

Abstract
The combination of the synthetic TLR9 ligand CpG and agnostic OX40 antibody can trigger systemic anti-tumor immune responses upon co-injection into the tumor microenvironment, eradicating simultaneous untreated sites of metastatic disease. Here we explore the application of this in situ immunotherapy to the neoadjuvant setting. Current neoadjuvant checkpoint blockade therapy is delivered systemically, resulting in off-target adverse effects. In contrast, intratumoral immunotherapy minimizes the potential for toxicities and allows for greater development of combination therapies. In two metastatic solid tumor models, neoadjuvant intratumoral immunotherapy generated a local T cell antitumor response that then acted systemically to attack cancer throughout the body. In addition, the importance of timing between neoadjuvant immunotherapy and surgical resection was established, as well as the increased therapeutic power of adding systemic anti-PD1 antibody. The combination of local and systemic immunotherapy generated an additional survival benefit due to synergistic inhibitory effect on tumor-associated macrophages. These results provide a strong rationale for translating this neoadjuvant intratumoral immunotherapy to the clinical setting, especially in conjunction with established checkpoint inhibitors.
Introduction
We have previously documented the synergistic immunotherapeutic effects of a CpG oligodeoxynucleotide (TLR9 ligand) and an antibody against OX40 (T cell activation target). This combination of immune stimulating agents co-injected at low doses directly into a single tumor site can eradicate disease throughout the body due to the induction of a specific anti-tumor T cell immune response (1). These preclinical results in models of metastatic disease have led to ongoing clinical trials in patients with lymphoma (NCT03410901) and solid tumors (NCT03831295).
In the present study we extended this immunotherapy modality to preclinical models representing the neoadjuvant setting. Neoadjuvant therapy, treatment prior to surgical resection, can reduce tumor burden, minimizing the extent of surgery required and thereby decrease the morbidity of the surgical procedure (2–4). In some cases, upfront treatment can make curative resection possible for patients who originally presented with unresectable or marginally resectable tumors (5). Neoadjuvant therapy also permits the examination of the resected tumor specimen to determine success of prior treatment. Evaluation of this pathologic response allows earlier determination of therapeutic success in individual patients and can help guide subsequent treatment decision-making (6). Furthermore, earlier therapy can improve systemic control in patients at high risk of having microscopic distant metastases at the time of surgery. In the neoadjuvant setting the presence of the primary tumor can serve as an antigenic reservoir for T cell priming (7,8). As cancer surgery remains the standard of care in a large proportion of patients with solid tumors, there is a large opportunity to apply a local immune priming maneuver prior to primary surgical resection.
Intratumoral immunotherapy, the direct inoculation of immune stimulating agents into the tumor itself, has several characteristics that readily translate to the neoadjuvant setting. First, as a limited immune activation model it avoids many of the off-target toxicities and adverse effects that accompany global immune stimulation (1,8). Furthermore, when compared to systemic administration, local administration requires a much lower doses of the agents to induce an antitumor response (9,10). Intratumoral delivery can potentially allow the use of agents or drug combinations that have poor systemic safety profiles (8). Direct injection at the tumor site also ensures access to tumor infiltrating T cells in the tumor microenvironment, which may already be enriched for tumor antigen recognition. As a result, local immunotherapy, compared to systemic immunotherapy, can more effectively elicit an immune response by leveraging the rich pool of antigens within the tumor to provide better priming of polyclonal antitumor response (1).
Despite the handful of on-going neoadjuvant systemic immunotherapy trials (11–15), only a few utilize intratumoral immunotherapy. Moreover, all the completed intratumoral trials have used autologous dendritic cell vaccines (NCT01347034, NCT00499083, NCT00365872). These approaches rely on a cell-based vaccine that is both laborious and expensive to produce (16). To realize the full potential of neoadjuvant intratumoral immunotherapy, we seek to develop a therapeutic modality that is not only capable of eliciting an effective local and systemic antitumor response against multiple types of solid tumors but is also cost-effective at scale.
Here we describe a preclinical study that applies our TLR9-anti OX40 intratumoral combination in two different solid tumor models of neoadjuvant therapy. These two preclinical tumor models allow us to demonstrate the abscopal effects of local immunotherapy on systemic metastatic disease in a setting that closely replicates surgical management of early-stage cancers.
Materials and Methods
Mouse Strains
BALB/c wild-type (WT) mice were purchased from Charles River (http://www.criver.com). Female mice greater than 9 weeks old were used in all experiments. Mice were housed in the Laboratory Animal Facility of the Stanford University Medical Center (Stanford, CA). To ensure statistical power experimental groups were typically composed of 10 animals each. For each experiment mice numbers, statistical tests and numbers of experimental replicates are described in the figure legends. Data include all outliers. Investigators were not blinded during evaluation of the in vivo experiments.
Cell Culture
CT26 colon carcinoma line was obtained from ATCC (Manassas, VA) and CT26-Luc cell line was generated in lab. 4T1-Luc breast carcinoma cell line was a gift from the S. Strober laboratory and the C. Contag laboratory (both at Stanford University). Tumor cells were cultured in complete medium (RPMI 1640; Cellgro) containing 10% fetal bovine serum (FBS; HyClone), 100 U/mL penicillin, 100μg/mL streptomycin, and 50μ M2-ME (Gibco). Cell lines were routinely tested for mycoplasma contamination.
Tumor inoculation and animal studies
CT26 tumor cells (5 × 105 cells/injection) were injected into the tail vein and a day later subcutaneously at the right side of the abdomen. 4T1 tumor cells (7.5 x 104 were injected orthotopically into the right abdominal mammary fat pad under direct visualization. In the preclinical 4T1 breast cancer tumor model mice develop metastases in the lungs, liver, bones, and brain, among other organs after tumor inoculation. When tumors size reached 0.7 cm in the largest diameter, mice were randomized to the experimental groups except in T cell depletion studies, in which animals were randomized prior to tumor cell inoculation. In groups receiving local treatments, either PBS or CpG and anti-OX40 were injected into the tumor in a volume of 50μL. In groups undergoing resection, the primary tumor is surgically resected 4 days after the last intratumoral treatment. During surgery, mice were anesthetized with ketamine/xylazine mixture before their primary tumors were resected, and their wounds closed with 4-0 Vicryl. Mice were monitored for symptoms of illness with changes to weight, posture, activity, and fur texture, and euthanized if clinical symptoms reached the cumulative limit outlined by animal ethics.
Primary tumor size was monitored in all animals with a digital caliper (Mitutoyo) every 2 to 3 days and expressed as volume (length × width × height). Systemic disease was evaluated using AMI HTX Imager (Spectral Instruments Imaging). For rechallenge experiments, long-term survivors were always injected subcutaneously with the indicated dose of tumor cells on the opposite flanks of the abdomen.
Lung metastasis count
To visualize and count lung metastases, mice were culled, and lungs and tracheas were exposed. Using a 10 mL syringe and 23 gauge needle, India ink (15% diluted in dH2O) was carefully injected through the trachea into the lungs until the whole lungs expanded and filled with India ink. Lungs were then removed, washed in water before incubating in Fekete’s solution (4.5% glacial acetic acid, 9% formalin, 64% ethanol in dH2O). Each lobe was sliced into half and metastases were counted under a dissection microscope with the numbers per half lobe added together for the total number of metastases per lung.
Suppression assay protocol
Mice bearing orthotopic 4T1 tumors were treated with either vehicle control, CpG/aOX40 or CpG/aOX40 and systemic aPD1 (n=3mice/group) every second day for 3 doses. 4 days after the last treatment, MDSCs were enriched from the blood using CD11b MicroBeads (Milteny). T regulatory cells were purified from the spleen using CD4+CD25+ Regulatory T Cell Isolation Kit (Milteny). Pan T cells were isolated from naïve mice spleen and were labeled with Violet Tracking dye (VTD- CellTrace, Thermo-Fischer) according to the manufacture protocol and were stimulated with mouse anti-CD3/anti-CD28 soluble antibodies (0.05 and 0.5 μg/mL respectively). Either MDSCs or Tregs were incubated with the T cells at 1:1 ratio (0.5x106 cells each) for 72 hours. Results showing dilution of VTD gated on live CD4/CD8 cells.
Flow cytometry
4T1 cells (7.5 x 104) were implanted orthotopically as described above. Once tumors reached 0.7cm in largest diameter mice were treated with IT injections for the following groups (n = 3 mice per group): vehicle, CpG and aOX40, CpG/aOX40 and aPD1.
Cell samples were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain in PBS for 30 min in the dark at 4°C and then washed twice. Cells were resuspended in 100 μL cold FACS buffer (1% BSA in PBS with 0.02% sodium azide) and incubated for 15 min at room temperature with mouse Fc block (anti-mCD16/32). Without washing, fluorescently-labeled antibodies against surface markers of interest were added directly to samples, followed by 50 μL of Brilliant Stain Buffer (BD Biosciences: 563794). Samples were incubated for 15 min at room temperature in the dark and then washed twice with FACS buffer. To enable intracellular antibody staining, samples were resuspended in 1 mL cold Fixation/Permeabilization buffer (eBioscience: 00-5521-00), incubated for 30 min at room temperature in the dark, and then washed twice with permeabilization buffer (eBioscience: 00-8333-56). Samples were resuspended in 100 μL permeabilization buffer, and fluorescently-labeled antibodies against intracellular markers of interest were added to the appropriate samples. Samples were incubated for 30 min at room temperature in the dark and then washed twice with permeabilization buffer. Samples were then resuspended in 250 μL fixation buffer (2% paraformaldehyde in PBS), incubated for 15 min at room temperature in the dark, and washed once with PBS. Samples were resuspended in 250 μL PBS and stored at 4°C in the dark until they were analyzed on a BD LSR II flow cytometer.
Data stored and analyzed using Cytobank (www.cytobank.org).
IFN-gamma assay
Single-cell suspensions were made from spleens of treated mice on day 4 after treatment, and red blood cells were lysed with ammonium chloride and potassium buffer (Quality Biological). T cells were isolated by negative selection from splenocytes (Pan T Cell Isolation Kit II; Miltenyi biotec) and then cocultured with media only or 0.5 × 106 tumor cells (A20 or 4T1) for 24 hours at 37°C and 5% CO2 in the presence of 0.5 μg/mL of purified NA/LE anti-mouse CD28 mAb (BD Pharmingen). For the positive stimulation control, T cells were treated with 0.5 μg/mL of purified NA/LE anti-mouse CD3 (BD Pharmingen) in addition to 0.5 μg/mL of anti-mouse CD28 mAb. The BD Cytofix/Cytoperm Plus Kit was used according to the manufacturer’s instructions for assessing intracellular IFN-γ. Monensin (0.3 μL/sample; GolgiStop; BD Biosciences) was added for the last 5 to 6 hours of incubation to inhibit protein transport. Extracellular staining was performed as described previously for immune infiltration studies. Samples were analyzed on a BD FACSCalibur flow cytometer. Data stored and analyzed using Cytobank.
Protein extraction and ELISA
Tumors were collected and ground in tissue protein extraction reagent (T-PERTM, Thermo Fisher Scientific) in the presence of 1% proteinase and phosphatase inhibitors (Thermo Fisher Scientific). The lysates were incubated at 4 °C for 30 min with slow rotation then centrifuged to remove debris. The supernatants were transferred to a clean tube for ELISA. Levels of IL-12, IFN-alpha and IFN-gamma in tumor tissue supernatants were measured by ELISA kits (R&D Systems), following the manufacturer’s instructions.
Antibodies and Reagents
CpG SD101 was provided by Dynavax Technologies. Anti-mouse CD8a, clone 2.43; anti-mouse CD4, clone GK1.5, aOX40 (CD134) antibodies were purchased from BioXcell. Anti-PD-1 mAb was obtained from Absolute Antibody.
Quantification and Statistical Analysis
The animal numbers used for all experiments are outlined in the corresponding figure legends. Statistical analysis was performed using GraphPad Prism software. P values <0.05 were considered significant. Significance was represented in figures as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns (not significant) for p > 0.05. The Kaplan-Meier method was employed for survival analysis, p values were calculated using the log-rank test (Mantel-Cox). Data include all outliers.
Study Approval
All animal experiments were approved by the Stanford administrative panel on laboratory animal care and conducted in accordance with Stanford University animal facility guidelines.
Results
Local injection of neoadjuvant immunotherapy into a primary tumor site improves systemic disease control
To test the concept of neoadjuvant immunotherapy, we first established a model in which CT26 colorectal carcinoma cells were inoculated intravenously into syngeneic mice to set up pulmonary metastases (Supplemental Figure 1) and also implanted the tumor subcutaneously to provide a site that could be injected and then resected. (Figure 1A). Once the subcutaneous tumors reached a standard size (0.7cm in largest diameter), they were treated with intratumoral injections of the immune stimulants. We performed two experiments, one comparing monotherapy immune stimulants (CpG alone vs aOX40 alone vs combination of CpG/aOX40) and one comparing neoadjuvant vs non-neoadjuvant combination CpG and aOX40 every second day for a total of 3 injections followed by complete resection of that subcutaneous tumor site four days after the last injection. In this neoadjuvant model surgery provides local disease control whereas neoadjuvant immunotherapy confers systemic control.
Figure 1.
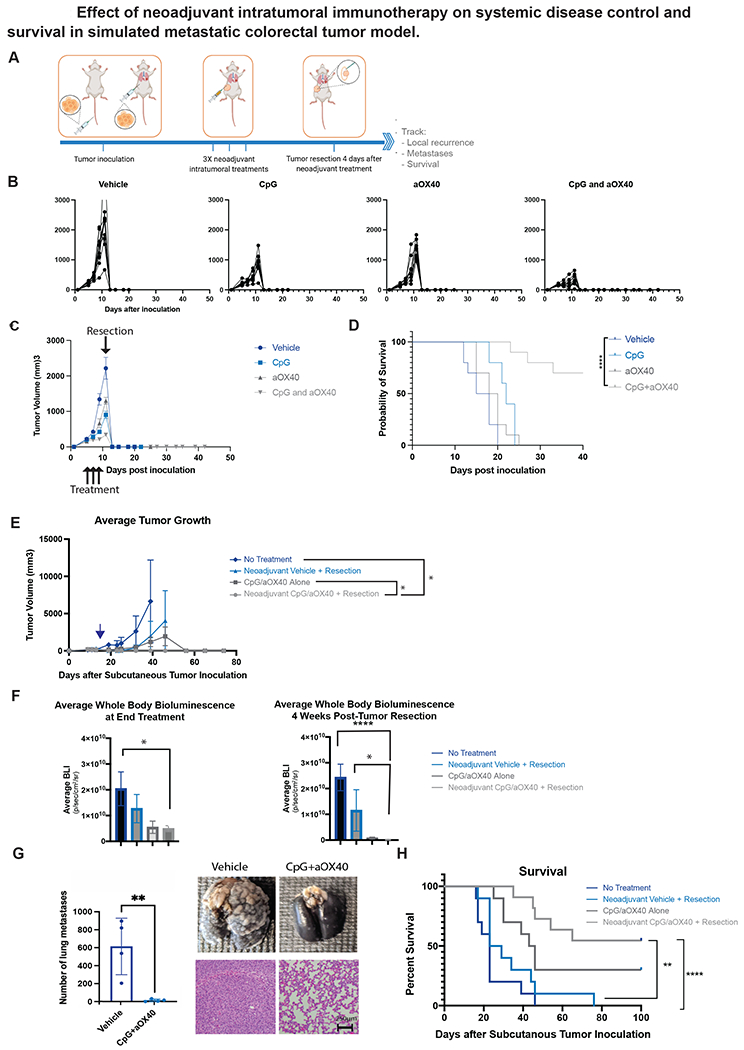
A-F, H Groups of BALB/c WT mice (n = 10) were inoculated with 5x105 CT26-Luc colorectal carcinoma tumor cells into tail vein followed a day later with inoculation of 5x105 CT26-Luc tumor cells subcutaneously into the right side of the abdomen to simulate metastatic and local disease respectively. Treatment started once local (subcutaneous) tumor reached 0.7cm in diameter. A. Schematic illustrating general experimental timeline. B-D. Groups of mice were treated with intratumoral administration of neoadjuvant immunotherapy (3X injections, 50ug CpG and 8ug aOX40 antibody per injection vs CpG alone vs aOX40 alone vs vehicle (PBS)) followed 4 days later by resection. B. Individual primary tumor growth curves for each treatment group. C. Average local tumor growth in different treatment groups. Data presented as mean tumor size +/− SEM. D. Kaplan-Meier curves for overall survival of each group shown. P values were calculated using the log-rank test (Mantel-Cox). p <0.001. E, F, H. Groups of mice were similarly treated with intratumoral administration of neoadjuvant immunotherapy (3X injections, 50ug CpG and 8ug aOX40 antibody per injection), immunotherapy alone (3X injections of 50ug of CpG and 8ug of aOX40 antibody per injection), neoadjuvant vehicle (PBS) followed by resection or no treatment. Groups that underwent resection did so on day 17 following subcutaneous tumor inoculation. E. Average local tumor growth in different treatment groups. Date of resection indicated by blue arrow. Data presented as mean tumor size +/− SEM. Difference in primary tumor size between groups were significant by unpaired t test. p = 0.03 (No treatment vs Neoadjuvant CpG/aOX40 + Resection). p = 0.02 (CpG/aOX40 alone/Neoadjuvant CpG vs aOX40 + Resection). F. Average systemic bioluminescent signal in different groups on last day of local immunotherapy treatment (n = 10) and at 4 weeks (n =2 in No Treatment group; n=3 in Resection Only; n = 6 in CpG/aOX40 group; n = 9 in Neoadjuvant CpG/aOX40 group) following resection of the primary tumor. Data presented as mean whole body bioluminescence (BLI) +/− SEM. Difference in average whole body bioluminescence at end treatment between groups were significant by unpaired t test. p = 0.03 (No Treatment vs Neoadjuvant CpG/aOX40 + Resection). Difference in average whole body bioluminescence at 4 weeks post tumor resection between groups were also significant by unpaired t test: p < 0.0001 (CpG/aOX40 alone vs Neoadjuvant CpG/aOX40 + Resection). p = 0.02 (CpG/aOX40 Alone vs Neoadjuvant CpG/aOX40 + Resection). G. In a separate experiment, mice in this group similarly inoculated with CT26 and lungs were harvested for examination after neoadjuvant treatment. Figure demonstrates gross burden of systemic disease in mice treated with neoadjuvant vehicle versus Cps/aOX40 at time of primary tumor resection. To detect metastatic pulmonary nodules, lungs were stained with either India ink (top) or H&E (bottom). Graph quantifies the numbers of the metastasis in the lungs. All experiments described above were performed at least twice to confirm findings p = 0.0087 by unpaired t test. H. Kaplan-Meier curves for overall survival of each group shown. P values were calculated using the log-rank test (Mantel-Cox). p < 0.003 (Neoadjuvant CpG/aOX40 + Resection vs Neoadjuvant Vehicle + Resection). p < 0.0001 (Neoadjuvant CpG/aOX40 + Resection vs No Treatment).
We found that the cohort of mice receiving combination intratumoral immunotherapy with both CpG and aOX40 had the best locoregional tumor control (Figure 1B and C) and survival (Figure 1D) when compared to monotherapy in the neoadjuvant setting. Again, when comparing neoadjuvant CpG and aOX40 to immunotherapy alone or neoadjuvant vehicle, mice that received combination neoadjuvant CpG and aOX40 had the best locoregional control of the subcutaneous tumor site, with no local recurrence in any of the animals undergoing resection (n=10) (Figure 1E). In contrast, 2/10 animals treated with vehicle control had local recurrence of the tumor following resection (Figure 1E). More striking was the improvement in systemic disease control in mice receiving the local intratumoral injections of CpG and aOX40 (Figure 1F). Mice treated with intratumoral injections of CpG and aOX40 were found to have significantly less disease in their lungs apparent even on gross examination (Figure 1G) compared to their vehicle treated cohorts. When the number of lung metastases were quantified, there was a significant difference between vehicle and CpG/aOX40 groups—attesting to the abscopal efficacy of the local immunotherapy combination. As expected, this dramatic improvement in systemic disease control from just local intratumoral immunotherapy translated directly into a significant improvement in long-term survival in the neoadjuvant immunotherapy group. The group treated with neoadjuvant local immunotherapy yielded the largest cohort of long-term survivors (6/10). Local immunotherapy treatment in mice that received no surgery had fewer long-term survivors (3/10) due to eventual outgrowth of the primary tumor (Figure 1H).
Efficacy of neoadjuvant immunotherapy is dependent on CD8+ T cells
To investigate the role of different immune cells during neoadjuvant immunotherapy treatment, we depleted CD4+ and CD8+ T cells prior to tumor inoculation and during neoadjuvant treatment. Mice were inoculated with tumor as described in Figure 1A. The groups underwent antibody mediated depletion of either CD4+ T cells, CD8+ T cells, both or neither. Specific T cell depletion was confirmed peripheral blood by flow cytometry (Supplemental Figure 2). All groups were treated with neoadjuvant intratumoral injections of CpG and aOX40, followed by resection 4 days after the last dose (Figure 2A). Depletion of CD8 T cells led to loss of local tumor control and this was further compounded when CD4 T cells were also depleted (Figure 2B). This loss of locoregional disease control in CD8 depleted mice was reflected in systemic disease burden as well—none of the CD8 or the double depleted mice survived due to heavy systemic disease burden (Figure 2B). In contrast, the majority of the control and the single CD4 depleted mice survived long term (Figure 2C).
Figure 2.
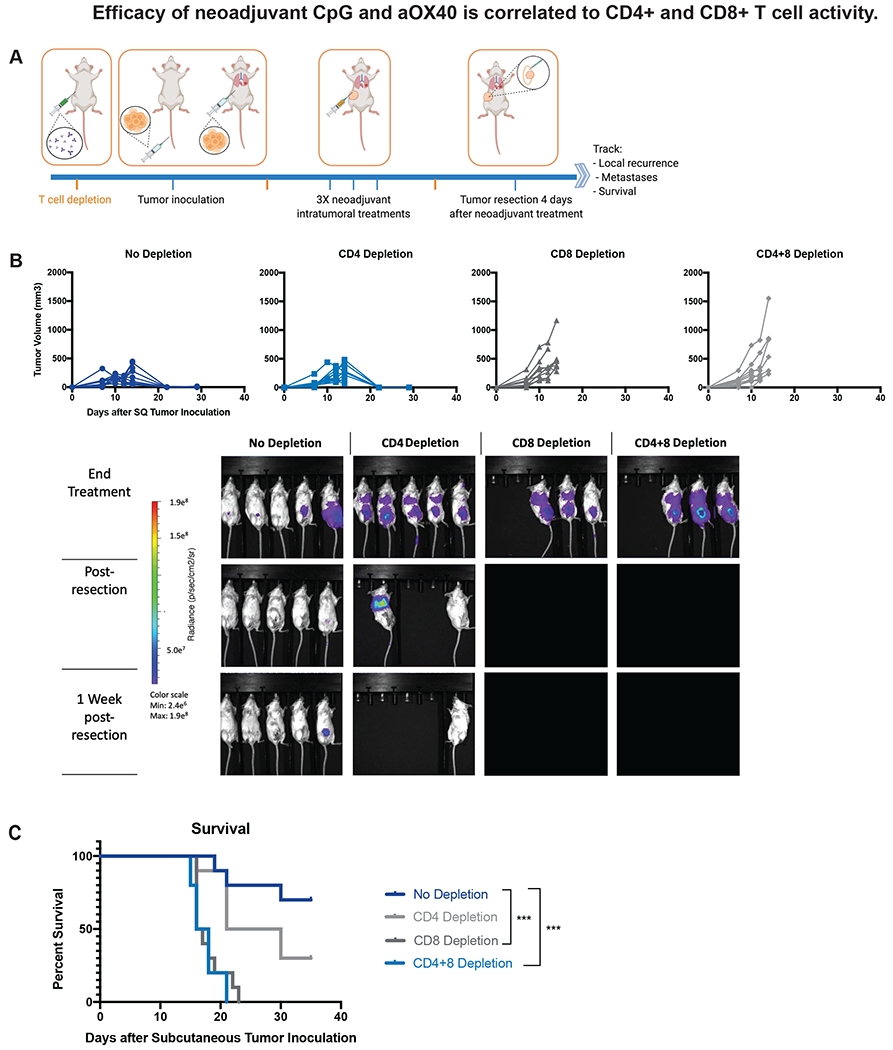
Efficacy of neoadjuvant immunotherapy dependent on CD4+ and CD8+ antitumor T cell activity. Groups of BALB/c WT mice (n = 10) were injected with either depleting anti-CD4, anti-CD8, anti-CD4 and anti-CD8 or no depleting antibodies one day prior to subcutaneous tumor inoculation and again on days 7 and 17 after tumor inoculation. Mice were inoculated with CT26-Luc tumors as described in Figure 1. All groups of mice were treated with intratumoral administration of neoadjuvant immunotherapy (3X injections, 50ug CpG and 8ug aOX40 antibody per injection). Neoadjuvant treatment started once local (subcutaneous) tumor reached 0.7cm in diameter. All groups underwent resection of the primary subcutaneous tumor four days after the last neoadjuvant intratumoral injection. A. Schematic illustrating general experimental timeline. B. Local tumor growth and recurrence in different groups. Resection occurred on day 20 following subcutaneous tumor inoculation. Also shown is metastatic dissemination following T cell depletion via BLI, images representing 5/10 mice in each group. C. Survival was monitored and investigated by Kaplan–Meier analysis. P values were calculated using the log-rank test (Mantel-Cox). p < 0.001 (No Depletion vs CD8 Depletion). p < 0.001 (No Depletion vs CD4+8 Depletion). None of the CD4+ and CD8+ depleted mice survived to resection due to heavy systemic disease burden and none of the CD8+ depleted mice survived to the post-resection assessment timepoint. In contrast, 9/10 CD4+ depleted mice survived to resection and 3/10 survived long term. In the group of mice that did not undergo T cell depletion, 7/10 mice treated with neoadjuvant immunotherapy survived long term.
Neoadjuvant local immunotherapy decreases local recurrence and improves survival in a model of spontaneous metastases
Having established the ability of neoadjuvant local immunotherapy to prolong survival by improving both locoregional and distant systemic disease control in a simulated model of metastatic cancer, we extended our study to the highly aggressive and spontaneously metastatic 4T1 triple negative breast cancer tumor. We inoculated 4T1 tumor cells orthotopically into one inguinal mammary fat pad of BALB/c WT mice to simulate a primary tumor. In this model, the tumors spontaneously metastasize to the lungs, liver and spleen (17). Once the primary tumors reach a standard size (0.7cm in largest diameter), the mice are treated with three intratumoral injections of immune stimulants CpG and aOX40 every second day. Mice in treatment groups undergoing resection underwent removal of the primary tumor four days after last intratumoral treatment (Figure 3A).
Figure 3.
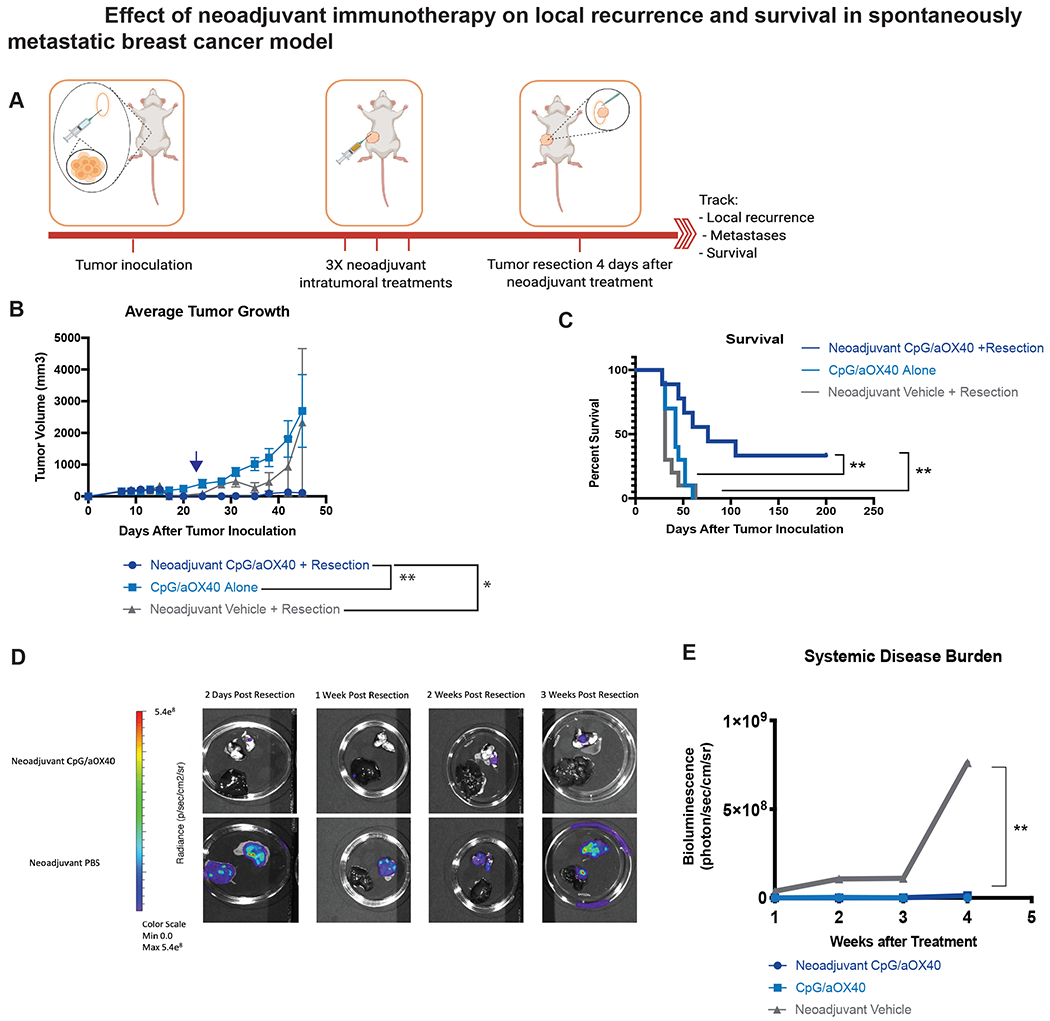
Neoadjuvant immunotherapy decreases local recurrence and improves survival in aggressively metastatic 4T1-Luc tumor model. A-C, Groups of BALB/c WT mice (n = 10) were inoculated with 7.5x104 mammary carcinoma 4T1-Luc tumor cells orthotopically into the right mammary fat pad. Groups of mice were treated with local administration of neoadjuvant immunotherapy (3X injections, 50ug CpG and 8ug aOX40 antibody per injection), immunotherapy alone (3X injections of 50ug of CpG and 8ug of aOX40 antibody per injection), or neoadjuvant vehicle. Treatment started once local (subcutaneous) tumor reached 0.7cm in diameter. Groups that underwent resection did so on day 15 (blue arrow) following subcutaneous tumor inoculation. A. Schematic illustrating experimental setup. B. Average local tumor growth in different treatment groups. Date of resection indicated by blue arrow. Data presented as mean tumor size +/− SEM. Differences in primary tumor growth between neoadjuvant vehicle followed by resection, CpG/aOX40 without resection and neoadjuvant CpG/aOX40 followed by resection were significant by unpaired t test. p = 0.008 (Neoadjuvant CpG/aOX40 + Resection vs CpG/aOX40 Alone). p = 0.04 (Neoadjuvant CpG/aOX40 + Resection vs Neoadjuvant Vehicle + Resection). C. Kaplan-Meier curves for overall survival of each group shown. P values were calculated using the log-rank test (Mantel-Cox). p = 0.006 (Neoadjuvant CpG/aOX40 + Resection vs CpG/aOX40 Alone). p = 0.002 (Neoadjuvant CpG/aOX40 + Resection vs Neoadjuvant Vehicle + Resection).
D-E. In a separate experiment, groups of BALB/c WT mice were inoculated with tumors as described above in A (n = 10). Mice from each group were euthanized at specific time points (2 days post resection, 1 week post resection, 2 weeks post resection and 3 weeks post resection) and their lungs and liver imaged ex vivo to evaluate systemic disease burden (n = 2 at each time point). The differences between the groups were statistically significant, p = 0.09 between neoadjuvant CpG/aOX40 vs Neoadjuvant Vehicle + Resection by paired t test.
We again noted that the cohort of mice receiving neoadjuvant local immunotherapy had the best locoregional control. 9/10 mice treated with neoadjuvant vehicle control had tumor recurrence following resection compared to 3/9 mice treated with neoadjuvant CpG and aOX40 (Figure 3B, Supplemental Figure 3). On average, mice treated with local immunotherapy without resection had the worst primary tumor control, though the difference between this group and the group treated with neoadjuvant vehicle followed by resection decreased over time. The group treated with neoadjuvant immunotherapy was the only group to yield a cohort of long-term survivors (3/9). In contrast, all mice from groups treated with neoadjuvant vehicle or immunotherapy without resection ultimately succumbed to metastatic disease (Figure 3C).
To examine the systemic disease burden in such mice we sacrificed a separate cohort of mice at timed intervals to evaluate tumor burden in lungs and liver via IVIS (Figure 3D). While metastatic disease could be detected in the lungs and liver of mice treated with either vehicle control or local immunotherapy at 2 days following resection, the average burden of systemic disease remained low in mice treated with CpG and aOX40, while increasing markedly in mice treated with just vehicle followed by resection (Figure 3E).
T cell dependence in spontaneously metastatic tumor
As in the colon cancer model previously described, we examined the immune cells involved in the protective effects of neoadjuvant local immunotherapy in this orthotopic and spontaneously metastatic 4T1 breast cancer model. We again depleted T cells either prior to or after tumor inoculation during neoadjuvant treatment. Groups of BALB/c WT mice were inoculated with tumor as described in Figure 3A. All groups were treated with neoadjuvant intratumoral injections of CpG and aOX40, followed 4 days later by resection (Figure 4A). Similar to before, we confirmed that depletion of CD8+ T cells led to loss of local tumor control and this was further compounded when CD4+ T cells were also depleted. In comparison, cohorts of mice that underwent no depletion or only CD4+ T cell depletion had better locoregional tumor control. In the more locally and systemically aggressive 4T1 model, mice that underwent no depletion of T cells had the best local tumor control whereas the cohort that underwent CD4 T cell depletion had significantly poorer local tumor control (Figure 4B). Systemic disease burden was once again heaviest in the double depleted and single CD8+ depleted groups (Figure 4C). The group that underwent no depletion had the most long-term survivors whereas all mice that underwent CD8 depletion ultimately succumbed to systemic metastases (Figure 4D).
Figure 4.
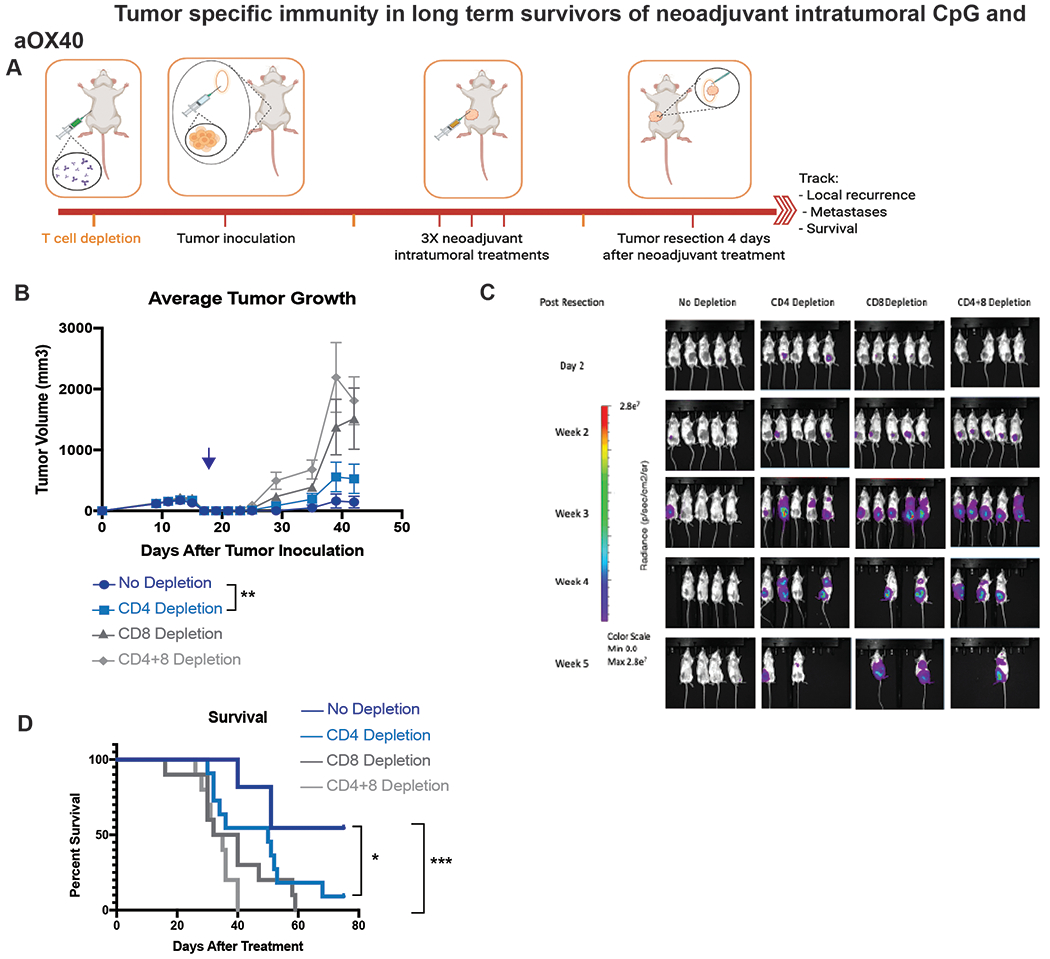
Tumor specific immunity in long term survivors of neoadjuvant intratumoral CpG and aOX40. A. Schematic illustrating experimental setup. B. Average local tumor growth in different treatment groups. Date of resection indicated by blue arrow. Data presented as mean tumor size +/− SEM. Differences in primary tumor growth between neoadjuvant vehicle followed by resection, CpG/aOX40 without resection and neoadjuvant CpG/aOX40 followed by resection were significant by unpaired t test. p = 0.004 (No Depletion vs CD4 Depletion). C. Metastatic dissemination following T cell depletion via BLI, images representing 5/10 mice in each group. D. Kaplan-Meier curves for overall survival of each group shown. P values were calculated using the log-rank test (Mantel-Cox). p = 0.02 (No Depletion vs CD4 Depletion). p < 0.001 (No Depletion vs CD4+8 Depletion).
Locally activated antigen specific T cells are present systemically in treated mice
Mice treated for CT26 or 4TI tumors by neoadjuvant immunotherapy as previously described were tested for antitumor T cells. Spleens from treated and vehicle mice were harvested and incubated with either homologous tumor cells or with various controls for 24 hours (Figure 5A). In both the CT26 and 4T1 treated mice specific tumor reactive CD8+ T cells could be detected by their upregulation of IFN-gamma and by their expression of the activation marker, CD44 (Figure 5B, Supplemental Figure 4). In addition, CD8+ T cells in the lungs and in the draining lymph node were more activated in the treated mice compared to vehicle mice as evidenced by proliferation and by their expression of granzyme B (Figure 5C). Of note, consistent with T cell activation, mice that received combination neoadjuvant immunotherapy had overall higher tumor microenvironment levels of IFN-gamma compared to monotherapy cohorts. (Supplemental Figure 5).
Figure 5.
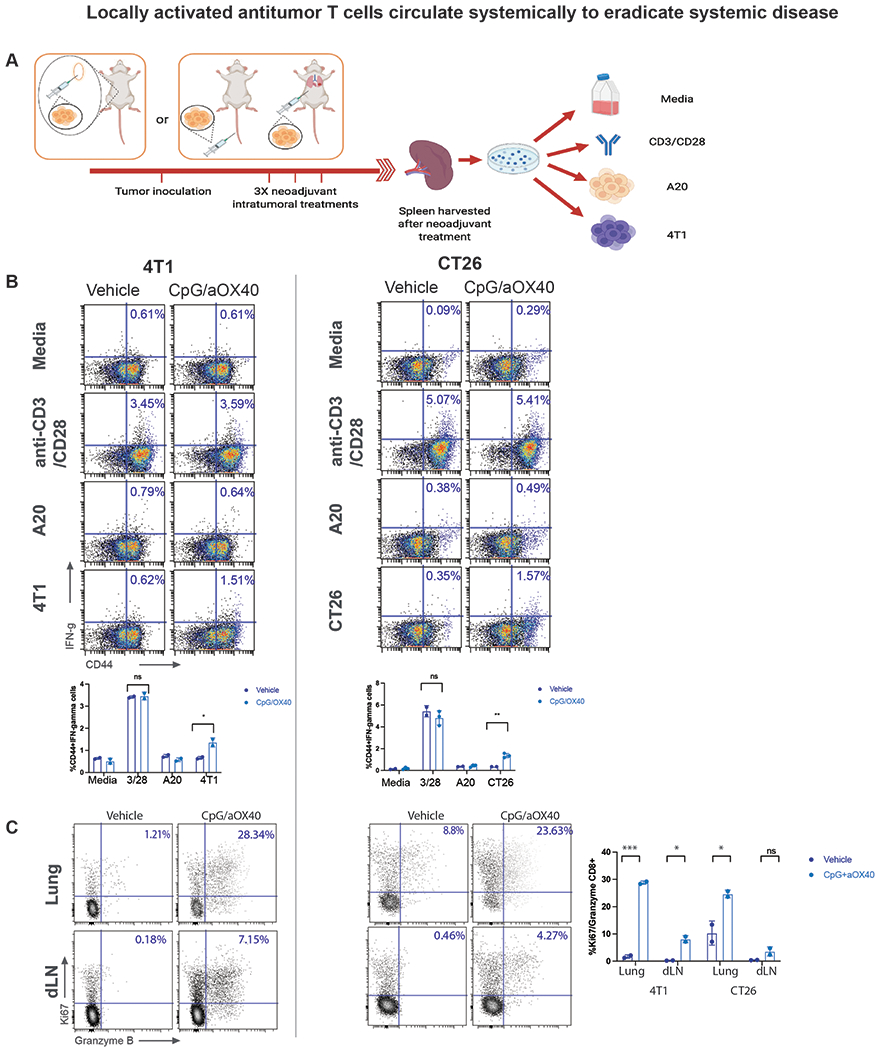
Locally activated antitumor T cells circulate systemically to eradicate systemic disease
A-C. Mice were implanted with either CT26 or 4T1 tumor cells as previously described in Figure 1 and 3 respectively and treated with either vehicle or intratumoral CpG and aOX40. Splenocytes from the indicated groups harvested on day 4 after treatment and cocultured with either media, CD3 and CD28 antibodies, A20 cells (unrelated control tumor), or homologous tumor cells for 24 hours. A. Schematic illustration of experiments. B. For 4T1, analyzed population was enriched for CD3+ cells. Intracellular IFN-γ was measured in CD8+ T cells by flow cytometry as a percentage of CD44hi (memory CD8) T cells shown in dot plots and bar graph n=3 mice/group, ns=not significant **p = 0.0018 (4T1) *p=0.044 (CT26), unpaired t test. C. Intratumoral injection of aOX40+ CpG induces production of Granzyme B and proliferation of CD8+ T cells in lungs and draining lymph nodes of treated mice by day 4 post treatment. Single cell suspensions of draining lymph nodes (dLN) and lungs were stained for Ki67 and Granzyme B, highlighted are percentages of the double positive cells. n=3 mice/group, ***p = 0.000557, *p = 0.0137 (4T1) *p=0.0489, ns=not significant (CT26), unpaired t test.
Critical timing of neoadjuvant immunotherapy
Clinically, a major concern with any neoadjuvant treatment is its potential to delay definitive cancer resection and to interfere with curative surgery (18). An additional concern is the requirement for the addition of an invasive procedure with intratumoral immunotherapy (19). To address these two issues, we designed an experiment to determine how varying the time between neoadjuvant immunotherapy and the resection of the primary tumor or varying the dosage and number of intratumoral injections impacted overall efficacy (Figure 6A). In our standard treatment protocol, we maintained a four-day window between the completion of neoadjuvant immunotherapy and surgery and used three doses of intratumoral immunotherapy. When we eliminated this 4 day window, we found that a much higher proportion of mice had local recurrence (5/10) (Figure 6B). In comparison, the group that received a single injection of CpG and aOX40 followed by a 4 day window prior to resection had local recurrence in only 2/10 mice. Furthermore, mice that did not have the 4 day window prior to resection lost much of the systemic disease control (Figure 6C) and survival benefit seen with neoadjuvant treatment (Figure 6D)
Figure 6.
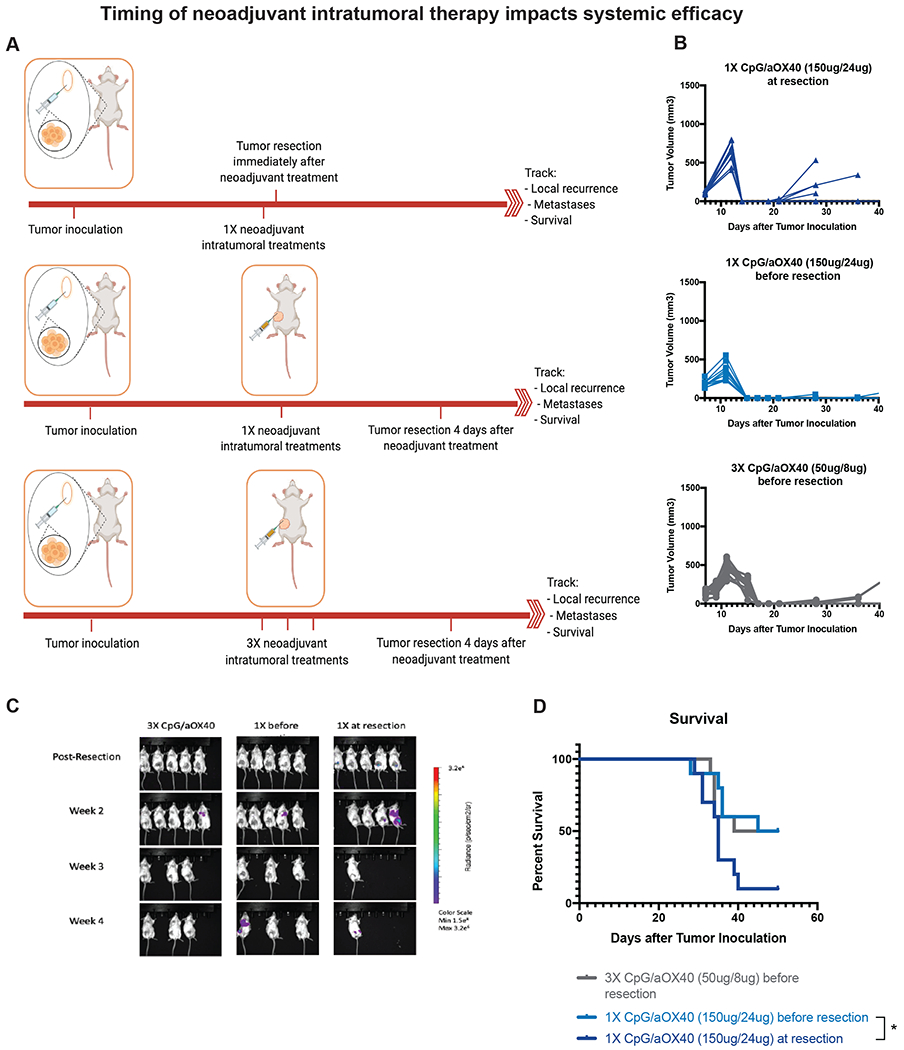
Duration between neoadjuvant immunotherapy and removal of primary tumor impacts long-term survival. A. Groups of BALB/c WT mice (n = 10 per group) were inoculated with 7.5x104 mammary carcinoma 4T1-Luc tumor cells orthotopically into the right mammary fat pad. As indicated on the schematic, one group of mice was treated intratumorally with 3X CpG (50ug/per injection) and aOX40 (8ug/injection) on days 7, 9, and 11, followed by resection of the primary tumor 4 days later on day 15. One group received 1X intratumoral injection of CpG (150ug) and aOX40 (24ug) on day 7 followed immediately by resection of the primary tumor. On group received 1X intratumoral injection of CpG (150ug) and aOX40 (24ug) on day 7 followed by resection of the primary tumor 4 days later on day 11. B. Growth curves of primary tumors in mice from each of the treatment groups. C. Metastatic dissemination following T cell depletion via BLI, images representing 5/10 mice in each group. D. Kaplan–Meier overall survival curve. P values were calculated using the log-rank test (Mantel-Cox). p = 0.03 (1X CpG/aOX40 (150ug/24ug) before resection vs 1X CpG/aOX40 (150ug/24ug) at resection).
An advantage of local immunotherapy is the ability to deliver higher doses of immune stimulants with less systemic toxicity (8). Therefore, we also tested combining the total doses of immunotherapy into a single injection. We found that mice that received a single higher neoadjuvant dose of CpG and aOX40 had comparable survival to mice that received the same cumulative doses in three separate injections (Figure 6D).
Enhanced efficacy of neoadjuvant intratumoral immunotherapy through addition of PD-1 blockade
When we analyzed the lungs of tumor bearing mice in both CT26 or 4T1 models that had been treated with intratumoral CpG and aOX40, we found those treated with CpG and aOX40 had significant upregulation of PD-1 on macrophages, T cells and CD11b+ c dendritic cells (Figure 7A). PD-1 plays an immunosuppressive role in effector T cells via inhibition of TCR and CD28 signals (20,21). F4/80/CD11b+ macrophages are known to suppress T cell proliferation and induce the development of inhibitory Treg cells (22). However, blockade of PD-1 on macrophages has been shown to increase their phagocytic activity for tumor cells (23). We hypothesized that adding systemic aPD-1 checkpoint inhibitor to CpG/aOX40 would relieve this inhibition of T cells and improve the therapeutic response. Indeed, we found that the addition of systemic aPD-1 resulted in increased proliferation and activity of CD8+ T cells in the lymph nodes of the treated mice (Figure 7B). MDSCs are a heterogeneous population that accumulates during cancer development (24). MDSCs have been shown to suppress the T cell proliferation as well as adaptive immune response to tumors (25). Regulatory T (Treg) cells are involved in tumor development and progression by inhibiting antitumor immunity. We therefore decided to analyze the impact of treatment with CpG/aOX40 and CpG/aOX40 and aPD-1on Tregs and MDSC function. Equal numbers of CD4+CD25+ Tregs cells or MDSCs were isolated from tumor-bearing mice treated either with vehicle control, CpG/aOX40 or CpG/aOX40 and aPD1. Purified naïve T cells were labeled with violet tracking dye (VTD) then stimulated to proliferate by beads coated with anti-CD3 and anti-CD28 mAbs. MDSCs and Tregs from CpG/aOX40 showed decreased suppressive activity that was further decreased by the addition of aPD-1 (Figure 7C). Taken together these results suggest that treatment with CpG/aOX40 and aPD1 mediates antitumor immune responses by reliving suppression on the T cell mediated by PD1 positive cells as well as Tregs and MDSCs.
Figure 7.
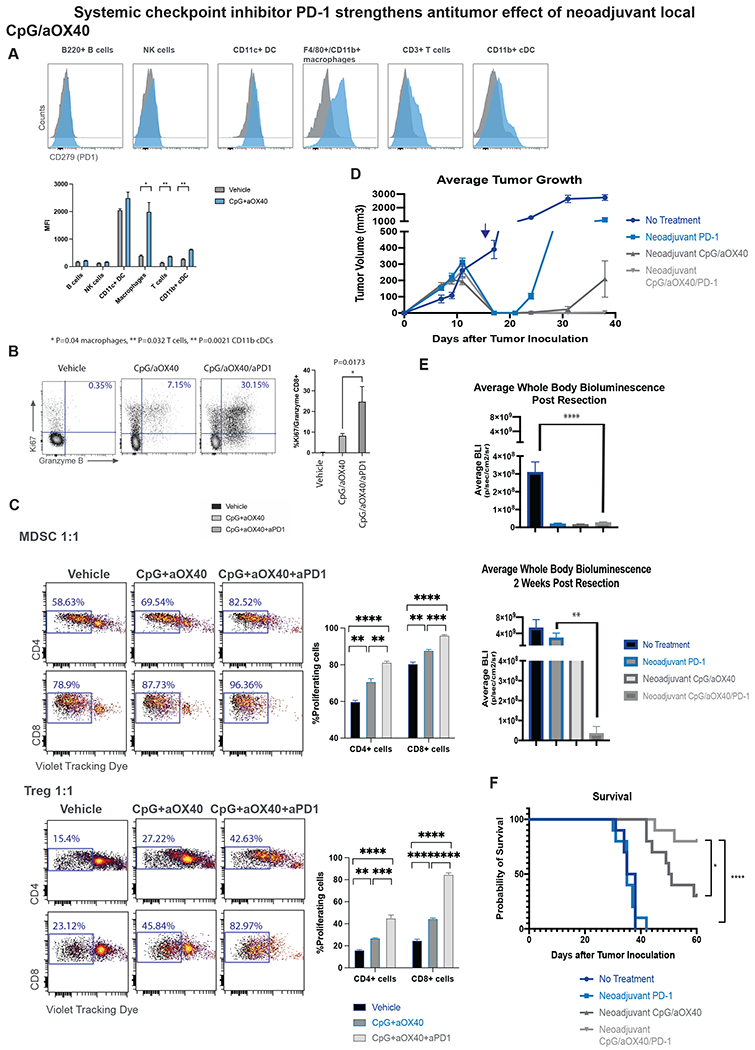
Combination of systemic and local neoadjuvant immunotherapy strengthens antitumor response. A. Intratumoral injection of CpG+aOX40 upregulates PD-1 on macrophages, T cells and CD11b cDCs. Groups of BALB/C WT mice (n = 3) were orthotopically inoculated with 4T1-Luc tumors as described in Figure 3. Mice were randomized into the following treatment groups: no treatment, and CpG/aOX40, (3X injections, 50ug CpG and 8ug aOX40 antibody per injection). Lungs were harvested 4 days after therapy and analyzed by flow cytometry. Histogram plots of the indicated populations for vehicle control (grey) or CpG/aOX40 (blue) shown here with data presented as a bar graph of n=3 mice, unpaired t test * p=0.04 macrophages, ** p=0.032 T cells, ** p=0.0021 CD11b cDCs. B. Addition of aPD-1 to the treatment combination resulted in increased T cell activation. Neoadjuvant treatment of aPD-1 (3X intraperitoneal injects, 10mg/kg per injection) was added to the CpG/aOX40 treatment and draining lymph nodes were analyzed by flow cytometry. Dot plots demonstrate Granzyme B and Ki67 of the CD8+ population, bar graph summarizing n=3 mice/group, unpaired t test. C. Suppression assays, MDSCs (top) and T regulatory cells (bottom) were incubated with VTD labeled T cells in 1:1 ratio for 72h. Dot plots describing VTD dilution of CD4 or CD8+ populations, graph bars summarizing n=3, unpaired t test. D-F. Groups of BALB/C WT mice (n = 10) were orthotopically inoculated with 4T1-Luc tumors. Mice were randomized into the following treatment groups: no treatment, neoadjuvant treatment of aPD-1 (3X intraperitoneal injects, 10mg/kg per injection), neoadjuvant CpG/aOX40 (3X injections, 50ug CpG and 8ug aOX40 antibody per injection) and neoadjuvant CpG/aOX40/aPD-1. D. Average local tumor growth and recurrence in different groups. Resection occurred on day 15 (blue arrow) following subcutaneous tumor inoculation. Data presented as mean tumor size +/− SEM. E. Average systemic bioluminescent signal in different groups. Data presented as mean whole body BLI +/− SEM. Systemic disease as denoted by systemic BLI was significantly different between No Treatment and triple therapy groups (p = 0.0001) at 2 days post-resection, and at 2 weeks post resection (p = 0.02) by unpaired t test. Systemic disease was also significantly lower in triple therapy group compared to aPD-1 alone (p = 0.01). F. Kaplan-Meier curves for overall survival of each group shown. P values were calculated using the log-rank test (Mantel-Cox). p = 0.02 between triple therapy vs CpG/OX40. p <0.0001 triple therapy vs No Treatment.
To further interrogate whether this interaction translates into an improvement in therapeutic efficacy, mice bearing the 4T1 tumor were treated as above with neoadjuvant immunotherapy with or without the addition of anti PD-1 antibody. The group that received systemic aPD-1 in addition to local CpG/aOX40 demonstrated improved local (Figure 7D) and systemic disease control (Figure 7E). Correspondingly, survival was improved in mice that received both systemic checkpoint inhibitor and intratumoral immunotherapy compared to mice that received only checkpoint inhibitor or only neoadjuvant immunotherapy (Figure 7F).
Discussion
In this report, we demonstrated that local administration of neoadjuvant immunotherapy with co-injection of TLR9 agonist and anti OX40 antibody improves survival and systemic disease control, and decreases local recurrence in two separate preclinical solid tumor models: a colorectal carcinoma model in which pulmonary metastases were established by intravenous injection and a breast carcinoma model in which the primary tumor metastasizes spontaneously (17). The systemic disease burden for both models can be quantified by bioluminescent imaging within 2 weeks of tumor implantation. If left untreated mice in both these solid tumor models die from their metastases within weeks of surgery. These findings are consistent with what is seen clinically in patients who undergo resection for early stage but high-risk cancers.
In our aggressively metastatic 4T1 model, neoadjuvant therapy consistently produced a cohort of long-term survivors. Local immunotherapy in particular may take advantage of the pre-existing T cell immune repertoire within the tumor microenvironment. Once activated these T cells are capable of inducing an abscopal anti-tumor effect at distant non-treated metastatic sites, resulting in better systemic disease control (1). The importance of CD8+ T cells was consistently underscored in our depletion experiments, in which the anti-tumor effect and survival benefit of neoadjuvant immunotherapy was abrogated in both solid tumor models.
Our experiments also established baseline parameters around timing for neoadjuvant treatment. The question of timing of neoadjuvant therapy is of great clinical importance as a major concern with any neoadjuvant therapy is the risk of delay of potentially curative surgery. With immunotherapy where the intact primary tumor acts as a source of antigens for T cell priming, the duration between immune stimulation and removal of this antigenic source may be critical in determining the success of the immunotherapy. A recent study by Liu et al suggested that the duration between systemic administration of immunotherapy and resection of the primary tumor had a profound effect on treatment efficacy in preclinical models of breast cancer (26). We similarly found that with neoadjuvant local immunotherapy an interval, albeit short, was needed between immunotherapy and surgical resection. When the neoadjuvant immunotherapy was given too close to resection the efficacy of neoadjuvant immunotherapy was lost. We further found that decreasing the number of intratumoral treatments but combining the doses into a single injection resulted in survival comparable to repeated deliveries of a lower dose of the immunotherapy cocktail. No animals treated with this single delivery of high dose intratumoral immunotherapy were lost to systemic toxicity or had resection of their primary tumor delayed due to ill effects, attesting to safety of the local treatment modality.
Lastly, while immune checkpoint inhibitors to date have generally been studied in advanced disease, the same agents may be more effective when used in a neoadjuvant setting for locoregionally advanced cancers that are resectable but are at high risk of relapse (27). Our data with CpG, aOX40 and aPD-1 provide compelling rationale to test this combination clinically in a neoadjuvant setting. As aPD-1 and CpG/aOX40 combinations are currently being tested separately in a number of clinical trials against solid tumors and lymphoma (NCT04025879, NCT03831295) it would be a logical next step to combine them in the neoadjuvant setting. For instance, aOX40 has been tested as a single neoadjuvant therapy in patients with HNSCC with biochemical evidence of increased T cell activation and proliferation, as well as correlation with increased disease-free survival (28). Furthermore, a clinical trial will give us an opportunity to evaluate major pathologic response (MPR) and pathologic complete response (pCR) as surrogate endpoints for improved survival after neoadjuvant therapy. In several solid tumors, such as NSCLC, MPR has been recognized as predictive of improvement in long term overall survival in patients receiving traditional chemotherapy (29). Establishing such surrogate endpoints for clinical benefit following neoadjuvant immunotherapy is of paramount importance as survival outcomes in clinical trials can take decades to complete.
In conclusion, by extending the use of local immune modulating treatment with a TLR9 agonist and an anti OX40 antibody to the neoadjuvant setting, we demonstrated effectiveness in eliminating distant pulmonary metastases and overall survival. and in conferring lasting tumor immunity. The two different preclinical solid tumor models utilized demonstrated the broad applicability of this neoadjuvant therapeutic modality to a diverse array of tumor types and its additional efficacy when combined regimen with systemic PD1 blockade.
Supplementary Material
Supplemental Figure 1. WT BALB/c mice (n =5) were inoculated with 500,000 CT26-Luc tumor cells intravenously into the tail vein. After a week pulmonary metastases were evaluated using IVIS. All mice that underwent tail vein injections had sufficient tumor burden in their lungs to be visible via IVIS. When untreated the pulmonary tumor burden increased and persisted in all inoculated mice.
Supplemental Figure 2. WT BALB/c mice underwent injections of anti-CD4 antibody, anti-CD8 antibody, both or neither prior to undergoing neoadjuvant immunotherapy. Depletion of CD4+ and CD8+ T cells were confirmed in the relevant groups via flow cytometry of peripheral blood.
Supplemental Figure 3. Individual primary tumor growth curves for neoadjuvant immunotherapy, immunotherapy alone and neoadjuvant vehicle in the 4T1 tumor model. Groups of BALB/c WT mice (n = 10) were inoculated with 7.5x104 mammary carcinoma 4T1-Luc tumor cells orthotopically into the right mammary fat pad. Groups of mice were treated with local administration of neoadjuvant immunotherapy (3X injections, 50ug CpG and 8ug aOX40 antibody per injection), immunotherapy alone (3X injections of 50ug of CpG and 8ug of aOX40 antibody per injection), or neoadjuvant vehicle.
Supplemental Figure 4. Flow cytometry gating hierarchy for T cell stimulation assay. Splenocytes were first positively selected for CD4 and CD8 positive cells, then for CD44 and IFN-gamma.
Supplemental Figure 5. Groups of BALB/c WT mice (n = 4) were inoculated with 5x105 CT26-Luc colorectal carcinoma tumor cells into tail vein followed a day later with inoculation of 5x105 CT26-Luc tumor cells subcutaneously into the right side of the abdomen to simulate metastatic and local disease respectively. Groups of mice were treated with intratumoral administration of neoadjuvant immunotherapy (3X injections, 50ug CpG and 8ug aOX40 antibody per injection vs CpG alone vs aOX40 alone vs vehicle) followed by resection. Primary tumors were harvested and levels of IL-12, IFN-alpha and IFN-gamma in tumor tissue supernatants measured by ELISA.
Statement of Significance.
This work demonstrates the ability of neoadjuvant intratumoral immunotherapy to target local and distant metastatic disease and consequently improve survival.
Acknowledgements:
This work was supported by grants from the National Institute of Health (5R35CA197353). Wan Xing Hong is supported by The National Institute of Health (5T32AI07290).
Footnotes
Conflict of Interest: Dr. Ronald Levy serves on the advisory boards for Five Prime, Corvus, Quadriga, BeiGene, GigaGen, Teneobio, Nurix, Dragonfly, Abpro, Apexigen, Spotlight, 47 Inc, XCella, Immunocore, Walking Fish.
The other authors have declared that no conflict of interest exists.
References
- 1.Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grossman HB, Natale RB, Tangen CM, Speights VO, Vogelzang NJ, Trump DL, et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med. 2003;349:859–66. [DOI] [PubMed] [Google Scholar]
- 3.von Minckwitz G, Eidtmann H, Rezai M, Fasching PA, Tesch H, Eggemann H, et al. Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N Engl J Med. 2012;366:299–309. [DOI] [PubMed] [Google Scholar]
- 4.van Hagen P, Hulshof MCCM, van Lanschot JJB, Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BPL, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366:2074–84. [DOI] [PubMed] [Google Scholar]
- 5.Sjölund K, Andersson A, Nilsson E, Nilsson O, Ahlman H, Nilsson B. Downsizing Treatment with Tyrosine Kinase Inhibitors in Patients with Advanced Gastrointestinal Stromal Tumors Improved Resectability. World J Surg. 2010;34:2090–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spring L, Greenup R, Niemierko A, Schapira L, Haddad S, Jimenez R, et al. Pathologic Complete Response After Neoadjuvant Chemotherapy and Long-Term Outcomes Among Young Women With Breast Cancer. J Natl Compr Canc Netw. 2017;15:1216–23. [DOI] [PubMed] [Google Scholar]
- 7.Brooks J, Fleischmann-Mundt B, Woller N, Niemann J, Ribback S, Peters K, et al. Perioperative, Spatiotemporally Coordinated Activation of T and NK Cells Prevents Recurrence of Pancreatic Cancer. Cancer Res. 2018;78:475–88. [DOI] [PubMed] [Google Scholar]
- 8.Marabelle A, Tselikas L, de Baere T, Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann Oncol. 2017;28:xii33–43. [DOI] [PubMed] [Google Scholar]
- 9.Marabelle A, Andtbacka R, Harrington K, Melero I, Leidner R, de Baere T, et al. Starting the fight in the tumor: expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann Oncol. 2018;29:2163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ascierto PA, Del Vecchio M, Robert C, Mackiewicz A, Chiarion-Sileni V, Arance A, et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2017;18:611–22. [DOI] [PubMed] [Google Scholar]
- 11.Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. New England Journal of Medicine. 2018;378:1976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24:1649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blank CU, Rozeman EA, Fanchi LF, Sikorska K, van de Wiel B, Kvistborg P, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nature Medicine. 2018;24:1655. [DOI] [PubMed] [Google Scholar]
- 14.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25:470–6. [DOI] [PubMed] [Google Scholar]
- 15.Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med. 2019;25:454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tacken PJ, Figdor CG. Targeted antigen delivery and activation of dendritic cells in vivo: Steps towards cost effective vaccines. Seminars in Immunology. 2011;23:12–20. [DOI] [PubMed] [Google Scholar]
- 17.Paschall AV, Liu K. An Orthotopic Mouse Model of Spontaneous Breast Cancer Metastasis. J Vis Exp [Internet]. 2016. [cited 2020 Apr 1]; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5091834/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen P, Wuthrick E, Chablani P, Robinson A, Simmons L, Wu C, et al. Does Delaying Surgical Resection After Neoadjuvant Chemoradiation Impact Clinical Outcomes in Locally Advanced Rectal Adenocarcinoma?: A Single-Institution Experience. Am J Clin Oncol. 2018;41:140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aznar MA, Tinari N, Rullán AJ, Sánchez-Paulete AR, Rodriguez-Ruiz ME, Melero I. Intratumoral Delivery of Immunotherapy—Act Locally, Think Globally. The Journal of Immunology. 2017;198:31–9. [DOI] [PubMed] [Google Scholar]
- 20.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science. 2017;355:1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31. [DOI] [PubMed] [Google Scholar]
- 23.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alicea-Torres K, Sanseviero E, Gui J, Chen J, Veglia F, Yu Q, et al. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat Commun. 2021;12:1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived Suppressor Cells Inhibit T Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010;70:68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, O’Donnell JS, Yan J, Madore J, Allen S, Smyth MJ, et al. Timing of neoadjuvant immunotherapy in relation to surgery is crucial for outcome. Oncoimmunology [Internet]. 2019. [cited 2020 Apr 1];8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6492961/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J, Blake SJ, Yong MCR, Harjunpää H, Ngiow SF, Takeda K, et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov. 2016;6:1382–99. [DOI] [PubMed] [Google Scholar]
- 28.Duhen R, Ballesteros-Merino C, Frye AK, Tran E, Rajamanickam V, Chang S-C, et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nat Commun. Nature Publishing Group; 2021;12:1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pataer A, Shao R, Correa AM, Behrens C, Roth JA, Vaporciyan AA, et al. Major pathologic response and RAD51 predict survival in lung cancer patients receiving neoadjuvant chemotherapy. Cancer Med. 2018;7:2405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. WT BALB/c mice (n =5) were inoculated with 500,000 CT26-Luc tumor cells intravenously into the tail vein. After a week pulmonary metastases were evaluated using IVIS. All mice that underwent tail vein injections had sufficient tumor burden in their lungs to be visible via IVIS. When untreated the pulmonary tumor burden increased and persisted in all inoculated mice.
Supplemental Figure 2. WT BALB/c mice underwent injections of anti-CD4 antibody, anti-CD8 antibody, both or neither prior to undergoing neoadjuvant immunotherapy. Depletion of CD4+ and CD8+ T cells were confirmed in the relevant groups via flow cytometry of peripheral blood.
Supplemental Figure 3. Individual primary tumor growth curves for neoadjuvant immunotherapy, immunotherapy alone and neoadjuvant vehicle in the 4T1 tumor model. Groups of BALB/c WT mice (n = 10) were inoculated with 7.5x104 mammary carcinoma 4T1-Luc tumor cells orthotopically into the right mammary fat pad. Groups of mice were treated with local administration of neoadjuvant immunotherapy (3X injections, 50ug CpG and 8ug aOX40 antibody per injection), immunotherapy alone (3X injections of 50ug of CpG and 8ug of aOX40 antibody per injection), or neoadjuvant vehicle.
Supplemental Figure 4. Flow cytometry gating hierarchy for T cell stimulation assay. Splenocytes were first positively selected for CD4 and CD8 positive cells, then for CD44 and IFN-gamma.
Supplemental Figure 5. Groups of BALB/c WT mice (n = 4) were inoculated with 5x105 CT26-Luc colorectal carcinoma tumor cells into tail vein followed a day later with inoculation of 5x105 CT26-Luc tumor cells subcutaneously into the right side of the abdomen to simulate metastatic and local disease respectively. Groups of mice were treated with intratumoral administration of neoadjuvant immunotherapy (3X injections, 50ug CpG and 8ug aOX40 antibody per injection vs CpG alone vs aOX40 alone vs vehicle) followed by resection. Primary tumors were harvested and levels of IL-12, IFN-alpha and IFN-gamma in tumor tissue supernatants measured by ELISA.