

Abstract
Antibody-mediated tumor delivery of cytokines can overcome limitations of systemic administration (toxicity, short half-lives). Previous work showed improved anti-tumor potency of anti-CD20-interferon alpha (IFNα) fusion proteins in preclinical mouse models of B-cell lymphoma. Although tumor targeting is mediated by the antibody part of the fusion protein, the cytokine component might strongly influence biodistribution and pharmacokinetics, as a result of its affinity, size, valency and receptor distribution.
Here, we used positron emission tomography (immunoPET) to study the in vivo biodistribution and tumor targeting of the anti-CD20 rituximab-murine IFNα1 fusion protein (Rit-mIFNα) and compared it to the parental mAb (rituximab, Rit). Rit-mIFNα and Rit were radiolabeled with zirconium-89 (89Zr, t1/2 78.4 h) and injected into C3H mice bearing syngeneic B-cell lymphomas (38C13-hCD20). Dynamic (2 h p.i.) and static (4, 24 and 72 h) PET scans were acquired. Ex vivo biodistribution was performed after the final scan.
Both 89Zr-Rit-mIFNα and 89Zr-Rit specifically target hCD20-expressing B-cell lymphoma in vivo. 89Zr-Rit-mIFNα showed specific uptake in tumors (7.6 ±1.0 %ID/g at 75 h p.i.), which was significantly lower than 89Zr-Rit (38.4 ±9.9 %ID/g, p<0.0001). ImmunoPET studies also revealed differences in the biodistribution, 89Zr-Rit-mIFNα showed rapid blood clearance and high accumulation in the liver compared with 89Zr-Rit. Importantly, immunoPET clearly revealed a therapeutic effect of the single 89Zr-Rit-mIFNα dose, resulting in smaller tumors and fewer lymph node metastases compared to mice receiving 89Zr-Rit. Mice receiving 89Zr-Rit-mIFNα had enlarged spleens, suggesting that systemic immune activation contributes to therapeutic efficacy in addition to the direct antitumoral activity of IFNα.
In conclusion, immunoPET allows the non-invasive tracking and quantification of the antibody-cytokine fusion protein and helps understand the in vivo behavior and therapeutic efficacy.
Keywords: Rituximab, Immunocytokine, B-cell lymphoma, Zirconium-89, Interferon alpha
Category: Large Molecule Therapeutics, Antibody-conjugates
INTRODUCTION
The anti-CD20 monoclonal antibody (mAb) rituximab (in combination with chemotherapy) has substantially improved therapy outcomes in a range of B-cell lymphomas and B-cell chronic lymphocytic leukemias (B-CLL) (1-3). However, a continuing challenge remains for finding effective therapies for “rituximab-refractory” patients who fail to respond to rituximab treatment or, more commonly, relapse (4).
One approach to improve the efficacy of CD20-targeted therapy is by combining the tumor-specific antibody with immune-modulating cytokines (5-7). Cytokines such as interferons and interleukins can activate and modulate both the innate and adaptive immune system (8-10). Recombinant IFNα (Intron®-A, Roferon®-A) and IL-2 (Proleukin®, aldesleukin) are approved for the treatment of malignancies and many more cytokines (interferons, interleukins, and members of the TNF family) are in clinical trials. Major limitations for the therapeutic use of cytokines, such as their short half-life and dose-limiting systemic toxicities, could be overcome by targeted delivery via combination with antibodies. Antibody-cytokine fusion proteins or immunocytokines accumulate the cytokine at the tumor site and lower systemic toxicity (7,11,12). Several immunocytokines, based on full-length antibodies or antibody fragments, and fused with a variety of cytokines, have entered clinical trials for cancer immunotherapy (7).
This work focuses on an immunocytokine composed of an antibody (rituximab-based) that binds to the CD20 antigen on B-cell lymphoma and the type-I interferon alpha (IFNα), genetically fused to the C-terminus of the IgGs heavy chain (Rit-IFNα). The type-I interferon families (both murine and human) comprise at least 13 functional IFNα subtypes that recognize the heterodimeric IFNα/β receptor (consisting of IFNAR1 and IFNAR2 subunits). Most cell types bind IFN, however, binding affinities (nM-pM), the number of binding sites (200-10,000 per cell), and the duration (t1/2) of activation can modulate the response. Binding of IFNα to tumor cells results in direct growth inhibition by inducing G2M cell cycle arrest and apoptosis. In addition, IFNα can have systemic effects by modulating the innate immune response (NK cells, macrophages, enhanced ADCC, antigen presentation) and by activating the adaptive immune system (antigen-specific T- and B-cell responses, immunological memory) (9).
Previous studies by Xuan et al., 2010 compared anti-CD20-mIFNα (murine IFNα1) and anti-CD20-hIFNα (human IFNα2a) to non-targeted immunocytokine and free IFNα, showing that both local tumor antigen and IFNAR expression are required for the potent anti-tumor activity against B-cell lymphoma (13). While the bioactivity of the fused IFNα is reduced compared with free IFNα, the anti-proliferative activity is higher than the non-targeted control immunocytokine. Importantly, efficient tumor eradication with no systemic toxicity was observed with repeated dosing of anti-CD20 Rit-IFNα (13,14).
The extended half-life of 6-8 h for Rit-IFNα lies between the reported half-life for recombinant IFNα (human or murine) in mice of less than 1 hour (15,16) and half-lives of 6-8 days for human IgG1 antibodies in adult mice (17). This raises several questions: for example, does IFN binding to its receptor affect the biodistribution and does the larger molecular size impair tumor targeting or uptake of Rit-IFNα. Non-invasive whole-body imaging using a residualizing radiometal could visualize differences in the in vivo behavior of these complex molecules. Therefore, this study uses immunoPET imaging with 89Zr-radiolabeled anti-CD20 immunocytokines (zirconium-89, t1/2=78.4 h) to assess in vivo pharmacokinetics, tumor targeting and biodistribution. Furthermore, we compared immunocytokines to species-specific variants and the parental antibody (human IgG1 vs mouse IgG2a and mouse IFNα1 vs human IFNα14).
MATERIALS AND METHODS
Cell lines and syngeneic mouse model
All procedures performed in studies involving animals were in accordance with the ethical standards of the University of California Los Angeles Animal Research Committee and were approved by the Institutional Animal Care and Use Committee.
The C3H syngeneic B-cell lymphoma cell line 38C13 expressing human CD20 (38C13-hCD20) was previously described (18). Briefly, the 38C13 cells transduced with a lentivirus encoding the gene sequence for human CD20 (gift from Josée Golay, Bergamo, Italy (19)), was sorted (UCLA Cancer Center Flow Cytometry Core) to yield the high-expressing line 38C13-hCD20. 38C13 and 38C13-hCD20 were cultured in RPMI 1640 (Life Technologies) supplemented with 10% FBS and 50 μmol/L 2-mercaptoethanol for up to 5 passages before tumor implantation. Cell authentication and mycoplasma testing was not performed.
Fluorescence quantitation was performed using the Quantum Simply Cellular (QSC) anti-Human IgG Kit (Bangs Laboratories, Inc) to determine the antibody binding capacity (ABC) of human CD20 expressing cells.
C3Hf/Sed/Kam mice were bred and housed in the UCLA Defined Pathogen Colony. Female mice (6-8 weeks old) were injected subcutaneously (s.c.) on the hind flank or in the shoulder area with 5 x 103 38C13-hCD20 cells in 200 μL HBBS (Hank’s balanced salt solution).
Antibodies and immunocytokines
Rituximab (Rit) was commercially obtained. Cloning, production and purification of anti-CD20 antibodies and anti-CD20 antibody fusion proteins were previously described (13,20,21). Briefly, DNA encoding for mature mouse IFNɑ1 (mIFNɑ1) or human IFNɑ14 (hIFNɑ14) was fused to the C-terminus of the IgG CH3 via a (G4S)-linker. Antibodies and immunocytokines were purified by protein A affinity chromatography.
DFO conjugation and biochemical characterization
Coupling of p-SCN-Bn-Deferoxamine (SCN-DFO, Macrocyclics, B-705) to lysine side chains was done by incubating protein at 2 mg/mL (adjusted to pH 9.0 using 0.1 M sodium carbonate buffer (Na2CO3)) with 5-fold molar excess chelator for 1 hour at room temperature. Free SCN-DFO was removed using size exclusion spin columns calibrated with PBS (Micro Bio-Spin® Columns with Bio Gel® P-6, Bio-Rad). Site-specific conjugation to free thiol-groups using Deferoxamine-maleimide (mal-DFO, Macrocyclics, B-772) was preceded by partial reduction of the interchain disulfide bonds (2.75-fold mole equivalents of TCEP, tris(2-carboxyethyl)phosphine), 0.5 h, room temperature. Mal-DFO was incubated with reduced protein at 5-fold molar excess for 2 h at room temperature and free mal-DFO was removed as described above.
Successful conjugation of chelator to the antibodies or immunocytokines was analyzed by SDS-PAGE under reducing and nonreducing conditions. Retained antibody integrity was confirmed by size exclusion chromatography (Superdex200 10/30 on an Äkta purifier) with PBS as mobile phase.
89Zr-radiolabeling and immunoreactivity
89Zr-radiolabeling was performed as described by Vosjan et al (22) using 300 μCi/11.1 MBq per 50-70 μg protein. Removal of free radiolabel and buffer exchange were performed as described above. Labeling efficiency and radiochemical purity were determined by instant thin layer chromatography (ITLC strips for monoclonal antibody preparation, Biodex Medical Systems) with 20 mM citric acid as solvent and strips were gamma counted (Wallac Wizard 1480 3” Automatic Gamma Counter, Perkin Elmer).
Immunoreactivity of 89Zr-labeled proteins was determined by incubating trace amounts of antibody with excess antigen expressing 38C13-hCD20 cells (1 h, room temperature) and measuring activity in supernatant vs cell bound activity. Binding of the IFNα portion and non-specific binding of the antibodies was assessed using hCD20-negative 38C13 cells.
ImmunoPET/CT imaging and ex vivo biodistribution
Imaging studies were initiated day 10-14 post tumor implantation. Groups of 4 mice were injected via the tail vein with 89Zr-Rit or 89Zr-anti-CD20-IFNα (10 μg protein/1.1-2.2 MBq). A third group received a higher protein dose of 89Zr-Rit-IFNα (50 μg/2.2 MBq). Mice were anesthetized (2% isoflurane) and 2-hour dynamic PET scans (Inveon, Siemens) were acquired for one mouse in each group, followed by 10-min static scans at 4, 24 and 75 h. Each PET scan was followed by a CT scan (CrumpCAT; UCLA in-house small-animal CT scanner). PET images were reconstructed using OSEM3D MAP algorithm and presented as whole-body maximum intensity projection (MIP) PET/CT overlays. Image analysis and display was conducted using AMIDE (23).
For ex vivo biodistribution studies, tissues were dissected and gamma counted. Percent injected dose per gram (%ID/g) was calculated using a decay-corrected standard and the tissue weight.
Statistical analysis
Data are reported as mean ± SD unless stated otherwise. Statistical significance was calculated using Two-Way ANOVA, corrected for multiple comparisons using the Tukey test (Prism 8 for macOS, GraphPad Software).
Data availability statement
The data generated in this study are available within the article and its supplementary data files.
RESULTS
Conjugation and radiolabeling of Rit-mIFNα fusion protein
Anti-CD20 antibody Rituximab (Rit) and antibody-murine interferon alpha fusion protein (Rit-mIFNα) were conjugated to the amine-reactive chelator isothiocyanabenzyl-deferoxamine (p-SCN-Bn-DFO) for subsequent 89Zr chelation. Conjugated antibodies were analyzed by SDS-PAGE and compared to their unconjugated counterparts (Fig1A). Under non-reducing (NR) conditions the DFO-conjugates migrate with slightly higher molecular weight. Under reducing (R) conditions the higher molecular weight is mostly associated with the heavy chain band. Size exclusion chromatography shows DFO-conjugated antibodies eluted as a distinct peak with similar elution times compared to the unconjugated antibodies (Fig1B).
Figure1. Conjugation of p-SCN-Bn-DFO to Rit and Rit-mIFNα fusion protein.
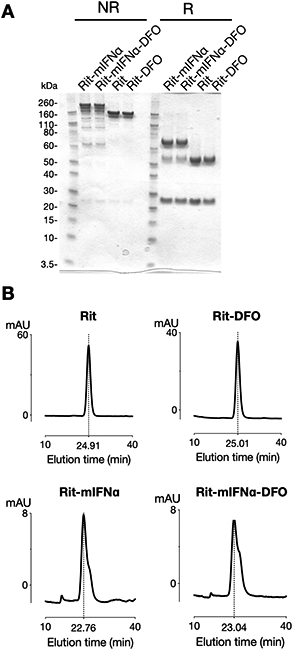
A) SDS-PAGE analysis of unconjugated and conjugated antibodies. Under non-reducing (NR) conditions the DFO-conjugates migrate with slightly higher molecular weight. Under reducing (R) conditions the higher molecular weight is mostly associated with the heavy chain band. B) Size exclusion chromatography of DFO-conjugated antibodies shows single peaks with slightly longer elution times.
DFO-conjugated Rit and Rit-mIFNα were radiolabeled with 89Zr with no significant differences in labeling efficiency, specific activity, radiochemical purity or immunoreactivity to hCD20-positive cells (38C13-hCD20, supplementary Table S1). While 89Zr-DFO-mRit showed minimal background binding to hCD20-negative 38C13 cells, the 89Zr-DFO-Rit-mIFNα fusion protein confirmed IFNα-mediated binding (44.6 ±3.3%) to IFN-receptor expressed by 38C13 cells.
Quantitative flow cytometry using standardize fluorescence intensity measurements (QSC anti-human IgG beads) determined an antibody binding capacity (ABC) value of 145 ± 13 x 103 for 38C13-hCD20 (n=3).
Differences in biodistribution of 89Zr-DFO-Rit and 89Zr-DFO-Rit-mIFNα fusion protein are visualized using immunoPET
Dynamic PET scans (0-120 min p.i.) of mice injected with 10 μg/2.2 MBq 89Zr-DFO-Rit-mIFNα or 89Zr-DFO-Rit showed profound differences in the in vivo biodistribution (Fig2). Fast clearance from the blood pool and accumulation in the liver and spleen was observed for 89Zr-DFO-Rit-mIFNα (Fig2A). In contrast, 89Zr-DFO-Rit remained in circulation, with activity visible in the blood pool (Fig2B) at all time points, as expected for a full-length IgG. Accumulation of 89Zr-DFO-Rit in the liver and clearance through the hepatobiliary route was lower compared with the cytokine-fusion protein.
Figure2. In vivo biodistribution of low dose 89Zr-DFO-Rit-mIFNα compared with 89Zr-DFO-Rit.

A) Dynamic (0-120 min) PET scan obtained post injection of 10 μg (2.2 MBq) 89Zr-DFO-Rit-mIFNα. B) Dynamic (0-120 min) PET scan obtained post injection of 10 μg (2.2 MBq) 89Zr-DFO-Rit.
Therapeutic efficacy of 89Zr-DFO-Rit-mIFNα against B-cell lymphoma are visualized using immunoPET
In addition to the low imaging dose (10 μg) of 89Zr-DFO-Rit-mIFNα and control 89Zr-DFO-Rit, a separate group of mice (s.c. 38C13-hCD20) was injected with a single therapeutic dose (50 μg) of 89Zr-DFO-Rit-mIFNα. Static 10-min PET scans were acquired at 4, 24 and 75 h p.i. (Fig3). Similar to the low dose group, accumulation of 89Zr-DFO-Rit-mIFNα was observed in the liver peaking at 4 h p.i. and decreasing at 24 h and 75 h p.i. (Fig3A). For both doses, clearance through the hepatobiliary route and excretion of activity with the feces is visible in the gastrointestinal tract. The higher dose (50 μg) of 89Zr-DFO-Rit-mIFNα resulted in more activity accumulating in the liver and being excreted through the intestines (Fig3B). Tumor uptake could be seen as early as 4 h p.i. for both the imaging (10 μg) and the therapeutic single dose (50 μg) and increased over time. In addition, tumor growth inhibition was evident for both dose levels of 89Zr-DFO-Rit-mIFNα (Fig3 and see below). In contrast, 89Zr-DFO-Rit (10 μg) immunoPET revealed higher tumor uptake compared with the IFN-fusion proteins (Fig3C). Despite a higher fraction of antibody accumulating in the tumor over time, no growth inhibition was observed. Furthermore, tumor-infiltrated lymph nodes are visible at 24 h post tracer injection (day 11 post injection of 38C13-hCD20 cells) and the number of lymph nodes showing uptake of 89Zr-DFO-Rit increased up to the last imaging time point with 11 positive LN detected at 75 h p.i. (compared to 0 and 1 positive lymph nodes detected in the 10 and 50 μg 89Zr-DFO-Rit-mIFNα groups, respectively). Blood pool activity (heart and vena cava) of 89Zr-DFO-Rit is higher and activity in the liver is lower compared with 89Zr-DFO-Rit-mIFNα.
Figure 3. 89Zr-DFO-Rit-mIFNα immuno-PET shows therapeutic efficacy in lymphoma bearing mice.

C3H mice bearing subcutaneous B-cell lymphoma (38C13-hCD20) tumors were injected with A) Low dose (2.2 MBq/10 μg) 89Zr-DFO-Rit-mIFNα; B) Therapeutic dose (2.2 MBq/50 μg) 89Zr-DFO-Rit-mIFNα; C) Low dose (2.2 MBq/10 μg) control 89Zr-DFO-Rit. Shown are coronal and sagittal whole-body MIP immuno-PET/CT overlays acquired 4, 24 and 75 h p.i. T=tumors, indicated white arrow, L=liver, GI=gastrointestinal tract, H=heart, white arrow=tumor infiltrated lymph nodes.
Ex vivo biodistribution and blood clearance
Ex vivo biodistributions were performed following the last immunoPET scan (75 h p.i.) and confirmed differential biodistribution for 89Zr-DFO-Rit-mIFNα (10 μg imaging dose and 50 μg therapeutic single dose) and parental control 89Zr-DFO-Rit (10 μg) (Fig4A and Table1). The percent injected dose per gram tissue (%ID/g) values in the liver were significantly higher for mice injected with 10 or 50 μg of 89Zr-DFO-Rit-mIFNα (12 ±0.3 and 13.5 ±0.6 %ID/g, respectively) compared with 89Zr-DFO-Rit (1.8 ±0.3 %ID/g, p<0.0001). Concordantly, retention in the blood was lower for 89Zr-DFO-Rit-mIFNα groups (1.0 ±0.1 and 1.2 ±0.0 %ID/g) compared with 4.0 ±0.8 %ID/g 89Zr-DFO-Rit (p=0.0465). The parental control 89Zr-DFO-Rit showed higher tumor uptake (38C13-hCD20, 38.4 ±5.0 %ID/g, p<0.0001) while no difference between the 89Zr-DFO-Rit-mIFNα dose groups was observed (10 μg, 7.6 ±0.5 %ID/g and 50 μg, 8.7 ±0.6 %ID/g, ns).
Figure 4. Ex vivo biodistribution.
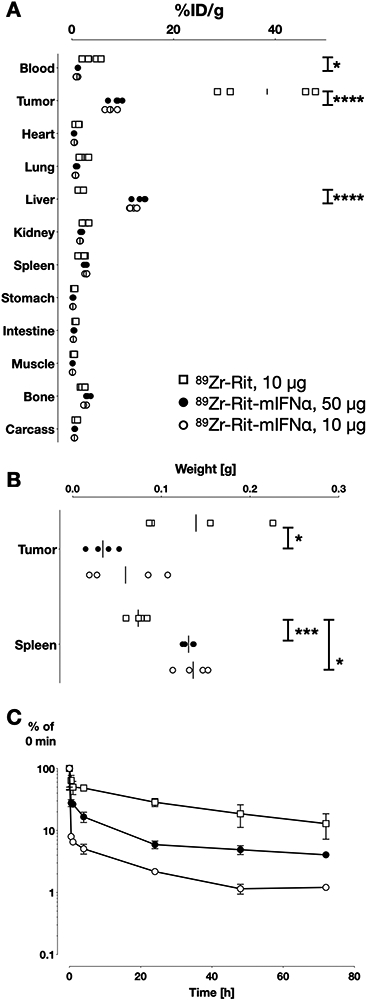
C3H mice bearing s.c. 38C13-hCD20 tumors were injected i.v. with 89Zr-Rit (10 μg, square), 89Zr-Rit-mIFNα (10 μg, circle or 50 μg, filled circle). A) Ex vivo biodistribution at 75 h p.i. %ID/g values are depicted for individual mice of n = 4 with a line at mean. Statistical significance determined using 2way-Anova, Holm-Sidak method with alpha = 0.05; P value: 0.1234 (ns), 0.0332 (*), 0.0021 (**), 0.0002 (***), <0.0001 (****).
B) Tumor (38C13-hCD20) and spleen weight at time of ex vivo biodistribution. C) Plasma activity of 89Zr-Rit (10 μg) and 89Zr-Rit-mIFNα (10 and 50 μg) after a single dose i.v. injection into C3H mice (n=4/group). Serum concentration of radiolabel were determined by gamma counting and normalized to the activity immediately after injection (0 min).
Table 1. Ex vivo biodistribution.
C3H mice bearing 38C13-hCD20 subcutaneous tumors. 75 h p.i., N=4 per group, %ID/g values are depicted as mean ± SEM.
| 89Zr-Rit-mIFNα, 10 μg | 89Zr-Rit-mIFNα, 50 μg | 89Zr-Rit, 10 μg | ||||
|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | |
| Blood | 1.02 | 0.08 | 1.20 | 0.02 | 3.97 | 0.83 |
| 38C13-hCD20 | 7.64 | 0.50 | 8.74 | 0.59 | 38.41 | 4.96 |
| Heart | 0.50 | 0.02 | 0.45 | 0.02 | 1.12 | 0.18 |
| Lung | 0.74 | 0.04 | 0.90 | 0.07 | 2.25 | 0.41 |
| Liver | 11.95 | 0.34 | 13.45 | 0.62 | 1.79 | 0.25 |
| Kidney | 1.61 | 0.03 | 1.87 | 0.10 | 2.65 | 0.27 |
| Spleen | 2.83 | 0.09 | 2.63 | 0.13 | 2.03 | 0.33 |
| Stomach | 0.20 | 0.01 | 0.25 | 0.04 | 0.48 | 0.06 |
| Intestine | 0.31 | 0.01 | 0.42 | 0.04 | 0.73 | 0.04 |
| Muscle | 0.14 | 0.01 | 0.16 | 0.01 | 0.41 | 0.07 |
| Bone | 2.61 | 0.11 | 3.13 | 0.21 | 2.19 | 0.22 |
| Carcass | 0.51 | 0.02 | 0.57 | 0.03 | 0.92 | 0.10 |
| Tissue:Blood | Mean | 95% CI | Mean | 95% CI | Mean | 95% CI |
| Tumor | 7.5 | 5.9-9.7 | 7.3 | 6.0-8.5 | 9.7 | 5.5-20.7 |
| Liver | 11.7 | 9.7-14.6 | 11.2 | 9.9-12.6 | 0.5 | 0.3-1.0 |
| Spleen | 2.8 | 2.3-3.5 | 2.2 | 1.9-2.5 | 0.5 | 0.3-1.1 |
Despite lower tumor uptake for 89Zr-DFO-Rit-mIFNα, tumor growth inhibition was observed in the 89Zr-DFO-Rit-mIFNα groups with tumor weights (75 h p.i.) of 60 ± 22 mg in the 10 μg dose group and 34 ± 8 mg in the 50 μg dose group compared with tumor weights of 139 ± 33 mg in the 89Zr-DFO-Rit 10 μg group (Fig4B). Further evidence of the bioactivity and the therapeutic effect of Rit-mIFNα was the observed increase in spleen weight (splenomegaly), which was significantly higher in mice injected with 10 or 50 μg of 89Zr-DFO-Rit-mIFNα (136 ± 9 mg, p=0.0332 and 131 ± 3 mg, p=0.0002) compared with the 89Zr-DFO-Rit control group (74 ± 5 mg). The increased spleen weight coincides with a more than two-fold higher uptake (%ID per organ) of the 89Zr-DFO-Rit-mIFNα groups (0.34 −0.38 %ID) compared with 89Zr-DFO-Rit (0.15 ± 0.02 %ID in the spleen).
Blood samples were collected at several time points (n≥3 per group) and analyzed for plasma levels of radioactivity (Fig4C). The pharmacokinetic profile of 89Zr-DFO-Rit showed a two-phase decay with a terminal half-life (t1/2β) of 33.9 h. The terminal half-life for 89Zr-DFO-Rit-mIFNα fusion proteins was 15.2 h for the low dose (10 μg) and 11.2 h for the high dose (50 μg). The fast half-life (t1/2α) of 89Zr-DFO-Rit-mIFNα could not be fitted with the two-phase decay model. The major fraction of 89Zr-DFO-Rit-mIFNα cleared in the t1/2α phase (>90% for the low dose (10 μg) and >70% for the high dose (50 μg), while for the parental 89Zr-DFO-Rit approximately 50% cleared in the t1/2α phase. These results indicate that while 89Zr-DFO-Rit is mainly cleared through antigen-antibody interaction, clearance of the immunocytokines might be accelerated by binding to IFN receptors in addition to antigen binding.
In vivo biodistribution of immunocytokines with varying species of IFN or IgG
To investigate if biodistribution of the immunocytokines is influenced by the species, isoform or subtype of either the interferon or the antibody portion, we radiolabeled anti-CD20 mIgG2a, anti-CD20 mIgG2a-mIFNα and anti-CD20 hIgG1 (Rit)-hIFNα14 in a site-specific manner using maleimide-DFO to chelate 89Zr. Anti-CD20 hIgG1 (Rit) was included to show that the different radiolabeling method did not influence biodistribution or tumor uptake.
Successful conjugation of the maleimide-DFO to reduced disulfide bonds in the antibody (hinge) was confirmed by size exclusion chromatography; the added molecular weight of the chelator led to slightly earlier elution of the conjugated proteins and showed that the integrity of the IgG portion was not impaired (Fig5A).
Figure 5. 89Zr-immuno-PET with mouse/human derivatives of anti-CD20-IFNα.

A) Anti-CD20 antibodies or anti-CD20 antibody-IFNα fusion proteins were site-specifically conjugated with mal-DFO and analyses by size exclusion chromatography. Slightly earlier elution times correspond with successful conjugation. B) Mal-DFO conjugated proteins were radiolabeled using 89Zr. 10 μg (1.11 - 1.85 MBq) were injected into C3H mice bearing s.c. 38C13-hCD20 tumors (right shoulder). Representative immunoPET/CT scans (20 h p.i., coronal and sagittal) of each group (n=4) are shown.
Radiolabeling was executed with similar labeling efficiencies (78-97%), radiochemical purities (82-98%) and resulted in comparable specific activities (3.0-5.6 μCi/μg, 0.111-0.207 MBq/μg) seen with the random conjugation (SCN-DFO). Immunoreactivity ranged from 57-79% using hCD20 positive B cells and antigen negative control cells.
Protein doses of 10 μg (1.1-2.1 MBq) were injected into mice bearing s.c. 38C13-hCD20 tumors (n=4 per group). Dynamic PET scans (0-120 min p.i.) revealed a biodistribution pattern similar to the one observed for Rit and Rit-mIFNα. While the antibody (89Zr-malDFO-mIgG2a) was mainly visible in the blood pool (heart), the immunocytokines (89Zr-malDFO-mIgG2a-mIFNα and 89Zr-malDFO-hIgG1-hIFNα14) displayed early accumulation in the liver and uptake in the spleen (Supplemental Fig1). Static scans acquired at 6 and 20 h p.i. showed higher tumor uptake and lower accumulation in liver, spleen and kidneys for the 89Zr-labeled anti-CD20 antibodies compared with the respective IFN-fusion proteins (Fig5B and Supplemental Table S2).
The results confirm that binding of the interferon portion of the immunocytokine influences the biodistribution and that systemic immune activation (spleen) might occur.
DISCUSSION
Antibody-cytokine fusion proteins (immunocytokines) comprising a variety of antibody formats, target antigens and cytokine payloads, have shown promising preclinical results and have the potential to significantly improve the therapeutic index of cytokines that promote anti-tumor immune responses. Some of these immunocytokines have entered clinical trials, with many urgent questions remaining regarding in vivo biodistribution and mechanism of action (reviewed in (24).
It is generally believed that the antibody portion of immunocytokines drives tumor targeting and determines biodistribution. However, conjugating or fusing a cytokine to an antibody adds molecular mass and a second binding interaction (to the cytokine receptor). Additionally, the charge of the cytokine portion can change the local and/or overall charge of the fusion protein and cytokines are usually glycosylated. All these factors could influence biodistribution, tumor accumulation, clearance route and plasma half-life of the immunocytokine. Non-invasive molecular imaging could verify targeted delivery of immunocytokines to the tumor, predict toxicity and efficacy and could help guide the further development of immunocytokines.
In this work, we used 89Zr-immunoPET imaging to visualize the whole-body biodistribution, anti-tumor efficacy, and pharmacokinetics of anti-CD20 interferon-alpha (Rit-mIFNɑ), a preclinical precursor of IGN002 (ImmunGene), currently in clinical trials (25). The residualizing characteristics of the radiometal 89Zr result in retention of signal in tissues where the tracer is internalized and degraded, which allows the monitoring of clearance and excretion route (26).
ImmunoPET studies with 89Zr-labeled Rit-mIFNɑ and the parental mAb (Rit) in a syngeneic rituximab-insensitive B-cell lymphoma expressing human CD20 (38C13-hCD20 in C3H mice) revealed significant differences in the in vivo behavior of these molecules.
The biodistribution and pharmacokinetics of 89Zr-Rit-mIFNɑ differed from the parental antibody 89Zr-Rit, with the fusion protein showing faster blood clearance rates (t1/2 11-15 h vs 34 h). This observation is in line with previously published half-lives for anti-CD20-IFNɑ fusion proteins of 6-8 h in mice (13). Other immunocytokines based on full-length IgG (with retained FcRn binding), but containing different cytokines, show similar characteristics and exhibit a much faster clearance rate compared with the respective intact IgG (27-29).
ImmunoPET confirmed specific accumulation of 89Zr-Rit-mIFNɑ in tumors, although at a significantly lower level than 89Zr-Rit (7.6 ±1.0 %ID/g vs 38.4 ±9.9 %ID/g at 75 h p.i.). Importantly, despite the lower tumor uptake, 89Zr-immunoPET and ex vivo biodistribution clearly showed a therapeutic effect for a single dose of 89Zr-Rit-mIFNɑ. Groups receiving 10 or 50 μg of 89Zr-Rit-mIFNɑ presented smaller tumors and fewer lymph node metastases compared to no observable antitumoral effect in mice receiving 89Zr-Rit. Because the 38C13 B-cell lymphoma model is rituximab insensitive, the observed antitumoral effect can be attributed to the cytokine portion of Rit-mIFNɑ (30). The lower tumor uptake is likely due to the shorter plasma half-live of the immunocytokine, although the higher molecular weight impairing tumor penetration and binding to receptor expressing cells elsewhere could be contributing factors (31).
Non-tumor tissue accumulation of 89Zr-Rit-mIFNɑ was highest in the liver and the spleen. Some activity is visible in the intestines indicating (at least partial) hepatobiliary clearance and excretion with the fecal matter. Other immunocytokines in preclinical development, e.g. 89Zr-labeled CEA-IL2v, have shown similar tissue distribution with high liver and spleen uptake (28).
A fraction of liver uptake is likely interferon receptor mediated, but further studies are needed to confirm this possibility. The liver is the major tissue responding to IFN-Is, followed by kidney and spleen, as was shown in a transgenic mouse model humanized for type-I interferon response (16).
The notable uptake of 89Zr-Rit-mIFNɑ in the spleen, which also significantly increased in size, resulted in up to 5-fold higher spleen-to-blood ratios of the immunocytokine 89Zr-Rit-mIFNɑ compared to 89Zr-Rit. This suggests that spleen uptake is driven by binding of the IFNɑ to its receptor on immune cells and that a systemic immune activation occurs and could contribute to the observed anti-tumor efficacy.
A major limitation of preclinical mouse models are species-specific differences and translation into clinical studies. Here, an immunocytokine containing murine IFNɑ1 was studied in a syngeneic mouse model because human interferons show only weak activation of murine IFNAR. The Fc portion could also affect biodistribution and immune activation (ADCC and CDC), however, the 38C13 B-cell lymphoma is rituximab insensitive and Fc-mediated effects were not expected. Mechanisms of rituximab action, as well as, mechanisms of rituximab resistance are challenging to study in vivo as they depend on the patient’s own immune system (32). 38C13-hCD20 cells express 145 ± 13 x 103 antigens per cell, which is in the range of CD20 expression levels described for most B cell lymphomas (65 - 300 x 103 per cell) in the literature (33). The high tumor uptake observed for 89Zr-Rit suggests that down-regulation or loss of human CD20 expression from the 38C13-hCD20 cells is not the cause for rituximab resistance.
In a validation study using species-specific derivatives of anti-CD20-IFNɑ similar biodistribution patterns were observed with anti-CD20 antibodies (human IgG1 or murine IgG2a) showing higher uptake in tumors and immunocytokines (murine IFNɑ1 or human IFNɑ14) resulting in higher uptake in liver and spleen, suggesting that the cytokine directs the tissue distribution.
Interferons not only have direct anti-proliferative effects on cell growth, they also enhanced communications between innate and adaptive immunity which could make them candidates/enhancers for combination therapies (chemotherapy, radiation, immunostimulatory drugs, checkpoint inhibitors, CAR T cells) (21,34,35).
In conclusion, zirconium-89 radiolabeling and immunoPET imaging enables non-invasive whole body monitoring and visualizes the biodistribution, tumor accumulation and clearance of novel immunocytokines. Serial immunoPET imaging corroborated the high potency of the anti-CD20-IFNɑ immunocytokine and showed delayed tumor growth and inhibition of lymph node invasion in a rituximab resistant tumor model. Furthermore, in vivo imaging showed that the cytokine portion determines tissue distribution of the fusion protein and suggests systemic immune activation, thereby providing some insight into possible mechanism of action of the immunocytokine. Radiolabeling with zirconium-89 for immunoPET imaging is applicable to other immunocytokines, targeting alternative antigens and carrying a variety of cytokines. ImmunoPET therefore has the potential to facilitate the development of targeted therapies, especially with multiple potential targeting interactions.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Crump Institute’s Preclinical Imaging Technology Center (UCLA) for technical support with small-animal PET imaging. Special thanks to Dr. Tove Olafsen for editing the manuscript.
Funding:
This work was supported by NIH grants CA149254 and the Jaime Erin Follicular Lymphoma Research Consortium (JM Timmerman) and CA162964 (SL Morrison) and by a generous gift from Ralph and Marjorie Crump to the Crump Institute for Molecular Imaging. Small-animal imaging was funded in part by the UCLA Jonsson Comprehensive Cancer Center (JCCC) Support Grant (NIH CA016042). AM Wu, SL Morrison and JM Timmerman are members of the JCCC.
Footnotes
Conflict of interest disclosure statement:
JM Timmerman reports receiving commercial research grants from Bristol-Myers Squibb, Kite Pharma, and Valor Biotherapeutics, and is a consultant/ advisory board member for Celgene and Seattle Genetics. SL Morrison holds ownership interest and is a consultant/advisory board member for Qwixel Therapeutics. AM Wu holds ownership interest in and is a consultant/advisory board member for ImaginAb, Inc.
All other authors declare no potential conflicts of interest.
All authors have approved the final version submitted for publication and affirm that no part of the manuscript is under consideration, in press, published or reported elsewhere.
REFERENCES
- 1.Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346(4):235–42 doi 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 2.Marcus R, Imrie K, Belch A, Cunningham D, Flores E, Catalano J, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 2005;105(4):1417–23 doi 10.1182/blood-2004-08-3175. [DOI] [PubMed] [Google Scholar]
- 3.Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376(9747):1164–74 doi 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- 4.Alduaij W, Illidge TM. The future of anti-CD20 monoclonal antibodies: are we making progress? Blood 2011;117(11):2993–3001 doi 10.1182/blood-2010-07-298356. [DOI] [PubMed] [Google Scholar]
- 5.Ortiz-Sanchez E, Helguera G, Daniels TR, Penichet ML. Antibody-cytokine fusion proteins: applications in cancer therapy. Expert Opin Biol Ther 2008;8(5):609–32 doi 10.1517/14712598.8.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kontermann RE. Antibody-cytokine fusion proteins. Arch Biochem Biophys 2012;526(2):194–205 doi 10.1016/j.abb.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Young PA, Morrison SL, Timmerman JM. Antibody-cytokine fusion proteins for treatment of cancer: engineering cytokines for improved efficacy and safety. Semin Oncol 2014;41(5):623–36 doi 10.1053/j.seminoncol.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trinchieri G Type I interferon: friend or foe? J Exp Med 2010;207(10):2053–63 doi 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14(1):36–49 doi 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016;5(6):e1163462 doi 10.1080/2162402X.2016.1163462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatt S, Parvin S, Zhang Y, Cho HM, Kunkalla K, Vega F, et al. Anti-CD20-interleukin-21 fusokine targets malignant B cells via direct apoptosis and NK-cell-dependent cytotoxicity. Blood 2017;129(16):2246–56 doi 10.1182/blood-2016-09-738211. [DOI] [PubMed] [Google Scholar]
- 12.Valedkarimi Z, Nasiri H, Aghebati-Maleki L, Majidi J. Antibody-cytokine fusion proteins for improving efficacy and safety of cancer therapy. Biomed Pharmacother 2017;95:731–42 doi 10.1016/j.biopha.2017.07.160. [DOI] [PubMed] [Google Scholar]
- 13.Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood 2010;115(14):2864–71 doi 10.1182/blood-2009-10-250555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trinh KR, Vasuthasawat A, Steward KK, Yamada RE, Timmerman JM, Morrison SL. Anti-CD20-interferon-beta fusion protein therapy of murine B-cell lymphomas. J Immunother 2013;36(5):305–18 doi 10.1097/CJI.0b013e3182993eb9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benson N, de Jongh J, Duckworth JD, Jones HM, Pertinez HE, Rawal JK, et al. Pharmacokinetic-pharmacodynamic modeling of alpha interferon response induced by a Toll-like 7 receptor agonist in mice. Antimicrob Agents Chemother 2010;54(3):1179–85 doi 10.1128/AAC.00551-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harari D, Abramovich R, Zozulya A, Smith P, Pouly S, Koster M, et al. Bridging the species divide: transgenic mice humanized for type-I interferon response. PLoS One 2014;9(1):e84259 doi 10.1371/journal.pone.0084259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vieira P, Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol 1988;18(2):313–6 doi 10.1002/eji.1830180221. [DOI] [PubMed] [Google Scholar]
- 18.Betting DJ, Yamada RE, Kafi K, Said J, van Rooijen N, Timmerman JM. Intratumoral but not systemic delivery of CpG oligodeoxynucleotide augments the efficacy of anti-CD20 monoclonal antibody therapy against B cell lymphoma. J Immunother 2009;32(6):622–31 doi 10.1097/CJI.0b013e3181ab23f1. [DOI] [PubMed] [Google Scholar]
- 19.Golay J, Cittera E, Di Gaetano N, Manganini M, Mosca M, Nebuloni M, et al. The role of complement in the therapeutic activity of rituximab in a murine B lymphoma model homing in lymph nodes. Haematologica 2006;91(2):176–83. [PubMed] [Google Scholar]
- 20.Vega GG, Franco-Cea LA, Huerta-Yepez S, Mayani H, Morrison SL, Bonavida B, et al. Overcoming rituximab drug-resistance by the genetically engineered anti-CD20-hIFN-alpha fusion protein: Direct cytotoxicity and synergy with chemotherapy. Int J Oncol 2015;47(5):1735–48 doi 10.3892/ijo.2015.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young PA, Yamada RE, Trinh KR, Vasuthasawat A, De Oliveira S, Yamada DH, et al. Activity of Anti-CD19 Chimeric Antigen Receptor T Cells Against B Cell Lymphoma Is Enhanced by Antibody-Targeted Interferon-Alpha. J Interferon Cytokine Res 2018;38(6):239–54 doi 10.1089/jir.2018.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vosjan MJ, Perk LR, Visser GW, Budde M, Jurek P, Kiefer GE, et al. Conjugation and radiolabeling of monoclonal antibodies with zirconium-89 for PET imaging using the bifunctional chelate p-isothiocyanatobenzyl-desferrioxamine. Nat Protoc 2010;5(4):739–43 doi 10.1038/nprot.2010.13. [DOI] [PubMed] [Google Scholar]
- 23.Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging 2003;2(3):131–7. [DOI] [PubMed] [Google Scholar]
- 24.Murer P, Neri D. Antibody-cytokine fusion proteins: A novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. N Biotechnol 2019;52:42–53 doi 10.1016/j.nbt.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Timmerman JM, Steward KK, Yamada RE, Young PA, Minning DM, Sachdev RK, et al. Antibody-Interferon-Alpha Fusion Protein Therapy for the Treatment of B-Cell Non-Hodgkin Lymphoma: Enhanced ADCC, Inhibition of Proliferation, and In Vivo Eradication of CD20+ Human Lymphomas. 57th ASH Annual Meeting. Orlando, FL: Blood (2015) 126 (23): 2762; . [Google Scholar]
- 26.Vivier D, Sharma SK, Zeglis BM. Understanding the in vivo fate of radioimmunoconjugates for nuclear imaging. J Labelled Comp Radiopharm 2018;61(9):672–92 doi 10.1002/jlcr.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ribba B, Boetsch C, Nayak T, Grimm HP, Charo J, Evers S, et al. Prediction of the Optimal Dosing Regimen Using a Mathematical Model of Tumor Uptake for Immunocytokine-Based Cancer Immunotherapy. Clin Cancer Res 2018;24(14):3325–33 doi 10.1158/1078-0432.CCR-17-2953. [DOI] [PubMed] [Google Scholar]
- 28.van Brummelen EMJ, Huisman MC, de Wit-van der Veen LJ, Nayak TK, Stokkel MPM, Mulder ER, et al. (89)Zr-labeled CEA-targeted IL-2 variant immunocytokine in patients with solid tumors: CEA-mediated tumor accumulation and role of IL-2 receptor-binding. Oncotarget 2018;9(37):24737–49 doi 10.18632/oncotarget.25343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, et al. First-in-Human Phase I Trial of a Tumor-Targeted Cytokine (NHS-IL12) in Subjects with Metastatic Solid Tumors. Clin Cancer Res 2019;25(1):99–109 doi 10.1158/1078-0432.CCR-18-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Betting DJ, Kafi K, Yamada RE, Steward KK, Olafsen T, Wu AM, et al. In Vivo Eradication of a Rituximab-Resistant Human CD20+ B Cell Lymphoma by Rituximab-CpG Oligodeoxynucleotide Conjugate Is Mediated by Natural Killer Cells and Complement. Blood 2009;114(22):723- doi 10.1182/blood.V114.22.723.723.19369228 [DOI] [Google Scholar]
- 31.Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci U S A 2015;112(11):3320–5 doi 10.1073/pnas.1416159112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierpont TM, Limper CB, Richards KL. Past, Present, and Future of Rituximab-The World's First Oncology Monoclonal Antibody Therapy. Front Oncol 2018;8:163 doi 10.3389/fonc.2018.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ginaldi L, De Martinis M, Matutes E, Farahat N, Morilla R, Catovsky D. Levels of expression of CD19 and CD20 in chronic B cell leukaemias. J Clin Pathol 1998;51(5):364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neri D Antibody-Cytokine Fusions: Versatile Products for the Modulation of Anticancer Immunity. Cancer Immunol Res 2019;7(3):348–54 doi 10.1158/2326-6066.CIR-18-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothschilds A, Tzeng A, Mehta NK, Moynihan KD, Irvine DJ, Wittrup KD. Order of administration of combination cytokine therapies can decouple toxicity from efficacy in syngeneic mouse tumor models. OncoImmunology 2019;8(5) doi 10.1080/2162402x.2018.1558678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files.