

Abstract
As a model to interrogate human macrophage biology, macrophages differentiated from human induced pluripotent stem cells (hiPSCs) transcend other existing models by circumventing the variability seen in human monocyte-derived macrophages, whilst epitomizing macrophage phenotypic and functional characteristics over those offered by macrophage-like cell lines ( Mukherjee et al., 2018 ). Furthermore, hiPSCs are amenable to genetic manipulation, unlike human monocyte-derived macrophages (MDMs) (van Wilgenburg et al., 2013 ; Lopez- Yrigoyen et al., 2020 ), proposing boundless opportunities for specific disease modelling.
We outline an effective and efficient protocol that delivers a continual production of hiPSC-derived-macrophages (iMACs), exhibiting human macrophage surface and intracellular markers, together with functional activity.
The protocol describes the resuscitation, culture, and differentiation of hiPSC into mature terminal macrophages, via the initial and intermediate steps of expansion of hiPSCs, formation into embryoid bodies (EBs), and generation of hematopoietic myeloid precursors.
We offer a simplified, scalable, and adaptable technique that advances upon other protocols, utilizing feeder-free conditions and reduced growth factors, to produce high yields of consistent iMACs over a period of several months, economically.
Keywords: Macrophages, Human induced pluripotent stem cells, Embryoid Bodies, Feeder-free, Differentiation
Background
Macrophages occupy multiple tissues and orchestrate both innate and adaptive immune responses. Research into macrophage function and characterization has been impeded by multiple limitations suffered by model systems, such as human primary monocyte-derived macrophages (MDMs) and leukaemia-derived human macrophage-like cell lines ( Hale et al., 2015 ; Alasoo et al., 2015 ; Mukherjee et al., 2018 ). The large volumes of blood required from donors presents ethical and logistical challenges, only exacerbated by the need for repeated venesection owing to the high variability observed within and between donors (van Wilgenburg et al., 2013 ). Human myeloid cell lines, such as THP-1, demonstrate karyotypical abnormalities and do not entirely represent human macrophages phenotypically or functionally (van Wilgenburg et al., 2013 ; Lopez- Yrigoyen et al., 2020 , Baldassarre, et al., 2021 ).
Furthermore, human macrophages are typically resistant to genetic manipulation, hampering disease-specific modelling ( Hale et al., 2015 ). Technology that employs human induced pluripotent stem cells (hiPSCs), boasting a self-renewing source of cells capable of differentiation into three germ layers, that are amenable to genetic manipulation, poses multiple attractive qualities as a model system over others ( Alasoo et al., 2015 ; Lachmann et al., 2015 ; Alasoo et al., 2018 ; Lee et al., 2018 , Nenasheva et al., 2020 , Mukhopadhyay et al., 2020 ).
This protocol improves upon predecessors by the utilisation of feeder-free culture conditions with reduced requirements for growth factors, whilst still generating high yields of consistent terminally differentiated macrophages, which share phenotypical and functional characteristics to primary human MDMs. Millions of hiPSC-derived-macrophages (iMACs) can be conveniently harvested every 5–7 days from generated embryoid bodies (EBs) maintained in culture for several months. The protocol is easy to follow, cost-effective, and can be scaled to the user’s requirements, creating reproducible results for repeated experiments over time.
Materials and Reagents
-
Human induced Pluripotent Stem cells culture and expansion
-
Human induced Pluripotent Stem Cells used in the development of this protocol:
NL9—a fibroblast-derived hiPSC line from a healthy individual, which is widely available ( Baghbaderani et al., 2016 ), obtained from the National Heart, Lung & Blood Institute (NHLBI) iPSC core (please see the Acknowledgments section for more information on the source of the NL9 hiPSC line)
-
KOLF_2—a skin tissue derived hiPSC line from a healthy individual, generated at the Sanger institute, as part of the HipSci initiative
Complete Essential 8 Basal Medium (Thermo Fisher, catalog number: A1517001)
Vitronectin (rhVTN-N) (Gibco, catalog number: A14700) 500 µg/mL
Dulbecco's phosphate-buffered saline (DPBS) without Ca2+/Mg2+ 500 mL (Thermo Fisher, catalog number: 14190144) (Storage conditions: 15–30°C. Shelf life: 36 months from date of manufacture)
ROCK inhibitor (ROCKi) (Y-27632 dihydrochloride) (Sigma, catalog number: Y0503-1MG)
10 cm or 6 well tissue culture treated plates (Corning, catalog number: 430167 or 3516)
0.22 µm filter steri-cups (Merck Millipore, catalog number: 15780319)
15 mL and 50 mL Falcon tubes (Falcon, catalog numbers: 352097 and 352098)
-
-
Embryoid body generation in a feeder-free system
0.5 M ultrapure Ethylenediaminetetraacetic acid (EDTA) (Invitrogen, catalog number: 15575020), 100 mL
Cryopreservation/freezing medium: KnockOut serum replacement (KSR) (Gibco, catalog number: 10828028), 500 mL
Dimethyl sulfoxide (DMSO) (Sigma, catalog number: D2438), 50 mL
Dulbecco's phosphate-buffered saline (DPBS) with Ca2+/Mg2+, 500 mL (Thermo Fisher, catalog number: 14040091) (Storage conditions: 2–8°C. Shelf life: 36 months from date of manufacture)
Bovine serum albumin (BSA) low endotoxin tissue culture-grade (Sigma, catalog number: A9543-5G)
Recombinant Human BMP-4 Protein 10 µg (R&D Systems, catalog number: 314-BP-010)
-
Embryoid Body Transfer, Culture and Macrophage Differentiation
Sterile water for embryo transfer (Sigma, catalog number: W1503)
Gelatin from porcine skin (Sigma, catalog number: G1890)
X-VIVO 15 Serum-free Hematopoietic Cell Medium 500 mL (Lonza, catalog number: LZBE02-060F)
L-Glutamine (200 mM), 100 mL (Gibco, catalog number: 25030181)
Penicillin-Streptomycin (10,000 U/mL), 100 mL (Gibco, catalog number: 15140122)
2-Mercaptoethanol (Sigma, catalog number: M3148)
Recombinant human M-CSF (Peprotech, catalog number: 300-25)
Recombinant human IL-3 (Peprotech, catalog number: 200-03)
RPMI 1640 Medium 500 mL (Sigma, catalog number: R0883)
Ultra-low adherent U bottom 96-well plates (Costar, catalog number: 7007)
10 cm tissue culture dishes or 6-well plates
70-100 µm cell strainers (Falcon)
Complete Essential 8 Basal Medium (see Recipes)
Vitronectin 1 mL vial (see Recipes)
ROCK inhibitor (ROCKi) (see Recipes)
0.5 mM Ethylenediaminetetraacetic acid (EDTA) solution (see Recipes)
Cryopreservation/freezing Medium (see Recipes)
Bovine serum slbumin (BSA) (see Recipes)
Recombinant Human BMP-4 Protein (see Recipes)
Sterile water for embryo transfer (see Recipes)
Gelatin from porcine skin (see Recipes)
EB-Myeloid precursor base medium (see Recipes)
Recombinant Human M-CSF (see Recipes)
Recombinant Human IL-3 (see Recipes)
-
Macrophage Differentiation base medium (see Recipes)
Flow Cytometry Antibodies used in this protocol
CD14 (BV605, Biolegend, catalogue number: 301834)
CD16 (APC, eBioscience, catalogue number: 17-0168-42)
CD80 (BV711, Biolegend, catalogue number: 305236)
CD86 (PE-Dazzle594, Biolegend, catalogue number: 374217)
CD206 (PerCP/Cyanine 5.5, Biolegend, Catelogue number: 321122)
-
CD204 (PE/Cyanine7 Biolegend, catalogue number: 371908)
Primer sequences used in this protocol
TATA-Binding Protein (TBP) (F: GGGAAGGGGCATTATTTG, R: CCAGATAGCAGCACGGTA).
CD68 (F: GGAAATGCCACGGTTCATCCA, R: TGGGGTTCAGTACAGAGATGC)
CSFR1 (F: TCCAACATGCCGGCAACTA, R: GCTCAAGTTCAAGTAGGCACTCTCT)
CD163 (F: TTTGTCAACTTGAGTCCCTTCAC, R: TCCCGCTACACTTGTTTTCAC)
SOX2 F: GCTACAGCATGATGCAGGACCA, R: TCTGCGAGCTGGTCATGGAGTT)
NANOG (F: CTCCAACATCCTGAACCTCAGC, R: CGTCACACCATTGCTATTCTTCG)
OCT4 (F: CCTGAAGCAGAAGAGGATCACC, R: AAAGCGGCAGATGGTCGTTTGG)
Equipment
Class 2 microbiological safety hood or cabinet using an aseptic technique
37°C incubator with 5% CO2
Centrifuge
Phase-contrast microscope (4×, 10×, 40× magnification)
Water bath set at 37°C
-80°C storage
Liquid Nitrogen storage
Cell freezing containers (“Mr Frosty”)
Pipette controller and selection of different volume stripettes (5 mL, 10 mL), which usually have large-bore orifices
Pipettes (P1000, P200, P20, P2) and corresponding sterile tips.
ParafilmTM
Procedure
General Considerations:
All cell culture, media preparation, tissue culture vessels, and other laboratory work is to be conducted inside a Class 2 microbiological hood, using aseptic technique.
The hood should be cleaned thoroughly before use with 3% of Distel (or equivalent disinfectant) and 70% ethanol.
Prepare all media, solutions, and reagents at room temperature (RT). See the Recipes section for more detailed information on media preparation.
-
Human induced Pluripotent Stem cell culture and expansion
Prepare all cell maintenance base media and reagents, as described in the Materials and Reagents or Recipes sections.
Prepare vitronectin plates. Vitronectin is a glycoprotein used to coat the surface of cell culture plates, to facilitate hiPSC attachment and spreading. Thaw one 60 μL aliquot to RT and dilute into 6 mL DPBS without Ca2+/Mg2+. If using a 6-well plate, add 1–1.5 mL per well; alternatively, add 6 mL to one 10 cm dish. Ensure the vitronectin covers the entire surface, and incubate in a hood for 1–2 h at RT. Vitronectin plates can be sealed with ParafilmTM and stored at 4°C for 5 days. Allow to warm to RT for 1–2 h prior to use.
Prepare the E8 basal medium by adding the thawed supplement and allowing the media to warm to RT. We recommend that thawed hiPSCs are cultured and expanded in E8 basal media, and that a master stock of hiPSC lines is frozen and stored early on.
Set up a 15 mL Falcon with 9 mL of E8 media plus 0.312 µM ROCKi for one frozen vial of 1 mL of hiPSC, i.e., 1 µL of ROCKi into a final volume to 10 mL. Although ROCKi allows attachment and recovery of hiPSC, whilst preventing apoptosis & spontaneous differentiation, it must be removed from the media after 24 h, to prevent interference with colony expansion.
-
Partially thaw the frozen hiPSCs quickly until just defrosted, with some ice remaining in the vial, using the water bath at 37°C (thawing should be quick, to avoid prolonged contact with DMSO; however, do not warm the entire vial to 37°C). Add 1 mL of E8 in a drop-wise fashion to the vial and gently transfer the cells to the 15 mL Falcon, using a wide-bore pipette tip or stripette. Invert the tube to mix and dilute out the DMSO.
Note: The exact cell number or density in a cryovial is usually not known nor determined at the time of freezing, to avoid creating a single cell suspension, and to improve recovery after thawing for subsequent culture. Between 6–10 cryovials are generated from one previously 70–80% confluent 10 cm plate of hiPSCs.
Spin at 290 × g and 20°C for 3 min. Discard the supernatant.
-
Aspirate the diluted vitronectin from the plate surface. One thawed cryovial should give a small 5–20 µL cell pellet in a 15 mL Falcon after centrifugation. Resuspend the pellet gently in the desired volume of E8 medium supplemented with 3.12 µM ROCKi (1 µL: 1,000 µL), avoiding excessive pipetting.
Note: ROCKi has been previously used in earlier adapted protocols at 10 µM, but we have titrated this down further, and have seen effective hiPSC attachment and recovery at a lower concentration of 3.12 µM.
Plate 2 mL of cell suspension per well of a 6-well plate or 8 mL per 10 cm dish; aim to use minimum volumes of media to enhance adherence. One frozen vial of hiPSC (equivalent to 1 well of a 6-well plate, or one-tenth of a 10 cm dish at 70–80% confluence), can be split into 2–3 wells of a 6-well plate or if the hiPSC line demonstrates good recovery, then cells can be seeded less densely straight into a 10 cm dish. Recovery is described as good when multiple small colonies have attached to the vitronectin plate 24 h after seeding (see Figure 1), and colony expansion occurs in the following days (see Figure 2).
Place the plate into a humidified 37°C incubator with 5% CO2. Ensure the plate is initially gently agitated to distribute the colonies evenly over the vitronectin.
Check for attachment under a phase-contrast microscope after 24 h (see Figure 1).
Change the medium every 24 h, remove the unattached colonies, dead cells, and spent media with a stripette or aspirator. If there are a lot of dead cells or debris, then wash the plate with DBPS before replacing the E8 media. Replace with fresh E8 basal media gently, using a stripette against the side wall of the vessel (ROCKi supplementation is not required after the first 24 h). hiPSCs are difficult to maintain in culture and are prone to differentiation, media cytokines, and factors that maintain pluripotency, necessitating daily media change.
-
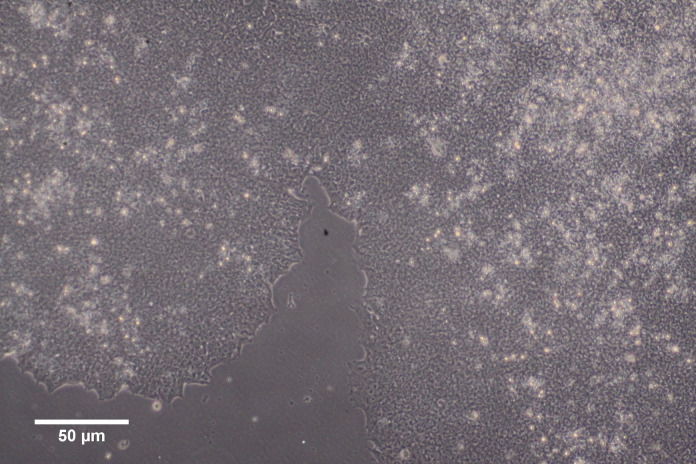
Continue to change media daily until the cells reach 70–80% confluency and ready for passaging. Do not allow the clusters/colonies to fuse together (see Figures 3, 4, and 5), which helps to avoid spontaneous differentiation. Once thawed hiPSCs are established, subsequent steps, culture systems, and further differentiation should take place after 1–2 passages.
Image taken using EVOSTM XL Core imaging system, with a 4× objective.
Figure 5. hiPSC at 70–80% confluency prior to passaging. Images obtained with a Olympus CKX41 Inverted Microscope, equipped with a 20× objective.
-
Passaging and freezing of hiPSC
Prepare vitronectin plates as before.
Remove the exhausted media from the plate with hiPSC at 70–80% confluency and wash the plate/wells twice with DPBS without Ca2+/Mg2+ (approximately 6 mL for 10 cm dish).
Remove the DPBS and add 6 mL of 0.5 mM PBS-EDTA solution, allowing full coverage of the plate surface.
Observe the cells under a phase-contrast microscope every 2 min for up to 4 min, to monitor cell morphology changes and detachment (bright halos and edge enhancement to the colonies) but do not to allow full detachment and separation of cells into single cells.
Gently remove and discard the PBS-EDTA with a stripette by tilting the plate, but do not to dislodge the colonies.
Using a stripette add 8 mL of E8 media to the 10 cm plate aiming to detach and wash the colonies in small clumps and clusters into suspension, by repeated aspiration and flushing of the media through the stripette up to four times with the same 8 mL volume. Collect the media and cells into a new 50 mL Falcon. Repeat this step up to twice more to maximise colony recovery, while avoiding excessive agitation and pipetting, which will result in single cells.
Spin cells at 290 × g and 20°C for 3 min. Discard the supernatant.
Resuspend the colonies in fresh E8 base media supplemented with 3.12 mM ROCKi at the desired splitting ratio (1:5–1:30, depending on the growth rate of the hiPSC line).
Aspirate the vitronectin solution from pre-prepared plates. Transfer and seed the split colonies into a new vitronectin plate/well.
-
Cryopreservation/freezing of hiPSC:
To freeze hiPSC cells, freshly prepare freezing medium (10% DMSO in KSR) at RT. After harvesting the hiPSCs (Steps B2–B7), resuspend the cell pellet in 10% DMSO-KSR and transfer 1 mL of cell suspension per cryovial. It is important that the cells spend minimal time in presence of DMSO at RT. Cryovials should be placed immediately into a Mr. Frosty or appropriate freezing container and into the -80°C freezer and then into liquid nitrogen after 24 h, for long term storage.
Note: The volume of freezing medium to prepare will depend on the number of cryovials to be stored. We advise 1 mL of freezing media per cryovial and 2 cryovials per well of a 6-well plate and 6–10 mL or 6–10 cryovials per 10 cm dish at 70–80% confluency.
-
Embryoid Body formation utilizing a feeder-free system
After harvesting the hiPSC (as described in Steps B2–B7), add 1 mL of fresh E8 media and pipette the hiPSCs up and down more than usual (3–4 pipetting strokes), to break up clumps and create a single-cell suspension.
-
Using a haemocytometer, count the cells in 10 µL of cell suspension, after adding 10 µL of Trypan blue (dilution factor = 2), and determine cell density:
(Number of cells in 3 squares / 3) × dilution factor × 10,000 = Number of cells/mL
-
Make individual, homogenous, and equally-sized EBs using an ultra-low adherent U bottom 96-well plate (1× EB per well). With the knowledge of the total cell number harvested above, both the desired EB size (cell number) and the overall total number of the individual EBs to be made, can be calculated. For example, if 3 × 106 hiPSCs are harvested = 100× individual EBs of 3 × 104 size can be made.
Note: The size of EB may need to be determined for each hiPSC line. However, in our hands, EB sizes of between 1 × 104–5 × 104 cells generate the highest yields of myeloid precursors for both the hiPSC lines used in the generation of this protocol (EBs up to 1 × 105 cells can also work well).
-
Each individual EB is made up in 100 µL of E8 media supplemented with 50 ng/mL recombinant BMP-4 and 3.12 µM ROCKi. Depending on the desired EB size, create a cell suspension of 1 × 105–10 × 105 cells per mL.
For the example calculated above, a cell suspension of 3 × 105 cell/mL should be created to generate individual EBs of 3 × 104 size in 100 µL of supplemented E8 per well of ultra-low adherent U bottom 96 well plate.
Transfer 100 µL of hiPSC cell suspension into each well of an ultra-low adherent U bottom 96-well plate.
Spin the plate at 800 rpm for 1 min and transfer to a humidified 37°C incubator with 5% CO2.
Leave these EBs undisturbed for 48 h and, on day 2 of culture, remove 50 µL of spent media and carefully replace with 50 µL of E8 media supplemented with 50 ng/mL BMP-4 only, aiming to not disturb the forming EB. Repeat for day 3 (see Figure 6). ROCKi can interfere with late EB and germ layer formation.
It is unusual to find more than one EB per well (see Figures 7 and 8); however, providing the appearance of each EB within one well is similar to that seen in Figure 6, with a clear rim and 3D structure, then all EBs within the well can be transferred. Whenever multiple small clumps or a broken and shattered EB appearance is found, the well and cellular material should be discarded.
On day 4, prepare gelatin-coated 10 cm dishes. Add 6 mL of 0.1% gelatine solution to a 10 cm dish and incubate in a hood for 1–2 h, allowing the gelatin to coat the entire surface and warm to RT (plates can be pre-prepared and stored at 4°C for 5 days).
Remove the gelatin solution from the plate prior to EB transfer.
-
Using wide bore P1000 filter tips (alternatively, P1000 filter tips can be cut higher up their length with a scalpel, to create a wide bore orifice), carefully aspirate the individual EBs which are easily visible, and transfer them to the gelatin-coated plate (Figure 9). The number of EBs per cm2 may need to be determined per hiPSC line, nevertheless, in our experience, up to 35× EBs per 10 cm dish provides a high yield of myeloid precursors.
This is an EB in one well of a 96-well ultra-low adherent plate, on day 4 prior to transfer into a gelatin plate. EB size is 3 × 104 hiPSCs. Images taken using EVOSTM XL Core imaging system with a 4× objective.
Once transferred, gently tilt the plate and remove the spent EB media from the plate, without removing EBs.
Slowly add 10–12 mL of fresh EB-Myeloid precursor media supplemented with 50 ng/mL M-CSF, which is critical for myeloid differentiation, and 25 ng/mL IL-3, which is critical for hematopoietic specification and proliferation.
Carefully move these plates to a humidified 37°C incubator with 5% CO2. And leave undisturbed for 1 week, to allow EBs to attach to the gelatin (see Figure 10).
At day 7, carefully remove spent EB media with a large bore stripette to avoid damaging any unattached EBs. Take care not to dislodge any attached EBs and pass the media through a 70–100 µm strainer over a 50 mL Falcon tube, to catch and retain all the unattached EBs that are in suspension.
Return the EBs in the strainer back into the original plate, by inverting the strainer over the 10 cm dish and gently adding fresh EB media supplemented with 50 ng/mL M-CSF and 25 ng/mL IL-3 warmed to RT over the inverted strainer. Continue to take care not to dislodge the attached EBs in the plate.
Once the EB media has been exchanged, and EBs returned to their dish, spin the 50 mL Falcon at 290 × g and 20°C for 3 min. At week 3, this tube will contain the harvested myeloid precursors in suspension. Discard supernatant.
Add 1 mL of macrophage differentiation media to the cell pellet and observe the appearance of the cells under a microscope, counting them with a haemocytometer (see Step C1).
EB media should continue to be exchanged as described in Steps C14–C17 each week. Depending on the hiPSC line, myeloid precursors should be harvested from weeks 3–4 of EB culture.
-
EBs can be maintained in culture for several months. In our hands, myeloid precursor yields begin to fall from 12–16 weeks.
Images taken using EVOSTM XL Core imaging system, with a 4× objective.
-
Macrophage differentiation
Harvest the myeloid precursors as described in Step C14–C17. Following counting, resuspend the harvested myeloid precursors in the desired volume of macrophage differentiation media supplemented with high concentration 100 ng/mL M-CSF.
Seed the cells into the desired culture plates, ideally 0.5 × 106–1 × 106 in approximately 8–10 mL of macrophage differentiation media per 10 cm dish, or alternatively 1.5 × 105 in 3 mL per well of a 6-well plate, or 1.5 × 104 in 150 µL per well on a 96-well plate (see Figure 11a).
Transfer the cells to a humidified 37°C incubator with 5% CO2 and leave undisturbed for 5–7 days (see Figure 11b).
-
Cells can then be lifted with 0.5 mM EDTA solution, with or without the help of a cell scraper. The 0.5 mM EDTA solution should be left covering the cells for 10 min. Cells should be recounted and phenotypically characterized by flow cytometry or qPCR, or functionally thereafter (see Figures 12 and 13).
Note: The number of macrophages should be similar to the number of myeloid precursors seeded onto the plate, but this number can vary between cell lines and also be dependent on EB size.
Cells left in culture for 10–14 days post harvest tend to become fibroblast-like (Figure 11c).

Figure 1. Day 1 hiPSC attachment and growing in small closely neighbouring colonies on vitronectin.

Images taken using an Olympus CKX41 Inverted Microscope with 10× objective.
Figure 2. Example of heterogenous hiPSC colonies growing and expanding on vitronectin.

Figure 3. A 70–80% confluent plate of hiPSCs, growing in colonies that are almost touching one another, on the screen of an EVOSTM XL Core imaging system, imaged with a 4× objective.

Figure 4. Close-up image of the 70–80% confluent plate, where multiple large colonies are visible to the naked eye.

Figure 6. Embryoid Bodies should have this appearance after 24 h in culture.

Figure 7. Single individual EBs in the centre of each well of a 96-well ultra-low adherent plate are visible to the naked eye.

Figure 8. Single EBs at close range and labelled with arrows.

Figure 9. Single EB aspirated into wide bore pipette for transfer into gelatin-coated plate below.

Figure 10. Embryoid Body attachment: 1 week post transfer onto the gelatin plate.

Figure 11. Myeloid precursor apperance.

(a) Myeloid precursors following harvest (b) differentiated attached macrophages 7 days later. Images taken using EVOSTM XL Core imaging system with a 20× objective. (c) Fibroblast-like cells at 14 days post harvest. Image taken using Olympus CKX41 Inverted Microscope with a 20× objective.
Figure 12. Representative flow cytometry plots demonstrating positivity for macrophage markers CD14 (BV605), CD16 (APC), CD80 (BV711), CD86 (PE-Dazzle594), CD206 (PerCP/Cyanine 5.5), and CD204 (PE/Cyanine7) in differentiated iMACS.

(Black = Unstained iMACs, Red = iMACs).
Figure 13. Representative qPCR data demonstrating macrophage characterization of iMACs. Relative expression is normalized to TATA-Bing Protein (TBP).

Data analysis
There is increased expression in pan-macrophage markers: CD68, CSFR1, and CD163 in iMACs over hiPSCs, and increased expression of pluripotent markers SOX2, NANOG, and OCT4 are seen in hiPSCs compared to iMACs.
Recipes
All media preparation and laboratory work to be conducted inside a Class 2 microbiological hood using aseptic techniques. Prepare all media, solutions and reagents at RT.
-
Complete Essential 8 Basal Medium
Made up of two components:
490 mL of media (store at 2–8°C and protect from light) with 10 mL of supplement (50×) (store at -5 to -20°C. Shelf life: 12 months).
To reconstitute, thaw supplement to RT and add to 490 mL of media.
Alternatively, aliquot supplement into smaller volumes for longer storage at -20 to -80°C and thaw as required.
Swirl or invert to mix.
Store reconstituted base media at 2–8°C, shelf-life 2 weeks.
Allow to warm to RT before use.
-
Vitronectin: 1 mL vial (store at -80°C. Shelf-life: 24 months)
Divide into 60 µL sterile aliquots.
To coat a plate: Dilute 60 µL into 6 mL of DPBS without Ca2+/Mg2+, or use at a concentration 2.5–10 µg/mL.
Invert to mix.
Ensure parafilm is applied to seal the edges of the plate, for any pre-prepared plates prior to storage.
-
ROCK inhibitor (ROCKi) (store at 2–8°C)
Prepare a 3.12 mM working concentration stock, i.e., reconstitute 1 mg with 1 mL of sterile ultrapure water.
Divide into 10 µL aliquots (store at -20°C).
For use at 0.312 µM or 3.12 µM final concentrations.
Once thawed, stock ROCKi can be stored at 2–8°C for 7 days.
-
0.5 mM Ethylenediaminetetraacetic acid (EDTA) solution (store at RT)
Dilute 50 µL in 50 mL of DPBS without Ca2+/Mg2+ for a 0.5 mM working concentration, store at RT, and use on the day of preparation.
-
Cryopreservation/freezing Medium (Store at -5 to -20°C and protect from light. Shelf-life 18 months)
Aliquot into small volumes.
Add 1 mL of DMSO to 9 mL of thawed KnockOut serum replacement (KSR), to make a final concentration of 10% DMSO for freezing.
Use on the day of preparation and for long term storage of hiPSC in liquid nitrogen.
-
Bovine serum slbumin (BSA) (store at 2–8°C)
Add 100 mg BSA to 100 mL of DPBS with Ca2+/Mg2+, mix, and leave in 37°C water bath until the BSA has dissolved.
Filter-sterilise (store at 2–8°C, shelf-life 4 months).
-
Recombinant Human BMP-4 Protein
(Store at -20 to -70°C. Shelf life 12 months)
Reconstitute to 50 µg/mL working concentration stock (10 µg in 200 µL of 4 mM HCl and 0.1% BSA).
Aliquot into small volumes.
For use at 50 ng/mL final concentration (store at -20 to -80°C).
Sterile water for embryo transfer (store at RT)
-
Gelatin from porcine skin (store at RT)
Make 0.1% gelatin solution: add 500 mg gelatin to 500 mL of water, heat to 56°C to dissolve, and filter-sterilise (Ssore at 4°C for up to 4 months).
-
EB-Myeloid precursor base medium
To 500mL of X-VIVO 15 Serum-free Hematopoietic Cell Medium, add:
2 mM (5 mL) L-Glutamine (store at -5 to -20°C. Shelf life 24 months),
100 IU/ mL (5 mL) Penicillin-Streptomycin (store at -5 to -20°C. Shelf life 12 months)
0.1 mM (3.5 µL) 2-Mercaptoethanol (store at RT)
Filter-sterilise (store at 2–8°C).
-
Recombinant Human M-CSF
Reconstitution: add 500 µL of 0.1% BSA solution to 0.5 mg lyophilised rhM-CSF, to achieve a 1 mg/mL stock concentration (store in 5 µL aliquots at -20 to -80°C. Shelf life 12 months).
Thaw to add to fresh media prior to use (for use at 50ng/mL final concentration).
Avoid repeated free-thaw cycles.
-
Recombinant Human IL-3
Reconstitution: add 1 mL of 0.1% BSA solution to 0.5 mg lyophilised rhIL-3, to achieve a 0.5 mg/mL stock concentration (store in 5 µl aliquots at -20 to -80°C. Shelf life 12 months).
Thaw to add to fresh media prior to use (for use at 25 ng/mL final concentration).
Avoid repeated free-thaw cycles.
-
Macrophage Differentiation base medium
To 500mL of RPMI 1640 Medium add:
10% (50 mL) of foetal bovine serum
2 mM (5 mL) L-Glutamine
Filter-sterilise (store at 2–8°C).
Acknowledgments
Funding for research provided by Medical Research Council, Kidney Research UK, National Institute for Health Research, The Royal Society, and The Wellcome Trust.
The NL9 hiPSC used in the development of this protocol were generously donated by Professor Claudia Kemper and the National Heart, Lung and Blood Institute (NHLBI) iPSC core (https://www.nhlbi.nih.gov/science/ipsc-core/research), Bathesda, Maryland, USA.
Thanks also go to Professor Anthony Dorling at King’s College London, for the patient guidance, supervision and enduring encouragement.
This protocol is derived and adapted from previously published protocols (van Wilgenburg et al., 2013 ; Mukherjee et al., 2018 ; Lopez- Yrigoyen et al., 2020 ).
Competing interests
There are no financial or non-financial competing interests to declare.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1. Alasoo K., Martinez F. O., Hale C., Gordon S., Powrie F., Dougan G., Mukhopadhyay S. and Gaffney D. J.(2015). Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Sci Rep 5: 12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alasoo K., Rodrigues J., Mukhopadhyay S., Knights A. J., Mann A. L., Kundu K., Consortium H., Hale C., Dougan G. and Gaffney D. J.(2018). Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet 50(3): 424-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baldassarre M., Solano-Collado V., Balci A., Colamarino R. A., Dambuza I. M., Reid D. M., Wilson H. M., Brown G. D., Mukhopadhyay S. and Dougan G. et al. (2021). The Rab32/BLOC-3-dependent pathway mediates host defense against different pathogens in human macrophages. Sci Adv 7(3): eabb1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lopez-Yrigoyen M., May A., Ventura T., Taylor H., Fidanza A., Cassetta L., Pollard J. W. and Forrester L. M.(2020). Production and Characterization of Human Macrophages from Pluripotent Stem Cells. J Vis Exp(158). DOI: 10.3791/61038-v. [DOI] [PubMed] [Google Scholar]
- 5. Mukherjee C., Hale C. and Mukhopadhyay S.(2018). A Simple Multistep Protocol for Differentiating Human Induced Pluripotent Stem Cells into Functional Macrophages. Methods Mol Biol 1784: 13-28. [DOI] [PubMed] [Google Scholar]
- 6. Mukhopadhyay S., Heinz E., Porreca I., Alasoo K., Yeung A., Yang H. T., Schwerd T., Forbester J. L., Hale C., Agu C. A., et al.(2020). Loss of IL-10 signaling in macrophages limits bacterial killing driven by prostaglandin E2. J Exp Med 217(2): e20180649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Wilgenburg B., Browne C., Vowles J. and Cowley S. A.(2013). Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS One 8(8): e71098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee C. Z. W., Kozaki T. and Ginhoux F.(2018). Studying tissue macrophages in vitro: are iPSC-derived cells the answer? Nat Rev Immunol 18(11): 716-725. [DOI] [PubMed] [Google Scholar]
- 9. Hale C., Yeung A., Goulding D., Pickard D., Alasoo K., Powrie F., Dougan G. and Mukhopadhyay S.(2015). Induced pluripotent stem cell derived macrophages as a cellular system to study salmonella and other pathogens. PLoS One 10(5): e0124307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lachmann N., Ackermann M., Frenzel E., Liebhaber S., Brennig S., Happle C., Hoffmann D., Klimenkova O., Luttge D., Buchegger T., et al.(2015). Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Reports 4(2): 282-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nenasheva T., Gerasimova T., Serdyuk Y., E. Grigor'eva, Kosmiadi G., Nikolaev A., Dashinimaev E. and Lyadova I.(2020). Macrophages Derived From Human Induced Pluripotent Stem Cells Are Low-Activated"Naive-Like" Cells Capable of Restricting Mycobacteria Growth. Front Immunol 11: 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baghbaderani B. A., Syama A., Sivapatham R., Pei Y., Mukherjee O., Fellner T., Zeng X. and Rao M. S.(2016). Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications. Stem Cell Rev Rep 12(4): 394-420. [DOI] [PMC free article] [PubMed] [Google Scholar]