

Abstract
Aims:
The causes of distinct patterns of reduced cortical thickness in the common human epilepsies, detectable on neuroimaging and with important clinical consequences, are unknown. We investigated the underlying mechanisms of cortical thinning using a systems-level analysis.
Methods:
Imaging-based cortical structural maps from a large-scale epilepsy neuroimaging study were overlaid with highly spatially-resolved human brain gene expression data from the Allen Human Brain Atlas. Cell-type deconvolution, differential expression analysis and cell-type enrichment analyses were used to identify differences in cell-type distribution. These differences were followed up in post-mortem brain tissue from humans with epilepsy using Iba1 immunolabelling. Furthermore, to investigate a causal effect in cortical thinning, cell-type specific depletion was used in a murine model of acquired epilepsy.
Results:
We identified elevated fractions of microglia and endothelial cells in regions of reduced cortical thickness. Differentially-expressed genes showed enrichment for microglial markers, and in particular, activated microglial states. Analysis of post-mortem brain tissue from humans with epilepsy confirmed excess activated microglia. In the murine model, transient depletion of activated microglia during the early phase of the disease development prevented cortical thinning and neuronal cell loss in the temporal cortex. Although the development of chronic seizures was unaffected, the epileptic mice with early depletion of activated microglia did not develop deficits in a non-spatial memory test seen in epileptic mice not depleted of microglia.
Conclusions:
These convergent data strongly implicate activated microglia in cortical thinning, representing a new dimension for concern and disease modification in the epilepsies, potentially distinct from seizure control.
Keywords: MRI, cortical thinning, gene expression, post mortem
Introduction
Significant progress is being made in understanding disease processes in the epilepsies. Many genetic variants causing or associated with rare and common epilepsies have been reported [1,2], with discovery continuing apace [3]. Numerous direct structural causes of epilepsy have been revealed by brain MRI. Several structural abnormalities are now themselves known to have a genetic basis [4]. As a result, the proportion of causally-explicable epilepsies is growing rapidly. Conversely, the mechanisms whereby these identified causes promote epileptogenesis and seizures remain obscure for most human epilepsies. Moreover, beyond causation and epileptogenesis, the epilepsies involve many other processes: some lead to clinically-apparent consequences, such as developmental delay or memory dysfunction, whereas others, without necessarily obvious symptoms, may be detected only on investigation - cerebellar atrophy is one example. The natural history of any given epilepsy need not be a single linear dynamic from causation to a unique, predictable, final outcome: for example, the epilepsies are associated with shortened longevity (even if seizures stop) [5] and increased risk of particular comorbidities [6]. Known causes per se may not explain all the observed outcomes, suggesting that many epilepsies could be conceptualised as intricate matrices of causation, processes and outcomes [7], with complex inter-dependencies, such as a likely link between reduction in cortical thickness and disease duration [8].
Through the ENIGMA-Epilepsy consortium, we recently showed that across a wide range of common human epilepsies, which are known to have both distinct and shared genetic architecture [2,3,9,10], there are also shared, pan-syndrome, and distinct, syndrome-specific, regional patterns of altered cortical thickness and altered subcortical grey matter volumes [8]. The causes of the structural changes in these epilepsies are not known. The findings suggest structural losses may reflect an initial insult, subsequent epileptogenesis or progressive neurodegeneration, or some combination, and show robustly that the common epilepsies cannot necessarily be considered entirely benign at the structural level. We sought to identify processes underlying the structural findings.
The pathophysiology of neurological disease has been successfully revealed using powerful combinations of brain MRI findings, regionalised brain-specific gene expression and gene co-expression networks, in a systems biology framework to implicate candidate genes [11–14]. Here, we used the findings from the in vivo ENIGMA-Epilepsy imaging study [8] in combination with the post mortem atlas of gene expression in the brain from healthy subjects, curated by the Allen Institute [15,16] to direct interrogation of regionalised brain cell-type composition and generic biological processes which might underlie thickness or volume reductions across the studied epilepsy syndromes. We hypothesised that this approach could suggest disease mechanisms causing the observed structural changes. We further explored the findings with a series of additional experiments in both human and animal tissues. We demonstrated experimentally in a murine model of acquired epilepsy that depletion of the implicated cell-type, microglia, can successfully avert cortical thinning and the concomitant neuronal cell loss and cognitive deficit, without modifying spontaneous seizures. Microglia have been shown to have various roles in a few rare, severe human epilepsies. Our new results implicate microglia in the widespread, but largely unstudied, reduction in cortical thickness that accompanies the numerically far more important common human epilepsies and point to the potential for prevention of such thinning by manipulation of microglia.
Materials and Methods
In order to explore mechanisms underlying cortical thinning in the epilepsies, we designed the study as shown in Fig. 1. We obtained statistical maps from a large structural MRI study comparing people with epilepsy to healthy controls conducted by the ENIGMA-Epilepsy consortium [8] (Table S1). To determine cell-type composition differences that spatially correlate with the reported structural changes on MRI, we used a healthy control dataset, the Allen Human Brain Atlas (AHBA), comprising densely-sampled gene expression across the cortex [15,16]. The statistical maps were mapped onto the AHBA using MNI-coordinates. This enabled us to investigate how the spatial distribution of gene expression in the healthy brain correlates with regional vulnerability in the diseased brain. Our primary hypothesis was that cell-types causing structural changes observed on human brain MRI could be identified using the design shown in Fig. 1A. Briefly, cortical brain regions were classified as vulnerable to, or relatively protected from, cortical thinning based on the previous imaging results [8]. Then two complementary approaches were used to identify cell-types with association to reduced cortical thickness: (i) AHBA microarray expression data were de-convolved into cell-type fractions and these fractions were tested for differences between brain regions; (ii) a standard differential expression analysis was carried out for each gene expression probe and cell-types were identified using enrichment analyses (for details on pre-processing, atlas mapping, quality control and the statistical approach see Supplementary Methods, section A). The analysis code for the gene expression analyses is available (https://github.com/andrealtmann/AHBA_Epilepsy).
Fig. 1. Analysis overview.
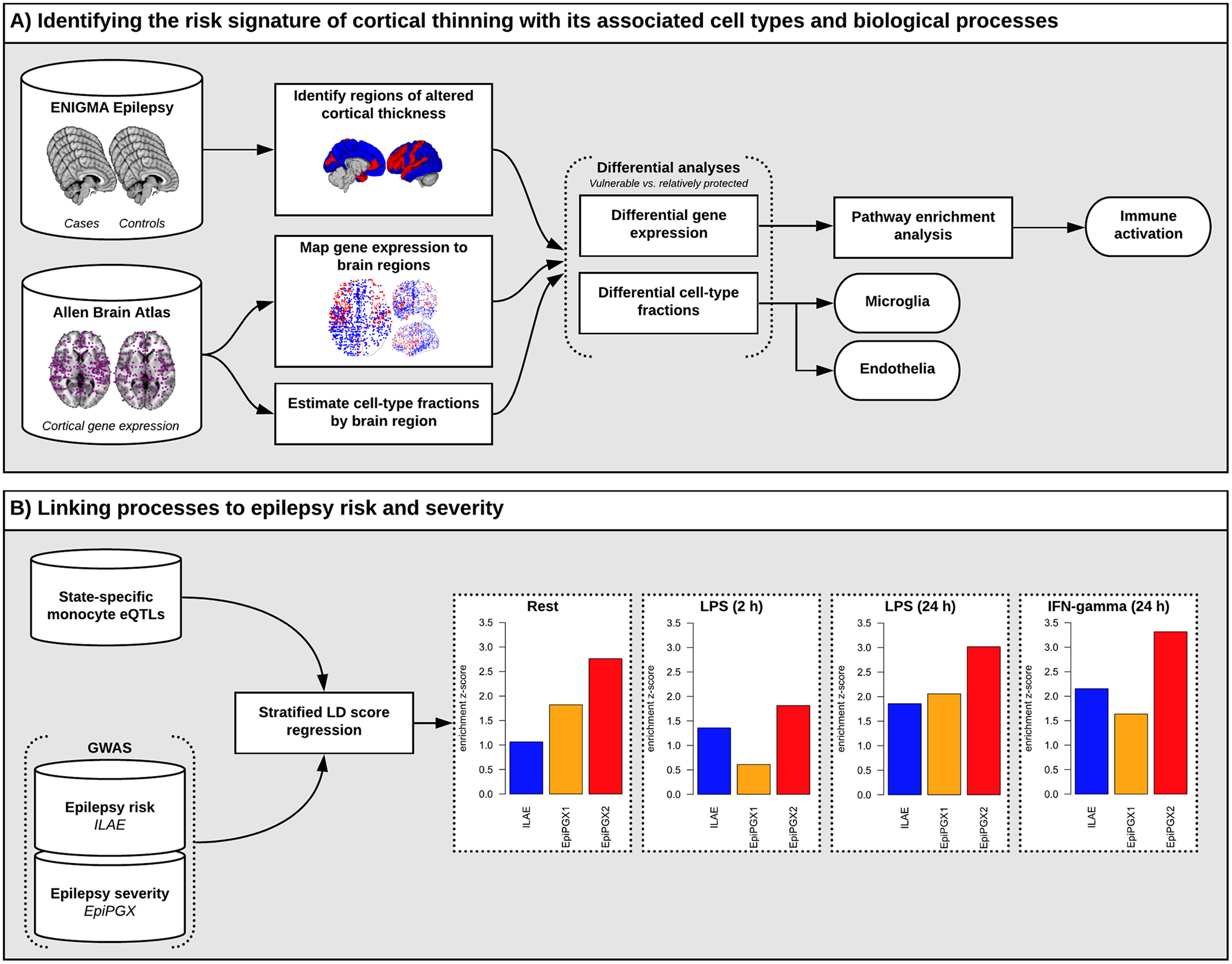
Panel A: The ENIGMA-Epilepsy study identified “vulnerable” and “relatively protected” brain regions indicated in red and blue, respectively (second column; top) [8]. Cortical samples of the AIBS dataset (purple dots; first column, bottom) were marked as either “vulnerable” (red dots) or “relatively protected” (blue dots) depending on their location (second column; middle). Brain cell-type fractions were estimated from the gene expression data and the differential analysis showed an increased fraction of microglia and endothelial cells in “vulnerable” compared to “relatively protected” regions. Differential gene expression analysis between the two groups followed by pathway analysis confirmed the enrichment for marker genes for microglia as well as immune activation related pathways.
Panel B: LD score regression estimating the enrichment of immune response eQTL signatures in different epilepsy GWAS finds strong enrichment in disease severity (drug-resistant vs drug-susceptible) but not in disease risk (cases vs controls).
We hypothesised that cell-types identified from the in silico analysis of data on the spatial distribution of gene expression in the brain from non-epileptic donors would also show compatible differences in spatial distribution in tissue samples obtained from people with epilepsy. Therefore, immunostaining of microglia in post mortem human brain tissue was carried out to substantiate the cell-type findings from the in silico work. The human post mortem cases were classified into different epilepsy groups and control groups according to clinical and pathology criteria. The final sample size was based on availability of tissues for certain epilepsies (as characterised by clinical data including EEG and MRI) and whether regional tissue paraffin-embedded block samples were available in each case from multiple brain regions (see Supplementary Methods, section C). Briefly, the labelling index (LI) sums overall microglia presence in terms of cell bodies as well as microglial processes (as cell bodies may not be present in every section, but processes are likely to be important in microglial roles), overcoming issues of microglial clustering, overlap and aggregation around vessels that can confound individual cell counts. LI also covers all microglial cell-types regardless of morphology, size and shape. LI was measured across 14 ROIs in post mortem brain tissue derived from 55 individuals, comprising individuals with non-lesional epilepsy (EP-NL, n=18), lesional epilepsy (EP-L, n=21) and non-epilepsy controls (NEC, n=16). No sample size calculations were performed since this was not a discovery sample, but a previous similar study of regional cortical pathology achieved a significant statistical result compared to controls with nine post mortem cases, whereas this study included 55 individuals [17]. Staining was repeated on the same cases in different immunohistochemistry runs for comparison.
Further, in order to assess whether cortical thinning can be prevented by manipulating microglia, we used a well-established murine model of epilepsy induced by convulsive status epilepticus (SE) provoked by intra-amygdalar injection of kainic acid in C57/BL6N adult male mice (30 g; Charles River, Italy)[18–21]. This model mimics features of MTLE with hippocampal sclerosis, with neuronal damage also observed in extrahippocampal areas [19–21]. To explore the importance of microglia in brain structural changes and pathologic outcomes, mice were treated with the CSF1R inhibitor PLX3397 in a controlled experiment. We assessed the effect of PLX3397 treatment on cognitive performance using the novel object recognition test (NORT), regional brain volume estimates from post mortem MRI, abundance of microglia and neurons quantified using immunohistochemistry and Nissl staining, respectively. The sample size was determined a priori based on previous experience with the same epilepsy model [18,20]. A simple random allocation was applied to assign a subject to a particular experimental group. Group 1: medicated diet (supplemented with PLX3397), n=5 and placebo (non-medicated) diet, n=5; Group 2: medicated diet (supplemented with PLX3397), n=6 and placebo (non-medicated) diet, n=5 (experimental design in Fig. S1A). Respective sham mice (n=8) were prepared for behavioural testing and post mortem analyses. These groups were run 15 days apart. Group 3: placebo diet mice exposed to SE, n=8 (run in parallel with group 1, n=4 and group 2, n=4) and respective sham mice, n=8. Group 3 mice were prepared to compensate for potential drop-outs during the longitudinal experiment (Fig. 3C). Limited PLX3397-supplemented diet availability prevented further experiments. To verify the reproducibility of the experimental findings, we compared several EEG measures between the different placebo-diet mouse cohorts, namely SE onset and duration, temporal distribution of spikes during SE, number and duration of spontaneous seizures. For all these measures, statistical analysis showed that replication was successful. The data were collected by anonymising samples and the assessor was blinded to diagnosis (for both human tissue and experimental mouse model data). For detailed Materials and Methods and mouse treatment protocols see Supplementary Methods (section D).
Fig. 3. Effects of microglia depletion in the early disease phase on entorhinal cortex thickness and neuronal cell loss, and on cognitive deficit in epileptic mice.
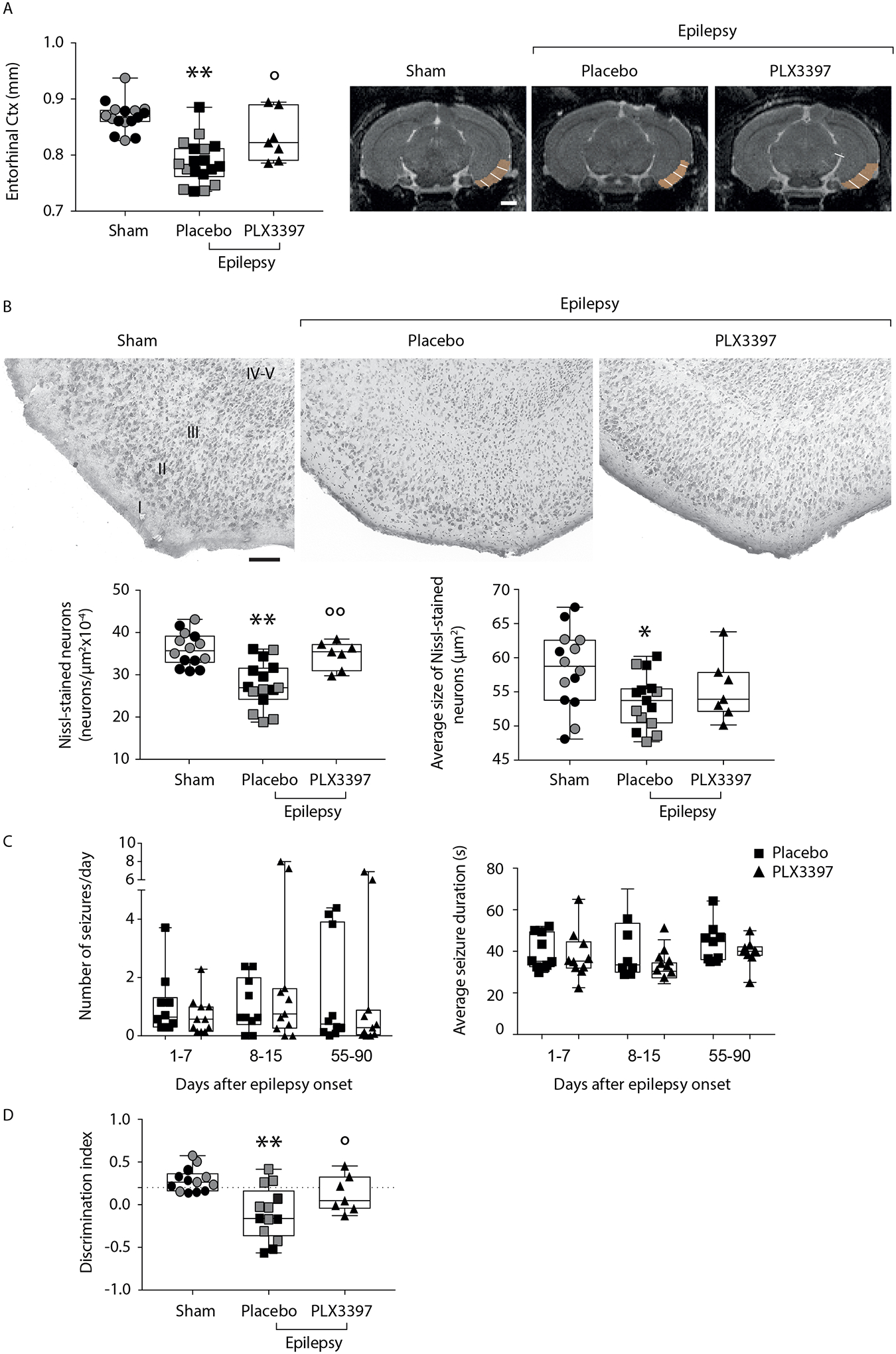
The experimental design is depicted in Fig. S1A. Grey symbols represent sham (n=6–8) and epileptic mice fed with placebo diet (n=8) run in parallel with experimental mice of Fig 3C (see text for details).
Panel A: box-and-whisker plots depicting median, minimum, maximum and single values related to the entorhinal cortex thickness, as assessed by quantitative post mortem MRI analysis performed in epileptic mice at the end of EEG monitoring (placebo are mice fed with non-medicated diet: n=18; PLX3397 are mice fed with medicated diet supplemented with PLX3397: n=7), and in sham mice (not exposed to status epilepticus; n=16). MRI images depict representative slices showing the ROI used to quantify the cortical thickness. Four mice in the PLX3397 group did not undergo MRI analysis and therefore they were not included in the subsequent histological (B) and behavioral analyses (D). The white line within the ROI was manually drawn to measure the cortical thickness. **P=0.0001 vs sham; °P=0.022 vs placebo by ANOVA followed by Tukey’s test. Scale bar: 1 cm.
Panel B: representative Nissl-stained sections (top row) of the entorhinal cortex in the experimental groups (top row; sham, n=14; placebo, n=15; PLX3397, n=7), and the relative quantification of the number and the average size of Nissl-stained neurons (bottom row). Two sham and three placebo mice were excluded from the analysis due to poor quality of Nissl staining. Data are shown by box-and-whisker plots depicting median, minimum, maximum and single values *P=0.0037, **P=0.0001 vs sham; °°P=0.006 vs placebo diet by ANOVA followed by Tukey’s test. Scale bars: 100 μm.
Panel C: box-and-whisker plots depicting median, minimum, maximum and single values of the number of spontaneous seizures/day and their average duration during days 1–7, 8–15 and 55–90 from epilepsy onset (day 1) in the placebo (n=10) and PLX3397-supplemented diet (n=11) experimental groups (protocol in Fig. S1A). Friedman’s two-way nonparametric ANOVA (p=0.041) followed by post-hoc multiple comparisons test with Bonferroni correction: P-values for Number of seizures/day: p=0.363, days 1–7; p=0.339, days 8–15; p=0.965, days 55–90; P-values for Seizures duration: p=0.799, days 1–7; p=0.325, days 8–15; p=0.262, days 55–90.
Outliers were identified only for the Number of seizures/day in the placebo group (n=1 in days 1–7) and in the PLX3397 group (n=2 in days 8–15 and n=2 in days 55–90), however, their omission did not change the results of the primary statistical analysis therefore the values were not removed from the corresponding data set (P-values for sensitivity analysis: p=0.831, days 1–7; p=0.375, days 8–15; p=0.084, days 55–90).
Panel D: Novel object recognition test (NORT) in epileptic mice fed with placebo- (n=13) or PLX3397-supplemented diet (n=7), and sham controls (n=13). Three sham and five placebo diet epileptic mice were excluded from the analysis since they showed a total exploration time <6 sec during the familiarisation phase. Memory was evaluated by measuring the discrimination index, which was calculated as time spent (sec) exploring the familiar (F) and the novel (N) object as follows: (N - F)/(N + F). Data are shown by box-and-whisker plots depicting median, minimum, maximum and single values, differences significant at **P=0.0004 vs sham by ANOVA followed by Tukey’s test; *P=0.049 vs placebo by Mann-Whitney test.
Results
Cortical regions at most risk of reduced thickness are characterised by higher density of microglia and endothelial cells
The AHBA provided gene expression profiles for all cortical regions of interest [15,16]. This gene expression atlas is unique in that from 158 to 348 different brain structures have been sampled from each control individual (n=6), none of whom had epilepsy, covering in total 414 different structures, and that sampling has been precisely mapped to MNI space, enabling linkage of the expression data to external MRI-brain maps in MNI space (see Supplementary Methods, section A). Thus, the gene expression profiles used in this analysis were highly regionally specified. However, only two out of the six brains have been sampled in both hemispheres: therefore, we restricted our analysis to the left hemisphere, which was available for all subjects [22].
We focussed on the regions where thickness was reduced and sought to identify mechanisms that underlie this cortical regional ‘vulnerability’ to damage; areas without significant loss of thickness compared to controls were considered ‘relatively protected’, remaining like normal cortex. To identify the molecular basis of this regional vulnerability in the broad spectrum of epilepsies, we focused our analyses on the shared pan-syndrome MRI findings [8]. Of the 34 cortical regions in the left hemisphere profiled within the ENIGMA-Epilepsy data set, eight were considered vulnerable to reduced cortical thickness, while 26 were considered relatively protected [8] (Table S1).
Using linear mixed effects models, we compared these two types of cortical regions for significant differences in cell-type fractions, inferred from gene expression using Scaden [23]. We identified increased ratios of endothelial cells (T=4.4; P=1×10−5) and microglia (T=4.1; P=4.5×10−5) in vulnerable regions, along with a decreased ratio of excitatory neurons (T=−3.9; P=1.2×10−4) in vulnerable regions (Table 1). We confirmed these cell-type enrichments by a complementary two-step approach. The first step was a gene-wise association screen, showing that, out of 14,138 tested genes, 3,182 genes were more highly expressed in vulnerable regions (at PFDR<0.05) and 2,223 genes were expressed significantly less in vulnerable regions (at PFDR<0.05; Dataset S1). In the second step, these gene-wise results were subject to threshold-free (i.e., not relying on lists of significantly differentially expressed genes) gene set enrichment analyses using cell-type-specific gene sets from different data sources (Dataset S2). This analysis confirmed the enrichment of endothelial cells and microglia in vulnerable regions, and neurons in protected regions. We noted that both analyses provided qualitatively the same result when using all 1,628 AHBA samples from both hemispheres, where 18 of 68 ROIs were considered vulnerable (Table S2; Figure S15). Thus, regions vulnerable to cortical thinning were characterized by elevated proportion of microglia and endothelial cells.
Table 1:
Association between inferred cell type fractions and reduced cortical thickness.
| Cell type | T-value | P-value | PFDR | Pperm |
|---|---|---|---|---|
| Astrocytes | 2.080608 | 0.037 | 0.043 | 0.17 |
| Endothelial | 4.426344 | 1.0×10 −5 | 7.67×10 −5 | 0.026 |
| ExNeurons | −3.855666 | 1.15×10−4 | 3.08×10−4 | 0.051 |
| InNeurons | 0.684069 | 0.49 | 0.49 | 0.37 |
| Microglia | 4.081655 | 4.5×10 −5 | 1.79×10 −4 | 0.032 |
| Oligodendrocytes | 2.456355 | 0.014 | 0.019 | 0.053 |
| OPC | 2.758717 | 5.8×10−3 | 0.014 | 0.068 |
| Unknown | 2.690987 | 0.007 | 0.011 | 0.066 |
Columns represent the cell type, the t-value from the association analysis, the corresponding P-value and the corrected P-value using FDR. The least column lists a P-value obtained from 1,000 permutations of the vulnerable/protected status of 34 ROIs in the left hemisphere.
Selected processes and microglial signatures are implicated amongst genes associated with reduced cortical thickness
Using our gene-level association results, we next investigated the biological processes that could underpin regional differences in vulnerability to reduced cortical thickness in epilepsy. In the first instance, we investigated enrichment of differentially expressed genes in Gene Ontology (GO) terms and KEGG and REACTOME pathways using a threshold-free approach based on the area under the receiver operator characteristics curve (AUC) [24]. Amongst genes with higher expression in relatively protected cortical regions, the most significant terms we identified related to RNA processing (REACTOME: RNA polymerase II transcription, AUC=0.429, PFDR=1.19×10−11) and synaptic function (GO: postsynaptic specialization organization, AUC=0.270, PFDR=3.95×10−4; Dataset S3). Conversely, amongst genes with higher expression in vulnerable cortical regions, the most significant terms related to electron transport (GO: electron transport chain, AUC=0.74, PFDR=3.45×10−18) and immune function and regulation (REACTOME: innate immune system, AUC=0.59, PFDR=1.46×10−15; GO: antigen processing and presentation, AUC=0.65, PFDR=7.98×10−13; Dataset S3), the latter being consistent with our results obtained using cell-specific gene sets. There was also strong enrichment for the KEGG pathways related to neurodegenerative diseases (Alzheimer’s disease: AUC=0.65 PFDR=3.53×10−6; Parkinson’s disease: AUC=0.75, PFDR=4.55×10−12; Huntington’s disease: AUC=0.66, PFDR=1.80×10−7). Moreover, this approach also enabled us to obtain more specific process-related information and suggested the importance of the interferon gamma signalling pathway (GO: Response to interferon gamma, AUC=0.597, PFDR=1.42×10−3; GO: interferon gamma mediated signalling pathway, AUC=0.621, PFDR=6.71×10−3; Dataset S3).
A number of cell-types showed enrichment in vulnerable regions. But both cell-type analysis and the observed pathway enrichments implicated microglia and immune processes, respectively, prompting us to investigate microglia in more detail, given existing evidence for a role for microglia in epilepsy [25] and in neurodegeneration in general [26]. More precisely, since microglia can exist in a range of activation states within the context of epilepsy [25], we sought to identify the microglial cell states of greatest importance in reduced cortical thickness. We collated gene signatures for distinct microglial states from the existing literature and also inferred signatures of microglial state through co-expression network analyses (see Supplementary Methods, section A). While there were significant overlaps in gene membership across the 16 microglial signatures used (Fig. S2), each of the gene lists was distinct. We identified a significant enrichment for 13 of the 16 signatures with genes overexpressed in vulnerable cortex. This included the microglial signature generated by Srivastava et al. [27] (AUC=0.68, PFDR=1.45×10−12; Dataset S4), which was positively correlated with seizure frequency in a particular mouse model of chronic epilepsy. Strong enrichments were also identified for an inferred human microglial signature enriched for type 1-like microglial markers (AUC=0.73, PFDR=7.27×10−18; grey60; Dataset S4), as well as signatures for aged, late activation, and de-activated microglia. However, we saw little evidence for enrichment within signatures of early activation (“Early Response” signature [28], AUC=0.53, PFDR=0.062). Similar microglial states have recently been implicated in various forms of chronic neurodegeneration [28–30], but more data are needed to definitively determine whether similar underlying processes and microglial states are indeed involved.
Genetic evidence supports immune activation as a modifying, but not causal, factor in the common epilepsies
In the ENIGMA-Epilepsy imaging study, region-specific reduced cortical thickness across the common epilepsies was correlated with disease duration and age of onset of epilepsy [8]. Given that the analyses of gene expression data from healthy donors described above suggest the importance of microglia responses in vulnerability to reduced cortical thickness, we hypothesized that genetic variants affecting microglial responses would also impact upon the severity of epilepsy. While no microglial eQTL data set exists to date, eQTL analyses have been performed using monocytes at rest, and also monocytes treated with IFN-γ or LPS [31]. We postulated that these eQTLs would be enriched for heritability of risk loci for epilepsy that is drug-resistant (one surrogate for disease severity) but not for epilepsy per se. To investigate this, we used GWAS data on epilepsy susceptibility [2] and separate GWAS data on the phenotype of drug-resistant epilepsy from the EpiPGX consortium (www.epipgx.eu). Using LD score regression, we sought enrichment in heritability for these phenotypes within state-specific monocyte eQTLs [31] (Supplementary Methods, section B). We found no significant enrichment in the heritability of epilepsy in this form of annotation, suggesting that susceptibility to common epilepsies is less likely to be driven by immune processes (Fig. 1B, Table 2). However, eQTLs regulating the response to IFN-γ were highly enriched for drug-resistant epilepsy vs drug-responsive epilepsy loci (P=0.00045; PFDR=0.0095, Table 2). This result was particularly striking given the small size of the EpiPGX drug-resistant epilepsy GWAS (2423 drug-resistant cases vs 1626 drug-responsive cases). Thus, we provide genetic evidence in support of microglial-mediated responses as a modifying factor in severity of epilepsy, but not its susceptibility.
Table 2:
Results from stratified LD score regression estimating the enrichment of immune response eQTL signatures (rows) in different epilepsy GWAS (columns).
| GWAS | ||||||
|---|---|---|---|---|---|---|
| ILAE (epilepsy vs HC) | Drug-resistant vs HC | Drug-resistant vs drug responders | ||||
| eQTL Type | P | PFDR | P | PFDR | P | PFDR |
| Naïve monocytes | 8.73×10−2 | 1.63×10−1 | 3.43×10−2 | 8.12×10−2 | 2.85×10−3 | 2.00×10 −2 |
| INF- γ-treated | 1.56×10−2 | 8.12×10−2 | 5.09×10−2 | 1.06×10−1 | 4.53×10−4 | 9.52×10 −3 |
| LPS-treated (2 hours) | 1.43×10−1 | 2.32×10−1 | 2.70×10−1 | 3.90×10−1 | 3.48×10−2 | 8.12×10−2 |
| LPS24–treated (24 hours) | 3.17×10−2 | 8.12×10−2 | 1.97×10−2 | 8.12×10−2 | 1.27×10−3 | 1.33×10 −2 |
GWAS, genome-wide association study; eQTL, expression quantitative trait locus; ILAE, International League Against Epilepsy study (The International League Against Epilepsy Consortium on Complex Epilepsies, 2014); HC, healthy controls; LPS, lipopolysaccharide. Bold font marks significant enrichments at PFDR<0.05.
Widespread regionalised over-representation of microglia is present in brain tissue from people with epilepsy
Based on the analyses of regional gene expression patterns in healthy brains above, we hypothesised that brain tissue from individuals with various forms of epilepsy would have regionalized higher densities of microglia as compared to tissue from non-epilepsy controls and that this would be apparent beyond the context of acute seizure activity. Using Iba-1 immunolabelling, we found that single ramified microglia and processes were found throughout the cortex; scattered perivascular macrophages were also labelled (Fig. 2A). Enlarged and more complex/branching microglia, focal aggregates and amoeboid/macrophage forms were noted in some ROIs (Fig. 2A). Consistent with our hypothesis, the Iba1 LI was significantly higher in all epilepsy (EP-NL and EP-L together) than NEC for all ROIs (P=4.0×10−13) and for both subgroup comparisons (EP-NL [P=3.7×10−13] and EP-L [P=3.5×10−13]) against NEC. Regional differences were noted within the epilepsy groups, and for individual ROIs compared between epilepsy and control groups (see Fig. 2, Fig. S16 and Table S3). We noted that the Iba1 LI was similar across EP-NL, EP-L and NEC in BA17, as compared to pulvinar and BA22 where the Iba1 labelling index was higher in EP-NL and EP-L groups than in NEC. Thus, these results are consistent with the view that there is an over-representation of microglial footprint in brain tissue from people with chronic epilepsy, and that such microglial responses may occur in a regionally-specific manner. This observation was supported by evidence of region-specific microglia expansion in epileptic mice as reflected by the increased number of Iba1-positive cells in entorhinal cortex but not in perirhinal cortex (Fig. S3). Moreover, in sham mice we also observed that the temporal cortices have a higher microglia density than the hippocampus, denoting regionalised microglia enrichment at baseline (Fig. S3).
Fig. 2. Presence of excess activated microglia in post mortem brain tissue from people with epilepsy.
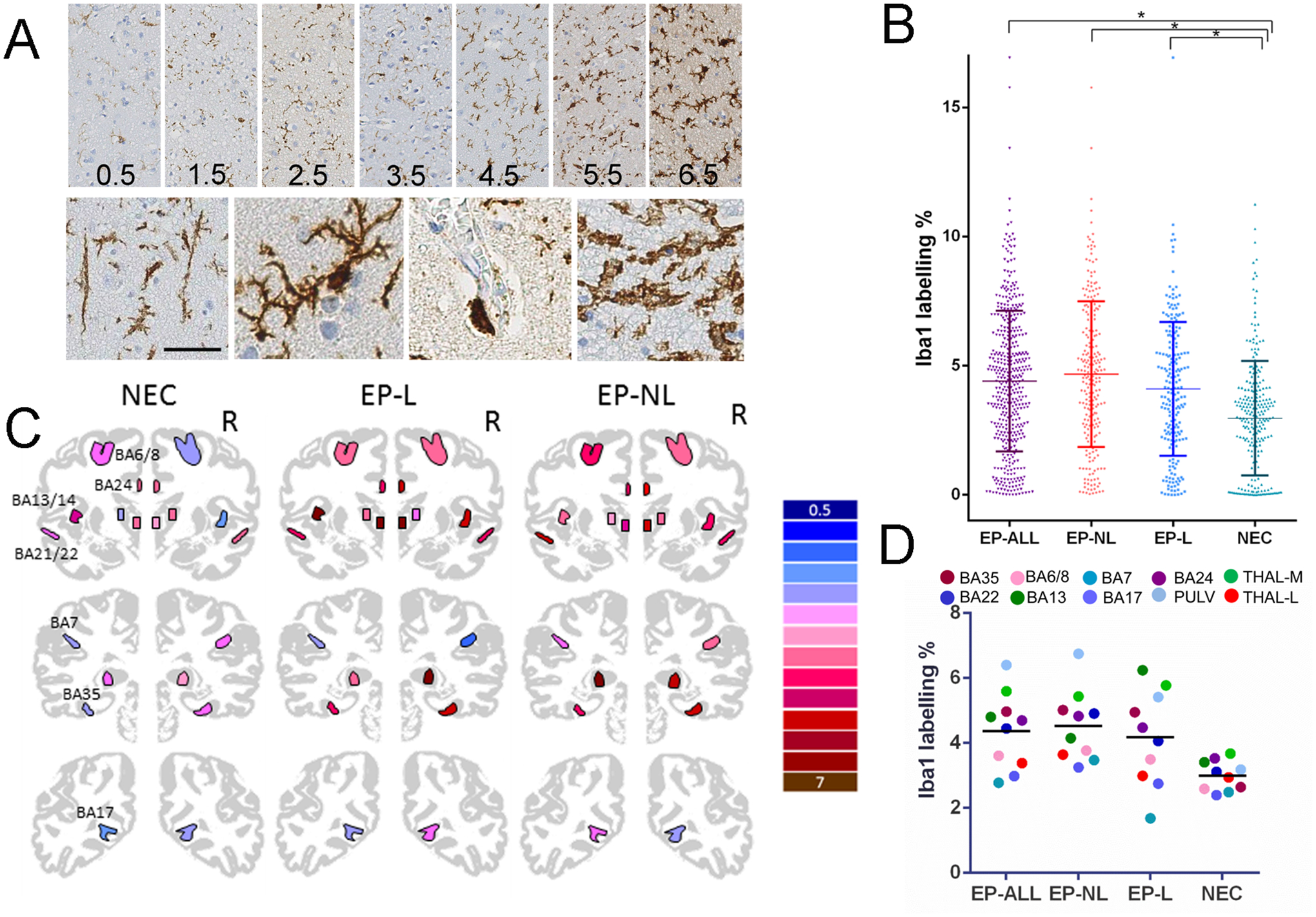
Panel A: High magnification of morphological types of Iba1-labelled cells including (a) ‘rod’ cells, (b) ramified microglia, (c) perivascular macrophage and (d) amoeboid forms. Fixation time in illustration of ramified microglia was 467 days (bar = 30 microns). Right column shows Iba1 labelling in randomly selected from cases in the study (representing all three groups) with a range of immunostaining quantified from 0.5 to 6.5% field fraction with progressive increase in complexity, number and size of ramified microglial (all taken at X20).
Panel B: Scatter graph of all data points from 709 sections including all brain regions showing mean and standard deviation for labelling index in the four main groups: Epilepsy-ALL (EP-ALL), Epilepsy Non-Lesional (EP-NL), Epilepsy-Lesional (EP-L) and non-epilepsy controls (NEC). EP-ALL, EP-NL, EP-L are all significantly greater than NEC (*respective P-values: 4.0×10−13, 3.7×10−13, 3.5×10−13).
Panel C. Iba1 immunolabelling shown in 10 Brodmann areas and thalamus in each hemisphere, colour coded for the mean percentage labelling index in the three groups as in B.
Panel D. Scatter graph of the mean Iba1 LI in the same Brodmann areas and thalamus (averaged over both hemispheres) in the three groups as in B.
Experimental evidence supports microglial activation as a modifying factor for cortical thickness in a mouse model of acquired epilepsy
The analyses so far have shown (i) elevated expression of microglia-related genes in brain regions, deemed ‘vulnerable’ from the ENIGMA-Epilepsy imaging study, in healthy brains and (ii) widespread regionalised over-representation of microglia in brain tissue from people with epilepsy compared to tissue from non-epileptic controls. To provide proof-of-concept evidence causally linking microglia activation to cortical thinning, we used a mouse model of acquired epilepsy where convulsive seizures originate and spread in the limbic system, and also involve the neocortex [18–21]. Spontaneous seizures develop a few days after the acute insult (mean±S.E.M., onset, 6.2±0.5 days, n=21 mice of Fig. 3C) and recur for months (Fig. S4B,C), and are drug-resistant [20]. Microglia are morphologically activated (CD11b-positive area, hippocampus: mm2, sham, 0.09±0.01; 1 week post-SE, 1.67±0.48, P=0.0002, Mann-Whitney test; n=10–11 mice) and proliferate by 2.0-fold on average within one week after SE as assessed in a different cohort of mice by analysis of Iba1-positive cells in the forebrain (number of cells, hippocampus: sham, 614.4±7.1; 1 week post-SE, 1347±68.6, P=0.0011, Mann-Whitney test; n=10–11 mice).
First, we studied whether the thickness and volume of selected cortical brain regions were reduced, and those of the lateral ventricles increased (directions of change as predicted by the ENIGMA-Epilepsy findings in humans) in placebo (non-medicated)-diet fed epileptic mice (n=10) vs sham mice (not exposed to SE, n=16) as assessed by post mortem MRI (experimental design in Fig. S1A). In the post mortem MRI analysis (Fig. 3A, Figs. S5 and S6), we also included additional placebo diet-fed epileptic mice (n=8) run in parallel with mice depicted in Fig. 3C (n= 10). These additional mice were video-EEG monitored only in the terminal disease phase (day 55–90), and their seizure frequency (0.91±0.26, n=8) was similar to mice depicted in Fig. 3C (1.65±0.75, n=10; p=0.41 by Mann-Whitney test). These additional epileptic mice were also included in the histopathological brain analysis (Fig. 3B) and for behavioural testing (Fig. 3D). We found that the lateral ventricles were enlarged by 2-fold (P=0.0006 by ANOVA followed by Tukey’s test; Fig. S5A): this effect was associated with a significant reduction in the volume of the entorhinal (P=0.043; Fig. S5E) and perirhinal (P=0.025 by ANOVA followed by Tukey’s test; Fig. S5F) cortices. No significant changes were observed in other brain areas such as the hippocampus, caudato-putamen and the thalamus, although their average volumes trended lower than the corresponding values in sham mice (Fig. S5B–D). Notably, a significant reduction in the thickness of entorhinal (P=0.0001 by ANOVA followed by Tukey’s test; Fig. 3A) and perirhinal (P=0.046 by ANOVA followed by Tukey’s test; Fig. S6) cortices was also measured in the same mice.
Next, we measured brain region volumes and cortical thickness in mice fed with medicated diet, supplemented with PLX3397, in order to deplete microglia by >90% (Fig. S7) during the initial disease development (i.e., until day 7 after spontaneous seizure onset; experimental design in Fig. S1A). Importantly, this decrease in microglia was induced transiently, since microglia re-populated the forebrain within one week after switching mice to a placebo non-medicated diet [32]. This protocol was followed in order to assess the impact of microglia activation in the initial phases of the disease on the structural brain changes detected in the epileptic mice. We found that the decrease in thickness of the entorhinal cortex of epileptic mice under placebo diet vs sham mice was prevented in mice that were depleted of microglia in the early disease phase (P=0.022 vs placebo by ANOVA followed by Tukey’s test; Fig. 3A). This indicates that early microglia depletion prevented the entorhinal cortical thinning determined in the chronic disease phase. Microglia depletion did not affect the thinning of the perirhinal cortex (Fig. S6) or the ventricle and subcortical volume changes occurring in epileptic mice (Fig. S5). Evaluation of neuronal cell number in the entorhinal cortex of epileptic mice receiving placebo diet showed a significant decrease in both neuronal cell density (P=0.0001 vs sham by ANOVA followed by Tukey’s test; Fig. 3B) and their average cell body size (P=0.037 vs sham by ANOVA followed by Tukey’s test; Fig. 3B). The reduction in cell density, but not of the average cell body size, in epileptic mice was prevented by microglia depletion in the early disease phase (P=0.006 vs placebo diet by ANOVA followed by Tukey’s test; Fig. 3B). Similarly, microglia depletion affected the reduction in discrimination index (a measure of non-spatial memory deficit dependent on entorhinal cortex function) [33] observed in epileptic mice fed with placebo diet (P=0.0004 epileptic mice vs sham by ANOVA followed by Tukey’s test; P=0.049 epileptic mice on the PLX3397 diet vs placebo diet by Mann-Whitney test) (Fig. 3D). The total exploration time of objects during the familiarisation phase did not differ in the various experimental groups (sham, 24.07±2.6 sec; SE+placebo, 28.4±4.7 sec; SE+PLX3397, 29.7±5.9 sec; P=0.25 sham vs placebo; P=0.5 SE+placebo vs SE+PLX3397). Notably, microglia depletion in the early disease phase did not modify the onset, duration and severity of SE (Fig. S4A). Moreover, both frequency and duration of spontaneous seizures was unaltered by early microglia depletion as compared to placebo non-medicated diet-fed mice (Fig. 3C; S4C vs B).
Discussion
The epilepsies are complex conditions with multiple facets including various causes, differing responses to treatment, and unpredictable outcomes. Most attention has been paid to causation and processes of epileptogenesis across the broad constituent spectrum of syndromes. In contrast, disease progression has not been a primary focus of research even though for some rare epilepsies (the DEE), the window of opportunity to ameliorate disease may be open for longer than expected, as disrupted patterns of gene expression last into adulthood [34]. Here, we show that across the broad spectrum of the more common epilepsies (specifically excluding DEE), gene expression-predicted microglia density spatially correlated with reduced cortical thickness; the related genes newly implicate innate immunity, and, particularly, microglial activation, as contributors to the underlying cause of cortical thinning. We also show that this molecular signature of innate immunity activation is significantly enriched for a gene set already causally linked to seizure frequency in a mouse model of chronic epilepsy [27], though we note our model differs from the model in that study. Further, our data add new evidence supporting the general concept that microglial activation is associated with at least some of the structural changes seen in brain areas involved in seizure circuitry, in that microglial depletion in mice early during disease development can directly prevent associated cortical thinning in the entorhinal cortex. Notably, microglia depletion in mice also prevented neuronal cell loss in the entorhinal cortex and this neuroprotective effect was associated with improvement of cognitive deficit measured by the non-spatial memory test, that is sensitive to entorhinal cortex function [33].
Furthermore, we tested the hypothesis that microglial/monocyte activation is a key modulator of the severity of epilepsy using both genetic and functional approaches. We used the availability of GWAS data for resistance to anti-epileptic drug treatment, a marker of disease severity, to investigate enrichment in heritability at genetic loci already known to influence the expression of genes involved in monocyte activation. We find a highly significant enrichment in the heritability of epilepsy severity amongst these immunomodulatory loci, despite the absence of a significant enrichment for heritability of epilepsy per se. Finally, in keeping with these observations, experimental microglial depletion timed to coincide with a period of epileptogenesis in a murine model of acquired epilepsy can prevent regionalised cortical thinning, but does not influence the eventual development of seizures themselves in our model. Data from the experimental model also demonstrate that the cortical thinning is at least partly due to reduced neuronal cell density and average neuronal size: reduction of neuronal density can be prevented by appropriately-timed microglial depletion while neuronal size changes were not rescued, which may explain the observation that entorhinal volume changes are not completely prevented by microglial depletion. We, and others [35–40], find activated microglia are present in excess in brain tissue from people with epilepsy, compared to brain tissue from healthy controls, providing evidence for translation to human epilepsies of our assertions from the experimental model data. We selected Iba1 as a robust immunomarker for microglia in formalin-fixed tissue [41]. Like HLA-DR and CD68, Iba1 labels all phenotypes of microglia from ramified and amoeboid forms to macrophages and is therefore suited to structural studies of normal cortex in the absence of focal pathology [42]. We recently reported increased Iba1 labelling in central autonomic cortical regions in SUDEP, which also associated with increased seizures prior to death [43]. Together, these findings separate important processes occurring in the course of the epilepsies, and incriminate potentially modifiable microglial activation states in the hitherto largely-ignored feature of cortical thinning in the common human epilepsies. Unsurprisingly, our results also suggest other factors are likely to be involved, which we have not explored further yet.
Importantly, we note as limitations that we assume a high degree of similarity in the genetics of gene expression in monocytes and microglia and that the clue to the possible role for microglial activation in cortical thinning came from a cross-sectional study of chronic human epilepsy: although reduced cortical thickness correlated with disease duration [8], we could not distinguish whether the structural difference had developed at disease onset (e.g., with causation), during epileptogenesis, during the course of the disease, or a combination of these epochs. Our multimodal data, and especially the experimental model results, allow us to begin to address this question. Notably, we used spatially-resolved whole-brain gene expression data from healthy controls, rather than from the brains of people with epilepsy specifically to avoid confounding by secondary effects: some such effects (e,g., compensatory changes) may be worth exploring, but that was not our purpose here. The murine model in our study relates to early processes in epileptogenesis, and shows a clear separation for microglial roles in cortical thinning and seizure occurrence, whilst data from other models relate to the chronic disease state, and shows an effect of microglial manipulation on seizure frequency in that chronic state [27,44] (cortical thinning was not assessed in those models). The experimental and human data are not directly compatible, and we cannot test hypotheses arising from the chronic mouse model in data from human ENIGMA-Epilepsy. However, taken together, the data suggest microglia may have multiple modifying roles during epileptogenesis and progression of disease across common human epilepsies, though we find no evidence that they contribute to the actual occurrence of these common forms of epilepsy (from either our human or murine data). That seizure frequency and cortical thinning may be separable processes adds to important epidemiological evidence that seizure frequency is not the only contributor to morbidity in people with a history of epilepsy [5]. Microglia have many roles in specific types of epilepsy, demonstrated clearly in a variety of animal models. Such roles include phagocytosis, which may link consumption of synapses with cognitive changes in long-term active epilepsy [45], providing another possible mechanism for actual loss of brain volume in epilepsy: ‘time is brain’ [46].
Dysregulation of innate immunity is considered to possibly contribute to brain pathology and seizures in some severe human epilepsies. Microglial activation is seen in Rasmussen’s encephalitis and in tissue from epilepsies due to hippocampal sclerosis and mesial temporal lobe sclerosis, focal cortical dysplasia and tuberous sclerosis [35–39]. The latter two conditions are known to have genetic, rather than inflammatory, causes, but the extent of microglial activation in the chronic disease phase correlates with severity (seizure frequency) and disease duration in these studies [35,39] and not just cause, pointing again to distinctions between processes related to the initial cause (e.g. genetic disorder) and others that manifest during active disease. Importantly, resected human brain tissue is only available from a few cases of a few types of epilepsies (mostly surgical specimens from MTLE and focal cortical dysplasia [47]), so that it is impossible to otherwise evaluate the role of microglia using neuropathological data in the majority of common human epilepsies, from which brain tissue cannot be obtained in life. Brain imaging in animal models using a label (TSPO) for microglial activation shows dynamic upregulation during epileptogenesis, with persistent, although declining, activity in the chronic phase and correlation with spontaneous seizures [48]; in chronic human temporal lobe epilepsy, there is increased TSPO binding ipsilateral and contralateral to seizure foci [49] and in the interictal phases [50]. Using immunolabelling, we demonstrate that there is over-representation of activated microglia in human and experimental brain tissue, compared to controls. The animal data also highlight that microglia expansion in epileptic mice occurs in the entorhinal but not in perirhinal cortex, and this effect is mirrored in the rescue of cortical thinning by microglia depletion (Fig. S3). However, neither TSPO imaging nor neuropathological study is realistically applicable to large numbers of people with epilepsy, especially common epilepsies, whilst MRI is, providing a readily-available means of evaluating clinical translation of the implications of our findings. We propose, using MRI-derived patterns and correlation with gene expression, that activated microglia-associated functions drive the important, but as yet largely neglected, phenotype of cortical thinning in a broad swathe of common human epilepsies. Subsequent experimental intervention in an animal model suggests that early manipulation of microglia has the capacity to rescue disease-related cortical thinning, neuronal cell loss and cognitive deficits, opening up new areas for attention and treatment in common human epilepsies. We note that other cell-types and processes are also implicated: these also need consideration, and suggest the possibility of multiple players in cortical thinning: we have focussed on one cell-type, which does not diminish the potential utility of its manipulation. Other processes and cell-types will be the subject of future investigations.
Our results point to important roles for neuroinflammatory pathways and potentially specific molecular actors, such as IFN-γ. However, the diversity of microglial states and functions, and the complex, dynamic, interactions between neurons, astroglia and microglia, that at the very least can promote epileptogenesis [25] have yet to be fully resolved. Clinical translation of our key observation of the widespread role of microglial activation across the breadth of types of epilepsy into therapeutic options to prevent irreversible loss of brain substance will require definition of the time course of thinning in the different types of epilepsy. Translation will also require the development of safe, effective and tolerable treatments that target precise mechanisms without compromising immune surveillance of brain tissue, a need across diverse neurological disorders [51].
Supplementary Material
Acknowledgements
We thank Angela Richard-Londt, Francesca Launchbury and Matthew Ellis for assistance with human neuropathology data collection, Mojgansadat Borghei, Ilaria Lagorio and Yi Yao for assistance with data collection and Rafal Kaminski and Jonathan van Eyll for helpful discussions. We thank Dr L Porcu (Department of Oncology, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano, Italy) for statistical advice.
Funding
A. Altmann holds a Medical Research Council eMedLab Medical Bioinformatics Career Development Fellowship. This work was supported by the Medical Research Council (grant number MR/L016311/1). M.R. holds an Medical Research Council Clinician Scientist Fellowship (grant number MR/N008324/1). R.H.R. was supported through the award of a Leonard Wolfson Doctoral Training Fellowship in Neurodegeneration. The work was supported by grants from the European Union (7th Framework Programme Grants 279062, EpiPGX and 602102, EPITARGET). This work was partly undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. The Epilepsy Society through the Katy Baggott Foundation supports the Epilepsy Brain and Tissue Bank at UCL and ERUK support the Corsellis Epilepsy brain collection. The work was also supported by the Epilepsy Society, UK (C.L., S.M.S.). Associazione Italiana Contro L’Epilessia (FIRE-AICE) and Fondazione Monzino (A.V.) and by NIH grants R01 NS097719 (M.E.M.) U54 EB020403 (P.T.) and NIH/NINDS R01 NS065838 (C.R.M.).
Abbreviations
- AHBA
Allen Human Brain Atlas
- BA
Brodmann Area
- DEE
developmental and epileptic encephalopathies
- eQTL
expression Quantitative Trait Locus
- EP-L
Lesional Epilepsy
- EP-NL
Non-Lesional Epilepsy
- FDR
False Discovery Rate
- GO
Gene Ontology
- GWAS
Genome Wide Association Study
- IFN-γ
Interferon
- KEGG
Kyoto Encyclopaedia of Genes and Genomes
- LD
Linkage Disequilibrium
- LI
Labelling Index
- LPS
Lipopolysaccharide
- MTLE
Mesial Temporal Lobe Epilepsy
- MNI
Montreal Neurological Institute
- NEC
Non-Epilepsy Controls
- ROI
region of interest
- SE
status epilepticus
Footnotes
Ethical Approval
The project has ethical approval (under NRES 17/SC/0573) and post mortem epilepsy cases had research consent, compliant with the UK Human Tissue Act 2014. Furthermore, in accordance with the ARRIVE guidelines, procedures involving animals and their care were conducted in conformity with the institutional guidelines that are in compliance with national (D.L. n.26, G.U. March 4, 2014) and international guidelines and laws (EEC Council Directive 86/609, OJ L 358, 1, December 12, 1987, Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996), and were reviewed and approved by the intramural ethical committee.
Conflict of Interest statements
C.D.W. is now an employee of Biogen Research and Early Development (Cambridge, Massachusetts, 02142, USA).
Data Availability Statement
The complete normalized microarray gene expression data from the Allen Institute for Brain Science that support the findings of this study are available from the institute’s website at http://human.brain-map.org/static/download. The eQTL summary statistics on state-specific monocytes that support the findings of this study are available in the supplementary tables at DOI: 10.1126/science.1246949. The summary results from the EpiPGX GWAS data that support the findings of this study are available from http://www.epigad.org. Data from the experimental model that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Orsini A, Zara F, Striano P. Recent advances in epilepsy genetics. Neurosci Lett. 2018;667:4–9. [DOI] [PubMed] [Google Scholar]
- 2.The International League Against Epilepsy Consortium on Complex Epilepsies. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. 2014;13(9):893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The International League Against Epilepsy Consortium On Complex Epilepsies. Genome-wide mega-analysis identifies 11 new loci and highlights diverse biological mechanisms in the common epilepsies. Nat Commun. 2018;9:5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barkovich AJ, Dobyns WB, Guerrini R. Malformations of Cortical Development and Epilepsy. Cold Spring Harb Perspect Med. 2015;5(5):a022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell GS, Neligan A, Giavasi C, Keezer MR, Novy J, Peacock JL, et al. Outcome of seizures in the general population after 25 years: A prospective follow-up, observational cohort study. J Neurol Neurosurg Psychiatry. 2016;87(8):843–50. [DOI] [PubMed] [Google Scholar]
- 6.Novy J, Bell GS, Peacock JL, Sisodiya SM, Sander JW. Epilepsy as a systemic condition: Link with somatic comorbidities. Acta Neurol Scand. 2017;136(4):352–9. [DOI] [PubMed] [Google Scholar]
- 7.Klein P, Dingledine R, Aronica E, Bernard C, Blümcke I, Boison D, et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia. 2018;59(1):37–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whelan CD, Altmann A, Botía JA, Jahanshad N, Hibar DP, Absil J, et al. Structural brain abnormalities in the common epilepsies assessed in a worldwide ENIGMA study. Brain. 2018;141(2):391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vadlamudi L, Milne RL, Lawrence K, Heron SE, Eckhaus J, Keay D, et al. Genetics of epilepsy: The testimony of twins in the molecular era. Neurology. 2014;83(12):1042–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corey LA, Pellock JM, Kjeldsen MJ, Nakken KO. Importance of genetic factors in the occurrence of epilepsy syndrome type: A twin study. Epilepsy Res. 2011;97(1–2):103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heidari M, Johnstone DM, Bassett B, Graham RM, Chua ACG, House MJ, et al. Brain iron accumulation affects myelin-related molecular systems implicated in a rare neurogenetic disease family with neuropsychiatric features. Mol Psychiatry. 2016;21(11):1599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153(3):707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson MR, Shkura K, Langley SR, Delahaye-Duriez A, Srivastava P, Hill WD, et al. Systems genetics identifies a convergent gene network for cognition and neurodevelopmental disease. Nat Neurosci. 2016;19(2):223–32. [DOI] [PubMed] [Google Scholar]
- 15.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hawrylycz M, Miller JA, Menon V, Feng D, Dolbeare T, Guillozet-Bongaarts AL, et al. Canonical genetic signatures of the adult human brain. Nat Neurosci. 2015;18(12):1832–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanc F, Martinian L, Liagkouras I, Catarino C, Sisodiya SM, Thom M. Investigation of widespread neocortical pathology associated with hippocampal sclerosis in epilepsy: A postmortem study. Epilepsia. 2011;52(1):10–21. [DOI] [PubMed] [Google Scholar]
- 18.Frigerio F, Pasqualini G, Craparotta I, Marchini S, van Vliet EA, Foerch P, et al. n-3 docosapentaenoic acid-derived protectin D1 promotes resolution of neuroinflammation and arrests epileptogenesis. Brain. 2018;141(11):3130–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jimenez-Mateos EM, Engel T, Merino-Serrais P, McKiernan RC, Tanaka K, Mouri G, et al. Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat Med. 2012;18(7):1087–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iori V, Iyer AM, Ravizza T, Beltrame L, Paracchini L, Marchini S, et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis. 2017. Mar;99:12–23. [DOI] [PubMed] [Google Scholar]
- 21.Gu B, Huang YZ, He XP, Joshi RB, Jang W, McNamara JO. A Peptide Uncoupling BDNF Receptor TrkB from Phospholipase Cγ1 Prevents Epilepsy Induced by Status Epilepticus. Neuron. 2015;88(3):484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.French L, Paus T. A FreeSurfer view of the cortical transcriptome generated from the Allen Human Brain Atlas. Front Neurosci. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menden K, Marouf M, Oller S, Dalmia A, Magruder DS, Kloiber K, et al. Deep learning--based cell composition analysis from tissue expression profiles. Sci Adv. 2020;6(30):eaba2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiner J 3rd, Domaszewska T. tmod: an R package for general and multivariate enrichment analysis. PeerJ. 2016;4. [Google Scholar]
- 25.Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013;36(3):174–84. [DOI] [PubMed] [Google Scholar]
- 26.Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava PK, van Eyll J, Godard P, Mazzuferi M, Delahaye-Duriez A, Van Steenwinckel J, et al. A systems-level framework for drug discovery identifies Csf1R as an anti-epileptic drug target. Nat Commun. 2018;9(1):3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017;21(2):366–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. 2015;3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. 2016;19(3):504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Sci (New York, NY). 2014;343(6175):1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson DIG, Langston RF, Schlesiger MI, Wagner M, Watanabe S, Ainge JA. Lateral entorhinal cortex is critical for novel object-context recognition. Hippocampus. 2013;23(5):352–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delahaye-Duriez A, Srivastava P, Shkura K, Langley SR, Laaniste L, Moreno-Moral A, et al. Rare and common epilepsies converge on a shared gene regulatory network providing opportunities for novel antiepileptic drug discovery. Genome Biol. 2016;17(1):245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boer K, Spliet WGM, Van Rijen PC, Redeker S, Troost D, Aronica E. Evidence of activated microglia in focal cortical dysplasia. J Neuroimmunol. 2006;173(1–2):188–95. [DOI] [PubMed] [Google Scholar]
- 36.Aronica E, Gorter JA, Redeker S, Ramkema M, Spliet WGM, Van Rijen PC, et al. Distribution, characterization and clinical significance of microglia in glioneuronal tumours from patients with chronic intractable epilepsy. Neuropathol Appl Neurobiol. 2005;31(3):280–91. [DOI] [PubMed] [Google Scholar]
- 37.Ravizza T, Gagliardi B, Noé F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29(1):142–60. [DOI] [PubMed] [Google Scholar]
- 38.Iyer A, Zurolo E, Spliet WGM, Van Rijen PC, Baayen JC, Gorter JA, et al. Evaluation of the innate and adaptive immunity in type i and type II focal cortical dysplasias. Epilepsia. 2010;51(9):1763–73. [DOI] [PubMed] [Google Scholar]
- 39.Ravizza T, Boer K, Redeker S, Spliet WGM, van Rijen PC, Troost D, et al. The IL-1beta system in epilepsy-associated malformations of cortical development. Neurobiol Dis. 2006;24(1):128–43. [DOI] [PubMed] [Google Scholar]
- 40.Morin-Brureau M, Milior G, Royer J, Chali F, LeDuigou C, Savary E, et al. Microglial phenotypes in the human epileptic temporal lobe. Brain. 2018;141(12):3343–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boche D, Perry VH, Nicoll JAR. Activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39(1):3–18. [DOI] [PubMed] [Google Scholar]
- 42.Hendrickx DAE, van Eden CG, Schuurman KG, Hamann J, Huitinga I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J Neuroimmunol. 2017;309:12–22. [DOI] [PubMed] [Google Scholar]
- 43.Somani A, El-Hachami H, Patodia S, Sisodiya S, Thom M. Regional microglial populations in central autonomic brain regions in SUDEP. Epilepsia. 2021;62(6):1318–28. [DOI] [PubMed] [Google Scholar]
- 44.Wu W, Li Y, Wei Y, Bosco DB, Xie M, Zhao M-G, et al. Microglia depletion aggravates the severity of acute and chronic seizures in mice. Brain Behav Immun. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinhard L, Di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun. 2018;9(1):1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caciagli L, Bernasconi A, Wiebe S, Koepp MJ, Bernasconi N, Bernhardt BC. A meta-Analysis on progressive atrophy in intractable temporal lobe epilepsy: Time is brain? Neurology. 2017;89(5):506–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blumcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG, et al. Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N Engl J Med. 2017;377(17):1648–56. [DOI] [PubMed] [Google Scholar]
- 48.Amhaoul H, Hamaide J, Bertoglio D, Reichel SN, Verhaeghe J, Geerts E, et al. Brain inflammation in a chronic epilepsy model: Evolving pattern of the translocator protein during epileptogenesis. Neurobiol Dis. 2015;82:526–39. [DOI] [PubMed] [Google Scholar]
- 49.Gershen LD, Zanotti-Fregonara P, Dustin IH, Liow J-S, Hirvonen J, Kreisl WC, et al. Neuroinflammation in Temporal Lobe Epilepsy Measured Using Positron Emission Tomographic Imaging of Translocator Protein. JAMA Neurol. 2015;72(8):882–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butler T, Li Y, Tsui W, Friedman D, Maoz A, Wang X, et al. Transient and chronic seizure-induced inflammation in human focal epilepsy. Epilepsia. 2016;57(9):e191–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta N, Shyamasundar S, Patnala R, Karthikeyan A, Arumugam TV., Ling EA, et al. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin Ther Targets. 2018;22(9):765–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The complete normalized microarray gene expression data from the Allen Institute for Brain Science that support the findings of this study are available from the institute’s website at http://human.brain-map.org/static/download. The eQTL summary statistics on state-specific monocytes that support the findings of this study are available in the supplementary tables at DOI: 10.1126/science.1246949. The summary results from the EpiPGX GWAS data that support the findings of this study are available from http://www.epigad.org. Data from the experimental model that support the findings of this study are available from the corresponding author upon reasonable request.