

In the title compound, the dihydroquinoline moiety is not quite planar and an intramolecular C—H⋯O hydrogen bond helps to establish the rotational orientation of the carboxyl group. In the crystal, sheets of molecules parallel to (10
 ) are generated by C—H⋯O and C—H⋯Cl hydrogen bonds.
) are generated by C—H⋯O and C—H⋯Cl hydrogen bonds.
Keywords: crystal structure, hydrogen-bonding, π-stacking, dihydroquinoline
Abstract
In the title compound, C12H10ClNO3, the dihydroquinoline moiety is not planar with a dihedral angle between the two ring planes of 1.61 (6)°. An intramolecular C—H⋯O hydrogen bond helps to establish the rotational orientation of the carboxyl group. In the crystal, sheets of molecules parallel to (10
) are generated by C—H⋯O and C—H⋯Cl hydrogen bonds, and are stacked through slipped π-stacking interactions between inversion-related dihydroquinoline units. A Hirshfeld surface analysis of the crystal structure indicates that the most important contributions for the crystal packing are from H⋯H (34.2%), H⋯O/O⋯H (19.9%), H⋯Cl/Cl⋯H (12.8%), H⋯C/C⋯H (10.3%) and C⋯C (9.7%) interactions. Computational chemistry indicates that in the crystal, the C—H⋯Cl hydrogen-bond energy is −37.4 kJ mol−1, while the C—H⋯O hydrogen-bond energies are −45.4 and −29.2 kJ mol−1. An evaluation of the electrostatic, dispersion and total energy frameworks revealed that the stabilization is dominated via the dispersion energy contribution. Density functional theory (DFT) optimized structures at the B3LYP/6–311 G(d,p) level are compared with the experimentally determined molecular structure in the solid state, and the HOMO—LUMO behaviour was elucidated to determine the energy gap.
Chemical context
Over the past few decades, heterocyclic chemistry has received increasing interest because of the pharmacological importance of the majority of heterocyclic compounds, especially N-containing heterocycles such as quinoline derivatives (Filali Baba et al., 2019 ▸; Hayani et al., 2021 ▸). Quinoline derivatives possess numerous biological properties, including antimicrobial (Katoh et al., 2004 ▸; Abdel-Wahab et al., 2012 ▸), anti-inflammatory (Leatham et al., 1983 ▸), antihypertensive (Muruganantham et al., 2004 ▸), antibiotic (Mahamoud et al., 2006 ▸), anti-HIV (Wilson et al., 1992 ▸; Strekowski et al., 1991 ▸) and corrosion-inhibitive activities (Filali Baba et al., 2016a
▸,b
▸). They are also considered to be important scaffolds for the development of new molecules of pharmaceutical interest (Filali Baba et al., 2020 ▸; Bouzian et al., 2018 ▸).

In a continuation of our research work devoted to the study of O- and N-alkylation reactions involving quinoline derivatives, we report here the synthesis and crystal structure of methyl 6-chloro-1-methyl-2-oxo-1,2-dihydroquinoline-4-carboxylate obtained by the alkylation reaction of 6-chloro-2-oxo-1,2-dihydroquinoline-4-carboxylic acid with an excess of methyl iodide as an alkylating reagent in phase transfer catalysis (PTC). The molecular and crystal structure as well as the Hirshfeld surface analysis of the title compound are reported. The results obtained using density functional theory (DFT) calculations, performed at the B3LYP/6-311G(d,p) level, are compared with the experimental results determined from the molecular and crystal structures in the solid state of the title compound, (I).
Structural commentary
The bicyclic molecular core is not planar as there is a dihedral angle of 1.61 (6)° between the mean planes of its constituent rings. The dihedral angle between the mean plane of the (C1/N1/C6–C9) ring and the plane defined by atoms C7, C11, O2 and O3 is 4.08 (8)° with the near coplanarity of the carboxyl group and the heterocyclic ring being caused, in part, by the intramolecular C5—H5⋯O2 hydrogen bond (Table 1 ▸, Fig. 1 ▸).
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2⋯O2iii | 0.95 | 2.57 | 3.5146 (16) | 178 |
| C5—H5⋯O2 | 0.95 | 2.19 | 2.8496 (16) | 126 |
| C8—H8⋯Cl1iv | 0.95 | 2.84 | 3.7786 (13) | 170 |
| C12—H12C⋯O1ii | 0.98 | 2.36 | 3.0016 (16) | 122 |
Symmetry codes: (ii)
 ; (iii)
; (iii)
 ; (iv)
; (iv)
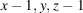 .
.
Figure 1.

The title molecule with labelling scheme and displacement ellipsoids drawn at the 50% probability level. The intramolecular C—H⋯O hydrogen bond is depicted by a dashed line.
Supramolecular features
In the crystal, C2—H2⋯O2iii hydrogen bonds (Table 1 ▸) form ribbons of molecules extending along [010], which are further linked into sheets parallel to (10
) by C12—H12C⋯O1ii and weak C8—H8⋯Cl1iv (H⋯Cl is 0.11 Å less than the sum of the van der Waals radii) hydrogen bonds (Table 1 ▸, Fig. 2 ▸). The sheets are stacked along the direction of the normal to (10
) by slipped π-stacking interactions between inversion-related dihydroquinoline moieties [centroid⋯centroid distance = 3.7140 (7) Å, dihedral angle = 1.61 (6)°, slippage = 1.63 Å] (Fig. 3 ▸).
Figure 2.

A portion of one layer projected on (10
) with C—H⋯O and C—H⋯Cl hydrogen bonds depicted, respectively, by black and green dashed lines.
Figure 3.

Packing of molecules viewed along [010] with slipped π-stacking interactions depicted by orange dashed lines.
Hirshfeld surface analysis
In order to visualize the intermolecular interactions in the crystal of (I), a Hirshfeld surface (HS) analysis (Hirshfeld, 1977 ▸) was carried out by using CrystalExplorer17.5 (Turner et al., 2017 ▸). In the HS plotted over d norm (Fig. 4 ▸ a), the white surface indicates contacts with distances equal to the sum of van der Waals radii, and the red and blue colours indicate distances shorter (in close contact) or longer (distinct contact) than the van der Waals radii (Venkatesan et al., 2016 ▸). Selected contacts are given in Table 2 ▸. The bright-red spots indicate their roles as the respective donors and/or acceptors; they also appear as blue and red regions corresponding to positive and negative potentials on the HS mapped over electrostatic potential (Spackman et al., 2008 ▸; Jayatilaka et al., 2005 ▸) shown in Fig. 4 ▸ b. The blue regions indicate a positive electrostatic potential (hydrogen-bond donors), while the red regions indicate a negative electrostatic potential (hydrogen-bond acceptors). The shape-index of the HS is a tool to visualize π–π stacking by the presence of adjacent red and blue triangles: if there are no adjacent red and/or blue triangles, then there are no π–π interactions. Fig. 4 ▸ c clearly suggests that there are π–π interactions in (I). The overall two-dimensional fingerprint plot is shown in Fig. 5 ▸ a, and those delineated into H⋯H, H⋯O/O⋯H, H⋯Cl/Cl⋯H, H⋯C/C⋯H, C⋯C, C⋯O/O⋯C, C⋯Cl/Cl⋯C, O⋯Cl/Cl⋯O, O·· O, H⋯N/N⋯H, N⋯Cl/Cl⋯N, C⋯N/N⋯C, Cl⋯Cl and N⋯O/O⋯N contacts (McKinnon et al., 2007 ▸) are illustrated in Fig. 5 ▸ b–o, respectively, together with their relative contributions to the Hirshfeld surface. The most important interaction is H⋯H, contributing 34.2% to the overall crystal packing, which is reflected in Fig. 5 ▸ b as widely scattered points of high density due to the large hydrogen content of the molecule with the tip at d e = d i = 1.24 Å. The pair of the scattered points of spikes in the fingerprint plot delineated into H⋯O/O⋯H contacts, Fig. 5 ▸ c, with a 19.9% contribution to the HS has a distribution of points with the tips at d e + d i = 2.28 Å. The H⋯Cl/Cl⋯H contacts, Fig. 5 ▸ d, with a 12.8% contribution to the HS have a symmetric distribution of points with the tips at d e + d i = 2.68 Å. In the absence of C—H⋯π interactions, the pair of characteristic wings in the fingerprint plot delineated into H⋯C/C⋯H contacts, Fig. 5 ▸ e, with a 10.3% contribution to the HS has the tips at d e + d i = 3.01 Å. The C⋯C contacts, Fig. 5 ▸ f, with a 9.7% contribution to the HS have a bullet-shaped distribution of points with the tip at d e = d i = 1.67 Å. The C⋯O/O⋯C contacts, Fig. 5 ▸ g, with a 3.4% contribution to the HS have the tips at d e + d i = 3.32 Å and d e + d i = 3.45 Å for sharp and tiny distributions of points. The symmetric distribution of points of the C⋯Cl/Cl⋯C contacts, Fig. 5 ▸ h, with a 3.0% contribution to the HS appear as scattered points with a tiny pair of spikes with the tips at d e + d i = 3.48 Å. Finally, the contributions of the remaining O⋯Cl/Cl⋯O, O⋯O, H⋯N/N⋯H, N⋯Cl/Cl⋯N, C⋯N/N⋯C, Cl⋯Cl and N⋯O/O⋯N contacts (Fig. 5 ▸ i–o) are smaller than 3.0% to the HS with low densities of points.
Figure 4.

(a) View of the three-dimensional Hirshfeld surface of the title compound, plotted over d norm in the range of −0.2172 to 0.9151 a.u.; (b) view of the three-dimensional Hirshfeld surface of the title compound plotted over electrostatic potential energy in the range −0.0500 to 0.0500 a.u. using the STO-3 G basis set at the Hartree–Fock level of theory; (c) Hirshfeld surface of the title compound plotted over shape-index.
Table 2. Selected interatomic distances (Å).
| Cl1⋯O3i | 3.1903 (10) | H2⋯O2iii | 2.57 |
| Cl1⋯H8i | 2.84 | O3⋯H8 | 2.25 |
| C12⋯O1ii | 3.0016 (16) | C2⋯H10C | 2.78 |
| O2⋯C5 | 2.8496 (17) | C2⋯H10B | 2.74 |
| O1⋯H10A | 2.26 | C10⋯H2 | 2.46 |
| H12C⋯O1ii | 2.36 | C11⋯H5 | 2.71 |
| O2⋯H12A | 2.64 | H2⋯H10B | 2.24 |
| O2⋯H5 | 2.19 | H2⋯H10C | 2.29 |
| O2⋯H12B | 2.55 |
Symmetry codes: (i)
 ; (ii)
; (iii)
.
; (ii)
; (iii)
.
Figure 5.

The full two-dimensional fingerprint plots for the title compound, showing (a) all interactions, and delineated into (b) H⋯H, (c) H⋯O/O⋯H, (d) H⋯Cl/Cl⋯H, (e) H⋯C/C⋯H, (f) C⋯C, (g) C⋯O/O⋯C, (h) C⋯Cl/Cl⋯C, (i) O⋯Cl/Cl⋯O, (j) O⋯O, (k) H⋯N/N⋯H, (l) N⋯Cl/Cl⋯N, (m) C⋯N/N⋯C, (n) Cl⋯Cl and (o) N⋯O/O⋯N interactions. The d i and d e values are the closest internal and external distances (in Å) from given points on the Hirshfeld surface.
The Hirshfeld surface representations with the function d norm plotted onto the surface are shown for the H⋯H, H⋯O/O⋯H, H⋯Cl/Cl⋯H, H⋯C/C⋯H and C⋯C interactions in Fig. 6 ▸ a–e, respectively.
Figure 6.

Hirshfeld surface representations with the function d norm plotted onto the surface for (a) H⋯H, (b) H⋯O/O⋯H, (c) H⋯Cl/Cl⋯H, (d) H⋯C/C⋯H and (e) C⋯C interactions.
The Hirshfeld surface analysis confirms the importance of H-atom contacts in establishing the packing. The large number of H⋯H, H⋯O/O⋯H, H⋯Cl/Cl⋯H and H⋯C/C⋯H interactions suggest that van der Waals interactions play the major role in the crystal packing (Hathwar et al., 2015 ▸).
Interaction energy calculations
The intermolecular interaction energies were calculated using the CE–B3LYP/6–31G(d,p) energy model available in CrystalExplorer17.5 (Turner et al., 2017 ▸), where a cluster of molecules would be needed by applying crystallographic symmetry operations with respect to a selected central molecule within the radius of 3.8 Å by default (Turner et al., 2014 ▸). The total intermolecular energy (E tot) is the sum of electrostatic (E ele), polarization (E pol), dispersion (E dis) and exchange-repulsion (E rep) energies (Turner et al., 2015 ▸) with scale factors of 1.057, 0.740, 0.871 and 0.618, respectively (Mackenzie et al., 2017 ▸). Hydrogen-bonding interaction energies (in kJ mol−1) were calculated to be −10.4 (E ele), −1.6 (E pol), −51.9 (E dis), 32.4 (E rep) and −37.4 (E tot) [for the C8—H8⋯Cl1iv hydrogen-bonding interaction], −0.9 (E ele), −2.8 (E pol), −84.0 (E dis), 49.9 (E rep) and −45.4 (E tot) (for C2—H2⋯O2iii) and −6.0 (E ele), −4.1 (E pol), −37.0 (E dis), 20.2 (E rep) and −29.2 (E tot) (for C12—H12C⋯O1ii).
Energy frameworks
Energy frameworks combine the calculation of intermolecular interaction energies with a graphical representation of their magnitude (Turner et al., 2015 ▸). Energies between molecular pairs are represented as cylinders joining the centroids of pairs of molecules with the cylinder radius proportional to the relative strength of the corresponding interaction energy. Energy frameworks were constructed for E ele (red cylinders), E dis (green cylinders) and E tot (blue cylinders) (Fig. 7 ▸ a–c). The evaluation of the electrostatic, dispersion and total energy frameworks indicates that the stabilization is dominated by the dispersion energy contributions in (I).
Figure 7.

Energy frameworks of (I).
DFT calculations
The geometrical parameters and energies of (I) in the gas phase were computed via density functional theory (DFT) using the standard B3LYP functional and 6–311G(d,p) basis-set calculations (Becke, 1993 ▸) as implemented in GAUSSIAN 09 (Frisch et al., 2009 ▸), see Table 3 ▸. The theoretical bond lengths and angles are in good agreement with those based on the X-ray analysis. However, a few differences exists in case of some dihedral angles (N1—C1—C6—C7; C10—N1—C1—C6; O1—C9—N1—C1; O2—C11—C7—C6; O3—C11—C7—C6; C12—O3—C11—C7), because in the DFT calculations there is only one molecule treated in the gas phase whereas in the solid state several molecules interact by hydrogen-bonding interactions (Fig. 2 ▸, Table 1 ▸). The torsion angles show that the conformation of the molecule in the gas phase has C 1 symmetry.
Table 3. B3LY/6–311G(d,p) equilibrium structural parameters (Å, °) and X-ray analysis of the title compound, (I).
| Bonds/angles | X-ray | B3LYP/6–311G(d,p) |
|---|---|---|
| C2—C1 | 1.4070 (17) | 1.4562 |
| C3—C2 | 1.3547 (18) | 1.3547 |
| C4—C3 | 1.3836 (17) | 1.3933 |
| C5—C4 | 1.3807 (17) | 1.3798 |
| C6—C5 | 1.4098 (16) | 1.4086 |
| C7—C6 | 1.4514 (16) | 1.4562 |
| C8—C7 | 1.3510 (16) | 1.3547 |
| C9—C8 | 1.4513 (17) | 1.4563 |
| N1—C1 | 1.3920 (15) | 1.3914 |
| C10—N1 | 1.4673 (16) | 1.4641 |
| H2—C2 | 0.95 | 1.07965 |
| H3—C3 | 0.95 | 1.0822 |
| Cl1—C4 | 1.7397 (13) | 1.7594 |
| H5—C5 | 0.9500 | 1.0777 |
| C11—C7 | 1.5022 (16) | 1.5077 |
| H8—C8 | 0.9500 | 1.0792 |
| O1—C9 | 1.2319 (15) | 1.2227 |
| O2—C11 | 1.2040 (16) | 1.2086 |
| O3—C11 | 1.3258 (15) | 1.3452 |
| C12—O3 | 1.4458 (15) | 1.4400 |
| H12A—C12 | 0.98 | 1.0907 |
| H12B—C12 | 0.98 | 1.0907 |
| H12C—C12 | 0.98 | 1.0872 |
| H10A—C10 | 0.98 | 1.0924 |
| H10B—C10 | 0.98 | 1.0859 |
| H10C—C10 | 0.98 | 1.0924 |
| C3—C2—C1 | 120.15 (11) | 121.15 |
| C4—C3—C2 | 120.29 (12) | 119.41 |
| C5—C4—C3 | 121.22 (12) | 121.07 |
| C6—C5—C4 | 120.00 (11) | 120.57 |
| C7—C6—C5 | 123.66 (11) | 123.69 |
| C8—C7—C6 | 119.52 (11) | 119.54 |
| C9—C8—C7 | 123.22 (11) | 123.86 |
| N1—C1—C6 | 120.32 (11) | 120.78 |
| C10—N1—C1 | 119.77 (10) | 120.37 |
| H2—C2—C1 | 119.9 | 120.30 |
| H3—C3—C4 | 119.9 | 120.32 |
| Cl1—C4—C5 | 118.82 (10) | 119.62 |
| H5—C5—C6 | 120.00 | 119.03 |
| C11—C7—C6 | 122.02 (10) | 122.01 |
| H8—C8—C9 | 118.40 | 114.79 |
| O1—C9—N1 | 122.17 (12) | 122.09 |
| O2—C11—C7 | 125.71 (11) | 125.86 |
| O3—C11—C7 | 111.59 (10) | 111.75 |
| C12—O3—C11 | 116.08 (11) | 115.75 |
| H12A—C12—O3 | 109.5 | 110.40 |
| H12B—C12—O3 | 109.5 | 110.40 |
| H12C—C12—O3 | 109.5 | 105.31 |
| H10A—C10—N1 | 109.5 | 110.62 |
| H10B—C10—N1 | 109.5 | 107.00 |
| H10C—C10—N1 | 109.5 | 110.40 |
| C4—C3—C2—C1 | 0.40 (19) | 0.00 |
| C5—C4—C3—C2 | 0.32 (19) | 0.00 |
| C6—C5—C4—C3 | −0.25 (19) | 0.00 |
| C7—C6—C5—C4 | −179.83 (11) | −180.00 |
| C8—C7—C6—C1 | −1.59 (17) | 0.00 |
| C9—C8—C7—C6 | 0.23 (18) | 0.00 |
| N1—C1—C6—C7 | 0.69 (17) | 0.00 |
| C10—N1—C1—C6 | −178.45 (12) | −180.0 |
| Cl1—C4—C5—C6 | −179.81 (9) | −180.0 |
| O1—C9—N1—C1 | 177.48 (12) | 179.99 |
| O2—C11—C7—C6 | −4.6 (2) | −0.01 |
| O3—C11—C7—C6 | 175.14 (11) | −179.99 |
| C12—O3—C11—C7 | −179.85 (11) | 179.99 |
The infrared spectrum of (I) on basis of the B3LYP/6-311G calculation is shown in the supporting information. All harmonic frequencies are positive, demonstrating the minimal signature of (I). The spectrum mainly constitutes 75 vibration modes. The CH3 torsion appears in the 17–119 cm−1 region, the ν C=C stretching mode is at 1363 cm−1, the vibrations of the aromatic N—CH3 appear at 1091 cm−1, and the O—CH3 and the C—Cl stretching bands are observed, respectively, at 1033 cm−1 and 1124 cm−1. The C—H stretch of the CH3 group appears at 3182 cm−1, however the aromatic C—H stretches appear in the 3208-3256 cm−1 region. The bending of CH3 appear between 1528 cm−1 and 1556 cm−1, Finally, the band positions of the bending of the HCC, HCN and HCO groups are respectively at 1169 cm−1, 1119 cm−1 and 1204 cm−1.
The HOMO and LUMO energies are predicted with the B3LYP method in combination of basis sets 6-31G(d,p). This molecule contains 65 occupied molecular orbitals and 309 unoccupied virtual molecular orbitals. The frontier molecular orbitals are shown in Fig. 8 ▸. The positive phase is shown in red and the negative phase is shown in green. The HOMO-LUMO energy gap of (I) reflects the chemical activity and was calculated by the DFT/B3LYP/6-31G(d,p) method (Table 4 ▸). The high value of the energy gap (3.68 eV) implies a high electronic stability and low reactivity. In general, low values mean that it will be easier to remove an electron from the HOMO orbital towards the LUMO orbital.
Figure 8.

The energy band gap of (I).
Table 4. Calculated energies.
| Molecular Energy (eV) | Compound (I) |
|---|---|
| Total Energy TE (eV) | −32759.86 |
| E HOMO (eV) | −6.50 |
| E LUMO (eV) | −2.82 |
| Gap, ΔE (eV) | 3.68 |
| Dipole moment, μ (Debye) | 0.6065 |
| Ionization potential, I (eV) | 6.50 |
| Electron affinity, A | 2.82 |
| Electronegativity, χ | 1.84 |
| Hardness, η | 3.68 |
| Softness, σ | 0.27 |
| Electrophilicity index, ω | −0.68 |
Molecular electrostatic potential (MESP) analysis
The study of MESP is a useful tool in the investigation of the molecular structure with its relation to physico-chemical properties. The MESP analysis of (I) was performed with the functional B3LYP and the basis set 6-311G (d,P). The different values of the electrostatic potential are represented by different colours (Seminario, 1996 ▸; Murray & Sen, 1996 ▸) such that red represents the region of the most negative electrostatic potential (electrophilic sites), blue represents the region of the most positive electrostatic potential (the nucleophilic reactivity) and green represents the region of zero potential. The potential increases in the following order: red < orange < yellow < green < blue. Fig. 9 ▸ reveals that the negative potential sites are on oxygen and chlorine atoms, as well as the positive potential site is around hydrogen atoms. From these results, we can deduce that the H atoms show the strongest attraction and the oxygen and chlorine atoms show the strongest repulsion in the density curve. The H atom of the methoxy and amine group has a higher positive value than the other H atoms.
Figure 9.

Contour surface of the electrostatic potential of (I).
Database survey
A search of the Cambridge Crystallographic Database (updated to Dec. 31, 2021; Groom et al., 2016 ▸) using the fragment shown in the scheme below yielded 20 hits of which 16 contained an ester group attached to C7 (the remainder contained an alkyl group at this position) and, of these, only two, ROKCIG (Filali Baba et al., 2019 ▸) and REYREV (Filali Baba et al., 2018 ▸) contain a halogen atom attached to the aromatic ring. The former is more closely related to the title molecule by having an ethyl group attached to nitrogen and also in the ester substituent. In contrast to the title molecule, that in ROKCIG forms inversion dimers through C—H⋯O hydrogen bonds (rather than ribbons), which are connected into layers approximately parallel to (10
 ), but there are no C—H⋯Cl hydrogen bonds or π-stacking interactions. In the non-halogenated analog of ROKCIG (ROKCOM; Filali Baba et al., 2019 ▸) C—H⋯O hydrogen bonds form ribbons of molecules along [001], which are connected by weak π-stacking interactions.
), but there are no C—H⋯Cl hydrogen bonds or π-stacking interactions. In the non-halogenated analog of ROKCIG (ROKCOM; Filali Baba et al., 2019 ▸) C—H⋯O hydrogen bonds form ribbons of molecules along [001], which are connected by weak π-stacking interactions.

Synthesis and crystallization
To a solution of 6-chloro-2-oxo-1,2-dihydroquinoline-4-carboxylic acid (1 g, 4.47 mmol) in 10 ml of DMF were added 3.30 ml (9.83 mmol) of methyl iodide, 3.17 g (22.36 mmol) of K2CO3 and 0.17 g (0.5 mmol) of tetra n-butylammonium bromide (TBAB). The reaction mixture was stirred at room temperature in DMF for 6 h. After removal of salts, the solvent was evaporated under reduced pressure and the residue obtained was dissolved in dichloromethane. The organic phase was dried over Na2SO4 and then concentrated in vacuo. A pure compound was obtained after recrystallization from dichloromethane/hexane (v/v 1/3).
Refinement
Crystal, data collection and refinement details are presented in Table 5 ▸. Hydrogen atoms were included as riding contributions in idealized positions with isotropic displacement parameters tied to those of the attached atoms. Two reflections obscured by the beamstop were omitted from the final refinement.
Table 5. Experimental details.
| Crystal data | |
| Chemical formula | C12H10ClNO3 |
| M r | 251.66 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 150 |
| a, b, c (Å) | 8.3515 (3), 16.6672 (6), 7.9705 (3) |
| β (°) | 107.191 (2) |
| V (Å3) | 1059.90 (7) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.36 |
| Crystal size (mm) | 0.35 × 0.19 × 0.04 |
| Data collection | |
| Diffractometer | Bruker D8 QUEST PHOTON 3 diffractometer |
| Absorption correction | Multi-scan (SADABS; Krause et al., 2015 ▸) |
| T min, T max | 0.94, 0.99 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 70718, 3544, 2977 |
| R int | 0.040 |
| (sin θ/λ)max (Å−1) | 0.737 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.041, 0.115, 1.06 |
| No. of reflections | 3544 |
| No. of parameters | 156 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.54, −0.30 |
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989022002912/wm5635sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989022002912/wm5635Isup3.hkl
Supporting information file. DOI: 10.1107/S2056989022002912/wm5635Isup4.cdx
the infrared spectrum of b3lyp/6-311g of the compound (I). DOI: 10.1107/S2056989022002912/wm5635sup5.tif
Supporting information file. DOI: 10.1107/S2056989022002912/wm5635Isup5.cml
CCDC reference: 2159047
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| C12H10ClNO3 | F(000) = 520 |
| Mr = 251.66 | Dx = 1.577 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 8.3515 (3) Å | Cell parameters from 9727 reflections |
| b = 16.6672 (6) Å | θ = 2.6–31.5° |
| c = 7.9705 (3) Å | µ = 0.36 mm−1 |
| β = 107.191 (2)° | T = 150 K |
| V = 1059.90 (7) Å3 | Plate, pale blue |
| Z = 4 | 0.35 × 0.19 × 0.04 mm |
Data collection
| Bruker D8 QUEST PHOTON 3 diffractometer | 3544 independent reflections |
| Radiation source: fine-focus sealed tube | 2977 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.040 |
| Detector resolution: 7.3910 pixels mm-1 | θmax = 31.6°, θmin = 2.8° |
| φ and ω scans | h = −12→12 |
| Absorption correction: multi-scan (SADABS; Krause et al., 2015) | k = −24→24 |
| Tmin = 0.94, Tmax = 0.99 | l = −11→11 |
| 70718 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: dual |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.041 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.115 | H-atom parameters constrained |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0614P)2 + 0.427P] where P = (Fo2 + 2Fc2)/3 |
| 3544 reflections | (Δ/σ)max = 0.001 |
| 156 parameters | Δρmax = 0.54 e Å−3 |
| 0 restraints | Δρmin = −0.30 e Å−3 |
Special details
| Experimental. The diffraction data were obtained from 12 sets of frames, each of width 0.5° in ω or φ, collected with scan parameters determined by the "strategy" routine in APEX3. The scan time was 15 sec/frame. |
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. H-atoms attached to carbon were placed in calculated positions (C—H = 0.95 - 0.98 Å). All were included as riding contributions with isotropic displacement parameters 1.2 - 1.5 times those of the attached atoms. Two reflections obscured by the beamstop were omitted from the final refinement. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.84517 (4) | 0.57446 (2) | 1.07652 (4) | 0.02841 (10) | |
| O1 | 0.11119 (15) | 0.35446 (6) | 0.34782 (14) | 0.0330 (2) | |
| O2 | 0.33909 (15) | 0.67466 (6) | 0.64348 (17) | 0.0415 (3) | |
| O3 | 0.12761 (13) | 0.64014 (6) | 0.41182 (12) | 0.0279 (2) | |
| N1 | 0.34559 (13) | 0.36878 (6) | 0.58343 (13) | 0.0213 (2) | |
| C1 | 0.46120 (15) | 0.41783 (7) | 0.70040 (16) | 0.0199 (2) | |
| C2 | 0.59973 (16) | 0.38341 (8) | 0.82548 (17) | 0.0236 (2) | |
| H2 | 0.612806 | 0.326763 | 0.831409 | 0.028* | |
| C3 | 0.71602 (16) | 0.43149 (7) | 0.93894 (17) | 0.0226 (2) | |
| H3 | 0.809737 | 0.407995 | 1.022678 | 0.027* | |
| C4 | 0.69735 (16) | 0.51402 (8) | 0.93189 (16) | 0.0223 (2) | |
| C5 | 0.56211 (15) | 0.54992 (7) | 0.81228 (16) | 0.0205 (2) | |
| H5 | 0.551223 | 0.606682 | 0.809742 | 0.025* | |
| C6 | 0.44019 (14) | 0.50236 (7) | 0.69390 (15) | 0.0187 (2) | |
| C7 | 0.29453 (15) | 0.53504 (7) | 0.56374 (15) | 0.0186 (2) | |
| C8 | 0.18725 (16) | 0.48494 (7) | 0.45164 (16) | 0.0213 (2) | |
| H8 | 0.092808 | 0.507382 | 0.367006 | 0.026* | |
| C9 | 0.20920 (16) | 0.39853 (8) | 0.45415 (16) | 0.0228 (2) | |
| C10 | 0.36647 (19) | 0.28134 (8) | 0.59449 (19) | 0.0288 (3) | |
| H10A | 0.272556 | 0.255765 | 0.506926 | 0.043* | |
| H10B | 0.369000 | 0.263150 | 0.712165 | 0.043* | |
| H10C | 0.471808 | 0.266706 | 0.571760 | 0.043* | |
| C11 | 0.25956 (15) | 0.62357 (7) | 0.54768 (16) | 0.0209 (2) | |
| C12 | 0.08323 (19) | 0.72398 (8) | 0.3841 (2) | 0.0304 (3) | |
| H12A | 0.174601 | 0.753257 | 0.357605 | 0.046* | |
| H12B | 0.064284 | 0.746230 | 0.490485 | 0.046* | |
| H12C | −0.019260 | 0.729287 | 0.285546 | 0.046* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.02388 (16) | 0.02942 (17) | 0.02545 (16) | −0.00297 (11) | −0.00271 (12) | −0.00145 (11) |
| O1 | 0.0406 (6) | 0.0229 (5) | 0.0279 (5) | −0.0084 (4) | −0.0016 (4) | −0.0034 (4) |
| O2 | 0.0370 (6) | 0.0184 (4) | 0.0512 (7) | 0.0009 (4) | −0.0147 (5) | −0.0053 (4) |
| O3 | 0.0309 (5) | 0.0208 (4) | 0.0250 (5) | 0.0054 (4) | −0.0028 (4) | 0.0003 (3) |
| N1 | 0.0261 (5) | 0.0153 (4) | 0.0205 (5) | 0.0001 (4) | 0.0041 (4) | −0.0008 (3) |
| C1 | 0.0217 (5) | 0.0175 (5) | 0.0201 (5) | −0.0002 (4) | 0.0056 (4) | −0.0002 (4) |
| C2 | 0.0249 (6) | 0.0198 (5) | 0.0251 (6) | 0.0033 (4) | 0.0061 (5) | 0.0024 (4) |
| C3 | 0.0222 (6) | 0.0223 (5) | 0.0222 (5) | 0.0027 (4) | 0.0048 (4) | 0.0022 (4) |
| C4 | 0.0202 (5) | 0.0242 (5) | 0.0200 (5) | −0.0009 (4) | 0.0023 (4) | −0.0003 (4) |
| C5 | 0.0205 (5) | 0.0186 (5) | 0.0210 (5) | 0.0002 (4) | 0.0039 (4) | 0.0001 (4) |
| C6 | 0.0189 (5) | 0.0178 (5) | 0.0182 (5) | 0.0002 (4) | 0.0035 (4) | −0.0002 (4) |
| C7 | 0.0196 (5) | 0.0162 (5) | 0.0188 (5) | 0.0000 (4) | 0.0041 (4) | 0.0002 (4) |
| C8 | 0.0221 (5) | 0.0188 (5) | 0.0206 (5) | −0.0012 (4) | 0.0027 (4) | 0.0004 (4) |
| C9 | 0.0262 (6) | 0.0202 (5) | 0.0203 (5) | −0.0025 (4) | 0.0043 (4) | 0.0001 (4) |
| C10 | 0.0364 (7) | 0.0153 (5) | 0.0323 (7) | 0.0006 (5) | 0.0063 (6) | 0.0007 (4) |
| C11 | 0.0206 (5) | 0.0176 (5) | 0.0221 (5) | 0.0008 (4) | 0.0027 (4) | 0.0008 (4) |
| C12 | 0.0334 (7) | 0.0221 (6) | 0.0314 (7) | 0.0077 (5) | 0.0030 (5) | 0.0055 (5) |
Geometric parameters (Å, º)
| Cl1—C4 | 1.7397 (13) | C4—C5 | 1.3807 (17) |
| O1—C9 | 1.2319 (15) | C5—C6 | 1.4098 (16) |
| O2—C11 | 1.2040 (16) | C5—H5 | 0.9500 |
| O3—C11 | 1.3258 (15) | C6—C7 | 1.4514 (16) |
| O3—C12 | 1.4458 (15) | C7—C8 | 1.3510 (16) |
| N1—C9 | 1.3825 (16) | C7—C11 | 1.5022 (16) |
| N1—C1 | 1.3920 (15) | C8—C9 | 1.4513 (17) |
| N1—C10 | 1.4673 (16) | C8—H8 | 0.9500 |
| C1—C2 | 1.4070 (17) | C10—H10A | 0.9800 |
| C1—C6 | 1.4189 (16) | C10—H10B | 0.9800 |
| C2—C3 | 1.3726 (18) | C10—H10C | 0.9800 |
| C2—H2 | 0.9500 | C12—H12A | 0.9800 |
| C3—C4 | 1.3836 (17) | C12—H12B | 0.9800 |
| C3—H3 | 0.9500 | C12—H12C | 0.9800 |
| Cl1···O3i | 3.1903 (10) | H2···O2iii | 2.57 |
| Cl1···H8i | 2.84 | O3···H8 | 2.25 |
| C12···O1ii | 3.0016 (16) | C2···H10C | 2.78 |
| O2···C5 | 2.8496 (17) | C2···H10B | 2.74 |
| O1···H10A | 2.26 | C10···H2 | 2.46 |
| H12C···O1ii | 2.36 | C11···H5 | 2.71 |
| O2···H12A | 2.64 | H2···H10B | 2.24 |
| O2···H5 | 2.19 | H2···H10C | 2.29 |
| O2···H12B | 2.55 | ||
| C11—O3—C12 | 116.08 (11) | C8—C7—C11 | 118.46 (11) |
| C9—N1—C1 | 122.97 (10) | C6—C7—C11 | 122.02 (10) |
| C9—N1—C10 | 117.27 (10) | C7—C8—C9 | 123.22 (11) |
| C1—N1—C10 | 119.77 (10) | C7—C8—H8 | 118.4 |
| N1—C1—C2 | 119.83 (11) | C9—C8—H8 | 118.4 |
| N1—C1—C6 | 120.32 (11) | O1—C9—N1 | 122.17 (12) |
| C2—C1—C6 | 119.85 (11) | O1—C9—C8 | 121.78 (12) |
| C3—C2—C1 | 120.15 (11) | N1—C9—C8 | 116.05 (11) |
| C3—C2—H2 | 119.9 | N1—C10—H10A | 109.5 |
| C1—C2—H2 | 119.9 | N1—C10—H10B | 109.5 |
| C2—C3—C4 | 120.29 (12) | H10A—C10—H10B | 109.5 |
| C2—C3—H3 | 119.9 | N1—C10—H10C | 109.5 |
| C4—C3—H3 | 119.9 | H10A—C10—H10C | 109.5 |
| C5—C4—C3 | 121.22 (12) | H10B—C10—H10C | 109.5 |
| C5—C4—Cl1 | 118.82 (10) | O2—C11—O3 | 122.70 (12) |
| C3—C4—Cl1 | 119.96 (10) | O2—C11—C7 | 125.71 (11) |
| C4—C5—C6 | 120.00 (11) | O3—C11—C7 | 111.59 (10) |
| C4—C5—H5 | 120.0 | O3—C12—H12A | 109.5 |
| C6—C5—H5 | 120.0 | O3—C12—H12B | 109.5 |
| C5—C6—C1 | 118.48 (11) | H12A—C12—H12B | 109.5 |
| C5—C6—C7 | 123.66 (11) | O3—C12—H12C | 109.5 |
| C1—C6—C7 | 117.86 (10) | H12A—C12—H12C | 109.5 |
| C8—C7—C6 | 119.52 (11) | H12B—C12—H12C | 109.5 |
| C9—N1—C1—C2 | −178.19 (12) | C1—C6—C7—C8 | −1.59 (17) |
| C10—N1—C1—C2 | 1.67 (18) | C5—C6—C7—C11 | −1.54 (18) |
| C9—N1—C1—C6 | 1.70 (18) | C1—C6—C7—C11 | 179.15 (11) |
| C10—N1—C1—C6 | −178.45 (12) | C6—C7—C8—C9 | 0.23 (18) |
| N1—C1—C2—C3 | 178.70 (11) | C11—C7—C8—C9 | 179.51 (11) |
| C6—C1—C2—C3 | −1.18 (19) | C1—N1—C9—O1 | 177.48 (12) |
| C1—C2—C3—C4 | 0.40 (19) | C10—N1—C9—O1 | −2.39 (19) |
| C2—C3—C4—C5 | 0.32 (19) | C1—N1—C9—C8 | −2.97 (17) |
| C2—C3—C4—Cl1 | 179.88 (10) | C10—N1—C9—C8 | 177.17 (11) |
| C3—C4—C5—C6 | −0.25 (19) | C7—C8—C9—O1 | −178.44 (13) |
| Cl1—C4—C5—C6 | −179.81 (9) | C7—C8—C9—N1 | 2.01 (18) |
| C4—C5—C6—C1 | −0.52 (18) | C12—O3—C11—O2 | −0.1 (2) |
| C4—C5—C6—C7 | −179.83 (11) | C12—O3—C11—C7 | −179.85 (11) |
| N1—C1—C6—C5 | −178.65 (11) | C8—C7—C11—O2 | 176.09 (14) |
| C2—C1—C6—C5 | 1.23 (18) | C6—C7—C11—O2 | −4.6 (2) |
| N1—C1—C6—C7 | 0.69 (17) | C8—C7—C11—O3 | −4.12 (16) |
| C2—C1—C6—C7 | −179.43 (11) | C6—C7—C11—O3 | 175.14 (11) |
| C5—C6—C7—C8 | 177.71 (12) |
Symmetry codes: (i) x+1, y, z+1; (ii) −x, y+1/2, −z+1/2; (iii) −x+1, y−1/2, −z+3/2.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···O2iii | 0.95 | 2.57 | 3.5146 (16) | 178 |
| C5—H5···O2 | 0.95 | 2.19 | 2.8496 (16) | 126 |
| C8—H8···Cl1iv | 0.95 | 2.84 | 3.7786 (13) | 170 |
| C12—H12C···O1ii | 0.98 | 2.36 | 3.0016 (16) | 122 |
Symmetry codes: (ii) −x, y+1/2, −z+1/2; (iii) −x+1, y−1/2, −z+3/2; (iv) x−1, y, z−1.
Funding Statement
This work was funded by Tulane University; Hacettepe University Scientific Research Project Unit grant 013 D04 602 004.
References
- Abdel-Wahab, B. F., Khidre, R. E., Farahat, A. A. & El-Ahl, A. S. (2012). Arkivoc, pp. 211–276.
- Becke, A. D. (1993). J. Chem. Phys. 98, 5648–5652.
- Bouzian, Y., Hlimi, F., Sebbar, N. K., El Hafi, M., Hni, B., Essassi, E. M. & Mague, J. T. (2018). IUCrData, 3, x181438.
- Brandenburg, K. & Putz, H. (2012). DIAMOND, Crystal Impact GbR, Bonn, Germany.
- Bruker (2020). APEX3 and SAINT. Bruker AXS, Inc., Madison, Wisconsin, USA.
- Filali Baba, Y., Elmsellem, H., Kandri Rodi, Y., Steli, H., Ouazzani Chahdi, F., Ouzidan, Y., Sebbar, N. K. & Essassi, E. M. (2016b). J. Mater. Environ. Sci. 7, 2424–2434.
- Filali Baba, Y., Elmsellem, H., Kandri Rodi, Y., Steli, H., Ouazzani Chahdi, F., Ouzidan, Y., Sebbar, N. K., Essassi, E. M., El-Hajjaji, F. & Hammouti, B. (2016a). Der Pharmacia Lettre. 8, 128–137.
- Filali Baba, Y., Gökce, H., Kandri Rodi, Y., Hayani, S., Ouazzani Chahdi, F., Boukir, A., Jasinski, J. P., Kaur, M., Hökelek, T., Sebbar, N. K. & Essassi, E. M. (2020). J. Mol. Struct. 1217, 128461.
- Filali Baba, Y., Kandri Rodi, Y., Mague, J. T., Ouzidan, Y., Ouazzani Chahdi, F. & Essassi, E. M. (2018). IUCrData, 3, x180288.
- Filali Baba, Y., Sert, Y., Kandri Rodi, Y., Hayani, S., Mague, J. T., Prim, D., Marrot, J., Ouazzani Chahdi, F., Sebbar, N. K. & Essassi, E. M. (2019). J. Mol. Struct. 1188, 255–268.
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A. Jr, Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V., Cioslowski, J. & Fox, D. J. (2009). GAUSSIAN09. Gaussian Inc., Wallingford, CT, USA.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hathwar, V. R., Sist, M., Jørgensen, M. R. V., Mamakhel, A. H., Wang, X., Hoffmann, C. M., Sugimoto, K., Overgaard, J. & Iversen, B. B. (2015). IUCrJ, 2, 563–574. [DOI] [PMC free article] [PubMed]
- Hayani, S., Sert, Y., Filali Baba, Y., Benhiba, F., Ouazzani Chahdi, F., Laraqui, F.-Z., Mague, J. T., El Ibrahimi, B., Sebbar, N. K., Kandri Rodi, Y. & Essassi, E. M. (2021). J. Mol. Struct. 1227, 129520.
- Hirshfeld, H. L. (1977). Theor. Chim. Acta, 44, 129–138.
- Jayatilaka, D., Grimwood, D. J., Lee, A., Lemay, A., Russel, A. J., Taylor, C., Wolff, S. K., Cassam-Chenai, P. & Whitton, A. (2005). TONTO – A System for Computational Chemistry. Available at: http://hirshfeldsurface.net/
- Katoh, M., Matsune, R., Nagase, H. & Honda, T. (2004). Tetrahedron Lett. 45, 6221–6223.
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst. 48, 3–10. [DOI] [PMC free article] [PubMed]
- Leatham, P. A., Bird, H. A., Wright, V., Seymour, D. & Gordon, A. (1983). J. Rheumatol. Inflamm. 6, 209–211. [PubMed]
- Mackenzie, C. F., Spackman, P. R., Jayatilaka, D. & Spackman, M. A. (2017). IUCrJ, 4, 575–587. [DOI] [PMC free article] [PubMed]
- Mahamoud, A., Chevalier, J., Davin-Regli, A., Barbe, J. & Pages, J. (2006). Curr. Drug Targets, 7, 843–847. [DOI] [PubMed]
- McKinnon, J. J., Jayatilaka, D. & Spackman, M. A. (2007). Chem. Commun. pp. 3814–3816. [DOI] [PubMed]
- Murray, J. S. & Sen, K. (1996). Molecular Electrostatic Potentials, Concepts and Applications, Elsevier, Amsterdam.
- Muruganantham, N., Sivakumar, R., Anbalagan, N., Gunasekaran, V. & Leonard, J. T. (2004). Biol. Pharm. Bull. 27, 1683–1687. [DOI] [PubMed]
- Seminario, J. M. (1996). Recent Developments and Applications of Modern Density Functional Theory, vol. 4, pp. 800–806. Amsterdam: Elsevier.
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Spackman, M. A., McKinnon, J. J. & Jayatilaka, D. (2008). CrystEngComm, 10, 377–388.
- Strekowski, L., Mokrosz, J. L., Honkan, V. A., Czarny, A., Cegla, M. T., Wydra, R. L., Patterson, S. E. & Schinazi, R. F. (1991). J. Med. Chem. 34, 1739–1746. [DOI] [PubMed]
- Turner, M. J., Grabowsky, S., Jayatilaka, D. & Spackman, M. A. (2014). J. Phys. Chem. Lett. 5, 4249–4255. [DOI] [PubMed]
- Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Spackman, P. R., Jayatilaka, D. & Spackman, M. A. (2017). CrystalExplorer17. The University of Western Australia.
- Turner, M. J., Thomas, S. P., Shi, M. W., Jayatilaka, D. & Spackman, M. A. (2015). Chem. Commun. 51, 3735–3738. [DOI] [PubMed]
- Venkatesan, P., Thamotharan, S., Ilangovan, A., Liang, H. & Sundius, T. (2016). Spectrochim. Acta A Mol. Biomol. Spectrosc. 153, 625–636. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Wilson, W. D., Zhao, M., Patterson, S. E., Wydra, R. L., Janda, L. & Strekowski, L. (1992). J. Med. Chem. Res, 2, 102–110.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989022002912/wm5635sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989022002912/wm5635Isup3.hkl
Supporting information file. DOI: 10.1107/S2056989022002912/wm5635Isup4.cdx
the infrared spectrum of b3lyp/6-311g of the compound (I). DOI: 10.1107/S2056989022002912/wm5635sup5.tif
Supporting information file. DOI: 10.1107/S2056989022002912/wm5635Isup5.cml
CCDC reference: 2159047
Additional supporting information: crystallographic information; 3D view; checkCIF report