

Abstract
目的
探讨转化生长因子β激活激酶1(TAK1)对甲苯二异氰酸酯(TDI)哮喘小鼠气道炎症的影响。
方法
建立TDI哮喘小鼠模型,为探讨TAK1在TDI哮喘模型中的作用,将32只小鼠随机分为4组(8只/组):AOO组,AOO+5Z-7-Oxozeaenol组,TDI组,TDI+5Z-7-Oxozeaenol组;为进一步探讨RIPK1在体内模型的作用,将另外32只小鼠随机分为4组(8只/组):AOO组,TDI组,TDI+5Z-7-Oxozeaenol组,TDI+5Z-7-Oxozeaenol+Necrostatin-1组。每次激发前使用TAK1抑制剂(5Z-7-Oxozeaenol,5 mg/kg)和/或RIPK1抑制剂(Necrostatin-1, 5 mg/kg)。检测各组小鼠气道反应性、气道炎症及气道重塑等指标。体外实验:配制TDI-人血清白蛋白(TDI-HSA)复合物联合TAK1抑制剂处理小鼠单核巨噬细胞(RAW264.7),通过CCK8检测各组细胞活力,通过Western blot检测各组TAK1、丝裂原活化蛋白激酶(MAPK)及受体相互作用丝/苏氨酸蛋白酶1(RIPK1)相关信号通路分子表达情况;使用RIPK1抑制剂进一步评估对细胞活力及TDI哮喘模型的影响。
结果
与单独TDI致敏激发哮喘小鼠相比,TAK1抑制剂加重了小鼠气道炎症、气道高反应性及气道重塑(P < 0.05)。抑制TAK1降低了RAW264.7细胞活力,与TDI-HSA共同处理进一步降低(P < 0.05)。抑制TAK1降低了TDI-HSA诱导的TAK1磷酸化水平及MAPK信号通路的活化(P < 0.05)。TAK1抑制剂与TDI-HSA共同处理后,RIPK1磷酸化水平升高,同时Caspase 8持续活化(P < 0.05);使用RIPK1抑制剂可以恢复TAK1抑制剂与TDI-HSA共同处理引起的细胞活力的减低(P < 0.05)。体内RIPK1抑制剂亦可以减轻TAK1抑制剂对TDI哮喘小鼠气道炎症的加重(P < 0.05)。
结论
抑制TAK1可能通过增加RIPK1的活性,引起Caspase 8的持续活化而增加巨噬细胞的死亡,参与促进TDI诱导的哮喘小鼠气道炎症。
Keywords: 甲苯二异氰酸酯, 哮喘, 转化生长因子β激活激酶1, 受体相互作用丝/苏氨酸蛋白酶1, 丝裂原活化蛋白激酶
Abstract
Objective
To explore the effect of transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) on toluene diisocyanate (TDI)-induced allergic airway inflammation in mice.
Methods
Thirty-two mice were randomly divided into AOO group, AOO+5Z-7-Oxozeaenol group, TDI group, and TDI+5Z-7-Oxozeaenol group. Another 32 mice were randomly divided into AOO group, TDI group, TDI +5Z-7-Oxozeaenol group, and TDI +5Z-7-Oxozeaenol + Necrostatin-1 group. TAK1 inhibitor (5Z-7-Oxozeaenol, 5 mg/kg) and/or RIPK1 inhibitor (Necrostatin-1, 5 mg/kg) were used before each challenge. Airway responsiveness, airway inflammation and airway remodeling were assessed after the treatments. We also examined the effect of TDI-human serum albumin (TDI-HSA) conjugate combined with TAK1 inhibitor on the viability of mouse mononuclear macrophages (RAW264.7) using CCK8 assay. The expressions of TAK1, mitogen-activated protein kinase (MAPK) and receptor interacting serine/threonine protease 1 (RIPK1) signal pathway in the treated cells were detected with Western blotting. The effects of RIPK1 inhibitor on the viability of RAW264.7 cells and airway inflammation of the mouse models of TDI-induced asthma were evaluated.
Results
TAK1 inhibitor aggravated TDI-induced airway inflammation, airway hyper responsiveness and airway remodeling in the mouse models (P < 0.05). Treatment with TAK1 inhibitor significantly decreased the viability of RAW264.7 cells, which was further decreased by co-treatment with TDI-HSA (P < 0.05). TAK1 inhibitor significantly decreased the level of TAK1 phosphorylation and activation of MAPK signal pathway induced by TDI-HSA (P < 0.05). Co-treatment with TAK1 inhibitor and TDI-HSA obviously increased the level of RIPK1 phosphorylation and caused persistent activation of caspase 8 (P < 0.05). RIPK1 inhibitor significantly inhibited the reduction of cell viability caused by TAK1 inhibitor and TDI-HSA (P < 0.05) and alleviated the aggravation of airway inflammation induced by TAK1 inhibitors in TDI-induced mouse models (P < 0.05).
Conclusion
Inhibition of TAK1 aggravates TDI-induced airway inflammation and hyperresponsiveness and may increase the death of macrophages by enhancing the activity of RIPK1 and causing persistent activation of caspase 8.
Keywords: toluene diisocyanate, asthma, transforming growth factor-β-activated kinase 1, receptor interaction serine/threonine protease 1, mitogen-activated protein kinase
哮喘具有多种临床表型和内因型,大部分哮喘患者在吸入性糖皮质激素等治疗后可以有效控制,但仍有5%~20%的哮喘患者即使用高剂量吸入性糖皮质激素治疗仍控制不佳,这些重度哮喘或激素抵抗型哮喘的发生机制尚未完全阐明[1-2]。其中,丝裂原活化蛋白激酶(MAPK)信号通路的过度激活可能在激素抵抗过程发挥着重要作用[2-4]。
本课题组长期关注甲苯二异氰酸酯(TDI)哮喘模型气道炎症发生机制[5, 6],有研究发现,TDI诱导的是一种激素抵抗型哮喘模型[7],我们前期研究发现MAPK信号通路在TDI哮喘模型明显活化,且阻断MAPK信号通路能有效减轻TDI哮喘小鼠的气道炎症、气道屏障破坏及气道高反应性[5, 6, 8],提示MAPK通路可能与激素抵抗有关。转化生长因子β激活激酶1(TAK1)是激活下游包括NF-κB和MAPK信号通路活化的关键酶,在调节各种细胞类型的炎症、免疫和细胞死亡发挥关键作用,为治疗炎症性疾病的潜在靶点[9, 10]。有小样本研究证实阻断TAK1可以恢复激素抵抗哮喘患者来源的巨噬细胞对激素的敏感性[4]。有文献报道TAK1可能通过影响Th17通路在中性粒细胞哮喘免疫调节中发挥重要作用[11]。然而,TAK1是否在TDI诱导的激素抵抗型哮喘模型扮演重要角色及如何发挥作用尚不清楚。因此,本研究旨在探讨TAK1对TDI哮喘小鼠气道炎症的影响。
1. 材料和方法
1.1. 试剂
丙酮、橄榄油、TDI(sigma);TAK1抑制剂5Z-7-Oxozeaenol(sigma),RIPK1抑制剂Necrostatin-1(MCE),CCK8(同仁),p-TAK1、TAK1、p-ERK、P38、p-P38、JNK、p-JNK、RIPK1、p-RIPK1等一抗(CST),ERK一抗(proteintech),Caspase8一抗(proteintech),GADPH一抗(proteintech),IRDye®荧光二抗(licor),ELISA试剂盒(赛默飞),苏木素染液,伊红染液,凯基全蛋白提取试剂盒等。
1.2. TDI哮喘模型建立及动物干预
SPF级雄性BALB/c小鼠64只(南方医科大学实验动物中心),6~8周,本实验经南方医院实验动物伦理委员会批准。
TDI哮喘模型构建:于造模第1、8天用0.3%TDI通过耳背皮肤致敏小鼠,第15、18、21天通过雾化吸入3% TDI激发小鼠,TDI溶于丙酮和橄榄油的混合物(AOO)中,空白对照小鼠只接受AOO致敏和激发,具体剂量参考先前研究[12]。
为探讨TAK1在TDI哮喘模型中的作用,将32只小鼠随机分为4组,8只/组:AOO组,AOO + 5Z-7-Oxozeaenol组,TDI组,TDI + 5Z-7-Oxozeaenol组。AOO +5Z-7-Oxozeaenol组、TDI +5Z-7-Oxozeaenol组于每次激发3 h前腹腔注射5 mg/kg TAK1抑制剂(5Z-7-Oxozeaenol)干预TAK1,AOO组、TDI组给予同等剂量的DMSO。
进一步探讨RIPK1在体内模型的作用,将另外32只小鼠随机分为4组,8只/组:AOO组,TDI组,TDI+5Z-7-Oxozeaenol组,TDI+5Z-7-Oxozeaenol+Necrostatin-1组。TDI + 5Z-7-Oxozeaenol组、TDI + 5Z-7-Oxozeaenol + Necrostatin-1组每次激发3 h前腹腔注射5 mg/kg TAK1抑制剂(5Z-7-Oxozeaenol)和/或RIPK1活性抑制剂(Necrostatin-1, 5 mg/kg),两种抑制剂给药时间间隔30 min。AOO组、TDI组给予同等剂量的DMSO。
按照上述方法造模后,于第22天检测气道反应性后,处死小鼠,取右肺进行石蜡包埋,常规脱蜡至水进行HE染色、PAS染色及MASSON染色。气道反应性及淋巴细胞培养详见文献[12]。
1.3. RAW264.7培养,刺激及细胞活力检测。
小鼠单核巨噬细胞株(RAW264.7,中科院),其采用含10%胎牛血清的DMEM培养基,在37 ℃、5% CO2培养箱中传代培养,细胞生长状态良好时用于实验,予不含血清的DMEM培养细胞12 h,即饥饿处理后,予不同浓度5Z-7-Oxozeaenol、1 μmol/L Necrostatin-1及80 μg/mL TDI-HSA刺激细胞一定时间,TDI-HSA复合物按文献配置[13]。细胞活力依照CCK8试剂盒说明书进行检测。
1.5. Western blot
采用凯基全蛋白提取试剂盒抽提细胞总蛋白,肺组织总蛋白,BCA法测定蛋白浓度,等量蛋白上样,使用BIO-RAD进行电泳及转膜,5%BSA封闭1 h,一抗4 ℃过夜,TBST洗膜30 min,二抗孵育1 h,LI-COR红外荧光扫描成像检测。
1.6. 统计学分析
采用SPSS 20.0统计软件分析处理,定量资料以均数±标准差表示,组间比较采用单因素方差分析;符合方差齐性检验者,组内比较使用LSD方法检验,不符合方差齐性检验者,组内比较使用Dunnett's T3分析,P < 0.05时认为差异具有统计学意义。
2. 结果
2.1. TAK1抑制剂降低TDI诱导的TAK1磷酸化水平的升高及MAPK信号通路的活化
结果显示,阻断TAK1不仅降低了TDI引起的肺组织TAK1磷酸化水平的升高,同时降低了其下游分子P38 MAPK,JNK及ERK的磷酸化水平(P < 0.05,图 1)。
图 1.

TAK1抑制剂对TDI哮喘小鼠TAK1及MAPK信号通路的影响
Effects of TAK1 antagonists on TAK1 and MAPK signaling pathway in mice with TDI-induced asthma. Western blotting was used to detect the expressions of p-TAK1 (A), p-P38, p-ERK and p-JNK (B) in different group with densitometric analysis of the blots for p-TAK1 (C), p-P38, p-ERK and p-JNK (D). *P < 0.05 vs AOO, #P < 0.05 vs TDI (n=3).
2.2. 阻断TAK1进一步加重了TDI诱导的气道高反应性及气道炎症
TDI致敏及激发建立了以中性粒细胞为主的混合粒细胞性气道炎症的哮喘模型(图 2E),同时伴有气道高反应性及气道重塑(图 2A,图 3D)。使用5Z-7-Oxozeaenol,进一步加重了小鼠的气道高反应性及增加了血清IgE浓度(P < 0.05,图 2A、B)。组织切片HE染色显示,阻断TAK1进一步加重了气道周围炎症细胞增多,上皮增生;进一步评估小鼠气道重塑指标,结果显示,TDI能显著引起小鼠气道粘液分泌的增加及上皮下胶原沉积(P < 0.05,图 3D、E)。PAS染色显示,阻断TAK1对于粘液分泌没有明显影响(P=0.850),MASSON染色则显示其能明显加重哮喘小鼠的胶原沉积(P < 0.05);而单独使用TAK1抑制剂,对对照组的气道炎症(P=0.147)、粘液分泌(P=0.85)及胶原下沉积(P= 0.63)没有明显影响。
图 2.

TAK1抑制剂对TDI哮喘小鼠气道炎症的影响
Inhibition of TAK1 exacerbates TDI-induced airway inflammation. A: Airway hyperresponsiveness assessed by RL. The results are shown as percentage of baseline. B: Total serum IgE were measured by ELISA. C, D: Total (C) and differential (D) inflammatory cells in BALF. Percentages of different inflammatory cells were calculated by counting a total of 200 cells in HE-stained samples. E: Representative HE-stained and PAS stained lung sections in different groups (Original magnification: × 200). Semi-quantification of airway inflammation (F) and PAS staining (G) was performed. *P < 0.05 vs AOO, #P < 0.05 vs TDI. (n=4-6).
图 3.

TAK1抑制剂对TDI哮喘小鼠气道炎症及气道重塑的影响
TAKI inhibitor exacerbates airway inflammation and remodeling in mice after TDI exposure. A-C: Levels of IL-4, IL-5 and IL-13 (A), IL-17A and IL-17 (B) in supernatants of cultured lymphocytes and IL-1βin BALF (C) measured by ELISA. D: Representative Masson trichrome-stained lung sections showing collagen deposition in each group (×200). E: Semi-quantification of Masson staining was performed using ImageJ software. *P < 0.05 vs AOO, #P < 0.05 vs TDI (n=4-6).
2.3. 阻断TAK1增加TDI哮喘小鼠淋巴上清Th2/Th17炎症水平
TDI哮喘小鼠Th2/Th17炎症因子升高(P < 0.05,图 3A、B),给予TAK1抑制剂增加了IL-4及IL-13水平(P < 0.05),而IL-5差异无统计学意义(P=0.21)。阻断TAK1升高了TDI哮喘小鼠淋巴上清IL-17F的水平(P < 0.05),降低了TDI引起IL-17A的水平的升高(P < 0.05)。同时,单纯的TDI致敏及激发组,未能在BALF中检测到IL-1β,而给予TAK1抑制剂,检测到IL-1β升高(P < 0.05,图 3C)。
2.4. TDI-HSA联合TAK1抑制剂对巨噬细胞RAW264.7活力的影响
TAK1抑制剂可以降低TDI哮喘小鼠肺泡灌洗液(BALF)分类计数细胞中巨噬细胞的比值(图 2D)。单独使用TDI-HSA(80 μg/mL)刺激细胞,未降低细胞活力(图 4)。而单独使用TAK1抑制剂,可以在较高浓度时(5000 nmol/L)时,在早期(2 h)时细胞活力降低(P < 0.05),而较低浓度(500 nmol/L)时到6 h时细胞活力出现降低(P < 0.05)。同时,与单独使用TAK1抑制剂相比,与TDI-HSA联合使用降低了细胞活力(P < 0.05)。为探讨抑制TAK1加重TDI诱导哮喘小鼠气道炎症的机制,以下实验中,选用500 nmol/L的抑制剂浓度处理细胞。
图 4.

不同浓度及时间5Z-7-Oxozeaenol联合TDI-HSA刺激对RAW264.7细胞活力的影响
Effect of different concentrations and time of 5Z-7-Oxozeaenol with or without TDI-HSA on RAW264.7 viability assessed by cell counting kit-8. *P < 0.05 vs control; #P < 0.05 vs 5Z-7-Oxozeaenol (5000 nmol/L), + P < 0.05 vs 5Z-7-Oxozeaenol (500 nmol/L), n=6.
2.5. TAK1抑制剂对TDI-HSA诱导的RAW264.7 TAK1磷酸化水平的影响
检测各组间TAK1水平及其磷酸化程度显示,单独TDI-HSA刺激RAW264.7后,TAK1的磷酸化水平在短时间内即升高(P < 0.05,图 5)。而使用TAK1抑制剂5Z-7-Oxozeaenol(500 nmol/L),可以降低TDI-HSA引起的TAK1磷酸化水平的升高(P < 0.05)。
图 5.
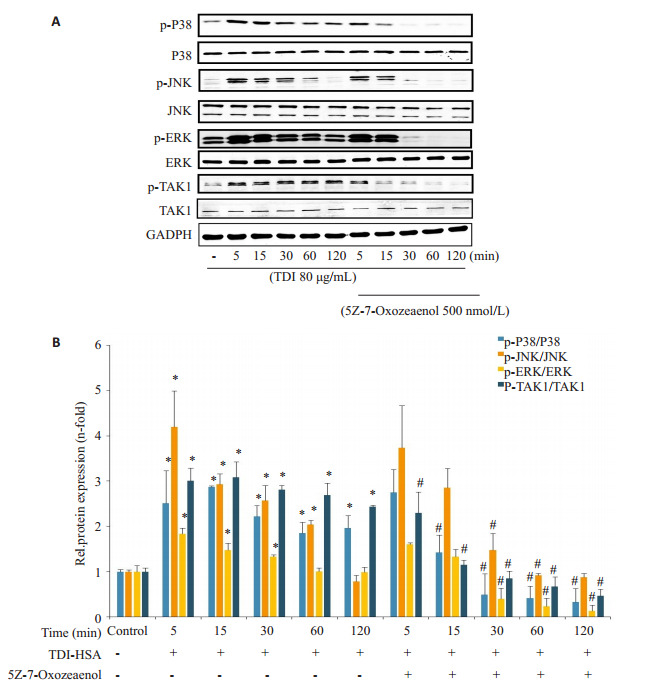
TDI-HSA及TAK1抑制剂对RAW264.7细胞MAPK通路的影响
Effects of TAK1 antagonists on TAK1 and MAPK signaling pathway in RAW264.7 cells. A: Western blots for p-P38, p-JNK, p-ERK and p-TAK1 in different groups. B: Densitometric analysis of the blots. *P < 0.05 vs control; #P < 0.05 vs TDI group with the treatment at the same time (n=3).
2.6. TAK1抑制剂对TDI-HSA诱导的MAPK信号通路活化的影响
TDI-HSA刺激RAW264.7细胞,5 min即引起pP38、p-JNK及p-ERK等蛋白表达水平明显升高(P < 0.05)。而给予TAK1抑制剂与TDI-HSA共同处理RAW264.7、JNK、P38及ERK蛋白的磷酸化水平都降低(P < 0.05)。
2.7. 阻断TAK1促进了RIPK1介导的细胞死亡
Western blotting结果显示,TDI-HSA刺激与TDIHSA联合TAK1抑制剂共同处理,未能明显影响RIPK1蛋白水平,而TDI-HSA联合TAK1抑制剂,在2 h时RIPK1磷酸化水平上升(P < 0.05),随后降低。单独使用TDI-HSA刺激RAW264.7,Caspase 8在2 h出现活化(P < 0.05),随后快速降低,而TDI-HSA联合TAK1抑制剂,其Caspase 8活性明显升高,且随后进一步升高(P < 0.05)。进一步使用RIPK1活性抑制剂Necrostatin-1,明显恢复TDI-HSA联合TAK1抑制剂引起的细胞活力的下降(P < 0.05),而对于单独TAK1抑制剂引起的细胞活力水平的下降没有明显影响(P=0.48)。
2.8. 使用RIPK1抑制剂可以恢复TAK1抑制剂引起的TDI哮喘小鼠气道炎症及气道重塑的加重
与单独使用TAK1抑制剂对TDI哮喘小鼠相比,联合使用RIPK1活性抑制剂可减轻气道炎症(P < 0.05,图 7),降低血清IgE浓度及淋巴上清中IL-4、IL-13及IL-17F水平及改善气道重塑(P < 0.05)。
图 7.

RIPK1抑制剂对TAK1抑制剂加重的TDI哮喘小鼠气道炎症的影响
RIPK1 inhibitors alleviate aggravation of airway inflammation in TDI-exposed mice treated with TAK1 inhibitors. A-C: Serum levels of IgE (A) and Th2-related cytokines (IL-4, IL-5 and IL-13) (B) and levels of Th17-related cytokines (IL-17A and IL-17F) (C) in supernatants of cultured lymphocytes. D: Representative HE-stained and Masson-stained lung sections in different groups (×200). Semi-quantification of airway inflammation (E) and Masson staining (F) was performed. *P < 0.05 vs AOO, #P < 0.05 vs TDI, +P < 0.05 vs TDI+ 5Z-7-Oxozeaenol, n=4-6.
图 6.

TAK1抑制剂对RAW264.7 RIPK1信号通路的影响
Effects of TAK1 antagonists on RIPK1 signaling pathway in RAW264.7 cells. A: Western blot for caspase 8 and p-RIPK1 in different groups. B: Densitometric analysis of the blots. *P < 0.05 vs control, n=3; C: RAW264.7 viability assessed by Cell Counting Kit-8. *P < 0.05 vs control, #P < 0.05 vs 5Z-7-Oxozeaenol+TDI, n=6.
3. 讨论
本研究首次发现在TDI哮喘模型中,抑制TAK1进一步加重了TDI哮喘小鼠的气道炎症,其机制可能与促进巨噬细胞RIPK1介导的细胞死亡有关。有研究报道,在小鼠颗粒物诱导的原代巨噬细胞模型中,敲除TAK1可以降低IL-6、IL-33的表达[14],与野生组型比,肺巨噬细胞TAK1条件敲除小鼠肺部炎症指标及相关细胞因子表达明显降低。本课题组既往的研究发现P38 MAPK、JNK、ERK等MAPK信号通路在TDI哮喘模型中明显活化,阻断JNK能明显减轻气道炎症[5, 6, 8]。TAK1作为MAPK信号通路的上游激酶,因此,我们设想TDI哮喘小鼠的气道炎症能被TAK1抑制剂所减轻。结果发现,TAK1抑制剂确实可以显著抑制TDI诱导的MAPK信号通路的活化。然而,非常有趣的是,TAK1抑制剂却进一步加重了TDI哮喘小鼠的气道炎症,上皮的脱落,气道的高反应性。这提示,TDI哮喘模型中,TAK1抑制剂加重哮喘气道炎症并不是通过促进下游的MAPK信号通路活化而发生。
与本研究抑制TAK1促炎作用相一致,在内毒素(LPS)诱导的小鼠模型中,与野生型相比,TAK1敲除小鼠的肺部炎症明显加重,伴IL-1β、IL-6、TNF-α水平明显增加,作者同时发现p38和JNK激酶的磷酸化以及活性氧ROS水平明显增加[15],进一步发现TAK1敲除小鼠来源的中性粒细胞IL-1β、IL-6、TNF-αmRNA表达明显增加。提示TAK1在不同的模型中发挥着不同的调节作用。
巨噬细胞是肺部最重要的免疫细胞之一,在变应原诱发的哮喘气道炎症中发挥重要作用,巨噬细胞可以通过分泌多种炎症介质,包括IL-1β、IL-33、TNF-α等在哮喘的发生发展中发挥着重要的作用[16-18],亦有研究发现,TDI哮喘中巨噬细胞活化[19]。因此,本研究假设在TDI哮喘模型中,TAK1抑制剂通过增加巨噬细胞的死亡,释放炎症介质,进一步加重TDI哮喘小鼠的气道炎症。与预期一致,本研究显示抑制TAK1后明显降低哮喘模型BALF中巨噬细胞比例,体外亦降低RAW264.7细胞活力,且在与TDI-HSA共同刺激时其细胞活力进一步下降,但其机制有待进一步阐明。
TAK1作为MAPK与NF-κB信号通路的上游激酶,通常参与促生存信号[20],研究发现当其被抑制或缺失时,细胞将发生PANoptosis[21, 22],细胞从促生存信号向促死亡信号方向转变[23]。PANoptosis包括焦亡、凋亡和坏死三种细胞死亡形式[24, 25],是由含有RIPK1、ASC和caspase 8的PANoptosome的形成触发的,其中RIPK1激酶活性对驱动焦亡、凋亡和坏死混合细胞死亡表型至关重要[21, 22]。本研究发现,联合使用TAK1抑制剂及TDI-HSA可以激活RIPK1的活性,同时持续裂解Caspase 8,促进巨噬细胞的死亡,使用RIPK1抑制剂可以降低RAW264.7的死亡,同时,体内实验也证实使用RIPK1抑制剂可以显著减轻TAK1抑制剂对于TDI哮喘小鼠气道炎症的加重。与本研究一致,近期有研究发现,衰老导致TAK1表达减少会导致RIPK1驱动的神经退行性变[26]。有研究提出耶尔森氏菌诱导细胞死亡模型,即部分巨噬细胞被耶尔森菌感染,当宿主细胞感知到TAK1被抑制时,形成由RIPK1、Caspase 8和FADD组成的细胞死亡复合体,导致细胞膜通透性增加和随后的细胞死亡,释放IL-1β,K+等,激活部分TAK1未被抑制的巨噬细胞,以促发更为迅速的炎症瀑布反应[27]。本研究设想在TDI哮喘模型中,TAK1磷酸化水平的增高,抑制了RIPK1活性,进而限制了Caspase 8的持续活化,从而降低了巨噬细胞的死亡。然而,在TAK1抑制剂处理的哮喘模型中,是否也发生了PANoptosis,其细胞死亡是否也为包括焦亡、凋亡和坏死多种细胞死亡形式的混合细胞死亡表型,需要进一步实验验证。
本研究发现,与其他炎症性疾病不同[14, 28-30],抑制TAK1出乎意料地加重了TDI哮喘小鼠的气道炎症,进一步发现其对于巨噬细胞死亡的影响。在不同细胞死亡途径之间存在着许多的联系[31],在特定刺激下,参与细胞焦亡、细胞凋亡和/或坏死的分子可以同时被激活。未来对细胞死亡之间各种联系和调节机制的深入研究,将进一步帮助理解其中的作用途径。
综上所述,在TDI哮喘小鼠模型中,TAK1阻断进一步加重了气道炎症,其机制可能与促进RIPK1信号通路及Caspase 8的持续活化,从而促发巨噬细胞的持续死亡,增加炎症介质的释放有关。
Biographies
杨淑銮,在读硕士研究生,E-mail: 1529776323@qq.com
赵文驱,硕士研究生,医师,E-mail: 398535173@qq.com
Funding Statement
国家自然科学基金(82070030,81770033,81900027);广东省科技计划(2017B020226006)
Supported by National Natural Science Foundation of China (82070030, 81770033, 81900027)
Contributor Information
杨 淑銮 (Shuluan YANG), Email: 1529776323@qq.com.
赵 文驱 (Wenqu ZHAO), Email: 398535173@qq.com.
赵 海金 (Haijin ZHAO), Email: 157975178@qq.com.
References
- 1.Schoettler N, Strek ME. Recent advances in severe asthma: from phenotypes to personalized medicine. Chest. 2020;157(3):516–28. doi: 10.1016/j.chest.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansbro PM, Kim RY, Starkey MR, et al. Mechanisms and treatments for severe, steroid-resistant allergic airway disease and asthma. Immunol Rev. 2017;278(1):41–62. doi: 10.1111/imr.12543. [DOI] [PubMed] [Google Scholar]
- 3.Lea S, Li J, Plumb J, et al. P38 MAPK and glucocorticoid receptor crosstalk in bronchial epithelial cells. J Mol Med (Berl) 2020;98(3):361–74. doi: 10.1007/s00109-020-01873-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goleva E, Jackson LP, Harris JK, et al. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am J Respir Crit Care Med. 2013;188(10):1193–201. doi: 10.1164/rccm.201304-0775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang GH, Su JW, Zhao WQ, et al. JNK modulates RAGE/β-catenin signaling and is essential for allergic airway inflammation in asthma. Toxicol Lett. 2021;336:57–67. doi: 10.1016/j.toxlet.2020.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Wang YH, Le YQ, Zhao WQ, et al. Short thymic stromal lymphopoietin attenuates toluene diisocyanate-induced airway inflammation and inhibits high mobility group box 1-receptor for advanced glycation end products and long thymic stromal lymphopoietin expression. Toxicol Sci. 2017;157(2):276–90. doi: 10.1093/toxsci/kfx043. [DOI] [PubMed] [Google Scholar]
- 7.Chen RC, Zhang QL, Chen SY, et al. IL-17F, rather than IL-17A, underlies airway inflammation in a steroid-insensitive toluene diisocyanate-induced asthma model. Eur Respir J. 2019;53(4):1801510–9. doi: 10.1183/13993003.01510-2018. [DOI] [PubMed] [Google Scholar]
- 8.熊 婧, 赵 文驱, 黄 国华, et al. 晚期糖基化终末产物受体促进甲苯二异氰酸脂哮喘小鼠MUC5AC表达及气道黏液高分泌. 南方医科大学学报. 2017;37(10):1301–7. doi: 10.3969/j.issn.1673-4254.2017.10.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. 2012;33(10):522–30. doi: 10.1016/j.tips.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Totzke J, Scarneo SA, Yang KW, et al. TAK1: a potent tumour necrosis factor inhibitor for the treatment of inflammatory diseases. Open Biol. 2020;10(9):200099–106. doi: 10.1098/rsob.200099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joetham A, Schedel M, Ning FK, et al. Dichotomous role of TGF-β controls inducible regulatory T-cell fate in allergic airway disease through Smad3 and TGF-β-activated kinase 1. J Allergy Clin Immunol. 2020;145(3):933–46.e4. doi: 10.1016/j.jaci.2019.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao L, Zhao H, Tang H, et al. Blockade of β-catenin signaling attenuates toluene diisocyanate-induced experimental asthma. Allergy. 2017;72(4):579–89. doi: 10.1111/all.13045. [DOI] [PubMed] [Google Scholar]
- 13.Zhao H, Peng H, Cai SX, et al. Toluene diisocyanate enhances human bronchial epithelial cells' permeability partly through the vascular endothelial growth factor pathway. Clin Exp Allergy. 2009;39(10):1532–9. doi: 10.1111/j.1365-2222.2009.03300.x. [DOI] [PubMed] [Google Scholar]
- 14.Cheng ZY, Chu HQ, Wang SQ, et al. TAK1 knock-down in macrophage alleviate lung inflammation induced by black carbon and aged black carbon. Environ Pollut. 2019;253:507–15. doi: 10.1016/j.envpol.2019.06.096. [DOI] [PubMed] [Google Scholar]
- 15.Ajibade AA, Wang QF, Cui J, et al. TAK1 negatively regulates NF-κB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity. 2012;36(1):43–54. doi: 10.1016/j.immuni.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fricker M, Gibson PG. Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J. 2017;50(3):1700196–205. doi: 10.1183/13993003.00196-2017. [DOI] [PubMed] [Google Scholar]
- 18.Laidlaw TM, Balestrieri B. Macrophages and acylcarnitines: new players in aspirin-exacerbated respiratory disease? J Allergy Clin Immunol. 2021;147(2):498–500. doi: 10.1016/j.jaci.2020.09.040. [DOI] [PubMed] [Google Scholar]
- 19.Xu CY, Chen SY, Deng Y, et al. Distinct roles of PI3Kδ and PI3Kγ in a toluene diisocyanate-induced murine asthma model. Toxicology. 2021;454:152747–58. doi: 10.1016/j.tox.2021.152747. [DOI] [PubMed] [Google Scholar]
- 20.Sanjo H, Nakayama J, Yoshizawa T, et al. Cutting edge: TAK1 safeguards macrophages against proinflammatory cell death. J Immunol. 2019;203(4):783–8. doi: 10.4049/jimmunol.1900202. [DOI] [PubMed] [Google Scholar]
- 21.Malireddi RKS, Gurung P, Mavuluri J, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med. 2018;215(4):1023–34. doi: 10.1084/jem.20171922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis) Front Cell Infect Microbiol. 2019;9:406–15. doi: 10.3389/fcimb.2019.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muendlein HI, Poltorak A. Flipping the switch from inflammation to cell death. Trends Immunol. 2020;41(8):648–51. doi: 10.1016/j.it.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christgen S, Zheng M, Kesavardhana S, et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis) Front Cell Infect Microbiol. 2020;10:237–43. doi: 10.3389/fcimb.2020.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis) Front Cell Infect Microbiol. 2020;10:238–45. doi: 10.3389/fcimb.2020.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu DC, Jin TJ, Zhu H, et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell. 2018;174(6):1477–91.e19. doi: 10.1016/j.cell.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarhan J, Liu BC, Muendlein HI, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis duringYersiniainfection. PNAS. 2018;115(46):E10888–97. doi: 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fechtner S, Fox DA, Ahmed S. Transforming growth factor β activated kinase 1: a potential therapeutic target for rheumatic diseases. Rheumatology (Oxford) 2017;56(7):1060–8. doi: 10.1093/rheumatology/kew301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones DS, Jenney AP, Swantek JL, et al. Profiling drugs for rheumatoid arthritis that inhibit synovial fibroblast activation. Nat Chem Biol. 2017;13(1):38–45. doi: 10.1038/nchembio.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu PF, Tao CR, Zhu YY, et al. TAK1 mediates neuronal pyroptosis in early brain injury after subarachnoid hemorrhage. J Neuroinflammation. 2021;18(1):188–95. doi: 10.1186/s12974-021-02226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christgen S, Tweedell RE, Kanneganti TD. Programming inflammatory cell death for therapy. Pharmacol Ther. 2021;162(10):108010–9. doi: 10.1016/j.pharmthera.2021.108010. [DOI] [PMC free article] [PubMed] [Google Scholar]