

Abstract
We investigated the feasibility of utilizing an exon-skipping approach as a genotype-dependent therapeutic for neurofibromatosis type 1 (NF1) by determining which NF1 exons might be skipped while maintaining neurofibromin protein expression and GTPase activating protein (GAP)-related domain (GRD) function. Initial in silico analysis predicted exons that can be skipped with minimal loss of neurofibromin function, which was confirmed by in vitro assessments utilizing an Nf1 cDNA-based functional screening system. Skipping of exons 17 or 52 fit our criteria, as minimal effects on protein expression and GRD activity were noted. Antisense phosphorodiamidate morpholino oligomers (PMOs) were utilized to skip exon 17 in human cell lines with patient-specific pathogenic variants in exon 17, c.1885G>A, and c.1929delG. PMOs restored functional neurofibromin expression. To determine the in vivo significance of exon 17 skipping, we generated a homozygous deletion of exon 17 in a novel mouse model. Mice were viable and exhibited a normal lifespan. Initial studies did not reveal the presence of tumor development; however, altered nesting behavior and systemic lymphoid hyperplasia was noted in peripheral lymphoid organs. Alterations in T and B cell frequencies in the thymus and spleen were identified. Hence, exon skipping should be further investigated as a therapeutic approach for NF1 patients with pathogenic variants in exon 17, as homozygous deletion of exon 17 is consistent with at least partial function of neurofibromin.
Keywords: MT: Oligonucletides: Therapies and Applications, neurofibromatosis type I, Ras/Raf/MAPK pathway, exon skipping, antisense oligo therapeutics, animal models
Graphical abstract
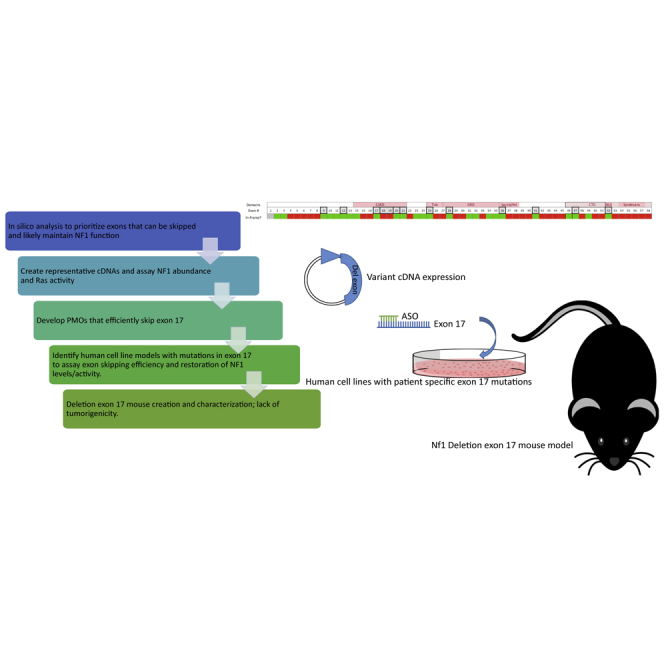
We evaluate neurofibromin sequence in silico; utilize an in vitro cDNA model system; design and test antisense oligos; and utilize them in vitro in human cell lines with exon-specific mutations. Additionally we model exon loss in vivo and demonstrate at least partial function of NF1, providing proof of concept.
Introduction
Neurofibromatosis type 1 (NF1) is one of the most common autosomal dominant neurological disorders and is caused by pathogenic variants in the neurofibromin (NF1) gene. The NF1 gene spans more than 282,000 bases of genomic DNA and encodes a large, multidomain protein named neurofibromin, containing a GTPase activating protein (GAP)-related domain (GRD).1 Haploinsufficiency and subsequent disruptive mutations on both alleles leads to a phenotype that can variably affect the skin (café-au-lait macules [CALMs], axillary freckling, hyperpigmentation, cutaneous neurofibromas), the eye (Lisch nodules and optic glioma), skeleton (dysplasias and scoliosis), and peripheral and central nervous systems (PNS and CNS) (cognitive disabilities, motor delays, gliomas, neurofibromas). Malignant peripheral nerve sheath tumors may also develop, with poor prognosis. Mutation analysis has been available for diagnostic purposes for the past 20 years. Almost 2,900 pathogenic variants have been reported in the Human Gene Mutation Database (HGMD, http://www.hgmd.org). While there are few hotspots, as pathogenic variants are found all along the gene, most lead to lack of full-length protein expression due to gene deletion, premature stop gain, frameshift, or abnormal splicing.
Despite its high prevalence (occurring in approximately 1 of 2,000–3,000 births),2,3 there are few effective therapeutics for NF1. Most currently available drugs being tested to treat NF1 are targeted at tumors and have focused on blocking Ras signaling or interfering with intercellular communication. MEK inhibitors such as selumetinib have demonstrated effectiveness for patients who respond and can tolerate treatment; however, not all patients benefit, plexiform neurofibromas do not completely disappear, and there can be significant side effects.4 Additional treatments that can be used alone or in conjunction with MEK inhibitors are therefore needed. One possible therapeutic class includes exon skipping, which utilizes specific antisense oligonucleotides (ASOs) to bind the target pre-mRNA through base pairing in a way that induces altered RNA splicing, causing the splicing machinery to “skip” one or more exons carrying a pathological variant. The resulting mRNAs are then translated into shortened proteins that—in the case of successful therapy—are able to compensate the loss of critical function as a consequence of the genetic change. Antisense-directed gene therapy for exon skipping has been successfully tested for the treatment of a number of diseases,5 most notably Duchenne muscular dystrophy (DMD).6 The Food and Drug Administration (FDA) has approved three exon-skipping therapies for DMD, eteplirsen (brand name Exondys 51) for ASO-based dystrophin exon 51 skipping,7,8 golodirsen (Vyondys 53) for dystrophin exon 53 skipping,9 and casimersen (Amondys 45) for dystrophin exon 45 skipping.
To evaluate the feasibility and potential utility of exon skipping as a therapeutic for NF1, we determined which regions, if any, could be removed or skipped while retaining normal expression and crucial GRD function of the neurofibromin protein. To this end, we evaluated NF1 (RefSeq: NM_000267.3) in silico, in vitro, and in vivo to test the effects of deletion/skipping of specific exons on neurofibromin function. As proof of concept, ASOs to skip exon 17 were evaluated in NF1 wild type (WT) and mutant human cell lines. ASOs demonstrating the highest fidelity in exon 17 skipping and restoration of neurofibromin expression and function were validated confirming the feasibility of this assay in a cell-based system. Validation on the organismal level was performed by deletion of exon 17 (DelE17) in vivo in a nullizygous mouse model to show that this exon is not required for at least partial neurofibromin function. DelE17 results in a viable adult mouse with normal lifespan and no evidence of tumors, but lymphoid proliferation was present in multiple organs. Collectively, our data provide a proof of concept that loss of exon 17 does not completely inhibit neurofibromin function but may alter its signaling properties in lymphoid subsets, requiring further investigation of targeted exon-skipping therapeutics in preclinical mouse models.
Results
In silico analysis and prioritization of exons
Figure 1 represents NF1 exons and various protein domains, including the GRD encoded by exons 27–35. Herein we label the exons consecutively 1 through 58, although other annotations have been used historically.10 Initial evaluation of the NF1 transcript identified 25 single and an additional 18 consecutive exon pairs that could be skipped while maintaining the translational reading frame (covering 43 of 58 exons). This represents a significant portion (74%) of the transcript that is potentially available for exon-skipping therapeutics.
Figure 1.

In silico analysis of NF1
Domain: exons that contribute to the formation of a known (or suspected) functional protein domain. Note that the C-terminal domain includes the nuclear localization signal and the binding region for Syndecans. Exon #: exon number. We utilized the exon-numbering nomenclature as outlined in LRG_214. Exons (E) that underwent in-depth in silico analysis and that were tested in vitro are marked with a dark border. In-Frame?: Single exons that, when skipped, produce an in-frame deletion (without producing a missense mutation) are marked in green. Single exons that, when skipped, produce a frameshift, leading to a truncated protein, are marked in red. The same holds for consecutive exon pairs that when skipped individually lead to a frameshift but when skipped together result in an in-frame deletion. Note that deletion of exon 1 (marked gray) is in frame. Skipping of exons 56 and 57 (marked gray) maintains reading frame but creates a missense mutation. Length: number of nucleotides contributed by each exon. The longer the exon, the higher the probability that it provides crucial functionality to the protein and the darker the color code. Patient: we mined the LOVD 3.0 (Build 21) for reports of genomic variants with individual exons deleted and report here the number of such entries. Moreover, we searched the literature for patients with individual exons skipped/deleted. Here, we mark an exon in dark red if a patient with NF1 has been reported that has that exon deleted in transcripts due to a mutation. PTM: exons with known, experimentally verified post-translational modifications (PTMs), in particular phosphorylation, ubiquitination, and acetylation (in human and/or murine tissue). We view phosphorylation as likely more important for NF1 function than other PTMs. Consequently, exons containing residues that have been experimentally verified to be phosphorylated are marked in dark red, while all others are marked in pink. Numbers refer to the number of modified residues in a given exon. Note that, with few exceptions, most were obtained through high-throughput proteomic mass spectrometry. Source: Phosphosite Plus, UniProtKB, and Kinexus. Accessibility: number of the amino acids (associated with a given exon) that are predicted to be exposed. Prediction obtained from PROFacc using PredictProtein. Disorder: number of disordered residues. Amino acids are counted toward the exon that provides at least two nucleotides. Prediction obtained from MD (MetaDisorder). MD results are provided as part of the PredictProtein output. MD includes four predictors, namely PROFbval, DISOPRED2, Ucon, and NORSnet. We summarize the output (MD2st; the two-state prediction by MD) for each exon. NORS: percentage of Non-ORdinary Secondary structure, i.e., unstructured loops, contributed by each exon. This is obtained using NORSnet. Avg. Cons.: average conservation score for each exon as calculated by ConSurf. # Max. Cons.: number of amino acids with the highest conservation score for each exon as obtained from ConSurf. Asterisk indicates that exon 31 is alternatively spliced and pathogenic variants have not been reported in databases. Black boxes ( ) indicate that exon 31 was not analyzed in silico. Bold outlined boxes (
) indicate that exon 31 was not analyzed in silico. Bold outlined boxes ( ) indicate exons selected for cDNA screen.
) indicate exons selected for cDNA screen.
We searched the literature and publicly available datasets (Leiden Open Variation Database [LOVD] and HGMD) for reports of NF1 patients with identified pathogenic variants that produced exon skipping or deleted exons in the mature mRNA. Analysis indicates that 49 out of the 58 individual exons can be found deleted or skipped in patient transcripts. From those that can be deleted while maintaining the reading frame, only four exons did not appear in our search, namely exons 17, 25, 31, and 52. As exon 31 also lacks pathogenic variants (none have been reported in databases), we excluded this exon from further analysis (denoted by black boxes in Figure 1), as it would not be a therapeutic target for exon skipping. Exon 31 is alternatively spliced and is present in isoform 2 but not in isoform 1. The remaining exons (17, 25, and 52) were prioritized for in vitro analysis. Of the consecutive exon pairs that might be skipped in combination and retain reading frame (6/7, 7/8, 15/16, 18/19, 29/30, 37/38, 42/43, 44/45, 50/51, 56/57), only 6/7 and 44/45 skipping/deletion have been reported in NF1 patients (documented in the University of Alabama at Birmingham [UAB] Medical Genomics Laboratory and University of Florida Laboratory, respectively; but not in public databases). Our findings for single exons are summarized in Figure 1 (row “Patients”) and Table S1A–S1C (“exon deletions—sources and phenotypes”).
Next, we evaluated exon length, since longer exons encode larger portions of the protein (see Figure 1, row “Length (nts)”). For instance, the longest exon is exon 21 with 441 nucleotides and was found deleted in NF1 patients, suggesting that it is essential. Additional long exons had already been excluded due to their skipping introducing a frameshift or known pathogenicity from reported NF1 patients. Prioritized exons 17, 25, and 52, for which no patients have been reported with skips or deletions, have similar lengths between 117 and 156 nucleotides, with a median of 135 nucleotides.
Since protein function is often associated with protein post-translational modifications (PTMs), we gathered information about the exon localization of experimentally verified PTMs, in particular phosphorylation, ubiquitination, and acetylation (sources: Phosphosite Plus, UniProtKB, and Kinexus). Given that phosphorylation is the most common PTM, we consider phosphorylation as likely more important for neurofibromin function than other PTMs. Consequently, exons containing residues that have been experimentally verified to be phosphorylated are highlighted (Figure 1, row “PTMs,” marked dark red), while numbers refer to the number of modified residues in the respective exon. Most phosphorylation data were obtained through high-throughput proteomic mass spectrometry, and neither the function of phosphorylated residues nor the responsible kinase are known. Six phosphorylation sites have been reported for exon 52, including positions T2554 by protein kinase A11 and Y255612 with known functional roles; pathogenic missense variants have not been reported at these sites. There are no known PTMs of residues in exon 25, and exon 17 carries one potential phosphorylation site.
PredictProtein results for neurofibromin complemented our first evaluation of exons for therapeutic exon skipping. Figure 1 summarizes the results of predicted features, including solvent accessibility, quantified in terms of number of residues predicted to be exposed (row “Accessibility”), disorder status in terms of number of residues predicted to be exposed (row “Disorder”), percentage of non-ordinary secondary structure contributed by each exon (row “NORS”), the average conservation score over all residues associated with an exon (row “Avg. Cons.”), and the number of maximally conserved residues (row “# Max. Cons.”). With respect to solvent accessibility, the three prioritized exon candidates (exons 17, 25, and 52) are very similar (between 9 and 13 residues are classified as exposed), while individual exon contributions to neurofibromin’s surface can be significantly higher (e.g., exon 21 is associated with 46 residues predicted to be exposed). While most exons are fully ordered (including exons 17 and 25), a few have residues classified as disordered. Among these, exons 13, 21, 51, 52, 57, and 58 generate more than 19 such residues, while the only two long (≥30 residues) intrinsically disordered regions (IDRs) are produced by exons 50–51 and 56–57. Of note, IDRs allow a protein to adopt an ensemble of different conformations, which are thought to be in dynamic equilibrium under physiological conditions.13 Only exons 50–53 and 57–58 contribute to a predicted non-ordinary secondary structure. Lastly, the obtained conservation scores strongly suggest that exon 25 is likely not suitable for exon skipping, with a high score of 8.1. In contrast, exons 17 and 52 have low conservation scores of 1.2 and 2.2, respectively.
Following this first round of analysis of NF1, we selected individual exons to model what might happen if they were deleted. All exons that produce a frameshift when deleted/skipped were discarded. Likewise, all exons reported as deleted in the mature transcript in at least one NF1 patient were deprioritized. This reduced the number of candidates for single exon-skipping-based therapy to three: exons 17, 25, and 52. For our additional in-depth in silico analysis, we chose an additional eight single exons, namely exons 9, 12, 20, 21, 28, 36, 41, and 47, all of which retain reading frame when being skipped but are found in patients with an NF1 phenotype. Our selection of the additional eight exon-skipping scenarios was partially based on providing controls, i.e., exons that, when skipped, are known to produce non-functional proteins, e.g., exon 28 that encodes part of the GRD and the critical R1276 “arginine finger” amino acid that binds Ras-GTP.14,15 Moreover, from the set of consecutive exon pairs we included exons 18/19 in our analysis. By further studying these proteins, we expected to gain additional information about the effect that exon skipping might have on neurofibromin function.
Our assessment of the likelihood that skipping an individual exon or exon pair may have a therapeutic effect is based on several factors. Data are summarized in Figure 2 and detailed in Table S2. Exon 17 appeared most promising as a therapeutic target for exon skipping, as changes to secondary structure, solvent accessibility, order, protein-binding sites, and PTMs would be minimal. Effects of exon 17 loss on the function of the cysteine serine-rich domain are unknown. Exon 52 may also be a good candidate due to minimal predicted changes in secondary structure, solvent accessibility, order, and protein-binding sites; however, the effects of the loss of the PTMs and its encoded nuclear localization signal (NLS)16 are unknown. All other exons have relatively high average conservation scores (in particular exons 25, 28, 36, and 41) and/or a large number of maximally conserved residues (such as 18/19, 21, 25, and 47), indicating crucial function. The secondary and tertiary structure of the protein may change dramatically when skipping exons 18/19, 20, 28, 41, and 47. Deletion of exon 21 would result in loss of PTMs and predicted protein-binding sites. Finally, the skipping of exons 9, 12, 20, and 21 may result in proteins with less flexibility, and hence some loss of function.
Figure 2.

PredictProtein results
Summary of in silico analysis for selected exons. Exon(s) skipped: number of exons skipped according to continuous 1…58 exon numbering outlined in LRG_214. Predicted secondary structure (%): the percentage of residues in the remaining protein (NF1delEX) that are predicted to undergo a change in secondary structure when compared with (full-length) human neurofibromin. Highest reliability (secondary structure): predictions of secondary structures have a reliability score assigned. Here we report the highest reliability reported for any such prediction. Predicted solvent accessibility (%): the percentage of residues in the remaining protein (NF1delEX) that are predicted to undergo a change in solvent accessibility when compared with predictions for (full-length) human neurofibromin. Highest reliability (solvent accessibility): predictions of solvent accessibility have a reliability score assigned. Here we report the highest reliability reported for any prediction. Surface contributed by exon (Å2): predicted solvent accessibility in squared angstroms attributed to the amino acids that have been translated from the exon(s). Change of surface area (Å2): predicted total solvent accessibility in squared angstroms of human full-length (fl) neurofibromin minus the predicted surface area contributed by the skipped exon(s) minus the predicted solvent accessibility of the protein with skipped exon. A positive value means that the surface of fl is larger than the surface of the protein with the skipped exon. Predicted O → D (#): number of residues in the shortened protein (NF1delEX) predicted to change status from ordered to disordered, when compared with full-length neurofibromin. Predicted D → O (#): number of residues in the shortened protein (NF1delEX) predicted to change status from disordered to ordered, when compared with full-length neurofibromin. Highest reliability (ordered/disordered): predictions of status (ordered versus disordered) have a reliability score assigned. Here we report the highest reliability reported for any prediction. Predicted P-P binding sites (#): number of predicted protein-protein binding sites as predicted by PROFisis (part of PredictProtein). Predicted PTMs: number of PTMs as predicted from Prosite as part of PredictProtein, CKSAAP_UbSite, and UbiProber (with score >0.8). This also includes PTMs that are not on residues formed by the exon but likely affected by the exon skipping, if the recognition sequence is directly adjacent to the skipped region. Avg. conservation score: average conservation score for each exon as calculated by ConSurf. Maximally conserved residues (#): number of amino acids with the highest conservation score for each exon as obtained from ConSurf.
Testing in cDNA assay system
To both evaluate the in silico predictions and determine the functional effects of exon skipping on neurofibromin, we created and tested Nf1 cDNAs coding for NF1 isoforms with deletions of exons 9, 12, 17, 20, 21, 25, 28, 36, 41, 47, and 52. We also evaluated deletion of both exons 18/19 consecutively. We used synthetic gene fragments to create these deletions and cloned them into a mouse Nf1 cDNA plasmid. All clones were validated by sequencing the entire cDNA region, and all isoforms representing the various exon skips were evaluated in four different functional assays of the NF1-Ras-mitogen activated protein kinase (MAPK) signaling pathway.
First, we determined the level of NF1 protein in NF1 null HEK293 cells when transiently transfected with a constant amount of cDNA (1 μg). A representative western blot probed with NF1 antibody is shown in Figure 3A, as are tubulin blots (used as loading control). A minimum of three separate experiments were quantified and are depicted in Figure 3B as NF1/tubulin; all data are normalized to the WT cDNA such that data can be combined across experiments and blots. Loss of exons 20, 21, 41, and 47 led to significant decreases in neurofibromin levels in comparison with WT control via t test (p < 0.05 and indicated with a red asterisk in Figure 3B), whereas loss of other exons, such as 9 and 17, had no significant effect on protein expression.
Figure 3.

NF1 protein expression and RAS activity
Functional analysis of mNf1 isoforms with selected exon skips. (A) Representative western blot of NF1 and tubulin levels. (B) Quantitation of NF1/tubulin ratios normalized to WT ratio; N ≥ 3. Error bars represent SEM. Resultant neurofibromin abundance for each cDNA was compared with WT levels by t test. Red asterisk indicates p < 0.05. (C) GTP-RAS levels normalized to WT and compared with EV control; N > 3. (D) Representative western blot of pERK/ERK ratios. (E) Quantitation of pERK/ERK ratios normalized to WT and compared with EV control; N ≥ 3. (F) ELK1 transcriptional activity normalized to WT and compared with EV control; N ≥ 3. Error bars represent SEM.
Second, we evaluated the ability of these truncated NF1 protein constructs to regulate levels of GTP-Ras (Figure 3C). GTP-Ras levels of all mutant protein isoforms were statistically compared by t test with that of empty vector (EV) plasmid with no cDNA insert. Mutant protein isoforms lacking exons 17, 25, 41, 47, or 52 were significantly more active (p < 0.05) than EV, as they displayed at least some ability to suppress levels of GTP-Ras. Those that are statistically less active than EV were isoforms lacking exons 18/19, 20, or 28 (p < 0.05).
Third, we evaluated downstream MAPK signaling focusing on pERK/ERK ratios (Figures 3D and 3E) as a second indication of function of NF1 GRD-mediated GAP function. All samples were normalized to the WT protein isoform, and pERK/ERK levels of all mutant isoforms were compared with that of EV by t test. Isoforms lacking exons 12, 17, 18/19, 20, 41, 47, or 52 retained the ability to suppress levels of pERK through GRD-mediated GTPase activity on upstream Ras, performing significantly better than EV (p < 0.05).
Lastly, we evaluated activity of the transcription factor ELK-1, which is downstream of Ras-MAPK signaling using luciferase activity (Figure 3F). All samples were normalized to WT protein isoform and evaluated in at least three experiments. Luciferase levels of all mutant isoforms were compared with that of EV plasmid by t test. Deletion of exons 9, 12, 17, 21, 25, 36, 41, 47, and 52 exhibited significantly lower luciferase levels than EV plasmid, indicating that exon loss did not inhibit the ability of these isoforms to suppress levels of ELK-1.
NF1 exon-specific human population data
We reviewed the UAB Medical Genomics Laboratory (MGL) dataset to verify the prevalence of pathogenic variants as well as to investigate possible genotype-phenotype correlations affecting those exons with no deletion reported in the public domain, i.e., exons 17, 25, and 52. In addition, we summarized the aggregated phenotypic features associated with these pathogenic variants (Table S3) and also gave detail on notable variants within each intron (Table S4). In the MGL dataset, 74 of 8,090 unrelated probands carry a truncating pathogenic variant in exon 17 (either nonsense, frameshift, or out-of-frame splice mutation), therefore ∼0.91% of the unrelated NF1 patients are expected to carry a truncating variant affecting exon 17. No variants that result in skipping or deletion of exon 17 only were detected. Missense and recurrent pathogenic variants are detailed in Table S4. Patients carrying a truncating pathogenic variant in exon 17 presented with a variable number of features typically associated with NF1 in an age-dependent manner, including pigmentary features (CALMs, skinfold freckling [Table S3]). A total of 76 out of 8,090 (∼0.94%) unrelated probands in the MGL dataset carry a truncating variant in exon 25, and another 26 carry one of 12 different missense variants that are either of uncertain significance, likely pathogenic or pathogenic according to recommendations by Richards et al.17 Splicing variants within exon 25 are detailed in Table S4. No variants resulting in skipping or single exon deletion of exon 25 were found. The phenotype in the individuals with the likely pathogenic/pathogenic missense variants was presence of CALMs with or without freckling, with or without learning disabilities, but we had only a single individual older than 19 years; therefore, the understanding of the potential phenotype associated with any of these missenses is limited. The phenotype associated with the presence of a truncating pathogenic variant is classic, predisposing to the full variety of features, as expected (Table S3). A total of 21 out of 8,090 (∼0.25%) unrelated probands carry a truncating pathogenic variant in exon 52: 14 probands carry a frameshift variant; 3 carry a nonsense; 4 carry a truncating splice variant. Splicing variants within exon 52 are detailed in Table S4. Probands (N = 21) with a truncating variant affecting exon 52 presented with pigmentary features (CALMs and/or freckling) and neurofibromas. No symptomatic optic pathway gliomas (but one asymptomatic) or malignancies were observed, but the dataset is limited (0/7 and 0/18). Only 6 of 21 probands with phenotypic data available were ≥18 years old at the time of data collection. Aggregated phenotypic data on the probands carrying a truncating variant in exon 52 are summarized in Table S4. Furthermore, we interrogated both Kaviar (http://db.systemsbiology.net/kaviar/cgi-pub/Kaviar.pl) and the 1,000 genome project (https://www.internationalgenome.org/data/) for NF1 exon 17, 25, and 52 deletions in healthy populations and were unable to find whole exon 17, 25, or 52 deletions.
ASO design and efficiency
We utilized ESEfinder to evaluate exon 17 and flanking intronic sequence to allow the design of ASOs to target exonic splice enhancer (ESE) motifs (Figures 4A and 4B). Pre-mRNA secondary structure of exon 17 and 250 bp of flanking introns were modeled using mFold software (Figure 4C, with the target sites of two of the designed ASOs highlighted). We selected and evaluated four 25mer and nine 28mer ASOs with phosphorodiamidate morpholino oligomer (PMO) chemistry, based upon positioning relative to ESEs, percent GC content, predicted Gibbs free energy of binding to the predicted secondary structure, percentage of ASO target sequence in open loop conformation, and number of ASO ends predicted to bind to single-stranded sequence (summarized in Figure 4A; Table 1). PMOs were transfected at a 2 μM dose using 6 μM Endoporter into WT HEK293 cells. RNA was harvested 24 h post treatment and cDNA generated and subjected to nested PCR. Amplicons were separated on 3% (w/v) agarose gels and quantified using densitometric analysis using ImageJ (Figures 4D–4F). The identity of both full-length and skipped amplicons was confirmed by sequencing (results not shown). The 25mers that produced the highest levels of exon 17 skipping were those targeting exon 17 bases [+70, +94] (24.1% ± 0.5%) and [+108, +132] (20.3% ± 1.1%), and the most efficacious 28mers targeted [+67, +94] (39.7% ± 0.5%), [+69, +96] (34.1% ± 0.9%), [+68, +95] (28.7% ± 0.1%), [+70, +97] (27.5% ± 1.291%), and [+108, +135] (20.2% ± 3.5%). All results were based upon N = 3 except [+69, +96] (N = 9), with standard error of the mean (SEM) indicated by error bars. These ASOs are predicted to bind with high energy to sequences within the pre-mRNA that has open conformation.
Figure 4.

Exon 17 PMO design and efficiency
(A) Table 1 summarizing designed ASO characteristics displaying percent GC content, number of ESE motifs covered, strength of binding, percent open conformation, and number of ends in open loop structures. (B) ESEfinder analysis of NF1 exon 17 with 50 nucleotides from each flanking intron. ESEfinder shows the ESE-binding motifs for indicated SR proteins that are above the threshold level in graphical format. The height of the bars indicates the motif scores generated by the tool. The width of the bar represents the length of the motif (6, 7, or 8 nucleotides). The different colors represent the different SR proteins. (C) mFold analysis of exon 17 NF1 pre-mRNA with 250 nucleotides of flanking intronic sequence, showing the most bioenergetically favorable folded structure, with two designed ASOs mapped as examples. (D and E) Gel electrophoresis and densitometric assessment of NF1 exon skipping, for four 25mer and nine 28mer ASOs with a phosphorodiamidate morpholino oligomer (PMO) chemistry. ASOs were transfected at a 2 μM dose using 6 μM Endoporter into WT HEK293 cells, and 24 h post-treatment cDNA was generated and subjected to nested PCR. Amplicons were separated on 3% (w/v) agarose gels, yielding full-length and exon 17 skipped amplicons of 365 bp and 209 bp, respectively (representative results shown in D) and quantified using densitometric analysis using ImageJ (E). (F) The same process described was repeated for three 28mer ASOs in the A15 HEK293T culture (mutation c.1885G>A) and the exon skipping quantified.
Table 1.
PMOs
| PMO name | Length | PMO 5′-3′ | % GC | ΔG | No. of ESE peaks covered | % in open conformation | No. of ends in open loop structures |
|---|---|---|---|---|---|---|---|
| hNF1.e17[+70;94] | 25 | GATCCATGGACATTTGACTGGTATT | 40 | −9.6 | 3 | 56 | 1 |
| hNF1.e17[+79;103] | 25 | ATTCTTCATGATCCATGGACATTTG | 36 | −5.15 | 5 | 36 | 2 |
| hNF1.e17[+85;109] | 25 | GTAGTAATTCTTCATGATCCATGGA | 36 | −1.4 | 6 | 28 | 0 |
| hNF1.e17[+108;132] | 25 | CCGGAGAGAGGCTCCAGGAGTACGT | 64 | −8.5 | 8 | 40 | 1 |
| hNF1.e17[+67;94] | 28 | GATCCATGGACATTTGACTGGTATTTCC | 42 | −10.2 | 4 | 50 | 1 |
| hNF1.e17 [+68;95] | 28 | TGATCCATGGACATTTGACTGGTATTTC | 39 | −10.5 | 4 | 50 | 0 |
| hNF1.e17[+69;96] | 28 | ATGATCCATGGACATTTGACTGGTATTT | 36 | −10.5 | 4 | 50 | 0 |
| hNF1.e17[+70;97] | 28 | CATGATCCATGGACATTTGACTGGTATT | 39 | −10.1 | 3 | 50 | 0 |
| hNF1.e17[+76;103] | 28 | ATTCTTCATGATCCATGGACATTTGACT | 36 | −8.4 | 5 | 43 | 2 |
| hNF1.e17[+79;+106] | 28 | GTAATTCTTCATGATCCATGGACATTTG | 36 | −6.4 | 5 | 39 | 1 |
| hNF1.e17[+81;+108] | 28 | CTTCCGGAGAGAGGCTCCAGGAGTACGT | 61 | −8.7 | 5 | 32 | 1 |
| hNF1.e17[+108;+135] | 28 | CTTCCGGAGAGAGGCTCCAGGAGTACGT | 61 | −8.7 | 8 | 43 | 0 |
| hNF1.e17[+115;142] | 28 | CTTTTCCCTTCCGGAGAGAGGCTCCAGG | 61 | −8.3 | 5 | 46 | 1 |
Creation and characterization of precision cell line models
Precision model systems were needed to evaluate the efficacy of each ASO’s ability to restore neurofibromin expression and functional ability to inhibit Ras signaling. We utilized two cell lines containing different patient-specific mutations. In the first, cell lines containing the recurrent and inactivating NF1 pathogenic variant c.1885G>A, which creates a cryptic splice acceptor site and deletion of r.1846–1886 resulting in a frameshift that is likely susceptible to nonsense-mediated decay within exon 17, were generated via CRISPR-Cas9 gene editing (Figure S1A). Multiple clones were isolated (A15, B6, B48, and B52) and characterized by subcloning and sequencing, evaluation of neurofibromin RNA, protein, and Ras signaling. Subclones had varying ratios of NF1 WT and variant alleles (Figure S1B). In particular, clone A15 showed only variant sequence. Based on presence of WT or variant alleles, RT-PCR products showed varying levels of WT and variant (deletion) transcripts in each clone (Figure S1C). Again, clone A15 showed only variant transcript. Neurofibromin protein levels based on western blot for each clone were as anticipated based on subcloning and RT results (Figures S1D and S1E), and clone A15 showed no neurofibromin protein. Readouts of Ras signaling including both GTP-Ras levels and pERK/ERK ratios were also as anticipated and elevated for clone A15 (Figures S1D, S1F, and S1G).
ASO efficacy
We began evaluating skipping efficiency in our NF1 mutant cell line clone A15 homozygous for c.1885G>A. PMOs include: hNF1.e17 [+79, +106], later replaced with more efficacious PMOs; hNF1.e17 [+67, +94]; and hNF1.e17 [+69, +96]. Our first analysis examined skipping efficiency at a single 2 μM dose (Figure 5F). In this assay, hNF1.e17 [+79, +106] leads to significantly more skipping than hNF1.e17 [+67, +94] and hNF1.e17 [+69, +96]. We then evaluated efficacy of the skipping in terms of restoration of neurofibromin expression and function in Ras-signaling inhibition. PMOs were utilized in dose response from 0.08 μM to 20 μM and included: Std Ctrl Oligo, hNF1.e17 [+79, +106] (Figures 6A, 6B, 6E, and 6F); hNF1.e17 [+67, +94] (Figures 6C and 6G); and hNF1.e17 [+69, +96] (Figures 6D and 6H). All ASOs showed a dose-response effect and were able to restore neurofibromin expression to up to 40% of WT levels (Figures 6A–6D) and activity in terms of repression of pERK/ERK ratios by 2- to 3-fold (Figures 6E–6H). To determine which ASO is most potent, we treated cells with each PMO at a single dose (2 μM) and ran assays simultaneously (Figures 6I–6L). The three PMOs have relatively similar efficacies at this 2 μM dose, but hNF1.e17 [+67, +94] can restore the most neurofibromin expression and yields the lowest pERK/ERK ratio, although this is not statistically significant. In conclusion, we have designed ASOs that are able to effect skipping of exon 17 to ameliorate the increased Ras signaling caused by the pathogenic variant.
Figure 5.

PMO efficacy in human cell lines
NF1 restoration efficacy was evaluated in HEK293 mutant cell line clone A15 with c.1885GA and patient-derived Schwann cell line icNF97.2a with c.1929delG. (A–H) Dose response for select PMOs. (A), (B), (E), and (F) were treated with PMO hNF1.e17 [+79, +106], (C) and (G) were treated with hNF1.e17 [+67, +94], and (D) and (H) were treated with hNF1.e17 [+69, +96]. (A–D) NF1/actin levels after treatment with PMOs. (A) Representative western blot showing restoration of neurofibromin expression (in comparison with actin expression) after treatment of cells with indicated doses of PMO. (B–D) Quantitation of neurofibromin/actin levels of at least three separate experiments after treatment of cells with indicated doses of PMOs. (E) Representative western blot showing restoration of suppression of pERK/ERK ratios after treatment of cells with indicated doses of PMOs. (F–H) Quantitation of pERK/ERK ratios of at least three separate experiments after treatment of cells with indicated doses of PMOs. (I–L) Comparison of PMOs at single 2 μM dose. (I) Representative western blot of NF1/actin levels. (J) Quantitation of neurofibromin/actin levels of at least three separate experiments after treatment of cells with indicated PMOs. (K) Representative western blot showing restoration of suppression of pERK/ERK ratios after treatment of cells with indicated PMOs. (L) Quantitation of pERK/ERK ratios of at least three separate experiments after treatment of cells with indicated PMOs. Error bars represent SEM. (M) Dose response for hNF1.e17 [+67,+94] in icNF97.2 cells. qRT-PCR products are depicted in the top band with the full-length product denoted with a red asterisk on the right and the shorter skipped product denoted with a green asterisk on the right. The second and third row depict representative western blots of NF1 and actin levels while the fourth and fifth rows depict representative western blots of pERK and total ERK. (N) Quantitation of NF1/actin ratios in three separate experiments after treatment of cells with indicated doses of PMO.
Figure 6.

Creation and phenotyping of Nf1 exon 17 deletion mice
(A) Schematic view of murine Nf1 genomic region with intron and exon boundaries. Blue bar represents exon 17. Top sequences depict WT allele. Exons are bolded and underlined with canonical splice sites in red text. Bottom sequences show the sequence of the deletion alleles. (B) RT-PCR products from WT and a DelE17 nullizygous brain. The DelE17 brain has a shortened RT-PCR product that corresponds to the loss of exon 17. (C) Open field activity of WT and DelE17 mice based on time spent in center of the field and outer edges. Left shows all mice, middle shows data from males, and right shows data from female mice. (D) Nest-building scores for all mice, male mice, and female mice. (E) Representative western blots showing neurofibromin levels and Ras signaling in brain tissue of three WT and three DelE17 mice. (F) Densitometric analysis of NF1 and Ras signaling in brain by WT and DelE17 genotype. Asterisks indicate p < 0.05. Error bars represent SEM.
We also evaluated the efficiency and efficacy of our best ASO, hNF1.e17 [+67, +94], on the second patient mutation cell line. This is an hTERT/mCdk4 immortalized patient-derived Schwann cell line, icNF97.2a, which contains the germline mutation c.233delA and the somatic mutation c.1929delG. c1929delG lies within exon 17 and could potentially be restored by ASOs targeting exon 17, but c233delA is not targeted and function cannot be restored with ASOs targeting exon 17. Treatment of cells with ASO in dose response (0.08–20 μM) restores RNA splicing and neurofibromin protein expression but is unable to lower pERK/ERK ratios (Figures 5M and 5N).
Mouse model DelE17
To test the impact of exon 17 loss on NF1 function in a complex biological system, we created mice that carry Nf1 alleles with exon 17 completely deleted (DelE17) (Figure 6). Two alleles were generated and the DNA sequence is depicted in Figure 6A, and RT-PCR with primers flanking the deletion indicates that the mutant transcript is shorter than the WT transcript (depicted in Figure 6B). When bred to homozygosity, mice with either of these deletions are viable. This contrasts with almost all other mouse models harboring Nf1 pathogenic variant alleles in which Nf1 nullizygosity is lethal by embryonic day 13.5. Likewise, the G848R nullizygous mouse is also viable.18,19 This provides proof of concept that exon 17 is not essential for neurofibromin function during embryogenesis or vital to maturation to adult mice, indicating that its loss does not inhibit critical functions of the NF1 protein that are essential for viability.
We established multiple independent cohorts of mice for cognitive evaluation. We evaluated baseline activity in an open field assay with six male and six female mice of both WT (+/+) and DelE17 (−/−) genotypes at 5–7 months of age and were unable to detect differences in overall activity or activity spent in the center or outer portions of the field (Figure 6C). Differences are undetectable even when evaluated by sex. We also evaluated nest-building behavior in nine WT females, nine WT males, nine null females, and eight null males at 6–14 months of age (Figure 6D). Overall, exon 17 null mice build significantly poorer nests than WT mice. Significance holds for male mice but not for female mice, since WT female mice show relatively poor nest building in this cohort.
Another cohort of three WT (two female, one male) and three null (two female, one male) animals at 11–15 months were utilized to investigate Ras signaling in whole brain (Figures 6E and 6F), which did not exhibit any histologic abnormalities in the cerebral cortex (Figures S2A and S3A), hippocampus (Figures S2C and S3C), cerebellum (Figures S2D and S3D), brainstem (Figures S2D and S3E), or spinal cord (Figure S2E). Neurofibromin levels in the brain are approximately equal for both genotypes, suggesting that despite loss of exon 17 the protein product remains stable. pERK/ERK ratios are not significantly different between genotypes, although the null mice may have slightly elevated levels. We see no differences in pAKT/AKT or pS6/S6 levels. Hence, there are no significant Ras-signaling differences. Thus, the protein sequence encoded by exon 17 is not involved in supporting the protein’s Ras-GAP activity in the brain.
Lastly, we aged a cohort of these DelE17 mice beyond 20 months (1.8 years) and have yet to observe any gross or histologic evidence of tumor development in the CNS (Figures S2 and S3), PNS, major organs, or hematopoietic system (N = 18; 7 females and 11 males). Additionally, we are following 60 additional nullizygous mice (30 males and 30 females) between 7 and 19 months of age for tumor development, but have yet to identify any. A separate cohort of 8-month-old mice (three male and three female nullizygous) were submitted for diagnostic necropsy and histology. We evaluated heart, lungs, kidney, pancreas, stomach and small intestine, large intestine, liver/spleen, reproductive tissue, and brain/spinal cord by hematoxylin and eosin staining. Initial examination was grossly unremarkable. Microscopic analysis resulted in observation of lymphoid hyperplasia in the spleen characterized by expansion of the white pulp with bridging between follicles (Figure 7A), increased presence of plasma cells in these bridging channels (Figure 7B), and germinal center formation (Figure 7C) at the interface between the follicular and periarteriolar region. Hyperplastic lymphoid nodules centered around vessels were also noted in the liver (Figure 7D), lungs (Figure 7E), kidney (Figure 7F), colon (Figure 7G), and oviduct (Figure 7H).
Figure 7.

Nf1 exon 17 deletion mice exhibit lymphoid hyperplasia in multiple organs
(A) Micrograph showing bridging of two lymphoid follicles in the white pulp of the spleen (10× magnification). (B) Higher magnification (40×) of bridging channel (orange dashed outline) between each follicle shows numerous plasma cells depicted (see inset) by their teardrop cellular shape, abundant basophilic cytoplasm, and acentric nuclei. (C) Micrograph showing two lymphoid follicles in the spleen with germinal centers depicted by the lighter basophilic staining (asterisk) and (inset) larger blast cells (20× magnification; inset, 40× magnification). (D) Perivascular lymphoid follicle in the portal region of the liver with foci of blast cells (asterisk), likely depicting a germinal center (20× magnification). (E) Foci of perivascular and peribronchiolar lymphoid aggregate in the lung (20× magnification). Within the bronchi lumen there are several eosinophilic crystals, which is a morphologic indication of eosinophilic crystalline pneumonia. (F) Foci of perivascular lymphoid aggregates in the kidney with a lighter basophilic area consisting of blast cells (asterisk), likely depicting a germinal center. (G) Lymphoid aggregate in the submucosa of the colon with a focal lighter basophilic area consisting of blast cells (asterisk), likely indicative of a germinal center (20× magnification). (H) Focal perivascular lymphoid aggregate in the oviduct (20× magnification).
A small cohort of three male and three female mice of each genotype (14 months, WT littermates and homozygous DelE17 mice) was utilized to explore the cellular subtype responsible for the lymphoid hyperplasia by examining T and B cell frequencies in the spleen, thymus, and lymph nodes by multiparameter flow cytometry. Grossly, spleen and thymus weights did not differ by genotype in this cohort (Figures S4A and S4B) nor did overall cellularity in the thymus, spleen, or mesenteric lymph nodes (Figures S4C–S4E). Single-cell suspensions of spleen, thymus, and lymph nodes were utilized for fluorescence-activated cell sorting (FACS) analysis of T and B cells (Figure 8A). In DelE17 mice significant differences in the proportions of thymic double-positive (CD3+ CD4+ CD8+) T cells (Figure 8B) and CD19+ B cells were noted (Figure 8B). In spleen a significant increase in the proportions of CD19+ B cells was noted (Figure 8C), and no significant differences in the proportions of T or B cells were noted in the mesenteric lymph nodes (Figure 7D). Furthermore, no differences in NK, NKT, or γδ T cells were noted in any of the lymphoid tissues (thymus, spleen, or lymph nodes) examined (data not shown).
Figure 8.

Nf1 exon 17 deletion mice exhibit increased percentage of CD3+, CD4+, and CD8+ double-positive thymocytes, and increased percentage of B cells
(A) Representative flow-cytometric gating scheme in thymus depicting: (1) lymphoid gate and evaluation of the percentages of CD3+ T and CD19+ B cells; (2) CD3+ gated T cells with several populations including (a) CD4−CD8− double-negative thymocytes, (b) CD4+CD8+ double-positive thymocytes, (c) CD4+ helper T cells, and (d) CD8+ cytotoxic T cells; and (3) gating on CD3+CD4+ T cells; Foxp3 expression is used to differentiate regulatory T cells. (B–D) Proportions of T and B cells in the (B) thymus, (C) spleen, and (D) mesenteric lymph node are represented as the percentages of total cells.
Discussion
Exon-skipping strategies have not been applied to NF1 to date. Two prior studies have shown that cryptic splice sites created by deep intronic mutations within NF1 (affecting 2%–3.5% of patients) can be silenced in vitro.20,21 ASOs were used to successfully target newly created 5′ splice sites, thereby restoring normal splicing in fibroblasts and lymphoblast cell lines with one of three different deep intronic pathogenic variants (c.288+2025T>G, c.5749+332A>G, and c.7908–321C>G).20 The first study showed antisense-dependent decrease in Ras-GTP levels, which is consistent with the restoration of neurofibromin function. The second study assessed c.3198-314G>A, and noted leakiness of the splicing mechanism that generated a proportion of correctly spliced transcripts and demonstrated correction of the splicing defect by using specific ASOs.21 Repression of a cryptic intronic splice site has the therapeutic advantage that no coding part of neurofibromin is removed; however, a new ASO therapy must be designed and tested for each pathogenic variant.
We sought to assess strategies to skip exons to mitigate intragenic NF1 pathogenic variants that reside in non-critical regions of neurofibromin. Production of even partially functional neurofibromin could help ameliorate phenotypes. Literature in this area is non-existent. In contrast to masking cryptic splice sites as discussed above, exon skipping has the possible benefit of a single therapy skipping over any pathogenic variant within the region of interest and potentially helping multiple patients with different pathogenic variants. We systematically evaluated NF1 in silico, in vitro, and in vivo to predict and prove which exons can be skipped with the resulting neurofibromin retaining GRD-related function as evaluated by multiple methods. Of note, we have previously been able to evaluate the functional consequences of specific variants using this in vitro approach.22,23 Our goal was not to define a precise level of activity for each variant such that they could be ranked, but instead to guide us in selection of exons for possible skipping. A limitation of our study is that the function assessed here is based only on GRD activity; it is possible that variants affect alternative neurofibromin functions in other ways. For instance, NF1 has recently been shown to bind to the estrogen receptor and act as a transcriptional co-repressor, independent of Ras.24
We showed that our series of in silico analyses strongly predicted the in vitro assays results. For example, exons predicted to have the least effect by PredictProtein (exons 17 and 52) have the highest levels of neurofibromin and ability to suppress Ras activity in vitro. The exons with the highest neurofibromin levels when skipped/deleted (9, 17, and 52) have the lowest percentage of residues predicted to undergo changes in secondary structure when compared with full-length neurofibromin. Conversely, exons 20, 41, and 47 were predicted to significantly alter secondary structure, and deletion of these exons led to the lowest neurofibromin expression. Those exon deletions that were unable to lower GTP-Ras levels (18/19, 20, and 28) were all predicted to have significant changes in secondary structure. Notably, exon 28 deletion retained the least function in Ras assays. This is likely because it encodes a portion of the GRD domain that physically interacts with Ras, including the “arginine finger” residue, R1276.14,15 Hence, we believe that our in silico model is predictive for Ras-GAP activity.
Our cDNA system enables us to routinely tease out differences in NF1 levels and GRD function. We have noted that cDNA deletions resulting in different protein isoforms result in variable levels of protein abundance. NF1 abundance may be the result of several potential mechanisms, as many factors and pathways can play a significant role. It is most likely that each variant has a different molecular effect on the mRNA and/or tertiary protein structure, stability, or degradation rate. However, even though some isoforms result in lower levels of neurofibromin expression, this does not equate to loss of GRD function. In fact, some of the isoforms with lower NF1 protein abundance maintain the ability to inhibit Ras activity. If these isoforms indeed lose stability but retain function, treatment with small molecules to stabilize NF1, such as protein correctors, similar to tezacaftor or lumacaftor used to stabilize the CFTR protein in cystic fibrosis, might be a promising therapy.
We also report here (and previously22) that some isoforms may lead to hyperactivation of GTP-Ras levels above what is seen with EV, e.g., without NF1. Deletion of exons 18/19, 20, and 28 leads to increased GTP-Ras levels (Figure 3C). This may imply that these changes could impede hydrolysis of Ras-GTP. While we do not see such evidence in our analysis of pERK/ERK levels for these clones (Figures 3D and 3E), exons 18/19, 20, and 28 are the only clones that were not able to suppress ELK1 transcriptional activity (Figure 3F). We note that all other isoforms are able to repress ELK1. We believe that this ELK1 data are a reflection of the response of a hypomorphic allele that when expressed at significantly high levels (as in this overexpression assay) is able to repress the most downstream signaling components. Hence, it is likely that exons 18/19, 20, and 28 encode regions of NF1 that are essential to its GRD function and should not be targeted for exon skipping.
We designed ASOs to efficiently skip exon 17 and evaluated their effect in cell lines with both WT and inactivating NF1 variants in exon 17. ASOs were able to skip mutant exons and restore neurofibromin protein expression. Protein expression is restored to within 40% of WT levels in HEK293 cells (Figure 5B) and 47% in Schwann cells (Figure 5N). In cells containing only mutant exon 17 (c.1885G>A), the restored protein was able to reduce pERK/ERK ratios, indicating that the restored protein is functional. However, in the compound heterozygous icNF97.2a cells containing both an exon 17 mutation and a second mutation outside of exon 17, restoration of protein was unable to completely compensate for the second mutation and pERK/ERK ratios were not lowered. This is not unanticipated, as NF1+/− cells characteristically have elevated pERK/ERK ratios. Hence, these ASOs restore expression/function specifically to variants within exon 17.
Comparison of efficacy of these NF1 PMOs with other genes/diseases is very challenging. There are a number of parameters that may influence the efficacy of PMOs, including the antisense oligonucleotide (AON) and pre-mRNA sequences, the dose administered, single or repeated administrations, and the delivery mechanism used within a given investigation. For example, eteplirsen is a 30mer PMO developed by Sarepta Therapeutics to achieve exon 51 skipping in DMD.25 Eteplirsen targets a distinct sequence, pre-mRNA secondary structure, and ESE profile. In addition, it is much further along in the translational pipeline, having gained accelerated approval from the FDA in 2016 following numerous preclinical and clinical investigations. Interestingly, however, the efficacy of the eteplirsen has been subject to scrutiny in the field, and in a recent screen it ranked 92nd of the 413 possible AON sequences trialed to promote DMD exon 51 skipping. The lead candidate identified in this study “Ac0,” demonstrated a 12-fold increase over eteplirsen.26 Furthermore, in clinical trials, limited restoration of dystrophin was observed in patients; however, the effect was deemed meaningful as it resulted in a slowing down or stabilization of disease pathology/progression. This serves to highlight the necessity of continuous screening and development of AONs to find the optimal candidate to take forward for clinical development.
The efficacy of the PMO in a clinical setting could also be improved by altering its delivery. It could be conjugated to a peptide to improve its cellular uptake and tissue-specific targeting. An important consideration, however, is that some peptide conjugated PMOs (PPMOs) can show toxicity. Therefore, judicious design and screening would be important to the success of this approach, and similar work has been performed by our group in collaboration with others.27,28 An alternative route could be to deliver AON sequences as an U7-SnRNA vector; this gene therapy approach has been utilized in the field of DMD29, 30, 31 and is associated with improved delivery and longevity of AON sequence expression. However, with adeno-associated virus (AAV)-capsid immunity it is limited to a single administration and would be best suited as part of a combinatorial therapeutic approach.
The safety and efficacy profiles of PMOs are well regarded. This is demonstrated by the clinical studies in both DMD and spinal muscular atrophy (SMA), and subsequently the FDA approval of eteplirsen, spinraza, and golodirsen. Notably, a preclinical study using SMN PMO25 in combination with myostatin expression delivered as an AAV showed not only prolonged survival in a mouse model but also improved neuromuscular junction maturation, innervation, and increases in both the size of sensory neurons in the dorsal ganglia and the preservation of proprioceptive synapses in the spinal cord.32 Taken together with the success of spinraza, being approved as the first antisense drug treatment for SMA by the FDA,33,34 this showcases that antisense treatments are considered safe and efficacious in the context of treating tissues of the nervous system and, indeed, neurological diseases.
Hence, PMOs are generally considered safe and well tolerated. This provides a promising benchmark for utilizing PMOs to treat NF1. The PMOs assessed and showcased in this paper provide a proof-of-principle demonstration that exon skipping could provide a therapeutic strategy for the treatment of NF1. However, further development of PMOs is ongoing in our lab with a focus on ongoing refinement of the PMO designs, screening sequences that could provide improved exon-skipping efficacy outcomes, modifications of AON chemistries, and delivery vehicles. All of these parameters are being considered to maximize the translational potential and impact of the research being undertaken in the context of NF1. While our investigations thus far have been in vitro in nature, when PMO screening transitions to a suitable NF1 animal model, efficacy and toxicity will be examined. It is anticipated that there is scope for translation in NF1.
As proof of concept, we deleted exon 17 in mouse models using CRISPR targeting. While tumor development was previously reported in the murine Nf1+/− model (pheochromocytoma, leukemia, and lymphoma),35 we do not observe this in the mice lacking both copies of exon 17. DelE17 mice are viable and grossly healthy with no tumor phenotypes; however, we did note generalized lymphoid hyperplasia in multiple organs including the primary and secondary lymphoid tissue of the thymus and spleen and alterations in behavior. Hence, this preliminary assessment validates the feasibility of our approach, but we have yet to determine the full scope of the biological significance of removing exon 17 from the NF1 protein. Additional phenotyping assessments will be performed to investigate these abnormalities at a later date. These relatively mild phenotypes suggest that loss of exon 17 creates a mild hypomorphic allele.
Some pre-existing Nf1 mutant mice may have similar phenotypes to DelE17; deletion of exon 31 (historically exon 23a, which is alternatively spliced) results in deficits in spatial learning, impaired contextual discrimination, and delayed acquisition of motor skills, but does not lead to tumorigenesis.36 In theory, exon 31 deletion simply results in lower GAP activity (in comparison with full-length NF1);37 however, altered Ras signaling in the brain or other tissues was not evaluated in these mice. Of note is the fact that one of the nullizygous exon 31 deletion mice showed splenic hyperplasia, with expansion of red pulp and increased extramedullary hematopoiesis. Furthermore, the data from the exon 31 deletion mouse indicates that the learning deficits caused by reduced GAP activity are not a result of developmental deficits (as mice have normal embryological development and tumor suppression), but instead suggest that they result from disruption of function in the adult brain, implying that treatment for these learning disabilities may be possible. Effects of Ras-inhibitory treatment on DelE17 mice remain to be characterized. If successful, these mice and the nest-building assay will represent a robust behavioral model in which to evaluate therapeutics to improve cognitive function in individuals with NF1.
In summary, our data suggest that clinical treatment by skipping NF1 exon 17 is feasible for individuals with pathogenic variants in this exon. Future studies will include the use of ASOs for exon skipping in precision mouse models with humanized exon 17 sequence.
Materials and methods
In silico analysis
Human NF1 cDNA sequence was downloaded. We utilized the exon-numbering nomenclature as outlined in LRG_214. Mutation nomenclature follows transcript NM_000267.3. Coding frames and exon boundaries were mapped to identify which exons could be skipped either as singletons or as two consecutive exons while still maintaining protein reading frame. Known and prospective protein domains were overlaid onto this map. Exon lengths were determined. We also evaluated the literature, the HGMD, and the LOVD for reports of patients with known NF1 mutations that result in skipping of exons during splicing (see Table S1 for summary of LOVD entries and list of publications mined for reports of NF1 patients with skipped/deleted exons and associated phenotype). In addition, we mapped experimentally verified PTMs of NF1 as reported on UniProtKB (https://www.uniprot.org/uniprot/P21359), PhosphoNet (http://www.phosphonet.ca), and Phosphosite Plus (https://www.phosphosite.org/). PTMs included phosphorylation, acetylation, methylation, and ubiquitination. Furthermore, neurofibromin (P21359-2) was analyzed using PredictProtein (https://www.PredictProtein.org),38 a web server that combines various (protein) sequence analysis and structure prediction tools. Specifically, PredictProtein returns predictions of protein secondary structure, solvent accessibility, disorder status, protein-protein binding, PTMs, and conservation, among others. Conservation scores were obtained via PredictProtein using ConSurf (https://consurf.tau.ac.il).39 For PTM predictions based on signature sequences, PredictProtein calls Prosite (https://prosite.expasy.org).40 Outputs were analyzed for all exons individually. Note that unlike other structure prediction tools, inputs to PredictProtein are not limited by their sequence length, which makes this tool particularly useful given neurofibromin’s length.
For a selection of 12 exons, 11 single and one pair of consecutive exons, we also ran PredictProtein on the amino acid sequences obtained by skipping the exon(s). Resulting predictions of various features were compared with those previously obtained for neurofibromin (see above) to assess the impact that individual exon deletions have on the protein structure and functionality. Residue-specific differences were quantified and summarized. In addition, we predicted ubiquitination sites in selected exons using the tools CKSAAP_UbSite (http://systbio.cau.edu.cn/cksaap_ubsite/)41 and UbiProber (http://bioinfo.ncu.edu.cn/UbiProber.aspx).42
cDNA expression system
We have established a heterologous cell-culture expression system using a full-length mNf1 and NF1-null human cell lines.22,23 Mouse and human NF1 sequences are extremely conserved with 92% sequence identity at the cDNA level and 98% amino acid identity. The full-length mNf1 cDNA produces a >250 kDa neurofibromin protein that is capable of modulating Ras signaling. We created mutant cDNAs encoding different protein isoforms representing various exon skips and assessed their ability to produce mature neurofibromin and restore Nf1 activity in NF1−/− cells. Ras activity data for isoforms included levels of GTP-Ras, pERK/ERK ratios, and ELK1 transcriptional activity normalized to WT for each experiment and in comparison with EV.
Cell culture
HEK293 (WT or NF1+/+) cells were obtained from the American Type Culture Collection (CRL-1573) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) + 10% fetal bovine serum and 1× penicillin-streptomycin using standard culture procedures. NF1−/− or null HEK293 cells were created through CRISPR-Cas9 targeting NF1 exon 2.22
Nf1 cDNA plasmid development
The Nf1 cDNA plasmid was developed by GeneCopoeia and is commercially available.
Transient transfections
HEK293 WT or NF1 null cells were transfected with LipoD293 (SignaGen Lab, cat. #SL100668) or Lipofectamine 3000 (Invitrogen, cat. #L30000008) and cDNA at 1 μg per 6-well dish seeded with 500,000 cells per well or 100 ng/96-well seeded with 50,000 cells. Assays were performed 48–72 h later. To control for possible variations in transfection efficiencies, we repeated experiments multiple times and used multiple independent plasmid preparations to control for quality of DNA.
Western blotting
Cells were lysed with RIPA buffer, and lysates were cleared by centrifugation at 20,000 rpm for 20 min at 4°C. Protein was quantitated with a Bradford assays and 50 μg of protein was loaded per well for NF1 blots and 10 μg of protein was loaded for other blots. SDS-polyacrylamide gels (8%) were run at 100 V for 2 h and transferred at 100 V for 2 h onto polyvinylidene fluoride. Blots were probed overnight at 4°C with primary antibody washed and probed 1 h at room temperature with secondary. Primary antibodies include N-Terminal NF1 (Cell Signaling Technology, cat. #D7R7D, 1:1,000), tubulin (Abcam, cat. #ab52866, 1:1,000), pERK (Cell Signaling, cat. #9101, 1:1,000), and total ERK (Cell Signaling, cat. #9102, 1:1,000). Secondary was horseradish peroxidase tagged from Santa Cruz Biotechnology. Chemiluminescent substrate from Bio-Rad was used as per manufacturer’s protocols.
RAS-G-LISA assay
The RAS-G-LISA assay was obtained from Cytoskeleton and was performed according to the manufacturer’s instructions.
ELK-1 Transcriptional repression assay
ELK1 is a major nuclear substrate for ERK, where phosphorylation of ELK1 by kinases results in the conformational change of ELK1 and triggers its DNA binding activity. Plasmids containing the ELK-1 transactivation domain fused to GAL-4 and UAS-Luciferase constructs were a kind gift from the Roger Davis laboratory. Together, they act as a reporter system to monitor ELK1-dependent transcriptional activity and MAPK signaling. In fact, ERK suppression has been measured by the ELK reporter assay in HEK293 cells to show that SPRED1 recruits NF1 to suppress Ras activation. Both NF1 and SPRED1 mutations in the GRD-EVH1 interaction domains reduce Ras-ERK suppression activity.43 A strong correlation among pathogenic mutations, disruption of the GRD-EVH1 interaction, and ERK suppression activity has been reported.43 Hence, NF1−/− HEK 293 cells were transfected with 25 ng of pGAL4 and pGal4-ELK1 plasmids and 1 ng of pNL1.1TK [Nluc/TK] transfection control, along with 100 ng of respective Nf1 mutation plasmids with Lipofectamine 3000 and plated on a 96-well plate such that each well received 50,000 cells. After 24 h, the medium was replaced with normal growth medium. The experiment was terminated at 48 h after transfection with reporter lysis buffer. After lysis, NanoLuc and firefly luciferase readings (relative light units) were obtained using Luciferase Assay Reagent (Promega, E1500) and a BioTek Synergy 2 plate reader. Readings were normalized to NanoLuc expression and percentage change in luciferase activity in comparison with NF1−/− cells transfected with WT cDNA vector. We evaluated statistical deviation of each cDNA from the EV clone to evaluate whether isoforms retained any function. Each mutation set was done in triplicate, and the entire experimental setup was repeated at least three times.
ASO design
PMOs were designed in accordance with previously published work.44 Multiple parameters were considered during PMO design; these included the PMO coverage of ESEs and the Gibbs free energy of PMO binding and association. In addition, in silico modeling of the pre-mRNA secondary structure of exon 17 and the immediately adjacent intron sequences was undertaken. Once a secondary structure was obtained, PMOs were mapped to identify whether their binding was within predicted “open” or “closed” confirmations of the pre-mRNA, which impacts PMO efficacy. In brief, NF1 exon 17 and 50 bp of flanking intron sequence was analyzed with ESE Finder to identify ESE motifs within the exon. Secondary structures of exon 17 and 250 bp of flanking sequence were then predicted with mfold, and the model with the most favorable energetic profile was selected.
PMOs were designed using sFold to assess the strength of binding to target. Candidates were selected for screening based upon the percentage of their GC content, their ability to mask predicted ESE motifs, and their Gibbs free energy of binding and position relative to “open” regions of the predicted secondary structure.
ASO treatment
A range of custom morpholinos spanning exon 17 were ordered from Gene Tools and reconstituted in 0.22 μM filtered ultrapure nuclease-free water, to make a 1 mM stock concentration, as per the manufacturer’s directions. Their efficacy was assessed in control WT HEK293 cell lines and those with a patient mutation in NF1 exon 17. Cells were cultured in 10% (v/v) fetal calf serum in DMEM (Gibco) with 1% penicillin-streptomycin (Gibco); once cells attained ∼70%–80% confluency they were treated with ASOs diluted in 1 mL of fresh culture medium at desired concentrations alongside 6 μM Endo-Porter (dimethyl sulfoxide) (Gene Tools). Cells were incubated at 37°C, 5% CO2 for 24 h prior to harvest. The PMOs identified with the best exon skipping efficacy were then selected and subject to a tiling screen, where PMOs in close proximity to the lead candidates were also assessed. This led to the identification of [+67, +94] (from [+69, 96]), which appeared to have better efficacy in the assessments undertaken. This PMO was then selected for onward study.
ASO efficiency assay
After treatment with PMOs, cells were lysed in RLT buffer and total RNA extracted using an RNeasy Miniprep Kit (Qiagen, UK). Extracted RNA was subject to a Quantitect cDNA synthesis reaction (Qiagen). In brief, 1 μg of RNA was added to 2 μL of genomic DNA wipeout and made up to 14 μL with nuclease-free water; this RNA-water mix was heated to 42°C for 2 min and held at 4°C for 5 min to remove secondary structures. Upon completion, 4 μL of 5× Quantiscript RT Buffer, 1 μL of Quantiscript reverse transcriptase, and 1 μL of primer mix was added, and the reaction was then heated to 42°C for 30 min and inactivated at 95°C for 3 min.
Resultant cDNA was then amplified via nested PCR. Amplification was achieved with GoTaq G2 Flex (Promega, UK). The standard 25 μL PCR reaction comprised 1.5 mM MgCl, 0.2 μM forward and reverse primers, 0.2 μM deoxynucleoside triphosphates, and 0.05 U/μL GoTaq G2 Flex Polymerase. For the first round, 3 μL of cDNA was added to 22 μL of master mix. Primers used were Ex17_1F 5′-AAT GGA GGC TCT GCT GGT TC-3′ and Ex17_1R 5′-ACA CTT CAT CCA CCC CAC AC-3′, yielding a full-length (unskipped) amplicon of 545 bp and an exon 17 skipped amplicon of 389 bp. Thermocycling conditions: 94°C for 2 min, 20× cycles (94°C for 30 s, 60°C for 30 s, 72°C for 30 s), and 72°C for 5 min.
For the second round, 1 μL of the first-round product was added to 24 μL of master mix. Primers used were Ex17_2F 5′-TCT CAA GTG GTT GCG GGA AA-3′ and Ex17_2R 5′-CTG CTT CCT CAC AGA GGT GG-3′, yielding a full-length (unskipped) amplicon of 365 bp and 209 bp. Thermocycling conditions: 94°C for 2 min, 30× cycles (94°C for 30 s, 63°C for 30 s, 72°C for 30 s), and 72°C for 5 min.
A 20 μL aliquot of the second-round product was resolved on a 3% (w/v) agarose in TAE buffer at 70 V for 2 h and imaged. The images were then subjected to densitometric analysis using ImageJ software. Arbitrary values assigned to amplicons based upon analysis were then subject to the following calculation:
Mutant cell line generation and characterization
c.1885G>A
CRISPR guides were designed using CRISPOR (crispor.tefor.org), and a repair template was designed to generate a known patient mutation and also introduce silent changes to obliterate the protospacer adjacent motif (PAM) site and generate a restriction site for screening as necessary. Exon 17 guide reverse strand: 5′-CAC ATC CTA CCC CGT AAA AAA GG-3′. Repair template coded for recurrent: c.1885G>A, which results in out-of-frame missplicing r.1846_1886del; p.Gln616GlyfrTer4 and found in 40 unrelated probands. The exon 17 repair template also coded for a silent c.1875C>T to obliterate the PAM sequence and a silent c.1890A>C to generate an Hpy 188I site for cell line screening.
Guides were cloned into pX459 and transfected into HEK293 cells. HEK293 cells were chosen because this cell line is well characterized, used historically in NF1 research, easily takes up exogenous DNA, and is easy to culture and scale. This cell line is derived from human embryonic kidney cells and carries a modal chromosome number of 64 in 30% of cells, and chromosome 17 (NF1) is present in 3–4 copies. Notably, this increased chromosome number does not affect any of the RAS or Ras-GAP genes. HEK293s have all three Ras isoforms. Cells were selected with puromycin, and single-cell cloning was used to isolate clonal lines for screening. DNA was isolated from each clone as screened for mutations of interest via restriction digest or direct sequencing of PCR products. PCR amplification primers include: exon 17, forward 5′-GGA AGA CAA CTC AAA TAA GTG TTT ATT CC-3′ and reverse 5′-AAT TTC ATT CAG AAA ACA AAC AGA GCA CAT AAA A-3′. Once clones of interest were identified, PCR products were cloned and individually sequenced to define alleles. Multiple clones containing the variant of interest (c.1885G>A) were identified and further characterized: A15, B6, B48, and B52.
icNF97.2a
Patient-derived Schwann cell cultures were established from NF1 cutaneous neurofibromas immortalized using lentiviral vectors containing WT murine Cdk4 cDNA and human telomerase (hTERT) cDNA (unpublished data, MRW), as previously done for NF1 plexiform neurofibroma Schwann cells.45 Sanger sequencing was used to identify the NF1 germline and somatic mutations. icNF97.2a contains germline mutation c.233delA, and the somatic mutation c.1929delG; c.1929delG is within exon 17.
Mutant mouse generation
CRISPR/single guide RNA design and synthesis
CRISPR guides were designed using CRISPOR (crispor.tefor.org) to excise exon 17 and the canonical AG splice receptor signal in the mouse Nf1 locus: Ex17 5′ G2 (forward strand), 5′-CGG ATA CGT CTT CTT TCC AGC-3′; Ex17 3′ G1 (reverse strand), 5′-ACA GAG GGA GTT ACC TAC CA-3′. Modified synthetic single guide RNAs (Synthego) were allowed to complex with Alt-R S.p. Cas9 Nuclease V3 (IDT) at room temperature for 15 min before dilution with DMEM to a final concentration of 100 ng/μL, 100 ng/μL, and 200 ng/μL of guide 1, guide 2, and Cas9, respectively.
Gonadotropins
Female C57B6J embryo donors from 3 to 4 weeks of age were administered 5 IU of pregnant mare serum gonadotropin (Sigma, St. Louis, MO, USA) on day −3 followed by 5 IU of human chorionic gonadotropin (Sigma) on day −1 to induce superovulation. Donor and recipient females were mated to stud and vasectomized males, respectively, on day −1.
Collection of embryos
At day 0.5 post conception, superovulated donor females with copulatory plugs were humanely sacrificed using CO2 followed by cervical dislocation. Oviducts were dissected into sterile medium and nicked to expose the cumulus masses containing fertilized embryos. Embryos were cultured in KSOM (Millipore, Darmstadt, Germany) under 5% blood gas prior to electroporation.
Electroporation
Fertilized embryos were placed in a 5 mm Petri dish parallel platinum plate electrode (Nepa Gene, Chiba, Japan) and electroporated using the NEPA21 Super Electoporator in batches of 25 using the following parameters: poring 150 V, 3 ms, No. 4, decay rate 10%, polarity +; transfer 20 V, 50 ms, No. 5, decay rate 40%, polarity +/−. Surviving embryos were transferred as previously described.
Animal identification
Animals were identified by cage card, sex, and unique “ear tags” consecutively numbered that were affixed at weaning.
Biopsies
Tail biopsies were collected at weaning. A 5–7 mm portion of the distal segment of the tail was cut for analysis and the remainder cauterized. Genomic DNA was purified from the lysed tail samples.
Identification of founders
Founder animals were identified by PCR using primers flanking the target loci that amplified a 519 bp fragment in WT animals (Ex17 F2: 5′-ACT TGG TTT GGA GGT TGG ACA-3′; Ex 17 R1: TTG CTC AGG GTT CCA CAG TG). PCR samples were run on 6% polyacrylamide/TBE gels at 100 V for 45 min before staining with ethidium bromide. Mutations were identified in three out of three pups born from electroporated embryos, and all appeared to harbor a large deletion allele. Positive samples were confirmed by modified Sanger sequencing, which confirmed all three founders had all of exon 17 deleted including the canonical AG splice site at the 3′ end of intron 16 flanking exon 17 as well as the +1 site of intron 17. At least two alleles were identified: c.1845-2_2007+1del and c.1845-9_2007+7insGACAC; both alleles result in p.Q616_M669del54.
RT-PCR for exon 17 in human and mouse
WT and c.1885G>A HEK293 cells were collected, and RNA was extracted using Qiagen RNeasy kits as per manufacturer’s directions. RNA was reverse transcribed and used in a PCR reaction with the following human primers: forward 5′-GGG AGA TTA GCT CAC AAA TGC-3′ and reverse 5′-GTG CTG CTC TTC CTG TTG ACA TC-3′. Transcripts containing exon 17 result in a PCR product of 552 bp. Transcripts not containing exon 17 result in a PCR product of 396 bp. Mouse brains were harvested from an age-matched WT and DelE17 null mice and stored immediately in RNAlater. RNA was extracted using Qiagen RNeasy kits as per manufacturer’s directions. RNA was reverse transcribed and used in a PCR reaction with the following primers: forward 5′-CTG ATT CAT GCA GAC CCA AA-3′ and reverse 5′-GAC ATG GGA CAT CCG TCT CT-3′. Transcripts containing exon 17 result in a PCR product of 972 bp. Transcripts not containing exon 17 result in a shorter PCR product. Sequencing of RT-PCR products reveals deletion of exon 17 RNA sequences. Identical results were obtained from both independent mouse lines; therefore, we combined them.
Mouse cohorts
At least four independent cohorts were generated for analysis: Cohort 1 for initial behavior studies (open field and nesting): 6 males and 6 females of WT and null genotypes aged 5–7 months. Cohort 2: for necropsy of 3 null males and 3 null females at 9 months. Cohort 3: for FACS analysis and additional nesting behavior of 3 males and 3 females of WT and null genotypes at 14 months. Cohort 4: 3 WT (2 females; 1 male) and 3 null (2 females; 1 male) mice for western blots at 11–15 months.
Open field activity
Assay was performed using standard operating procedures at the UAB Small Animal Behavioral Assessment Core as previously described with the exception that we tested only one animal at a time.46 Animals were tracked in an open field (square box) for 4 min. The amount of time spent in the center of the arena versus the side was recorded as a measure of anxiety. Other measures included: ambulation time, rearing, self-grooming, and fecal droppings.
Nest-building assay
Nest building was performed and scored on a 1–5 scale as previously described.47 In brief, mice were given cotton nestlets overnight and scoring was performed on a 1–5 scale, with score 1 having more than 90% of the nestlet intact and score 5 having a near perfect nest.
Necropsy
Animals were examined prior to euthanasia and exhibited no remarkable abnormalities. Animals were euthanized following standard protocol using barbital prior to blunt dissection. All organ systems were examined succeeding ventral dissection and noted as grossly unremarkable. Abdominal organs were excised by pluck dissection and placed in formalin. Brain and spinal column were placed in Formical for 24 h and then transferred to 70% EtOH. Tissues were dissected from pluck and placed into individual cassettes to be submitted to the Comparative Pathology Laboratory at UAB where 5-μm sections were cut from paraffin-embedded tissue specimens and stained with hematoxylin and eosin. Samples were blinded, and histologic lesion spectra were evaluated by a board-certified veterinary anatomic pathologist. Representative lesions were imaged using Nikon Elements D software and a Nikon Eclipse Ci light microscope.
FACS analysis
Animals were euthanized according to a standard protocol using isofluorane inhalation followed by cervical dislocation. Cardiac puncture was performed to collect blood for plasma analysis. Spleen, thymus, and mesenteric lymph nodes were collected and weighed, then single-cell suspensions were isolated from fresh tissues and cells were counted manually using a hemocytometer. Zombie Aqua (cat. #423101, dilution 1:1,000) was used to stain for dead cells. Cell-surface stains were applied and FACS analysis was performed using LSRII (Flow Cytometry Core, UAB). Primary antibodies were all from Biolegend and include: CD3 PE (cat. #100307, dilution 1:200), CD4 PacBlue (cat. #100427, dilution 1:200), CD8 PECy7 (cat. #100721, dilution 1:400), TCRγδ PerCP Cy5.5 (Cat # 118117, dilution 1:400), NK1.1 FITC (cat. #108705, dilution 1:100), Foxp3 AF647 (cat. #126407, dilution 1:100), and CD19 FITC (cat. #152403, dilution 1:200). Data were gated and analyzed using FlowJo software.
NF1 genotype-phenotype correlations
Comprehensive NF1 mutation analysis, interpretation of variant pathogenicity, and collection of clinical data were performed as previously described.48,49 To describe the phenotypes, we used the same approach as previously reported.48, 49, 50, 51 For the current study, the aggregated phenotypic dataset was derived solely from the phenotypic checklists originally submitted by the referring physicians when genetic testing was requested. Individuals with missing data for a particular sign and/or symptom were classified as “unknown” or “not specified” and consequently excluded from that part of the aggregated phenotypic data. Most features were identified by physical examination; ophthalmologic examination for Lisch nodules and imaging to detect asymptomatic optic pathway gliomas and spinal neurofibromas was not performed in most individuals.
Ethics statement
All procedures were conducted with the approval of the UAB Institutional Animal Care and Use Committee (IACUC) and the UAB Animal Resources Program and follow guidelines for the care and use of laboratory animals and rodent survival surgery under approval number APN 20300. Animals are sacrificed in full accordance with the IACUC guidelines. This includes CO2 gas inhalation followed by cervical dislocation or decapitation for rodents. These methods are consistent with the recommendation of the Panel on Euthanasia of the American Veterinary Association.
Acknowledgments
This work was supported through the Gilbert Family Foundation’s Gene Therapy Initiative grant number 563624 to D.W. and funds from the Neurofibromatosis Therapeutic Acceleration Program (NTAP) to M.R.W. The authors would like to thank members of the University of Alabama at Birmingham Transgenic and Genetically Engineered Models (TGEMS) facility for creating the DelE17 mice, the Behavioral Assessment Core, and the Comprehensive Flow Cytometry Core. TGEMS is supported by NIH NCI grant P30CA13148 and NIH NIDDK grants P30 DK074038 and P60 DK079626 (to R.A.K.).
Author contributions
Concept or design of the article: A.L., B.R.K., R.A.K., G.D., L.P., and D.W. Acquisition of data: M.M., H.L., M.D., A.M.H., L.M., J.S., L.C., L.L., J.F., and D.W. Analysis and interpretation of data: A.L., L.M., M.M., H.L., M.D., A.M.H., L.M., J.S., L.C., J.F., R.A.K., L.P., M.R.W., and D.W. Drafted the article: A.L., M.M., L.M., B.R.K., J.F., R.A.K., L.P., and D.W. Revised the article critically: A.L., M.M., L.P., L.M., J.F., B.R.K., M.R.W., R.A.K., and D.W.
Declaration of interests
Conflicts of interest are as follows; the remaining authors do not have any competing interests.
B.R.K. is Chair, Children’s Tumor Foundation Medical Advisory Committee; Chair, External Advisory Committee for NTAP and also for NF Research Initiative; and Member, advisory committees for AstraZeneca, Springworks Therapeutics, and Infixion. G.D. has consultancies with Sarepta, Biopharma, AskBio, and RegenX. R.A.K. is a lead adviser for Infixion.
D.W., R.A.K., L.L., B.R.K., A.L., L.P., and G.D. are inventors on US Patent (PCT/US20/51827)—Exon skipping to treat neurofibromatosis type 1.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.03.011.
Supplemental information
References
- 1.Gutmann D.H., Collins F.S. The neurofibromatosis type 1 gene and its protein product, neurofibromin. Neuron. 1993;10:335–343. doi: 10.1016/0896-6273(93)90324-k. [DOI] [PubMed] [Google Scholar]
- 2.Friedman J.M., Gutmann D.H., MacCollin M., Riccardi V.M. Johns Hopkins University Press; 1999. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. [Google Scholar]
- 3.Kallionpää R.A., Uusitalo E., Leppävirta J., Pöyhönen M., Peltonen S., Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2018;20:1082–1086. doi: 10.1038/gim.2017.215. [DOI] [PubMed] [Google Scholar]
- 4.Dombi E., Baldwin A., Marcus L.J., Fisher M.J., Weiss B., Kim A., Whitcomb P., Martin S., Aschbacher-Smith L.E., Rizvi T.A., et al. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N. Engl. J. Med. 2016;375:2550–2560. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siva K., Covello G., Denti M.A. Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic Acid Ther. 2014;24:69–86. doi: 10.1089/nat.2013.0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarmin S., Kymalainen H., Popplewell L., Dickson G. New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2014;14:209–230. doi: 10.1517/14712598.2014.866087. [DOI] [PubMed] [Google Scholar]
- 7.Walsh S. FDA: News Release; 2016. FDA Grants Accelerated Approval to First Drug for Duchenne Muscular Dystrophy. [Google Scholar]
- 8.Aartsma-Rus A., Krieg A.M. FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017;27:1–3. doi: 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aartsma-Rus A., Corey D.R. The 10th oligonucleotide therapy approved: golodirsen for Duchenne muscular dystrophy. Nucleic Acid Ther. 2020;30:67–70. doi: 10.1089/nat.2020.0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anastasaki C., Le L.Q., Kesterson R.A., Gutmann D.H. Updated nomenclature for human and mouse neurofibromatosis type 1 genes. Neurol. Genet. 2017;3:e169. doi: 10.1212/NXG.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng L., Yunoue S., Tokuo H., Ozawa T., Zhang D., Patrakitkomjorn S., Ichimura T., Saya H., Araki N. PKA phosphorylation and 14-3-3 interaction regulate the function of neurofibromatosis type I tumor suppressor, neurofibromin. FEBS Lett. 2004;557:275–282. doi: 10.1016/s0014-5793(03)01507-2. [DOI] [PubMed] [Google Scholar]
- 12.Jørgensen C., Sherman A., Chen G.I., Pasculescu A., Poliakov A., Hsiung M., Larsen B., Wilkinson D.G., Linding R., Pawson T. Cell-specific information processing in segregating populations of Eph receptor ephrin-expressing cells. Science. 2009;326:1502–1509. doi: 10.1126/science.1176615. [DOI] [PubMed] [Google Scholar]
- 13.Babu M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016;44:1185–1200. doi: 10.1042/BST20160172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheffzek K., Ahmadian M.R., Kabsch W., Wiesmüller L., Lautwein A., Schmitz F., Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 15.Scheffzek K., Welti S. In: Neurofibromatosis Type 1: Molecular and Cellular Biology. Upadhyaya M., Cooper D.N., editors. Springer; 2012. Neurofibromin: protein domains and functional characteristics; pp. 305–325. [Google Scholar]
- 16.Vandenbroucke I., Van Oostveldt P., Coene E., De Paepe A., Messiaen L. Neurofibromin is actively transported to the nucleus. FEBS Lett. 2004;560:98–102. doi: 10.1016/S0014-5793(04)00078-X. [DOI] [PubMed] [Google Scholar]
- 17.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li K., Turner A.N., Chen M., Brosius S.N., Schoeb T.R., Messiaen L.M., Bedwell D.M., Zinn K.R., Anastasaki C., Gutmann D.H., et al. Mice with missense and nonsense NF1 mutations display divergent phenotypes compared with human neurofibromatosis type I. Dis. Model. Mech. 2016;9:759–767. doi: 10.1242/dmm.025783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toonen J.A., Anastasaki C., Smithson L.J., Gianino S.M., Li K., Kesterson R.A., Gutmann D.H. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum. Mol. Genet. 2016;25:1703–1713. doi: 10.1093/hmg/ddw039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernández-Rodríguez J., Castellsagué J., Benito L., Benavente Y., Capellá G., Blanco I., Serra E., Lázaro C. A mild neurofibromatosis type 1 phenotype produced by the combination of the benign nature of a leaky NF1-splice mutation and the presence of a complex mosaicism. Hum. Mutat. 2011;32:705–709. doi: 10.1002/humu.21500. [DOI] [PubMed] [Google Scholar]
- 21.Pros E., Fernández-Rodríguez J., Canet B., Benito L., Sánchez A., Benavides A., Ramos F.J., López-Ariztegui M.A., Capellá G., Blanco I., et al. Antisense therapeutics for neurofibromatosis type 1 caused by deep intronic mutations. Hum. Mutat. 2009;30:454–462. doi: 10.1002/humu.20933. [DOI] [PubMed] [Google Scholar]
- 22.Wallis D., Li K., Lui H., Hu K., Chen M.J., Li J., Kang J., Das S., Korf B.R., Kesterson R.A. Neurofibromin (NF1) genetic variant structure-function analyses using a full-length mouse cDNA. Hum. Mutat. 2018;39:816–821. doi: 10.1002/humu.23421. [DOI] [PubMed] [Google Scholar]
- 23.Long A., Liu H., Liu J., Daniel M., Bedwell D.M., Korf B., Kesterson R.A., Wallis D. Analysis of patient-specific NF1 variants leads to functional insights for Ras signaling that can impact personalized medicine. Hum. Mutat. 2022;43:30–41. doi: 10.1002/humu.24290. [DOI] [PubMed] [Google Scholar]
- 24.Zheng Z.Y., Anurag M., Lei J.T., Cao J., Singh P., Peng J., Kennedy H., Nguyen N.C., Chen Y., Lavere P., et al. Neurofibromin is an estrogen receptor-alpha transcriptional Co-repressor in breast cancer. Cancer Cell. 2020;37:387–402.e387. doi: 10.1016/j.ccell.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dzierlega K., Yokota T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 2020;27:407–416. doi: 10.1038/s41434-020-0156-6. [DOI] [PubMed] [Google Scholar]
- 26.Echigoya Y., Lim K.R.Q., Trieu N., Bao B., Miskew Nichols B., Vila M.C., Novak J.S., Hara Y., Lee J., Touznik A., et al. Quantitative antisense screening and optimization for exon 51 skipping in Duchenne muscular dystrophy. Mol. Ther. 2017;25:2561–2572. doi: 10.1016/j.ymthe.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang J.K., Malerba A., Popplewell L., Foster K., Dickson G. Antisense-induced myostatin exon skipping leads to muscle hypertrophy in mice following octa-guanidine morpholino oligomer treatment. Mol. Ther. 2011;19:159–164. doi: 10.1038/mt.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malerba A., Sharp P.S., Graham I.R., Arechavala-Gomeza V., Foster K., Muntoni F., Wells D.J., Dickson G. Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol. Ther. 2011;19:345–354. doi: 10.1038/mt.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goyenvalle A., Vulin A., Fougerousse F., Leturcq F., Kaplan J.C., Garcia L., Danos O. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306:1796–1799. doi: 10.1126/science.1104297. [DOI] [PubMed] [Google Scholar]
- 30.Goyenvalle A., Wright J., Babbs A., Wilkins V., Garcia L., Davies K.E. Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Mol. Ther. 2012;20:1212–1221. doi: 10.1038/mt.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aupy P., Zarrouki F., Sandro Q., Gastaldi C., Buclez P.O., Mamchaoui K., Garcia L., Vaillend C., Goyenvalle A. Long-term efficacy of AAV9-U7snRNA-mediated exon 51 skipping in mdx52 mice. Mol. Ther. Methods Clin. Dev. 2020;17:1037–1047. doi: 10.1016/j.omtm.2020.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou H., Meng J., Malerba A., Catapano F., Sintusek P., Jarmin S., Feng L., Lu-Nguyen N., Sun L., Mariot V., et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J. Cachexia Sarcopenia Muscle. 2020;11:768–782. doi: 10.1002/jcsm.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E., et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017;377:1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 34.Mercuri E., Darras B.T., Chiriboga C.A., Day J.W., Campbell C., Connolly A.M., Iannaccone S.T., Kirschner J., Kuntz N.L., Saito K., et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018;378:625–635. doi: 10.1056/NEJMoa1710504. [DOI] [PubMed] [Google Scholar]
- 35.Jacks T., Shih T.S., Schmitt E.M., Bronson R.T., Bernards A., Weinberg R.A. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 36.Costa R.M., Yang T., Huynh D.P., Pulst S.M., Viskochil D.H., Silva A.J., Brannan C.I. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat. Genet. 2001;27:399–405. doi: 10.1038/86898. [DOI] [PubMed] [Google Scholar]
- 37.Andersen L.B., Ballester R., Marchuk D.A., Chang E., Gutmann D.H., Saulino A.M., Camonis J., Wigler M., Collins F.S. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol. Cell Biol. 1993;13:487–495. doi: 10.1128/mcb.13.1.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yachdav G., Kloppmann E., Kajan L., Hecht M., Goldberg T., Hamp T., Hönigschmid P., Schafferhans A., Roos M., Bernhofer M., et al. PredictProtein--an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014;42:W337–W343. doi: 10.1093/nar/gku366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sigrist C.J., Cerutti L., Hulo N., Gattiker A., Falquet L., Pagni M., Bairoch A., Bucher P. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3:265–274. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- 41.Chen Z., Zhou Y., Song J., Zhang Z. hCKSAAP_UbSite: improved prediction of human ubiquitination sites by exploiting amino acid pattern and properties. Biochim. Biophys. Acta. 2013;1834:1461–1467. doi: 10.1016/j.bbapap.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Chen X., Qiu J.D., Shi S.P., Suo S.B., Huang S.Y., Liang R.P. Incorporating key position and amino acid residue features to identify general and species-specific Ubiquitin conjugation sites. Bioinformatics. 2013;29:1614–1622. doi: 10.1093/bioinformatics/btt196. [DOI] [PubMed] [Google Scholar]
- 43.Hirata Y., Brems H., Suzuki M., Kanamori M., Okada M., Morita R., Llano-Rivas I., Ose T., Messiaen L., Legius E., Yoshimura A. Interaction between a domain of the negative regulator of the Ras-ERK pathway, SPRED1 protein, and the GTPase-activating protein-related domain of neurofibromin is implicated in legius syndrome and neurofibromatosis type 1. J. Biol. Chem. 2016;291:3124–3134. doi: 10.1074/jbc.M115.703710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Popplewell L.J., Trollet C., Dickson G., Graham I.R. Design of phosphorodiamidate morpholino oligomers (PMOs) for the induction of exon skipping of the human DMD gene. Mol. Ther. 2009;17:554–561. doi: 10.1038/mt.2008.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H., Chang L.J., Neubauer D.R., Muir D.F., Wallace M.R. Immortalization of human normal and NF1 neurofibroma Schwann cells. Lab. Invest. 2016;96:1105–1115. doi: 10.1038/labinvest.2016.88. [DOI] [PubMed] [Google Scholar]
- 46.Seibenhener M.L., Wooten M.C. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp. 2015 doi: 10.3791/52434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deacon R.M. Assessing nest building in mice. Nat. Protoc. 2006;1:1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- 48.Koczkowska M., Callens T., Chen Y., Gomes A., Hicks A.D., Sharp A., Johns E., Uhas K.A., Armstrong L., Bosanko K.A., et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276 and p.Lys1423: genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2019;41:299–315. doi: 10.1002/humu.23929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koczkowska M., Chen Y., Callens T., Gomes A., Sharp A., Johnson S., Hsiao M.C., Chen Z., Balasubramanian M., Barnett C.P., et al. Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844-848. Am. J. Hum. Genet. 2018;102:69–87. doi: 10.1016/j.ajhg.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koczkowska M., Callens T., Gomes A., Sharp A., Chen Y., Hicks A.D., Aylsworth A.S., Azizi A.A., Basel D.G., Bellus G., et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): an update of genotype-phenotype correlation. Genet. Med. 2018;21:867–876. doi: 10.1038/s41436-018-0269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rojnueangnit K., Xie J., Gomes A., Sharp A., Callens T., Chen Y., Liu Y., Cochran M., Abbott M.A., Atkin J., et al. High incidence of noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum. Mutat. 2015;36:1052–1063. doi: 10.1002/humu.22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.