

Abstract
Background:
Calcium (Ca2+) is a key regulator of energy metabolism. Impaired Ca2+ homeostasis damages mitochondria, causing cardiomyocyte death, pathological hypertrophy, and heart failure. This study investigates the regulation and the role of the mitochondrial Ca2+ uniporter (MCU) in chronic stress-induced pathological cardiac remodeling.
Methods:
MCU knockout or transgenic mice were infused with isoproterenol (ISO, 10 mg/kg/day, 4 weeks). Cardiac hypertrophy and remodeling were evaluated by echocardiography and histology. Primary cultured rodent adult cardiomyocytes were treated with ISO (1 nM, 48 hr). Intracellular Ca2+ handling and cell death pathways were monitored. Adenovirus-mediated gene manipulations were used in vitro.
Results:
Chronic administration of the β-adrenergic receptor (β-AR) agonist ISO increased the levels of the MCU and the MCU complex in cardiac mitochondria, raising mitochondrial Ca2+ concentrations, in vivo and in vitro. ISO also upregulated MCU without affecting its regulatory proteins in adult cardiomyocytes. Interestingly, ISO-induced cardiac hypertrophy, fibrosis, contractile dysfunction, and cardiomyocyte death were exacerbated in global MCU knockout (KO) mice. Cardiomyocytes from KO mice or mice overexpressing a dominant negative MCU exhibited defective intracellular Ca2+ handling and activation of multiple cell death pathways. Conversely, cardiac-specific overexpression of MCU maintained intracellular Ca2+ homeostasis and contractility, suppressed cell death, and prevented ISO-induced heart hypertrophy. ISO upregulated MCU expression through activation of Ca2+/calmodulin kinase II δB (CaMKIIδB) and promotion of its nuclear translocation via calcineurin-mediated dephosphorylation at serine 332. Nuclear CaMKIIδB phosphorylated cAMP-response element binding protein (CREB), which bound the MCU promotor to enhance MCU gene transcription.
Conclusions:
The β-AR/CaMKIIδB/CREB pathway upregulates MCU gene expression in the heart. MCU upregulation is a compensatory mechanism that counteracts stress-induced pathological cardiac remodeling by preserving Ca2+ homeostasis and cardiomyocyte viability.
Keywords: Animal Models of Human Disease, Basic Science Research, Cell Biology/Structural Biology, Metabolism, Heart Failure, Mitochondrial Ca2+ uniporter, Calcium calmodulin kinase II δB, β-adrenergic receptor, Cardiomyocyte death, Pathological cardiac hypertrophy
Introduction
Impaired mitochondrial energy metabolism is a key contributor to human disease, including heart failure.1 Mitochondrial Ca2+ has a critical role in regulating energy metabolism, and dysregulated Ca2+ can cause mitochondrial damage and even cell death.2,3 The mitochondrial Ca2+ uniporter (MCU), which forms a complex with its regulatory proteins, is responsible for mitochondrial Ca2+ uptake across the inner membrane.4–6 Although, MCU may be dispensable for heart function and energetics under normal conditions, it participates in metabolic adaptation during acute β-adrenergic receptor (β-AR) activation and ischemia-reperfusion injury in the heart.7–9 Until now, the regulation and role of MCU in chronic stress-induced pathological cardiac remodeling had remained poorly understood.6 MCU is upregulated in the heart by exercise and pressure overload, suggesting that its function is complicated and depends on the context.10 In addition, either excessive or absent MCU-mediated Ca2+ uptake can cause cell death and mitochondrial damage.11,12 For instance, MCU knockout (KO) increases Ca2+ sensitivity of the mitochondrial permeability transition pore (mPTP).11 However, transcriptional, translational, and post-translational regulation of MCU under chronic pathological conditions have not been thoroughly investigated.10,12–14
β-adrenergic receptor (β-AR) signaling is the most powerful regulatory mechanism in the heart, as it underlies both the acute fight-or-flight response and chronic stress-induced cardiac pathology.15 β1-AR is the major β-AR subtype that stimulates multiple signaling pathways, including cyclic adenosine monophosphate / protein kinase A (cAMP/PKA), exchange protein directly activated by cAMP, calcineurin, and Ca2+/calmodulin kinase II (CaMKII).16,17 Chronic β1-AR activation leads to heart hypertrophy and dysfunction mainly through CaMKII.18 One major CaMKII subtype is CaMKIIδC, which induces cardiomyocyte death and mitochondrial dysfunction.19 However, CaMKIIδB, another major CaMKII subtype, may benefit the heart through mechanisms that are not fully established.20
Here, we demonstrated that the β-AR/CaMKIIδB/cAMP-response element binding protein (CREB) pathway increases MCU gene transcription in the stressed heart. By using gain- or loss-of-function approaches, we found that MCU upregulation maintains intracellular Ca2+ and energy homeostasis, prevents cell death, limits cardiac hypertrophy, and preserves heart function during chronic stress. Therefore, MCU is a stress-responsive gene underlying the beneficial effects of CaMKIIδB in pathological cardiac remodeling.
Methods
Detailed methods are in the Online Supplemental Materials. The authors declare that all supporting data are available within the article and the Online Data Supplement. The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. Raw data are available from the corresponding author on reasonable request.
Animals
All animal procedures were performed in accordance with the approved protocol by the Institutional Animal Care and Use Committee (IACUC) of the University of Washington. The experiments included in this study used both male and female mice.
Statistical Analysis
Data are shown as mean ± standard error (SEM). When multiple experiments using different numbers of animals were pooled for statistical analysis, the range of animal numbers was indicated in the figure legend. Statistical analyses used GraphPad Prism 7.02 software. Shapiro-Wilk normality test was performed to determine the data distribution. Non-normally distributed data were analyzed by nonparametric tests: Kruskal-Wallis test followed by Dunn’s post hoc analysis for >2 groups or Mann-Whitney test for 2 groups. Normally distributed data were analyzed by using parametric tests: one-way analysis of variance (ANOVA) followed by Tukey test for >2 groups or unpaired Student’s t-test for 2 groups. Multiple testing adjustment was included in the Dunn’s test and Tukey test through controlling the Type I error for the family of comparisons. Experiments consisting of two categorically independent variables and one dependent variable were analyzed by two-way ANOVA. A P value <0.05 was considered statistically significant.
Results
Chronic β-AR activation upregulated MCU and the MCU complex in cardiac mitochondria
The β-AR agonist isoproterenol (ISO, 10 mg/kg/day for 1–4 weeks) increased protein levels of MCU and MCU complex in the mitochondrial fraction and tissue homogenates of mouse heart (Figure 1A and Supplemental Figure 1A). Meanwhile, MCU regulatory proteins were unchanged, except for MCUb and EMRE, which decreased during weeks 1 and 4, respectively (Supplemental Figure 1A). In cultured mouse cardiomyocytes, ISO dose- and time-dependently elevated the level of MCU protein but had mild or no effects on MCU regulatory proteins (Figure 1B–C, Supplemental Figure 1B–C). ISO also upregulated MCU in adult rat cardiomyocytes and H9C2 cardiomyoblasts (Supplemental Figure 1D–E). Functionally, upregulation of MCU and MCU complex was accompanied by higher Ca2+ concentrations in cardiac mitochondria, both in vivo and in vitro (Figure 1D–E), and accelerated Ca2+ uptake by isolated mitochondria after each bolus Ca2+ addition (Figure 1F). These results suggest that chronic β-AR activation specifically targets MCU and promotes mitochondrial Ca2+ uptake in cardiomyocytes.
Figure. 1. Chronic β-AR activation upregulated MCU in the heart and in cardiomyocytes.
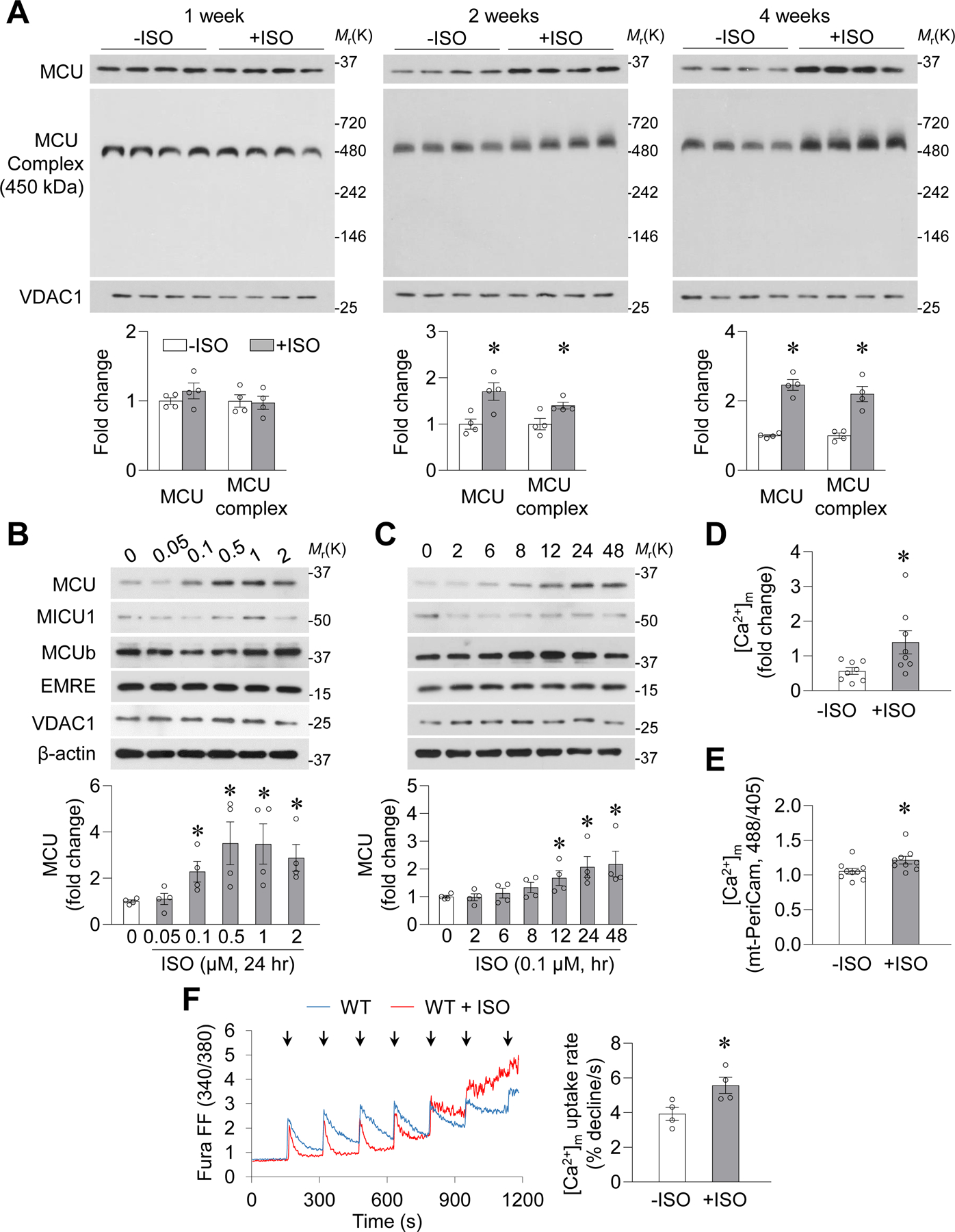
A, MCU and MCU complex measured in cardiac mitochondria after isoproterenol (+ISO, 10 mg/kg/day) or vehicle (−ISO) administration in mice for 1, 2 (*: P=0.018 or 0.0313 vs. −ISO for MCU or MCU complex, n=4), and 4 weeks (*: P=8.6E-05 or 0.0021 vs. −ISO for MCU or MCU complex, n=4). B-C, Dose- and time-dependent changes in MCU and associated proteins in adult mouse cardiomyocytes after ISO incubation. In B, *: P=0.0294, 0.0352, 0.0296, or 0.0176 vs. −ISO for 0.1, 0.5, 1 or 2 μmol/L ISO groups, respectively. In C, *: P=0.046, 0.0309 or 0.0438 vs −ISO for 12, 24 or 48 hr ISO groups, respectively. n=4. D, Mitochondrial matrix free Ca2+ concentrations ([Ca2+]m) determined by rhod-2 and FCCP-induced mitochondrial Ca2+ release in freshly isolated and permeabilized adult mouse cardiomyocytes after 4-week ISO administration. *: P=0.032 vs. −ISO. n=8 mice. E, Steady state mitochondrial Ca2+ concentrations measured by mt-PeriCam in adult rat cardiomyocytes after ISO (0.1 μmol/L, 24 hr) incubation. *: P=0.036 vs. −ISO. n=9 rats. F, Representative traces and summarized data showing Ca2+ uptake by freshly isolated mitochondria from the hearts of WT mice with or without in vivo ISO stimulation (ISO, 10 mg/kg/day, 4 weeks). Arrows: additions of Ca2+ (25 μmol/L each time). *: P=0.0337 vs. −ISO, n=4 mice.
MCU KO exacerbated ISO-induced pathological cardiac hypertrophy
To explore the consequences of MCU upregulation in the heart, we used global MCU KO mice.7 MCU and MCU complex were absent from KO mitochondria; however, MCU regulatory proteins were not significantly changed (Figure 2A and Supplemental Figure 2A). MCU KO mitochondria did not take up Ca2+ and had lower levels of free Ca2+ in the matrix (Supplemental Figure 2B–C). Unexpectedly, ISO (10 mg/kg/day, 4 weeks)-induced heart hypertrophy (heart weight to body weight ratio, heart weight to tibia length ratio, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) mRNA levels, and cross-sectional area of cardiomyocytes), contractile dysfunction (ejection fraction and fractional shortening), and fibrosis (Picrosirius red staining and Col1a2 mRNA level) were all exacerbated in KO heart (Figure 2B–H and Supplemental Figure 2D–G). In addition, ISO induced apoptosis (caspase 3/7 activity) in KO heart tissue and higher levels of cell death in cultured adult cardiomyocytes from KO mice (Figure 2I and Supplemental Figure 2H). These results indicate that the lack of MCU potentiates, rather than ameliorates, pathological cardiac remodeling induced by chronic β-AR activation.
Figure. 2. MCU KO exacerbated cardiac hypertrophy and dysfunction during chronic β-AR activation in mice.
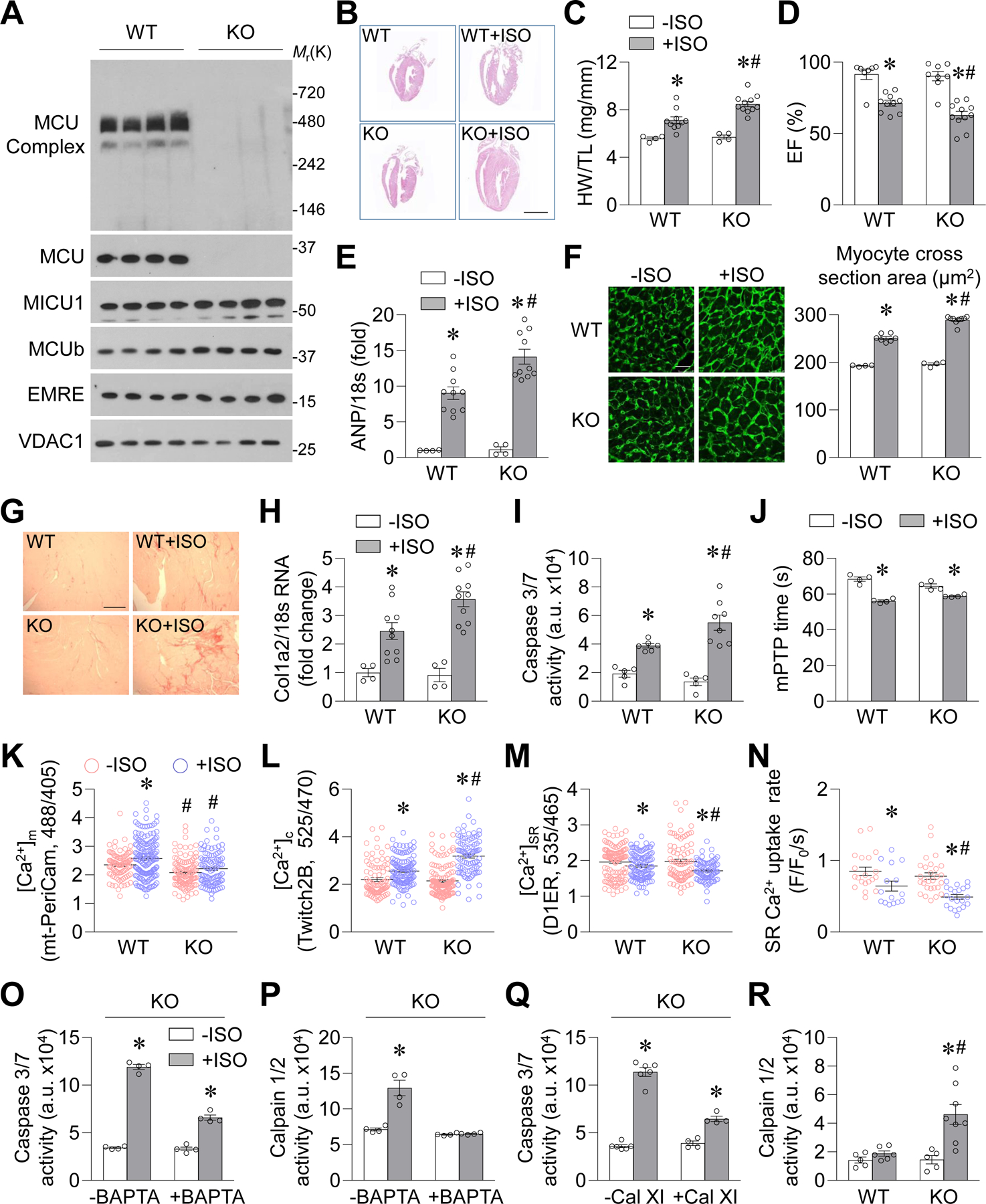
A, Western blotting images showing MCU complex, MCU, and MCU regulatory proteins in mitochondrial fractions from wild type (WT) or MCU knockout (KO) mouse heart. n=4. B, Representative coronal sections of the heart with (+ISO) or without (−ISO) 4-week ISO administration (10 mg/kg/day). Scale bar = 5 mm. C, Heart weight to tibia length ratio (HW/TL). *: P=0.0053 WT+ISO vs. WT−ISO, *: P=9.57E-06 KO+ISO vs. KO−ISO, #: P=0.0013 KO+ISO vs. KO−ISO. n=5–6 for −ISO groups and n=10–11 for +ISO groups. D, Left ventricular ejection fraction. *: P=0.0001 WT+ISO vs. WT−ISO, *: P=5.84E-06 KO+ISO vs. KO−ISO, #: P=0.0246 KO+ISO vs. KO−ISO. n=7–11. E, mRNA levels of atrial natriuretic peptide (ANP) in mouse heart. *: P=0.0006 WT+ISO vs. WT−ISO, *: P=5.17E-05 KO+ISO vs. KO−ISO, #: P=0.0025 KO+ISO vs. WT+ISO. n=4 for −ISO groups and n=10 for +ISO groups. F-G, Wheat germ agglutinin staining for cardiomyocyte cross-sectional area (F, *: P=5.97E-07 WT+ISO vs. WT−ISO, *: P=5.73E-09 KO+ISO vs. KO−ISO, #: P=1.17E-06 KO+ISO vs. WT+ISO) and Picrosirius red staining for fibrosis (G). n=4 for −ISO groups and n=7–9 for +ISO groups. Scale bars = 100 μm. H, mRNA level of collagen type I α2 chain (Col1a2) in mouse heart. *: P=0.0441 WT+ISO vs. WT−ISO, *: P=0.0004 KO+ISO vs. KO−ISO, #: P=0.0119 KO+ISO vs. WT+ISO. n=4 for −ISO groups and n=10 for +ISO groups. I, Caspase 3/7 activity in mouse heart. *: P=0.0003 WT+ISO vs. WT−ISO, *: P=0.0004 KO+ISO vs. KO−ISO, #: P=0.0322 KO+ISO vs. WT+ISO. n=5 for −ISO groups and n=6–8 for +ISO groups. J, mPTP time in cardiomyocytes after ISO (0.1 μmol/L, 24 hr) incubation. *: P=0.0003 WT+ISO vs. WT−ISO, *: P=0.0061 KO+ISO vs. KO−ISO. n=4 mice. K-N, Mitochondrial Ca2+ concentrations (K, [Ca2+]m, measured by mt-PeriCam, *: P=0.0048 WT+ISO vs. WT−ISO, #: P=7E-05 KO−ISO vs. WT−ISO, #: P=2.66E-050.0637 KO+ISO vs. WT+ISO, n=97–157 cells from 4–5 mice), cytosolic Ca2+ concentrations (L, [Ca2+]c, measured by Twitch2B, *: P=9.37E-05 WT+ISO vs. WT−ISO, *: P=2.6E-22 KO+ISO vs. KO−ISO, #: P=6.5E-10 KO+ISO vs. WT+ISO, n=87–152 cells from 4–5 mice), SR Ca2+ content (M, [Ca2+]SR, measured by D1ER, *: P=0.0485 WT+ISO vs. WT−ISO, *: P=0.0002 KO+ISO vs. KO−ISO, #: P=0.0054 KO+ISO vs. WT+ISO, n=78–126 cells from 4–5 mice), and SR Ca2+ uptake rate (N, *: P=0.0291 WT+ISO vs. WT−ISO, *: P=1.31E-05 KO+ISO vs. KO−ISO, #: P=0.0357 KO+ISO vs. WT+ISO, n=15–28 cells from 4–5 mice) in adult cardiomyocytes from WT or KO mice after 4-week ISO administration. For K-M, cells in +ISO groups were cultured with ISO (0.1 μmol/L) for 2 days for indicator expression. For N, freshly isolated cells were used. O-Q, Effects of BAPTA-AM (0.1 μmol/L, O-P) or calpain inhibitor XI (Cal XI, 1 μmol/L, Q) on ISO (0.1 μmol/L, 24 hr)-induced caspase 3/7 activity (O, Q) or calpain1/2 activity (P) in adult cardiomyocytes from KO mice. In O, *: P=7.723E-08 +ISO vs. −ISO, *: P=7.01E-05 BAPTA+ISO vs. BAPTA−ISO. In P, *: P=0.0019 +ISO vs. −ISO. In Q, *: P=1.58E-08 +ISO vs. −ISO, *: P=0.0004 BAPTA+ISO vs. BAPTA−ISO. n=4. R, Calpain1/2 activity in mouse heart. *: P=0.0007 KO+ISO vs. KO−ISO, #: P=0.0357 KO+ISO vs. WT+ISO. n=5 for −ISO groups and n=6–8 for +ISO groups.
To determine whether the above phenotypes in global MCU KO hearts are cardiomyocyte-specific, we crossed MCUfl/fl mice with αMHC-MerCreMer (MCM) mice to generate cardiomyocyte-specific MCU KO mice (cKO, Supplemental Figure 3A).8,9 ISO (10 mg/kg/day, 4 weeks)-induced heart hypertrophy, contractile dysfunction, fibrosis, and apoptosis were also exacerbated in cKO hearts (Supplemental Figure 3B–I), suggesting that the lack of MCU in cardiomyocytes facilitates pathological cardiac remodeling.
MCU KO impaired intracellular Ca2+ handling and activated cell death pathways
To investigate why MCU KO exacerbated pathological cardiac remodeling, we first found that ISO increased the sensitivity of laser-induced mitochondrial permeability transition pore (mPTP) openings similarly in cultured wild-type (WT) and KO cardiomyocytes (Figure 2J), suggesting that MCU KO does not further activate the mitochondrial cell death pathway. Next, we explored Ca2+ handling in cardiomyocytes after the 4-week period of ISO administration. As expected, ISO elevated mitochondrial Ca2+ concentrations (measured by mt-PeriCam) in WT, but not in KO, cardiomyocytes (Figure 2K and Supplemental Figure 4A). Interestingly, ISO increased diastolic cytosolic Ca2+ concentrations (measured by Twitch2B), decreased sarcoplasmic reticulum (SR) Ca2+ content (measured by D1ER), and suppressed the SR Ca2+ uptake rate (decay of caffeine-induced Ca2+ transients) significantly more in MCU KO cardiomyocytes compared to WT (Figure 2L–N and Supplemental Figure 4B–D), indicating that MCU KO magnified intracellular Ca2+ dysregulation. Because SR stress markers and sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) 2 levels were comparable in WT and KO hearts (Supplemental Figure 4E–F), the decreased SR Ca2+ uptake rate in MCU KO cardiomyocytes might be due to compromised SERCA function.
Subsequently, we determined whether the elevated cytosolic Ca2+ concentrations were responsible for increased cell death in KO mouse heart tissue. Chelation of cytosolic Ca2+ with BAPTA-AM (0.1 μmol/L) suppressed ISO-induced caspase 3/7 activation in both WT and MCU KO cardiomyocytes (Supplemental Figure 4G and Figure 2O). However, ISO increased the activity of calpain,21,22 a protease activated by high levels of Ca2+, only in KO cardiomyocytes, and BAPTA treatment abolished calpain activation (Supplemental Figure 4H and Figure 2P). Moreover, calpain inhibitor XI (Cal XI) blocked ISO-induced activation of calpain and caspase 3/7 in KO cardiomyocytes (Supplemental Figure 4I–J and Figure 2Q). Finally, increased calpain activity was found in KO or cKO, but not control, hearts after the 4-week period of ISO administration (Figure 2R and Supplemental Figure 4K). These results suggest that cytosolic Ca2+ overload and calpain activation may mediate the increased cell death observed in MCU KO cardiomyocytes.
MCU maintained Ca2+ and energy homeostasis in cardiomyocytes
To explore how MCU-mediated mitochondrial Ca2+ uptake regulates intracellular Ca2+ homeostasis and cell viability, we inhibited or enhanced MCU function in cultured adult rat cardiomyocytes. First, overexpressing a dominant negative MCU (DNMCU, Supplemental Figure 5A)5 recapitulated the dysregulated intracellular Ca2+ handling found in MCU KO cardiomyocytes. DNMCU attenuated mitochondrial Ca2+ uptake in permeabilized cardiomyocytes, reduced matrix Ca2+ levels at baseline, and abolished ISO (0.1 μmol/L, 24 hr)-induced mitochondrial Ca2+ elevation (Supplemental Figure 5B and Figure 3A). In cultured rat cardiomyocytes, ISO (0.1 μmol/L, 24 hr) had no effect on cytosolic or SR Ca2+ content but decreased the amplitude and decay velocity of pacing-induced Ca2+ transients (Figure 3B–D and Supplemental Figure 5C–D). In DNMCU-overexpressing cells, ISO elevated cytosolic Ca2+ concentrations, lowered SR Ca2+ content, and further decreased the amplitude and decay velocity of pacing-induced Ca2+ transients (Figure 3B–D and Supplemental Figure 5C–D). Conversely, overexpression of FLAG-tagged MCU in adult rat cardiomyocytes elevated mitochondrial Ca2+ concentrations, lowered cytosolic Ca2+ concentrations, and increased SR Ca2+ content at baseline and after ISO incubation (Figure 3E–H).
Figure. 3. MCU modulated Ca2+ and energy homeostasis in rat cardiomyocytes during chronic ISO incubation.
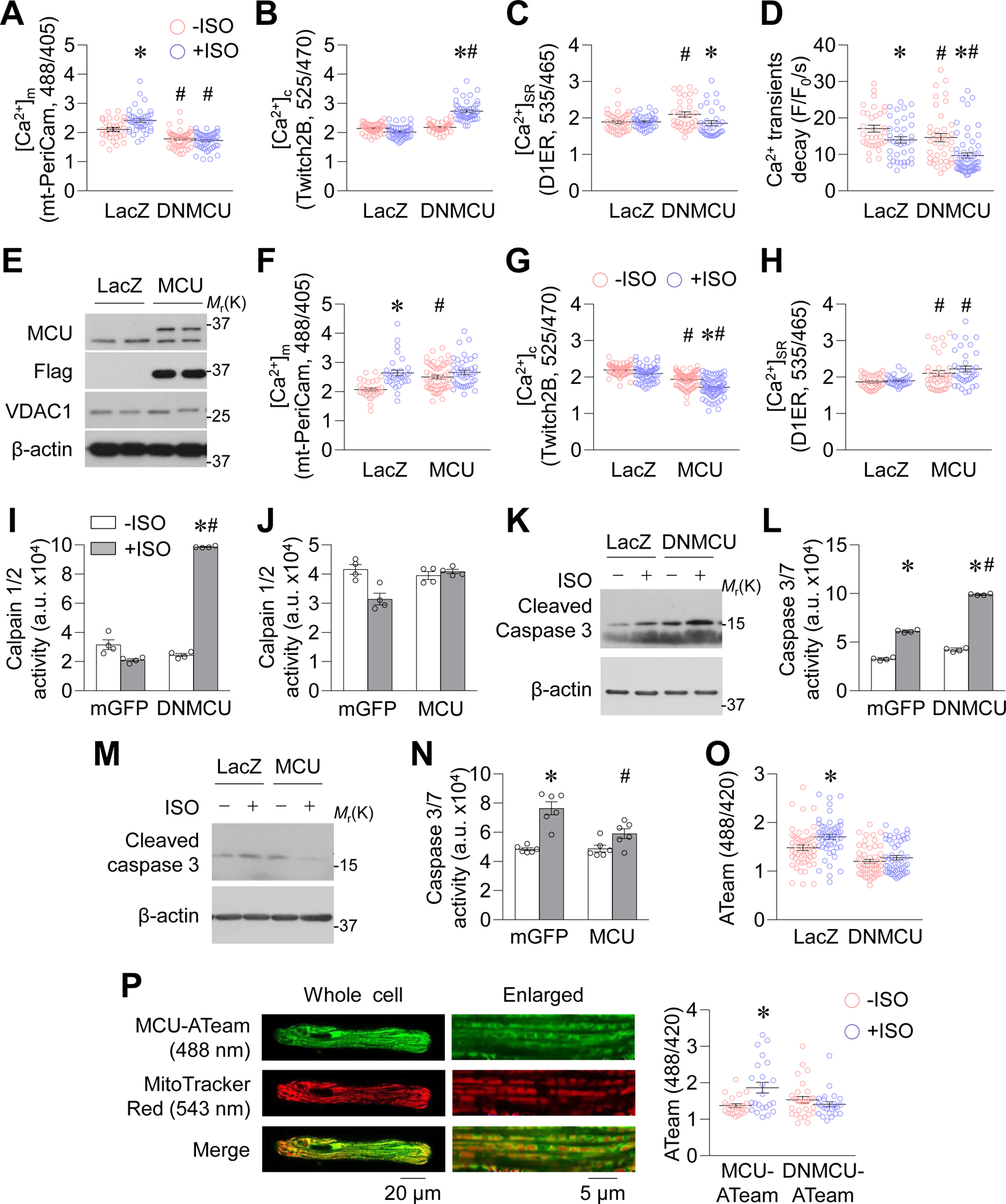
A-D, Mitochondrial Ca2+ concentrations (A, [Ca2+]m, measured by mt-PeriCam, *: P=0.0013 LacZ+ISO vs. LacZ−ISO, #: P=2.979E-05 DNMCU−ISO vs. LacZ−ISO, #: P=2E-15 DNMCU+ISO vs. LacZ+ISO, n=48–71 cells from 5–7 rats), cytosolic Ca2+ concentrations (B, [Ca2+]c, measured by Twitch2B, *: P=8.32E-12 DNMCU+ISO vs. DNMCU−ISO, #: P=2.71E-23 DNMCU+ISO vs. LacZ+ISO, n=36–69 cells from 4–7 rats), SR Ca2+ content (C, [Ca2+]SR, measured by D1ER, #: P=0.0121 DNMCU−ISO vs. LacZ−ISO, *: P=0.0155 DNMCU+ISO vs. DNMCU−ISO, n=33–41 cells from 4 rats), and the decay rate of pacing-induced cytosolic Ca2+ transients (D, measured by GCaMP6f, *: P=0.0202 LacZ+ISO vs. LacZ−ISO, #: P=0.0113 DNMCU−ISO vs. LacZ−ISO, *: P=0.0002 DNMCU+ISO vs. DNMCU−ISO, #: P=0.0003 DNMCU+ISO vs. LacZ+ISO, n=33–60 cells from 5 rats) in adult rat cardiomyocytes overexpressing dominant negative MCU (DNMCU) and incubated with ISO (0.1 μmol/L, 24 hr). E, Western blotting images showing the protein levels of overexpressed flag-tagged MCU in adult rat cardiomyocytes. F-H, [Ca2+]m, [Ca2+]c, and [Ca2+]SR in adult rat cardiomyocytes overexpressing MCU and with ISO (0.1 μmol/L, 24 hr) incubation. In F, *: P=3.86E-06 LacZ+ISO vs. LacZ−ISO, #: P=0.0964 MCU+ISO vs. LacZ+ISO, n=48–71 cells from 5–7 rats. In G, #: P=1.5E-05 MCU−ISO vs. LacZ−ISO, *: P=8.9E-08 MCU+ISO vs. MCU−ISO, #: P=3.27E-16 MCU+ISO vs. LacZ+ISO, n=36–69 cells from 4–7 rats. In H, #: P=0.0238 MCU−ISO vs. LacZ−ISO, #: P=0.0006 MCU+ISO vs. LacZ+ISO, n=33–41 cells from 4 rats. I-J, Calpain 1/2 activity in adult rat cardiomyocytes. In I, *: P=8.4E-10 DNMCU+ISO vs. DNMCU−ISO, #: P=2.70E-10 DNMCU+ISO vs. mGFP+ISO. n=4. For J, n=6. K-N, Cleaved caspase 3 (K, M) and caspase 3/7 activity (L, N) in adult rat cardiomyocytes. n=3 for K and M. In L, *: P=1.13E-07 mGFP+ISO vs. mGFP−ISO, *: P=2.44E-09 DNMCU+ISO vs. DNMCU−ISO, #: P=6.89E-09 DNMCU+ISO vs. mGFP+ISO, n=4. In N, *: P=0.0077 mGFP+ISO vs. mGFP−ISO, #: P=0.0232 MCU+ISO vs. mGFP+ISO, n=6. O, Cytosolic ATP levels measured by ATeam in adult rat cardiomyocytes. *: P=0.0048 LacZ+ISO vs. LacZ−ISO. n=51–53 cells from 5 rats. For A-O, LacZ or mGFP was expressed in Control cells. P, Representative confocal images and summarized data showing mitochondrial ATP measurements in adult rat cardiomyocytes overexpressing MCU-ATeam or DNMCU-ATeam and with ISO incubation. *: P=0.003 MCU-ATeam +ISO vs. MCU-ATeam −ISO. n=23–30 cells from 4–6 rats.
DNMCU overexpression activated calpain and further enhanced caspase 3/7 activity; however, MCU overexpression exhibited no calpain activation and attenuated caspase 3 activation after ISO incubation (Figure 3I–N). Finally, we used an ATP indicator (ATeam) to monitor steady-state ATP levels in cultured rat cardiomyocytes.23 ISO elevated cytosolic ATP levels in control, but not in DNMCU-overexpressing, cardiomyocytes (Figure 3O). Moreover, the free mitochondrial ATP level (mt-ATeam) was increased by ISO, while DNMCU abolished this effect (Supplemental Figure 5E and Figure 3P). Mitochondrial respiration trended higher after ISO incubation or MCU overexpression, and lower after DNMCU overexpression in adult rat cardiomyocytes (Supplemental Figure 5F). Taken together, our results suggest that inhibition of MCU impairs, whereas overexpression of MCU benefits, energy and Ca2+ homeostasis in cardiomyocytes during chronic stress.
MCU transgenic mice were protected during chronic β-AR activation
To determine the significance of MCU upregulation in stress-induced heart dysfunction in vivo, we generated inducible, heart-specific MCU transgenic (TG) mice, which express FLAG-tagged MCU (MCU-Flag) after tamoxifen (5 mg/kg/day for 2 days) induction (Supplemental Figure 6A–B). MCU TG mice had increased levels of MCU complex and decreased phosphorylation of pyruvate dehydrogenase in cardiac mitochondria, but they had no differences in MCU regulatory proteins, mitochondrial Na+/Ca2+ exchanger (NCLX), and SERCA2a levels (Figure 4A and Supplemental Figure 6C–D), which is consistent with a central role of MCU in MCU complex assembly and mitochondrial energy metabolism. Notably, mitochondrial Ca2+ uptake assays revealed that isolated mitochondria from MCU TG heart tissue took up more Ca2+ after each bolus Ca2+ addition and at a faster rate than WT mitochondria (Figure 4B).
Figure. 4. MCU TG ameliorated heart dysfunction induced by chronic β-AR activation.
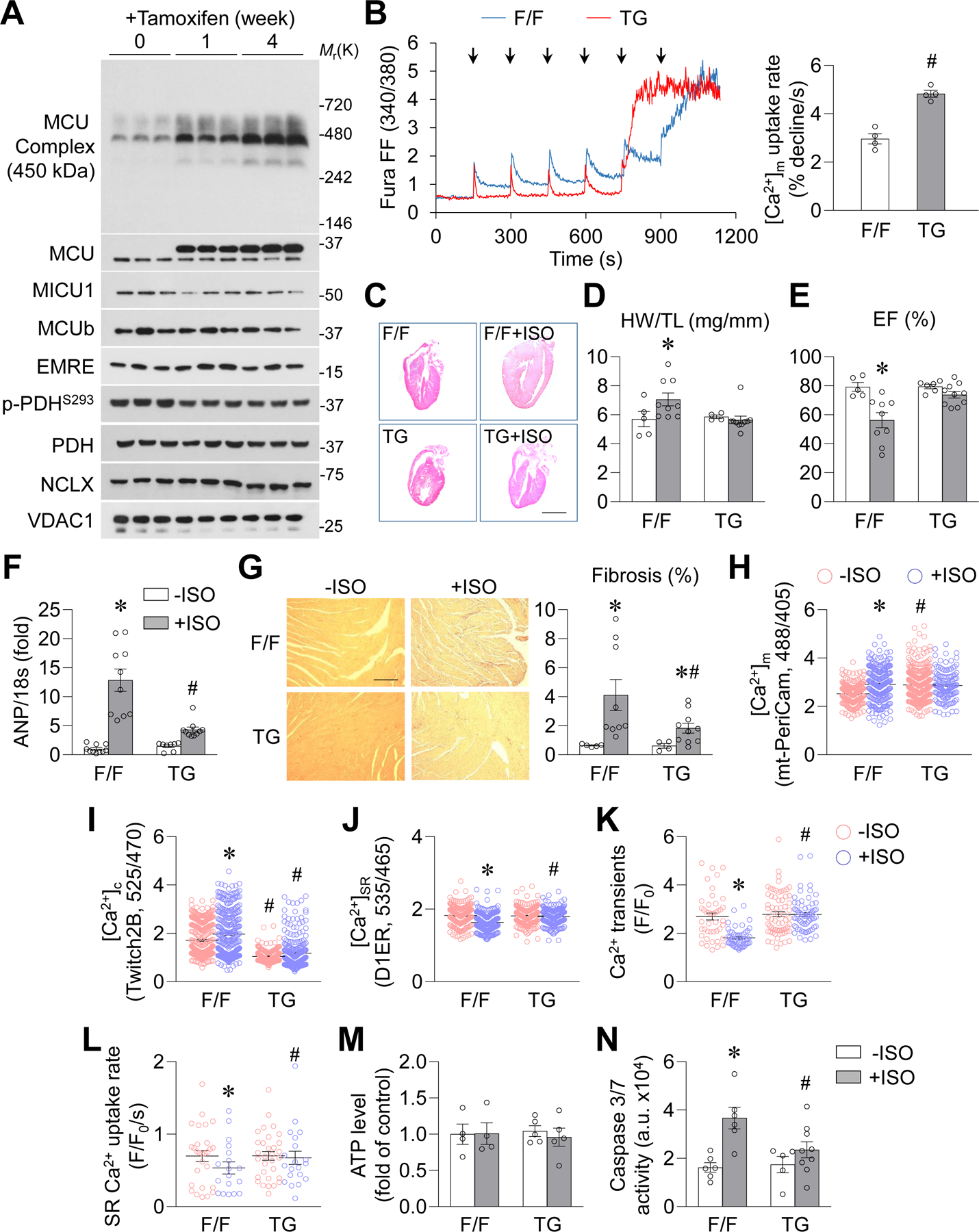
A, Time-dependent changes in MCU, MCU complex, MCU regulatory proteins, NCLX and PDH phosphorylation in cardiac mitochondria from MCU floxed/floxed (F/F) mice after tamoxifen induction. n=3. B, Representative traces and normalized data showing Ca2+ uptake by freshly isolated mitochondria from the heart of MCU TG (TG) or F/F mouse. #: P=0.0003 TG vs. F/F. n=4. Arrows indicate the additions of Ca2+ (25 μmol/L each time). C, Representative H&E staining images of the coronal section of whole heart from F/F or TG mouse after ISO administration (10 mg/kg/day, 4 weeks). Scale bar = 5 mm. D, Heart weight to tibia length ratio (HW/TL) in F/F or TG mouse. *: P=1.13E-07 F/F+ISO vs. F/F−ISO. n=5 for −ISO groups and n=9–10 for +ISO groups. E, Left ventricular ejection fraction in F/F or TG mouse. *: P=0.0087 F/F+ISO vs. F/F−ISO. n=5–6 for −ISO groups and n=9–10 for +ISO groups. F, mRNA level of ANP in mouse heart. *: P=8.76E-06 F/F+ISO vs. F/F−ISO, #: P=0.0005 TG+ISO vs. F/F+ISO. n=8–10. G, Picrosirius red staining for fibrosis in mouse heart. *: P=0.0357 F/F+ISO vs. F/F−ISO, *: P=0.0499 TG+ISO vs. TG−ISO, #: P=0.0364 TG+ISO vs. F/F+ISO. n=5–6 for −ISO groups and n=9–10 for +ISO groups. Scale bar = 100 μm. H-L, Mitochondrial Ca2+ concentrations (H, [Ca2+]m, measured by mt-PeriCam, *: P=1.84E-05 F/F+ISO vs. F/F−ISO, #: P=0.0263 TG−ISO vs. F/F−ISO, n=132–329 cells from 4–5 mice), cytosolic Ca2+ concentrations (I, [Ca2+]c, measured by Twitch2B, *: P=0.0232 F/F+ISO vs. F/F−ISO, #: P=5.64E-11 TG−ISO vs. F/F−ISO, #: P=2.79E-06 TG+ISO vs. F/F+ISO, n=191–243 cells from 4–5 mice), SR Ca2+ content (J, [Ca2+]SR, measured by D1ER, *: P=3.18E-09 F/F+ISO vs. F/F−ISO, #: P=0.0002 TG+ISO vs. F/F+ISO, n=162–257 cells from 4–5 mice), pacing-induced cytosolic Ca2+ transients (K, fluo-8, *: P=1.95E-08 F/F+ISO vs. F/F−ISO, #: P=1.62E-17 TG+ISO vs. F/F−ISO, n=53–74 cells from 4–5 mice), and SR Ca2+ uptake rate (L, *: P=0.0265 F/F+ISO vs. F/F−ISO, #: P=0.0424 TG+ISO vs. F/F−ISO, n=22–36 cells from 4 mice) in adult cardiomyocytes from F/F or MCU TG mouse after 4-week ISO administration. For H-K, the cells in +ISO groups were cultured with ISO (0.1 μmol/L) for 2 days after isolation for indicator expression. For L, freshly isolated cells were used. M, ATP levels measured in whole heart homogenates. n=4–5. N, Caspase 3/7 activity in mouse hearts. *: P=0.0019 F/F+ISO vs. F/F−ISO, #: P=0.0319 TG+ISO vs. F/F+ISO. n=5–9.
ISO (10 mg/kg/day, 4 weeks)-induced cardiac hypertrophy, reduced contractility, and fibrosis were all attenuated in MCU TG mouse hearts compared to the control floxed/floxed Stop polyA MCU (F/F) or αMHC-MCM (MCM) mouse hearts (Figure 4C–G, Supplemental Figure 7A–E, and Supplemental Figure 8A–H). At baseline, adult cardiomyocytes from TG mice exhibited higher mitochondrial Ca2+ and lower cytosolic Ca2+ concentrations, as well as unchanged SR Ca2+ content, pacing-induced Ca2+ transients, and SR Ca2+ uptake rate (Figure 4H–L). After the 4-week period of ISO administration, mitochondrial Ca2+ concentration remained high and cytosolic Ca2+ concentration remained low in TG cardiomyocytes (Figure 4H–I). The SR Ca2+ content, pacing-induced Ca2+ transients, and SR Ca2+ uptake rate were unchanged in TG but decreased in F/F cardiomyocytes after ISO administration (Figure 4J–L). These results are consistent with the observations from MCU-overexpressing cardiomyocytes, indicating that upregulating MCU maintains intracellular Ca2+ homeostasis, prevents cytosolic Ca2+ overload, and facilitates SR Ca2+ uptake. Finally, the total ATP level was unchanged, calpain was not activated, and caspase 3/7 activity was suppressed in TG compared to F/F or MCM hearts after the 4-week ISO administration (Figure 4M–N, Supplemental Figure 7F, and Supplemental Figure 8I–J). In addition, whereas mPTP opening was suppressed in the TG heart, NCLX and SR stress markers were comparable in TG and F/F heart after ISO administration (Supplemental Figure 7G–I). In summary, overexpression of MCU protects the heart from stress-induced pathological hypertrophy through enhancing mitochondrial Ca2+ uptake and cytosolic Ca2+ cycling, which suppresses cell death pathways.
CaMKIIδB and calcineurin upregulated MCU
Next, we explored the signaling pathway responsible for MCU upregulation. First, we screened downstream signaling pathways of β-AR. Overexpressing PKA inhibitory peptide (PKI) did not affect, whereas CaMKII inhibitor KN93 (0.5 μmol/L) abolished, ISO-induced MCU upregulation in adult mouse cardiomyocytes (Figure 5A–B and Supplemental Figure 9A). In order to bypass the receptor, a constitutively active Gsα (caGsα),24 which stimulates downstream signaling pathways of β-AR, was over expressed. caGsα activated CaMKII through autophosphorylation of threonine 287 (T287), resulting in upregulation of MCU in a KN93-dependent manner (Figure 5C and Supplemental Figure 9B). Furthermore, shRNA-mediated CaMKII knockdown (shCaMKII) inhibited ISO-induced MCU upregulation (Figure 5D and Supplemental Figure 9C). These results suggest that CaMKII, but not PKA, is responsible for ISO-induced MCU upregulation.
Figure. 5. Chronic β-AR activation upregulated MCU through CaMKIIδB.
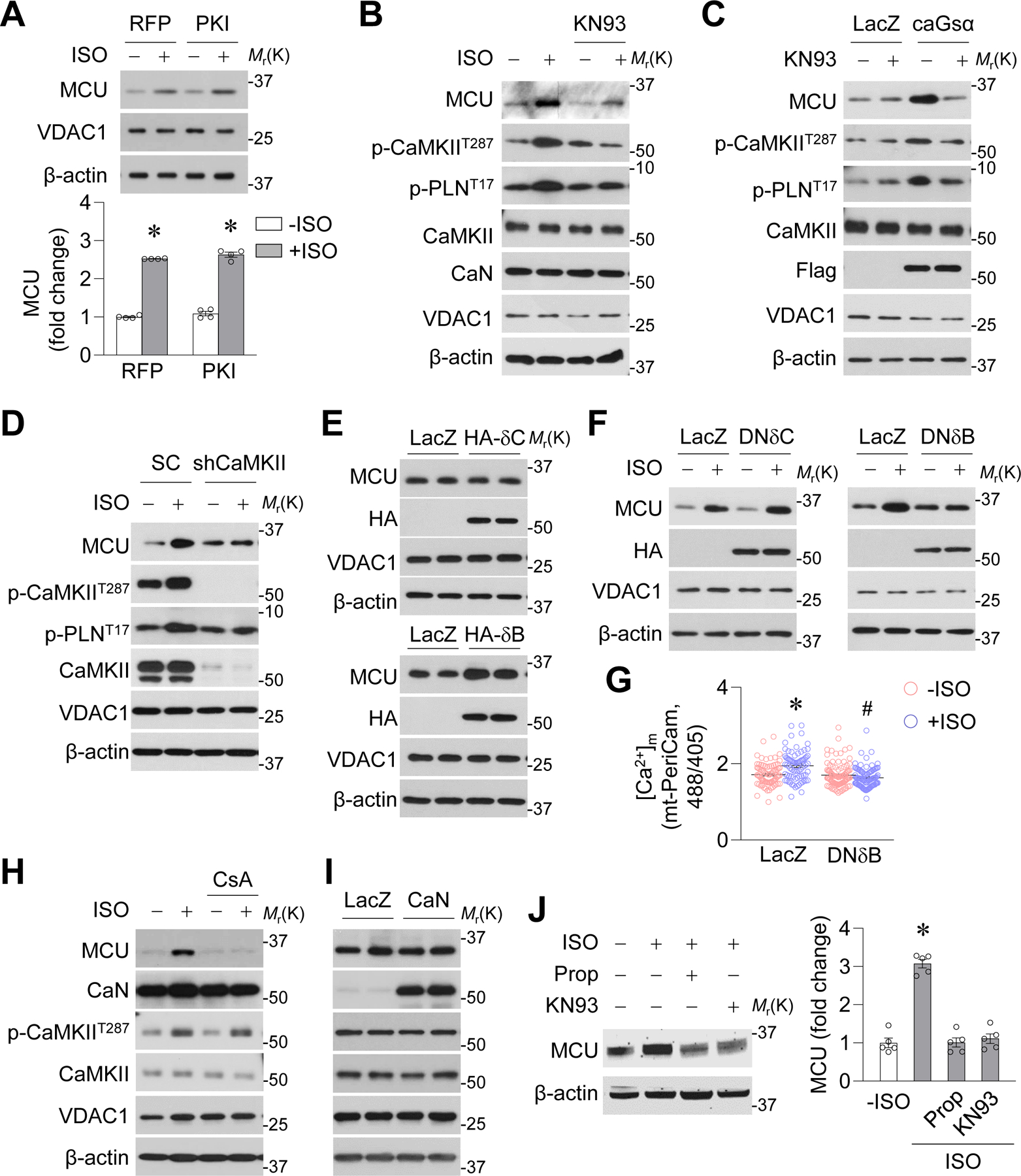
A-B, Effects of PKA inhibitory peptide (PKI, A, *: P=2.72E-10 RFP+ISO vs. RFP−ISO, *: P =2.04E-06 PKI+ISO vs. PKI−ISO) or CaMKII inhibitor (KN93, 0.5 μmol/L, B) on ISO (0.1 μmol/L, 24 hr) induced MCU upregulation in adult mouse cardiomyocytes. n=4. C, Effects of KN93 on constitutively active Gsα (caGsα) induced MCU upregulation. n=4. D, Effects of CaMKIIδ shRNA (shCaMKII) on ISO-induced MCU upregulation. n=4. E, Effects of overexpressing HA-tagged CaMKIIδC (δC) or δB on MCU expression. n=4. F, Effect of overexpressing HA-tagged dominant negative CaMKIIδC (DNδC) or DNδB on ISO-induced MCU upregulation. n=4. G, Effects of DNδB on mitochondrial Ca2+ concentrations (measured by mt-PeriCam) after ISO administration. *: P=3.04E-05 LacZ+ISO vs. LacZ−ISO, #: P=4.89E-10 DNδB+ISO vs. LacZ+ISO. n=79–98 cells from 4 mice. H, Effects of calcineurin (CaN) inhibitor cyclosporine A (CsA, 1 μmol/L) on ISO-induced MCU upregulation. n=4. I, Effects of CaN overexpression on MCU protein levels. n=4. J, Effects of β blocker propranolol (Prop, 10 mg/kg/day) or KN93 (10 μmol/kg/day) on ISO-induced MCU upregulation in mouse hearts *: P=1.529E-06 +ISO vs. −ISO. n=6.
Next, we determined which CaMKII subtype upregulates MCU. Adenovirus-mediated overexpression of HA-tagged CaMKIIδB, but not CaMKIIδC, increased MCU protein and mRNA levels in adult cardiomyocytes (Figure 5E and Supplemental Figure 9D–E). Conversely, HA-tagged dominant negative CaMKIIδB (DNδB), but not DNδC, blocked ISO-induced MCU upregulation (Figure 5F and Supplemental Figure 9F). Meanwhile, DNδB also abolished the elevated mitochondrial Ca2+ concentrations induced by ISO (Figure 5G). Finally, we overexpressed δB or δC in adult cardiomyocytes isolated from CaMKIIδ KO mice25 and found that δB, but not δC, elevated MCU expression (Supplemental Figure 10). Taken together, our results suggest that CaMKIIδB, but not δC, is necessary and sufficient for ISO-induced MCU upregulation.
Calcineurin, also known as protein phosphatase 2B, was activated by ISO (Supplemental Figure 11A). Calcineurin inhibitor cyclosporine A (CsA, 1 μmol/L) blocked ISO-induced MCU upregulation, but overexpressing WT calcineurin did not affect MCU protein levels in adult cardiomyocytes (Figure 5H–I and Supplemental Figure 11B–C), indicating that calcineurin is necessary but not sufficient for MCU upregulation. In addition, CsA or WT calcineurin did not affect CaMKII phosphorylation at T287 (Figure 5H–I and Supplemental Figure 11B–C). Lastly, β blocker propranolol (Prop, 10 mg/kg/day) or CaMKII inhibitor KN93 (10 μmol/kg/day) abolished ISO-induced MCU upregulation in mouse heart tissue in vivo (Figure 5J).
Nuclear translocation of activated CaMKIIδB upregulated MCU expression
CaMKIIδB and δC are alternative splicing products of the CaMKIIδ gene. They share an identical sequence, except that δB has an 11-amino acid nuclear localization sequence (NLS).26 Because δB is distributed in different compartments of cardiomyocytes,27 we first determined whether nuclear δB regulates MCU expression. Targeting a green fluorescent protein (GFP)-tagged autocamtide-2-related inhibitory peptide (AIP, a CaMKII inhibitor) to the cytosol (c-GFP-AIP) or nucleus (n-GFP-AIP) abolished ISO-induced CaMKII phosphorylation at T287 in the corresponding compartments (Supplemental Figure 12A). However, only n-GFP-AIP, but not c-AIP, blocked ISO-induced MCU upregulation in adult mouse cardiomyocytes (Supplemental Figure 12B). Similarly, targeting a red fluorescent protein (RFP)-tagged CaMKII inhibitory protein to the cytosol (c-CaMKIIN) or nucleus (n-CaMKIIN) attenuated ISO-induced CaMKII activation (T287 phosphorylation) in the corresponding compartments (Figure 6A and Supplemental Figure 12C), but only n-CaMKIIN prevented ISO-induced MCU upregulation (Figure 6B). Taken together, our results suggest that nuclear CaMKIIδB regulates MCU gene expression.
Figure. 6. Chronic β-AR activation promoted nuclear translocation of CaMKIIδB via calcineurin-mediated S332 de-phosphorylation.
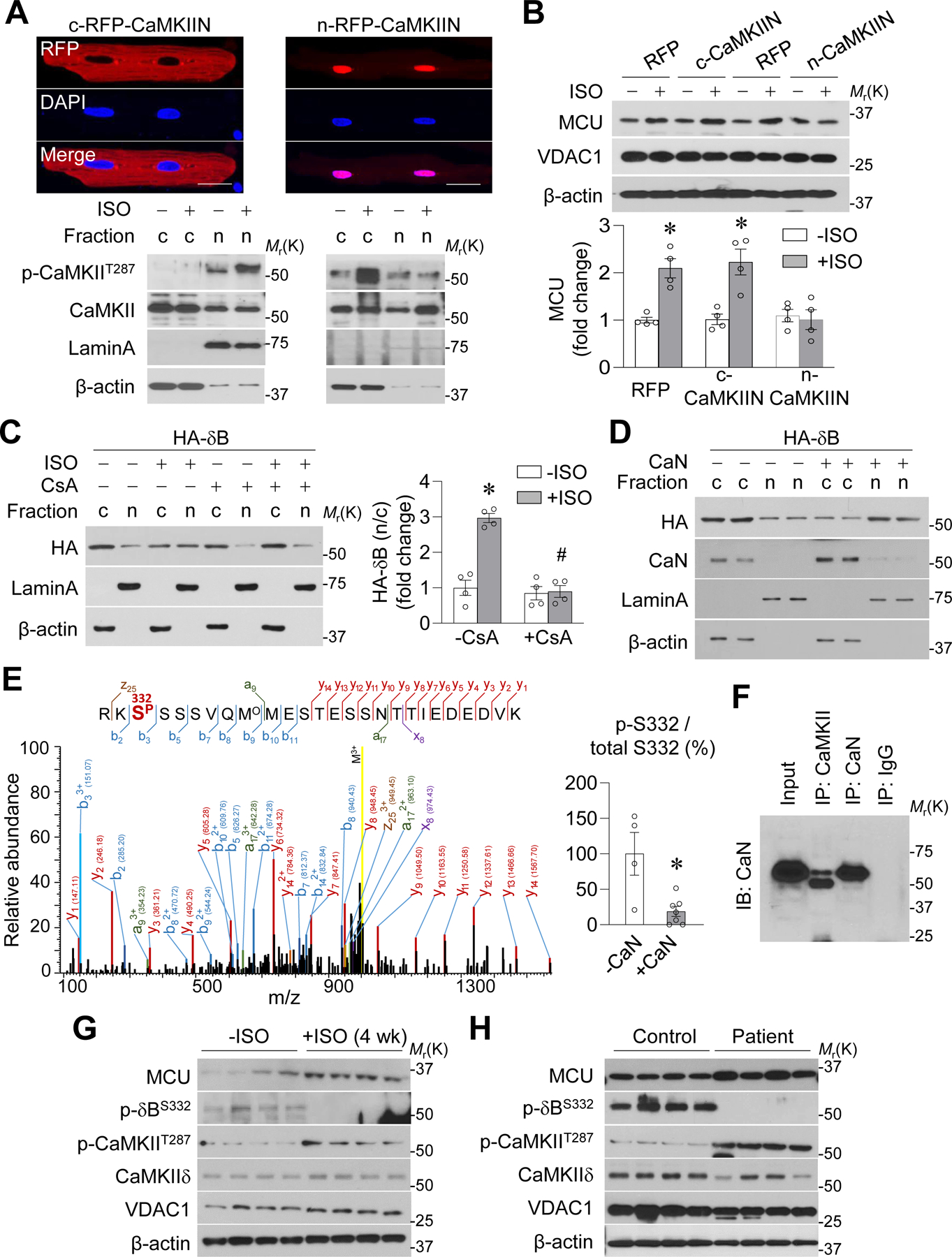
A-B, Effects of overexpressing cytosol-targeted and RFP-tagged CaMKII inhibitory protein (c-CaMKIIN) or nucleus-targeted CaMKIIN (n-CaMKIIN) on CaMKII T287 phosphorylation in cytosol or nucleus (A) and on ISO-induced MCU upregulation (B) in adult mouse cardiomyocytes. *: P=0.002 RFP+ISO vs. RFP−ISO, *: P=0.0061 c-CaMKIIN+ISO vs. c-CaMKIIN−ISO. n=4. C, Effects of ISO (0.1 μmol/L) and cyclosporine A (CsA, 0.1 μmol/L) on intracellular location of HA-tagged CaMKIIδB (δB) in adult mouse cardiomyocytes. *: P=0.0002 +ISO vs. −ISO, #: P=5.859E-05 ISO+CsA vs. ISO-CsA. n=4. D, Effects of overexpressing calcineurin (CaN) on intracellular location of δB in adult cardiomyocytes. n=4. E, Representative mass spectrum of a peptide in CaMKIIδB containing S332 phosphorylation. The peptide sequence and phosphorylation site were confidently identified based on the matched b- or a- and y- or x- ion series indicated by the spectrum annotations. Right panel showed that the abundance of peptide containing phosphorylated S332 was significantly lower after CaN incubation. *: P=0.0077 +CaN vs. -CaN. n=4–6. F, Co-IP showing the binding between CaMKIIδB and CaN in adult cardiomyocytes. n=3. G-H, Western blotting images showing the detection of δB phosphorylation at S332 or T287 and MCU protein level in mouse hearts after ISO administration for 4 weeks (G) or in human failing heart samples (H). n=4.
Because the phosphorylation status of serine 332 (S332), which is located near the NLS, controls intracellular localization of CaMKIIδB,28 we tested whether S332 dephosphorylation promotes its nuclear translocation and regulation of MCU expression. First, ISO-induced nuclear translocation of HA-tagged CaMKIIδB was blocked by CsA (Figure 6C). Conversely, overexpressing calcineurin promoted nuclear translocation of CaMKIIδB (Figure 6D and Supplemental Figure 12D). Second, we overexpressed HA-tagged and phosphorylation mimetic (S332E) or unphosphorylated (S332A) mutated CaMKIIδB in adult cardiomyocytes. S332E stayed in the cytosol with or without ISO incubation, whereas S332A was located inside the nucleus even in the presence of CsA (Supplemental Figure 12E). Importantly, S332E abolished ISO-induced MCU upregulation, whereas S332A upregulated MCU despite the presence of CsA (Supplemental Figure 12F). These results support our hypothesis that calcineurin dephosphorylates CaMKIIδB at S332 to facilitate its nuclear translocation for regulation of MCU expression.
Next, we determined whether calcineurin directly dephosphorylates CaMKIIδB at S332. First, S332 phosphorylation was abundantly detected by mass spectrometry in purified CaMKIIδB but not after calcineurin incubation (Figure 6E). In addition, calcineurin bound CaMKIIδB (Figure 6F) and dephosphorylated it at S332 (detected by using an antibody against phosphorylated S332) in a cell-free system and in adult cardiomyocytes after ISO incubation or calcineurin overexpression (Supplemental Figure 13A–B). Furthermore, CsA blocked ISO- or calcineurin-induced S332 dephosphorylation (Supplemental Figure 13C). CaMKIIδB phosphorylation at S332 was detected only in the cytosol (Supplemental Figure 13D). Overexpressing a dominant negative calmodulin attenuated ISO-induced MCU upregulation (Supplemental Figure 13E), consistent with the role of calmodulin in activating both CaMKII and calcineurin. In whole-heart samples from mice after 4 weeks of ISO administration or from heart failure patients, we confirmed the decrease in CaMKIIδB S332 phosphorylation, increase in CaMKIIδB T287 phosphorylation, and elevated MCU protein levels (Figure 6G–H and Supplemental Figure 13F–G). Taken together, these results suggest that calcineurin dephosphorylates CaMKIIδB at S332 to facilitate its nuclear translocation and MCU upregulation.
CaMKIIδB phosphorylated CREB to promote MCU transcription
ISO increased MCU mRNA levels in a KN93-dependent manner and enhanced the activity of the MCU promoter (Supplemental Figure 14A–B). Bioinformatics analysis revealed four sequences in the MCU promoter region that are highly homologous to the consensus cAMP response element (CRE I, II, III, and IV) (Supplemental Figure 14C–D). A Luciferase reporter system containing a truncated MCU promotor or CRE mutants showed that CRE IV was responsible for ISO-induced MCU gene transcription (Figure 7A–B). Next, chromatin immunoprecipitation assays and electrophoretic mobility shift assays, respectively, revealed that CREB bound CRE IV in adult cardiomyocytes and in vitro (Figure 7C–E). Moreover, ISO increased CREB phosphorylation at its activating site (S133) in a KN93-dependent manner (Figure 7F and Supplemental Figure 14E). Co-immunoprecipitation detected that endogenous CREB and CaMKII bound each other in adult cardiomyocytes, and purified CaMKIIδB bound and phosphorylated CREB at S133 in vitro (Figure 7G–H and Supplemental Figure 14F–G). A dominant negative mutation (K43A, in the ATP binding domain) of CaMKIIδB, but not S332E or S332A, failed to phosphorylate CREB (Figure 7I), suggesting that the phosphorylation status of S332 does not affect the kinase activity of CaMKIIδB and the kinase activity is needed for CREB phosphorylation. Finally, overexpression of a dominant negative CREB (S133A) abolished ISO or CaMKIIδB’s effects on MCU mRNA and protein levels and on mitochondrial Ca2+ concentrations in adult cardiomyocytes (Figure 7J–L and Supplemental Figure 14H), suggesting that CREB phosphorylation mediates ISO/CaMKIIδB-induced MCU expression. These results indicate that CaMKIIδB phosphorylates CREB at S133 to modulate MCU gene transcription.
Figure. 7. CaMKIIδB phosphorylated CREB to modulate MCU gene expression.
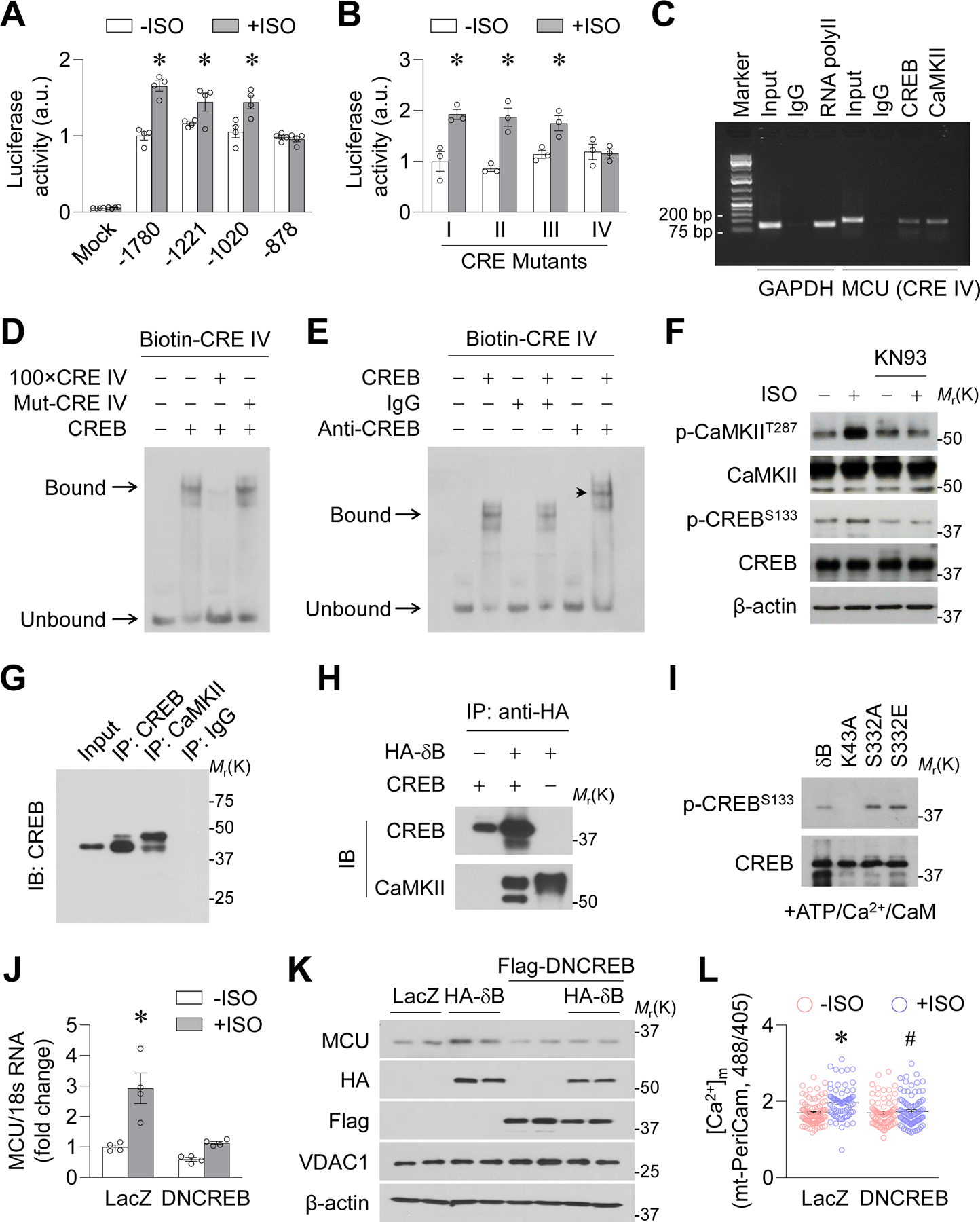
A-B, Luciferase activity assay showing the activity of truncated MCU promoter (A, *: P=0.0003, 0.0149 or 0.0414 +ISO vs. −ISO for −1780, −1221 or-1020 groups, respectively) or the MCU promoter fragments that contain mutated sequences of CRE I-IV (B, *: P=0.0131, 0.0053 or 0.022 +ISO vs. −ISO for I, II or III groups, respectively) in H9C2 myoblasts. n=4. C, Chromatin immunoprecipitation assay showing the binding of CRE IV with CREB and CRE IV with CaMKII in adult cardiomyocytes. The binding between RNA polymerase II and a sequence in GAPDH promoter was used as positive control. n=3. D, Electrophoretic mobility shift assay (EMSA) showing the binding between CREB and double-stranded and biotin-labeled oligonucleotides corresponding to CRE IV sequence (Biotin-CRE IV) in vitro, which was blocked by 100 fold unlabeled CRE IV sequence (100xCRE IV) but not by mutated biotin-CRE IV (Mut-CRE IV). E, EMSA showing that addition of CREB antibody caused a shift of the band toward higher molecular weight (arrow head). n=3. F, Effects of KN93 on ISO-induced CREB phosphorylation at S133 in adult mouse cardiomyocytes. n=4. G, Co-IP showing the binding between endogenous CaMKII and CREB in adult mouse cardiomyocytes. Representative of 4 repeats. H, Pull down analysis showing the binding between HA-CaMKIIδB and CREB in vitro. Representative of 3 repeats. I, In vitro phosphorylation assay showing the effects of wild type (δB), S332A, S332E or K43A mutations of CaMKIIδB on CREB phosphorylation at S133. Representative of 3 repeats. J, Effects of dominant negative CREB (DNCREB) on ISO-induced MCU mRNA upregulation in adult mouse cardiomyocytes. *: P=0.0087 LacZ+ISO vs. LacZ−ISO. n=6. K, Effects of DNCREB on HA-δB-induced MCU upregulation. n=4. L, Effects of DNCREB on ISO-induced changes in mitochondrial Ca2+ concentrations ([Ca2+]m, measured by mt-PeriCam. *: P=5.440E-06 LacZ+ISO vs. LacZ−ISO. #: P=0.0003 DNCREB+ISO vs. LacZ+ISO, n=76–93 cells from 4 mice.
Discussion
In this study, we discovered a novel mechanism, which is centered on the MCU, to limit pathological cardiac remodeling (Figure 8). During chronic β-AR stimulation, the CaMKIIδB/CREB pathway upregulates MCU gene transcription, leading to increased formation of the MCU complex and mitochondrial Ca2+ uptake. MCU-mediated Ca2+ uptake orchestrates Ca2+ handling among mitochondria, SR, and cytosol to ameliorate cytosolic Ca2+ overload-induced cardiomyocyte death. MCU KO worsens, whereas MCU transgenic overexpression prevents, heart hypertrophy and dysfunction. Therefore, enhancing MCU-mediated mitochondrial Ca2+ uptake could be a new approach to treat heart failure.
Figure. 8. Schematic illustration.
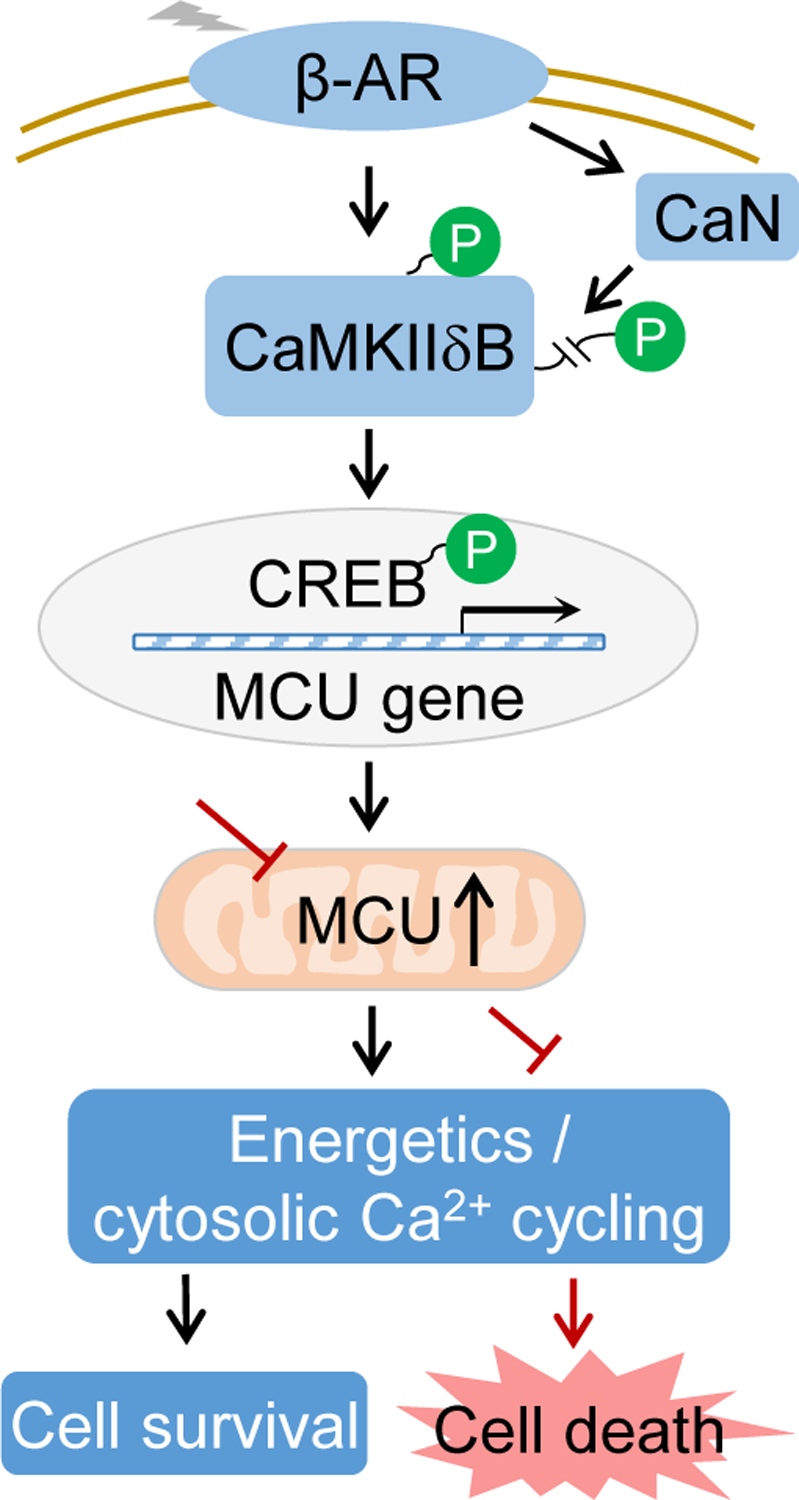
MCU expression is upregulated by the β-AR/CaMKIIδB/CREB pathway to maintain cytosolic Ca2+ and energy homeostasis in the heart. Phosphorylation at T287 increased the kinase activity of CaMKIIδB and de-phosphorylation at S332 triggered its nuclear translocation, both of which are needed for MCU gene upregulation.
Pathophysiological functions of MCU, the major mitochondrial Ca2+ uptake channel,4,5 in the heart remain elusive, because genetic manipulations of MCU have yielded conflicting results.6 For instance, mitochondrial Ca2+ overload is known to mediate acute ischemia-reperfusion injury in the heart through boosting respiration, reactive oxygen species production, and mPTP opening.29 However, reperfusion injury of the heart is not ameliorated in all MCU KO models.7–9 Chronic stress-induced heart dysfunction is linked to either mitochondrial Ca2+ overload or reduced Ca2+ uptake.3,30 Consistent with this notion, a recent report showed that increasing MCU levels rescues heart failure in guinea pigs.31 However, MCU KO does not prevent pressure overload-induced heart hypertrophy and failure in mice,32 casting doubts on the role of endogenous MCU in the failing heart. Here, we showed that MCU KO or TG oppositely impacted ISO-induced heart remodeling, providing explicit evidence to support a critical role of MCU in the chronically stressed heart. Future studies are needed to elucidate whether MCU plays similar or distinct roles in different heart failure models and/or at different disease stages.
Our results highlight that regulation of cytosolic Ca2+ is a new mechanism by which mitochondrial Ca2+ participates in heart failure development. Unlike in other cell types, such as skeletal muscle and some cell lines,33,34 mitochondria in cardiomyocytes play a negligible role in directly buffering cytosolic Ca2+ under physiological conditions.7 We found that during chronic adrenergic stress, increased mitochondrial Ca2+ uptake limits cytosolic Ca2+ overload indirectly through boosting energy metabolism and SR Ca2+ reuptake. Importantly, when mitochondrial Ca2+ uptake is further and persistently enhanced, as in the MCU TG mice, cytosolic Ca2+ overload and pathological heart hypertrophy are completely prevented. Interestingly, we detected increased ATP levels in cultured cells but not in heart tissue when mitochondrial Ca2+ uptake is enhanced. This is likely because that increased energy production is fully and promptly matched by increased workload in the beating heart but not in the quiescent cells. Mitochondrial respiration was not compromised by ISO incubation, suggesting the lack of significant mitochondrial dysfunction, which could be induced by more robust adrenergic stress35.
Our study also revealed a central role of MCU in the assembly of the MCU complex in cardiomyocytes. The MCU complex is the functional unit for mitochondrial Ca2+ uptake.5 A noteworthy finding is that the levels of MCU and MCU complex, as well as the mitochondrial Ca2+ uptake rate, are modulated concomitantly by ISO stimulation and in MCU TG or KO hearts. In contrast, the levels of MCU regulatory proteins are mostly unchanged. Therefore, MCU and its regulatory proteins are independently regulated by ISO stimulation in the heart and manipulating MCU is sufficient to influence the formation of functional MCU complex. Thus, MCU could be a stress response gene whose upregulation quickly leads to enhanced mitochondrial Ca2+ uptake. It should be noted that ISO induces different changes in some of the MCU regulatory proteins in cultured cells and the heart. Future studies are needed to explore whether this is due to the different dosages and/or duration of ISO stimulation or the different conditions of cardiomyocytes (in vitro versus in vivo).
Finally, this study delineated a molecular mechanism by which CaMKIIδB controls MCU gene expression in the heart. Chronic β-AR stimulation activates both the δB and δC subtypes of CaMKII, which have unique functions in the heart.36 In particular, CaMKIIδB regulates the expression of genes associated with hypertrophic cardiomyopathy and leads to compensated hypertrophy, whereas CaMKIIδC causes pathological hypertrophy with impaired heart function.20,37,38 The exact mechanisms or targets of CaMKIIδB responsible for maintaining cardiac function in the hypertrophic heart are unknown. Previous reports showed that CaMKII regulates gene expression through CREB in neurons and that CREB modulates MCU gene expression in cancer cells.14,39 Our results suggest that CaMKIIδB regulates MCU gene transcription through phosphorylating CREB at S133 in cardiomyocytes. Consistent with our findings, mice expressing dominant-negative CREB (S133A) in the heart exhibited dilated cardiomyopathy with mitochondrial dysfunction.40,41 Moreover, we found that both nuclear translocation through S332 dephosphorylation and kinase activation through T287 phosphorylation are needed for CaMKIIδB to upregulate MCU gene expression (Figure 8). Previous reports showed that CaMKI or CaMKIV phosphorylates CaMKII at S332, keeping it in the cytosol, whereas phosphatases (PP1α and PP1γ1) dephosphorylate CaMKII and promote its nuclear translocation.28,42 We found that calcineurin can also dephosphorylate CaMKIIδB at S332 to facilitate its nuclear translocation. Importantly, the dual mechanisms occur independently and act synergistically. Because both calcineurin and CaMKII are Ca2+- and calmodulin-dependent, elevated cytosolic Ca2+ concentrations might be the initial trigger of the CaMKIIδB/CREB/MCU pathway, which serves as a negative feedback mechanism to limit cytosolic Ca2+ overload, maintain cell viability, and prevent pathological remodeling.
In summary, the MCU is transcriptionally upregulated by the β-AR/CaMKIIδB/CREB pathway to maintain intracellular Ca2+ and energy homeostasis as well as cell viability (Figure 8). Targeting the CaMKIIδB/CREB/MCU axis may be a novel approach to counteract chronic stress-induced pathological cardiac remodeling.
Supplementary Material
Clinical Perspective.
What is new?
MCU is upregulated in the stressed heart to orchestrate mitochondrial, sarcoplasmic reticulum, and cytosolic Ca2+ handling, preventing cytosolic Ca2+ overload-induced cardiomyocyte death.
Lack of MCU-mediated mitochondrial Ca2+ uptake is detrimental, whereas transgenic overexpression MCU is beneficial, to the heart during chronic β adrenergic stimulation.
The nuclear translocation of CaMKIIδB, via calcineurine-mediated dephosphorylation of serine 332, activates CREB to promote MCU gene expression in adult cardiomyocytes.
What are the clinical implications?
Our study indicates that enhancing mitochondrial Ca2+ uptake could be a new approach to prevent chronic β adrenergic stimulation-induced heart remodeling.
Targeting the CaMKIIδB/CREB/MCU pathway could be a therapeutic opportunity for pathological cardiac remodeling associated with chronic adrenergic stress.
Acknowledgments
We thank Drs. Toren Finkel, Elisabeth Murphy, Joan Heller Brown, John Elrod, Dan Shao, Stephen C. Kolwicz Jr, Sergio De la Fuente, and Matthew Walker for technical support and helpful suggestions. P.W. performed majority of the experiments with help from S.X., J.X., Y.X., H.Z. and B.Z.. Y.L. and H.X. performed echocardiography. S.S.S and R.T. provided technical help and conceptual advices. P.W. and W.W. designed experiments and wrote the manuscript with inputs from coauthors. W.W. coordinated and oversaw the whole project.
Sources of Funding
This work was partially supported by NIH (HL114760 to W.W., HL137266 to S.S.S. and W.W., and HL110349 to R.T.), American Heart Association (18EIA33900041 to W.W. and 19CDA34660311 to H.Z.) and Mitochondria and Metabolism Center, UW (Research Seed Grant to P.W.).
Non-standard Abbreviations and Acronyms
- ANP
atrial natriuretic peptide
- β-AR
β-adrenergic receptor
- BNP
brain natriuretic peptide
- BW
body weigh
- caGsα
constitutive active G protein sα
- Cal XI
calpain inhibitor XI
- CaM
Calmodulin
- CaMKII
Ca2+ calmodulin kinase II
- cAMP
cyclic adenine monophosphate
- CaN
Calcineurin
- ChIP
chromatin immunoprecipitation
- cKO
cardiomyocyte-specific MCU knockout mice
- Col1a2
collagen type I α 2 chain
- CRE
cAMP response element
- CREB
cAMP-response element binding protein
- CsA
cyclosporine A
- DNCaM
dominant negative calmodulin
- DNCREB
dominant negative CREB
- DNMCU
dominant negative MCU
- EMRE
essential mitochondrial calcium uniporter regulator
- EMSA
electrophoretic mobility shift assay
- F/F
MCU floxed/floxed
- GFP
green fluorescent protein
- HW
heart weight
- ISO
Isoproterenol
- KHB
Krebs-Henseleit Buffer
- KO
global MCU knockout mice
- MCM
αMHC-MerCreMer mice
- MCU
mitochondrial calcium uniporter
- MCUb
mitochondrial calcium uniporter regulatory subunit b
- MICU1
mitochondrial calcium uptake 1
- mPTP
mitochondrial permeability transition pore
- MS
mass spectrometry
- NLS
nuclear localization sequence
- PKA
protein kinase A
- PKI
PKA inhibitory peptide
- RFP
red fluorescent protein
- SERCA2a
sarco/endo plasmic reticulum Ca2+ ATPase 2a
- SR
sarcoplasmic reticulum
- TG
heart specific and inducible MCU transgenic mice
- TL
tibia length
- WT
wild type mice
Footnotes
Disclosures
None.
Supplemental Materials
Expanded Methods
Reference
- 1.Tian R, Colucci WS, Arany Z, Bachschmid MM, Ballinger SW, Boudina S, Bruce JE, Busija DW, Dikalov S, Dorn GW, II, et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System: Path to a Cure in Heart Failure-A Report from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation. 2019;140:1205–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:11389–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang P, Fernandez-Sanz C, Wang W, Sheu SS. Why don’t mice lacking the mitochondrial Ca(2+) uniporter experience an energy crisis? J Physiol. 2020;598:1307–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015;12:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015;12:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaglia T, Ceriotti P, Campo A, Borile G, Armani A, Carullo P, Prando V, Coppini R, Vida V, Stolen TO, et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E9006–E9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parks RJ, Menazza S, Holmstrom KM, Amanakis G, Fergusson M, Ma H, Aponte AM, Bernardi P, Finkel T, Murphy E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc Res. 2019;115:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fieni F, Johnson DE, Hudmon A, Kirichok Y. Mitochondrial Ca2+ uniporter and CaMKII in heart. Nature. 2014;513:E1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, Hines KJ, Ragheb J, Jog NR, Caricchio R, et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal. 2015;8:ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. [DOI] [PubMed] [Google Scholar]
- 16.Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc’h F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res. 2008;102:959–965. [DOI] [PubMed] [Google Scholar]
- 17.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol. 2010;48:322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng N, Anderson ME. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J Mol Cell Cardiol. 2017;103:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem. 2007;282:10833–10839. [DOI] [PubMed] [Google Scholar]
- 20.Gray CB, Suetomi T, Xiang S, Mishra S, Blackwood EA, Glembotski CC, Miyamoto S, Westenbrink BD, Brown JH. CaMKIIdelta subtypes differentially regulate infarct formation following ex vivo myocardial ischemia/reperfusion through NF-kappaB and TNF-alpha. J Mol Cell Cardiol. 2017;103:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida K, Inui M, Harada K, Saido TC, Sorimachi Y, Ishihara T, Kawashima S, Sobue K. Reperfusion of rat heart after brief ischemia induces proteolysis of calspectin (nonerythroid spectrin or fodrin) by calpain. Circ Res. 1995;77:603–610. [DOI] [PubMed] [Google Scholar]
- 22.Matsumura Y, Saeki E, Otsu K, Morita T, Takeda H, Kuzuya T, Hori M, Kusuoka H. Intracellular calcium level required for calpain activation in a single myocardial cell. J Mol Cell Cardiol. 2001;33:1133–1142. [DOI] [PubMed] [Google Scholar]
- 23.Imamura H, Nhat KP, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, Noji H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zachary I, Masters SB, Bourne HR. Increased mitogenic responsiveness of Swiss 3T3 cells expressing constitutively active Gs alpha. Biochem Biophys Res Commun. 1990;168:1184–1193. [DOI] [PubMed] [Google Scholar]
- 25.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. The Journal of clinical investigation. 2009;119:1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srinivasan M, Edman CF, Schulman H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J Cell Biol. 1994;126:839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishra S, Gray CB, Miyamoto S, Bers DM, Brown JH. Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ Res. 2011;109:1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heist EK, Srinivasan M, Schulman H. Phosphorylation at the nuclear localization signal of Ca2+/calmodulin-dependent protein kinase II blocks its nuclear targeting. J Biol Chem. 1998;273:19763–19771. [DOI] [PubMed] [Google Scholar]
- 29.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Yang N, Sidor A, O’Rourke B. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca(2+) Leak. Circ Res. 2021;128:1191–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmstrom KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol. 2015;85:178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao H, Li T, Wang K, Zhao F, Chen J, Xu G, Zhao J, Chen L, Li L, Xia Q, et al. AMPK-mediated activation of MCU stimulates mitochondrial Ca(2+) entry to promote mitotic progression. Nat Cell Biol. 2019;21:476–486. [DOI] [PubMed] [Google Scholar]
- 34.Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 2015;10:1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garrido-Moreno V, Diaz-Vegas A, Lopez-Crisosto C, Troncoso MF, Navarro-Marquez M, Garcia L, Estrada M, Cifuentes M, Lavandero S. GDF-11 prevents cardiomyocyte hypertrophy by maintaining the sarcoplasmic reticulum-mitochondria communication. Pharmacol Res. 2019;146:104273. [DOI] [PubMed] [Google Scholar]
- 36.Gray CB, Heller Brown J. CaMKIIdelta subtypes: localization and function. Front Pharmacol. 2014;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Little GH, Saw A, Bai Y, Dow J, Marjoram P, Simkhovich B, Leeka J, Kedes L, Kloner RA, Poizat C. Critical role of nuclear calcium/calmodulin-dependent protein kinase IIdeltaB in cardiomyocyte survival in cardiomyopathy. J Biol Chem. 2009;284:24857–24868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng W, Zhang Y, Zheng M, Cheng H, Zhu W, Cao CM, Xiao RP. Cardioprotection by CaMKII-deltaB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ Res. 2010;106:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–2539. [DOI] [PubMed] [Google Scholar]
- 40.Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest. 1998;101:2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watson PA, Birdsey N, Huggins GS, Svensson E, Heppe D, Knaub L. Cardiac-specific overexpression of dominant-negative CREB leads to increased mortality and mitochondrial dysfunction in female mice. Am J Physiol Heart Circ Physiol. 2010;299:H2056–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shioda N, Sawai M, Ishizuka Y, Shirao T, Fukunaga K. Nuclear Translocation of Calcium/Calmodulin-dependent Protein Kinase IIdelta3 Promoted by Protein Phosphatase-1 Enhances Brain-derived Neurotrophic Factor Expression in Dopaminergic Neurons. J Biol Chem. 2015;290:21663–21675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W, Yoon Y, Tian R, Wang W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat Commun. 2016;7:13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Q, Zhang H, Gutierrez Cortes N, Wu D, Wang P, Zhang J, Mattison JA, Smith E, Bettcher LF, Wang M, et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ Res. 2020;126:456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai B, Wang N, Mao W, You T, Lu Y, Li X, Ye B, Li F, Xu H. Deletion of FoxO1 leads to shortening of QRS by increasing Na(+) channel activity through enhanced expression of both cardiac NaV1.5 and beta3 subunit. J Mol Cell Cardiol. 2014;74:297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106:1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong G, Liu X, Wang W. Regulation of metabolism in individual mitochondria during excitation-contraction coupling. J Mol Cell Cardiol. 2014;76:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thestrup T, Litzlbauer J, Bartholomaus I, Mues M, Russo L, Dana H, Kovalchuk Y, Liang Y, Kalamakis G, Laukat Y, et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat Methods. 2014;11:175–182. [DOI] [PubMed] [Google Scholar]
- 51.Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17404–17409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol. 1992;453:591–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marcu R, Neeley CK, Karamanlidis G, Hawkins BJ. Multi-parameter measurement of the permeability transition pore opening in isolated mouse heart mitochondria. J Vis Exp. 2012;67:4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H, Wang P, Bisetto S, Yoon Y, Chen Q, Sheu SS, Wang W. A novel fission-independent role of dynamin-related protein 1 in cardiac mitochondrial respiration. Cardiovasc Res. 2017;113:160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, et al. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest. 2003;111:1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6:6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ji Y, Li B, Reed TD, Lorenz JN, Kaetzel MA, Dedman JR. Targeted inhibition of Ca2+/calmodulin-dependent protein kinase II in cardiac longitudinal sarcoplasmic reticulum results in decreased phospholamban phosphorylation at threonine 17. The Journal of biological chemistry. 2003;278:25063–25071. [DOI] [PubMed] [Google Scholar]
- 59.Ishida A, Fujisawa H. Stabilization of calmodulin-dependent protein kinase II through the autoinhibitory domain. The Journal of biological chemistry. 1995;270:2163–2170. [DOI] [PubMed] [Google Scholar]
- 60.Braun AP, Schulman H. A non-selective cation current activated via the multifunctional Ca(2+)-calmodulin-dependent protein kinase in human epithelial cells. J Physiol. 1995;488 ( Pt 1):37–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10890–10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE Jr., Thiel W, Guatimosim S, Song LS, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. [DOI] [PubMed] [Google Scholar]
- 63.Glass DB, Cheng HC, Mende-Mueller L, Reed J, Walsh DA. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J Biol Chem. 1989;264:8802–8810. [PubMed] [Google Scholar]
- 64.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.