

Abstract
The human proteins used in most biochemical studies are commonly obtained using bacterial expression. Owing to its relative simplicity and low cost, this approach has been extremely successful, but is inadequate for many proteins that require the mammalian folding machinery and posttranslational modifications (PTMs) for function. Moreover, the expressed proteins are typically purified using N- and/or C-terminal affinity tags, which are often left on proteins or leave non-native extra amino acids when removed proteolytically. Many proteins cannot tolerate such extra amino acids for function. Here we describe a protein production method that resolves both these issues. Our method combines expression in human Expi293F cells, which grow in suspension to high density and can process native PTMs, with a chitin-binding domain (CBD)-intein affinity purification and self-cleavable tag, which can be precisely removed after purification. In this protocol, we describe how to clone a target gene into our specifically designed human cell expression vector (pJCX4), and how to efficiently transfect the Expi293F cells and purify the expressed proteins using a chitin affinity resin.
Graphic abstract:
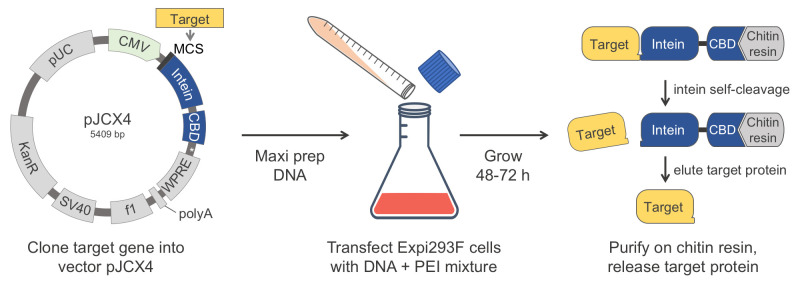
Keywords: Expi293F cells, Protein expression, Post-translational Modification (PTM), Affinity purification, Intein
Background
The study of mammalian proteins in vitro often requires large amounts of pure proteins, which are typically expressed in bacteria and purified via the addition of N- and/or C-terminal affinity tags. These tags are often left on proteins or leave non-native extra amino acids when removed proteolytically (Waugh, 2011). The use of bacterial protein expression offers many advantages; it is a well-established system available in many laboratories, its adoption is relatively straightforward, and the overall cost of protein production is low. For these reasons, this will continue to be the method of choice for most applications. However, there are many cases in which bacterial expression is inadequate. A major disadvantage is the inability of bacteria to process posttranslational modifications (PTMs), which are often crucial for function ( Brown et al., 2017 ). Even in cases where the role of PTMs is well established, they tend to be ignored, or their absence considered to be an unavoidable drawback of the method. Many mammalian proteins also have specific folding requirements that the bacterial expression system cannot account for (Rosano and Ceccarelli, 2014; Georgiou et al., 1994 ). The N- and C-termini of proteins are often important for function, such that many proteins cannot tolerate extra amino acids from purification tags at the ends ( Brault et al., 1999 ; Wu and Filutowicz, 1999; Chant et al., 2005 ; Sliwinska et al., 2008 ; Suh-Lailam and Hevel, 2009). Here, we describe a method that resolves these concerns and can be broadly applied to protein expression. Our method combines expression in human Expi293F cells, which grow in suspension to high density and can process native PTMs, with a chitin-binding domain (CBD)-intein affinity purification and self-cleavable tag, which can be precisely removed after purification. The vector generated here to implement this method (pJCX4) has elements necessary to drive high protein expression in human cells, including the cytomegalovirus (CMV) promoter, the Woodchuck Hepatitis Virus (WHV) Posttranscriptional Regulatory Element (WPRE), and the Simian Virus 40 (SV40) PolyA sequence. This vector also supports cloning and plasmid production in E. coli, using the pUC origin of replication and the kanamycin resistance gene. Our goal here is to provide a detailed description of the method, to facilitate its adoption by new users. We have also deposited our vector with Addgene for the convenience of users that would like to adopt this method.
Materials and Reagents
-
Preparation of plasmid DNA
Polystyrene Petri dish, 10-cm (Stellar Scientific, catalog number: TC-WB-7452)
pJCX4 vector (Addgene, catalog number: 170756)
NEBNext High-Fidelity 2× PCR Master Mix (New England BioLabs, catalog number: M0541L)
SYBR Safe DNA Gel Stain (Thermo Fisher Scientific, Invitrogen, catalog number: S33102)
Tris base (Fisher Scientific, catalog number: BP152-500)
Glacial acetic acid (Millipore Sigma, catalog number: A6283-1L)
Ethylenediaminetetraacetic acid (EDTA) (Thermo Fisher Scientific, Invitrogen, catalog number: 15576028)
NucleoSpin Gel and PCR Cleanup, Mini kit for gel extraction and PCR clean up (Macherey-Nagel, catalog number: 740609.50)
NucleoSpin Plasmid, Mini kit for plasmid DNA (Macherey-Nagel, catalog number: 740588.50)
High-Fidelity restriction enzymes (New England BioLabs)
SeaKem Gold agarose (Lonza, catalog number: 50152)
Quick Ligation Kit (New England BioLabs, catalog number: M2200S)
XL-10 Gold competent cells (Agilent, catalog number: 200317)
LB Agar (Thermo Fisher Scientific, catalog number: 22700025)
Kanamycin monosulfate (Gold Biotechnology, catalog number: K-120-5)
PureLink HiPure Plasmid Filter Maxiprep Kit (Thermo Fisher Scientific, Invitrogen, catalog number: K210017)
0.5 M EDTA (see Recipes)
50× Tris-acetate-EDTA (TAE) (see Recipes)
1× TAE (see Recipes)
1% agarose gel with SYBR Safe DNA gel stain (see Recipes)
-
Expression in Expi293F Cells
125-mL and 500-mL polycarbonate, disposable, sterile, vent-cap Erlenmeyer shaker flasks (TriForest Labware, catalog numbers: FPC0125S and FPC0500S)
15-mL and 50-mL conical tubes (Fisher Scientific, Falcon, catalog numbers: 14-959-53A and 14-432-22)
10-mL serological pipettes (Corning, catalog number: 357551)
500-mL centrifuge bottles (Corning, catalog number: CLS431123-6EA)
Expi293F Expression System Kit (Thermo Fisher Scientific, Gibco, catalog number: A14635)
Expi293F Cells (Thermo Fisher Scientific, Gibco, catalog number: A14527)
Expi293F Expression Medium (Thermo Fisher Scientific, Gibco, catalog number: A1435102)
Opti-MEM Reduced-Serum Medium (Thermo Fisher Scientific, Gibco, catalog number: 31985070)
PEI MAX, Transfection Grade Linear Polyethylenimine Hydrochloride (Polysciences, catalog number: 24765-1)
Dulbecco’s Phosphate-Buffered Saline (PBS) (Thermo Fisher Scientific, catalog number: MT21031CV)
1 mg/mL PEI transfection reagent (see Recipes)
-
Purification of the intein-tagged protein
50-mL polycarbonate bottles with caps (Beckman Coulter, catalog number: 361693)
Chitin affinity resin (New England BioLabs, catalog number: S6651S)
KONTES chromatography Flex-Column (Avantor, VWR, catalog number: KT420401-2510)
HEPES (Gold Biotechnology, catalog number: H-400-1)
Sodium Chloride (NaCl) (Thermo Scientific, catalog number: J21618-A9)
DL-Dithiothreitol (DTT) (Gold Biotechnology, catalog number: DTT100)
Phenylmethylsulfonyl fluoride (PMSF) (Gold Biotechnology, catalog number: P-470-25)
β-mercaptoethanol (βME) (Santa Cruz Biotechnology, catalog number: sc-202966)
Adenosine-5’-triphosphate (ATP) (Gold Biotechnology, catalog number: A-081-1)
Coomassie Brilliant Blue R-250 (Gold Biotechnology, catalog number: C-461-5)
Methanol (Ibis Scientific, catalog number: MX0486-1)
Acetic acid (Ibis Scientific, catalog number: CP-M1010S)
12% acrylamide SDS-PAGE gels (Bio-Rad, catalog number: 4561046)
Coomassie stain solution for acrylamide gels (see Recipes)
Lysis buffer (see Recipes)
ATP wash buffer (see Recipes)
Intein cleavage buffer (see Recipes)
Equipment
-
Preparation of plasmid DNA
PTC-200 thermocycler (MJ Research, catalog number: MJ-G200)
Horizontal electrophoresis system (Fisherbrand, catalog number: FB-SBR-2025)
Eppendorf tabletop centrifuge (Eppendorf, catalog number: EP-5415D)
Water bath (Thomas Scientific, Humboldt, catalog number: 1196X13)
Imperial-III 37°C incubator (Lab-Line, catalog number: 305)
QIArack (Qiagen, catalog number: 19015)
-
Expression in Expi293F Cells
Isotemp CO2 Incubator (Fisherbrand, catalog number: 11-676-604)
CO2 resistant orbital shaker (Thermo Scientific, catalog number: 88881101)
Biological safety cabinet (Fisher Scientific, catalog number: 13-261-222)
Refrigerated benchtop centrifuge (Beckman Coulter, model: Allegra X-15R)
-
Purification of the intein-tagged protein
Dounce glass piston homogenizer (VWR, Kontes Glass, catalog number: KT885300-0040)
Beckman Avanti J-20-I centrifuge (Beckman Coulter, catalog number: 8043-30-1169)
JA25.50 rotor (Beckman Coulter, catalog number: 363055)
Peristaltic pump (Bio-Rad, catalog number: 7318140)
SDS-PAGE gel running apparatus (Bio-Rad, catalog number: 1658025FC)
Procedure
-
Preparation of plasmid DNA
Design PCR primers to amplify the target gene for insertion into the multicloning site (MCS) of plasmid pJCX4 ( Figure 1A ). Include a 5’ overhang on each primer according to the design guide ( Figure 1B ).
-
Mix the following components for the PCR reaction.
50 μL of PCR reaction volume:
25 μL of NEBNext 2× PCR master mix
1 μL of target gene template DNA (1–10 ng DNA)
2.5 μL of forward primer at 10 μM (final concentration = 0.5 μM)
2.5 μL of reverse primer at 10 μM (final concentration = 0.5 μM)
19 μL of ddH2O
-
Carry out the PCR reaction in a thermocycler
Initial denaturation (30 s, 98°C)
Denaturation (10 s, 98°C)
Annealing (30 s, 52–65°C)
-
Extension (25 s/kb, 72°C)
Repeat steps b–d for 30 cycles.
Final extension (2 min, 72°C)
Run the PCR product on a 1% agarose gel with SYBR Safe, in a horizontal electrophoresis system at 80 V for 20 min.
Extract the PCR product band from the gel and recover the DNA following the manufacturer’s protocol, using the NucleoSpin Gel and PCR cleanup kit. Elute the recovered PCR product in a volume of 25 μL.
-
Digest the recovered PCR product with restriction enzymes corresponding to those included in the PCR primer overhang.
30 μL of reaction volume:
25 μL of recovered PCR product, about 1 μg of DNA
1 μL of High-Fidelity enzyme 1 (incorporated on 5’ overhang of forward primer)
1 μL of High-Fidelity enzyme 2 (incorporated on 5’ overhang of reverse primer)
3 μL of 10× buffer (CutSmart buffer if using NEB High-Fidelity enzymes)
Incubate in a water bath at 37°C for 1 h.
-
Digest the plasmid pJCX4 with the same restriction enzymes incorporated on the primer overhangs of the PCR product.
30 μL of reaction volume:
variable volume of plasmid pJCX4 (1 μg of DNA)
1 μL of High-Fidelity enzyme 1
1 μL of High-Fidelity enzyme 2
3 μL of 10× buffer (CutSmart buffer if using NEB High-Fidelity enzymes)
ddH2O up to a total volume of 30 μL
Incubate in a water bath at 37°C for 1 h.
Run the digested plasmid on a 1% agarose gel with SYBR Safe at 80 V for 20 min. Extract the plasmid band from the gel and recover the DNA following the manufacturer’s protocol, using the NucleoSpin Gel and PCR cleanup kit. Elute the digested plasmid DNA in volume of 20 μL.
Recover the digested PCR product following the manufacturer’s protocol, using the NucleoSpin Gel and PCR cleanup kit. Elute the digested plasmid DNA in a volume of 20 μL.
-
Mix the following components for a ligation reaction, using a plasmid to insert ratio of 2:8.
20 μL of reaction volume:
10 μL of Quick Ligase 2× buffer
8 μL of digested PCR product
2 μL of digested plasmid
1 μL of Quick Ligase enzyme
Incubate in a water bath at 25°C for 10 min.
Transform 5 μL of ligation mixture into XL-10 Gold competent cells, following the manufacturer’s protocol. Plate 200 μL of the transformation on an LB agar plate containing 50 µg/mL kanamycin and grow in an incubator at 37°C for 12–18 h.
Scrape half of a single colony to inoculate a 5 mL culture of LB containing 50 µg/mL kanamycin and grow in an incubator with agitation (220 rpm) at 37°C for 12–18 h. Indicate on the plate the culture used for inoculation, and store the LB agar plate at 4°C.
Isolate plasmid DNA from the culture using the NucleoSpin Plasmid miniprep kit, following the manufacturer’s protocol.
-
Confirm the presence and location of the target gene in the plasmid by Sanger sequencing with CMV-for (forward) and Mxe-rev (reverse) sequencing primers. See Table 1 for primer sequences.
Table 1. Primers used to verify the insertion of the target gene into the MCS of pJCX4.
Primer name Primer sequence CMV-for 5’- CGCAAATGGGCGGTAGGCGTG Mxe-rev 5’- GATTGCCATGCCGGTCAAGG From the LB agar plate, use the same colony that was sequence-verified to contain the target gene to inoculate a 100 mL culture of LB containing 50 µg/mL kanamycin.
Isolate plasmid DNA from the culture using the PureLink HiPure Plasmid Filter Maxiprep Kit and a QIArack following the manufacturer’s protocol.
Note: The miniprep and maxiprep kits are both used to isolate plasmid DNA from bacterial cultures. The maxiprep is used once the plasmid is sequence-verified, to produce large amounts of DNA needed for transfection of Expi293F cells.
-
Expression in Expi293F Cells
Follow the manufacturer’s protocol for the Expi293F Expression System Kit to thaw, establish, and culture cells. Grow the cells in Expi293F media and sterile vent-cap Erlenmeyer shaker flasks, on an orbital shaker (125 rpm) at 37°C using an incubator with a humidified atmosphere of 8% CO2. No antibiotic is required for selection.
Prepare cultures for transfection at a density of 2.5 × 106 cells/mL in sterile vent-cap Erlenmeyer shaker flasks. We typically transfect a 250 mL-culture. Keep the cultures inside the incubator at 37°C with 8% CO2 to maximize their health.
Aliquot 5 mL of Opti-MEM at 25°C into each of two 15-mL conical tubes under a sterile cabinet.
Into one conical tube of Opti-MEM, add 1 µg of pure plasmid DNA from the maxiprep for each 1 mL of Expi293F culture to transfect.
Into the other conical tube of Opti-MEM, add 3 µL of 1 mg/mL polyethylenimine (PEI) for each 1 mL of Expi293F culture to transfect.
Incubate the DNA and PEI mixtures separately for 5 min at 25°C under the sterile cabinet, then combine both mixtures into one 15-mL conical tube and incubate for another 25 min.
Briefly transfer the Expi293F culture under the cabinet to add the DNA and PEI mixture, then return it to the incubator to grow for 48 to 72 h, as was optimized for each protein. To optimize the growth time, perform small-scale purifications or Western blot to verify protein expression levels at various times post-transfection (Figute 2).
Harvest the cells by centrifugation at 4,000 × g and 4°C for 10 min. To wash residual media, resuspend the cell pellet in 10 mL of PBS at 4°C using a serological pipette, transfer to a 50-mL conical tube, then pellet the cells again by centrifugation. Store at -80°C until purification.
Figure 2. Schematic of Expi293F transfection.
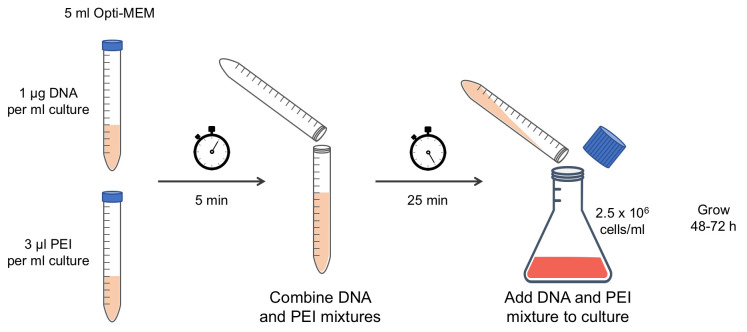 Pure plasmid DNA and polyethylenimine (PEI) are incubated separately in Opti-MEM media for 5 min, then combined and incubated an additional 25 min. The DNA and PEI mixture is poured into an Expi293F culture at a density of 2.5 × 106 cells/mL and the culture is grown for 48 to 72 h after transfection.
Pure plasmid DNA and polyethylenimine (PEI) are incubated separately in Opti-MEM media for 5 min, then combined and incubated an additional 25 min. The DNA and PEI mixture is poured into an Expi293F culture at a density of 2.5 × 106 cells/mL and the culture is grown for 48 to 72 h after transfection. -
Purification of the intein-tagged protein
Prepare fresh lysis buffer (see Recipes) the day of purification and store at 4°C.
Prepare a Flex-Column with chitin affinity resin, by pipetting ~15 mL of the manufacturer’s resin mixture into the column, then packing the resin at the bottom by allowing the storage solution to drain through. The packed resin volume should be 7–10 mL.
Equilibrate the chitin affinity column by flowing 10 column volumes of lysis buffer through. For this and all subsequent steps, either flow by gravity or by peristaltic pump at a rate of 1 mL/min.
Thaw the conical tube containing the pellet of transfected Expi293F cells in a water bath at 25°C.
Resuspend the cells in a volume of lysis buffer equal to two times the volume of the cell pellet, by pipetting up and down using a serological pipette.
Lyse the cells with 20 strokes of a Dounce glass piston homogenizer, using a tight-fitting piston. Avoid introducing bubbles while using the homogenizer to lyse the cells.
Transfer the lysate to centrifuge tubes, and clarify by centrifugation at 20,000 × g and 4°C for 20 min.
Load the clarified lysate onto the equilibrated chitin affinity column.
Wash the column with 20 column volumes of lysis buffer.
Wash the column with 10 column volumes of ATP wash buffer (see Recipes). ATP helps to remove unwanted chaperone proteins bound to the target protein.
Remove excess buffer from the resin, then add an amount of intein cleavage buffer (see Recipes) equal to two times the volume of the resin. Gently stir the resin to evenly distribute the cleavage buffer.
Incubate the resin in intein cleavage buffer at 25°C for 24 h.
Elute the cleaved target protein by draining the liquid from the chitin column. Protein storage will depend on the stability of each protein. Generally, proteins can be stored for several days on ice or long-term in a -80°C freezer.
-
Example
The procedure described here has been used for the expression-purification of several proteins in our laboratory. One example is tropomyosin (Tpm), a parallel coiled-coil protein that binds symmetrically on each side of the actin filament, forming a long rope-like polymer of head-to-tail interacting Tpm molecules. The head-to-tail interaction of Tpm depends on the integrity of both its N- and C-terminal ends, as well as native N-terminal acetylation (Heald and Hitchcock-DeGregori, 1988; Maytum et al., 2000 ; Palm et al., 2003 ), and it is also affected by other forms of PTMs, such as phosphorylation ( Rao et al., 2009 ; Lehman et al., 2015 ; Palani et al., 2019 ; Rajan et al., 2019 ). Numerous biochemical studies on Tpm had relied on proteins expressed in bacteria that used N-terminal acetylation mimics which, as we have shown ( Carman et al., 2021 ), do not accurately account for the native PTMs of some Tpm isoforms. The expression system described here allowed us to obtain fully native Tpm proteins for the first time, free of extra amino acids from tags, and featuring native PTMs, such as N-terminal acetylation ( Figure 3 ).
-
Considerations
If the target protein cannot tolerate incubation at 25°C, intein self-cleavage may be performed at 4°C; however, to achieve full cleavage, the incubation time will likely be longer (48 to 72 h). While we have had success using this method to obtain proteins featuring disulfide bonds, such as nanobodies, the reducing agent needed to trigger intein cleavage may make this method unsuitable for some disulfide bond-containing proteins.

Figure 1. Instructions for cloning a target gene into vector pJCX4.

(A) Schematic representation of the expression cassette of vector pJCX4 for mammalian cell expression, including a restriction enzyme map of the multicloning site for the insertion of a target gene. The recognition site of each restriction enzyme is highlighted in grey, and the digestion site is indicated by magenta lines. (B) Two examples of forward (5’) and reverse (3’) primer designs to amplify a target gene for insertion into vector pJCX4. The underlined sequence is the recognition site of the restriction enzyme. Note that the three bases preceding the recognition site are included to ensure efficient enzyme cleavage. In both 5’ primers, the Kozak consensus sequence GCCACC follows the restriction enzyme site. NNN NNN indicates the nucleotide sequence of the target gene. In the 3’ primers, the bases following the restriction enzyme site are required to ensure precise ligation with the intein. Do not include a stop codon in the annealing region of the 3’ primer.
Figure 3. SDS-PAGE analysis Tpm3.1 obtained using the method described here.

Data analysis
Protein expression and purification may be analyzed by SDS-PAGE. Acrylamide gels (12%) may be loaded with 10 μL of protein samples and run at 200 V for 50 min. Protein bands can be visualized by staining with Coomassie stain and destaining (see Recipes).
Recipes
-
0.5 M EDTA
Add 18.61 g of EDTA to 80 mL of ddH2O and stir with a magnetic stirrer.
Adjust the pH to 8.0 using sodium hydroxide pellets. The EDTA will not be completely dissolved until the solution is close to pH 8.0.
Autoclave or sterile-filter through a 0.22 μm vacuum membrane.
-
50× TAE
242 g of Tris base
57.1 mL of glacial acetic acid
100 mL of 0.5 M EDTA, pH 8.0
ddH2O up to 1 L
Store at 25°C.
-
1× TAE
20 mL of 50× TAE
ddH2O up to 1 L
Store at 25°C.
-
1% agarose gel with SYBR Safe DNA gel stain
Add 0.5 g of agarose into a 250-mL beaker and suspend in 50 mL of 1× TAE.
Microwave in 30 s increments until the solution is clear. Avoid letting the solution boil over the edge of the beaker.
Allow the solution to cool to approximately 50°C. Add 5 mL of 1,000× SYBR Safe DNA gel stain to the solution and swirl to mix.
Pour the agarose solution into a gel casting apparatus and allow it to solidify.
-
1 mg/mL PEI transfection reagent
Add 100 mg of PEI-MAX into a beaker and suspend in 90 mL of ddH2O. Stir with a stir bar.
Add drops of hydrochloric acid until pH is <2.0.
Cover the top of the beaker and stir until the powder fully dissolves.
Add drops of sodium hydroxide until pH is 7.0.
Transfer solution to 100-mL graduated cylinder and add ddH2O up to 100 mL.
Sterile-filter through a 0.22 μm vacuum membrane. Aliquot into 1.5-mL Eppendorf tubes under a sterile cabinet, and store at 4°C.
-
Coomassie stain solution for acrylamide gels
40% methanol (v/v)
10% acetic acid (v/v)
1 g Coomassie Brilliant Blue R-250
-
Destain solution for acrylamide gels
40% methanol (v/v)
10% acetic acid (v/v)
-
Lysis buffer
20 mM HEPES pH 7.5
500 mM NaCl
1 mM DTT
1 mM phenylmethanesulfonyl fluoride (PMSF)
-
ATP wash buffer
20 mM HEPES pH 7.5
200 mM NaCl
5 mM MgCl2
5 mM ATP
-
Intein cleavage buffer
20 mM HEPES pH 7.5
200 mM NaCl
50 mM β-mercaptoethanol (βME)
Acknowledgments
This protocol was adapted from a recently published study ( Carman et al., 2021 ). This work was supported by National Institutes of Health Grants R01 GM073791 and RM1 GM136511 to R.D. and a Blavatnik Family Foundation fellowship to P.J.C.
Competing interests
The authors declare no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1. Brault V., Reedy M. C., Sauder U., Kammerer R. A., Aebi U. and Schoenenberger C.(1999). Substitution of flight muscle-specific actin by human(beta)-cytoplasmic actin in the indirect flight muscle of Drosophila . J Cell Sci 21): 3627-3639. [DOI] [PubMed] [Google Scholar]
- 2. Brown C. W., Sridhara V., Boutz D. R., Person M. D., Marcotte E. M., Barrick J. E. and Wilke C. O.(2017). Large-scale analysis of post-translational modifications in E. coli under glucose-limiting conditions . BMC Genomics 18(1): 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carman P. J., Barrie K. R. and Dominguez R.(2021). Novel human cell expression method reveals the role and prevalence of posttranslational modification in nonmuscle tropomyosins. J Biol Chem 297(4): 101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chant A., Kraemer-Pecore C. M., Watkin R. and Kneale G. G.(2005). Attachment of a histidine tag to the minimal zinc finger protein of the Aspergillus nidulans gene regulatory protein AreA causes a conformational change at the DNA-binding site. Protein Expr Purif 39(2): 152-159. [DOI] [PubMed] [Google Scholar]
- 5. Georgiou G., Valax P., Ostermeier M. and Horowitz P. M.(1994). Folding and aggregation of TEM beta-lactamase: analogies with the formation of inclusion bodies in Escherichia coli . Protein Sci 3(11): 1953-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heald R. W. and Hitchcock-DeGregori S. E.(1988). The structure of the amino terminus of tropomyosin is critical for binding to actin in the absence and presence of troponin. J Biol Chem 263(11): 5254-5259. [PubMed] [Google Scholar]
- 7. Lehman W., Medlock G., Li X. E., Suphamungmee W., Tu A. Y., Schmidtmann A., Ujfalusi Z., Fischer S., Moore J. R., Geeves M. A., et al.(2015). Phosphorylation of Ser283 enhances the stiffness of the tropomyosin head-to-tail overlap domain. Arch Biochem Biophys 571: 10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maytum R., Geeves M. A. and Konrad M.(2000). Actomyosin regulatory properties of yeast tropomyosin are dependent upon N-terminal modification. Biochemistry 39(39): 11913-11920. [DOI] [PubMed] [Google Scholar]
- 9. Palani S., Köster D. V., Hatano T., Kamnev A., Kanamaru T., Brooker H. R., Hernandez-Fernaud J. R., Jones A. M. E., Millar J. B. A., Mulvihill D. P., et al.(2019). Phosphoregulation of tropomyosin is crucial for actin cable turnover and division site placement. J Cell Biol 218(11): 3548-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palm T., Greenfield N. J. and Hitchcock-DeGregori S. E.(2003). Tropomyosin ends determine the stability and functionality of overlap and troponin T complexes. Biophys J 84(5): 3181-3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rajan S., Jagatheesan G., Petrashevskaya N., Biesiadecki B. J., Warren C. M., Riddle T., Liggett S., Wolska B. M., Solaro R. J. and Wieczorek D. F.(2019). Tropomyosin pseudo-phosphorylation results in dilated cardiomyopathy. J Biol Chem 294(8): 2913-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rao V. S., Marongelli E. N. and Guilford W. H.(2009). Phosphorylation of tropomyosin extends cooperative binding of myosin beyond a single regulatory unit. Cell Motil Cytoskeleton 66(1): 10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosano G. L. and Ceccarelli E. A.(2014). Recombinant protein expression in Escherichia coli: advances and challenges . Front Microbiol 5: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sliwinska M., Skorzewski R. and Moraczewska J.(2008). Role of actin C-terminus in regulation of striated muscle thin filament. Biophys J 94(4): 1341-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suh-Lailam B. B. and Hevel J. M.(2009). Efficient cleavage of problematic tobacco etch virus(TEV)-protein arginine methyltransferase constructs. Anal Biochem 387(1): 130-132. [DOI] [PubMed] [Google Scholar]
- 16. Waugh D. S.(2011). An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif 80(2): 283-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu J. and Filutowicz M.(1999). Hexahistidine(His6)-tag dependent protein dimerization: a cautionary tale. Acta Biochim Pol 46(3): 591-599. [PubMed] [Google Scholar]