitant alterations of various organ functions.1 An excessive and uncontrolled inflammatory response and associated metabolic derangements increases susceptibility to organ failure, infection, and ultimately death in these patients.2 Many inflammatory meditators and pathways are elevated following injury, among the most consistent of which is interleukin-6 (IL-6).3,4 Studies have shown that IL-6 mRNA and protein are produced in the bone marrow of burn patients and mice subject to thermal injury.5 In fact, the magnitude of plasma IL-6 elevation has been shown to correlate with the extent of wound severity, organ dysfunction, and poor outcomes in both experimental and clinical studies.6,7
Interleukin-6 is characterized as a pleiotropic cytokine with both immune and metabolic regulatory functions in health and disease. For instance, in the context of immunology it regulates the acute phase response, stimulates macrophage recruitment and polarization, and chemokine production.8 Conversely, IL-6 has also been shown to function like a neuroendocrine hormone that can regulate cardiovascular, lipid, and glucose metabolism. Indeed, studies have shown in obese mice that brown adipose tissue (BAT)-derived IL-6 regulates both energy metabolism and glucose homeostasis.9 Our group has also previously demonstrated that IL-6 regulates whole-body energy metabolism in mice subjected to thermal injury by promoting the vast remodeling of white adipose tissue (WAT) in a process termed “browning”.10,11 Specifically, we showed that, under prolonged adrenergic stress, energy storing white adipocytes convert into energy releasing “beige” adipocytes by adopting brown fat characteristics. This phenotypic switch, characterized by the expression of uncoupling protein 1 (UCP-1), has been shown to be a key mediator of the adverse burn-induced hypermetabolic response.12
To date, WAT browning has been investigated in a variety of conditions, including obesity, cancer, and burns.13 In the former, enhanced thermogenesis in conditions of energy surplus (ie, obesity) serves an advantageous purpose including improved insulin sensitivity and enhanced weight loss.14 While beneficial in obesity, these attributes can be detrimental in cancer and burns. In fact, in these hypermetabolic conditions, WAT browning has been shown to contribute to the progression of cancer-associated cachexia, a condition characterized by severe weight loss and muscle catabolism.15 Similarly, a previous study from our group has shown that WAT browning-mediated lipolysis facilitated hepatic steatosis and dysfunction in post-burn mice, thereby increasing morbidity and mortality.15,16 Interestingly, in both conditions of cancer and burns where WAT browning has been implicated in persistent hypermetabolism and poor outcomes, IL-6 was identified as the initiator of this pathological cascade of events. Thus, dampening WAT browning by targeting IL-6 should theoretically improve post-burn hypermetabolism.
The therapeutic potential of IL-6 pathway inhibition has long been studied as early as the 1990s when IL-6 was implicated in conditions such as rheumatoid arthritis, Castleman’s disease, and giant cell arteritis.17 Indeed, these early studies pioneered the development of several drugs that block the IL-6 pathway, including the well-known drug tocilizumab, a humanized anti-IL-6 receptor antibody.18 Clinically, IL-6 signaling blockade has been shown to be efficacious for treating rheumatoid arthritis, a condition characterized by chronic IL-6 mediated inflammation that causes permanent joint destruction and deformity.19 These promising findings support the prevailing concept that IL-6 blockade can also be employed to treat patients in cancers and burns, where persistent IL-6 signaling increases susceptibility to metabolic complications and poor outcomes by driving the pathological browning response. However, the use of anti-IL-6 agents for indications other than those discussed above has yet to be studied.
The goal of the current study was to investigate the therapeutic potential of anti-IL-6 treatment for post-burn hypermetabolism and its associated pathology. We recently demonstrated that genetic deletion of IL-6 protects mice from many of the adverse alterations that occur after a burn injury like cachexia, WAT browning, and hepatic steatosis. Based on this compelling evidence, we postulate that pharmacological IL-6 blockade will exert similar beneficial effects after injury and ultimately improve outcomes. To test this, we evaluated the safety, tolerability and efficacy of a validated mouse anti-IL-6 monoclonal antibody (mAb), which neutralizes mouse IL-6 activities similar to clinically approved agents like tocilizumab, in our murine model of thermal injury. Our findings support the possibility of anti-IL-6 interventions as a potentially powerful adjunct therapy in burn care.
2 |. MATERIALS AND METHODS
2.1 |. Animals and model
Animal experiments were conducted in accordance and approved by the Sunnybrook Research Institute Animal Care Committee (Toronto, Ontario, Canada). Male wild-type C57BL/6J (WT) mice (8–10 weeks old, n = 6–7 per group) were purchased from Jackson Laboratories (Bar Harbor, Maine) and housed at ambient temperature and cared in accordance with the Guide for the Care and Use of Laboratory Animals. All mice were anesthetized with 2.5% isoflurane and shaved along the dorsal spine region. Ringers lactate (2–3 mL) was injected in the dorsal region subcutaneously in all mice to protect the spine and buprenorphine (0.05–0.1 mg/kg body weight) was injected for pain management. A full-thickness, third degree scald burn encompassing 30% total body surface area (TBSA) was achieved by immersing the dorsum of the mice in 98°C water for 10 seconds and the ventral region for 2 seconds. Burned mice were subsequently housed individually in sterile cages and food and water were given ad libitum. Sham mice (control) underwent identical experimental procedures, with the exception of the burn injury. The in vivo anti-mouse IL-6 receptor monoclonal antibody (clone 15A7) was purchased from BioXCell. Anti-IL-6 mAb treatment (50 mg/kg) was administered intraperitoneally once every 24 hours and continued until sacrifice at 7- and 14-day post-burn. Mice not receiving anti-IL-6 treatment were given vehicle (saline) injections. All injured rodents were health scored by both certified veterinarians and laboratory staff daily to minimize animal pain and distress. Health scoring was based on a scale of 15, with up to 3 points given for eyes and nose, activity, food intake, grooming, and hydration. The liver, dorsal skin, and inguinal adipose depot were harvested upon sacrifice and blood collected after cardiac puncture. All tissues were stored in −80°C until further analysis.
2.2 |. Histology and immunohistochemistry
Inguinal WAT, liver, and dorsal skin tissue were collected and immediately fixed in 10% formalin and then maintained in 70% ethanol before paraffin embedding. Subsequently, tissues were sectioned and stained with hematoxylin and eosin (H&E) or incubated with UCP1 antibody (Abcam, #155117) followed by DAB staining. For Oil Red O staining, tissues were coated with optimal cutting temperature (OCT) compound, placed on dry ice and stored at −80°C until further analysis. Frozen tissue blocks were sectioned 10 μm thick, mounted on slides, and fixed in formaldehyde (40%) for 1 minutes. The slides were stained with Oil Red O for 10 minutes. at room temperature, rinsed with water and stained using Gill’s hematoxylin for 1 min. For picrosirius red staining, paraffin-embedded slides were heated for 30 minutes at 60°C. The slides were then deparaffinized with citrosol, followed by rehydration through 100% × 2, 95%, 70%, and washed in distilled water. Slides were placed in Bouin’s solution (26 367–01; EMS, Hatfield, PA, USA) for 1 hour at 56°C and washed. Hematoxylin stain (HHS16; Sigma, Saint Louis, MO, USA) and Biebrich scarlet-acid fuchsin solution were applied sequentially for 10 minutes. After each stain, the slides were washed. Next, slides were differentiated in phosphomolybdic-phosphotungstic acid for 15 minutes, and transferred to aniline blue for 5 minutes. All slides were washed in distilled water and then differentiated in 1% acetic acid for 2 minutes. Slides were dehydrated through 95% ethanol and absolute ethanol followed by clearing in citrosol and were mounted with SHUR/Mount xylene-based liquid mounting media (Triangle Biomedical Sciences, Durham, NC, USA). Imaging was performed on a LSM confocal microscope (Zeiss, Germany). For quantification of adipocyte size, sections were imaged at a 20X magnification. The smallest and largest diameters of each adipocyte were manually measured using Leica Application Suite Version 4.3.0 (Leica Microsystems, Switzerland), and the average of the two values was used for subsequent analyses.
2.3 |. Inflammation and organ damage markers
Rodent plasma was collected and circulating levels of serum amyloid A (SAA ELISA Kit, Abcam#65336), alanine aminotransferase (ALT Colorimetric Assay Kit, Abcam#105134), aspartate transaminase (AST ELISA Kit, Abcam #263882) and creatinine (Creatinine Colorimetric Assay Kit, Abcam#65339) were measured using assay kits according to the manufacturer’s instructions. Intracellular interleukin-6 levels were determined using a mouse IL-6 ELISA kit (Abcam, #46100). All samples were normalized to total tissue protein concentration.
2.4 |. Lipid and catecholamine measurements
Plasma-free fatty acids (Abcam, #65341), plasma glycerol (Abcam, #65337), and tissue triglycerides (Abcam, #65336) were quantified using colorimetric assay kits according to the manufacturer’s instructions. Tissue norepinephrine measurements were taken for each sample in duplicate using a 2-Cat ELISA kit (2-Cat ELISA Fast Track, BA E-6500: Rocky Mountain Diagnostics, Inc, CO, USA). All samples were normalized to total tissue protein concentration.
2.5 |. Gene expression using RT-PCR
Total RNA isolated from adipose tissue was analyzed by quantitative real-time polymerase chain reaction (RT-PCR). RNA was isolated from tissue and cells using TRIzol-chloroform (Life Technologies) with subsequent purification using the RNeasy Kit (Qiagen) according to the manufacturer’s instructions. RNA (2 μg) was transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems). RNA was extracted from rodent adipose tissue using Trizol (Invitrogen, CA, USA). Reverse transcription was performed with high-capacity cDNA reverse transcription kit (ABI, MA, USA). RT-PCR was performed using the Applied Biosystems Step One Plus Real-Time PCR System. Gene expression was expressed relative to β-actin, and gene expression levels were determined using the following formula: 2^(-ΔCt). Housekeeping gene expression was stable between groups, and samples with Ct values >25 were not included in our analyses. Primer sequences used are available upon request.
2.6 |. Statistical analysis
All data are represented as mean ±SEM. Statistical differences between three or more groups were evaluated using a one and two-way ANOVA followed by Tukey’s post hoc tests for multiple comparisons. All graphs were created using Graphpad Prism 6.0 (San Diego, CA) and analyzed statistically using SPSS 20 (IBM Corp., NY, NY), with significance accepted at P < .05 (*), P < .01 (**), P < .001 (***), P < .0001 (****).
3 |. RESULTS
3.1 |. Daily anti-IL-6 mAb administration diminishes indices of burn-induced hypermetabolism independent of changes in mortality
We have previously implicated IL-6 in mediating many of the pathological alterations caused by the hypermetabolic response to burns.9 Therefore, we decided to investigate the therapeutic effect of inhibiting IL-6 signaling on post-burn hypermetabolism. To achieve this, mice were subjected to a 30% total body surface area (TBSA) burn injury and treated with anti-IL-6 mAb daily for 7 consecutive days. We first evaluated the safety of daily anti-IL-6 mAb administration by assessing its effect on post-burn survival; no significant difference in fatalities was observed between anti-IL-6-treated and non-treated burn mice over the course of 7 days (Figure S1). It is important to emphasize, though, that mice have a more resistant mortality response to a 30% TBSA thermal injury compared to what is clinically observed in burn patients with a similar TBSA, and thus conducting a more severe burn injury in mice is ethically unfeasible to fully assess mortality effects. Nevertheless, these findings suggest that daily administration of anti-IL-6 mAb is safe and does not cause drug toxicity-related mortality in mice after burn.
While administering the IL-6R blocker did not alter post-burn mortality rates, it was effective in protecting mice against the systemic effects of burn-induced hypermetabolism, namely severe body and fat wasting. As illustrated in Figure 1A, untreated burn mice lost 3.4% of total body weight at day 7 post-injury, while anti-IL-6-treated burn mice gained 0.85% at this time point (P < .0001). When comparing adipose tissue mass (Figure 1B), daily administration of the IL-6 blocker prevented the wasting of iWAT after 7 consecutive days of treatment (0.96% vs 0.62%, P < .05). Similarly, eWAT weight was significantly decreased in the untreated burn group after injury, whereas no change was observed in treated burn mice after injury (0.88% vs 1.27% P < .01). Surprisingly, the absence of burn-induced weight loss in anti-IL-6-treated burn mice was accompanied by an increase in food consumption at 7 days post-injury relative to their control counter parts (35.3% vs 28.7%, P < .05) (Figure 1C). To confirm anti-IL-6 mAb’s beneficial effects were attributed to a reduction in IL-6 signaling, we compared plasma concentrations of Serum amyloid A protein (SAA), an acute-phase target of IL-6 that is increased during hyperinflammatory states (Figure 1D). Consistent with our previous reports, circulating levels of SAA were drastically elevated at 7 days post-burn injury (42.37 vs 18.36 μg/mL, P < .0001). These levels were reduced in burn mice treated with the IL-6 blocker (38.95 vs 42.37 μg/mL, P < .05), however only slightly. Plasma SAA concentrations were still elevated in anti-IL-6-treated burn mice relative to controls at this time point (38.95 vs 18.36 μg/mL, P < .0001), suggesting a key role for other proinflammatory cytokines in mediating SAA production following burn injury. Nonetheless, our findings demonstrate that post-burn treatment with anti-IL-6 mAb not only effectively diminishes the development of systemic hypermetabolism (weight loss, fat wasting) after injury, but is also safe without any adverse effects on mortality.
FIGURE 1.
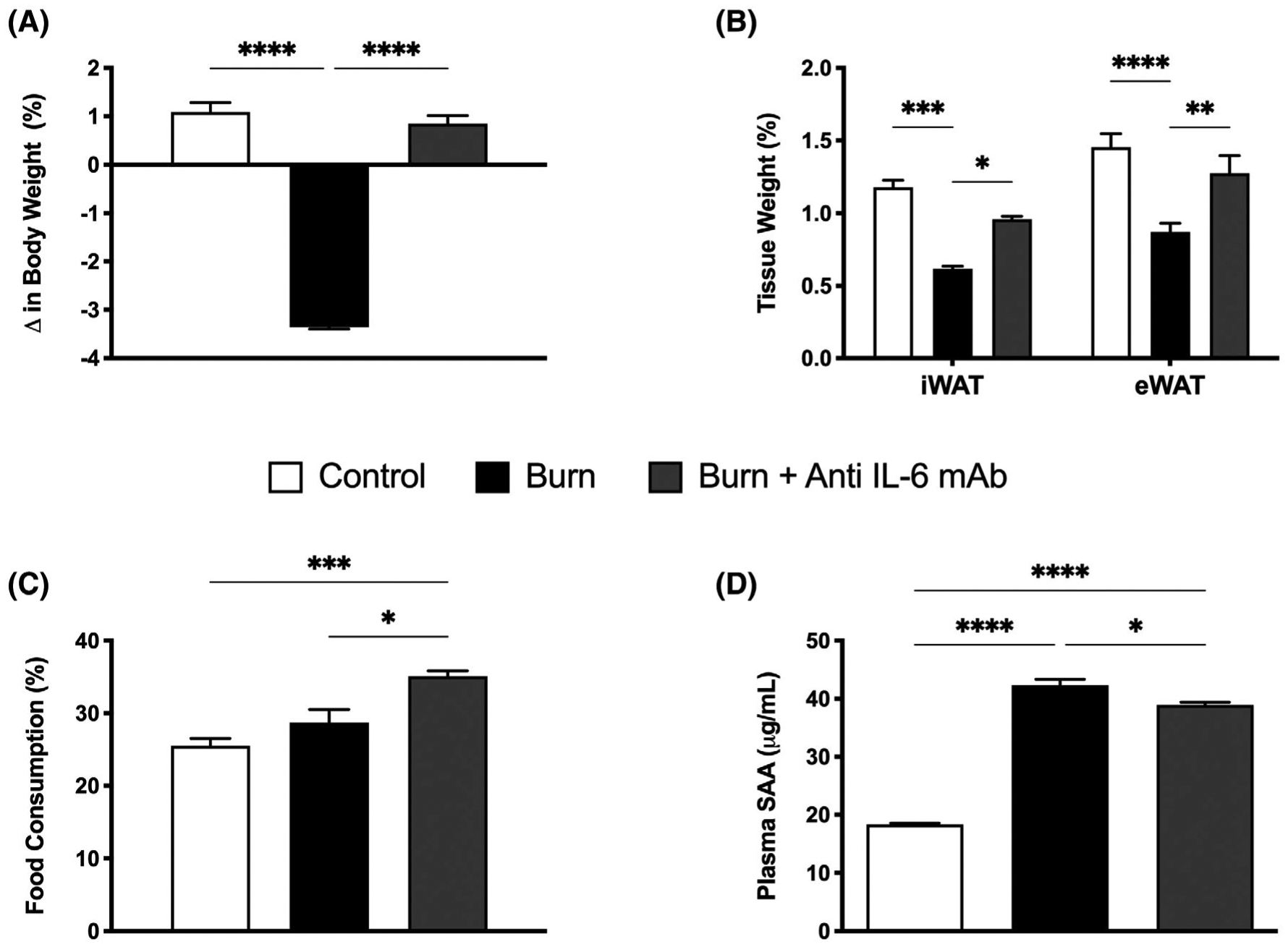
Effect of anti-IL-6 mAb treatment on post-burn body weight and composition. A, Change in total body weight in post-burn mice treated with vehicle or anti-mouse IL-6R monoclonal antibody daily (anti-IL-6 mAb) daily for 7 days. B, Adipose tissue weights expressed as a percentage of body weight demonstrating alterations in inguinal WAT (iWAT) and epidydimal WAT (eWAT). C, Increased dietary intake as a percentage of total food consumed in anti-IL-6-treated burn mice compared with controls and untreated burn mice. D, Increased circulating levels of serum amyloid A (SAA) in untreated burn mice relative to control and anti-IL-6-treated burn mice. Values are presented as mean ±standard error. *P < .05; **P < .01; ***P < .001; ****P < .0001, n = 7/group
3.2 |. IL-6 receptor blockade mitigates burn-induced beige adipocyte formation and lipolysis
Thus far having shown that anti-IL-6 mAb attenuated the adverse systemic hypermetabolic response after in burn mice, we wanted to determine whether this drug’s protective effects extended to key metabolic organs critical to post-burn outcomes. Our group and others have previously reported WAT browning as one of the key metabolic responses to a burn injury, and that genetic deletion of IL-6 protects mice from this adverse response.10 Therefore, we hypothesized that the IL-6 blocker would similarly attenuate adipose browning and its associated wasting. To investigate this, we performed immunohistochemical staining for the key browning gene UCP-1 in the subcutaneous iWAT depot from control, burn, and burn mice injected with anti-IL-6 mAb for 7 days. As illustrated in Figure 2A, histological examination of the iWAT of untreated burned mice at 7 days showed the presence of multilocular UCP-1 + adipocytes—key indicators of the phenotypic switch from white to beige adipose. Consistent with our hypothesis, post-burn anti-IL-6 mAb treatment effectively mitigated this pathological browning response, indicated by diminished UCP-1 protein staining and decreased presence of multilocular cells in the iWAT of treated mice at the same time point. This was further corroborated by our gene expression findings, which showed significantly decreased expression of several key browning genes including UCP-1 (231.7 vs 2992.5, P < .0001 for UCP-1; 0.40 vs 9.09, P < .0001 for CIDEA; 0.19 vs 2.18, P < .05 for PGC1⍺; 0.40 vs 2.31, P < .05 for PPARγ) after post-burn anti-IL-6 treatment (Figure 2B). Consistent with reduced WAT browning and remodeling, treated mice also maintained a larger lipid droplet size at 7 days post-injury (41.4 vs 15.3 μm, P < .0001) (Figure 2C). In fact, intracellular IL-6 levels were also substantially lower in adipocytes of anti-IL-6-treated burn mice at 7 days post-injury (194.1 vs 410.3 pg/μg of protein, P < .05) (Figure 2D). Norepinephrine, a key hormone involved in activating WAT browning, was also decreased in burn mice after anti-IL-6 mAb treatment, further corroborating an inhibitory effect of IL-6 blockade on the adipose browning process (48.73 vs 84.16 pg/μg of protein, P < .01) (Figure 2E).
FIGURE 2.
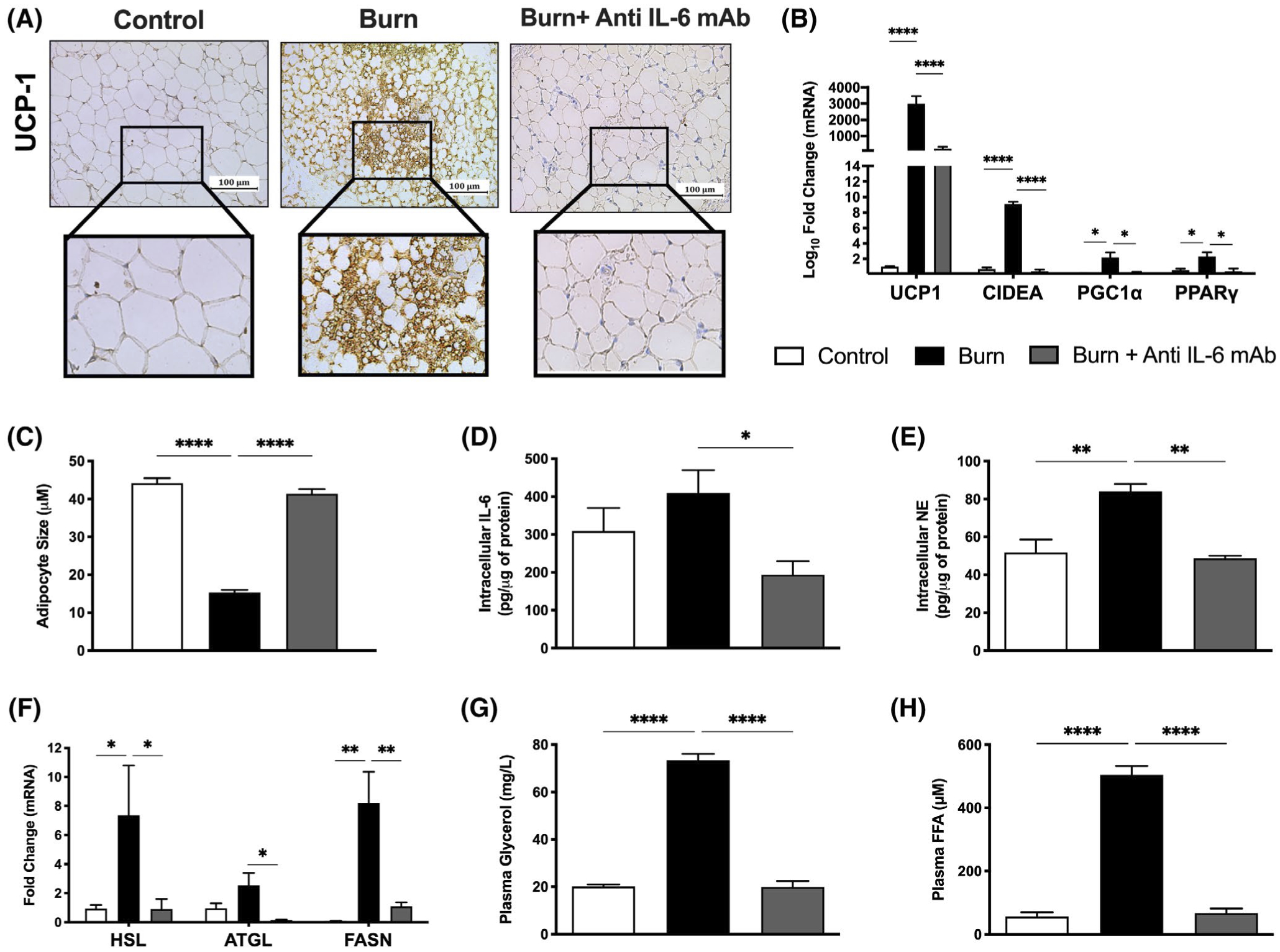
IL-6 receptor blockade mitigates burn-induced browning and lipolysis. A, Immunohistochemical staining for Uncoupling protein 1 (UCP1) in inguinal WAT isolated from control, untreated and anti-IL-6-treated burn mice at 7 days post-injury. B, Quantitative RT-PCR analysis of browning genes in inguinal WAT at 7 days post-injury. C, Post-burn changes in adipocyte diameter (n = 150 for anti-IL-6-treated burn mice, n = 241 for untreated burn mice) at 7 days post-injury. D, Intracellular IL-6 and (E) norepinephrine levels in inguinal WAT at 7 days post-injury. F, Quantitative RT-PCR analysis of genes involved in lipid metabolism in inguinal WAT at 7 days post-injury. G, Plasma free fatty acid (FFA) and (H) glycerol concentrations at 7 days post-injury. Values are presented as mean ±standard error. *P < .05; **P < .01; ****P < .0001, n = 7/group
Given that burn-induced browning is associated with an increased lipolytic state, we next assessed whether administering the IL-6 blocker had any effect on lipid metabolism by profiling the expression of key genes involved in lipid synthesis and breakdown (Figure 2F). Compared to untreated burn mice, anti-IL-6-treated mice exhibited lower gene expression of hormone sensitive lipase (HSL), adipose triglyceride lipase (ATGL) and fatty acid synthase (FASN) at 7 days post-injury (0.90 vs 7.36, P < .05 for HSL, 0.15 vs 2.54, P < .05 for ATGL, 1.10 vs 8.21, P < .01 for FASN). To confirm the metabolic significance of this reduced expression, we compared circulating levels of glycerol and free fatty acids (FFA), the key metabolic by-products of fat catabolism (Figure 2G,H). Surprisingly, post-burn treatment restored plasma glycerol and FFA concentrations back to baseline levels, indicating it also effectively suppressed elevated rates of lipolysis after injury (19.9 vs 73.4 mg/L, P < .0001 for glycerol; 67.3 vs 56.0 μM, P < .0001 for FFAs). Overall, these results provide compelling evidence that blocking IL-6 signaling with anti-IL-6 mAb likely attenuates systemic post-burn hypermetabolism by specifically targeting key browning enzymes in the iWAT, ultimately suppressing lipolysis and the excessive release of FFAs, thereby preventing whole-body wasting after injury.
3.3 |. Anti-IL-6 mAb-mediated inhibition of browning blocks the development of post-burn hepatic steatosis
Another key consequence of the post-burn hypermetabolic response includes browning-induced lipotoxicity and its impact on essential organs, such as the liver and kidney. Indeed, we recently implicated WAT browning and the associated efflux of lipids during this remodeling period in facilitating ectopic lipid deposition and dysfunction in the liver.12 Sadly, hepatic steatosis, or fatty liver, remains a leading cause of morbidity and mortality in burn patients.20 Given that post-burn IL-6 receptor blockade suppressed the pathological browning response in post-burn mice, we next sought to determine if anti-IL-6 mAb could also counteract its adverse lipotoxic effects on the liver after injury. To assess this, we examined liver sections from control, burn, and burn mice treated with anti-IL-6 mAb using Oil Red O staining for lipid droplets. As shown in Figure 3A, the livers of untreated burn mice showed both increased lipid infiltration and fat deposition at 7 days post-burn, as demonstrated by increased Oil Red O staining relative to control mice. In contrast, treatment with anti-IL-6 mAb effectively minimized the extent of ectopic fat accumulation in post-burn mice, which was further confirmed by the decrease in liver weight at this time point (4.68% vs 5.73%, P < .01; Figure 3B). Indeed, hepatic triglyceride content was also significantly reduced in anti-IL-6-treated burn mice relative to the untreated burn group after injury (247.2 vs 118.4 mg/dL, P < .01) (Figure 3C). Given that increased lipid accumulation in the liver is associated with augmented apoptosis and ER stress, we next assessed the effects of post-burn anti-IL-6 mAb treatment on key markers of hepatic damage. Consistent with our findings, we observed a robust activation of the ER stress markers Bip, CHOP, PERK, and IRE1a in the livers of mice at 7 days post-burn, which was not observed in the anti-IL-6-treated burn mice (4.21 vs 1.48, P < .05 for Bip; 7.35 vs 1.81, P < .0001 for PERK; 9.36 vs 1.64, P < .01 for CHOP; 7.49 vs 1.5, P < .0001 for IRE1a) (Figure 3D). To assess how this altered liver functions after injury, we then measured circulating levels of hepatic damage markers, alanine transaminase (ALT) and aspartate aminotransferase (AST). As expected, both ALT and AST levels were drastically elevated in untreated burn mice indicating organ dysfunction and the activation of hepatic apoptotic and regenerative pathways after severe injury (Figure 3E,F). Remarkably, anti-IL-6-treated mice showed decreased plasma ALT and AST levels at 7 days post-burn (6821.8 U/I vs 7422.2 U/I P < .05 for AST, 82.2 U/I vs 15.5 U/I, P = .678), suggesting that the IL-6 blocker effectively mitigated the effects of lipid-induced hepatic dysfunction. Moreover, the changes we observed in markers of organ damage were limited to the liver, as circulating levels of the kidney damage marker, creatinine, was unaffected both by the treatment and burn injury (Figure 2). These results are consistent with our previous findings that inhibition of IL-6 signaling either through genetic knock out or in this case through anti-IL-6 mAb treatment, has the synergistic effect of both attenuating WAT browning and hepatic steatosis to minimize post-burn hypermetabolism.
FIGURE 3.
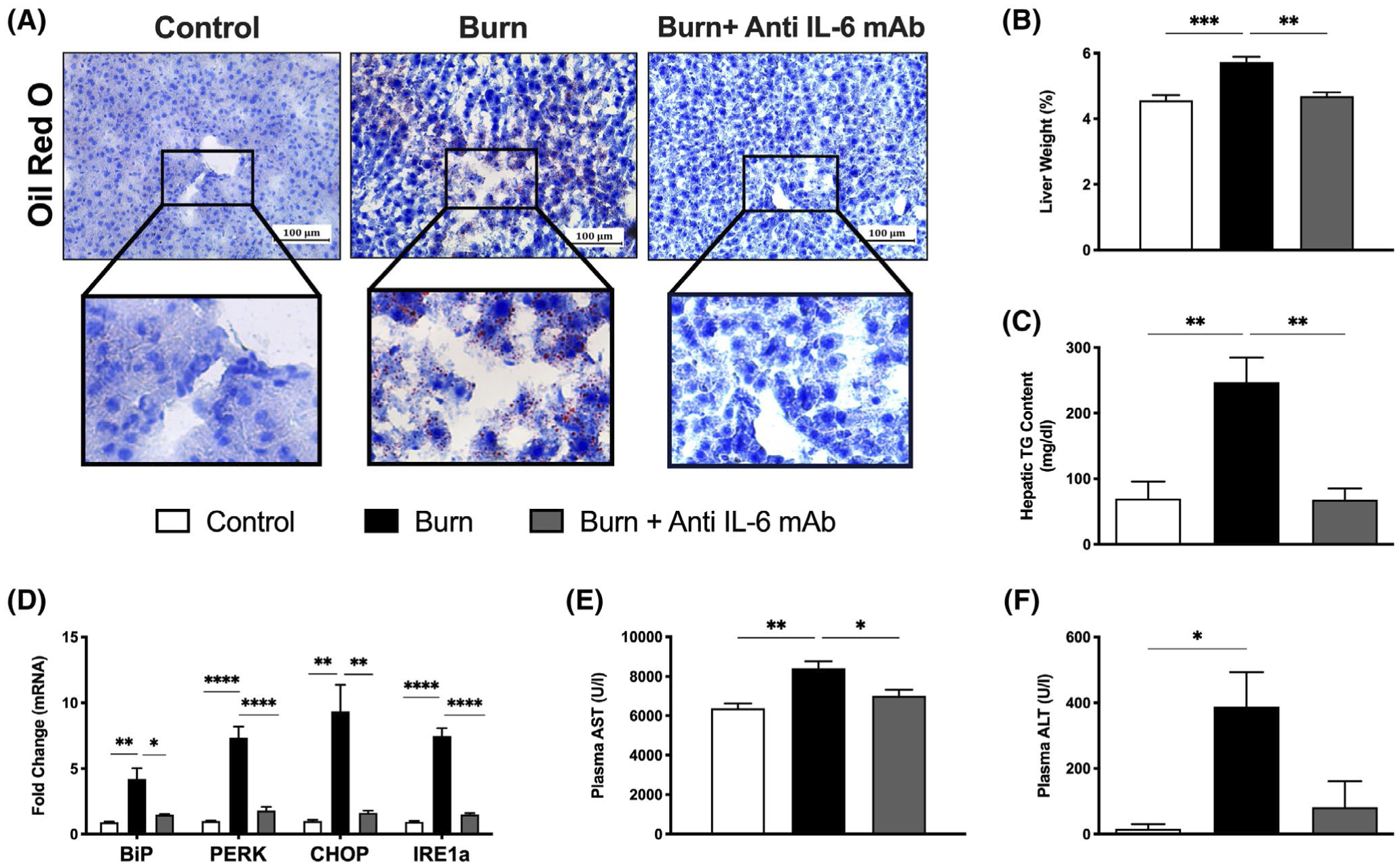
Anti-IL-6 mAb-mediated inhibition of browning attenuates lipid-induced hepatic dysfunction. A, Oil Red O staining for lipid droplets in liver sections from control, untreated, and anti-IL-6-treated burn mice at 7 days post-injury. B, Liver weights normalized to body weight at 7 days post-injury. C, Liver triglyceride (TG) content at 7 days post-injury. D, Quantitative RT-PCR analysis of key ER stress gene markers in livers from control, untreated, and anti-IL-6-treated burn mice. E, Plasma aspartate transaminase (AST) and (F) alanine aminotransferase (ALT) concentrations at 7 days post-injury. Values are presented as mean ±standard error. *P < .05; **P < .01; ***P < .001; ****P < .0001, n = 7/group
3.4 |. Inhibition of IL-6 signaling via anti-IL-6 mAb after a burn injury exerts antifibrotic effects in the skin
The complications of persistent burn-induced hypermetabolism and hyperinflammation extend far beyond the adipose and liver, but also impede skin wound healing after injury. Indeed, IL-6 is critical for the switch of the healing wound bed from a hyperinflammatory state to a reparative state. In fact, dysregulated IL-6 signaling has been shown to lead to a constitutively active proliferative phase, increased collagen deposition, and hypertrophic scar formation.21 Unfortunately, up to 70% of burn patients now develop hypertrophic scars that are disfiguring, limit normal movement, and prevent patient recovery.22 Therefore, we next wanted to elucidate whether blocking IL-6 signaling exerts beneficial effects on aberrant wound healing responses post-burn injury. As illustrated in Figure 4A, we first profiled the expression of pro-fibrotic growth factors and found they were drastically upregulated in skin excised from untreated burned mice even at 14 days post-injury. Compared to control mice, burned mice showed elevated expression of VEGF and FGF2, key regulators of extracellular matrix deposition and fibroblast proliferation. This was also accompanied by an increase in TGF-β expression, a critical mediator of fibroblast differentiation, collagen deposition and eventually, scar formation in the wound bed. Surprisingly, gene expression of these pro-fibrotic markers was substantially reduced in skin excised from anti-IL-6-treated burn mice at this time point (0.17 vs 7.29, P < .05 for FGF2; 0.43 vs 1.95, P < .05 for TGF-β; 0.46 vs 4.02, P < .05 for VEGF). This is noteworthy as previous studies have demonstrated that full wound healing is completed by day 14 in burn mice,23 therefore persistent elevations in growth factors post-injury likely suggests an abnormal response that can potentially lead to skin fibrosis and scar formation if left unmitigated.
FIGURE 4.
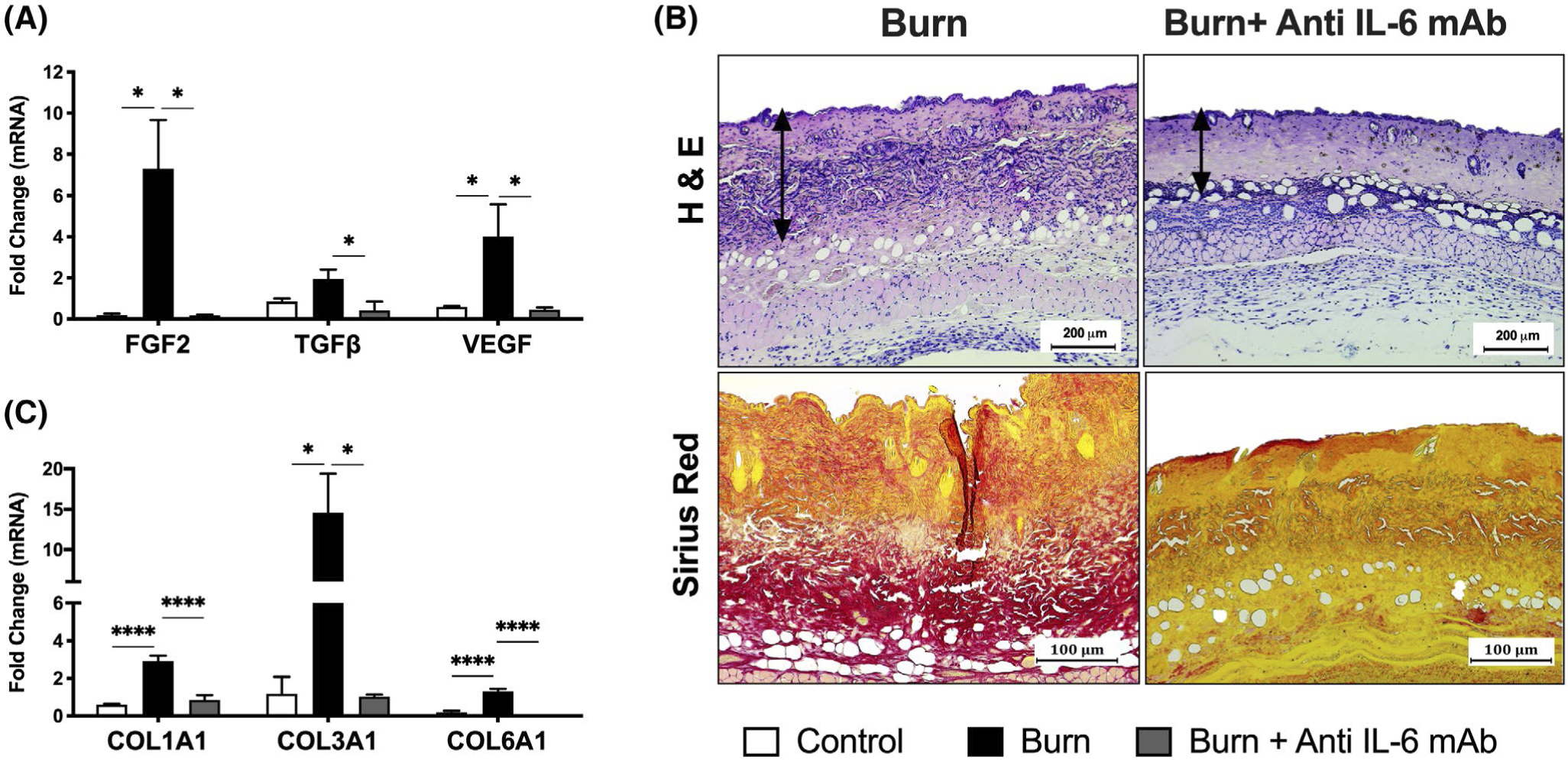
Blocking IL-6 signaling via anti-IL-6 mAb post-burn injury exerts antifibrotic effects in the skin. A, H&E (top panel) and picrosirius red staining for collagen distribution (bottom panel) in skin excised from control, untreated, and anti-IL-6-treated burn mice at 14 days post-injury. C, Quantitative RT-PCR analysis of pro-fibrotic growth factors in skin excised from control, untreated, and anti-IL-6-treated burn mice at 14 days post-injury. D, Quantitative RT-PCR analysis of collagen genes in skin excised from control, untreated, and anti-IL-6-treated burn mice at 14 days post-injury. Values are presented as mean ±standard error. *P < .05; ***P < .001; ****P < .0001, n = 7/group
Indeed, differences in dermal collagen distribution between the two burn groups were demonstrated histologically and further confirmed with picrosirius red staining for collagen fibers. As shown in Figure 4B, picrosirius red staining revealed an excess accumulation of collagen in skin excised from burn mice at 14 days post-injury, which was not observed in anti-IL-6-treated burn mice. This was corroborated by our PCR data, which demonstrated significantly decreased gene expression of collagen types 1, 3, and 6 in the skin of anti-IL-6-treated relative to untreated burn mice after injury (2.93 vs 0.853, P < .0001 for Col1A1; 14.58 vs 1.03, P < .05 for Col3A1; 1.29 vs 0.0041, P < .0001 for Col6A1) (Figure 4C). Taken together, we show that anti-IL-6 mAb treatment can effectively control hyperactive skin fibroblasts after burn injury by modulating levels of key pro-fibrotic growth factors and collagen distribution. Given that fibroblast proliferation, growth factor production, and collagen synthesis play a central role in wound healing, these findings suggest that pharmacological inhibition of IL-6 signaling in the acute phase of post-burn care should perhaps be carefully tempted. Nevertheless, delayed administration of the IL-6R antagonist may prove beneficial for halting pathologic skin fibrosis and scarring after severe injury.
4 |. DISCUSSION
Over the last three decades, there has been a lack of major breakthroughs in burn care treatments despite intensive investigation of the hypermetabolic response and its mechanisms.1 Genetic predisposition, environmental factors and complicated interactions between the immune system and key immunometabolic organs all contribute to the development of post-burn hypermetabolism—a long lasting stress response characterized by hyperglycemia, increased lipid and protein catabolism causing significant increases in resting energy expenditure.2 Unfortunately, prolonged hypermetabolism becomes detrimental and is associated with vast catabolism, multi-organ failure, and death. While great strides have been made in dealing with components of this hypermetabolic response, such as insulin to treat burn-induced hyperglycemia and propranolol to improve cardiac function, burn patients still experience unacceptably higher rates of morbidity and mortality. It is now appreciated that the post-burn hypermetabolic response is more complex than previously once thought, and that new therapeutic agents that target the alterations that occur in keyorganslike the adipose tissue and liver have are needed to further improve outcomes for burn patients.
Indeed, it was recently discovered that a key feature of the hypermetabolic response to burn injury is WAT browning, a process by which the adipose tissue undergoes vast morphological and lipolytic changes to dissipate energy in the form of heat. Generally, the former is thought to promote the latter, and vice versa, establishing adipose tissue as a critical post-burn driver of whole body energy metabolism.3 With the characterization of several animal models, investigators have highlighted a critical role for bone marrow derived IL-6 in mediating post-burn WAT browning.10,11 Increased levels of this inflammatory cytokine have been shown to correlate with the magnitude of injury and organ failure, and genetic deletion of IL-6 in mice attenuates post-burn hypermetabolism via reductions in WAT browning and whole-body energy metabolism.4,12 Despite all these findings that have implicated both WAT browning and IL-6 as major drivers of post-burn hypermetabolism, therapeutic agents that target these responses have lagged behind. Thus, we reasoned that inhibition of post-burn IL-6 signaling using an anti-IL-6 monoclonal antibody would have a more potent effect in altering post-burn hypermetabolism through its effects on both the WAT and liver. We have taken several steps to address this hypothesis in this study.
First, we studied the effect of anti-IL-6 mAb treatment on post-burn WAT browning and lipid metabolism. Administration of anti-IL-6 mAb in post-burn mice reduced the production of IL-6 and its downstream target of SAA, confirming blockade of IL-6 signaling. Post-burn anti-IL-6 mAb treatment also protected mice from body weight loss and adipose wasting relative to untreated burn mice. WAT browning and the associated increased lipid metabolism have been recognized as one of the most important drivers of increased resting energy expenditure and the associated wasting in burns.24 In line with protection against burn-induced wasting, anti-IL-6 mAb suppressed WAT browning and the corresponding increase in lipolysis in burned mice. Consistent with our findings, a recent study in cancer which evokes a similar hypermetabolic response to burns, demonstrated that IL-6 signaling blockade protected mice from cachexia via inhibition of WAT browning.25 Additionally, another study showed anti-IL-6 treatment in advanced non-small cell lung cancer patients attenuated cancer-induced cachexia and recovered body weight in these patients.26 However, our study is the first to characterize the effects of anti-IL-6 mAb treatment on post-burn hypermetabolism and WAT browning. We hope these findings spur efforts to further study the therapeutic potential of anti-IL-6 interventions for use clinically in burn patients to inhibit burn-induced cachexia and wasting.
Another important aspect of burn-induced WAT browning, and remodeling is that it induces excessive adipose lipolysis that is mobilized into the liver. Indeed, increased fat infiltration and hepatomegaly are two widely reported complications of the hypermetabolic response to burn injury.20 Furthermore, increased hepatic steatosis and impairments in liver function have been correlated with increasing mortality risk from 6% (total population) to 27% in burn patients.27 In the present study, anti-IL-6-treated burned mice showed reductions in plasma glycerol and FFAs relative to non-treated burned mice. Consistent with these findings, anti-IL-6-treated mice were also protected from burn-induced hepatic steatosis as their livers showed reductions in both fat infiltration and TG content relative to untreated burned mice. We and others have previously shown that fat infiltration in the liver if left unabated can induce ER stress mediated hepatic apoptosis and damage.28 Remarkably, anti-IL-6-treated burned mice also showed reductions in key markers of ER stress and liver damage, suggesting that the beneficial effects of anti-IL-6 mAb were not limited to just inhibiting lipid infiltration. These hepatic findings regarding IL-6 blockade might seem contradictory in the context of studies that have associated IL-6 signaling in accelerating liver regeneration.29 For instance, IL-6−/−mice have severe liver necrosis which can be ameliorated with the addition of recombinant IL-6 and activation of the anti-apoptotic factor Mcl-1.30 To that effect, it’s feasible that the administration and the dose of anti-IL-6 mAb we used in this study was both beneficial in mitigating changes to adipose morphology post-burn, while keeping other IL-6 related pathways involved in liver regeneration intact.
The chronic hyperinflammatory and hypermetabolic response that ensues after a burn injury has also been implicated in skin fibrotic complications like hypertrophic scars and keloids, which affect more than 70% of the burn patient population.22 Normal wound healing is characterized by three distinct phases: inflammation, proliferation, and remodeling.8 Dysfunction and adverse modulation of these three phases can result in a switch from normal skin healing without a scar to excessive skin fibrosis.8 Burn patients are especially prone to skin fibrosis, as burn injuries activate deep dermal fibroblasts that are larger in size, proliferate slower, and produce large quantities of collagen and inflammatory cytokines.22 At the center of all these fibrotic changes in the wound bed is IL-6, which has been shown to regulate the differentiation of fibroblasts to myofibroblasts and its maintenance in hypertrophic scars.31 Particularly, IL-6 signaling has been shown to regulate the expression of both TGF-β and VEGF, key regulators of Type-1 collagen synthesis, fibroblast proliferation, and extracellular matrix deposition and persistence in the wound bed.31 In this study we found that relative to burned mice, anti-IL-6-treated burned mice showed decreased expression of TGF-β and VEGF in the skin. In corroboration of these findings, skin from anti-IL-6-treated post-burn mice also showed reduced expression of collagen subtypes 1,3, and 6, which have been implicated in skin fibrosis. These two key effects of anti-IL-6 mAb treatment are significant because hallmarks of burn skin fibrosis (hypertrophic scars, keloids) are increased TGF-β mediated perturbed collagen production, and increased VGEF expression to sustain fibroproliferative growth. In fact, similar to our findings an RCT in patients with systemic sclerosis found that subcutaneous skin anti-IL-6 mAb injections effectively reversed TGF-β-induced molecular and genetic alterations in systemic sclerosis fibroblasts that results in fibrosis.32 Paradoxically, intact IL-6 signaling has also been shown to be critical in wound healing of diabetic and obese patients, with inhibition of IL-6 signaling or genetic KO of the IL-6 gene resulting in delayed and poorly healed wounds.33 In this regard, the injury etiology might provide clues as to why in diabetes IL-6 blockade impairs wound healing, while in systemic sclerosis and burns it mitigates skin fibrosis. Unlike in diabetes and obesity, both burns and systemic sclerosis are characterized by hyperactive IL-6 signaling and distinct deep dermal wound injuries that make them prone to skin fibrosis.34 However, given that mice primarily heal through wound contraction making them less prone to post-burn fibrosis, further studies are needed using an animal model similar to human skin architecture and healing to fully study the effects of IL-6 inhibition on this process.
In summary, the data in this study clearly show that inhibition of IL-6 signaling through anti-IL-6 mAb has beneficial effects on several organs to attenuate many of the pathological alterations caused by the post-burn hypermetabolic response. We hope this study spurs further efforts to study this drug more in depth and in the context of other hypermetabolic conditions like cancer. While the potential for sex differences to affect outcomes following burn injury has not been well explored, a limitation of this study is that only male mice were used. This can be rectified in future studies with female mice to ensure IL-6 blockade has no detrimental impacts on organ function and survival. Future studies should also consider the plethora of other organs regulated by IL-6 signaling including cardiac and muscle tissue, targets not covered in this study. Notwithstanding this limitation, the positive effects of anti-IL-6 mAb on the adipose and liver are still important as these two organs have been implicated as major drivers of post-burn outcome relative to the others. Furthermore, whether the effects of anti-IL-6 mAb on the key regulators of skin healing are protective against fibrosis or are detrimental to wound healing will have to be further experimentally tested—specifically in a more clinically relevant animal model that mimics the complexities and intricacies of human skin wound healing. Thus, it is likely that resolving and addressing all the pathologies associated with the hypermetabolic response to burns will require a combination of therapeutic agents and thus other drugs in conjunction with anti-IL-6 agents should be further explored.
Supplementary Material
ACKNOWLEDGMENTS
AA is a recipient and supported by the Banting Postdoctoral Fellowships program through the Canadian Institutes of Health Research. RV is a recipient of the Frederick Banting and Charles Best Canada Graduate Scholarship (CGS-D). This work was supported by grants from the Canadian Institutes of Health Research (#123336), the Canada Foundation for Innovation Leaders Opportunity Fund (#25407) and National Institutes of Health (2R01GM087285-05A1).
Funding information
This study was supported by Canadian Institutes of Health Research: #123336; CFI Leaders Opportunity Fund: Project #25407; NIH: R01GM087285, 5R01GM133961-02
Abbreviations:
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ATGL
adipose tissue triglyceride lipase
- BAT
brown adipose tissue
- BiP
immunoglobulin heavy chain-binding protein
- CHOP
C/EBP homologous protein
- CIDEA
cell death-inducing DNA fragmentation factor-alpha-like effector A
- Col1A1
collagen type I α1 chain
- Col3A1
collagen type III α1 chain
- Col6A1
collagen type VI α1 chain
- ER
endoplasmic reticulum
- eWAT
epididymal white adipose tissue
- FASN
fatty acid synthase
- FFA
free fatty acids
- FGF2
fibroblast growth factor-2
- HSL
hormone-sensitive lipase
- IL-6
interleukin-6
- IRE1α
inositol-requiring enzyme 1α
- iWAT
inguinal white adipose tissue
- mAb
monoclonal antibody
- NE
norepinephrine
- PERK
protein kinase R-like endoplasmic reticulum kinase
- PGC1α
peroxisome proliferator-activated receptor-γ coactivator 1-α
- PPARγ
peroxisome proliferator-activated receptor γ
- RT-PCR
real-time polymerase chain reaction
- SAA
serum amyloid A
- TBSA
total body surface area
- TG
triacylglycerides
- TGF-β
transforming growth factor β
- UCP-1
uncoupling protein-1
- VEGF
vascular endothelial growth factor
- WAT
white adipose tissue
Footnotes
CONFLICT OF INTEREST
The authors have no competing financial interests to declare.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Jeschke MG, Gauglitz GG, Kulp GA, et al. Long-term persistence of the pathophysiologic response to severe burn injury. PLoS One. 2011;6:e21245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brigham PA, McLoughlin E. Burn incidence and medical care use in the United States: estimates, trends, and data sources. J Burn Care Rehabil. 1996;17:95–107. [DOI] [PubMed] [Google Scholar]
- 3.Abdullahi A, Jeschke MG. White adipose tissue browning: a double-edged sword. Trends Endocrinol Metab. 2016;27:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeschke MG, Gauglitz GG, Finnerty CC, et al. Survivors versus nonsurvivors postburn: differences in inflammatory and hypermetabolic trajectories. Ann Surg. 2014;259:814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hager S, Foldenauer AC, Rennekampff HO, et al. Interleukin-6 serum levels correlate with severity of burn injury but not with gender. J Burn Care Res. 2018;39:379–386. [DOI] [PubMed] [Google Scholar]
- 6.Biffl WL, Moore EE, Moore FA, Peterson VM. Interleukin-6 in the injured patient. Marker of injury or mediator of inflammation? Ann Surg. 1996;224:647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gebhard F, Pfefsch H, Steinbach G, Strecker W, Kinzl L, Bruckner UB. Is interleukin 6 an early marker of injury severity following major trauma in humans? Arch Surg. 2000;135:291–295. [DOI] [PubMed] [Google Scholar]
- 8.Rose-John S, Winthrop K, Calabrese L. The role of IL-6 in host defence against infections: immunobiology and clinical implications. Nat Rev Rheumatol. 2017;13:399–409. [DOI] [PubMed] [Google Scholar]
- 9.Yeh FL, Lin WL, Shen HD, Fang RH. Changes in circulating levels of interleukin 6 in burned patients. Burns. 1999;25:131–136. [DOI] [PubMed] [Google Scholar]
- 10.Abdullahi A, Chen P, Stanojcic M, Sadri AR, Coburn N, Jeschke MG. IL-6 signal from the bone marrow is required for the browning of white adipose tissue post burn injury. Shock. 2017;47:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdullahi A, Auger C, Stanojcic M, et al. Alternatively activated macrophages drive browning of white adipose tissue in burns. Ann Surg. 2019;269:554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdullahi A, Samadi O, Auger C, et al. Browning of white adipose tissue after a burn injury promotes hepatic steatosis and dysfunction. Cell Death Dis. 2019;10:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzik TJ, Skiba DS, Touyz RM, Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc. Res 2017;113:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villarroya F, Cereijo R, Gavalda-Navarro A, Villarroya J, Giralt M. Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J Intern Med. 2018;284:492–504. [DOI] [PubMed] [Google Scholar]
- 15.Petruzzelli M, Schweiger M, Schreiber R, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014;20:433–447. [DOI] [PubMed] [Google Scholar]
- 16.Barayan D, Vinaik R, Auger C, Knuth CM, Abdullahi A, Jeschke MG. Inhibition of lipolysis with acipimox attenuates postburn white adipose tissue browning and hepatic fat infiltration. Shock. 2020;53:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6:a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohsugi Y Recent advances in immunopathophysiology of interleukin-6: an innovative therapeutic drug, anti-IL-6 mAb (recombinant humanized anti-human interleukin-6 receptor antibody), unveils the mysterious etiology of immune-mediated inflammatory diseases. Biol Pharm Bull. 2007;30:2001–2006. [DOI] [PubMed] [Google Scholar]
- 19.Mihara M, Ohsugi Y, Kishimoto T. Anti-IL-6 mAb, a humanized anti-interleukin-6 receptor antibody, for treatment of rheumatoid arthritis. Open Access Rheumatol. 2011;3:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeschke MG. The hepatic response to thermal injury: is the liver important for postburn outcomes? Mol Med. 2009;15:337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kondo T, Ohshima T. The dynamics of inflammatory cytokines in the healing process of mouse skin wound: a preliminary study for possible wound age determination. Int. J. Legal Med 1996;108:231–236. [DOI] [PubMed] [Google Scholar]
- 22.Finnerty CC, Jeschke MG, Branski LK, Barret JP, Dziewulski P, Herndon DN. Hypertrophic scarring: the greatest unmet challenge after burn injury. Lancet. 2017;388:1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdullahi A, Amini-Nik S, Jeschke MG. Animal models in burn research. Cell Mol Life Sci. 2014;71:3241–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sidossis LS, Porter C, Saraf MK, et al. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab. 2015;22:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narsale AA, Carson JA. Role of interleukin-6 in cachexia: therapeutic implications. Curr Opin Support Palliat Care. 2014;8: 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ando K, Takahashi F, Kato M, et al. Anti-IL-6 mAb, a proposed therapy for the cachexia of Interleukin6-expressing lung cancer. PLoS One. 2014;9:e102436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price LA, Thombs B, Chen CL, Milner SM. Liver disease in burn injury: evidence from a national sample of 31,338 adult patients. J Burns Wounds. 2007;12:e1. [PMC free article] [PubMed] [Google Scholar]
- 28.Abdullahi A, Barayan D, Vinaik R, Diao L, Yu N, Jeschke MG. Activation of ER stress signalling increases mortality after a major trauma. J Cell Mol Med. 2020;24:9764–9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters HPF, Wiersma WC, Akkermans LMA, et al. Gastrointestinal mucosal integrity after prolonged exercise with fluid supplementation. Med Sci Sports Exerc. 2000;32:134–142. [DOI] [PubMed] [Google Scholar]
- 30.Cressman DE, Greenbaum LE, DeAngelis RA, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–1383. [DOI] [PubMed] [Google Scholar]
- 31.Gallucci RM, Sugawara T, Yucesoy B, et al. Interleukin-6 treatment augments cutaneous wound healing in immunosuppressed mice. J Interferon Cytokine Res. 2001;21:603–609. [DOI] [PubMed] [Google Scholar]
- 32.Koch AE, Kronfeld-Harrington LB, Szekanecz Z, et al. In situ expression of cytokines and cellular adhesion molecules in the skin of patients with systemic sclerosis. Their role in early and late disease. Pathobiology. 1993;61:239–246. [DOI] [PubMed] [Google Scholar]
- 33.Duncan MR, Berman B. Stimulation of collagen and glycosaminoglycan production in cultured human adult dermal fibroblasts by recombinant human interleukin 6. J Invest Dermatol. 1991;97:686–692. [DOI] [PubMed] [Google Scholar]
- 34.Grossman RM, Krueger J, Yourish D, et al. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci. 1989;86:6367–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.