

Abstract
Targeting cyclin-dependent kinases 4 and 6 (CDK4/6) is a successful therapeutic approach against breast and other solid tumors. Inhibition of CDK4/6 halts cell cycle progression and promotes antitumor immunity. However, the mechanisms underlying the antitumor activity of CDK4/6 inhibitors are not fully understood. We found that CDK4/6 bind and phosphorylate the p53 family member p73 at threonine 86, which sequesters p73 in the cytoplasm. Inhibition of CDK4/6 led to dephosphorylation and nuclear translocation of p73, which transcriptionally activated death receptor 5 (DR5), a cytokine receptor and key component of the extrinsic apoptotic pathway. p73-mediated induction of DR5 by CDK4/6 inhibitors promoted immunogenic cell death (ICD) of cancer cells. Deletion of DR5 in cancer cells in vitro and in vivo abrogated the potentiating effects of CDK4/6 inhibitors on immune cytokine TNF-related apoptosis-inducing ligand (TRAIL), 5-fluorouracil (5-FU) chemotherapy, and anti-PD-1 immunotherapy. Together, these results reveal a previously unrecognized consequence of CDK4/6 inhibition, which may be critical for potentiating the killing and immunogenic effects on cancer cells.
Keywords: CDK4/6 inhibitors, DR5, p73, colorectal cancer, apoptosis, anti-PD-1
Introduction
Cyclin-dependent kinases (CDKs) are serine/threonine kinases that play a key role in cell cycle regulation. CDKs 4 and 6 (CDK4/6) bind to D-type cyclins to form active kinases, which phosphorylate retinoblastoma 1 (RB1) to relieve its inhibition on E2F transcription factors leading to G1/S transition (1). A hallmark of cancer is sustained cell proliferation due to abnormal cell cycle regulation (2). The CDK4/6-cyclin D-RB1-E2F axis is one of the most frequently dysregulated cell cycle pathways in cancer cells (3).
Pharmacological inhibition of CDK4/6 has emerged as a promising anticancer strategy. To date, three small-molecule CDK4/6 inhibitors (CDK4/6i), including Palbociclib, Ribociclib, and Abemaciclib, have been approved for the treatment of hormone receptor (HR) positive and advanced breast cancer (4). CDK4/6i have several anticancer effects including inhibition of the cell cycle and modulation of cellular senescence, tumor cell metabolism, and tumor microenvironment (5). Recent studies have shown that CDK4/6 inhibition triggers antitumor immunity and potentiates anti-PD-1 immunotherapy (6,7). Despite these studies, the anticancer mechanisms of CDK4/6i in different types of cancer cells, the molecular markers for predicting response and resistance to CDK4/6i, and the rationale for combining CDK4/6i with other anticancer agents, have yet to be determined.
CDK4/6i combined with other anticancer agents enhance apoptosis of cancer cells (8). Nonetheless, it is unclear how CDK4/6i promote cell death, and if such an effect is essential for the anticancer and immunogenic effects of CDK4/6i. Apoptosis is initiated via the extrinsic and/or intrinsic pathways. The extrinsic pathway is engaged upon activation of the tumor necrosis factor (TNF) family receptors such as Death Receptor 5 (DR5; TRAILR2), leading to activation of caspase 8 and effector caspases (9). DR5 is a receptor of immune cytokine TRAIL (10). DR5 can also be transcriptionally activated by p53, TAp73 (p73 thereafter), NF-κB, and C/EBP Homologous Protein (CHOP) in response to various stresses (11,12). The intrinsic pathway is controlled by the Bcl-2 proteins via mitochondrial dysfunction (13,14). Different forms of cell death have distinct immunological consequences (15,16). For example, DR5-mediated apoptosis has characteristics of immunogenic cell death (ICD) (10), which stimulates an immune response against dead-cell antigens (15,16).
CDK4/6i have shown promising activity against different cancer types including colorectal cancer (CRC) (17), a leading cause of cancer-related deaths in the United States (18). CRC patients are treated with 5-fluorouracil (5-FU)-based chemotherapy, targeted therapy, and recently, anti-PD-1 immunotherapy (19,20). In this study, we analyzed the effects of CDK4/6i in CRC cells and found that CDK4/6 inhibition leads to induction of DR5 via a novel p73-dependent, but p53- and RB1-independent mechanism. Our results suggest a critical role of DR5 induction in mediating the antitumor and immunogenic effects of CDK4/6i in CRC cells.
Materials and Methods
Cell culture and drug treatment
Parental cell lines, including human CRC cell lines HCT116, RKO, DLD1, HT29, and LoVo, human breast cancer cell lines MCF-7, T47D, and MDA-MB-231, and mouse CRC cell line CT26, were purchased from the American Type Culture Collection (ATCC). Lim2405 was a gift from Dr. Alberto Bardelli (University of Torino, Italy). NCM356 non-transformed human colonic epithelial cells were from INCELL. Isogenic derivative cell lines, including DR5-knockout (KO) HCT116, DLD1, RKO, and CT26, and p73-KO HCT116 and DLD1, were generated by homologous recombination or CRISPR/Cas9 as described in Supplemental Methods. FADD-KO, Bid-KO, Caspase 8-knockdown (KD) and Flag-p73 knock-in (Flag-KI) HCT116 cell lines were previously described (21,22). p53-KO HCT116 was from Dr. Bert Vogelstein (Johns Hopkins University).
Parental cancer cell lines were authenticated by analysis of known oncogenic drivers including DNA mismatch repair (MMR) status and p53, KRAS, BRAF, PIK3CA, FBW7 and other mutations (23–25). Breast cancer cell lines were also verified by the expression of estrogen receptor (ER), progesterone receptor (PR), and/or HER2 (25). NCM356 was verified by the normal function of APC tumor suppressor (26). Isogenic derivative cell lines were authenticated by sequencing of the targeted genomic regions and analysis of protein expression. All cell lines were routinely checked for Mycoplasma contamination by PCR. Cell lines with fewer than 15 passages from the original stocks were used.
Cell lines, chemicals, cell culture media, and supplements are listed in Table S1. All cell lines were maintained at 37°C and 5% CO2 atmosphere, and cultured in McCoy’s 5A modified media, except for NCM356, which was cultured in M3 media (INCELL). All media were supplemented with 10% defined FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin. For drug treatment, cells were plated in 12-well plates at 20–30% density 24 hours before treatment. DMSO stocks of agents used, including Palbociclib (Palbo; Apexbio) and Ribociclib (Ribo; Apexbio), were diluted to appropriate concentrations with cell culture media. TRAIL (Pepro Tech) was diluted with distilled water.
Analysis of cell viability and apoptosis
MTS assays were performed using the MTS Assay Kit (Promega) according to the manufacturer’s instructions. Chemiluminescence was measured by a Wallac Victor 1420 Multilabel Counter (Perkin Elmer). Apoptosis was analyzed by staining adherent and floating cells with Hoechst 33258 (Invitrogen) and counting cells with condensed and fragmented nuclei (27). A minimum of 300 cells were counted for each sample. Apoptosis was also analyzed through annexin V/propidium iodide (PI) (Invitrogen) staining followed by flow cytometry as described (27). Long-term cell survival was analyzed by colony formation assays as described (27). Each assay was conducted in triplicate and repeated three times.
Transfection and small interfering RNA (siRNA) knockdown
Expression constructs and siRNAs are described in Supplemental Methods and Table S1. Transfection of expression constructs and siRNA was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. siRNA transfection was performed 24 hours prior to drug treatment using 200 pmole of siRNA.
Recombinant proteins and in vitro kinase assay
Purified recombinant proteins, including human CDK4, CDK6, TRAIL, wildtype (WT) p73, and T86A p73, are described in Supplemental Methods. In vitro kinase assay was performed as described in Supplemental Methods.
Western blotting
Western blotting was performed as previously described (28). Antibodies include those for DR5 (ProSci), DR4 (EMD Millipore), CDK4, CDK6, FLIP, FADD, FAS, phospho-RB1 (p-RB1; Ser780), p53, hemagglutinin (HA), Lamin A/C, p65 (Santa Cruz), RB1, Flag, p-p65 (Ser536), CHOP, cleaved caspases 3, 8, 9, Bid (Cell Signaling), p73 (Bethyl), p-p73 (Thr86) (Affinity Biosciences), Noxa, p21, β-Actin (Sigma), V5, phospho-Thr (Invitrogen), p16 (Abcam) and DNA methyltransferase 1 (DNMT1) (Novus). Additional information on antibodies is in Table S1.
Real-time Reverse Transcriptase (RT) PCR
Total RNA was isolated from cells using the Mini RNA Isolation II kit (ZYMO Research) according to the manufacturer’s protocol. Total RNA (1μg) was used to generate cDNA using SuperScript II reverse transcriptase (Invitrogen). PCR was performed using previously described conditions (27) and primers listed in Table S2.
Gene targeting by homologous recombination and CRISPR/Cas9
Targeting DR5 in HCT116 cells by homologous recombination, and DR5 in DLD1 and RKO cells, p73 in HCT116 and DLD1 cells, and DR5 in CT26 cells by CRISPR/Cas9 are described in Supplemental Methods.
Analysis of cell line and patient derived xenografts
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Mice were housed in a sterile environment with micro isolator cages and allowed access to water and chow ad libitum. Cell line xenografts were established by subcutaneously injecting 4×106 WT or DR5-KO HCT116 cells into both flanks of 5–6-week-old female Nu/Nu mice (Charles River).
Patient derived xenograft (PDX) tumors were propagated in 5–6-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory). For PDX passage, tumor tissues were cut into 25-mg pieces and implanted subcutaneously into both flanks of NSG mice as described (29,30). PDX models analyzed include PDX1 and PDX2, which were established using treatment-naïve primary tumors resected from patients with newly diagnosed CRCs under written informed consent (29). The tissue samples and de-identified clinical information were obtained from the Biospecimen Core at the University of Pittsburgh. PDX1 was from a KRAS-mutant (G13D), NRAS-mutant (G12D), FBW7-mutant (R505C), and MMR-proficient tumor (T4N0M1) in the sigmoid colon of a 77-year-old male (29,30). PDX2 was from an MMR-proficient tumor (T2N0) in the right colon of a 69-year-old female. Tumors passaged for two generations (P2) were analyzed.
Cell line xenograft or PDX tumors reached ~60 mm3 in size before treatment. Mice bearing tumors were randomized into different groups and treated with Palbo (oral gavage; 150 mg/kg daily), TRAIL (i.p.; 100 μg/mice every other day), 5-FU (i.p.; 25 mg/kg every other day), or a combination of Palbo with TRAIL or 5-FU for 10 days. Palbo was dissolved in sodium lactate buffer (pH 4.0). Tumor growth was monitored by calipers and tumor volume was calculated according to the formula 1/2×length×width2. Ethical endpoint represents a timepoint when tumors reached 2 cm or more in any dimension.
Tumor tissues were dissected and fixed in 10% formalin and embedded in paraffin. Immunostaining was performed for Terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling (TUNEL), active caspase 3, active caspase 8, and Ki67 on 5-μm paraffin-embedded sections using kit and primary antibodies listed in Table S1 as described (27). Signals were detected using AlexaFluor 488-conjugated secondary antibody (Invitrogen) with nuclear counter staining by 4’6-Diamidino-2-phenylindole (DAPI).
Analysis of ICD
Induction of ICD in WT, p73-KO, and DR5-KO HCT116 cells treated with 1 μM Palbo or Ribo for 24 hours was determined by analyzing cell-surface calreticulin (CRT) and phagocytosis of tumor cells by DCs as described in Supplemental Methods.
Analysis of syngeneic tumors and tumor-infiltrating lymphocytes
Syngeneic tumors were established by subcutaneously injecting CT26 cells into the flanks of 5–6-week-old BABL/cJ mice (Jackson Laboratory). Each mouse was inoculated with 5×105 WT or DR5-KO CT26 cells to establish a single tumor. Tumor-bearing mice were randomized and treated with Palbo (oral gavage; 100 mg/kg), PD-1 blockade by InVivoPlus anti-mouse PD-1 (BioXcell; i.p.; 200 μg/dose), or their combination as indicated in Fig. 8A. Some mice also received CD8 blockade by InVivoPlus anti-mouse CD8α (BioXcell; i.p.; 400 μg/dose) as in Fig. 8A. Tumor growth and tumor volume were analyzed as done for cell line and PDX models. Tumor infiltrating lymphocytes were analyzed by immunostaining followed by flow cytometry on tumors dissected 24 hours after the third treatment as described in Supplemental Methods. Antibodies included those for CD3, CD4, CD8α, CD11c, CD25, CD45, CD86, FoxP3, Granzyme B (GzmB), interferon γ (IFNγ), and MHC II (BioLegend). Tumor sections were also stained using antibodies for CD3 (Thermo Fisher) and CD8α (BioLegend). Additional information on antibodies is in Table S1.
Figure 8. DR5 is required for the potentiation effect of Palbo on anti-PD-1 immunotherapy.

(A)-(C) Syngeneic tumors were established by implanting 5×105 WT or DR5-KO CT26 cells in BALB/cJ mice. When tumors reached 50~100 mm3, mice were treated with Palbo (oral gavage; 100 mg/kg), anti-mouse PD-1 (i.p.; 200 μg/dose), or their combination (N=8 in each group). Mice were sacrificed before tumor volume reached 2000 mm3. Some mice also received anti-mouse CD8α (i.p.; 400 μg/dose). (A) Schematic diagram of treatment schedules. (B) Growth of individual tumors in each group. (C) Survival curves showing mice bearing WT tumors with statistical significance for indicated comparisons (left panel) and mice bearing DR5-KO tumors (right panel). (D)-(L) WT or DR5-KO CT26 tumors treated as in (A) and resected at day 10 were analyzed by flow cytometry for infiltrating immune cells (N=6 in each group): (D) CD3+/CD45+, (E) CD4+/CD45+, (F) CD8+/CD45+, (G) CD25+/FoxP3+/CD4+, (H) GzmB+/CD8+, (I) IFNγ+/CD8+, (J) CD11c+/CD45+, (K) CD86+/CD11c+, and (L) MHCII+/CD11c+. (M) Growth curves of CT26 tumors treated with Palbo/anti-PD-1 combination with or without CD8+ cell depletion by anti-CD8α as shown in Fig. 8A (N=6 in each group). In (C)-(M), NS, not significant (P > 0.05); *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Statistical Analysis
Statistical analysis was performed using Prism 9 software (GraphPad). For cell culture and immunostaining experiments, P values were calculated using Student’s t-test. Means ± standard deviation (SD) are indicated in the figures. For animal treatment experiments, P values were calculated by ANOVA with Fisher’s LSD post-hoc test for tumor volume analysis, and by Log-Rank test for survival analysis. Means ± standard error of the mean (SEM) are indicated in the figures. Differences were considered significant if P < 0.05.
Results
Induction of DR5 in response to CDK4/6 inhibition.
We analyzed RNA-Seq data from HCT116 CRC cells transfected with CDK4 siRNA or treated with Palbociclib (Palbo). Upregulation of DR5 was identified as a prominent and consistent change in the apoptotic pathways (Fig. S1, A–D). DR5 mRNA and protein were markedly induced in HCT116 cells upon CDK4/6 knockdown (KD) (Fig. 1, A and B), or inhibition by Palbo or Ribociclib (Ribo) (Fig. 1, C–E). In contrast, other apoptosis regulators were unchanged or only slightly induced (Fig. 1, A and C; Fig. S2A). Induction of DR5 by CDK4/6 KD or inhibition was observed in CRC cell lines with wildtype (WT) or mutant p53 (Fig. 1, F and G; Fig. S2B), and in MCF-7, MDA-MB-231, and T47D breast cancer cells (Fig. S2, C–E). Interestingly, DR5 was not induced by Palbo or Ribo in NCM356 normal colonic epithelial cells (Fig. S2F). CDK4/6 inhibition suppressed RB1 phosphorylation (Ser780) and downregulated RB1 (Fig. 1H). However, RB1 KD did not induce DR5 or affect DR5 induction and G1 cycle arrest following Palbo treatment (Fig. 1I and Fig. S2G). Furthermore, ectopic expression of p16 in HCT116 cells induced DR5 (Fig. S2H). CDK4/6 inhibition downregulated DNMT1 (Fig. S2I) (6), but did not affect CpG methylation in the DR5 promoter (Fig. S2J). These results suggest that DR5 induction by CDK4/6i in CRC cells is mediated by transcriptional activation independent of p53, RB1, and DNMT1.
Figure 1. DR5 is induced following CDK4/6 depletion or inhibition.
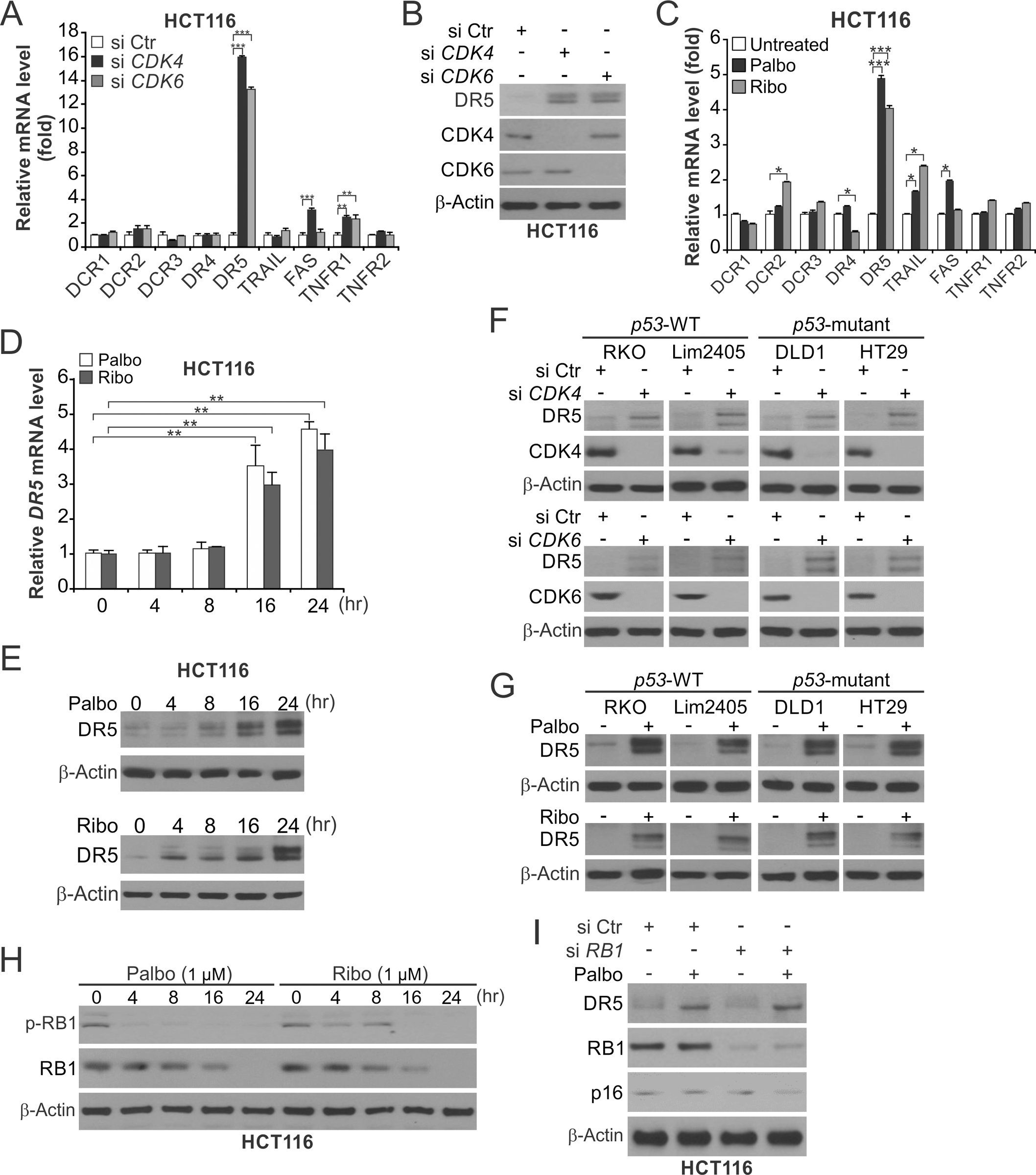
(A) RT-PCR analysis of indicated genes in HCT116 cells transfected with control scrambled (Ctr), CDK4, or CDK6 siRNA for 24 hours. (B) Western blotting of indicated proteins in HCT116 cells transfected with indicated siRNA as in (A). (C) RT-PCR analysis of indicated genes in HCT116 cells treated with 1 μM Palbociclib (Palbo) or Ribociclib (Ribo) for 24 hours. (D) RT-PCR analysis of DR5 in HCT116 cells treated with 1 μM Palbo or Ribo at indicated time points. (E) Western blotting of DR5 in HCT116 cells treated as in (D). (F), (G) Western blotting of indicated proteins in CRC cell lines with WT or mutant p53 (F) transfected as in (A) or (G) treated as in (C). (H) Western blotting of total and phosphorylated RB1 (Ser780) in HCT116 cells treated as in (D). (I) Western blotting of indicated proteins in HCT116 cells transfected with indicated siRNA or treated with Palbo as in (C). Bar graphs show results of 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
CDK4/6 inhibition upregulates DR5 via p73-mediated transcriptional activation.
We then investigated the transcription factor responsible for DR5 induction. CDK4/6 KD or inhibition did not affect the levels of p53, p73, p65, p16 and CHOP (Fig. S3, A and B). DR5 induction was abrogated in p73-knockout (KO) HCT116 cells (Fig. 2, A–C), p73-KO DLD1 cells (Fig. S3, C and D), and p73-KD RKO cells (Fig. S3E). In contrast, DR5 induction was reduced in p53-KO cells (Fig. 2B) and remained intact in p65-KD and CHOP-KD cells.
Figure 2. DR5 induction upon CDK4/6 inhibition is mediated by p73.
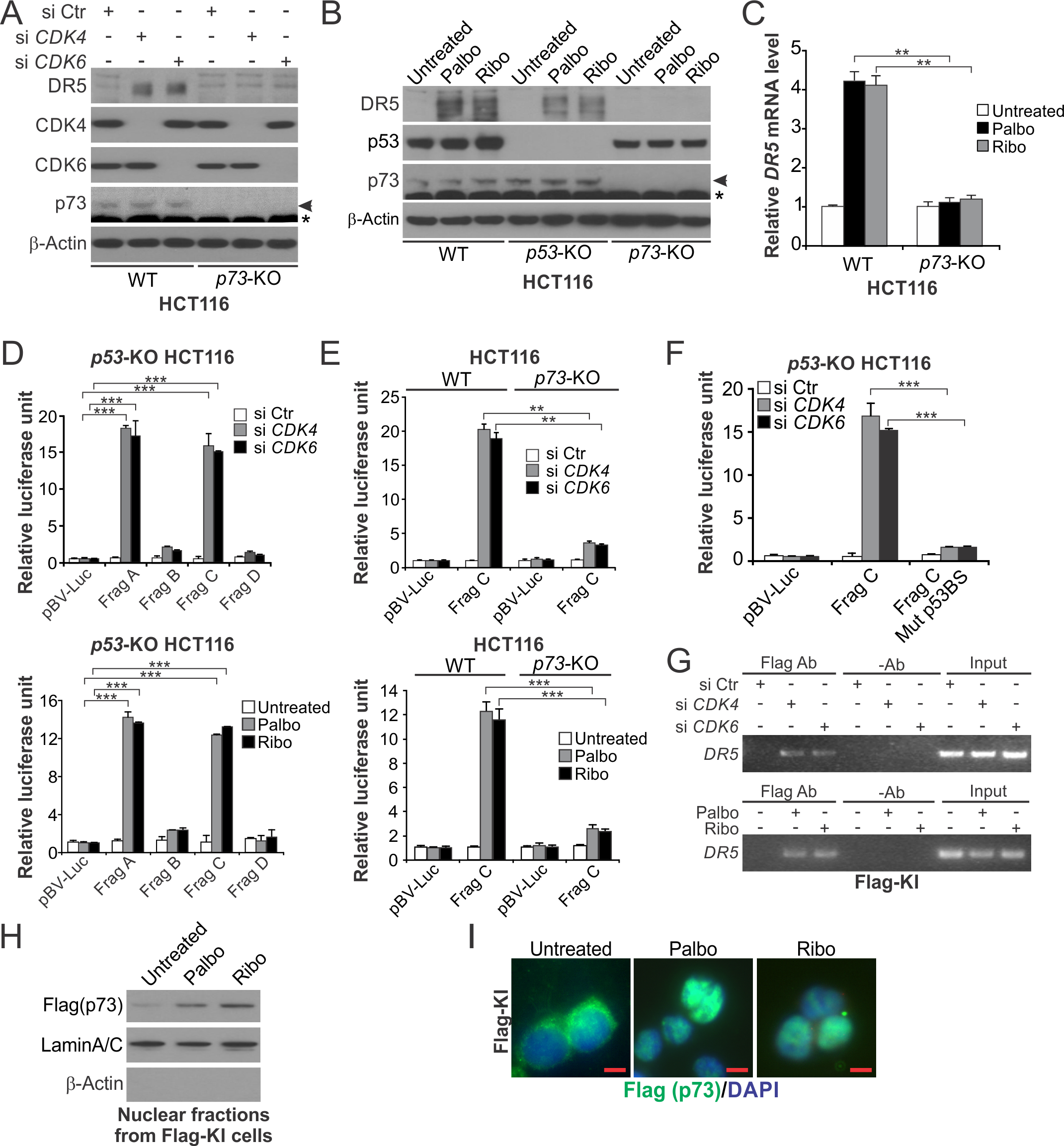
(A) Western blotting of indicated proteins in wildtype (WT) and p73-KO HCT116 cells transfected with indicated siRNA for 24 hours. (B) Western blotting of indicated proteins in WT, p53-KO and p73-KO HCT116 cells treated with 1 μM Palbo or Ribo for 24 hours. In (A) and (B), arrows indicate p73 and stars indicate non-specific bands. (C) RT-PCR analysis of DR5 mRNA expression in WT and p73-KO HCT116 cells treated as in (B). (D) Relative luciferase activities of indicated DR5 reporters in p53-KO HCT116 cells with CDK4/6 knockdown as in (A) (upper panel) or inhibition as in (B) (lower panel). (E) Relative luciferase activities of indicated DR5 promoter reporters in WT and p73-KO HCT116 cells treated and analyzed as in (D). (F) Relative luciferase activities of indicated DR5 promoter reporters with or without p53 binding site mutation (Mut p53BS) in p53-KO HCT116 cells with CDK4/6 knockdown as in (D). (G) HCT116 cells with knock-in of Flag-tagged p73 (Flag-KI) were transfected with siRNA as in (A) for 24 hours (upper panel) or treated with 1 μM Palbo or Ribo for 12 hours (lower panel). The binding of Flag-p73 to the DR5 promoter was analyzed by ChIP followed by PCR analysis of the DR5 promoter region. (H) Nuclear fractions isolated from Flag-KI cells treated with 1 μM Palbo or Ribo for 24 hours were analyzed for p73 by western blotting. Lamin A/C (nuclear) and β-actin (cytoplasmic) were used as loading and fractionation controls. (I) Immunofluorescence analysis of Flag-p73 (green) in Flag-KI cells treated with 1 μM Palbo or Ribo for 3 hours with DAPI (blue) for nuclear counter staining. Scale bars: 10 μm. Bar graphs show results of 3 independent experiments. **, P < 0.01; ***, P < 0.001.
We then analyzed luciferase reporters containing different regions of the DR5 promoter (Fig. S3F). CDK4/6 KD or inhibition strongly activated the DR5 reporters containing the p53 binding site (Frag A and C; Fig. S3F) in a p73-dependent manner (Fig. 2, D and E). Transfection of exogeneous p73 also induced DR5 and activated the DR5 promoter (Fig. S3, G and H). These effects were abolished by mutating key residues in the p53 binding site (Fig. 2F and Fig. S3, F and H). CDK4/6 inhibition also upregulated p73 targets p21 and Noxa in a p73-dependent manner (Fig. S3I). Chromatin immunoprecipitation (ChIP) was performed by using HCT116 cells with knock-in (KI) of Flag-tagged p73 (Flag-KI) (22). CDK4/6 KD or inhibition markedly enhanced the binding of p73 to the DR5 promoter (Fig. 2G). p73 is regulated by nuclear import to transcriptionally activate p53 targets (31,32). CDK4/6 inhibition increased nuclear accumulation of p73 in Flag-KI cells (Fig. 2, H and I). These results demonstrate that p73 directly binds to the p53 binding site in the DR5 promoter to activate its transcription upon CDK4/6 inhibition.
Inhibition of CDK4/6-mediated p73 Thr86 phosphorylation leads to DR5 induction.
p73 subcellular localization and activity are regulated by post-translational modifications such as tyrosine (Tyr) and threonine (Thr) phosphorylation (33,34). CDK4/6 inhibition had no effect on the most common p73 modification, Tyr99 phosphorylation (p-Y99) (34), which was undetectable in HCT116 cells (Fig. S4A). In contrast, CDK4/6 KD or inhibition abrogated Thr phosphorylation of p73 in Flag-KI cells (Fig. 3, A and B). Transfection of WT CDK4 or CDK6, but not dominant negative (DN) mutants, enhanced Thr phosphorylation (Fig. 3C and S4B), which was suppressed by Palbo treatment (Fig. 3D and S4C). CDK4/6 transfection also retarded migration of Flag-p73 in gel electrophoresis (Fig. 3E), which was inhibited by λ protein phosphatase (λPPase) or Palbo treatment (Fig. 3, E and F). Immunoprecipitation (IP) analysis detected direct binding of CDK4 to endogenous p73 in Flag-KI cells (Fig. S4D), which was markedly enhanced upon Palbo or Ribo treatment (Fig. 3G). In an in vitro kinase assay, incubating recombinant human CDK4 or CDK6 with purified p73 (Fig. S4E) strongly stimulated kinase reaction (Fig. 3H).
Figure 3. CDK4/6 inhibition suppresses p73 Thr86 phosphorylation.

(A)-(E) Flag-KI cells were transfected with different constructs or treated with CDK4/6i. p73 threonine (Thr) phosphorylation (p-Thr) was analyzed by immunoprecipitation (IP) using anti-Flag, followed by western blotting using an anti-p-Thr antibody. (A) p-Thr of p73 in cells transfected with indicated siRNA for 24 hours. (B) p-Thr of p73 in cells treated with 1 μM Palbo or Ribo for 8 hours. (C) p-Thr of p73 in cells transfected with WT or dominant negative (DN) CDK4 for 24 hours. (D) Western blotting of p-Thr of p73 in cells transfected with CDK4 with or without Palbo (1 μM) treatment for 24 hours. (E) Western blotting of indicated proteins using cell lysates isolated from Flag-KI cells transfected with indicated constructs for 24 hours, with or without incubation with lambda Protein Phosphatase (λPPase) for 30 minutes. (F) Western blotting of Flag-p73 in Flag-KI cells transfected with CDK4 or CDK6 with or without 1 μM Palbo treatment for 24 hours. Arrows in (E) and (F) indicate upper shifted bands. (G) IP western blotting for analyzing the binding between Flag-p73 and CDK4 in Flag-KI cells treated as in (B). (H) In vitro kinase assay was performed by incubating recombinant human CDK4 or CDK6 with purified WT p73 or T86A mutant, followed by measurement of ADP product concentrations. Results were from 3 independent experiments. ***, P < 0.001. (I) IP western blotting of p-Thr in p73-KO HCT116 cells co-transfected with V5-tagged WT or T86A p73, along with HA-tagged CDK4. (J) Western blotting of HA-p73 in p73-KO HCT116 cells co-transfected with HA-tagged WT or T86A p73 along with CDK4 or CDK6. Arrows indicate upper shifted bands. (K) Western blotting of V5-p73 in cytosolic (Cyto) and nuclear (Nuc) fractions isolated from p73-KO HCT116 cells transfected with V5-tagged WT, T86A or T86D p73 for 24 hours. Whole cell extracts (WCE) were analyzed for comparison. Lamin A/C (nuclear) and β-actin (cytoplasmic) were used as loading and fractionation controls. (L) Western blotting of indicated proteins in p73-KO HCT116 cells transfected with V5-tagged WT, T86A or T86D p73.
We then determined the specific Thr residue of p73 that is phosphorylated by CDK4/6. Thr86 of p73 was shown to undergo CDK1/2-mediated phosphorylation (35,36). We therefore analyzed Thr86 by testing phospho-dead T86A and phospho-mimic T86D mutants. In contrast to WT p73, CDK4/6 transfection had no effect on Thr phosphorylation and gel migration of T86A expressed in p73-KO cells (Fig. 3I, 3J and S4F). Incubating CDK4 or CDK6 with purified T86A protein did not stimulate kinase reaction (Fig. 3H). Compared to WT p73, T86A mutant had increased nuclear accumulation and more strongly induced DR5 (Fig. 3, K and L). Conversely, T86D mutant was defective in nuclear accumulation and DR5 induction (Fig. 3, K and L), resistant to CDK4/6 inhibition (Fig. S4G), and unable to restore DR5 induction in p73-KO cells (Fig. S4H). Together, these results demonstrate that CDK4/6 inhibition suppresses Thr86 phosphorylation and leads to nuclear translocation of p73, which activates DR5 transcription.
DR5 is critical for the anticancer activity of CDK4/6i in CRC cells.
We tested if CDK4/6i sensitize CRC cells to the DR5 ligand TRAIL and other anticancer agents. CDK4/6 KD or inhibition markedly potentiated HCT116 and DLD1 cells to TRAIL, as shown by a lower IC50 and enhanced growth suppression, apoptosis induction, and caspase activation (Fig. S5, A–H). Similar effects of CDK4/6i were observed in breast cancer cells (Fig. S5, I–K), but not in NCM356 cells (Fig. S5L). CDK4/6i also sensitized CRC cells to commonly used anticancer drugs, including 5-FU, oxaliplatin, CPT11, cetuximab, and cisplatin, with enhanced growth suppression, apoptosis induction, and caspase activation (Fig. S6, A–F).
DR5 KO by homologous recombination in HCT116 cells abolished the potentiating effects of CDK4/6i on growth suppression, apoptosis induction, caspase activation, and inhibition of colony formation by TRAIL (Fig. 4, A–D; Fig. S7, A–D) and by 5-FU (Fig. 4, E–H; Fig. S7E). DR5 KO in DLD1 and RKO cells by CRISPR/Cas9 confirmed the requirement of DR5 for the potentiating effects of CDK4/6i (Fig. S7, F–I). Furthermore, perturbation of DR5 downstream apoptosis regulators, including FADD, caspase 8, and Bid, suppressed the potentiating effects of CDK4/6i on TRAIL and 5-FU (Fig. S8, A–F). In contrast, DR5 KO did not affect the inhibition of RB1 phosphorylation by CDK4/6i (Fig. S8G), and RB1 KD did not change TRAIL sensitivity (Fig. S8H). Together, these findings demonstrate a critical role of DR5-dependent apoptosis in mediating the anti-CRC activity of CDK4/6i.
Figure 4. CDK4/6 inhibition requires DR5 to potentiate the killing effects of TRAIL and 5-FU.

(A) MTS analysis of WT and DR5-KO HCT116 cells treated with TRAIL at indicated concentrations +/− 1 μM Palbo or Ribo for 72 hours. (B) Apoptosis in WT and DR5-KO HCT116 cells treated with 10 ng/mL TRAIL +/− 1 μM Palbo or Ribo for 24 hours was analyzed by counting cells with condensed and fragmented nuclei after nuclear staining. (C) Western blotting of indicated proteins in WT and DR5-KO HCT116 cells treated as in (B). (D) Colony formation of WT and DR5-KO HCT116 cells treated as in (B). (E) MTS analysis of WT and DR5-KO HCT116 cells treated with 5-FU at indicated concentrations +/− 1 μM Palbo or Ribo for 72 hours. (F) Apoptosis in WT and DR5-KO HCT116 cells treated with 15 μg/mL 5-FU +/− 1 μM Palbo or Ribo for 24 hours was analyzed as in (B). (G) Western blotting of indicated proteins in WT and DR5-KO HCT116 cells treated as in (F). (H) Colony formation of WT and DR5-KO HCT116 cells treated as in (F) was analyzed as in (D). Bar graphs show results of 3 independent experiments. **, P < 0.01; ***, P < 0.001.
DR5 mediates the in vivo antitumor effects of Palbo in isogenic and PDX models.
We then analyzed Palbo-induced therapeutic sensitization in vivo. WT and DR5-KO HCT116 xenograft tumors growing in athymic nude mice were treated with Palbo alone or in combination with TRAIL or 5-FU. At relatively low doses used, Palbo or TRAIL alone had little effect on WT HCT116 xenografts (Fig. 5A), despite DR5 induction and inhibition of p73 Thr86 phosphorylation by Palbo (Fig. 5B). Combining Palbo and TRAIL more strongly inhibited tumor growth than Palbo alone (P < 0.01, ANOVA with Fisher’s LSD post-hoc test) (Fig. 5A) and enhanced TUNEL and active caspase 3 and 8 immunostaining (P < 0.001, two tailed Student’s t test) (Fig. 5C; Fig. S9, A and B). Similarly, combining Palbo and 5-FU at non-toxic doses significantly improved antitumor efficacy (Fig. 5D), and enhanced DR5 induction (Fig. 5E) and tumor cell apoptosis (Fig. 5F; Fig. S9, C and D). Importantly the antitumor and apoptotic effects of Palbo/TRAIL and Palbo/5-FU combinations were abolished in DR5-KO tumors (Fig. 5, A–F; Fig. S9, A–D). We also generated stable p73-KO HCT116 cells with reconstituted WT or T86D mutant p73 (Fig. S10A). p73 KO phenocopied DR5 KO in suppressing the antitumor and apoptotic effects of Palbo combined with TRAIL (Fig. S10, B–E). Reintroducing WT p73, but not T86D mutant, restored these effects in p73-KO tumors (Fig. S10, B–E).
Figure 5. DR5 is required for the antitumor activity of combining Palbo and TRAIL or 5-FU in mice.

(A)-(C) Nude mice with established WT or DR5-KO HCT116 xenografts were treated with TRAIL (i.p.; 100 μg/mice every other day), Palbo (oral gavage;150 mg/kg daily), or their combination as indicated. (A) Tumor volume at indicated time points with statistical significance for indicated comparisons (N=6 in each group). (B) Western blotting of DR5 and p-p73 (Thr86) in randomly selected WT tumors treated as in (A) for 4 consecutive days. (C) Tumor sections from mice treated as in (B) were analyzed for apoptosis by active caspase 3 staining (left panel) and quantification (right panel). Scale bars: 25 μm. (D)-(F) Nude mice with established WT or DR5-KO HCT116 xenografts were treated with 5-FU (i.p.; 25 mg/kg every other day), Palbo (oral gavage;150 mg/kg daily), or their combination as indicated. (D) Tumor volume at indicated time points with statistical significance for indicated comparisons (N=6 in each group). (E) Western blotting of DR5 in randomly selected WT tumors treated as in (D) for 4 consecutive days. (F) Tumor sections from mice treated as in (E) were analyzed for apoptosis by active caspase 3 staining (left panel) and quantification (right panel). Scale bars: 25 μm. In (C) and (F), arrows indicate examples of positive signals. Bar graphs show results of 300 cells in each mouse (N=3 in each group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We further analyzed the antitumor effects of CDK4/6i on 2 PDX models established from treatment-naïve primary CRC tumors, including PDX1 (T4N0M1; KRAS-G13D; NRAS-G12D; MMR-proficient) and PDX2 (T2N0; MMR-proficient) (29). Palbo combined with TRAIL, but not either agent alone, markedly suppressed the growth of PDX1 and PDX2 tumors without affecting animal weight (Fig. 6, A and B; Fig. S11A). PDX growth inhibition was associated with enhanced tumor cell loss, DR5 induction, p73 Thr86 dephosphorylation, apoptosis induction, and proliferation inhibition (Fig. 6, C–E; Fig. S11, B and C). Combining Palbo and 5-FU also strongly suppressed PDX1 and PDX2 growth and enhanced DR5 induction, apoptosis, and proliferation inhibition (Fig. 6, F–J; Fig. S11, D–F). These results suggest that the in vivo antitumor effects of CDK4/6i are mediated by p73 and DR5 via enhanced apoptosis.
Figure 6. DR5 is involved in suppression of PDX growth by Palbo combined with TRAIL or 5-FU.

(A), (B) NSG mice subcutaneously implanted with PDX1 or PDX2 tumors were treated with TRAIL (i.p.; 100 μg/mice every other day), Palbo (oral gavage;150 mg/kg daily), or their combination as indicated. Volume of (A) PDX1 and (B) PDX2 tumors at indicated time points was plotted with statistical significance for indicated comparisons (N=4 in each group). (C) H&E staining of representative PDX1 tumors from mice treated as in (A) for 10 consecutive days. Scale bars: 100 μm. (D) Western blotting of DR5 and p-p73 (Thr86) in tumors from mice treated as in (C). (E) Active caspase 3 staining (left panel) and quantification (right panel) of tumor sections from mice treated as in (C). Scale bars: 25 μm. (F), (G) NSG mice subcutaneously implanted with PDX1 or PDX2 tumors were treated with 5-FU (i.p.; 25 mg/kg every other day), Palbo (oral gavage;150 mg/kg daily), or their combination as indicated. Volume of (F) PDX1 and (G) PDX2 tumors at indicated time points was analyzed as in (A) (N=4 in each group). (H) H&E staining of representative PDX1 tumors from mice treated as in (F) for 10 consecutive days. Scale bars: 100 μm. (I) Western blotting of DR5 in PDX1 and PDX2 tumors from mice treated as in (H). (J) Active caspase 3 staining (left panel) and quantification (right panel) of tumor sections from mice treated as in (H). Scale bars: 25 μm. In (E) and (J), arrows indicate examples of positive signals. Bar graphs show results of 300 cells in each mouse (N=3 in each group). *, P < 0.05; **, P < 0.01.
DR5 mediates CDK4/6i-induced ICD.
The induction of DR5 by CDK4/6i prompted us to test if the immunogenic effects of CDK4/6i are mediated by ICD, which is characterized by cell-surface exposure of the ER chaperone calreticulin (CRT) and dendritic cell (DC) maturation (15,16). Treating HCT116 cells with Palbo or Ribo markedly increased cell-surface CRT, which was blocked in DR5-KO and p73-KO cells (Fig. 7, A and B). To test if CDK4/6i-induced dying CRC cells can be recognized and phagocytized by DCs, we labelled DCs differentiated from peripheral blood mononuclear cells (PBMCs) of healthy donors with Far Red (red), and Palbo- or Ribo-treated HCT116 cells with carboxyfluorescein succinimidyl ester (CFSE; green). Upon co-culturing labelled DCs and HCT116 cells, we detected by fluorescence microscopy and flow cytometry a significant increase in double positive fused cells following Palbo or Ribo treatment, which was suppressed by DR5 or p73 KO (Fig. 7, C–E). These results suggest a key role of p73-dependent DR5 induction in mediating the immunogenic effects of CDK4/6i.
Figure 7. DR5 and p73 mediate CDK4/6i-induced immunogenic cell death (ICD).

(A), (B) WT, DR5-KO, and p73-KO HCT116 cells treated with 1 μM Palbo or Ribo for 24 hours were analyzed for CRT cell-surface translocation by (A) CRT immunostaining and (B) flow cytometry. (C)-(E) HCT116 cells treated as in (A) and dendritic cells (DCs) differentiated from healthy donors’ monocytes were labelled with CFSE (green) and Far Red (red), respectively, and co-incubated at 1:1 ratio for 2 hours. DC phagocytosis was analyzed by detecting phagocytic DCs fused with HCT116 cells by (C) fluorescence microscopy with arrows indicating examples (scale bars: 10 μm), (D) flow cytometry (upper right quadrant), and (E) quantification of flow cytometry results. Bar graphs show results of 3 independent experiments. **, P < 0.01.
DR5 is required for the potentiation effects of Palbo on anti-PD-1 immunotherapy.
We then used immuno-competent CT26-BALB/cJ syngeneic tumor model to determine if CDK4/6i enhance anti-PD-1 immunotherapy through DR5-dependent ICD. BALB/cJ mice bearing WT or isogenic DR5-KO (generated by CRISPR/Cas9) CT26 tumors were treated with anti-mouse PD-1 antibody at a previously described dose (i.p.; 200 μg/dose) and schedule (3× every 2 days followed by 2× weekly; Fig. 8A) (37), with or without combination with Palbo at a reduced dose (100 mg/kg) and frequency (3 days on, 4 days off) (Fig. 8A). Although either agent alone had little or no efficacy, the combination group showed striking therapeutic efficacy with complete inhibition of WT CT26 tumor growth in 4 out of 8 mice at the endpoint on day 28 (Fig. 8B and Fig. S12A), along with significant extension of animal survival (Fig. 8C). The combination was well tolerated without affecting body weight (Fig. S12B). In contrast, the therapeutic effect was almost completely lost in DR5-KO CT26 tumors (Fig. 8, B and C; Fig. S12A).
The combination treatment significantly increased infiltration of CD3+ and CD8+ T cells, and decreased infiltration of CD25+/FoxP3+ immunosuppressive regulatory T cells (Tregs) in WT tumors (Fig. 8, D–G; Fig. S12, C–F). The treatment also increased granzyme-B (GzmB) and interferon γ (IFNγ) production by tumor-infiltrating CD8+ T cells (Fig. 8, H and I), and expanded infiltrating CD11c+ DCs expressing increased CD86 and MHC class II (MHCII) (Fig. 8, J–L). Remarkably, all of the observed changes were suppressed in DR5-KO CT26 tumors (Fig. 8, D–L), indicating a critical role of DR5-dependent cell death in mediating the antitumor immune response to this combination. Furthermore, depleting CD8+ cytotoxic T cells (Fig. 8A) abolished the effects of combination treatment on WT tumors (Fig. 8M), confirming the role of CD8+ T cells in tumor suppression.
Discussion
We identified p73-dependent and RB1-independent induction of DR5 as a novel antitumor mechanism of CDK4/6i in CRC cells. CDK4/6 inhibition suppresses p73 Thr86 phosphorylation, which leads to p73 nuclear translocation and increased DR5 promoter binding and transcription. Thr86 of p73 is highly conserved among different species. Thr86 phosphorylation is mediated by CDK1/2 in lung cancer cells (35) but by CDK4/6 in CRC cells. A non-canonical function of CDKs seems to regulate p73 Thr86 phosphorylation in cancer cells (38). DR5 induction by CDK4/6i is largely p53-independent, reflecting the distinct functions of p73 and p53 in regulating common target genes. However, p53 seems to impact DR5 induction (Fig. 2B), likely via its effects on p73 (39). Interestingly, our recent study showed that CDK4/6 inhibition in mice protects against radiation-induced intestinal injury by abolishing p53 and PUMA induction (40). The distinct functional roles and complex interplay of p53, p73, and other p53 family members in coordinating response to CDK4/6i in cancer and normal cells remain to be further elucidated. Some of our mechanistic experiments are limited by a single cell line and need to be repeated using other cell lines.
Our results suggest that rational combinations of CDK4/6i with other anticancer agents can markedly improve therapeutic efficacy through DR5-dependent ICD. The striking therapeutic effects of CDK4/6i combined with TRAIL via DR5 induction provide a compelling rationale for developing the TRAIL-CDK4/6i combination for treating advanced and therapy-resistant CRCs. DR5 induction mediates the enhanced apoptotic and antitumor effects of combining 5-FU and CDK4/6i in CRC cells. DR5 induction may lower the apoptotic threshold to sensitize CRC cells to different stimuli, including TRAIL, chemotherapy, or activated immune cells. The enhanced DR5 induction in CRC cells treated by Palbo combined with 5-FU may be explained by simultaneous activation of DR5 transcription by p73, p53, NF-κB, CHOP, and/or other transcription factors. DR5 induction may be a useful indicator for therapeutic efficacy and effective drug combinations.
Recent studies have demonstrated multiple effects of CDK4/6i in antitumor immunity, such as activating innate immunity by inducing endogenous retroviral elements, intracellular double-stranded RNA, and interferon response (6), upregulating PD-L1 by dephosphorylating the E3 ubiquitin ligase SPOP (7), and stimulating T cell activation by derepressing NFAT family proteins and targets (41). As DR5 functions as a nodule point of different apoptotic pathways, including cytokine-induced apoptosis in immune response, these immunogenic effects of CDK4/6i may converge and culminate in DR5 induction required for robust killing of tumor cells. DR5-mediated tumor cell death may function as a key link between innate and adaptive immune responses through increased phagocytosis by DCs.
The clinical benefit of immune checkpoint inhibitors (ICIs) is limited to 10–15% of CRCs that are MMR-deficient (42). A burning issue is to convert immunologically “cold” tumors into “hot” tumors (43). CDK4/6 inhibition represents a promising strategy for targeting immune escape mediated by aberrant CDK signaling in cancer cells, especially for those that are insensitive to ICIs. DR5 induction may be useful for predicting effective combinations and responsiveness to ICI combinations.
Collectively, our results reveal a novel, on-target mechanism of CDK4/6i through p73-dependent DR5 induction, which is crucial for the apoptotic, chemo-sensitizing, and immunogenic effects of CDK4/6i in CRC cells.
Supplementary Material
Significance.
This work demonstrates how inhibition of CDK4/6 sensitizes cancer cells to chemotherapy and immune checkpoint blockade and may provide a new molecular marker for improving CDK4/6-targeted cancer therapies.
Acknowledgments
This work was supported by the U.S. National Institutes of Health grants (R01CA203028, R01CA217141, R01CA236271, R01CA247231, and R01CA248112 to L. Zhang; U19AI068021 and R01CA215481 to J. Yu; R01CA260357 to Y. Huang; T32GM133332 to K. Ermine). This project used Animal Facility, Cytometry Facility, and Tissue and Research Pathology Services supported by P30CA047904.
Footnotes
Conflict of interest: The authors have no conflict of interest.
References:
- 1.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999;13:1501–12. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 3.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153–66. [DOI] [PubMed] [Google Scholar]
- 4.Du Q, Guo X, Wang M, Li Y, Sun X, Li Q. The application and prospect of CDK4/6 inhibitors in malignant solid tumors. J Hematol Oncol 2020;13:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 2018;34:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017;548:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018;553:91–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Zhou L, Zhao S, Dicker DT, El-Deiry WS. The CDK4/6 inhibitor palbociclib synergizes with irinotecan to promote colorectal cancer cell death under hypoxia. Cell Cycle 2017;16:1193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashkenazi A Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 2008;7:1001–12. [DOI] [PubMed] [Google Scholar]
- 10.Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev 2018;37:733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu GS, Burns TF, McDonald ER 3rd, Jiang W, Meng R, Krantz ID, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 1997;17:141–3. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 2004;279:45495–502. [DOI] [PubMed] [Google Scholar]
- 13.Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends Biochem Sci 2014;39:101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhola PD, Letai A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol Cell 2016;61:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017;17:97–111. [DOI] [PubMed] [Google Scholar]
- 16.Wang YJ, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis 2018;5:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov 2016;6:740–53. [DOI] [PubMed] [Google Scholar]
- 18.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 19.Chu E An update on the current and emerging targeted agents in metastatic colorectal cancer. Clin Colorectal Cancer 2012;11:1–13. [DOI] [PubMed] [Google Scholar]
- 20.Lee JJ, Chu E. Recent Advances in the Clinical Development of Immune Checkpoint Blockade Therapy for Mismatch Repair Proficient (pMMR)/non-MSI-H Metastatic Colorectal Cancer. Clin Colorectal Cancer 2018;17:258–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leibowitz B, Qiu W, Buchanan ME, Zou F, Vernon P, Moyer MP, et al. BID mediates selective killing of APC-deficient cells in intestinal tumor suppression by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A 2014;111:16520–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D, Ming L, Zou F, Peng Y, Van Houten B, Yu J, et al. TAp73 promotes cell survival upon genotoxic stress by inhibiting p53 activity. Oncotarget 2014;5:8107–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong J, Tan S, Zou F, Yu J, Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene 2017;36:787–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tong J, Tan S, Nikolovska-Coleska Z, Yu J, Zou F, Zhang L. FBW7-Dependent Mcl-1 Degradation Mediates the Anticancer Effect of Hsp90 Inhibitors. Mol Cancer Ther 2017;16:1979–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2019;38:390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fletcher R, Tong J, Risnik D, Leibowitz BJ, Wang YJ, Concha-Benavente F, et al. Non-steroidal anti-inflammatory drugs induce immunogenic cell death in suppressing colorectal tumorigenesis. Oncogene 2021;40:2035–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong J, Wang P, Tan S, Chen D, Nikolovska-Coleska Z, Zou F, et al. Mcl-1 Degradation Is Required for Targeted Therapeutics to Eradicate Colon Cancer Cells. Cancer Res 2017;77:2512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong J, Zheng X, Tan X, Fletcher R, Nikolovska-Coleska Z, Yu J, et al. Mcl-1 Phosphorylation without Degradation Mediates Sensitivity to HDAC Inhibitors by Liberating BH3-Only Proteins. Cancer Res 2018;78:4704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan X, Tong J, Wang YJ, Fletcher R, Schoen RE, Yu J, et al. BET inhibitors potentiate chemotherapy and killing of SPOP-mutant colon cancer cells via induction of DR5. Cancer Res 2019;79:1191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song X, Shen L, Tong J, Kuang C, Zeng S, Schoen RE, et al. Mcl-1 inhibition overcomes intrinsic and acquired regorafenib resistance in colorectal cancer. Theranostics 2020;10:8098–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue T, Stuart J, Leno R, Maki CG. Nuclear import and export signals in control of the p53-related protein p73. J Biol Chem 2002;277:15053–60. [DOI] [PubMed] [Google Scholar]
- 32.Fontemaggi G, Kela I, Amariglio N, Rechavi G, Krishnamurthy J, Strano S, et al. Identification of direct p73 target genes combining DNA microarray and chromatin immunoprecipitation analyses. J Biol Chem 2002;277:43359–68. [DOI] [PubMed] [Google Scholar]
- 33.Rosenbluth JM, Pietenpol JA. The jury is in: p73 is a tumor suppressor after all. Genes Dev 2008;22:2591–5. [DOI] [PubMed] [Google Scholar]
- 34.Knickelbein K, Tong J, Chen D, Wang YJ, Misale S, Bardelli A, et al. Restoring PUMA induction overcomes KRAS-mediated resistance to anti-EGFR antibodies in colorectal cancer. Oncogene 2018;37:4599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaiddon C, Lokshin M, Gross I, Levasseur D, Taya Y, Loeffler JP, et al. Cyclin-dependent kinases phosphorylate p73 at threonine 86 in a cell cycle-dependent manner and negatively regulate p73. J Biol Chem 2003;278:27421–31. [DOI] [PubMed] [Google Scholar]
- 36.Melino G, Lu X, Gasco M, Crook T, Knight RA. Functional regulation of p73 and p63: development and cancer. Trends Biochem Sci 2003;28:663–70. [DOI] [PubMed] [Google Scholar]
- 37.Selby MJ, Engelhardt JJ, Johnston RJ, Lu LS, Han M, Thudium K, et al. Preclinical Development of Ipilimumab and Nivolumab Combination Immunotherapy: Mouse Tumor Models, In Vitro Functional Studies, and Cynomolgus Macaque Toxicology. PLoS One 2016;11:e0161779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol 2016;17:280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lokshin M, Li Y, Gaiddon C, Prives C. p53 and p73 display common and distinct requirements for sequence specific binding to DNA. Nucleic Acids Res 2007;35:340–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei L, Leibowitz BJ, Wang X, Epperly M, Greenberger J, Zhang L, et al. Inhibition of CDK4/6 protects against radiation-induced intestinal injury in mice. J Clin Invest 2016;126:4076–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 2018;8:216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lizardo DY, Kuang C, Hao S, Yu J, Huang Y, Zhang L. Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: From bench to bedside. Biochim Biophys Acta Rev Cancer 2020;1874:188447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.