

Abstract
Accurate prioritization of immunogenic neoantigens is key to developing personalized cancer vaccines and distinguishing those patients likely to respond to immune checkpoint inhibition. However, there is no consensus regarding which characteristics best predict neoantigen immunogenicity, and no model to date has both high sensitivity and specificity and a significant association with survival in response to immunotherapy. We address these challenges in the prioritization of immunogenic neoantigens by 1) identifying which neoantigen characteristics best predict immunogenicity, 2) integrating these characteristics into an immunogenicity score, the NeoScore, and 3) demonstrating a significant association of the NeoScore with survival in response to immune checkpoint inhibition. One thousand random and evenly split combinations of immunogenic and non-immunogenic neoantigens from a validated dataset were analyzed using a regularized regression model for characteristic selection. The selected characteristics, the dissociation constant and binding stability of the neoantigen:MHC class I complex and expression of the mutated gene in the tumor, were integrated into the NeoScore. A web application is provided for calculation of the NeoScore. The NeoScore results in improved, or equivalent, performance in four test datasets as measured by sensitivity, specificity, and area under the receiver operator characteristics curve compared to previous models. Among cutaneous melanoma patients treated with immune checkpoint inhibition, a high maximum NeoScore was associated with improved survival. Overall, the NeoScore has the potential to improve neoantigen prioritization for the development of personalized vaccines and contribute to the determination of which patients are likely to respond to immunotherapy.
Introduction:
Cancers arise through mutations in the genome of healthy human cells. As these mutations occur, some will produce mutated proteins, which have the potential to be processed into neoantigens that bind MHC class I and are presented on the cell surface. These neoantigens then act as tumor-specific targets with the potential to elicit a cytotoxic CD8+ T cell response (1–4). Tumor-specific neoantigens have strong potential to be targets of T cell-mediated destruction, because they are not subject to immune tolerance or non-reactivity to self. However, there are two key ways in which the above mechanism may fail in tumor destruction. For one, recent evidence suggests that most neoantigens do not elicit an immune response in their natural state, termed immunologic ignorance (5). Second, once a T cell response is mounted to a neoantigen, that response may become exhausted over time due to inhibitory signals from the tumor microenvironment (6).
Circumventing these limitations to re-invigorate the host immune response has been the goal of many recent cancer therapies. To address immunologic ignorance, personalized cancer vaccines have been created and have demonstrated early success (7–12). These vaccines have taken several forms, including direct exposure to neoantigens (11), neoantigen-encoding RNA vaccines (12), neoantigen-loaded dendritic cell vaccines (7), and adoptive transfer of neoantigen-specific T cells (2, 13). Each of these methods requires accurate knowledge of the neoantigens presented by the tumor cell with the potential to elicit an immune response. In silico prioritization methods have been used to prioritize which set of neoantigens should be experimentally tested, but the ability to prioritize the immunogenicity of each neoantigen, with high sensitivity and specificity, is still limited (14–18).
Exhaustion of T cells and attenuation of T cell activation can be overcome by immune checkpoint inhibition, such as monoclonal antibodies against PD-1 or CTLA-4. Immune checkpoint inhibition blocks inhibitory signals to the T cells to enhance T cell-mediated tumor destruction (19–21). However, immune checkpoint inhibition is only effective in a subset of patients (6), and there is no consensus on how to prioritize which patients will respond (18, 21–26). In a pan-cancer analysis, Yarchoan et al. demonstrated that cancer types with a higher mutational burden, such as melanoma, had improved response to anti-PD-1 therapy compared to cancer types with a lower mutational burden (27). There are limitations to mutational burden as a predictor of response to immune checkpoint inhibition. First, in multiple myeloma, there was an association of increased tumor mutational burden with decreased response to immune checkpoint inhibition (28). Second, in melanoma, the association between the tumor mutational burden and response to immune checkpoint inhibition was confounded by the melanoma subtype (29). Finally, in lung cancers that progressed after treatment with immune checkpoint inhibitors, there was an increase in the tumor mutational burden compared to the pretreatment state (30), thus, contradicting the expectation that tumor cells resistant to immune checkpoint inhibition would have a low number of neoantigens. Together, these findings suggest that tumor mutational burden is not sufficient for predicting response to immune checkpoint inhibition.
Several recent papers have been dedicated to predicting neoantigen immunogenicity based on the characteristics of validated immunogenic neoantigens (16–18, 23, 31). However, there is no consensus regarding which neoantigen characteristics are important for the prioritization of immunogenic neoantigens or the best way to combine the characteristics into an overall immunogenicity score. To identify the characteristics that best prioritize the immunogenicity of the neoantigens, a model-based approach was applied to evaluate neoantigen characteristics encompassing expression, processing, presentation, and T cell recognition in prioritizing neoantigen immunogenicity. The selected characteristics were then combined into an overall logistic regression model called the “NeoScore.” The development of the NeoScore has largely focused on melanoma, except for one lung cancer patient included in the Tumor Neoantigen Selection Alliance (TESLA) consortium dataset (16).
Immune checkpoint inhibition and personalized neoantigen vaccines are particularly effective in mutation-rich melanoma (25, 32–34). However, even in melanoma, a positive outcome from these interventions is not assured (6). To assess the clinical utility of the NeoScore and its ability to improve assessment of outcome in melanoma, the relationship of the NeoScore to survival in response to immunotherapy was tested using the datasets of Van Allen et al. 2015 and Liu et al. 2019 (21, 29).
Materials and Methods:
Datasets
For training of the neoantigen prioritization model, whole-exome sequencing (WES) and RNA sequencing (RNAseq) data were obtained from the TESLA consortium database on Synapse (16). The TESLA consortium data came from four patients with melanoma and a single lung cancer patient. The TESLA consortium provided RNAseq data, WES data, and clinically determined HLA types for each of these patients to 28 teams and used the neoantigen rankings from 25 of the teams to prioritize which neoantigens were experimentally validated. The neoantigens validated for their ability to elicit a T cell response were selected by two guidelines: 1) the top 5 neoantigens ranked by each team were tested and 2) the neoantigens that came up most frequently in the top 50 ranked neoantigens for each team were selected. The neoantigens tested were also constrained by HLA restriction requirements for the validation experiments. The final available dataset includes 5 patients with a total of 347 neoantigens that had been tested for their ability to elicit a T cell response using multimer staining. The TESLA consortium found that 26 of the 347 tested neoantigens elicited a T cell response in an unvaccinated state (16). Neoantigens were used for model creation in this manuscript if they were 1) tested for immunogenicity by the TESLA consortium, 2) identified by either GATK Mutect2 or Strelka, and 3) had expression data. Accession information for the TESLA consortium dataset is included in Table I.
Table I:
Summary of datasets used, materials used, and accession information
| Dataset | Materials used | Accession |
|---|---|---|
| TESLA Consortium (16) | Raw RNAseq and WES data | https://www.synapse.org/#!Synapse:syn21048999/wiki/603788 |
| Lists of validated neoantigens and the T cell response | Supplementary Table S4 | |
| Cohen (35) | Raw RNAseq data | https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP062169 |
| Lists of validated neoantigens and the T cell response | Supplementary Table 2 | |
| Strønen (36) | Lists of validated neoantigens and the T cell response | Supplementary Table S8 |
| Carreno (7) | Lists of validated neoantigens and the T cell response | Supplementary Tables S1-S3 |
| Ott (11) | Lists of validated neoantigens and the T cell response | Supplementary Table 4 |
| Van Allen (21) | Raw RNAseq and WES data | dbGaP accession number (phs000452.v3.p1) https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000452.v3.p1 |
| Liu (29) | Raw RNAseq and WES data | |
| Rizvi (25) | Raw WES data | dbGaP accession number (phs000980.v1.p1) https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000980.v1.p1 |
For testing the neoantigen prioritization model, lists of tested neoantigens from melanoma tumors were obtained (7, 11, 35, 36). These datasets contain 357 tested neoantigens (n=7 immunogenic neoantigens) (35), 149 tested neoantigens (n=18 immunogenic neoantigens) (11), 57 tested neoantigens (n=11 immunogenic neoantigens) (36) and 21 tested neoantigens (n=9 immunogenic neoantigens) (7). The neoantigens were tested for immunogenicity with tetramer staining and cytokine secretion (35), ELISPOT (11), and multimer staining (7, 36). Strønen, Ott, and Carreno also provided the expression data quantified as either fragments per kilobase of transcript per million mapped reads (FPKM), reads per kilobase of transcript per million mapped reads (RPKM), or transcripts per million (TPM) (7, 11, 35, 36). For the Cohen dataset, no expression data was provided. The RNAseq data from the Cohen dataset was obtained and analyzed as described below for read count quantification. Accession information is in Table I.
Finally, for survival analysis, WES and RNAseq data were obtained from the Van Allen et al., Liu et al., and Rizvi et al. cohorts (21, 25, 29) (all accession information is in Table I). The inclusion criteria for the Van Allen dataset were as follows: 1) both WES and RNAseq data available, 2) cutaneous melanoma as the primary lesion. All patients in the Van Allen cohort underwent treatment with an anti-CTLA-4 monoclonal antibody (ipilimumab). Inclusion criteria for the Liu et al dataset were as follows: 1) both WES and RNAseq data available, 2) cutaneous melanoma as the primary lesion, 3) no prior treatment with an anti-CTLA-4 monoclonal antibody (37). All patients in the Liu cohort underwent treatment with an anti-PD-1 monoclonal antibody (nivolumab or pembrolizumab). All samples from the Rizvi et al. dataset were included in the analysis including adenocarcinoma, squamous cell carcinoma, and non-small cell lung cancer. All patients in the Rizvi cohort underwent treatment with an anti-PD-1 monoclonal antibody (pembrolizumab only).
Data Preparation
WES and RNAseq FASTQ files from the TESLA consortium, Liu et al., and Van Allen et al., RNAseq FASTQ files from Cohen et al., and WES FASTQ files from Rizvi et al. were visualized for quality using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). FASTQ files were trimmed for quality using Trimmomatic (38) IlluminaClip with the following parameters: seed_mismatches = 2, palindrome_clip_threshold = 30, simple_clip_threshold = 10, leading = 10, trailing = 10, winsize = 4, winqual = 15. Quality was then re-visualized after trimming. Trimmed WES reads were mapped to the GRCh38.p7 reference genome, from 1000 genomes (39), and read group labels were added using BWA-mem (40). SAM files were converted to BAM and coordinate sorted (41). The BAM files were then converted to pileup format using SAMtools v. 1.4 (41).
Identification of somatic mutations and neoantigen generation
Single nucleotide variants (SNVs) and small insertions/deletions (indels) were identified using four programs, along with their recommended filters: GATK Mutect2 version 4.1.7.0 with default parameters, Varscan2 version 2.3.9 with minimum coverage of 10, minimum variant allele frequency of 0.08, and somatic p-value of 0.05, Strelka version 2.9.2 with default parameters, and LoFreq version 2.1.1 with default parameters (42–45). Results for GATK Mutect2 were filtered with the recommended FilterMutectCalls, and Varscan2 results were filtered using the Perl false-positive filter (https://github.com/ckandoth/variant-filter). Results from all four programs were examined with and without their respective filters. LoFreq results were not filtered to allow maximal potential to identify the missing mutations. Matched normal samples were used as the reference for each sample. The overlap of GATK Mutect2 and Strelka was used for the identification of SNVs and indels in the Liu, Van Allen, and Rizvi datasets for assessing the association of the NeoScore model with survival in response to immune checkpoint inhibition in melanoma.
Somatic mutations were separated from those SNVs that fell within 1 bp of an indel position, as these are likely to be false positives due to alignment errors. The mutations were annotated using the Variant Effect Predictor (VEP) tool from Ensembl version 90.9 (46). Then, 21mer amino acid sequences were generated for each mutation using pVAC-Seq tools version 3.0.5 (47). Finally, the 21mers were split into all possible 9mers and 10mers where the mutation of interest was in every location. The full pipeline from read mapping through to the identification of somatic mutations is available at https://github.com/SexChrLab/Cancer_Genomics.
Because the clinical samples for Liu et al. and Van Allen et al. were comprised of formalin-fixed, paraffin embedded (FFPE) samples, we considered applying an FFPE filter to remove false positive mutations introduced by FFPE storage. However, the characteristic G→A and C→T mutations introduced by FFPE processing overlap with the mutational signature introduced by ultraviolet radiation. Removal of the FFPE signature also removed the characteristic bias towards G→A and C→T mutations in the samples, and therefore, an FFPE filter was not applied.
Calculation of neoantigen characteristics
For each of the validated neoantigens, neoantigen characteristics with potential significance in predicting expression, processing, MHC binding, and T cell receptor (TCR) recognition probability were calculated as described here. The full pipeline for calculation and processing of the neoantigen characteristics can be found at https://github.com/ElizabethBorden/Process_peptide_lists. A log10 transformation was applied if the distribution of the characteristic had a large degree of skewness. The difference in the values for each of the neoantigen characteristics between immunogenic and non-immunogenic neoantigens was assessed using a two-sample, two-sided t-test. Correlation coefficients were calculated using Spearman correlation coefficients. P-values below 0.05 were considered statistically significant.
Expression:
For the datasets from Cohen et al., the TESLA consortium, and Liu et al., transcriptome assembly and read count quantifications were completed with Salmon version 0.11.3, using the Ensembl GRCh38.p7 reference genome (48, 49). mRNA expression in units of TPM was log10-transformed, and a constant of 0.1 was added to all values before the transformation to avoid taking the log of zero. To account for the different units used across each dataset, expression values were centered and normalized by subtracting the mean and then dividing by the standard deviation.
Clonality:
Copy number variation was calculated with sequenza (50), and clonality was calculated using the deconvolution software, FastClone (51).
Variant allele frequency:
Variant allele frequency (VAF) as calculated by GATK Mutect2 (42). Since the VAF was only calculated by GATK Mutect2, but not for Strelka, 14 missing data points were estimated as the average of the rest of the data. No statistically significant difference in the VAF was observed between immunogenic and non-immunogenic neoantigens, either with or without these data points.
Processing:
Cleavage and TAP transport potentials were calculated for each of the available neoantigens using NetCTLpan1.0 (52).
Dissociation constants:
Dissociation constants of the neoantigen:MHC class I complex were calculated in nanomolar (nM) units using NetMHCpan4.0 (53). These values were log10-transformed before inclusion in the model.
Binding stability:
Binding stability of the neoantigen:MHC class I complex was then calculated as the half-life in units of hours using NetMHCstabpan1.0 (53, 54). These values were log10-transformed and adjusted by a factor of 0.01 to avoid taking the log of zero before inclusion in the model.
Hydrophobicity method from the TESLA consortium:
The number of hydrophobic residues was divided by the total number of residues in the neoantigen to create a “hydrophobicity fraction” (16). In keeping with the methods of the TESLA consortium, hydrophobic residues here were isoleucine, leucine, phenylalanine, methionine, tryptophan, valine, and cysteine.
Hydrophobicity with empirical prevalence:
The hydrophobicity fraction was calculated as described for the TESLA consortium using the amino acids found to be empirically prevalent by Chowell et al. (55). Proline, leucine, and methionine were considered to have a high probability and given a score of +2. Glycine, tryptophan, phenylalanine, isoleucine, and valine were found to have a medium probability and given a score of +1. All others were given a score of 0.
Hydrophobicity Łuksza method:
A neoantigen was given a score of zero if the mutation at the anchor residue changed from a hydrophobic to a hydrophilic residue (23). All other neoantigens were given a score of one. Hydrophobic neoantigens here were defined as alanine, isoleucine, leucine, methionine, phenylalanine, tryptophan, tyrosine, and valine.
TCR recognition:
Prediction of TCR recognition probability, R, was calculated as described (23). A BLOSUM62 similarity matrix was used to assess the sequence similarity between a neoantigen and the closest matched known T cell epitope from the Immune Epitope Database (IEDB) (56). The sequence similarity was then used in place of binding energies, and the TCR recognition probability was calculated as:
Where is the sequence similarity, is the horizontal displacement of the binding curve, and sets the steepness of the curve at . Based on the model fit by Łuksza et al., the parameters and were set to 26 and 4.87 respectively (23). These parameters were optimized for both melanoma and lung cancer patients, the two cancer populations included in our training and test datasets. Finally, Z(k) is the partition function over the unbound state and all bound states, calculated as follows:
Sequence Similarity:
The closest matched human peptide was identified using Blast v. 2.10.1 (57), and the sequence similarity of the potential neoantigen to the closest matched human peptide was calculated using a BLOSUM62 matrix, as described (17). The sequence similarity was normalized across neoantigen length by dividing by the number of amino acids.
Amplitude:
Dissociation constants for the neoantigen:MHC class I complex were then adjusted by the dissociation constants for the closest matched normal human peptide:MHC class I complex, a characteristic called amplitude. The ratio was taken of the dissociation constant of the closest matched human peptide:MHC class I () to the dissociation constant of the neoantigen:MHC class I () as follows (23):
Analysis of anchor vs. non-anchor residue mutations:
Neoantigens were separated by their mutation position (anchor vs. non-anchor residues). Then, both the amplitude and the dissociation constant of the neoantigen:MHC class I complex were compared between the immunogenic and non-immunogenic neoantigens within each group. Comparisons were done using a two-sample, two-sided t-test. P-values below 0.05 were considered statistically significant.
HLA typing
HLA types were identified on normal tissue samples for each patient in the Liu et al. dataset using HLA-LA (58). HLA types provided in the supplementary data were used for each patient in the Van Allen et al. and Rizvi et al. datasets (21, 25).
Elastic net regression modeling
The TESLA consortium data was split into 1000 random subgroups containing all the immunogenic neoantigens (n=26) and an equal number of randomly selected non-immunogenic neoantigens. An elastic net regression was performed using the glmnet package in R for each of these splits (59). The selected coefficients and area under the receiver operator characteristics curve (AUC) from each model were tabulated across the 1000 splits. An optimal threshold for the model was selected in the TESLA dataset to optimize both sensitivity and specificity by the Youden Index and was implemented through the optimal cutpointr package from R statistical software (https://github.com/thie1e/cutpointr).
Logistic regression modeling
Logistic regression modeling was performed, and optimism values were calculated with the RMS package in R statistical software (https://cran.r-project.org/web/packages/rms/rms.pdf).
Survival analysis
To avoid scaling expression values on a patient-level basis, coefficients were determined on the TESLA consortium data for log10-transformed, non-scaled TPM expression data. The intercept changed to −1.7951 and the expression coefficient to 1.2868, but there was no change in the coefficients of the dissociation constant or binding stability and no effect on the performance of the model. The optimal threshold for each of the mutational burden, neoantigen burden, and maximum NeoScore were determined by optimizing based on an adjusted log rank test implemented in the MaxStat package in R (60). Then, the survival analysis was performed between scores above the selected threshold and below, using Kaplan Meier estimation and the log-rank test statistic. P-values below 0.05 were considered statistically significant.
Results:
Overlap of Strelka and GATK Mutect2 identifies the maximum number of validated immunogenic mutations
The first step in effective prioritization of neoantigens is to identify a high-fidelity list of somatic mutations. Isolating somatic mutations in cancers is more difficult than variant calling in normal tissue since cancers do not follow the typical rules of copy number or heterozygosity and often consist of multiple clonal populations with normal tissue contamination. The TESLA consortium validated neoantigens derived from mutations identified by 25 teams. The methods used for identifying the somatic mutations were not reported, making it difficult to reproduce all neoantigens (16). To maximize the immunogenic mutations identified, three highly rated programs were used to identify single nucleotide variants (SNVs) and small insertions and deletions (indels) for the data from the TESLA consortium: VarScan2, GATK Mutect2, and Strelka (42–44). A large degree of overlap was found in the ability of each program to identify the 34 validated immunogenic mutations. As shown in Figure 1, GATK Mutect2 and Strelka successfully identified 27/34 mutations, while VarScan2 identified 24/34. The mutations from each of these programs overlapped, such that 27 was the maximum number of immunogenic mutations identified. To ensure that the success of VarScan2 in identifying the immunogenic mutations was not hindered by over-filtering, the unfiltered output was assessed and only one immunogenic mutation had been eliminated by filtration steps (data not shown). GATK Mutect2 and Strelka identified the same number of immunogenic mutations with or without their respective filters (data not shown). LoFreq was also tested for the identification of somatic mutations, as LoFreq is optimized to call low-frequency mutations (45). However, LoFreq did not identify any additional immunogenic mutations (data not shown). The neoantigens derived from unidentified immunogenic mutations may be due to the reference genome version, peptide generation steps, or mutations identified by programs that were not tested. Overall, the combination of GATK Mutect2 and Strelka identified greater than or equal to the number of validated immunogenic neoantigens as in all other reported pipelines (16), while simultaneously decreasing the total number of potential neoantigens by 89.44%.
Figure 1. Maximum Number of Validated Immunogenic Mutations Identified by the Overlap of Strelka and GATK Mutect2.
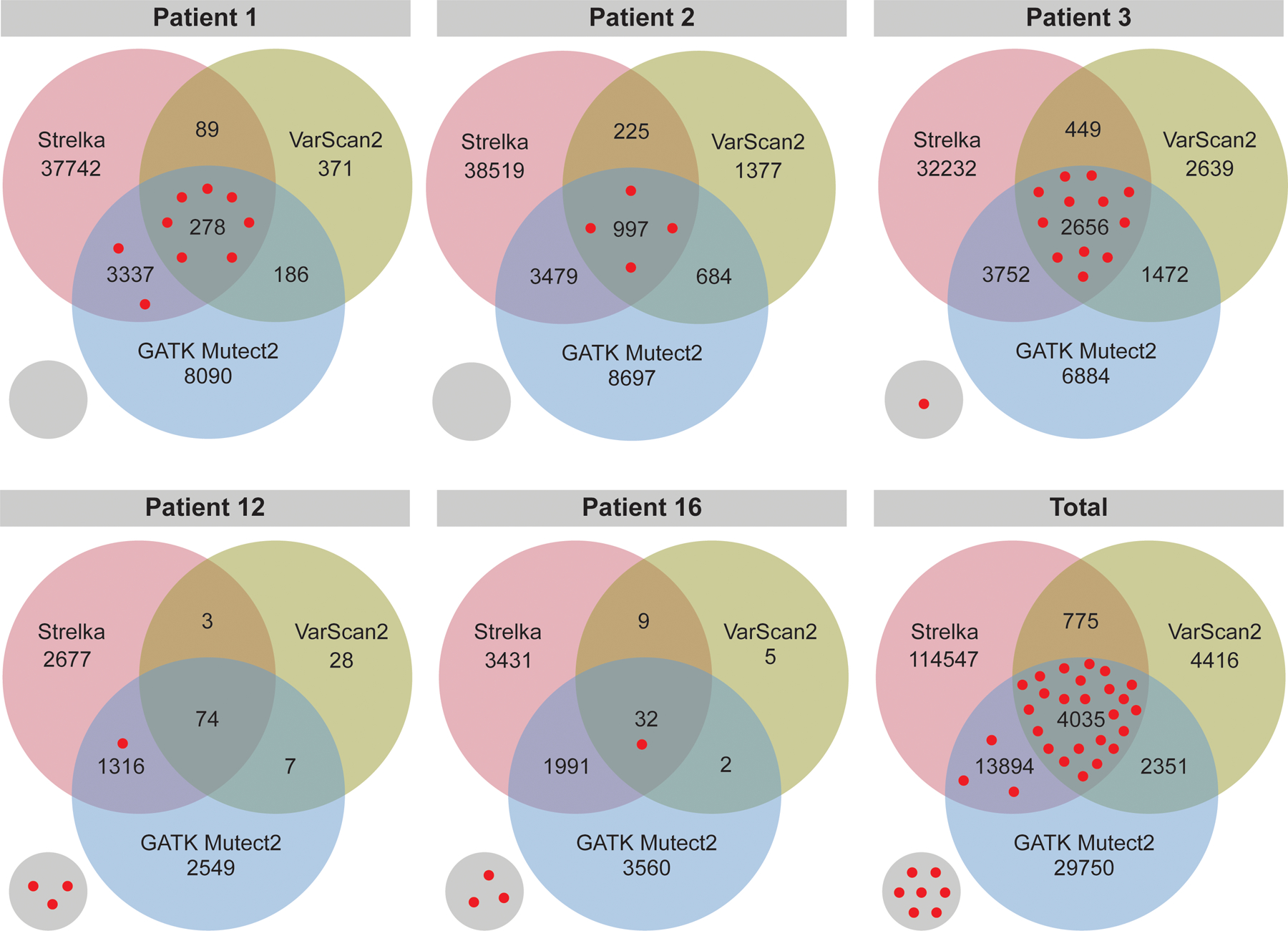
Comparison of the number of neoantigens derived from single nucleotide variants (SNVs) and small insertions and deletions (indels) by each of three different programs (Varscan2, GATK Mutect2, and Strelka) in the TESLA consortium dataset. Red dots represent the validated immunogenic neoantigens from each patient. The small circle represents validated immunogenic neoantigens that were not identified by the three programs.
Dissociation constant and binding stability of the neoantigen:MHC class I complex as well as expression are significantly different between immunogenic and non-immunogenic neoantigens
A set of computationally predicted neoantigen characteristics were calculated for the expression, processing, presentation, and T cell receptor (TCR) recognition of SNV and small indel-derived neoantigens (Figure 2A). Each of these characteristics was included in the development of the NeoScore model. Figure 2B demonstrates that the set of characteristics included in model development is inclusive of all of those considered by the three models to which the NeoScore is compared. To begin, the distribution of each of these characteristics was assessed for immunogenic and non-immunogenic neoantigens from the TESLA consortium dataset.
Figure 2. Expression, Dissociation Constant, and Stability are Significantly Different Between Immunogenic and Non-Immunogenic Neoantigens.

(A) Steps in the formation of an MHC class I-restricted immunogenic neoantigen. (B) Computationally predicted neoantigen characteristics included in development of the NeoScore model in comparison to three other models. Expression consists of the gene-level mRNA expression and clonality or variant allele frequency of the mutation. Processing consists of proteasomal cleavage and TAP transport potential. MHC class I binding is assessed through the MHC class I dissociation constant, binding stability, and hydrophobicity (Figure 3) of the neoantigen. The potential to stimulate a T cell response is assessed by the similarity to known T cell epitopes, the sequence similarity to normal human peptides, and the relative MHC class I dissociation constant compared to the dissociation constant of the closest matched normal peptide. All ten characteristics listed under the NeoScore model were considered for inclusion, and those in bold (expression, dissociation constant, and binding stability) were the characteristics selected for inclusion in the final model. (C-K) Comparison of the distribution of the characteristics for immunogenic and non-immunogenic neoantigens. (C) Log10-transformed distribution of mRNA expression. (D) Distribution of variant allele frequency (VAF) calculated by GATK Mutect2. (E-F) Distribution of proteasomal cleavage and TAP transport potential calculated by NetCTLpan. (G) Log10-transformed distribution of the MHC class I to neoantigen dissociation constant calculated by NetMHCpan. (H) Log10-transformed distribution of the MHC class I to neoantigen binding stability calculated by NetMHCstabpan. (I) T cell recognition (TCR) recognition probability calculated using the model from Łuksza et al. (J) Log10-transformed distribution of the BLOSUM62 sequence similarity between the closest matched human peptide and the neoantigen of interest, divided by the length of the neoantigen to normalize. (K) Log10-transformed distribution of the amplitude calculated as the ratio of the dissociation constant of the closest matched human peptide to MHC class I to the dissociation constant of the neoantigen to MHC class I. **:p<0.001, ***:p<10−5
Expression was included in the NeoScore model development in two ways: 1) mRNA expression level and 2) variant allele frequency (VAF). Expression level from RNAseq data was calculated as the transcripts per million (TPM) expression of the gene from which the neoantigen is derived. Immunogenic neoantigens had a significantly higher expression level than non-immunogenic neoantigens (p=2.875×10−4, Figure 2C). The clonality of the neoantigen was calculated by FastClone (51), but FastClone did not converge for 4/6 of the samples, so the VAF calculated by GATK-mutect2 was included in the development of the NeoScore model instead. No significant difference in the VAF was observed between immunogenic and non-immunogenic neoantigens (p=0.359, Figure 2D).
Processing steps for the neoantigen were included in the development of the NeoScore model as both the proteasomal cleavage potential and TAP (transporter associated with antigen processing) potential scores. While there was a higher average for both characteristics in the immunogenic than non-immunogenic neoantigens, no statistically significant difference was observed (p=0.817 and p=0.836, respectively) (Figure 2E–F). The binding to the MHC class I molecule was then considered by both the dissociation constant and stability of the neoantigen:MHC class I interaction. NetMHCpan was selected as the software for predicting the MHC class I dissociation constants due to its enhanced performance compared to other binding prediction methods (53). NetMHCpan achieved over a 0.90 AUC across 6 independent test datasets (61). The MHC class I dissociation constants were significantly lower in immunogenic than non-immunogenic neoantigens, indicating a higher binding affinity of immunogenic neoantigens to MHC class I (p=2.088×10−7, Figure 2G). The binding stability showed significantly higher values for immunogenic over non-immunogenic neoantigens. (p=3.895×10−5, Figure 2H).
The ability of the neoantigen to stimulate a T cell response was included in the NeoScore model development using the model created by Łuksza et al. to calculate a characteristic referred to as the TCR recognition probability (23). The TCR recognition probability is a probabilistic model that considers the sequence similarity between the neoantigen and a known T cell epitope from the Immune Epitope Database (IEDB) as a proxy for the binding affinity of the neoantigen:MHC class I-TCR interaction. The TCR recognition probability showed no statistically significant difference between the immunogenic and non-immunogenic neoantigens (p=0.636, Figure 2I).
Two methods to account for T cell development were also included in NeoScore development. During maturation in the thymus, T cells expressing TCRs with high avidity to normal human peptides undergo apoptosis. Thus, a neoantigen with a high degree of sequence similarity to a normal human peptide is less likely to elicit a T cell response. The first method used to account for T cell development was the sequence similarity to the closest matched normal human peptide. No statistically significant difference was observed in the sequence similarity for immunogenic and non-immunogenic neoantigens (p=0.511, Figure 2J). The second method considered was the amplitude, where the dissociation constant of the neoantigen:MHC class I complex is adjusted by the dissociation constant of the closest matched human peptide:MHC class I complex. The amplitude adjusts for the regulation of TCR specificities during T cell maturation but also considers that only a normal human peptide capable of binding an MHC class I molecule will significantly impact the immunogenicity of the neoantigen. The amplitude was not significantly different between immunogenic and non-immunogenic neoantigens (p=0.209, Figure 2K).
One final characteristic considered in the development of the NeoScore model was the hydrophobicity of the neoantigen. The hydrophobicity of the neoantigen has been proposed to be associated with greater neoantigen immunogenicity because of the hydrophobicity of key binding pockets in the MHC class I binding groove (55). Mixed results have been reported for the association of hydrophobicity and immunogenicity to date (16, 18, 23, 55). One reason is the use of different methods for hydrophobicity, three of which are considered here. In the first method, the hydrophobicity is calculated as a fraction of the neoantigen residues that are hydrophobic (16). The TESLA consortium reported a significantly lower hydrophobicity of immunogenic neoantigens compared to non-immunogenic neoantigens, which is in the opposite direction as expected. While there was not a statistically significant difference in the neoantigens included in our analysis, the hydrophobicity fraction still had a lower average for immunogenic than non-immunogenic neoantigens (Figure 3, p=0.204). No difference in hydrophobicity was seen in additional datasets from Carreno or Strønen et al., and a significantly higher hydrophobicity of immunogenic neoantigens was observed in the dataset from Ott et al. (Figure 3, p=0.0168) (7, 11, 36). The second method calculated the hydrophobicity fraction using the empirical observations from Chowell et al. to determine which amino acids would increase the likelihood of immunogenicity (55). The method using the observations from Chowell et al. considers both hydrophobicity and other chemical properties such as side chain bulkiness and polarity. No differences in hydrophobicity were seen across the four datasets using the empirical observations from Chowell et al. (Figure 3). The final method considered the hydrophobicity at the anchor residues. A mutation that changed a previously hydrophobic anchor residue to a hydrophilic residue was given a score of zero, while all other changes or no change were given a score of one (23). While there was no statistically significant difference in hydrophobicity for any of the four datasets using the method of Łuksza et al., three out of four datasets showed a greater percentage of immunogenic neoantigens without a loss of hydrophobicity at an anchor residue (Figure 3). Overall, since the method from Łuksza showed the greatest consistency, only this method was included in development of the NeoScore model.
Figure 3. No Method for Calculating Neoantigen Hydrophobicity is Consistently Associated with Immunogenicity.

Comparison of the distribution of hydrophobicity values for the TESLA consortium, Carreno, Strønen, and Ott datasets between immunogenic and non-immunogenic neoantigens. Hydrophobicity was calculated by three methods. The TESLA consortium method is calculated as the number of hydrophobic amino acids divided by the total number of amino acids. The method based on the Chowell et al. empirical data uses the same calculation scheme but considers both hydrophobicity and other chemical properties such as side chain bulkiness and polarity in ranking the neoantigens. The Łuksza et al. method considers the change of an amino acid at the anchor residues compared to the closest matched human peptide. *:p<0.05
Given the inconsistent association of hydrophobicity with immunogenicity, we explored the association of hydrophobicity and immunogenicity when the data was separated out by HLA allele. Published motifs of the peptides that bind to different HLA alleles have demonstrated distinct differences in the conserved amino acids, suggesting that hydrophobicity may play a greater role in determining binding depending on the HLA allele. For example, published motifs for peptides that bind HLA-A01:01 and HLA-A03:01 contain conserved polar and charged amino acids whereas motifs for peptides that bind HLA-A02:01 contain several conserved hydrophobic amino acids (62). When we assessed the association of hydrophobicity and immunogenicity for each HLA allele independently, immunogenic neoantigens were significantly less hydrophobic than immunogenic neoantigens for HLA-A01:01 using the hydrophobicity method from the TESLA consortium, consistent with the polar and charged conserved amino acids in the peptides that bind this allele (Supplementary Table 1). This data suggests that there might be an advantage to separately evaluating the role of the hydrophobicity characteristic in predicting neoantigens for different HLA alleles. However, due to the small sample sizes available, we were not able to incorporate the effect of hydrophobicity on predicting immunogenicity based on the HLA allele.
Next, the degree of correlation between the characteristics calculated for each neoantigen was assessed. Only two characteristics were significantly correlated: the dissociation constant and the binding stability (Figure 4A). Despite their correlation, there is evidence that the two characteristics both contribute to accurate neoantigen prioritization. While the dissociation constant assesses the affinity of the interaction between the neoantigen and the MHC class I molecule, the stability predicts the length of time that the neoantigen will remain bound. The importance of binding stability is demonstrated in Figure 4B, where clustering of the immunogenic neoantigens in the upper left-hand corner can be observed, indicating the influence of both characteristics in determining immunogenicity.
Figure 4. Binding Stability and Dissociation Constant are Significantly Correlated but Both Contribute to Immunogenicity.
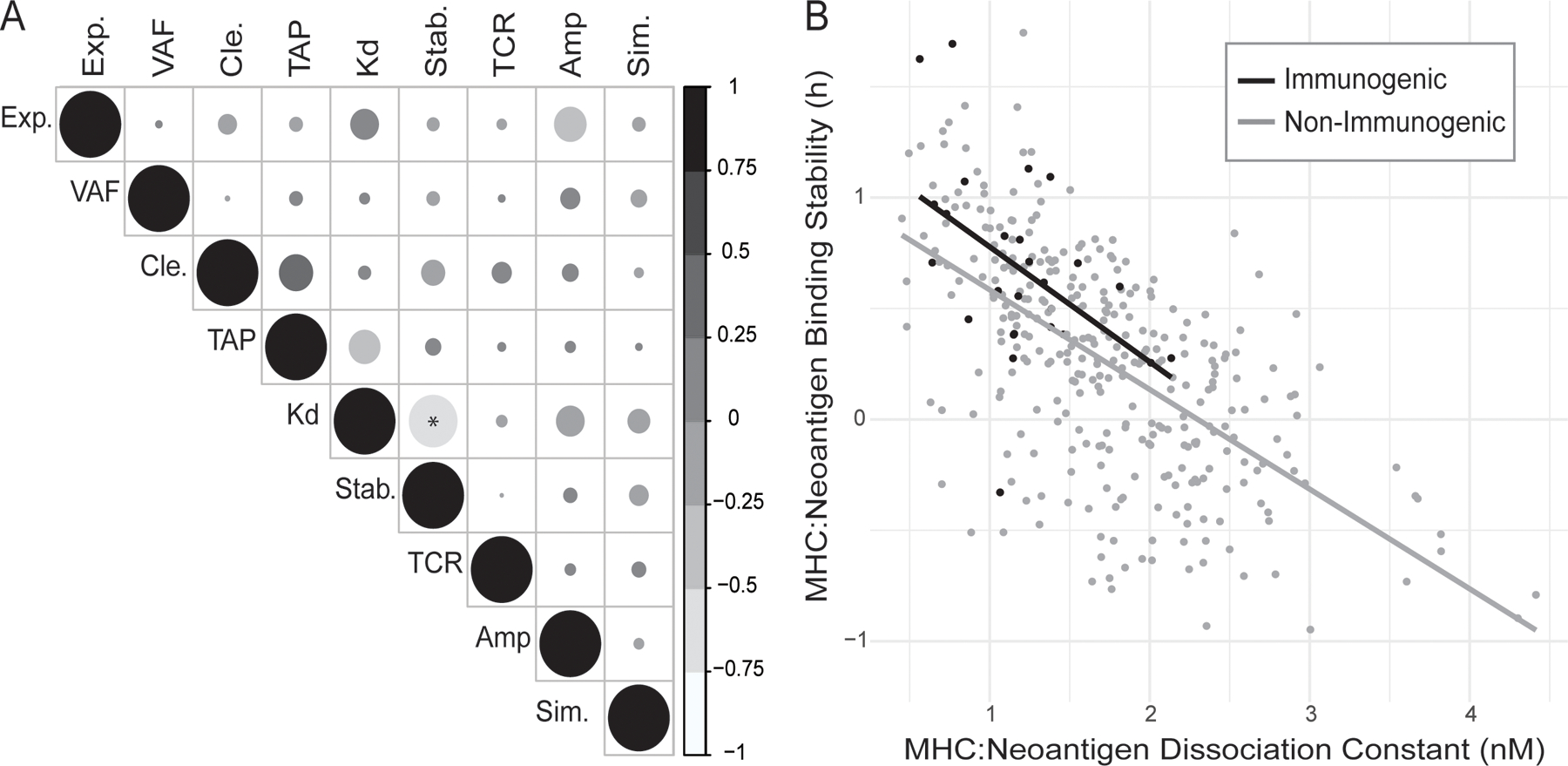
(A) Correlation between all calculated neoantigen characteristics: mRNA expression (Exp.), Variant Allele Frequency (VAF), proteasomal cleavage potential (Cle.), TAP transport potential (TAP), MHC class I to neoantigen dissociation constant (Kd), MHC class I to neoantigen binding stability (Stab.), TCR recognition probability (TCR), amplitude, defined as the ratio of the MHC class I binding of the closest matched human peptide and the neoantigen (Amp.), and normalized sequence similarity score for closest matched human peptide and the neoantigen (Sim.). The size of the circles indicates the absolute magnitude of the correlation. (B) Correlation of MHC class I dissociation constant with MHC class I binding stability of the neoantigen. Lines show the linear relationship between the characteristics for immunogenic (black) and non-immunogenic (gray) neoantigens, respectively. *:p<0.05
Capietto et al. recently found that a different set of neoantigen characteristics will influence immunogenicity, if the mutation occurs in an anchor residue compared to mutations in non-anchor residues (63). They demonstrated that, if a mutation occurred in an anchor residue, the amplitude had a greater predictive value than the dissociation constant of the neoantigen:MHC class I complex alone. To assess the influence of the mutation position, the distribution of the amplitude and the unadjusted dissociation constant for neoantigens with a mutation in an anchor or a non-anchor residue were analyzed. To maximize the chances of detecting a significant difference with the relatively low number of immunogenic neoantigens derived from mutations in anchor residues (n=13), the analysis of the impact of mutation’s position was performed across the combination of four datasets (7, 11, 16, 36). As shown in Supplementary Figure 1, no statistically significant difference was observed for the amplitude with either anchor or non-anchor residue mutations. While the anchor-residue mutations did have a higher average amplitude in immunogenic neoantigens, the difference was not statistically significant. In contrast, the dissociation constant was significantly lower in immunogenic neoantigens than non-immunogenic neoantigens. Therefore, there was not a compelling reason to fit separate models for neoantigens with mutations in anchor residues and those with mutations in non-anchor residues. Furthermore, because the amplitude is mathematically dependent on the dissociation constant, the amplitude was not included in the subsequent steps of model development.
Regularized regression approach creates a neoantigen prioritization model, NeoScore, that outperforms existing models that score each neoantigen
While analysis of the distribution of each characteristic determined those with a statistically significant difference between immunogenic and non-immunogenic neoantigens, we next assessed the full set of characteristics to determine which influenced the ability to optimally prioritize SNV and small indel-derived immunogenic neoantigens. A regularized regression is a model-based approach to determine the group of characteristics that are important for discriminating between immunogenic and non-immunogenic neoantigens, whether or not each characteristic is statistically significant. A logistic model using an elastic net-based regularization was unable to identify individual characteristics that are most predictive of immunogenicity by optimizing shrinkage penalties; a consequence that is often seen with a small effective sample size. The effective sample size for these regularized regression methods using models from the binomial family is based on the class with the smallest number of observations, which is the immunogenic neoantigens (n=26). Consequently, a cross-validation approach was applied to select the best subset of neoantigen characteristics. One thousand combinations of 26 non-immunogenic and 26 immunogenic neoantigens were randomly selected, and the elastic net regularized regression was fit on each combination (Figure 5A). Performing 1000 random samples allowed for the examination of the impact of neoantigen characteristics on immunogenicity while adjusting for the small effective sample size. The number of times each characteristic was selected out of the 1000 random combinations was tracked. The dissociation constant, binding stability, and mRNA expression were each selected over 700 of the 1000 times, while no other characteristic was selected over 500 times (Figure 5B). These fits were consistently able to distinguish immunogenic and non-immunogenic neoantigens, as demonstrated by the high density of area under the receiver operator characteristics curve (AUC) values around the mean of 0.861 (25th percentile 0.828, 75th percentile 0.895) (Figure 5C).
Figure 5. Regularized Regression Approach Selects Dissociation Constant, Expression, and Stability as the Characteristics of Greatest Importance in Prioritizing Immunogenicity.
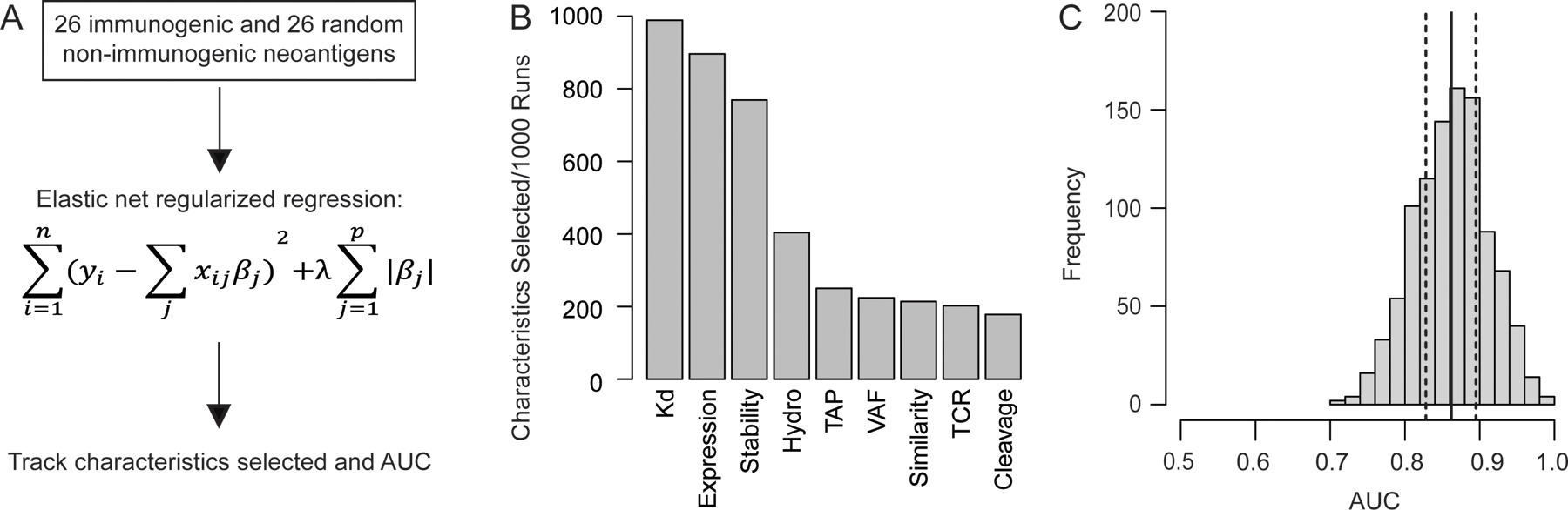
Regularized (elastic net) regression approach to the selection of neoantigen characteristics. (A) Process used for characteristic selection. 26 immunogenic and 26 randomly selected non-immunogenic neoantigens were isolated 1000 times and fit with an elastic net regression. For every fit, the characteristics selected by the model as well as its performance were tracked. (B) Number of times each characteristic was selected out of 1000 runs. The characteristics included are as follows: MHC class I to neoantigen dissociation constant (Kd), mRNA expression (Expression), MHC class I to neoantigen binding stability (Stability), Variant Allele Frequency (VAF), hydrophobicity of the anchor residues (Hydro), TAP transport potential (TAP), T cell recognition potential (TCR), normalized sequence similarity score for closest matched human peptide and the neoantigen (Similarity), and proteasomal cleavage potential (Cleavage). (C) Distribution of the area under the receiver operator characteristics curve (AUC) values for each of the 1000 fits. The solid black line indicates the mean AUC, and the dashed black lines represent the 25th and 75th percentile, respectively.
Based on the results of the model-based regression approach, a logistic regression model was fit with the dissociation constant, binding stability, and expression in the TESLA consortium data. The final logistic regression model will be called the “NeoScore.” The equation for the model is as follows:
Where is the scaled, log10-transformed expression value, is the log10-transformed dissociation constant in units of nanomolar (nM), and is the log10-transformed stability measured as the half-life of the binding interaction in units of hours. The coefficients for expression and stability were both positive, as expected since higher expression and longer binding times likely increase the chance of a neoantigen to elicit an immune response. The coefficient for the dissociation constant was negative, as expected since a lower dissociation constant indicates a higher binding affinity. These coefficients match the observed directions of change from Figure 2. Raw data from NetMHCpan, NetMHCstabpan, and Salmon can be processed and combined to return a set of neoantigens prioritized by their NeoScore using the following web application: https://bordene.shinyapps.io/MHCI_neoantigen_prioritization/.
Given the large discrepancy between the number of immunogenic and non-immunogenic neoantigens, we assessed the impact of changing the ratio of immunogenic to non-immunogenic neoantigens on the performance of the model. This was done by fitting the logistic regression model on 100 random down-sampled datasets from the TESLA consortium data at 10 different ratios of immunogenic to non-immunogenic neoantigens. Each of the logistic regression models was then applied to the Cohen dataset, and the optimism was calculated by subtracting the AUC on the Cohen dataset from the AUC on the TESLA dataset. The data demonstrated that decreasing the sample size of either immunogenic or non-immunogenic neoantigens increased the optimism, suggesting a less generalizable model. The lowest optimism was obtained when the maximal number of both immunogenic and non-immunogenic neoantigens were used, even though there were not equivalent numbers of immunogenic to non-immunogenic neoantigens. (Supplementary Figure 2).
Once the NeoScore model was fit in the TESLA dataset, model performance was assessed in both the TESLA training dataset and four independent test datasets. In the TESLA consortium dataset, the NeoScore had an AUC of 0.845, which exceeds the AUC of 0.70 needed to be considered a discriminatory model (64). The NeoScore also outperformed the AUC of the Łuksza model (AUC=0.615) (Table II; Figure 6A). The NeoScore was then tested in four additional datasets. The NeoScore outperformed the Łuksza model in the Cohen (0.832 AUC vs. 0.689 AUC) and Strønen datasets (0.681 AUC vs. 0.620 AUC) (35, 36) (Table II; Figure 6B,C). In the Carreno dataset, the NeoScore slightly outperformed Łuksza and the pTuneos hydrophobicity model (0.704 AUC for NeoScore, 0.696 AUC for pTuneos hydrophobicity, and 0.657 AUC for Łuksza) (Table II; Figure 6D). Published results of the immunogenicity scores from the pTuneos model were used (18), as the model was not able to be successfully run with the other datasets. Both the hydrophobicity-only model and the full model provided by pTuneos are included, as the hydrophobicity model outperformed the full model. Similarly, in the Ott dataset, the NeoScore slightly outperformed the Łuksza model (0.609 AUC for NeoScore and 0.575 AUC for Łuksza) (11) (Table II; Figure 6E).
Table II:
Comparison of the AUC and optimal threshold sensitivities and specificities for the prediction of immunogenic neoantigens using NeoScore vs. other models.
| Dataset | Model | Sensitivity [95% C.I.] | Specificity [95% C.I.] | AUC |
|---|---|---|---|---|
| TESLA consortium (n = 347) | NeoScore | 0.846 [0.769–1.00] | 0.738 [0.523–0.844] | 0.845 |
| Abbreviated NeoScore 1 | 0.923 [0.731–1.00] | 0.567 [0.530–0.760] | 0.772 | |
| Łuksza | 0.692 [0.462–0.885] | 0.561 [0.439–0.763] | 0.615 | |
| TESLA consortium | 0.385 | 0.941 | -------- | |
| Cohen (n = 357) | NeoScore | 0.857 [0.571–1.00] | 0.546 [0.494–0.597] | 0.832 |
| Abbreviated NeoScore 1 | 1.00 [1.00–1.00] | 0.363 [0.311–0.414] | 0.744 | |
| Łuksza | 1.00 [1.00–1.00] | 0.254 [0.211–0.303] | 0.689 | |
| TESLA consortium | 0.571 | 0.857 | --------- | |
| Strønen (n = 57) | NeoScore | 0.364 [0.091–0.636] | 0.889 [0.800–0.978] | 0.681 |
| Abbreviated NeoScore 1 | 0.909 [0.727–1.00] | 0.644 [0.511–0.778] | 0.794 | |
| Łuksza | 0.727 [0.455–1.00] | 0.422 [0.289–0.556] | 0.620 | |
| TESLA consortium | 0.364 | 0.867 | --------- | |
| Carreno (n = 21) | NeoScore | 0.444 [0.111–0.778] | 0.750 [0.500–1.00] | 0.704 |
| Abbreviated NeoScore 1 | 0.778 [0.556–1.00] | 0.833 [0.583–1.00] | 0.935 | |
| Łuksza | 0.778 [0.553–1.00] | 0.583 [0.333–0.833] | 0.657 | |
| TESLA consortium | 0.222 | 1.00 | -------- | |
| pTuneos hydrophobicity | 0.500 [0.125–1.00] | 0.857 [0.429–1.00] | 0.696 | |
| pTuneos full model | 0.250 [0.125–1.00] | 1.00 [0.143–1.00] | 0.536 | |
| Ott (n = 165) | NeoScore | 0.222 [0.056–0.389] | 0.878 [0.823–0.932] | 0.609 |
| Abbreviated NeoScore 1 | 0.389 [0.167–0.611] | 0.782 [0.714–0.844] | 0.597 | |
| Łuksza | 0.626 [0.544–0.701] | 0.500 [0.278–0.722] | 0.575 | |
| TESLA consortium | 0.056 | 0.959 | -------- |
The abbreviated NeoScore omits expression.
Figure 6. NeoScore Outperforms the Model by Łuksza and Performs Equivalently to the Model by the TESLA Consortium.
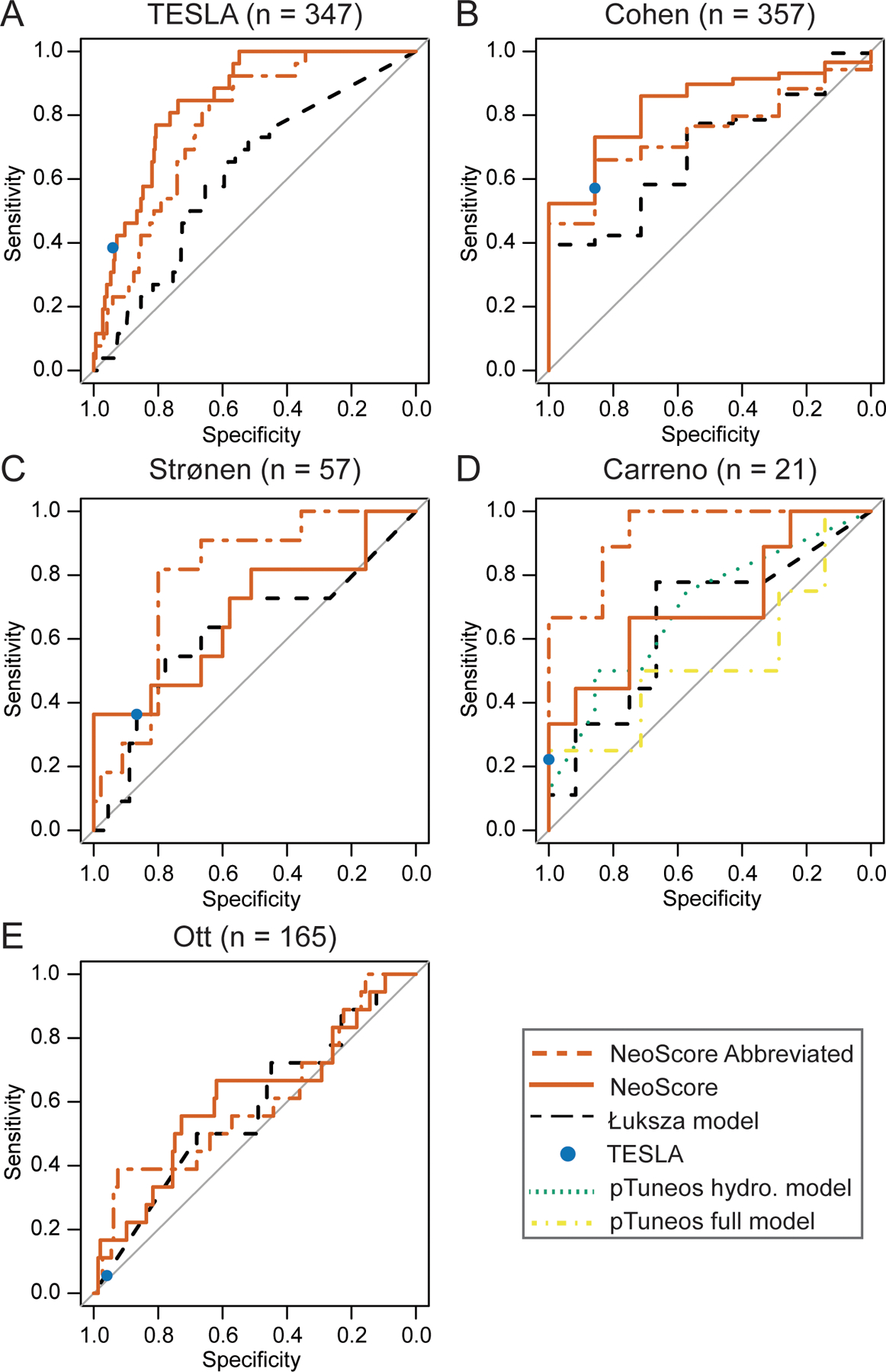
Comparison of the area under the receiver operator characteristics curves (AUC) for the NeoScore and abbreviated NeoScore compared to the models by Łuksza, the TESLA consortium, and pTuneos. A) Performance of the NeoScore and abbreviated NeoScore in the TESLA consortium dataset (the dataset in which the model was fit). Comparison of the NeoScore and abbreviated NeoScore with existing models in the (B) Cohen dataset, (C) Strønen dataset, (D) Carreno dataset, and (E) Ott dataset.
Since the model by the TESLA consortium consists of a single set of recommended thresholds for the neoantigen:MHC class I dissociation constant, neoantigen:MHC class I binding stability, and expression, the NeoScore could not be compared to the TESLA consortium model in terms of the AUC. Therefore, an optimal threshold for the NeoScore was selected that maximized the sum of the sensitivity and specificity in the TESLA dataset and classified the NeoScore into high and low immunogenicity. The reported sensitivity and specificity for each dataset are based on the threshold optimized in the TESLA dataset (−2.478). In the TESLA and Cohen datasets, the NeoScore obtained a greater sensitivity with a lower specificity at the optimal cutpoint compared to the model by the TESLA consortium. Across all the remaining datasets, the sensitivity and specificity of the NeoScore were statistically equivalent to that achieved by the TESLA consortium, as demonstrated by the overlap of the 95% confidence interval (Table II).
Since only a subset of neoantigens were tested for immunogenicity by the TESLA consortium, we assessed the full set of neoantigens predicted to be immunogenic by the NeoScore model. All possible 9 and 10mer neoantigens were generated from each mutation across the five tumors in the TESLA dataset, and the immunogenicity of each neoantigen was prioritized by the NeoScore model. To make the analysis comparable to that done by the TESLA consortium, the neoantigens were assessed for their predicted immunogenicity in the context of the HLA types that were tested by the TESLA consortium. The sensitivity and specificity for the validated immunogenic neoantigens were as reported in Table I. The NeoScore predicted 740 additional immunogenic neoantigens across the five tumors that had not been tested by the TESLA consortium (range 54–310 per patient, data not shown). The large number of untested neoantigens is consistent with the low overlap observed between groups in the original TESLA consortium submissions. Among the 25 submissions, a median of 13% overlap was observed between the neoantigens predicted to be immunogenic (16). The low overlap and large number of untested candidates supports the need for further validation of neoantigen prioritization models.
Despite the high performance of the NeoScore in the TESLA and Cohen datasets, there is a marked decrease in the performance in the Carreno, Strønen, and Ott datasets. A likely cause for the decreased performance is how the immunogenicity was tested in these datasets. TESLA and Cohen both tested for reactive T cells present in the patient with no additional T cell stimulation. In contrast, Carreno et al. administered a dendritic cell vaccine with each of the predicted neoantigens and subsequently tested for an immune response to each neoantigen (7). Strønen et al. exposed PBMCs from healthy patients to dendritic cells transfected with the neoantigen of interest. They then tested for an immune response to those neoantigens (36). Ott et al. immunized patients with pools of long synthetic peptides and then tested for an immune response to each neoantigen that could be generated from the long peptides (11). None of these three methods rely on the expression of the neoantigen in the tumor to activate neoantigen-specific T cells. Therefore, a logistic regression model was fit to the TESLA dataset using only the neoantigen:MHC class I binding stability and dissociation constant. The following abbreviated NeoScore model was obtained:
The coefficients obtained for stability and dissociation constant are comparable to those obtained in the full NeoScore model. The threshold for the abbreviated NeoScore was optimized in the TESLA dataset (−2.856) and then tested in the Cohen, Strønen, Carreno, and Ott datasets. As expected, the abbreviated NeoScore underperformed compared to the full NeoScore model in Cohen and TESLA but outperformed the full NeoScore model in both Carreno and Strønen (Table II; Figure 6). These results suggest that expression predicts neoantigen immunogenicity when priming of T cell responses is dependent on expression of the neoantigen by the tumor. However, when the T cells are stimulated independently of expression by the tumor, the predictive benefit of expression is undermined. Similarly, the performance of the TESLA consortium model, which relies on neoantigen expression, distinctly drops in the Carreno and Strønen datasets (Table II; Figure 6). The poor performance in Ott dataset across models remains unexplained. The Ott dataset did not significantly differ from the other datasets in terms of the distribution of the location of the mutations (anchor vs. non-anchor residues) or the general distribution of the characteristics (data not shown). Overall, elimination of expression enhanced the performance of the NeoScore model when considering the immune response stimulated independently of the tumor.
To further reaffirm the subset of neoantigen characteristics selected, a logistic regression model was fit using all nine neoantigen characteristics from Figure 5B, which did not notably improve the AUC (0.853 for the model with all nine characteristics compared to 0.845 for the NeoScore, data not shown). Furthermore, the optimism is much higher for the model that included all neoantigen characteristics (8.64%) than the NeoScore (2.47%). The optimism indicates the likelihood that the model is overfitting the training data, which would make a less generalizable model. The lack of benefit from the additional characteristics adds support that the subset of characteristics selected is optimal for prioritizing immunogenic neoantigens. Overall, the model with the neoantigen:MHC class I dissociation constant and binding stability, and mRNA expression showed the best potential to consistently separate immunogenic and non-immunogenic neoantigens in validated datasets.
A high maximum NeoScore has a significant association with improved survival in cutaneous melanoma patients treated with immunotherapy
Once the ability of the NeoScore to discriminate immunogenic from non-immunogenic neoantigens with high sensitivity and specificity was established, the association of a single, strongly immunogenic neoantigen with survival in response to immune checkpoint inhibition was assessed. The survival analysis was performed across two datasets, one with treatment with an anti-CTLA-4 monoclonal antibody (21) and the other with anti-PD-1 monoclonal antibodies (29). In both datasets, the cohort was restricted to immunotherapy-naive patients with cutaneous melanoma (21, 29), which allowed us to assess the factors that drive response to immune checkpoint inhibition in melanoma with a high tumor mutational burden and no previous immunoediting. While the range of mutational burden in cutaneous melanoma is wide (1,368–33,591 for the Van Allen dataset and 864–24,292 for the Liu dataset), all samples have a high mutational burden due to UV-induced, DNA mutations. The final cohort sizes were 34 for Van Allen and 53 for Liu. However, the statistical power of the survival analysis is limited by the number of deaths observed in each dataset, resulting in an effective sample size of 22 for Van Allen and 20 for Liu. The NeoScore for each individual neoantigen per tumor was calculated. Since each patient has many neoantigens with a wide range of NeoScores, there are several potential ways to summarize the neoantigen profile for the sake of comparison between patients. Three ways were selected here: 1) the mutational burden, 2) the neoantigen burden, and 3) the highest NeoScore for a neoantigen from the patient, referred to as the “maximum NeoScore.” The mutational burden and neoantigen burden were attempted for the sake of comparison to the literature, while the maximum NeoScore was used in accordance with the principle that a single immunogenic neoantigen can drive the immune response. Survival analysis was then performed separately for the mutational burden, neoantigen burden, and the maximum NeoScore.
Although an optimal NeoScore threshold was determined for the prediction of neoantigen immunogenicity, every patient had at least one neoantigen that exceeded the optimized threshold, indicating that the presence of a predicted immunogenic neoantigen was not sufficient to differentiate the response to immune checkpoint inhibition. Therefore, unique optimal thresholds for survival analysis were determined for the maximum NeoScore using maximally ranked statistics implemented in the MaxStat package and R statistical software (60). Maximally ranked statistics methods implement a search over all possible log-rank test statistics based on thresholds of a predictor variable (here the maximum NeoScore) for the largest standardized log rank test statistic. Since maximally ranked statistic methods optimizes the threshold to detect a difference, performance of the NeoScore and optimal threshold will need to be validated in an independent dataset.
Next, the association of mutational burden and neoantigen burden with survival in response to immune checkpoint inhibition were evaluated. For the sake of consistency, maximally ranked statistics were also used to select the optimal threshold for the mutational burden and neoantigen burden. In the Van Allen dataset, there was no association between mutational burden and progression-free survival in response to immune checkpoint inhibition (p=0.37) (Figure 7A). In the Liu dataset, a high tumor mutational burden was significantly associated with poor progression-free survival (p=0.047) (Figure 7B), which is consistent with the finding of Liu et al. 2019 (29). The neoantigen burden was strongly correlated with the mutational burden in both datasets (Figure 7C and D). On survival analysis, the same results were observed for neoantigen burden as for mutational burden where a high neoantigen burden was not associated with improved progression-free survival in the Van Allen dataset and was associated with decreased progression-free survival in the Liu dataset (data not shown).
Figure 7. High Maximum NeoScore has Improved Association with Survival Compared to Mutational Burden.
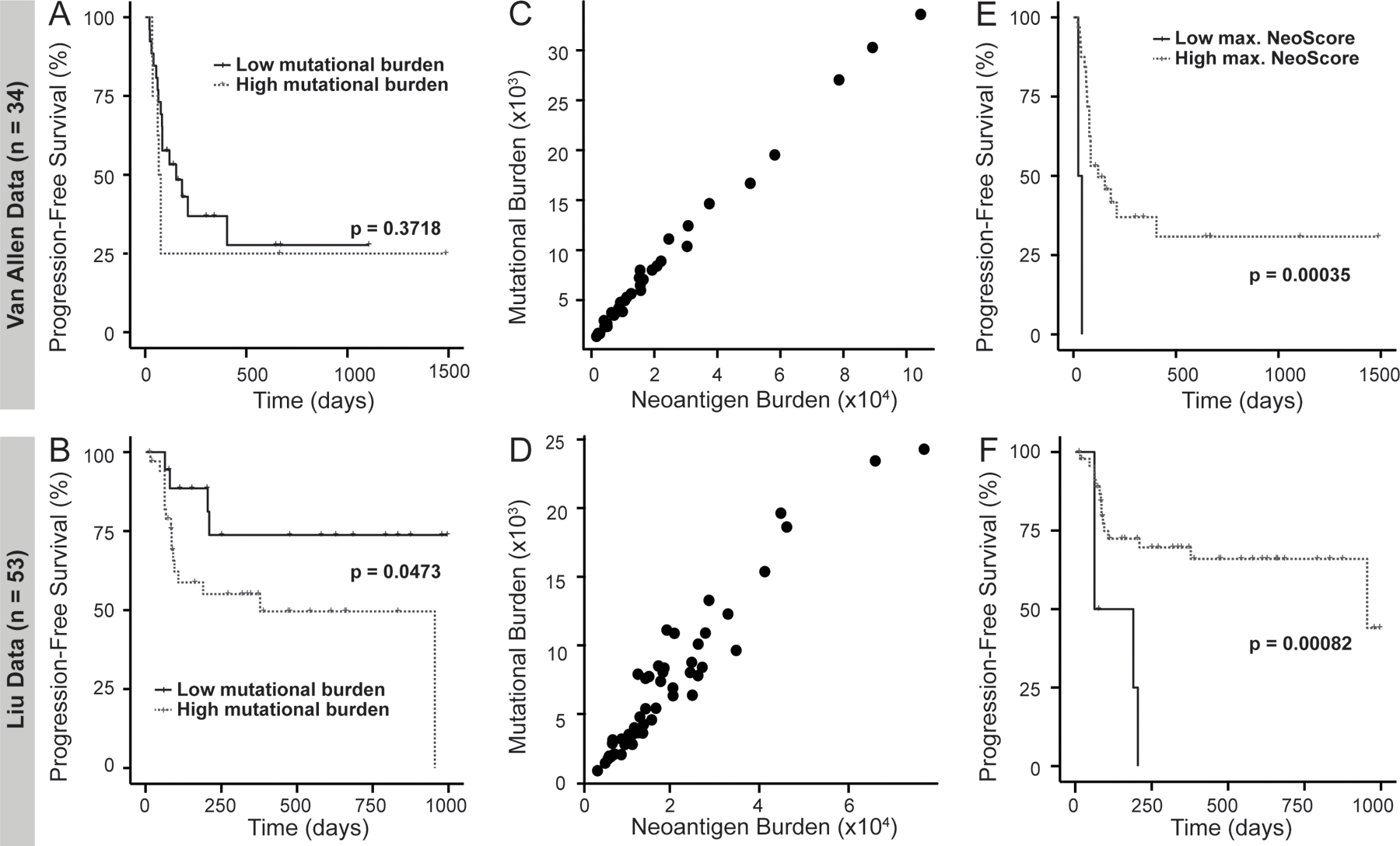
Comparison of progression-free survival probability within treatment-naive, cutaneous melanoma patients. All splits between the groups were determined using maximally ranked statistics, and p-values were calculated using a log-rank test. A high tumor mutational burden is (A) not significantly associated with survival in the Van Allen data and (B) significantly associated with decreased survival in the Liu data. Mutational burden directly correlates with neoantigen burden in (C) the Van Allen data and (D) the Liu data. Patients with a high maximum (max.) NeoScore have (E) significantly increased survival in the Van Allen data and (F) significantly increased survival in the Liu dataset.
When tested with the optimal threshold, patients in the Van Allen dataset with a high maximum NeoScore (above −0.525) had significantly improved progression-free survival (p = 3.5 × 10−4) (Figure 7E). Similarly, in the Liu dataset, patients with a high maximum NeoScore (above −0.152) had significantly improved progression-free survival (p = 8.2 × 10−4) (Figure 7F). In both the Van Allen and Liu datasets a high maximum NeoScore was associated with significantly improved overall survival at the same thresholds (p = 0.002 and p = 0.013, respectively, data not shown). Overall, these results suggest an improved association of the NeoScore with survival following treatment with immunotherapy, compared with tumor mutational burden, in tumors with high tumor mutational burden.
Given the added expense of RNAseq data in a clinical setting, we assessed whether the abbreviated NeoScore would have a similar association with progression-free survival in response to immunotherapy. Therefore, we analyzed the association of the maximum abbreviated NeoScore with progression-free survival in response to treatment in the Liu and Van Allen dataset, as well as an additional lung cancer dataset with no available RNAseq data from Rizvi et al. While the abbreviated NeoScore demonstrated a significant association with progression-free survival in the Van Allen dataset, there was no significant association of the abbreviated NeoScore with progression-free survival in either of the other test datasets (Supplementary Figure 3). The loss of a significant association in the absence of the expression characteristic further emphasizes the importance of expression to the clinical relevance of the NeoScore model.
Discussion:
Prioritization of immunogenic neoantigens is critical for applications to both the development of personalized vaccines and the identification of patients that are likely to benefit from treatment with immune checkpoint inhibition. However, many neoantigen characteristics have been suggested in the literature to date with no consensus on which characteristics impact whether the neoantigen generates a T cell response. Additionally, no model has demonstrated both the ability to score each neoantigen with high sensitivity and specificity and significant association with survival in response to immune checkpoint inhibition. The successes of this study are as follows: 1) identification of those neoantigen characteristics of greatest importance in determining neoantigen immunogenicity, 2) the combination of these characteristics into a single overall immunogenicity score, the NeoScore, with practical applications to personalized vaccine development, 3) integration of the NeoScore into a web application for easy use, and 4) demonstration of the clinical significance of the NeoScore in melanoma.
A model-based statistical prediction approach was used to select the characteristics of SNV and small indel-derived neoantigens that were most predictive of immunogenicity. The dissociation constant and binding stability of the neoantigen:MHC class I complex, and the expression were the combination of neoantigen characteristics best able to discriminate between immunogenic and non-immunogenic neoantigens. While the identification of these three characteristics is consistent with the recent findings of the TESLA consortium (16), it is important to note that the approaches taken by our group and the original group differed in several key ways. First, a completely agnostic approach was applied to the selection of characteristics, including all characteristics that have been suggested in the literature to date; whereas the TESLA consortium began with those shown to have statistical significance. Taking a completely agnostic approach ensured the greatest potential to select the combination of characteristics that maximized the separation of immunogenic and non-immunogenic neoantigens. Second, additional characteristics were included that were not considered by the TESLA consortium, including variant allele frequency and sequence similarity to the closest matched human peptide. Finally, these results were expanded by combining the characteristics into an overall immunogenicity score, the NeoScore. A web application has been made available to calculate the NeoScore or the abbreviated NeoScore and provide a list of neoantigens prioritized by their predicted immunogenicity. The web application is expected to streamline the application of NeoScore for research purposes.
One of the key advantages of the NeoScore is that it allows for the prioritization of neoantigens that may not exceed the single set of thresholds provided by the TESLA consortium. While a threshold for the NeoScore was optimized in the TESLA dataset and demonstrated strong performance across the test datasets, the optimized threshold is necessarily conservative for two reasons. First, all datasets used to build and test the NeoScore consisted of neoantigens that had already been prioritized by the original group. Thus, the NeoScore is trained to discriminate between top candidates. Second, the NeoScore is trained on the natural T cell responses to neoantigens with no stimulation by a therapeutic agent. Treatment with immunotherapy or personalized vaccines may be able to elicit an immune response to neoantigens with a lower NeoScore, Therefore, all neoantigens can be ranked in order of their NeoScore. Researchers applying NeoScore to a new dataset can first rank the predicted neoantigens, then decide on the optimal number of neoantigens to test for a patient based on the unique neoantigen profile of the given patient.
One consideration for future applications of the NeoScore is that it is based on a combination of predictive tools that each come with their own sensitivity and specificity. While we have selected highly rated tools for MHC class I dissociation constants and binding stability, these tools are constantly improving. One example is that the MHC class I binding stability tool that we employed is trained on stability data for 25,000 peptides, which is over an order of magnitude less than the training data available for MHC class I dissociation constant predictions (54). As expanded training data becomes available, the predictive value of each tool is likely to increase, which in turn, will further enhance the predictive value of integrated models such as the NeoScore. Additionally, some recent work has suggested the potential for combining predictions from multiple tools in either a consensus or aggregate approach to further enhance the predictive value of each individual tool (65). Further research is needed into how to optimally aggregate the scores from multiple tools to enhance their application. However, the high performance of the NeoScore in predicting immunogenicity of individual neoantigens and significant association with survival in response to immune checkpoint inhibitors indicates the high performance of each of the individual tools combined to create the NeoScore.
A surprising finding is the lack of association between mutational burden and progression-free survival in the Van Allen dataset and the association of increased mutational burden with decreased progression-free survival in the Liu dataset. These results are inconsistent with the association of increased mutation burden with increased response rate to immune checkpoint inhibition observed across cancer types (27). An explanation for the lack of association of mutation burden with survival following treatment with immune checkpoint inhibition is that the cutaneous melanoma subset consists of all tumors with a high mutational burden. A tumor with a particularly low mutational burden may not have any neoantigens, causing it to have a poor response to immune checkpoint inhibition. However, a high mutational burden alone does not guarantee a good response to immune checkpoint inhibition. As demonstrated by our work, a high maximum NeoScore has an improved association with progression-free survival compared to mutational burden.
The literature to date supports that mutational burden is associated with response to therapy across cancers (27) or across cancer sub-types, but not within tumors with a high mutational burden. Within non-small cell lung cancer, there was a significant increase in response rates and survival in patients with a higher mutational burden than those with a lower mutational burden (25). These results reflect a split between the patients with a mutational signature from smoking carcinogens and those with no evidence of exposure to smoking carcinogens. In contrast, within small cell lung cancers, which are nearly universally associated with smoking, there was a weaker association of mutational burden with response to therapy (22). Three independent studies demonstrated a significant association between high mutational burden and improved response to immune checkpoint inhibition with either anti-CTLA-4 or anti-PD-1 monoclonal antibody treatment in melanoma patients (21, 26, 29). However, these studies included cutaneous, occult, acral, and mucosal melanoma, which may have different mutational profiles. As demonstrated here, there is no association between high mutational burden and improved progression-free survival in the Van Allen and Liu datasets when restricted to cutaneous melanoma. As noted by Snyder et al., the patient in their dataset with the highest number of mutations had minimal or no benefit from anti-CTLA-4 monoclonal antibody treatment. Overall, in combination with prior studies, our results highlight the importance of considering the immunogenicity of the neoantigen in predicting the response to immune checkpoint inhibition.
Despite the successes of our work to date, there are several limitations that highlight areas for future research. For one, the maximum immunogenicity score is not able to account for the response to therapy of all patients. One possible reason that the NeoScore is not able to fully predict treatment response is that the model did not include neoantigens derived from gene fusions, products of noncanonical open reading frames, canonical open reading frames with a frameshift, or large indels. To our knowledge, there is no available dataset that has validated neoantigens derived from these sources. Given that recent work has demonstrated that a single neoantigen from a gene fusion product can drive complete tumor regression (66) and non-canonical proteins disproportionately generate MHC class I binding neoantigens (67), patients misclassified by our model may have had neoantigens from one of these classes of mutations. Additionally, since each of the validated datasets tested the immunogenicity of neoantigens that had already been prioritized by the original group, there may be classes of neoantigens that are immunogenic but were not included in any test dataset. Consideration of additional classes of neoantigens is particularly important given recent evidence that there are classes of neoantigens that have very low binding to MHC class I (68). The inclusion of neoantigens derived from a broader set of mutations may alter the neoantigen characteristics selected as important by our regularized regression approach. For example, neoantigens derived from gene fusions, large indels, or frameshifts are likely to have less sequence similarity to normal human peptides than an SNV or small indel-derived neoantigen, causing characteristics such as the sequence similarity or amplitude to be of greater importance. The need to consider additional types of mutations underscores the importance of generating a validated dataset, including these additional mutations, and repeating characteristic selection.
A second reason that the NeoScore may not be able to account for the response to therapy of all patients is that the model does not consider whether the neoantigens ranked as immunogenic were able to elicit a CD4+ T cell response. Studies have observed that the most effective vaccines are those with neoantigens that elicit combined CD4+ and CD8+ T cell responses (69–73). Additionally, work by Alspach et al. demonstrated the need for MHC class II-restricted neoantigen expression by tumor cells to elicit CD4+ T cell responses and generate effective anti-tumor immune responses both in the absence of therapy and in response to immunotherapy. What is still unknown is if there is any advantage to having a single neoantigen that elicits both a CD8+ and CD4+ T cell-mediated response compared to two independent neoantigens that elicit CD8+ and CD4+ T cell responses, independently. If the ideal circumstance is a single neoantigen capable of stimulating a combined response, an efficient approach may be to test the top MHC class I-restricted neoantigens for their potential to elicit a CD4+ T cell response. However, if two separate neoantigens are equally effective, a separate model for the prioritization of MHC class II-restricted neoantigens may be of greater clinical utility. Overall, there is a need for improved understanding of the interplay between MHC class I- and II-restricted neoantigens in stimulating an effective anti-tumor immune response.
This work has successfully identified the key neoantigen characteristics associated with neoantigen immunogenicity using an agnostic approach that considered all the characteristics suggested by the literature to date. These characteristics have been integrated into a single, overall score, the NeoScore, that predicts the immunogenicity of each neoantigen with high sensitivity and specificity. Finally, a high maximum NeoScore has a significant association with improved survival in response to treatment with immune checkpoint inhibition in cutaneous melanoma. The NeoScore is anticipated to improve neoantigen prioritization for the development of personalized vaccines and the determination of which patients are likely to respond to immunotherapy.
Supplementary Material
Key Points:
MHC class I binding, stability, and expression predict neoantigen immunogenicity.
These characteristics were integrated into the NeoScore to rank immunogenicity.
A high NeoScore is associated with improved response to melanoma immunotherapy.
Acknowledgments:
We thank Dr. Tanya N. Phung for her advice and assistance in comparing the software for the identification of SNVs and indels and integrating the pipeline from raw whole-exome sequencing data to potential neoantigens. We thank Dr. Chi Zhou for his assistance with the pTuneos software. We thank Anngela Adams for her assistance in polishing figures.
Financial Support:
This work was supported in part by the Springboard Initiative from the University of Arizona College of Medicine-Phoenix (K.T.H), the University of Arizona College of Medicine-Phoenix M.D./Ph.D. Program (E.S.B), and a Medical Student Award from the Melanoma Research Foundation Medical Student Award (E.S.B.). This work benefited from support for a related project by the Merit Review Award I01-BX005336 from the United States Department of Veterans Affairs (VA), Biomedical Laboratory Research and Development Service (K.T.H.). The contents do not represent the views of the VA or the United States Government.
Footnotes
Declaration of interests: The authors declare no competing interests.
References:
- 1.Schumacher TN, and Schreiber RD. 2015. Neoantigens in cancer immunotherapy. Science 348: 69–74. [DOI] [PubMed] [Google Scholar]
- 2.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, and Rosenberg SA. 2014. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344: 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward JP, Gubin MM, and Schreiber RD. 2016. The Role of Neoantigens in Naturally Occurring and Therapeutically Induced Immune Responses to Cancer. Adv Immunol 130: 25–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, and Jaffee EM. 2017. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 17: 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linette GP, Becker-Hapak M, Skidmore ZL, Baroja ML, Xu C, Hundal J, Spencer DH, Fu W, Cummins C, Robnett M, Kaabinejadian S, Hildebrand WH, Magrini V, Demeter R, Krupnick AS, Griffith OL, Griffith M, Mardis ER, and Carreno BM. 2019. Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. PNAS 116: 23662–23670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rausch MP, and Hastings KT. 2017. Immune Checkpoint Inhibitors in the Treatment of Melanoma: From Basic Science to Clinical Application. In Cutaneous Melanoma: Etiology and Therapy. Ward WH, and Farma JM, eds, Brisbane (AU) Chapter 9, Codon Publications. [PubMed] [Google Scholar]
- 7.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, and Linette GP. 2015. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348: 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L, and K.−. Investigators. 2015. Pembrolizumab for the treatment of non-small-cell lung cancer. Europe PMC 372: 2018–2028. [DOI] [PubMed] [Google Scholar]
- 9.Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V, van der Burg SH, Thor Straten P, Martinez-Ricarte F, Ponsati B, Okada H, Lassen U, Admon A, Ottensmeier CH, Ulges A, Kreiter S, von Deimling A, Skardelly M, Migliorini D, Kroep JR, Idorn M, Rodon J, Piro J, Poulsen HS, Shraibman B, McCann K, Mendrzyk R, Lower M, Stieglbauer M, Britten CM, Capper D, Welters MJP, Sahuquillo J, Kiesel K, Derhovanessian E, Rusch E, Bunse L, Song C, Heesch S, Wagner C, Kemmer-Bruck A, Ludwig J, Castle JC, Schoor O, Tadmor AD, Green E, Fritsche J, Meyer M, Pawlowski N, Dorner S, Hoffgaard F, Rossler B, Maurer D, Weinschenk T, Reinhardt C, Huber C, Rammensee HG, Singh-Jasuja H, Sahin U, Dietrich PY, and Wick W. 2019. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565: 240–245. [DOI] [PubMed] [Google Scholar]
- 10.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, Shukla SA, Hu Z, Li L, Le PM, Allesoe RL, Richman AR, Kowalczyk MS, Abdelrahman S, Geduldig JE, Charbonneau S, Pelton K, Iorgulescu JB, Elagina L, Zhang W, Olive O, McCluskey C, Olsen LR, Stevens J, Lane WJ, Salazar AM, Daley H, Wen PY, Chiocca EA, Harden M, Lennon NJ, Gabriel S, Getz G, Lander ES, Regev A, Ritz J, Neuberg D, Rodig SJ, Ligon KL, Suva ML, Wucherpfennig KW, Hacohen N, Fritsch EF, Livak KJ, Ott PA, Wu CJ, and Reardon DA. 2019. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565: 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, and Wu CJ. 2017. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Muller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Bruck AK, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Holler C, Utikal J, Huber C, Loquai C, and Tureci O. 2017. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547: 222–226. [DOI] [PubMed] [Google Scholar]
- 13.Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, Gartner J, Jia L, Trebska-McGowan K, Somerville RP, Robbins PF, Rosenberg SA, Goff SL, and Feldman SA. 2018. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med 24: 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjerregaard AM, Nielsen M, Hadrup SR, Szallasi Z, and Eklund AC. 2017. MuPeXI: prediction of neo-epitopes from tumor sequencing data. Cancer Immunol Immunother 66: 1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim S, Kim HS, Kim E, Lee MG, Shin EC, Paik S, and Kim S. 2018. Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann Oncol 29: 1030–1036. [DOI] [PubMed] [Google Scholar]
- 16.Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, Rech AJ, Zaretsky JM, Comin-Anduix B, Ng AHC, Chour W, Yu TV, Rizvi H, Chen JM, Manning P, Steiner GM, Doan XC, Alliance TTNS, Merghoub T, Guinney J, Kolom A, Selinsky C, Ribas A, Hellmann MD, Hacohen N, Sette A, Heath JR, Bhardwaj N, Ramsdell F, Schreiber RD, Schumacher TN, Kvistborg P, and Defranoux NA. 2020. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 183: 818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood MA, Paralkar M, Paralkar MP, Nguyen A, Struck AJ, Ellrott K, Margolin A, Nellore A, and Thompson RF. 2018. Population-level distribution and putative immunogenicity of cancer neoepitopes. BMC Cancer 18: 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou C, Wei Z, Zhang Z, Zhang B, Zhu C, Chen K, Chuai G, Qu S, Xie L, Gao Y, and Liu Q. 2019. pTuneos: prioritizing tumor neoantigens from next-generation sequencing data. Genome Med 11: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, and Schreiber RD. 2014. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515: 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lommatzsch M, Bratke K, and Stoll P. 2018. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med 379: e14. [DOI] [PubMed] [Google Scholar]
- 21.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, and Garraway LA. 2015. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, Rizvi NA, Hirsch FR, Selvaggi G, Szustakowski JD, Sasson A, Golhar R, Vitazka P, Chang H, Geese WJ, and Antonia SJ. 2019. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 35: 329. [DOI] [PubMed] [Google Scholar]
- 23.Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, Rizvi NA, Merghoub T, Levine AJ, Chan TA, Wolchok JD, and Greenbaum BD. 2017. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551: 517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukla SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, and Swanton C. 2016. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351: 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, and Chan TA. 2015. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, and Chan TA. 2014. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371: 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yarchoan M, Hopkins A, and Jaffee EM. 2017. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 377: 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller A, Asmann Y, Cattaneo L, Braggio E, Keats J, Auclair D, Lonial S, Network MC, Russell SJ, and Stewart AK. 2017. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J 7: e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Arnon L, Zimmer L, Gutzmer R, Satzger I, Loquai C, Grabbe S, Vokes N, Margolis CA, Conway J, He MX, Elmarakeby H, Dietlein F, Miao D, Tracy A, Gogas H, Goldinger SM, Utikal J, Blank CU, Rauschenberg R, von Bubnoff D, Krackhardt A, Weide B, Haferkamp S, Kiecker F, Izar B, Garraway L, Regev A, Flaherty K, Paschen A, Van Allen EM, and Schadendorf D. 2019. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 25: 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, Hruban C, Guthrie VB, Rodgers K, Naidoo J, Kang H, Sharfman W, Georgiades C, Verde F, Illei P, Li QK, Gabrielson E, Brock MV, Zahnow CA, Baylin SB, Scharpf RB, Brahmer JR, Karchin R, Pardoll DM, and Velculescu VE. 2017. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov 7: 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt J, Smith AR, Magnin M, Racle J, Devlin JR, Bobisse S, Cesbron J, Bonnet V, Carmona SJ, Huber F, Ciriello G, Speiser DE, Bassani-Sternberg M, Coukos G, Baker BM, Harari A, and Gfeller D. 2021. Prediction of neo-epitope immunogenicity reveals TCR recognition determinants and provides insight into immunoediting. Cell Reports Medicine 2: 100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, Lao CD, Schadendorf D, Wagstaff J, Dummer R, Ferrucci PF, Smylie M, Hill A, Hogg D, Marquez-Rodas I, Jiang J, Rizzo J, Larkin J, and Wolchok JD. 2018. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 19: 1480–1492. [DOI] [PubMed] [Google Scholar]
- 33.Shemesh CS, Hsu JC, Hosseini I, Shen BQ, Rotte A, Twomey P, Girish S, and Wu B. 2021. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol Ther 29: 555–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solomon BJ, Beavis PA, and Darcy PK. 2020. Promising Immuno-Oncology Options for the Future: Cellular Therapies and Personalized Cancer Vaccines. Am Soc Clin Oncol Educ Book 40: 1–6. [DOI] [PubMed] [Google Scholar]
- 35.Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, Li YF, El-Gamil M, Rosenberg SA, and Robbins PF. 2015. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest 125: 3981–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strønen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Boschen ML, Lund-Johansen F, Olweus J, and Schumacher TN. 2016. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352: 1337–1341. [DOI] [PubMed] [Google Scholar]
- 37.Orenbuch R, Filip I, Comito D, Shaman J, Pe’er I, and Rabadan R. 2020. arcasHLA: high-resolution HLA typing from RNAseq. Bioinformatics 36: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolger AM, Lohse M, and Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fairley S, Lowy-Gallego E, Perry R, and Flicek P. 2019. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res 48: D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. [Google Scholar]
- 41.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome S Project Data Processing. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, and Lichtenstein L. 2019. Calling Somatic SNVs and Indels with Mutect2. bioRxiv. [Google Scholar]
- 43.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, and Cheetham RK. 2012. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28: 1811–1817. [DOI] [PubMed] [Google Scholar]
- 45.Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, and Nagarajan N. 2012. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40: 11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F. 2016. The Ensembl Variant Effect Predictor. Genome Biol 17: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, and Griffith M. 2016. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med 8: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Kahari AK, Keenan S, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Overduin B, Parker A, Patricio M, Perry E, Pignatelli M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Aken BL, Birney E, Harrow J, Kinsella R, Muffato M, Ruffier M, Searle SM, Spudich G, Trevanion SJ, Yates A, Zerbino DR, and Flicek P. 2015. Ensembl 2015. Nucleic Acids Res 43: D662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, and Eklund AC. 2015. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol 26: 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao Y, Wang X, Zhang H, Ulintz PJ, Li H, and Guan Y. 2020. FastClone is a probabilistic tool for deconvoluting tumor heterogeneity in bulk-sequencing samples. Nature Communications 11: 4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stranzl T, Larsen MV, Lundegaard C, and Nielsen M. 2010. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62: 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reynisson B, Alvarez B, Paul S, Peters B, and Nielsen M. 2020. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res 48: W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rasmussen M, Fenoy E, Harndahl M, Kristensen AB, Nielsen IK, Nielsen M, and Buus S. 2016. Pan-Specific Prediction of Peptide-MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J Immunol 197: 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chowell D, Krishna S, Becker PD, Cocita C, Shu J, Tan X, Greenberg PD, Klavinskis LS, Blattman JN, and Anderson KS. 2015. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. PNAS 112: E17540E11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, and Peters B. 2015. The immune epitope database (IEDB) 3.0. Nucleic Acids Res 43: D405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dilthey AT, Mentzer AJ, Carapito R, Cutland C, Cereb N, Madhi SA, Rhie A, Koren S, Bahram S, McVean G, and Phillippy AM. 2019. HLA*LA-HLA typing from linearly projected graph alignments. Bioinformatics 35: 4394–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Engebretsen S, and Bohlin J. 2019. Statistical predictions with glmnet. Clin Epigenetics 11: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hothorn T 2007. MaxStat: maximally selected rank statistics.
- 61.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, and Nielsen M. 2017. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol 199: 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bassani-Sternberg M, Chong C, Guillaume P, Solleder M, Pak H, Gannon PO, Kandalaft LE, Coukos G, and Gfeller D. 2017. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput Biol 13: e1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Capietto A-H, Jhunjhunwala S, Pollock SB, Lupardus P, Wong J, Hänsch L, Cevallos J, Chestnut Y, Fernandez A, Lounsbury N, Nozawa T, Singh M, Fan Z, de la Cruz CC, Phung QT, Taraborrelli L, Haley B, Lill JR, Mellman I, Bourgon R, and Delamarre L. 2020. Mutation position is an important determinant for predicting cancer neoantigens. Journal of Experimental Medicine 217: e20190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hosmer DW, Lemeshow S, and Sturdivant RX. 2013. Applied logistic regression, third edition. John Wiley & Sons, Inc. [Google Scholar]
- 65.Bonsack M, Hoppe S, Winter J, Tichy D, Zeller C, Kupper MD, Schitter EC, Blatnik R, and Riemer AB. 2019. Performance Evaluation of MHC Class-I Binding Prediction Tools Based on an Experimentally Validated MHC-Peptide Binding Data Set. Cancer Immunol Res 7: 719–736. [DOI] [PubMed] [Google Scholar]
- 66.Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D, Makarov V, Hoen D, Dalin MG, Wexler L, Ghossein R, Katabi N, Nadeem Z, Cohen MA, Tian SK, Robine N, Arora K, Geiger H, Agius P, Bouvier N, Huberman K, Vanness K, Havel JJ, Sims JS, Samstein RM, Mandal R, Tepe J, Ganly I, Ho AL, Riaz N, Wong RJ, Shukla N, Chan TA, and Morris LGT. 2019. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med 25: 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruiz Cuevas MV, Hardy M-P, Hollý J, Bonneil É, Durette C, Courcelles M, Lanoix J, Côté C, Staudt LM, Lemieux S, Thibault P, Perreault C, and Yewdell JW. 2021. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Reports 34: 108815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, Blanchard T, McMahon D, Sidney J, Sette A, Baker BM, Mandoiu II, and Srivastava PK. 2014. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med 211: 2231–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, Meng W, Lichti CF, Esaulova E, Vomund AN, Runci D, Ward JP, Gubin MM, Medrano RFV, Arthur CD, White JM, Sheehan KCF, Chen A, Wucherpfennig KW, Jacks T, Unanue ER, Artyomov MN, and Schreiber RD. 2019. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574: 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bennett SR, Carbone FR, Karamalis F, Miller JF, and Heath WR. 1997. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med 186: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC, Tadmor AD, Schoenberger SP, Huber C, Tureci O, and Sahin U. 2015. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520: 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ossendorp F, Mengede E, Camps M, Filius R, and Melief CJ. 1998. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med 187: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu M, Kallinteris NL, and von Hofe E. 2012. CD4+ T-cell activation for immunotherapy of malignancies using Ii-Key/MHC class II epitope hybrid vaccines. Vaccine 30: 2805–2810. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.