

A noninvasive drug delivery approach that targets the urinary system overcomes the current barriers facing effective treatment of bladder cancer.
Abstract
The standard treatment of nonmuscle invasive bladder cancer (NMIBC) is transurethral resection of the tumors, followed by intravesical therapy (IT), which comprises a direct instillation of a solution of Bacillus Calmette-Guérin vaccine or chemotherapy into the bladder. However, the recurrence rate in this disease remains unacceptably high. IT is a local treatment that fails to reach tumors developed in the upper urinary tract (ureter and renal pelvis). The catheterization procedure required for IT is invasive, painful, and poses an increased infection risk, resulting in poor patient quality of life and compliance. There is an unmet need for a potent, comprehensive, and noninvasive option. Without chemical modifications, peptides are rapidly removed by renal clearance. This “shortcoming” can be advantageous when used as a drug carrier for directing therapy to NMIBC. Here we develop a urinary drug-disposing (UDD) approach to improve NMIBC treatment. A 12-amino acid bio-inert peptide (Bdd) that can be exclusively eliminated via renal filtration was generated for delivering the microtubule inhibitor DM1 to NMIBC with minimal nonspecific accumulation in other organs. The UDD approach prolonged survival of mice bearing human bladder tumors. Unlike IT, the treatment was given noninvasively (intravenously). Furthermore, it was more effective at suppressing tumor growth than clinically used IT (mitomycin) and safer than free DM1. The application of this UDD approach to treat kidney tumors and deliver other drugs such as doxorubicin was also demonstrated. Overall, the rapid renal clearance of peptides can be exploited to direct cancer therapies to the urinary system.
Significance:
A noninvasive drug delivery approach that targets the urinary system overcomes the current barriers facing effective treatment of bladder cancer.
Graphical Abstract
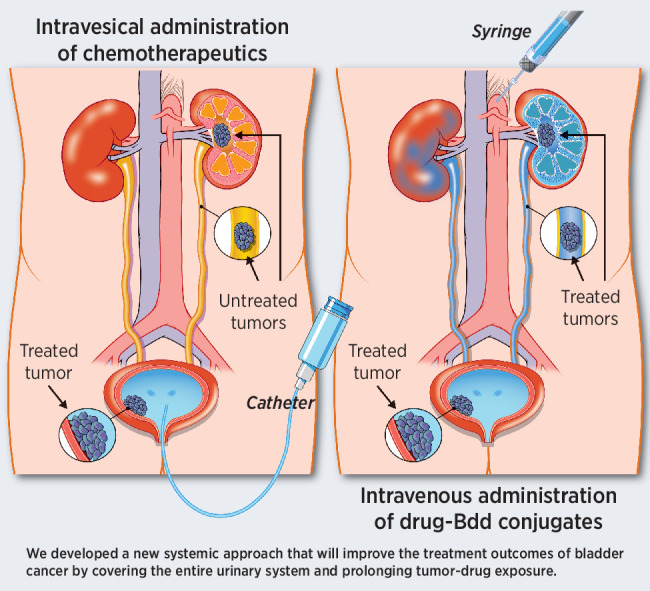
Introduction
Urothelial carcinoma, also known as bladder cancer, is one of the most expensive life-time cancers to treat due to the high recurrence rate, repeated surgeries, and constant monitoring and treatment (1). More than 75% to 80% of bladder cancer are nonmuscle invasive (NMIBC; ref. 2). The standard treatment is transurethral surgical resection, followed by intravesical therapy (IT). IT is a direct instillation of solutions of Bacillus Calmette-Guérin (BCG) immunotherapy or chemotherapy into the bladder, via a catheter, to prevent disease recurrence (3, 4). Because the treatment is local, IT possesses the advantage of minimizing systemic absorption and off-target toxicity (5). However, the recurrence rate of NMIBC remains unacceptably high (up to 80%; refs. 6–8). The physiologic need to urinate makes it difficult for patients, especially the elderly, to hold the instilled drug solution for longer than 1 to 2 hours. This limits the drug–tumor contacting time (9, 10). Patient compliance is also poor, as the catheterization procedure is invasive (11). IT can cause pain, infection, significant urinary symptoms, or complications such as bladder perforation and perivesical fat necrosis (12), leading to discontinuation of treatment. Importantly, IT is unable to dispense drugs to tumors that may occur in more proximal portions of the upper urinary tract, such as the ureter and the renal pelvis. There is also a significant shortage of BCG due to a global increase in demand and limited supply (13–15). A critical result: surgeons are often compelled to remove the kidney and ureter, even in the case of less aggressive cancers. There is an unmet clinical need for a more effective drug delivery approach that is noninvasive, can access the entire urothelium, has a longer duration of drug–tumor exposure, and improves patient compliance by eliminating IT-associated morbidity. The goal of this study is to develop a urinary drug-disposing (UDD) approach to overcome the drug delivery barrier for NMIBC treatment.
Different approaches have been proposed to improve intravesical chemotherapy (ITC) treatment. To prolong treatment duration, a thermal-sensitive hydrogel, UGN-102, was designed to convert into a semisolid drug depot inside the bladder and slowly release mitomycin (MIT). A gemcitabine (GEM)-containing semipermeable silicon tube, GemRis, which functions as an osmotic pump, has also been developed to control the release of GEM (16). Other treatments are currently being tested in clinical trials for patients who do not respond to BCG. Oportuzumab monatox is an antibody–protein conjugate that targets tumor cells expressing EpCAM (17). Adstiladrin is a nonreplicating adenovirus vector that encodes the human IFNα-2b gene (18). The resulting IFNα-2b proteins, synthesized and expressed in a large quantity, display an antitumor activity through inhibition of angiogenesis and induction of apoptosis in human bladder cancer cells. However, all the aforementioned approaches are invasive, requiring catherization and/or surgical procedures. Alternatively, renal-clearable nanoparticles can be used as drug carriers (19–22), but they are known to be nonspecifically captured by the reticuloendothelial system, leading to a high off-target accumulation in the liver.
Advances in phage display have led to the discovery of many bioactive peptides that target the urinary system (URS; refs. 23–25). For example, a galectin-3 targeting peptide, G3-C12, has been used for delivering captopril, an angiotensin-converting enzyme inhibitor (25). Another peptide, (KKEEE)3K, has been used to carry ciprofloxacin (26–28). These peptides were pharmacologically active. They primarily targeted the kidneys by binding to cell-surface receptors, and prolonged the post-delivery local retention. They have not been applied to bladder cancer treatment. Further, without chemical modifications, a peptide is not a good drug candidate or carrier. It displays unfavorable pharmacokinetics (PK), is rapidly degraded by protease enzymes (29–31), and can be eliminated by renal filtration. In the present studies, we exploited a peptide's rapid renal clearance to our advantage for disposing treatments to the URS. Specifically, we generated a small, 12-amino acid, negatively charged peptide (Bdd) that can bypass the recticuloendothelial system and other organs, and is exclusively excreted into the urine with minimal reabsorption. We propose using it as an alternative to ITC for minimizing off-target accumulation in other organs, promoting drug delivery to the URS, and prolonging bladder retention time, resulting in a comprehensive and more effective bladder cancer treatment.
Materials and Methods
Chemicals and supplies
2(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphate (HBTU) and the N-hydroxybenzotriazole (HOBt) were purchased from Vivitide. All protected amino acids, rink amide MBHA resin, and N-methylmorpholine (NMM) were supplied by Gyros Protein Technologies. Polyethylene glycol (PEG3) was obtained from Creative PEGWorks. Trifluoroacetic acid (TFA), piperidine, thioanisole, anisole, 1,2-ethanedithiole, methyl-tert-butyl ether, N,N-diisopropylethylamine (DIPEA), dimethylformamide (DMF), acetonitrile (ACN), N-methyl-2-pyrrolidone (NMP), sodium carbonate, Sephadex G25, L-glutathione reduced (GSH), lipopolysaccharide (LPS), and cis-dichlorodiammine platinum (II) (cisplatin) were purchased from Sigma-Aldrich. Succinimidyl 3-(2-pyridyldithio)propionate (SPDP) was supplied by Invitrogen. p-SNC-deferoxamine (DFO) was from Macrocyclics Inc. N2'-deacetyl-N2'-(3-mercapto-1-oxopropyl)-maytansine (DM1) and aldoxorubicin HCl (aldox) were from MedKoo Biosciences. Doxorubicin (DOX) and GEM were obtained from LC Laboratories. MIT was supplied by Selleckchem. Cyanine5.5 NHS ester was purchased from Lumiprobe Corporation and luciferin was from Caliper LifeScience.
Peptide synthesis
All peptides were synthesized on a solid-phase peptide synthesizer (PS3, Gyros Protein Technologies) using the N-α-Fmoc methodology on Rink amide resin, as described previously (32–35). The side-chain protected amino acids (0.4 mmol) were attached to the resin (385 mg, 0.1 mmol) by stepwise elongation using NMM as a base, HBTU/HOBt as coupling reagents, and piperidine (20% in DMF v/v) as a deprotecting agent. A β-alanine was incorporated at the peptide N-terminal as a spacer for further conjugation of the desired moieties, including fluorophore (Cyanine5.5), chelator (DFO), or linker (SPDP) prior to peptide cleavage.
Immunogenicity assay
Female BALB/cJ mice (catalog no. 000651; The Jackson Laboratory) were treated with a single dose of Bdd or LPS (5 mg/kg) via tail-vein injection (n = 3/group). Mice administered with PBS were used as negative controls. Blood samples were collected 4 hours later via retro-orbital sinus puncture technique, and the concentration of the innate immune and inflammatory cytokines IL1β, IL2, IL6, IL10, TNFα, and INFγ in plasma was measured with commercial sandwich ELISA kits. ELISA assays were performed according to the manufacturer's instructions (Invitrogen).
Methods to incorporate different functionalities to peptide
Cyanine5.5 NHS ester (50 mg, 1.4 eq) in DMF (4 mL), DFO (25 mg, 1 eq) in DMSO (4 mL), or SPDP (25 mg, 1 eq) in NMP (4 mL), were added to the resin (0.05 mmoL, 1 eq) and allowed to react overnight at room temperature. For fluorophore and chelator conjugation, the reactions were performed in the presence of an organic base (DIPEA, 1 mL). The peptides were then removed from the resin using a cleaving cocktail (5 mL) containing TFA/thioanisole/1,2-ethandithiol/anisole (90:5:3:2) for 4 hours and precipitated in methyl-tert-butyl ether. The resulting peptides (Cy-peptide, DFO-peptide, and SPDP-peptide) were purified to >98% purity using reverse-phase high-performance liquid chromatography (rp-HPLC; Agilent) and were characterized by MALDI-TOF analysis (Tufts Medical School) to confirm their molecular weights.
Synthesis of cleavable drug–peptide conjugates
DM1 (1 mg, 1 eq) was added to SPDP-peptide (10 mg, 3 eq) in a cosolvent of NMP (100 μL) and PBS (10 mmol/L, pH 7, 100 μL), and allowed to react for 2 days at room temperature. The DM1-peptide was then purified by rp-HPLC. To conjugate aldox, a thiol-reactive side (cysteine) was introduced at the peptide N-terminal. Aldox (2 mg, 1 eq) was added to the peptide (10 mg, 3 eq) in PBS (10 nmol/L, 1 mL, pH 7.4), and allowed to react for 30 minutes. The drug–peptide obtained was pH-sensitive and thereby was purified by rp-HPLC in neutral conditions (mobile phase A: PBS, mobile phase B: 90% ACN in PBS). Size exclusion chromatography (TipTop C-18 column) was used to remove the salt content. All the final drug–peptide conjugates were characterized using MALDI-TOF analysis and were quantified with UV absorbance, according to the predetermined extinction coefficient of DM1 (ε = 3,700 cm–1 M–1) or aldox (ε = 13,000 cm–1 M–1) in 5% (v/v) PBS in methanol.
Radiochemistry
89Zirconium (89Zr) was supplied by 3DImaging LLC. The 89Zr-oxalate (500 μCi) was first neutralized with an equivalent volume of sodium carbonate solution (2 M), and then added to the DFO-peptides (0.2 mg, 250 μL). After incubation for 1.5 hours at room temperature, the radiolabeled peptides (89Zr-peptides) were purified by size exclusion chromatography using Sephadex G-25 gel to remove the free zirconium.
PK study
BALB/cJ mice (catalog no. 000651; The Jackson Laboratory) were administered with 89Zr-peptide or free 89Zr (20 μCi, 100 μL) via tail-vein injections (n = 4/condition). Blood samples (20 μL) were collected at various time intervals, using the retro-orbital sinus puncture technique. The radioactivities were measured on Wallac Wizard 2 gamma counter (Perkin-Elmer). The PK models and parameters of 89Zr-peptides and free 89Zr, including the serum half-life and the plasma clearance, were estimated by fitting the data for compartmental model selection using PKSolver 2.0 software.
μPET/CT imaging and biodistribution study
BALB/cJ mice (catalog no. 000651; The Jackson Laboratory) were intravenously injected with different 89Zr-peptide analogues (20 μCi, 100 μL). μPET/CT imaging was performed in animals with and without urine collection prior to the first imaging (n = 4/analogue/condition). Whole-body images were acquired 1, 4, and 24 hours after injection, using an Inveon μPET/CT scanner (Siemens Medical Solutions). μPET/CT maximum energy projections were processed using Amide v1.0.4 and Inveon Research Workplace software. The radioactivities at different regions of interest (ROI) were determined. For endpoint biodistribution studies, the mice were euthanized 0.17, 1, 2, 5, and 7 days after treatment with 89Zr-peptides (n = 3/peptide analogue/time point). The radioactivities of the harvested organs were measured using the Wallac Wizard 2 gamma counter. The results were corrected from radioactive decay, and were expressed as a percentage of the injected dose (%ID) or percentage of injected dose per gram of tissue (%ID/g).
Fluorescence imaging
Female SHO mice (catalog no. 474; Charles River Laboratories) were administered with free Cyanine5.5 or Cy-peptide analogues (0.5 nmol of Cyanine5.5 content measured by UV absorbance at 680 nm) in a PBS solution (150 μL) via tail-vein injections (n = 4/group). Real-time fluorescence imaging was performed using an In Vivo Xtreme imaging system (Bruker). Whole body fluorescence images were acquired 1 and 4 hours postinjection using the appropriate excitation (670 nm) and emission (750 nm) filters. The animals were then euthanized. The organs were excised to perform ex vivo fluorescence imaging. Imaging was also performed on urine samples (20 μL) collected from separate animals 1, 4, and 6 hours after intravenous injection of free dye or Cy-peptide analogues (n = 4/treatment). Bruker MI software was used to process the fluorescence/bright light images and measure the fluorescence intensity in different ROIs. All the data were corrected to eliminate the organ or fluid auto-fluorescence.
Cell lines
MB49 murine bladder carcinoma cell line (catalog no. SCC148) was supplied by EMD Millipore Corporation. UMUC-3 and T24 human urinary bladder cancer cell lines (catalog nos. CRL-1749 and HTB-4), and Renca murine kidney adenocarcinoma cell line (catalog no. CRL-2947), were obtained from ATCC. Each cell line was cultured and maintained (no more than 10 passages after initial thawing) according to the company's instructions. Mycoplasma tests (Lonza) were performed periodically to ensure that there was no contamination. Both UMUC-3 and Renca cell lines were further transduced with GlowCell 16 FLuc-F2A-GFP lentivirus (Biosettia) carrying both firefly luciferase and GFP genes. Briefly, the cells were seeded in six-well plates (0.25 × 106 cells/well) and incubated for 3 days with the virus (2 × 107 IU/well) in the presence of polybrene (10 μg/mL). To ensure more than 95% of cell purity, the cell lines were analyzed and sorted for GFP expression using flow cytometry. The successful transduction was further confirmed by imaging, using an EVOS FL Auto Fluorescence microscope (Life Technology).
Cell viability and cytotoxicity assay
Cancer cells (3 × 103/well) were seeded on a flat bottom 96-well plate overnight. Different concentrations of DM1, GEM, MIT, CIS, DOX, aldox, or drug-loaded peptides (DM1-peptide and aldox-peptide) were then added to the cells for 72 hours and then washed twice with PBS (400 μL). CellTiter Glo reagent (Promega) was added to each well (50 μL). The luminescence generated was recorded using a microplate reader (Tecan US Inc.). The dose response curves were plotted, and the IC50 values were calculated using Graph Pad Prism 6.0 software.
In vitro drug release study
DM1–peptide conjugates (10 μmol/L of drug content) were incubated in the presence of the reducing agent GSH (1 mmol/L) in PBS buffer (800 μL). At different time intervals (0, 2, 4, 6, 8, 12, 24, and 48 hours), a small amount of solution (100 μL) was taken out for rp-HPLC analysis using a C18 analytical column. The amount of DM1 active metabolites released over time was measured (absorbance detected at 254 nm) and then quantified. The experiment was performed independently in triplicate. A similar experiment was performed to quantify the release of aldox. Aldox–peptide conjugates (100 μmol/L of drug content) were incubated in PBS buffer (800 μL) of different pH levels (7.4 and 5.5) in glass vials coated with silica to avoid nonspecific adsorption on the reaction vessel surfaces (36, 37). The amount of drug released over time was then measured as described above (absorbance detected at 480 nm). For accurate quantification, all the results were normalized compared with a DOX control incubated in similar conditions.
Animal care
All animals used for this project were housed in a pathogen-free barrier room, maintained at a controlled temperature (72 ± 2°F) with daily 12 hours cycles of light and dark. All the procedures conducted on mice were approved by the Weill Cornell Medical Center Institutional Animal Care and Use Committee (protocol No. 2019–0003), and were consistent with the recommendations of the American Veterinary Medical Association and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The mice were allowed to acclimate for at least 7 days before performing any experiment. Immunocompromised NSG mice are susceptible to infection. Thus, the animals were fed a diet containing sulfatrim antibiotic (Envigo). Animals used for fluorescence imaging were fed with AING93 nonfluorescence diet (Envigo).
Therapeutic efficacy of drug–Bdd conjugates for treating bladder cancer
Animals were orthotopically implanted with tumors as described previously (38, 39). Briefly, the urine of 7- to 9-week-old female NSG mice (catalog no. 005557; The Jackson Laboratory) was removed through a sterile 26 G pediatric venous catheter (Covidien). A trypsin solution (0.125%, 80 μL) was then delivered into the bladder. Subsequently, UMUC-3/GFP-Luc cells (4 × 104 cells) in culture media (50 μL) were transferred into the bladders and allowed for seeding. Tumor progression was monitored by bioluminescence imaging using an In Vivo Xtreme imaging system (Bruker). Luciferin (3 mg) in PBS (100 μL) was given to the animals (via intraperitoneal injection) 15 minutes prior to performing imaging. Once tumor progression was confirmed in bladders (based on bioluminescence signal), the animals were randomly assigned for weekly treatment with PBS, DM1, or DM1–Bdd (0.75 mg/kg of drug content) in saline (150 μL) via tail-vein injection for 3 weeks (n = 14/group). Separate groups of animals were assigned for intravesical DM1, DM1–Bdd (0.75 mg/kg of drug content), or MIT (1 mg/mL) in saline (50 μL) treatments (n = 14/group). The therapeutic efficacy was evaluated on the basis of tumor growth inhibition (bioluminescence imaging) and long-term survival. Additional mice were recruited for histopathologic analysis (n = 3/group). These animals were immediately euthanized 1 week after the completion of the treatment schedule. The bladders were harvested and preserved in neutral-buffered formalin (10%). The same experimental conditions were used for comparing the treatment outcomes among animals treated with PBS, aldox, or aldox-Bdd via tail-vein injection or intravesical administration (5 mg/kg of drug content).
Evaluation of DM1–Bdd for kidney cancer treatment
Renca/GFP-Luc cells (4 × 103 cells) in PBS (3 μL) were orthotopically implanted into the kidney capsules of 7- to 9-week-old female BALB/cJ mice (catalog no. 000651, The Jackson Laboratory) as described previously (40). Tumor progression was confirmed by bioluminescence imaging using an In Vivo Xtreme imaging system (Bruker). The animals were randomly assigned into four groups for weekly treatment with PBS, DM1, or DM1–Bdd (0.75 mg/kg of drug content, 150 μL) through tail-vein injection, or DM1–Bdd (0.75 mg/kg of drug content, 50 μL) through intravesical administration (n = 14/treatment). The animals were monitored for tumor growth inhibition and survival as described above. Additional mice were used for histopathologic analysis (n = 4/group).
Histologic analysis
The tissue samples were fixed in 10% neutral-buffered formalin, dehydrated with ethanol, and embedded in paraffin. The tissue sections (5 μm) were stained with hematoxylin and eosin (H&E). For IHC, the bladder sections were deparaffined and then rehydrated before incubation with anti-Ki67 or anti-GFP antibodies (Abcam) overnight. Slides were then counterstained with hematoxylin. High-resolution images were acquired using Aperio 9 Digital Pathology slide scanner (Leica Biosystems).
Necropsy
Seven- to 9-week-old female BALB/cJ mice (catalog no. 000651; The Jackson Laboratory) were treated weekly with PBS, DM1, DM1-peptide (0.75 mg/kg of drug content, 150 μL), or cisplatin (10 mg/kg, 150 μL) via tail-vein injection (n = 4/group). At the end of a 3-week treatment, mice were euthanized. The organs/tissues were harvested, fixed in 10% neutral-buffered formalin for 2 days. Bones were decalcified in a formic acid solution (Surgipath Decalcifier I; Leica Biosystems). The samples were then embedded in paraffin, sectioned (5 μm), and stained with H&E for examination by an ACVP board certified anatomic pathologist.
Nephrotoxicity studies
PBS, DM1, DM1-peptide, MIT, GEM, CIS (0.75 mg/kg of drug content, 150 μL), or a high dose of CIS (10 mg/kg, 150 μL), was administered to BALB/cJ mice (catalog no. 000651; The Jackson Laboratory) via the tail veins (n = 3/group). The level of acute renal injury biomarkers, neutrophil gelatinase-associated lipocalin (NGAL), and kidney injury molecule-1 (KIM-1) in the urine samples was measured using ELISA assays according to the manufacturer's instructions (R&D Systems). The complementary histopathologic analyses were performed on the kidneys of animals treated with PBS, DM1, DM1–Bdd, (0.75 mg/kg of drug content), or a high dose of CIS (10 mg/kg). All the animals (n = 3/group) were euthanized at the end of the treatment and the kidneys were collected and fixed in 10% neutral buffered formalin. The tissues were then processed, embedded in paraffin, sectioned, and stained with H&E. IHC for kidney injury (NGAL and KIM-1) was also performed.
Hematology and biochemistry
Blood samples were collected via cardiac puncture. A complete blood count, including red blood cell, white blood cell, reticulocyte, and platelet, with an automated differential leukocyte count, was performed using an IDEXX Procyte DX hematology analyzer (iDEXX). Blood smears were prepared, stained with modified Wright's stain, examined in a blinded fashion by a board-certified veterinary clinical pathologist (T. Stokol), and imaged using a Nikon Eclipse TE2000-U fluorescent microscope equipped with a photometrics CoolSNAP HQ2 camera (Nikon Corporation). The blood was also centrifuged (1,500 × g) for 15 minutes to obtain the serum for biochemical analysis using a Beckman Coulter AU680 analyzer (Beckman Coulter).
Statistical analysis
Statistical analyses were performed using Graph Pad Prism 6.0 software and R v4.0.5 (R Foundation for Statistical Computing) software. All data were presented as mean ± SD and significances were assigned at *, P < 0.05; **, P < 0.01; ***, P < 0.001. Significant differences between groups were determined using a two-tailed Student t test. For survival evaluation, the Mantel–Cox log-rank test was performed to compare the survival curves of animals treated with intravenous DM1–Bdd or aldox-Bdd to other treatments. P values were adjusted for multiple comparisons using the Benjamini and Hochberg method. All P values were two-sided with statistical significance evaluated at the 0.05 α level. Ninety-five percent (exact) confidence intervals for all parameters were calculated to assess the precision of the obtained estimates.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Bdd can be exclusively eliminated via renal clearance
Bdd was designed in D-configuration to avoid degradation by protease enzymes in the blood circulation (41). It was composed of multiple D-aspartic acid (d) and β-alanine (B) residues (Fig. 1A). The aspartic acids contributed to the overall negative charge, preventing nonspecific uptake by major organs, and promoting renal clearance of the peptide (42, 43). The B residues served as linkers to avoid the formation of a secondary structure. To investigate how the charges were critical for governing Bdd's in vivo behavior, we also synthesized a panel of Bdd analogues that were in L-configuration (BDD), neutral (BKD), carrying positive charges (BKK), and in pegylated form (PEG3DD) for comparative studies. The peptides were labeled with 89Zircronium (89Zr), a long-lived radioisotope (t1/2 = 78 hours), which allowed us to study the PKs and long-term biodistribution (BD) using μPET/CT imaging. The radiolabeled peptides (89Zr-peptides) were synthesized by first conjugating a deferoxamine (DFO) chelator to the peptide in solid phase after amino acid elongation. We then complexed 89Zr with the resulting DFO-peptide conjugates in solution under basic conditions (Fig. 1B).
Figure 1.

Bdd shows remarkable urinary disposing properties. A, A table showing the components and the net charges of BDD as well as its D-configuration (Bdd), neutrally charged (BKD), positively charged (BKK), and pegylated (PEG3DD) counterparts. B, A synthetic scheme showing radiolabeling of the peptides with 89Zr for comparative PK and biodistribution studies. C, Representative μPET/CT images of BALB/cJ mice acquired 1, 4, and 24 hours after administration of different 89Zr-radiolabeled peptides (20 μCi, 20 μg, in 100 μL PBS) via tail-vein injections (n = 4/group). The whole body images were acquired with or without emptying the animal's bladders. D, Bar chart showing the amount of radioactivity in urine samples (20 μL) collected from animals (n = 5) at different time intervals after administration of 89Zr-Bdd. E, Comparing the renal clearance of the radiolabeled peptides. Plots showing the percentage of injected dose (%ID) in kidneys of the animals over time. The percentage of ID was calculated according to the radioactivity measured at the ROI of the acquired PET images. F, Comparing the PK profiles of the radiolabeled peptides (n = 4/analogue). Blood samples (20 μL) were collected at various time intervals after injection of the peptides or free 89Zr to the animals. The results (radioactivity measured in blood samples) were fit into a two-compartmental model for determining the t1/2α (half-life of distribution phase) and t1/2β (half-life of elimination phase). The other PK parameters are available in Supplementary Fig. S1A. G, Endpoint biodistribution study of 89Zr-Bdd. The animals were euthanized at different time intervals (n = 4/time point) after administration of the peptide (20 μCi, 20 μg, in 100 μL PBS). The amount of peptide (radioactivity) in the harvested organs was determined. The results (corrected from decay) were expressed as %ID. Student t test; **, P < 0.01; ***, P < 0.001.
To compare the in vivo biodistribution of the 89Zr-peptides, we injected them into BALB/cJ mice and acquired the whole-body μPET/CT images. Our results showed that 89Zr-Bdd and 89Zr-BDD displayed minimal off-target delivery (Fig. 1C). We could not detect any radioactivity (peptide) in the major organs, except the kidneys. Both peptides were rapidly excreted into the urine within 1 hour after intravenous administration, as confirmed by the absence of radioactivity in the bladders, which were catheterized and emptied prior to imaging. Our results also showed that as much as 80% of the total injected dose (ID) appeared in the first urine sample collected from the animals injected with 89Zr-Bdd (Fig. 1D), suggesting that the peptide was filtered via the glomerulus with minimal reabsorption. On the other hand, the positively charged 89Zr-BKK was delivered to the liver in addition to the URS (Fig. 1C). We also compared renal uptake among the peptide analogues. Except for an initial increase in PEG3DD, they all showed reduced accumulation in kidneys over time (Fig. 1E). To determine the PK profile, we fit our experimental data into a two-compartment model. Our results showed that all the peptide analogues displayed shorter half-lives compared with the free 89Zr (Fig. 1F). 89Zr-Bdd had a terminal half-life of 0.53 hour and rapid plasma clearance (Supplementary Fig. S1A). We further performed an endpoint biodistribution study of 89Zr-Bdd. The results were in good agreement with the imaging and PK studies. As expected, we did not detect any peptide in major organs or blood circulation 4 hours after intravenous injection (Fig. 1G; Supplementary Fig. S1B). Despite the rapid clearance, 15.1% of 89Zr-Bdd's ID was found in kidneys, which decreased to 2.1% after 7 days.
As a carrier of hydrophobic molecules
Most chemotherapeutics are hydrophobic molecules that display unfavorable PK and BD, leading to off-target delivery and undesired toxicity. We foresaw that the Bdd's rapid renal clearance could be beneficial for promoting drug delivery to the URS. To demonstrate that, we covalently attached a hydrophobic Cyanine5.5 fluorophore (Cy) as a drug model to the peptide analogues (Fig. 2A). We then injected the resulting conjugates (Cy-peptides) into hairless SHO mice for comparative delivery studies using optical imaging. Both Cy-Bdd and Cy-BDD could be rapidly eliminated via renal clearance. They reached the animals' bladders 1 hour after intravenous injections (Fig. 2B), with as much as 70–75% ID present in the urine (Fig. 2C). On the other hand, Cy-BKD, Cy-BKK, Cy-PEG3DD, and free Cy were mainly taken up by liver and/or other organs, and slowly excreted from the body through renal clearance. We also performed ex vivo imaging of the harvested organs (Fig. 2D). As expected, the accumulation of Cy-Bdd or Cy-BDD in kidneys was minimal (Fig. 2E). Cy-PEG3DD showed a prolonged retention in kidneys (Figs. 2D and E), which might explain the delayed renal excretion profile compared with the other peptide conjugates (Fig. 2C). The results were also in good agreement when using PEG3DD to deliver 89Zr (Fig. 1E). Overall, Bdd could be used for delivering hydrophobic molecules, such as Cy, to the URS while maintaining the comprehensive UDD properties. Unlike 89Zr-Bdd, trace amounts of Cy-Bdd and Cy-BDD were taken up by the stomach, liver, and intestines. The biodistribution differences were likely attributed to the replacement of a negatively charged 89Zr-DFO by the hydrophobic Cy. Fortunately, the Cy-Bdd's off-target accumulation was quickly eliminated by hepatic clearance within 24 hours (data not shown). However, further studies are needed to fine-tune the physicochemical properties (such as the surface charges) of Bdd and improve the urinary disposing efficiency.
Figure 2.

Bdd effectively distributes the conjugated Cyanine5.5 fluorophore to the URS. A, A synthetic scheme of the Cyanine5.5-labeled peptide (Cy-peptide) analogues. B, Representative merged fluorescence/bright light images of SHO mice acquired 1 and 4 hours after tail-vein injection of the different Cy-peptide analogues (0.5 nmol, 150 μL) or free Cyanine5.5 (n = 4/group). C, Plots comparing the amount of fluorophore in urine (% of injected dose) based on the measured fluorescence. Bottom, fluorescence image of the urine samples (20 μL) collected from the same animals 1, 4, and 6 hours after the Cy-peptides or fluorophore administration (n = 4/group). For each time point, the bladders of the animals were completely emptied using a sterile 26 G pediatric venous catheter. D, Representative ex vivo merged fluorescence/bright light images of the organs harvested from animals 4 hours after tail-vein injection of different Cy-peptides (n = 4/group). E, Bar chart comparing peptide distribution in the harvested organs (n = 4/group), based on the total fluorescence intensity. Student t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., nonsignificant.
As carrier of chemotherapeutics
The Bdd's excellent UDD properties prompted us to further investigate it for delivering chemotherapeutics for NMIBC treatment. Many peptides are immunogenic (44, 45). We first confirmed that the Bdd peptide did not trigger innate immune responses. There was no increase of inflammatory cytokine levels (IL1β, IL2, IL6, IL10, TNFα, and INFγ) in the plasma of BALB/cJ mice 4 hours after intravenous injection (Fig. 3A). Using the fluorescence Cy-Bdd, we further confirmed that the peptide could be taken up by human BC (UMUC-3) and murine kidney cancer (Renca) cells (Fig. 3B). We observed overlapping of the peptide (red) and lysosome (green) fluorescence, suggesting that cellular uptake of the peptide mainly occurred via endocytosis. As proof of principle, we selected emtansine (DM1), a potent microtubule inhibitor, as a drug candidate. Compared with other conventional chemotherapeutics, DM1 was more potent than CIS, DOX, and MIT, and as effective as GEM against UMUC-3, with IC50 values at the nanomolar range (Fig. 3C). The drug was also effective against the Renca cell line. Next, we synthesized a DM1–Bdd conjugate by attaching DM1 to Bdd via a cleavable disulfide linker (Fig. 3D), enabling drug release in a reducing environment, such as in the presence of intracellular glutathione (GSH; Fig. 3E; refs. 46, 47). We also established the chemistry for conjugating other chemotherapeutics, such as aldox, to Bdd (Fig. 3F). Aldox is a DOX derivative modified with a hydrazone linker, which is known to be sensitive to the acidic lysosome and tumor microenvironment. The drug release from the aldox–Bdd conjugate was pH-dependent (Fig. 3G). In terms of cytotoxicity, both DM1–Bdd and aldox–Bdd exhibited a similar potency to the corresponding free drugs against different murine (MB49) and human (UMUC-3 and T24) bladder cancer cell lines, as well as murine (Renca) kidney cancer cell line (Fig. 3H and I).
Figure 3.

Bdd as a carrier of chemotherapeutics. A, Bdd does not trigger any innate immune response. No increase in the inflammatory cytokines concentrations was detected in the plasma of female BALB/cJ mice (n = 3/group) 4 hours after intravenous administration of the Bdd peptide (5 mg/kg, 150 μL). LPS was used as a positive control and concentrations of each cytokine were measured by ELISA Kit. B, Cellular uptake of Cyanine5.5-labeled Bdd (Cy-Bdd). Representative fluorescence microscopic images of human UMUC-3 BC cells and murine Renca renal adenocarcinoma cells incubated for 6 and 24 hours with Cy-Bdd (0.5 nmol). Dapi (9 μmol/L) and LysoTracker-GFP (1 μmol/L) were used for nuclear (blue) and organelle (green) staining, respectively, and were added to the cells 30 minutes prior to imaging. Scale bar, 25 μm. C, Comparing the potency of different chemotherapeutics (DM1, GEM, MIT, CIS, and DOX). UMUC-3 and Renca cells were incubated with the drugs at various concentrations for 72 hours prior to measuring the cell viability. The dose–response curves were plotted and the IC50 values of each drug calculated using Graph Pad Prism 6.0 software. D, Conjugation of DM1 to Bdd. The cleavable linker SPDP was first conjugated to the peptide N-terminal in solid phase. DM1 was then added to the cleaved peptide in a solution mixture of PBS and NMP. E, Plot showing the percentage of accumulated DM1 released from the DM1–Bdd over time in PBS in the absence and presence of GSH (1 mmol/L). The amount of drug released was quantified using rp-HPLC analysis (absorbance detected at 254 nm). F, Conjugation of aldox to Bdd. The peptide, supplemented with a N-terminal cysteine, was incubated with aldox in PBS (pH 7.4) for 30 minutes prior to purification by rp-HPLC in neutral conditions. G, Plots showing the percentage of the accumulated DOX active metabolite released from aldox–Bdd (100 μmol/L) over time in PBS buffers with different pH values. The amount of drug released was quantified using rp-HPLC analysis (absorbance detected at 480 nm). H, DM1–Bdd displays a similar cytotoxicity compared with free drug against murine bladder (MB49), human bladder (UMUC-3 and T24), and murine kidney (Renca) cancer cell lines. Plots of relative cell viability against the drug concentration. I, Aldox–Bdd is more potent than free aldox. Plots of the relative cell viability against the drug concentration. *, P < 0.05; **, P < 0.01; n.s., nonsignificant.
A more effective alternative to conventional ITC
Unlike with intravesical administration, animals did not need to void shortly after intravenous injection (Fig. 4A). DM1–Bdd, administered through intravenous injection, should prolong the drug's bladder-dwelling time. This, together with the unique UDD properties, should offer a more effective (compared with ITC) and safer (compared with systemic chemotherapy) therapeutic option when using Bdd as a drug carrier. However, Bdd could temporarily accumulate in kidneys (Fig. 1C and G). Therefore, prior to evaluating the therapeutic efficacy of DM1–Bdd, we first assessed the tolerability of the kidneys to a single injectable dose in healthy BALB/cJ mice. Our results showed that DM1–Bdd slightly increased levels of urinary tubular injury markers, KIM-1, and NGAL, in the urine 1 day after the injection (Fig. 4B). However, the increases were minimal and transient, returning to basal levels within 3 days. Histologic examination at 3 days after drug administration did not reveal any abnormalities in the kidneys, with no elevation of the immunoreactivity of the tissue sections for KIM-1 and NGAL, when compared with PBS (Fig. 4C). In contrast, other intravenous chemotherapeutics, including DM1, MIT, CIS, and GEM, showed a continued induction of KIM-1 and/or NGAL at this time point. We also found dilation and degeneration of tubular epithelium in animals treated with a high dose of intravenous CIS (positive control; Fig. 4C).
Figure 4.

Therapeutic efficacy of DM1–Bdd in treating bladder cancer. A, Bar chart comparing the time when BALB/cJ mice (n = 20/group) needed to void naturally following intravenous or intravesical administration of PBS (80 μL). The bladders were emptied prior to starting the experiment. Each animal was isolated for monitoring the urination pattern. B, Comparing the nephrotoxicity of DM1–Bdd to other chemotherapeutics. Bar chart showing the concentrations of renal injury biomarkers, NGAL and KIM-1, in urine collected from animals 1, 3, and 7 days after treatment with PBS, DM1 (0.75 mg/kg), DM1–Bdd (0.75 mg/kg of drug content), MIT (0.75 mg/kg), CIS (0.75 mg/kg), CIS (10 mg/kg), or GEM (0.75 mg/kg) via tail-vein injection (150 μL). Student t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001. C, Immunochemical staining for NGAL and KIM-1 was also performed. Representative microscopic images of kidney sections from BALB/cJ mice intravenously administered with PBS (150 μL), DM1 (0.75 mg/kg, 150 μL), DM1–Bdd (0.75 mg/kg of drug content, 150 μL), or CIS (10 mg/kg, 150 μL) as a positive control. The organs were harvested 3 days after the drug treatments and stained with H&E. Black arrows indicate the multifocal degeneration of the tubular epithelium after treatment with CIS. Red and green arrows indicate renal tubular epithelial cells immunoreactive for NGAL and KIM-1, respectively, following CIS treatment. Scale bar, 50 μm. D, Brightfield and fluorescence images of UMUC-3/GFP-Luc cells that were stably transduced with a lentivirus carrying both GFP and firefly luciferase genes. Scale bar, 80 μm. E, Orthotopic xenograft model. Representative image of bladders collected from female NSG mice (n = 3) 1 week after implantation of UMUC-3/GFP-Luc cells (4 × 104 cells/animal). Black arrows, tumors growing in the lamina propia. Scale bar, 500 μm. F, Representative merged bioluminescence/brightfield images of the tumor-bearing NSG mice after weekly treatments with intravenous PBS (150 μL), intravenous DM1 (0.75 mg/kg, 150 μL), intravenous DM1–Bdd (0.75 mg/kg of drug content, 150 μL), intravesical MIT (1 mg/mL, 50 μL), intravesical DM1 (0.75 mg/kg, 50 μL), or intravesical DM1–Bdd (0.75 mg/kg, 50 μL) for 3 weeks (n = 10/group). Images were acquired every week to monitor and compare tumor growth in each treatment group. G, Representative pictures of bladders excised from each animal group (additional recruitment of n = 3/treatment group) 1 week after completing the treatment cycles. H, Kaplan–Meier cumulative survival plot of animals administered with different drugs (n = 14/group). The significant differences in survival between the animals treated with intravenous DM1–Bdd and the other groups was evaluated using the Mantel–Cox log-rank test and the Benjamini and Hochberg adjusted P values. I and J, Representative image(s) of bladder sections from the animals in each treatment group (n = 3/group). The organs were harvested at the end of the 3-week treatment and then paraffin-embedded, sectioned, and stained with H&E (I) and Ki67 (proliferation marker; J).
We then evaluated DM1–Bdd for treating NSG mice bearing orthotopic human UMUC-3 tumors. Prior to implanting UMUC-3 cells into the animals' bladders, we stably transduced them with a dual luciferase and GFP reporter (Fig. 4D). This allowed us to monitor the disease progression with bioluminescence imaging (39). We also confirmed that the established tumors, growing in the lamina propia, were noninvasive in nature and limited to the urinary bladder submucosa (Fig. 4E). Compared with the clinically used ITC (i.t. MIT), both intravenous DM1 and DM1–Bdd were more effective in inhibiting tumor growth (Fig. 4F–G; Supplementary Figs. S2A–S2E) and prolonged animal survival (Fig. 4H). An intravenous injection of DM1–Bdd provided a better treatment outcome when compared with intravesical administration. According to the imaging data acquired during the 3 once per week treatments course, the antitumor activity of intravenous DM1–Bdd and DM1 was similar (Fig. 4F). However, those animals treated with intravenous DM1–Bdd showed a significant improvement in overall survival (36% vs. 0% survived after 100 days). In a separate experiment, we confirmed the therapeutic effect on tumor reduction by histology. Tumors of the animals treated with intravenous DM1–Bdd were smaller (Fig. 4I). IHC revealed fewer GFP-positive cells and lower proportion of neoplastic cells expressing the proliferation marker (Ki67), confirming the inhibition of tumor cell growth and proliferation (Fig. 4J; Supplementary Fig. S2F). More importantly, DM1–Bdd treatment was curative for 21% of the animals. The survivor mice lacked gross and histologic evidence (using GFP and Ki67 immunostaining) of tumors, suggesting they were disease-free after 210 days (Supplementary Fig. S2G). Bdd is a versatile delivery platform that can carry different chemotherapeutics. Aldox–Bdd was also able to prolong the animal survival compared with PBS and free DOX (Supplementary Figs. S3A–S3C). However, it was not as effective as DM1–Bdd in improving overall survival (Supplementary Fig. S3C). Surprisingly, mice treated with free DOX had a shorter life expectancy compared with the PBS control, with the animals continuously losing weight during DOX treatment (Supplementary Figs. S3D and S3E). They eventually died prior to receiving the final dose, suggesting drug-induced toxicity and mortality.
Anatomic flexibility
Next, we evaluated DM1–Bdd for treating renal carcinoma. We used a syngeneic mouse model for the studies, which involved a surgical implantation of Renca cells (stably transduced with GFP and Luc) into the capsule of the right kidney of BALB/cJ mice (Fig. 5A). The tumor growth was aggressive. A tumor mass of a substantial size developed as early as 1 week after implantation (Fig. 5B). Our imaging studies showed that intravenous DM1–Bdd was not only able to inhibit tumor progression (Fig. 5C and D) but also reduce tumor size, as shown by decreased bioluminescence signals in most of the animals during the treatment (Fig. 5E). There was 50% of survival after 160 days (Figs. 5F and G). On the other hand, all animals treated with either intravenous or intravesical DM1 died. In a separate experiment, we performed histologic examination of the animals' kidneys 1 week after completing the treatment course (Fig. 5H). Minimal tumor was present in animals treated with intravenous DM1–Bdd, whereas tumors of animals treated with DM1 or PBS control were large and accompanied with an infiltration of mononuclear cells. Overall, our UDD approach was anatomically flexible and could be used for treating tumors located at the upper urinary tract.
Figure 5.

Therapeutic efficacy of DM1–Bdd in treating renal carcinoma. A, Brightfield and fluorescence images of Renca cells that were stably transduced with a lentivirus carrying both GFP and firefly luciferase genes. Scale bar, 80 μm. B, Syngeneic xenograft model. Representative image of the histologic analysis of kidneys collected from female BALB/cJ mice (n = 3) 1 week after implantation of murine Renca cells (4 × 103 cells/animal) in the renal capsules (black arrow). Scale bar, 1 mm. C, Representative merged bioluminescence/brightfield images of BALB/cJ mice bearing Renca/GFP-Luc tumors after treatment with intravenous PBS (150 μL), intravenous DM1 (0.75 mg/kg, 150 μL), intravenous DM1–Bdd (0.75 mg/kg of drug content, 150 μL), or intravesical DM1 (0.75 mg/kg, 50 μL) weekly for 3 weeks (n = 10/group). D, Representative photos of the kidneys excised from the animals after completion of the treatment cycle (additional n = 4/group). E–G, Longitudinal comparisons of bioluminescence signals at the region of interest (ROI = kidney; E), body weight (F), and survival (G) among animals receiving different treatments (n = 14/group). The significant differences in survival between animals treated with intravenous DM1–Bdd and the drugs was evaluated using the Mantel–Cox log-rank test and the Benjamini and Hochberg adjusted P values. ***, P < 0.001. H, Representative kidney sections from animals of each treatment group (additional n = 4/group). The sections were stained with H&E. The green, yellow, black, and blue arrows indicate the presence of pigment-laden macrophage, focal mineralization, interstitial fibrosis, and mononuclear cell infiltrates, respectively. Scale bars, 2 mm and 50 μm.
Toxicity profile
Finally, we assessed the toxicity profile of DM1–Bdd in healthy BALB/cJ mice after completing 3 weeks of weekly treatments. DM1–Bdd did not affect the red blood cell, leukocyte, or platelet counts or morphologic features (Fig. 6A and B; Supplementary Fig. S4). On the other hand, DM1-induced reticulocytosis without apparent anemia, and an inflammatory response characterized by increased proportions of neutrophils, monocytes, and platelets. The positive control CIS was also toxic, as evidenced by thrombocytopenia and lymphopenia. We also performed serum biochemical analysis to assess for liver injury and renal function. DM1–Bdd was not hepatotoxic, with no significant alteration in the release of ALP, ALT, and AST enzymes (Fig. 6C; Supplementary Fig. S5). Importantly, the urea nitrogen/creatinine ratio was normal, suggesting that DM1–Bdd did not affect renal clearance of nitrogenous waste. Histopathologic studies further confirmed that there was no morphologic evidence of injury in the liver, spleen, heart, lungs, or kidneys of the animals treated with DM1–Bdd (Fig. 6D; Supplementary Fig. S6). In contrast, the biochemical testing showed that both DM1 and CIS induced hepatic and renal toxicity, as shown by increases in ALT and AST activities and BUN/CREA ratios (Fig. 6C). They also induced muscle injury (CK activity), which would partly explain the increases in the AST activity. Histologic examination also revealed that CIS caused degeneration and necrosis of the proximal renal tubules, lung inflammation, and depletion of both erythrocytes and extramedullary hematopoiesis (EMH) in spleen (Fig. 6D; Supplementary Fig. S6). Animals treated with DM1 showed similar renal damage but to a lesser degree. We also observed increased splenic and hepatic EMH with DM1, which may explain the observed reticulocytosis in the treated animals.
Figure 6.

DM1–Bdd displays a safe toxicity profile. A, Representative microscopic images of blood smears collected from female BALB/cJ mice after intravenous administration of PBS (150 μL), DM1 (0.75 mg/kg, 150 μL), DM1–Bdd (0.75 mg/kg of drug content, 150 μL), or CIS (10 mg/kg, 150 μL) as a positive control, weekly for 3 weeks. Black arrows, polychromatophilic macrocytes. Scale bar, 10 μm. B, Selected hematologic results obtained 1 week after completing the different treatment courses. RBC, red blood cells; WBC, white blood cells. Student t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001. C, Comparison of selected serum biochemical analytes, including liver enzyme activity (ALP, ALT, and AST), muscle enzyme activity (AST and CK), and clearance of nitrogenous waste (BUN/CREA ratio). ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK, creatine kinase; BUN, blood urea nitrogen; CREA, creatinine. D, Histopathologic analysis of the major organs (liver, spleen, heart, lungs, and kidneys) from animals administered with the different drug treatments. Black arrows indicate the increased of hepatocyte mitotic activity in liver. Blue arrows show the enhanced hepatic and splenic EMH. The area in between the white arrows indicates depletions of erythrocytes and EMH elements in the red pulp of the spleen. Red arrows highlight the presence of large and foamy macrophages in the alveoli. Yellow and green arrows indicate flattened renal tubular cells and necrotic sloughed debris of dying cells contained within the lumen, respectively. Scale bar, 30 μm.
Discussion
Chemotherapeutics gain some selectivity by targeting tumor cells that proliferate rapidly (48). However, most of them are still toxic to both healthy and cancer cells. They are given by infusion so that they can be continuously administered at a higher total dose over a longer period of time. The goal is to achieve more effective treatment to improve patient tolerance to off-target toxicities by maintaining the drug's plasma concentration at a certain level and prolonging the tumor exposure to the drug. In this study, we aimed to promote, rather than reduce, a drug's clearance as a noninvasive alternative of ITC. Many bioactive peptides have been approved for treating various diseases, including cancer, diabetes, and cardiovascular disease (49). Without chemical modifications, peptides have a short circulating half-life of several minutes. They are rapidly degraded by protease enzymes (29–31) and eliminated by renal filtration (50). We recognized that a peptide's rapid renal clearance could be advantageous as a drug carrier to dispose most systemically administered drugs in urine for treating NMIBC and reducing the unwanted systemic side-effects. We introduced a UDD approach using a bio-inert, negatively charged peptide (Bdd) that was uptaken minimally by the recticuloendothelial system and other organs, and was exclusively excreted into the urine. We employed Bdd for delivering DM1, a microtubule inhibitor. We selected DM1 as it was 100-fold more potent than commonly used bladder cancer drugs, including MIT, DOX, and CIS, against a panel of bladder cancer cell lines. Further, conjugating the drug to the Bdd peptide did not compromise cytotoxicity.
In terms of therapeutic efficacy, intravenous administered DM1-peptide conjugate (DM1–Bdd) improved overall survival in mice with bladder cancer compared with conventional intravesical MIT (Fig. 4H). It was also more effective compared with the same treatment given intravesically (Figs. 4F–I). The improved efficacy was expected given that animals did not need to void shortly after intravenous injection (Fig. 4A). Promoting renal clearance could reduce a drug's off-target toxicity. DM1–Bdd did not induce unwanted toxicity (Fig. 6). In contrast, animals treated with DM1 or CIS showed evidence of hepatic and renal injury. We also highlighted the flexibility of the UDD approach in reducing drug-induced toxicity by using aldox–Bdd for bladder cancer treatment. Unlike free DOX, which caused mortality, we observed improved survival in animals treated with aldox–Bdd (Supplementary Fig. S3). However, compared with DM1–Bdd, the aldox–Bdd treatment only slowed down the cancer progression and did not eliminate tumors in individual animals. The result was expected because DM1 is more potent than DOX (Fig. 3C).
ITC is a local treatment that only covers tumors in the bladder as the drug solution cannot reach the upper urinary tract. DM1–Bdd is administered through intravenous injection. This, together with the rapid renal clearance, should allow drugs to flush the entire URS. We applied DM1–Bdd to renal carcinomas and found that DM1–Bdd offered a significant survival benefit compared with the free drug. In fact, approximately 50% of the animals lacked gross or histologic evidence of the tumor 1 week after completing the treatment course. Intravenous drug administration will thus allow more comprehensive coverage of the URS when used for treating bladder cancer, as tumors can extend, migrate into, or recur throughout the entire urothelium, including the renal pelvis and the ureter from which 8% to 12% of the urothelial carcinomas originate (51). Currently, when treating patients with renal pelvic or ureteral tumors, surgeons are often left with no choice but to remove the entire kidney and ureter (even when the tumors are noninvasive) to prevent disease recurrence for URS tumors. We foresee that DM1–Bdd can potentially be a kidney-sparing treatment option for patients with upper tract urothelial cancers.
A drawback of IT is its poor patient compliance rate (16%–30%; ref. 11). Patients receiving ITC need to be catheterized weekly by trained personnel in hospital/clinic. Our intravenous DM1–Bdd treatment is minimally invasive, so can avoid complications associated with catheterization procedures and improve patient's quality of life and compliance. The lifetime management of bladder cancer is costly (52), and requires repeated treatments due to a high recurrence rate. Systemic administration of a chemotherapeutic, such as DM1–Bdd, will likely reduce bladder cancer treatment costs.
Overall, we developed a UDD approach that could minimize nonspecific accumulation in other organs, and offer a comprehensive treatment by supplying drug to the entire URS, as a more effective alternative to ITC. The developed DM1–Bdd is clinically translatable. The FDA has approved many peptides for treating different cancers. The employed DM1 is an active pharmacophore already used in antibody–drug conjugates, such as Herceptin-DM1 (T-DM1), for breast cancer treatment. Furthermore, the same approach of drug to peptide conjugation can be applied to treating other diseases, such as kidney and bladder infections, simply by replacing cytotoxic drugs with antibiotics.
Supplementary Material
Acknowledgments
Research reported in this publication was supported in part by the NCI (CA222802 to B. Law) and the Radiology Department, WCM. The authors acknowledge the Laboratory of Comparative Pathology Core Facility (MSKCC), partially funded by NCI grant P30 CA008748, for the preparation of the organ sections for histologic and fluorescence analyses, as well as the Department of Pathology and Laboratory Medicine (WCM) for their help with the slice scanning of the histopathologic samples. They also thank Elizabeth Sweeney and Arindam Roy Choudhury for assisting with the statistical analyses of the survival studies. The authors express their gratitude to those anonymous patients for their financial support of the studies. Finally, the authors thank Cynthia Fox for the editing of this manuscript.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
V. Bellat reports a patent for Peptide-linked drug delivery system pending. A.O. Michel reports employment with Regeneron Pharmaceuticals Inc. C. Thomas reports grants from Clinical and Translational Science Center at Weill Cornell Medical College outside the submitted work. B.B. Choi reports a patent for Peptide-linked drug delivery system pending. B. Law reports a patent for Peptide-linked drug delivery system pending. No disclosures were reported by the other authors.
Authors' Contributions
V. Bellat: Conceptualization, data curation, formal analysis, validation, investigation, methodology, writing–original draft, writing–review and editing. A.O. Michel: Investigation, methodology. C. Thomas: Formal analysis. T. Stokol: Investigation, methodology. B.B. Choi: Conceptualization. B. Law: Conceptualization, resources, supervision, funding acquisition, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Williams SB, Howard LE, Foster ML, Klaassen Z, Sieluk J, De Hoedt AM, et al. Estimated costs and long-term outcomes of patients with high-risk non-muscle-invasive bladder cancer treated with Bacillus Calmette Guerin in the veterans affairs health system. JAMA Netw Open 2021;4:e213800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lenis AT, Lec PM, Chamie K, Mshs MD. Bladder cancer: a review. JAMA 2020;324:1980–91. [DOI] [PubMed] [Google Scholar]
- 3. Sylvester RJ, Oosterlinck W, Holmang S, Sydes MR, Birtle A, Gudjonsson S, et al. Systematic review and individual patient data meta-analysis of randomized trials comparing a single immediate instillation of chemotherapy after transurethral resection with transurethral resection alone in patients with stage pTa-pT1 urothelial carcinoma of the bladder: which patients benefit from the instillation? Eur Urol 2016;69:231–44. [DOI] [PubMed] [Google Scholar]
- 4. Huncharek M, Geschwind JF, Witherspoon B, McGarry R, Adcock D. Intravesical chemotherapy prophylaxis in primary superficial bladder cancer: a meta-analysis of 3703 patients from 11 randomized trials. J Clin Epidemiol 2000;53:676–80. [DOI] [PubMed] [Google Scholar]
- 5. Griffin JG, Holzbeierlein J. Side effects of perioperative intravesical treatment and treatment strategies for these side effects. Urol Clin North Am 2013;40:197–210. [DOI] [PubMed] [Google Scholar]
- 6. Addeo R, Caraglia M, Bellini S, Abbruzzese A, Vincenzi B, Montella L, et al. Randomized phase III trial on gemcitabine versus mytomicin in recurrent superficial bladder cancer: evaluation of efficacy and tolerance. J Clin Oncol 2010;28:543–8. [DOI] [PubMed] [Google Scholar]
- 7. Messing EM, Tangen CM, Lerner SP, Sahasrabudhe DM, Koppie TM, Wood DP Jr, et al. Effect of intravesical instillation of gemcitabine vs saline immediately following resection of suspected low-grade non-muscle-invasive bladder cancer on tumor recurrence: SWOG S0337 randomized clinical trial. JAMA 2018;319:1880–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cambier S, Sylvester RJ, Collette L, Gontero P, Brausi MA, van Andel G, et al. EORTC nomograms and risk groups for predicting recurrence, progression, and disease-specific and overall survival in non-muscle-invasive stage Ta-T1 urothelial bladder cancer patients treated with 1–3 years of maintenance Bacillus Calmette-Guerin. Eur Urol 2016;69:60–9. [DOI] [PubMed] [Google Scholar]
- 9. Schmittgen TD, Wientjes MG, Badalament RA, Au JL. Pharmacodynamics of mitomycin C in cultured human bladder tumors. Cancer Res 1991;51:3849–56. [PubMed] [Google Scholar]
- 10. De Bruijn EA, Sleeboom HP, van Helsdingen PJ, van Oosterom AT, Tjaden UR, Maes RA. Pharmacodynamics and pharmacokinetics of intravesical mitomycin C upon different dwelling times. Int J Cancer 1992;51:359–64. [DOI] [PubMed] [Google Scholar]
- 11. Tapiero S, Helfand A, Kedar D, Yossepowitch O, Nadu A, Baniel J, et al. Patient compliance with maintenance intravesical therapy for nonmuscle invasive bladder cancer. Urology 2018;118:107–13. [DOI] [PubMed] [Google Scholar]
- 12. Filson CP, Montgomery JS, Dailey SM, Crossley HS, Lentz H, Tallman CT, et al. Complications associated with single-dose, perioperative mitomycin-C for patients undergoing bladder tumor resection. Urol Oncol 2014;32:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tully KH, Cole AP, Krimphove MJ, Friedlander DF, Mossanen M, Herzog P, et al. Contemporary treatment patterns for non-muscle invasive bladder cancer: has the use of radical cystectomy changed in the BCG shortage era? Urology 2020;20:30983–3. [DOI] [PubMed] [Google Scholar]
- 14. Desouky E. BCG versus COVID-19: impact on urology. World J Urol 2021;39:823–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Packiam VT, Werntz RP, Steinberg GD. Current clinical trials in non-muscle-invasive bladder cancer: heightened need in an era of chronic BCG shortage. Curr Urol Rep 2019;20:84. [DOI] [PubMed] [Google Scholar]
- 16. Grimberg DC, Shah A, Inman BA. Overview of Taris GemRIS, a novel drug delivery system for bladder cancer. Eur Urol Focus 2020;6:620–2. [DOI] [PubMed] [Google Scholar]
- 17. Kowalski M, Guindon J, Brazas L, Moore C, Entwistle J, Cizeau J, et al. A phase II study of oportuzumab monatox: an immunotoxin therapy for patients with noninvasive urothelial carcinoma in situ previously treated with Bacillus Calmette-Guerin. J Urol 2012;188:1712–8. [DOI] [PubMed] [Google Scholar]
- 18. Shore ND, Boorjian SA, Canter DJ, Ogan K, Karsh LI, Downs TM, et al. Intravesical rAd-IFNalpha/Syn3 for patients with high-grade, Bacillus Calmette-Guerin-refractory or relapsed non-muscle-invasive bladder cancer: a phase II randomized study. J Clin Oncol 2017;35:3410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Du BJ, Jiang XY, Das A, Zhou QH, Yu MX, Jin RC, et al. Glomerular barrier behaves as an atomically precise bandpass filter in a sub-nanometre regime. Nat Nanotechnol 2017;12:1096–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ni D, Jiang D, Im HJ, Valdovinos HF, Yu B, Goel S, et al. Radiolabeled polyoxometalate clusters: kidney dysfunction evaluation and tumor diagnosis by positron emission tomography imaging. Biomaterials 2018;171:144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burns AA, Vider J, Ow H, Herz E, Penate-Medina O, Baumgart M, et al. Fluorescent silica nanoparticles with efficient urinary excretion for nanomedicine. Nano Lett 2009;9:442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J, Yu M, Zhou C, Yang S, Ning X, Zheng J. Passive tumor targeting of renal-clearable luminescent gold nanoparticles: long tumor retention and fast normal tissue clearance. J Am Chem Soc 2013;135:4978–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Audige A, Frick C, Frey FJ, Mazzucchelli L, Odermatt A. Selection of peptide ligands binding to the basolateral cell surface of proximal convoluted tubules. Kidney Int 2002;61:342–8. [DOI] [PubMed] [Google Scholar]
- 24. Odermatt A, Audige A, Frick C, Vogt B, Frey BM, Frey FJ, et al. Identification of receptor ligands by screening phage-display peptide libraries ex vivo on microdissected kidney tubules. J Am Soc Nephrol 2001;12:308–16. [DOI] [PubMed] [Google Scholar]
- 25. Geng Q, Sun X, Gong T, Zhang ZR. Peptide-drug conjugate linked via a disulfide bond for kidney targeted drug delivery. Bioconjug Chem 2012;23:1200–10. [DOI] [PubMed] [Google Scholar]
- 26. Wischnjow A, Sarko D, Janzer M, Kaufman C, Beijer B, Brings S, et al. Renal targeting: peptide-based drug delivery to proximal tubule cells. Bioconjug Chem 2016;27:1050–7. [DOI] [PubMed] [Google Scholar]
- 27. Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol 2002;3:256–66. [DOI] [PubMed] [Google Scholar]
- 28. Lenhard SC, McAlexander A, Virtue A, Fieles W, Skedzielewski T, Rambo M, et al. In vivo imaging of small molecular weight peptides for targeted renal drug delivery: a study in normal and polycystic kidney diseased mice. J Pharmacol Exp Ther 2019;370:786–95. [DOI] [PubMed] [Google Scholar]
- 29. Bottger R, Hoffmann R, Knappe D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS One 2017;12:e0178943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barrett TM, Chen XS, Liu C, Giannakoulias S, Phan HAT, Wang J, et al. Studies of thioamide effects on serine protease activity enable two-site stabilization of cancer imaging peptides. ACS Chem Biol 2020;15:774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aneja R, Grigoletto A, Nangarlia A, Rashad AA, Wrenn S, Jacobson JM, et al. Pharmacokinetic stability of macrocyclic peptide triazole HIV-1 inactivators alone and in liposomes. J Pept Sci 2019;25:e3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bellat V, Lee HH, Vahdat L, Law B. Smart nanotransformers with unique enzyme-inducible structural changes and drug release properties. Biomacromolecules 2016;17:2040–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bellat V, Ting R, Southard TL, Vandat L, Molina H, Fernandez J, et al. Functional peptide nanofibers with unique tumor targeting and enzyme-induced local retention properties. Adv Funct Mater 2018;28:1803969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Law B, Weissleder R, Tung CH. Peptide-based biomaterials for protease-enhanced drug delivery. Biomacromolecules 2006;7:1261–5. [DOI] [PubMed] [Google Scholar]
- 35. Law B, Weissleder R, Tung CH. Protease-sensitive fluorescent nanofibers. Bioconjug Chem 2007;18:1701–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benvenuto JA, Anderson RW, Kerkof K, Smith RG, Loo TL. Stability and compatibility of antitumor agents in glass and plastic containers. Am J Hosp Pharm 1981;38:1914–8. [PubMed] [Google Scholar]
- 37. Curry D, Scheller H, Lu M, Mkandawire M, Servos MR, Cui S, et al. Prevention of doxorubicin sorptive losses in drug delivery studies using polyethylene glycol. RSC Adv 2015;5:25693–8. [Google Scholar]
- 38. Huebner D, Rieger C, Bergmann R, Ullrich M, Meister S, Toma M, et al. An orthotopic xenograft model for high-risk non-muscle invasive bladder cancer in mice: influence of mouse strain, tumor cell count, dwell time and bladder pretreatment. BMC Cancer 2017;17:790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kasman L, Voelkel-Johnson C. An orthotopic bladder cancer model for gene delivery studies. J Vis Exp 2013;82: 50181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murphy KA, James BR, Wilber A, Griffith TS. A syngeneic mouse model of metastatic renal cell carcinoma for quantitative and longitudinal assessment of preclinical therapies. J Vis Exp 2017;122:55080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feng Z, Xu B. Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol Concepts 2016;7:179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol 2015;33:941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang Y, Jiang K, Zhang X, Chung EJ. The effect of size, charge, and peptide ligand length on kidney targeting by small, organic nanoparticles. Bioeng Transl Med 2020;5:e10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Flower DR. Designing immunogenic peptides. Nat Chem Biol 2013;9:749–53. [DOI] [PubMed] [Google Scholar]
- 45. Van Regenmortel MH. Antigenicity and immunogenicity of synthetic peptides. Biologicals 2001;29:209–13. [DOI] [PubMed] [Google Scholar]
- 46. Wagh A, Law B. Methods for conjugating antibodies to nanocarriers. Methods Mol Biol 2013;1045:249–66. [DOI] [PubMed] [Google Scholar]
- 47. Pillow TH, Sadowsky JD, Zhang D, Yu SF, Del Rosario G, Xu K, et al. Decoupling stability and release in disulfide bonds with antibody-small molecule conjugates. Chem Sci 2017;8:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell 2012;23:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Usmani SS, Bedi G, Samuel JS, Singh S, Kalra S, Kumar P, et al. THPdb: database of FDA-approved peptide and protein therapeutics. PLoS One 2017;12:e0181748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu H, Huang J. Optimization of protein and peptide drugs based on the mechanisms of kidney clearance. Protein Pept Lett 2018;25:514–21. [DOI] [PubMed] [Google Scholar]
- 51. Transitional cell cancer of the renal pelvis and ureter treatment (PDQ(R)): Health Professional Version. PDQ Cancer Information Summaries. Bethesda (MD)2002. [Google Scholar]
- 52. Kaye DR, Min HS, Herrel LA, Dupree JM, Ellimoottil C, Miller DC. Costs of cancer care across the disease continuum. Oncologist 2018;23:798–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.