

Abstract
Human immunodeficiency virus type 1 (HIV-1) is known to provoke microglial immune responses which likely play a paramount role in the development of chronic neuroinflammatory conditions and neuronal damage related to HIV-1 associated neurocognitive disorders (HAND). In particular, HIV-1 Tat protein is a proinflammatory neurotoxin which predisposes neurons to synaptodendritic injury. Drugs targeting the degradative enzymes of endogenous cannabinoids have shown promise in reducing inflammation with minimal side effects in rodent models. Considering that markers of neuroinflammation can predict the extent of neuronal injury in HAND patients, we evaluated the neurotoxic effect of HIV-1 Tat-exposed microglia following blockade of fatty acid amid hydrolyze (FAAH), a catabolic enzyme responsible for degradation of endocannabinoids, e.g. anandamide (AEA). In the present study, cultured murine microglia were incubated with Tat and/or a FAAH inhibitor (PF3845). After 24 h, cells were imaged for morphological analysis and microglial conditioned media (MCM) was collected. Frontal cortex neuron cultures (DIV 7–11) were then exposed to MCM, and neurotoxicity was assessed via live cell calcium imaging and staining of actin positive dendritic structures. Results demonstrate a strong attenuation of microglial responses to Tat by PF3845 pretreatment, which is indicated by 1) microglial changes in morphology to a less proinflammatory phenotype using fractal analysis, 2) a decrease in release of neurotoxic cytokines/chemokines (MCP-1/CCL2) and matrix metalloproteinases (MMPs; MMP-9) using ELISA/multiplex assays, and 3) enhanced production of endocannabinoids (AEA) using LC/MS/MS. Additionally, PF3845’s effects on Tat-induced microglial-mediated neurotoxicity, decreased dysregulation of neuronal intracellular calcium and prevented the loss of actin-positive staining and punctate structure in frontal cortex neuron cultures. Interestingly, these observed neuroprotective effects appeared to be independent of cannabinoid receptor activity (CB1R & CB2R). We found that a purported GPR18 antagonist, CID-85469571, blocked the neuroprotective effects of PF3845 in all experiments. Collectively, these experiments increase understanding of the role of FAAH inhibition and Tat in mediating microglial neurotoxicity in the HAND condition.
Keywords: Anandamide, Endocannabinoid, Fatty acid amide hydrolase, G protein coupled receptor, HIV-1 Tat, Matrix metalloproteinase, Microglia, Monocyte chemoattractant protein 1, Neuroinflammation, Neuroprotection
1. Introduction
Combined antiretroviral therapy (cART) has drastically improved the outlook for patients infected with human immunodeficiency virus type 1 (HIV-1); however, many display neurocognitive impairments related to prefrontal cortex function (Cysique et al., 2004; Heaton et al., 2011), referred to as HIV-associated neurocognitive disorders (HAND) (Ellis et al., 2007; Heaton et al., 2011). A significant correlate of HAND progression is synaptodendritic damage, suggesting that loss of viable structures underlie deficits found in HIV-1 infected patients (Masliah et al., 1997). The HIV-1 transactivator of transcription (Tat) is detectable in cART treated individuals (Henderson et al., 2019) and acts as a potent neurotoxin contributing to the pathogenesis of HAND (Carroll and Brew, 2017; King et al., 2006; Rao et al., 2014). Tat directly impacts neuronal function through dysregulation of AMPA/NMDA function leading to enhanced excitability, and dendritic simplification and loss of function (Brailoiu et al., 2008; Fitting et al., 2014; Haughey et al., 2001; Longordo et al., 2006; Prendergast et al., 2002). In addition, microglial activation by HIV-1 virotoxins drives immune signaling that contributes to neuronal damage (Jin et al., 2012; Sheng et al., 2000). Considering the innate immune response in the brain occurs shortly after HIV-1 infection, reducing microglial activity offers a promising strategy for altering the progression of HAND.
Microglia contribute to brain disease through chronic immune activation, proinflammatory signaling cascades, and immune quiescence (Dheen et al., 2007; Perry and Teeling, 2013). Specifically, microglia exposed to HIV-1 and/or viral proteins undergo proinflammatory activation and promote neurotoxic brain conditions (Jin et al., 2012; Liu et al., 2013; Sheng et al., 2000). Thus, anti-inflammatory drugs may prove useful for treating neurodegenerative conditions, in part, through microglia. Cannabinoids elicit anti-inflammatory effects in several disease models (Nagarkatti et al., 2009) and specifically block proinflammatory signaling in microglia (Ashton and Glass, 2007; Ehrhart et al., 2005). Therefore, cannabinoids may be an effective treatment strategy for slowing and/or blocking neurotoxic inflammation and HAND progression.
Levels of the endogenous cannabinoid ligands, anandamide (N-arachidonoylethanolamine, AEA) and 2-arachidonoylglycerol (2-AG), are heightened in individuals diagnosed with neurodegenerative disorders and negatively correlate with severity of disease progression (Chen et al., 2011; More and Choi, 2015; Pryce et al., 2003). These ligands exert their effects primarily via cannabinoid receptors type 1 and 2 (CB1R and CB2R), but also through other G protein-coupled receptors (GPCRs) such as GPR55 and GRP18 (Harkany et al., 2008; Henstridge, 2012; McHugh, 2012; Ross et al., 2012). Activated microglia express CB2Rs, which regulate microglial immune function (Ashton and Glass, 2007; Ehrhart et al., 2005; Malek et al., 2015), and stimulation of this receptor blunts neurotoxic cytokine release mediated by HIV-1 gp120 protein (Hu et al., 2013). Moreover, GPR55 and GPR18 reduce neurotoxicity in organotypic hippocampal cultures likely through modulation of microglial activity (Grabiec et al., 2019; Kallendrusch et al., 2013). Therefore, the present study proposes to explore further microglial cannabinoid receptors and their function in regulating neuroinflammation.
Currently available cannabinoid receptor agonists have limited therapeutic use because of their undesirable cannabimimetic side effects (Cooper and Haney, 2009; Justinova et al., 2003; Lichtman et al., 1995). Conversely, drugs that elevate endogenous cannabinoid signaling show promise for use as therapeutics with limited cannabimimetic side effects (Booker et al., 2012; Ignatowska-Jankowska et al., 2015; Ignatowska-Jankowska et al., 2014). Because endogenous cannabinoids are synthesized and released “on-demand” (Marsicano et al., 2003), inhibiting their degradation leads to increased cannabinoid receptor signaling locally where their production is elevated (e.g. at sites of brain injury). A fatty acid amide hydrolase (FAAH) inhibitor, PF3845, enhances AEA content in the central and peripheral nervous systems (Booker et al., 2012), and supports neuronal survival and attenuate neuroinflammation in vivo (Tchantchou et al., 2014). In addition, microglia express both FAAH (Tham et al., 2007) and AEA (Carrier et al., 2004; Stella, 2009), indicating they are viable cellular targets for FAAH inhibition to increase AEA signaling at cannabinoid receptors. Enzyme inhibitors that block endocannabinoid degradation, such as PF3845, affect microglial activity. Thus, we hypothesize that FAAH inhibition will elicit microglia-mediated protective effects against HIV-1 mediated neurotoxicity.
To test this hypothesis, the present study evaluates the effects of PF3845 on murine microglial-mediated neurotoxicity via live cell calcium imaging and staining of actin positive dendritic structures and characterizes microglial changes in morphology using fractal analysis. In addition, ELISA/multiplex assays are employed to assess release of neurotoxic cytokines/chemokines and matrix metalloproteinases (MMPs). Finally, mass spectrometry (LC/MS/MS) was used to quantify the levels of endocannabinoids in cultured microglia. Collectively, these experiments provide insight into the role of FAAH inhibition and Tat in mediating microglial neurotoxicity.
2. Materials and methods
2.1. Experimental design
The design of each experiment is provided in detail in the method sections, including the between-subjects factors and a full description of critical variables required for independent replication. All experiments were approved by the University of North Carolina at Chapel Hill and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Council, 2011).
2.2. Treatments
Cultured microglia were incubated with HIV-1 Tat1–86 (100 nM; IIIB, #1002, ImmunoDx, MA), lipopolysaccharide (LPS; 1 μg/mL; L2880, Sigma-Aldrich, MO), PF3845 (100 nM, Dr. Benjamin Cravatt; Scripps, CA), the endogenous GPR18 agonist NAGly (100 nM – 10 μM, Tocris, UK), the CB2R agonist HU-308 (100 nM; #90086, Cayman, MI), the CB1R antagonist SR141716A (SR141, 100 nM, Tocris, UK), the CB2R antagonist SR144528 (SR144, 100 nM, Tocris, UK), the selective GPR18 antagonist CID-85469571 (CID, 100 nM, AOB9837, Aobious, MA), the selective GPR55 antagonist ML193 (100–1 μM, Tocris, UK), the MCP-1 inhibitor bindarit (300 μM; #11479, Cayman, MI), and/or the MMP-9 inhibitor I (150 nM; #444278, Sigma-Aldrich, MO). Tat 100 nM concentration was selected as it produces cellular deficits in vitro comparable to those found in post-mortem tissue samples collected from individuals with HAND (El-Hage et al., 2008; El-Hage et al., 2011; Kruman et al., 1998; Perry et al., 2010). Tat concentrations in human brain tissue have not been determined yet but have been detected immunohistochemically and in protein extracts from the brains of simian immunodeficiency virus (SIV)-infected rhesus macaques with encephalitis (Hudson et al., 2000). Even though the concentrations of Tat in the cerebral spinal fluid (CSF) has been reported at around 1.14 nM (~16 ng/mL), higher concentrations of Tat could be attainable locally within close proximity of HIV-1 infected cells (Westendorp et al., 1995). At DIV 14 microglial cultures were incubated with PF3845 or NAGly for 1 h prior to Tat treatment. Tat was added to cultures and allowed to incubate for 24 h before media was collected. Treatment with selective inhibitors or receptor antagonists was conducted 1 h or 30 min prior to PF3845 treatment, respectively, and were present throughout the duration of Tat incubation. GPR18 antagonist CID-85469571 (CID) was selected because it has been shown to have 36-fold selectivity compared to GPR55 (Rempel et al., 2014). For a positive control experiment DIV 14 microglial cultures were treated with LPS and 1 h later HU-308 was added to the cultures, similar to what has been published previously (Presley et al., 2015). After 24 h, MCM was collected for ELISA analysis to determine MCP-1/CCL2 and pro-MMP-9/MMP9 levels.
2.3. Cell culture and conditioned media
Primary microglial cultures were derived from whole brain punches taken from C57BL/6 (Charles River, NC) neonatal mouse pups at postnatal day 0–1. In addition, for calcium imaging experiments, both FAAH wildtype (WT) and FAAH knockout (KO) animals were used to derive cultures. These animals were provided by Dr. Aron Lichtman (Virginia Commonwealth University, VA). Punches were suspended in DMEM (Invitrogen, CA) with fetal bovine serum, and 7.5% sodium bicarbonate, in six well ultra-low attachment plates (Sigma-Aldrich, MO) to prevent neuronal and astroglial growth. Cells were grown to confluency over 2 wks (97.5 ± 17.9 cells/mm2) and used for experiments on days in vitro (DIV) 14–17. Media was completely changed one day after plating and partial media replacement was carried out every 3 d after initial change. All drug treatments were diluted in Brainphys culture media (Stemcell Technologies, Canada) and collected 24 h following Tat administration. Microglia conditioned medium (MCM) was collected through 0.2 μm filter before being immediately frozen. Samples were stored at −20 °C for no longer than 4 wks and never freeze-thawed more than twice before experimental use. Whole MCM was used for ELISA, multiplex, and mass spectrometry while 1:4 dilution was used for calcium imaging and synaptic integrity experiments.
Primary neuronal cultures were derived from dissociated frontal cortex tissue of embryonic day 16–17 C57BL/6C mice. Frontal cortex tissue was dissected from the rostral portion of the neocortex similar to the frontal cortex size that has been shown with gene expression markers (Cholfin and Rubenstein, 2007). Cultures for calcium imaging were prepared as previously described (Hermes et al., 2018). In brief, collected tissue was minced and incubated (30 min, 37 °C) with trypsin (2.5 mg/mL) and DNase (0.015 mg/mL) in HBSS (Invitrogen, CA) and centrifuged 2× for 5 min. Cells were then plated on MatTek 35 mm glass bottom dishes, coated with poly-L-lysine (Sigma-Aldrich, MO), at a concentration of 1 × 105 cells per dish and maintained in neurobasal medium (NBM) supplemented with B27 (Invitrogen, CA), 0.5 mM L-glutamine, 0.025 mM glutamate at 37 °C in a humidified atmosphere containing 5% CO2. Calcium imaging experiments were performed on neuronal cultures at DIV 7–11 ensuring that rudimentary dendritic/axonal structures were established. Neurons used for synaptic integrity experiments were collected in HBSS using papain (0.5 mg/mL, Stemcell Technologies, Canada) for dissociation with no centrifugation. Cells were plated on glass coverslips (1 × 105 cells per coverslip) and maintained in NBM Plus (Invitrogen, CA) with B27 Plus (Invitrogen, CA), 0.5 mM L-glutamine, 0.025 mM glutamate for DIV 9 and changed into NBM Plus without glutamate. Synaptic integrity experiments were carried out on DIV 21 so that fine synaptic structures were present. All cell media was supplemented with antibiotic mixture (Invitrogen, CA). Media was completely changed one day after plating and partially changed every 2–3 d.
2.4. Calcium imaging
Live cell imaging was conducted on frontal cortex neuronal cultures using a Zeiss Axio Observer Z.1 inverted microscope (20× objective) with an automated, computer-controlled stage encoder with environmental control (37 °C, 95% humidity, 5% CO2). The cell permeant acetoxymethyl (AM) ester-linked [Ca2+]i indicator fura-2 AM (Kd = 145 nM; 2.5 uM, Molecular Probes, Eugene, OR) was used for measuring [Ca2+]i and diluted in Brainphys culture media. MCM was added 1 min after experiment start. Neurons were imaged for a 30 min period. Using ZEN 2010 blue Edition software, images were acquired with a MRm digital camera (Zeiss, MN) at a frame rate of 0.2 Hz during the first 5 min, and 0.033 Hz from 5 min to 30 min. Relative fluorescence ratio images were acquired at 340/380 nm excitation and 510 nm emission wavelengths. Conversion to [Ca2+]i was calculated according to an equation described previously (Grynkiewicz et al., 1985). Regions of interest (ROIs) were manually assigned to neuronal somata. Due to the heterogeneity among neurons, the mean standard error of the mean (SEM) values for changes in [Ca2+]i were computed comparing individual neurons before and at specific intervals during treatment using a repeated-measure analysis of variance (ANOVA). Quantitative analyses of [Ca2+]i levels in neuronal somata were performed on 10–15 randomly selected neurons per treatment per experiment. At least three independent experiments were run for each treatment group.
2.5. Immunocytochemistry
DIV 21 neuronal cultures grown on glass coverslips were fixed in 4% paraformaldehyde for 10 min. Dendritic structures were visualized using Acti-stain 488 phalloidin (Cytoskeleton, Denver, CO) to label mature and immature pre-synaptic structures. Neurons were then counter-stained with Hoechst 33342 for 3 min and mounted using ProLong Gold (Molecular Probes, OR). PBS 1× washes were applied between each step. Cells were visualized as immunofluorescent image z-stacks acquired using a Zeiss LSM 700 laser scanning confocal microscope equipped with a 20× objective (Zeiss, MN). Randomly selected regions containing phalloidin-positive neuronal processes were selected and orthogonal projections were taken from each image stack. Phalloidin-positive (F-actin) staining of neuronal dendrites has been shown to generate discrete “hot spots” or bright “puncta”, which represent a variety of dendritic structures, including mature spines, non-spiny synapses and immature spines (Li et al., 2016). Thus, puncta, as the analyzed particles, were defined as objects between 0.3 and 1.2 μm, similar to what has been described previously (Li et al., 2016; Roscoe et al., 2014). Selection threshold criterion was determined by the size of punctate structures found in the randomly collected images (minimal pixel threshold: 20). Square ROIs were randomly assigned to areas containing positive fluorescent signal using ImageJ. Mean gray pixel values and number of phalloidin-positive punctate structures contained in each ROI were computed using built-in ImageJ functions. 10 images were analyzed per treatment group, with 22 ROIs randomly assigned to each. Based on the calcium imaging data only the four main treatment groups were included (MCM Control, MCM Tat 100 nM, MCM PF3845 100 nM+Tat, MCM CID 100 nM+PF3845+Tat), with excluding drug only conditions. Gray pixel values and puncta count were used as dependent factors.
2.6. ELISA and multiplex quantification of cytokines/chemokines and MMP-9
Levels of cytokines/chemokines and MMPs in MCM were quantified using the following products, CYTOMAG-70 K (cytokine panel, Millipore, MA), MMMP3MAG-79 K (proMMP-9, Millipore, MA), and Pico-Kine EK0466 (MMP-9, Boster Bio, CA). Levels of both the inactive proMMP-9 and cleaved/activated MMP-9 were quantified. Experiments were carried out as detailed in the manufacturer’s instructions except all standards were run in Brainphys to match the MCM samples. For ELISAs a BioTek Synergy HT multi-mode plate reader (BioTek, VT) was used to measure absorbance and for multiplexes a Bio-Rad Bio-Plex 200 (Bio-Rad, CA) reader was used to measure mean fluorescence intensity. At least three independent experiments were run per treatment group. Concentrations of MCP-1/CCL2 and MMPs were used as dependent factors.
2.7. Fractal analysis of microglial morphology
As microglial function is well-correlated with microglia morphology (i.e. ramified/resting compared to unramified/activated microglia) we employed fractal analysis as a quantitative method to assess the morphology of microglia based on complexity (Karperien and Jelinek, 2015; Morrison et al., 2017). Phase images of DIV 14 live microglial cells were acquired using Zeiss Axio Observer Z.1 inverted microscope equipped with a 20× objective. 10 randomly selected regions, per treatment group, containing microglia were selected for analysis. All images were processed using ImageJ as previously described (Young and Morrison, 2018). In brief, a bandpass and unmask filter were applied to images followed by the despeckle function. The threshold function was used to make cells into solid, single object representations. Cells that were difficult to delineate from bordering cells were excluded from analysis. Gaps in the borders of cells were filled in manually. Fractal analysis of individual microglial cells was carried out using the box-counting method provided with the FracLac plug-in for ImageJ. Fractal dimension (DF) and lacunarity (Λ) are measures of microglial morphological complexity. Fractal dimension quantifies the number of repeating complex pattern within the cell’s shape (Karperien and Jeli- nek, 2015). Thus, activated (more circular shaped) microglia are less complex and have a lower fractal dimension compared to ramified/resting microglia. On the other hand, lacunarity describes the degree of heterogeneity that exists in a cell’s shape. Activated ameboid microglia are expected to be more homogenous with more similarly sized gaps and little rotation variance in their morphology, and thus have low lacunarity. Ramified/less activated microglia tend to show more heterogeneity with many different sized gaps and notable rotational variance, and thus have higher lacunarity (Karperien and Jelinek, 2015). The ROIs used for analysis were identical for all cells. Around 15 cells were analyzed per image. Fractal dimension and lacunarity were used as dependent factors.
2.8. Liquid chromatography tandem mass spectrometry
Microglia (post 24 h drug treatment) were aspirated and washed with PBS 1× to remove residual culture media. Cells were suspended in 250 μL of 70% methanol and flash frozen in liquid nitrogen. Details regarding sample preparation can be found in the Supplementary Methods section. In brief, solid phase extraction was performed to enrich for molecules of interest followed by drying with N2 and reconstitution in 200 μL of resuspension buffer (7:1.5:1.5 0.1% acetic acid:acetonitrile: isopropanol). LC/MS/MS analysis of endocannabinoids was performed as described previously (Gouveia-Figueira and Nording, 2015) with some modifications (for details see Supplementary Methods). Briefly, mass spectrometric analysis was performed on an Agilent 6490 triple quadrupole mass spectrometer in positive ionization mode. Quantitation of endocannabinoids was achieved using a general isotope dilution strategy. Calibration standards were analyzed over a range of concentrations from 0.2–40 pg on column for all of the ethanolamides. At least three independent experiments were run per treatment group. Concentrations of AEA (pg/mL) was used as the dependent factor.
2.9. Statistical analysis
Data were analyzed using analysis of variances (ANOVAs) followed by Bonferroni’s post hoc test as appropriate (SPSS Statistics, Version 25, IBM). Violations of compound symmetry in repeated-measures ANOVAs for the within-subjects factors (i.e., comparing time points) were addressed by using the Greenhouse-Geisser degrees (pGG) of freedom correction factor (Greenhouse and Geisser, 1959). An alpha level of p < 0.05 was considered significant for all statistical tests. Statistics not shown in the text were found to be non-significant. ω2 was provided as a measure of effect size. Data are expressed as the mean ± SEM. All experiments and data analyses were carried out by experimenters blind to treatment condition.
3. Results
3.1. Calcium imaging
3.1.1. Tat-treated microglia produce MCM that provokes neurotoxic calcium responses in neurons and this effect is blocked by PF3845 pretreatment
To understand the role of FAAH inhibition and Tat in mediating microglial neurotoxicity, we investigated [Ca2+]i responses of frontal cortex neuron cultures to MCMs derived from PF3945- and/or Tat-treated microglia in the presence or absence of different cannabinoid or non-cannabinoid receptor antagonists (Fig. 1). Application of MCM Tat onto neurons caused a significant increase in [Ca2+]i and this effect was not present when applying MCM derived from microglia co-treated with PF3845 and Tat or when applying MCM heat inactivated Tat (Fig. 1A & B). A two-way ANOVA showed a significant main effect for both, time [F(35, 7315) = 16.0, pGG < 0.001] and treatment [F(4, 209) = 105.6 p < 0.001], and a significant treatment x time interaction [F(140, 7315) = 12.1, pGG < 0.001] (Fig. 1B). A one-way ANOVA conducted on the last 10 min revealed a significant treatment effect [F(4, 209) = 35.4, p < 0.001, ω2 = 0.404] with MCM Tat 100 nM significantly increasing [Ca2+]i compared to all other groups (p < 0.001; Fig. 1B′). Note, Tat’s neurotoxic and PF3845’s neuroprotective effects are specific to their action on microglia and not on neurons treated with MCM as PF3845 100 nM or Tat 100 nM incubated in culture media for 24 h at 37 °C did not show significant changes in [Ca2+]i levels of frontal cortex neurons (Supplementary Fig. 1).
Fig. 1.

(A) Pseudocolor images of neuronal ratiometric calcium imaging comparing frontal cortex neurons (DIV 7–11) treated with MCM Control or MCM Tat conditions over 30 min. (B-B′) MCM Tat 100 nM caused significant increases in [Ca2+]i levels while MCM PF3845 + Tat caused no significant changes over 30 min (B), which is also supported by the last 10 min of calcium assessment (B′), indicating that PF3845 100 nM blocked MCM Tat effects. Additionally, MCM heat inactivated Tat (100 nM) did not elicit any significant [Ca2+]i response. (C) MCM Tat-induced increases in [Ca2+]i levels are significantly downregulated by PF3845 indicated by the MCM PF3845 + Tat condition. (D) Incubation with the CB1R antagonist, SR141 (100 nM), failed to block PF3845’s neuroprotective effect as seen in the MCM SR141 + PF3845 + Tat condition for the last 10 min of the time course data. (E) Likewise, the CB2R antagonist, SR144 (100 nM), failed to block PF3845’s neuroprotective effect as seen in the MCM SR144 + PF3845 + Tat condition for the last 10 min of the time course data. (F) Interestingly, the GPR18 antagonist, CID (100 nM), significantly blocked the neuroprotective effects of PF3845 in the MCM CID + PF3845 + Tat condition for the last 10 min of the time course data where the change in [Ca2+]i was similar to the MCM Tat condition, suggesting that GPR18-related activity was necessary for the observed protective effect. Statistical significance was assessed by ANOVAs followed by Bonferroni’s post hoc tests. Bonferroni’s post hoc test were conducted on the last 10 min of the time course data (bar graphs); *p < 0.001 vs. MCM Control, #p < 0.05 vs. MCM Tat, $p < 0.05 vs. MCM antagonists + Tat; φp < 0.001 vs. MCM PF3845 + Tat (at least three independent experiments). Scale bars = 50 um. MCM, microglia conditioned media; hiTat, heat inactivated Tat; SR141, SR141716A (CB1R antagonist); SR144, SR144528 (CB2R antagonist); CID, CID-85469571 (GPR18 antagonist). Please see Supplementary Fig. 2 for the actual time course data of the CB1R, CB2R and GPR18 antagonists experiments over a 30 min time period.
3.1.2. GPR18 mediates PF3845 ablation of neurotoxic effects of Tat treated MCM on neuronal [Ca2+]i
Using selective CB1R and CB2R antagonists (SR141716 and SR144528, respectively), we assessed if these cannabinoid receptors mediated PF3845 blockade of MCM neurotoxicity (Fig. 1D & E; also see Supplementary Fig. 2 for time course data). As shown in Fig. 1D, the CB1R was not necessary to block MCM-mediated neurotoxicity. A one-way ANOVA conducted on the last 10 min revealed a significant treatment effect [F(3,196) =45.5, p < 0.001, ω2 = 0.400], with SR141716 100 nM failing to significantly block the effect of PF3845 (MCM SR141 + PF3845 + Tat vs. MCM Control, MCM PF3845 + Tat; p > 0.05). Please see Supplementary Fig. 2A for the time course data.
Further, results also show the lack of CB2R involvement in mediating PF3845’s neuroprotective effects (Fig. 1E). A one-way ANOVA conducted on the last 10 min revealed a significant treatment effect [F (3,196) = 46.2, p < 0.001, ω2 = 0.405], with results mirroring what was found for CB1R antagonism. Please see Supplementary Fig. 2B for the time course data.
We next assessed the role of GPR18 using the selective antagonist CID (Fig. 1F). A one-way ANOVA conducted on the last 10 min revealed a significant treatment effect [F(3,195) = 38.0, p < 0.001, ω2 = 0.358], with CID 100 nM blocking the protective effect of PF3845 (MCM CID + PF3845 + Tat vs. MCM Control, MCM PF3845 + Tat; p < 0.001). Please see Supplementary Fig. 2C for the time course data. These findings suggest that the neuroprotective effect of PF3845 is GPR18-dependent, which is also confirmed when further testing the role of GPR18 by incubating microglia with increasing concentrations of the GPR18 selective agonist, NAGly (Supplementary Fig. 3). NAGly significantly blunted MCM Tat increases in neuronal [Ca2+]i levels in a concentration-dependent manner, with the highest concentration completely blocking this effect. Additionally, using the selective GPR55 receptor antagonist ML193, data indicated that 100 nM ML193 failed to block PF3845’s effect and although ML193 1 μM did block the neuroprotective effects of PF3845, this concentration also significantly enhance neuronal [Ca2+]i levels in isolation, making it difficult to clearly conclude a role of GPR55 in PF3845 microglial neuroprotection (Supplementary Fig. 4).
3.1.3. FAAH KO microglia show a GPR18-mediated neuroprotective phenotype
We compared the neurotoxicity of MCM derived from FAAH KO microglia to FAAH WT microglia to further assess the role of FAAH in regulating Tat-mediated microglial neurotoxicity (Fig. 2). A two-way ANOVA revealed a significant main effect for time [F(35, 7000) = 48.2, pGG < 0.001] and treatment [F(4, 200) = 66.0, p < 0.001], and a significant treatment x time interaction [F(140, 7000) 10.9, pGG < 0.001]. A one-way ANOVA conducted on the last 10 min revealed a significant treatment effect [F(4,200) = 71.7, p < 0.001, ω2 = 0.535]. MCM Tat 100 nM significantly increased neuronal [Ca2+]i levels compared to control conditions (MCM FAAH WT vs. MCM FAAH WT + Tat, p < 0.001; MCM FAAH WT vs. MCM FAAH KO + Tat, p = 0.003; MCM FAAH WT vs. MCM FAAH KO + CID + Tat, p < 0.001). Particularly noteworthy, the MCM FAAH WT + Tat and MCM FAAH KO + CID + Tat groups elicited significantly higher neuronal [Ca2+]i levels than MCM FAAH KO and MCM FAAH KO + Tat groups (p < 0.001), suggesting the neuroprotective effect of FAAH KO in microglia is GPR18 dependent.
Fig. 2.

Live cell calcium imaging experiments of frontal cortex neurons (DIV 7–11) treated with MCM derived from FAAH WT and FAAH KO microglia over a 30 min time period. The MCM FAAH KO + Tat condition induced significantly less [Ca2+]i levels than the MCM FAAH WT + Tat condition indicating that decreased FAAH activity reduced the neurotoxicity of the MCM. Likewise, the MCM FAAH KO + CID + Tat condition displayed significantly higher levels of neuronal [Ca2+]i compared to the MCM FAAH KO + Tat condition suggesting that this observed neuroprotective effect was dependent on GPR18 activity. Statistical significance was assessed by ANOVAs followed by a Bonferroni’s post hoc test that was conducted on the last 10 min of the time course data (bar graph); *p < 0.05 vs. MCM FAAH WT, #p < 0.05 vs. MCM FAAH WT + Tat, $p < 0.05 vs. MCM FAAH KO + CID + Tat (at least three independent experiments). MCM, microglia conditioned media; WT, wildtype; KO, knockout.
3.2. Quantification of dendritic punctate structure following MCM challenge
3.2.1. PF3845 blocks MCM-mediated degeneration of dendritic spine punctate structures through a GPR18-dependent mechanism
As PF3845 significantly blocked neuronal [Ca2+]i production of MCM derived from Tat treated microglia over a 30 min time period via GPR18-mediated mechanisms, we were interested in whether this effect would replicate for long-term events, i.e. when looking at synaptodendritic injury, which plays an essential role in HAND pathogenesis. Thus, we examined the effects of PF3845 on murine microglial-mediated Tat neurotoxicity and potential GPR18-mechanisms involvement after 24 h by staining for phalloidin positive (F-actin) dendritic structures (Fig. 3). As numbers of phalloidin-positive puncta reflects a balance among active synapses (excitatory and inhibitory), actin dynamics, and synapse stability (Zhang and Benson, 2001), changes in dendritic F-actin rich structures suggest alterations in synaptic integrity and connectivity. Using ImageJ we randomly assigned ROIs to quantify the number of punctate structures present on frontal cortex neurons stained with green fluorescent phalloidin (Fig. 3A & B). A one-way ANOVA assessing changes in mean gray pixel value [F(3,876) = 56.1, p < 0.001, ω2 0.158] and puncta count [F(3,876) = 208.0, p < 0.001, ω2 = 0.414] revealed a significant treatment effect for both measurements (Fig. 3C & D). MCM Tat treatment significantly reduced both gray pixel value and puncta count compared to the control condition (MCM Control vs. MCM Tat, p < 0.001), while PF3845 blocked this neurotoxic effect (MCM Tat vs. MCM PF3845 + Tat, p < 0.001). Administration of CID prevented both of these protective effects (MCM PF3845 + Tat vs. MCM CID + PF3845 + Tat, p < 0.001). It is also noteworthy that incubation with PF3845 significantly enhanced both mean gray pixel value and puncta count compared to control (MCM Control vs. MCM PF3845, p ≤ 0.001). Altogether these data suggest that PF3845 blocks Tat toxicity by decreasing overall F-actin structure and specifically dendritic puncta number.
Fig. 3.
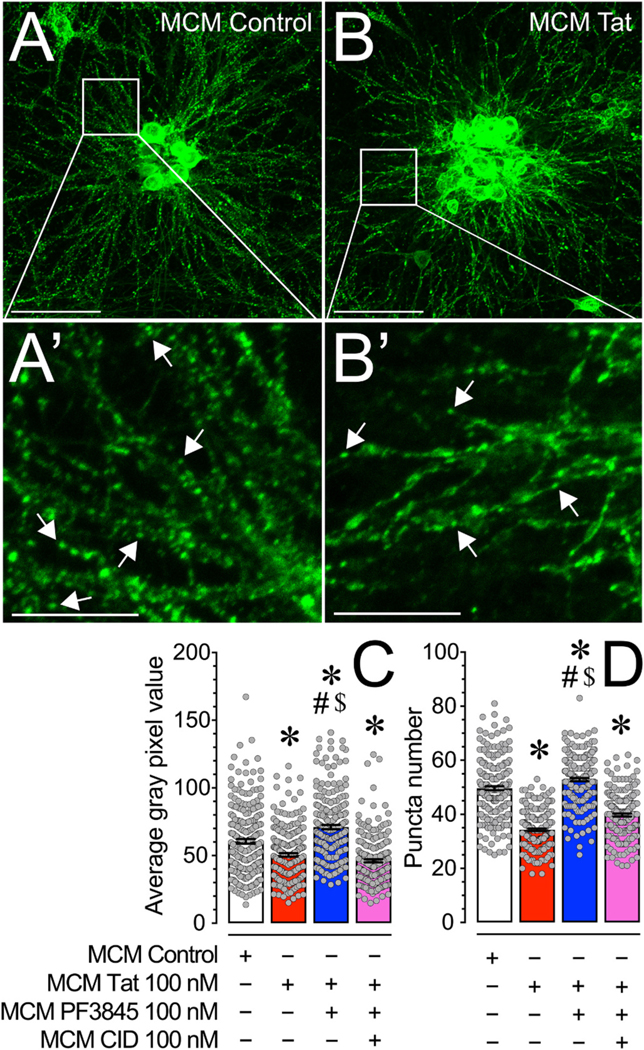
(A & B) Pseudocolor images of phalloidin-stained frontal cortex neuronal cultures (DIV 21) treated with various MCM conditions for 24 h. (A′ & B′) Inset images are depicted on the main image by white boxes and the white arrows denote phalloidin stained punctate structures. (C) MCM Tat treatment significantly decreased average gray pixel value of phalloidin staining while MCM PF3845 + Tat enhanced average gray pixel value of phalloidin staining, relative to MCM Control treatment. MCM CID + PF3845 + Tat did not cause the protective effect seen in MCM PF3845 + Tat treatment, suggesting that PF3845 treatment blocks the loss of actin in dendritic structures induced by Tat in MCM via a GPR18-related mechanism. (D) MCM Tat treatment significantly decreased phalloidin labelled puncta number while MCM PF3845 + Tat enhanced phalloidin labelled puncta number, relative to MCM Control treatment. This suggests that PF3845 protected and increased the number of actin-containing synaptic structures against Tat-related neurotoxicity through microglia. MCM CID + PF3845 + Tat did not cause the protective effect seen in MCM PF3845 + Tat treatment, indicating that GPR18 appears to be a necessary receptor in this PF3845 neuroprotective effect. Statistical significance was assessed by ANOVAs followed by Bonferroni’s post hoc tests; *p < 0.001 vs. MCM Control, #p < 0.001 vs. MCM Tat, $p ≤ 0.001 vs. MCM CID + PF3845 + Tat. Scale bars: Primary = 100 μm, Inset = 30 μm. MCM, microglial conditioned media; CID, CID-85469571.
3.3. ELISA and multiplex
In order to assess the presence and concentration of neurotoxic agents in our experimental MCM treatments we used multiplex assays to examine a range of cytokine and chemokine related molecules, while an ELISA was used to specifically quantify levels of MMP-9 (Fig. 4). All experiments were conducted on MCM harvested from microglial cultures 24 h following treatment with Tat 100 nM. Significant levels of IFNγ, IL-1β, IL-4, IL-6, IL-10, and TNF-α were not found above the detection threshold of the assay (data not shown).
Fig. 4.

(A) MCM Tat caused a significant increase in MCP-1/CCL2 levels compared to MCM Control treatment while incubation with PF3845 blocked this increase and further decreased MCP-1/CCL2 levels compared to control. Antagonizing GPR18 with CID blocked these PF3845-mediated decreases in MCP-1/CCL2 as seen in the MCM CID + PF3845 + Tat condition. (B) MCM Tat caused a significant increase in proMMP-9 levels compared to control and incubating with PF3845 blocked this effect. Antagonizing GPR18 with CID blocked this PF3845-mediated decrease in proMMP-9 as seen in the MCM CID + PF3845 + Tat condition. (C) MCM Tat caused a significant increase in MMP-9 levels compared to control and incubation with PF3845 blocked this effect. Antagonizing GPR18 with CID blocked the PF3845-mediated decrease in MMP-9 as seen in the MCM CID + PF3845 + Tat condition. Collectively, these data suggest that Tat enhanced the excretion of neurotoxins, while PF3845 blunted this effect at least partially through GPR18-related activity. Statistical significance was assessed by ANOVAs followed by Bonferroni’s post hoc tests; *p < 0.05 vs. MCM Control, #p < 0.05 vs. MCM Tat, $p < 0.05 vs. MCM CID + PF3845 + Tat. MCM, microglial conditioned media; CID, CID-85469571.
3.3.1. PF3845 with Tat significantly downregulates levels of MCP-1/CCL2 found in MCM
We found that PF3845 pretreatment alongside Tat significantly decreased levels of MCP-1/CCL2 compared to both MCM Control and MCM Tat conditions (Fig. 4A). A one-way ANOVA revealed a significant main effect of treatment [F(3,20) = 30.5, p < 0.001, ω2 = 0.787]. MCP- 1/CCL2 levels were significantly decreased in the MCM PF3845 + Tat group compared to MCM Control and MCM Tat groups (p < 0.001). Treatment with CID 100 nM blocked this effect (MCM CID + PF3845 + Tat vs. MCM Control, MCM Tat; p > 0.05). Note, that pretreating microglia with the MCP-1 inhibitor bindarit 300 μM before Tat exposure shows attenuation of MCM Tat-induced [Ca2+]i levels in frontal cortex neurons (Supplementary Fig. 5).
3.3.2. PF3845 blunts Tat protein enhancement of proMMP-9 and MMP-9 levels in MCM in a GPR18-dependent manner
Tat significantly enhanced levels of inactive proMMP-9 and cleaved/activated MMP-9 compared to the control condition and PF3845 blunted this effect via a GPR18 based mechanism (Fig. 4B & C). A one-way ANOVA revealed a significant main effect of treatment for proMMP-9 [F(3,20) = 209.5, p < 0.001, ω2 = 0.963] and MMP-9 [F(3,15) = 91.4, p < 0.001, ω2 = 0.935]. MCM levels of proMMP-9 and MMP-9 were significantly increased by Tat incubation and this effect was attenuated by PF3845 (MCM Control vs. MCM Tat vs. MCM PF3845 + Tat; p < 0.001). Incubation with CID showed that PF3845’s effect on MMP levels was tied to the GPR18 receptor (MCM Tat vs. MCM CID + PF3845 + Tat, p > 0.05). Similar to the effects seen with bindarit 300 μM, pretreating microglia with MMP-9 inhibitor I 150 nM before Tat exposure demonstrated an attenuation of MCM Tat-induced [Ca2+]i levels in frontal cortex neurons, suggesting that both MCP-1/CCL2 and MMP-9 at least partially mediate MCM Tat-induced toxicity (Supplementary Fig. 5).
3.4. Microglial fractal analysis
3.4.1. PF3845 treatment significantly increases fractal dimension (DF) and significantly decreases lacunarity (Λ) of primary murine microglia
Microglia have been shown to take on a wide range of morphological features which have been associated with distinct functions, thus we were interested in whether morphological changes can be observed in cultured microglia. As significant upregulated levels of MCP-1/CCL2, inactive proMMP-9, and cleaved/activated MMP-9 were noted in MCM Tat, which was significantly downregulated by MCM PF3845 pretreatment, we were interested in whether changes can be observed in microglia morphology as they have been associated with distinct functions. Thus, using live phase images of 24 h treated microglial cells at DIV 14 we conducted fractal analysis using ImageJ to determine fractal dimension (DF) and lacunarity (Λ) as measures of microglial morphological complexity (Fig. 5). A one-way ANOVA assessing DF and Λ revealed a significant treatment effect for both measurements [F(5,599) = 19.4, p < 0.001, ω2 = 0.132 and F(5,600) = 18.5, p < 0.001, ω2 = 0.126, respectively]. Whereas no effect was noted for Tat treatment, PF3845 significantly enhanced DF compared to control (Control vs. PF3845, PF3845 + Tat; p < 0.001) and significantly lowered Λ compared to control (Control vs. PF3845, PF3845 + Tat; p < 0.001). CID blocked both of these effects.
Fig. 5.

(A) An overlay of a phase image of cultured microglia with outlines of randomly selected microglia processed for ImageJ FracLac analysis. (A′) Phase image of cultured microglia showing a range of amoeboid and branched morphologies. (A″) Outlines of randomly selected microglia processed for ImageJ FracLac analysis with an inset image that is represented on the primary image by a black box showing microglial ruffles. (C) Microglia treated with PF3845 and PF3845 + Tat treatment conditions demonstrated a significant increase in fractal dimension (DF) compared to other treatment conditions, indicating microglia morphology following PF3845 treatment is more associated with an intermediate activation state and less with the activated amoeboid state. (D) Microglia treated with PF3845 and PF3845 + Tat treatment conditions demonstrated a significant decrease in lacunarity (Λ) compared to other treatment conditions, suggesting that the PF3845 treated microglia have more overall morphological homogeneity. No effects were noted for Tat treatment potentially as microglia grown in culture in vitro under control conditions reside in a more amoeboid phenotype/shape. Statistical significance was assessed by ANOVAs followed by Bonferroni’s post hoc tests; *p < 0.05 vs. Control. Scale Bars: Primary = 100 μm, Inset = 30 μm. CID, CID-85469571.
3.5. LC/MS/MS
3.5.1. PF3845 significantly enhances levels of AEA in primary murine microglia
To confirm the effects of PF3845 as a FAAH enzyme inhibitor, LC/MS/MS was used to determine the expression of AEA and other pre-dominant substrates of FAAH in MCM conditions derived from microglia treated with PF3845 100 nM + Tat 100 nM for 24 h (Fig. 6).
Fig. 6.

Concentrations of AEA in MCM were assessed using LC/MS/MS analysis following 24 h of PF3845, Tat and/or CID exposure to cultured microglia. All MCM conditions treated with PF3845 had significantly enhanced levels of AEA compared to MCM Control and MCM Tat. This suggests that PF3845 was effective in inhibiting microglial FAAH’s ability to degrade AEA. No effects on AEA levels were noted when CID was added to MCM PF3845 + Tat, indicating that blocking AEA’s activity on GPR18 receptors had no effect on AEA levels itself. Statistical significance was assessed by ANOVAs followed by a Bonferroni’s post hoc test; *p < 0.05 vs. MCM Control, #p < 0.05 vs. MCM Tat. MCM, microglial conditioned media; CID, CID-85469571.
As expected PF3845 led to a significant elevation of AEA levels in MCM conditions (Fig. 6). A two-way ANOVA detected a significant treatment effect of MCM PF3845 [F(1,11) = 270.3, p < 0.001], and MCM Tat [F(1,11) = 7.2, p = 0.200], but no significant interaction effect [F(1,11) = 1.4, p > 0.05]. A one-way ANOVA assessing treatment effect [F(4,10) = 72.8, p < 0.001, ω2 = 0.950] found a significant difference between all PF3845 treatment groups and all non-PF3845 treated groups (MCM Control, MCM Tat vs. all MCM PF3845 conditions). PF3845 100 nM, regardless of treatment group, caused a roughly two-fold increase of AEA levels in MCM conditions. FAAH inhibition also led to significant increases in other fatty acids (i.e., docosahexaenoyl ethanolamide (DHEa) and oleoylethanolamide (OEA); Supplementary Fig. 7).
4. Discussion
MCM derived from Tat-treated microglia significantly increased neuronal [Ca2+]i levels, comparable to the increase seen with direct Tat treatment (Brailoiu et al., 2008; Hermes et al., 2018) and with HIV-1 conditioned media (Meeker et al., 2016). The 24 h time point for MCM collection was selected to avoid the effect of drugs/proteins applied to stimulate the microglia from being carried from microglial treatment to neuronal cultures. Considering that Tat-induced MCM was a mixture of different factors excreted from microglia, we were unable to conclude on specific mechanisms of neuronal calcium dysregulation, nonetheless, upregulated MCP-1/CCL2 and MMP-9 might be a contributing factor to the upregulation of intracellular calcium in our neuronal cultures. MCP- 1/CCL2 upregulates NMDA receptor function (Zhou et al., 2016) in a fashion similar to what has been described in earlier reports of Tat-mediated neurotoxicity (Fitting et al., 2014; Haughey et al., 2001). Likewise, MMP-9 enhances NMDA receptor surface trafficking, which may alter receptor functioning (Michaluk et al., 2009). Remarkably, PF3845 pretreatment of microglia completely abolished Tat-induced MCM’s changes in neuronal [Ca2+]i. The current study examined receptor mechanism of action through the employment of selective antagonists for CB1R, CB2R, GPR18, and GPR55. Here we show that this neuroprotective effect was independent of CB1R, CB2R, and GPR55, but reliant on GPR18 activity. In addition to showing that the GPR18 antagonist, CID, blocked this neuroprotective effect of PF3845, incubation of microglia with increasing concentrations of the GPR18 selective agonist, NAGly, confirmed the role of this receptor in MCM Tat-induced neurotoxicity, with the highest concentration of NAGly completely blocking this effect (Supplementary Fig. 3). It is also important to consider that NAGly can inhibit FAAH, particularly at high μM concentrations (Grazia Cascio et al., 2004). A simplified schematic summarizes our findings in Fig. 7.
Fig. 7.

(A) Shows the effects of treatment conditions on microglia morphology. Tat does not show any effects on fractal dimention (DF) and lacunarity (Λ) whereas PF3845 via GPR18 increases DF and decreases Λ, which is associated with a less activated microglia state and more overall morphological homogeneity, respectively. (B) Shows the soluble factors released from treated microglia into the microglial conditioned media (MCM). MCM conditions derived from Tat treated microglia (MCM Tat) show upregulated proinflammatory responses with increased levels in MCP-1/CCL2, MMP-9, and proMMP-9. In contrast, MCMs derived from microglia pretreated with PF3845 (MCM PF3845) show increased AEA levels by inhibiting the catabolic FAAH enzyme via GPR18-related mechanisms, which results in anti-inflammatory responses by decreasing MCP-1/CCL2, MMP-9, and proMMP-9 levels. (C) Shows neuronal changes that occur when frontal cortex neurons are treated with MCM conditions derived from treated microglia. MCM Tat increases neuronal Ca2+ dysregulation and synaptodendritic damage whereas MCMs derived from microglia pretreated with PF3845 alongside Tat (MCM PF3845 + Tat) results in reduction of Ca2+ dysregulation and synaptodendritic damage via GPR18-related mechanisms. Λ, lacunarity; Ca2+, calcium; CID-85469571, GPR18 antagonist; DF, fractal dimension.
Even though a range of cannabinoid receptor ligands such as AEA, 2- AG, Δ9-tetrahydrocannabinol (THC), and cannabidiol (CBD) bind to both GPR55 and GPR18 (Lauckner et al., 2008; McHugh et al., 2012), GPR55 did not show a clear role in PF3845 microglial neuroprotection (Supplementary Fig. 4). It is also noteworthy that GPR55 antagonism alone could be anti-inflammatory (Saliba et al., 2018) in some models of microglial signaling, though the present study did not directly assess this action. Further, a GPR18-dependent neuroprotective effect similar to PF3845 was found in FAAH KO microglia, implying that higher fatty acid amide content, such as AEA, ameliorated microglial neurotoxicity (Fig. 2). The findings that the neuroprotective effects were based on GPR18, but not CB2R, were particularly surprising given the prominent role of CB2R activation on neuroprotective and anti-inflammatory activation in microglia (Ehrhart et al., 2005; Malek et al., 2015; Maresz et al., 2005). A positive control experiment using lipopolysaccharide (LPS) to increase MCP-1/CCL2 levels in our microglia cultures demon- strated MCM MCP-1/CCL2 downregulation when the CB2R agonist HU-308 was used in combination with LPS 1 μg/mL challenge (Chhor et al., 2013; Presley et al., 2015, Supplementary Fig. 6) suggesting that our microglia express CB2Rs. An important factor to keep in mind when comparing these different studies is that variations in cannabinoid receptor expression can be tied to microglial activation states and signaling. Differences in CB2R expression have been shown previously, with LPS-activated macrophages having low CB2R expression, while interferon gamma (IFNγ) treatment greatly enhances microglial CB2R (Ashton and Glass, 2007). Also, a recent study has shown that CB2R and GPR18 readily form heterodimers and display negative-crosstalk and cross-antagonism, suggesting that extricating the individual roles of these receptors in modulating microglial activity may be more difficult than originally expected (Reyes-Resina et al., 2018).
Glia-based neuroinflammation has been linked to neuronal damage/death and specifically to the progression of synaptodendritic injury in HIV-related disease (Block et al., 2007; Bruce-Keller, 1999; Green et al., 2019; Liu et al., 2013; Lu et al., 2011; Nath et al., 1999; Takeuchi, 2010), which in turn strongly correlates with HAND symptom severity (Ellis et al., 2007; Masliah et al., 1992; Masliah et al., 1997). Dendritic spines are small, actin rich structures that are paramount in healthy neuronal signaling and are highly sensitive to changes in intracellular calcium (Higley and Sabatini, 2008; Hotulainen and Hoogenraad, 2010; Maha- jan and Nadkarni, 2019). We have previously shown that Tat directly disrupts healthy dendritic morphology and PF3845 partially blocked this effect (Hermes et al., 2018). The loss of overall synaptic connectivity in DIV 21 MCM-treated neurons was quantified via overall actin staining and number of phalloidin-positive punctate structures. Based on our calcium findings, it was not surprising that MCM Tat significantly reduced overall actin staining and puncta count, while treating microglia with PF3845 blocked this effect (Fig. 3). Specifically, it has been shown previously that a 2–4-fold upregulation from baseline [Ca2+]i levels at around 100 nM to ~200–400 nM for an extended amount of time (>5 min) precede profound changes in neuronal morphology as well as neuronal loss in primary CNS neuron cultures (Fitting et al., 2014; Kouzoukas et al., 2013; Tolar et al., 1999). Calcium dysregulation is a common theme in several neurodegenerative diseases (Imamura et al., 2016; Verma et al., 2018; Wojda et al., 2008; Zündorf and Reiser, 2011) and chronic neuroinflammation correlates with loss of neuronal calcium equilibrium (Oliveira et al., 2014; Sama and Norris, 2013; Zündorf and Reiser, 2011). Previous studies have shown that modulating NMDA receptor activity can prevent Tat-induced synapse loss (Raybuck et al., 2017) and cannabinoids can achieve this through similar mechanisms (Kim et al., 2008).
Tat appears to be a major component of HAND progression as it is expressed early in HIV-1 infection (Fisher et al., 1986; Nicoli et al., 2013) and is detectable in appreciable levels in diagnosed patients (Henderson et al., 2019). Further, Tat has been shown to drive proinflammatory signaling and immune dysregulation (Ben Haij et al., 2015; Gandhi et al., 2009). Microglia exposed to Tat become activated and release various signaling molecules that can be ultimately deleterious to neuronal survival and proper functioning (Jin et al., 2012; Lu et al., 2011; Thangaraj et al., 2018). As the proinflammatory cytokines TNF-α, IL-1β and IL-6 were below the lower detection threshold in the present study, no conclusions could be made on the involvement of these mediators. However, suprathreshold levels of MCP-1/CCL2 and pro-form/cleaved MMP-9 were detected in MCM, significantly increased following Tat treatment (Fig. 4), and partially mediated Tat-induced toxicity (Supplementary Fig. 5). MCP-1/CCL2 is a key mediator of microglial migration following activation (Park et al., 2001), is upregulated following Tat treatment (Eugenin et al., 2005, Fig. 4A), and is capable of enhancing neuronal excitability via CCR2 (Zhou et al., 2011). Disruptions in the balance of MMP expression have been linked with a wide range of neurodegenerative and neurodevelopmental diseases (Reinhard et al., 2015) and MMP-9, in particular, plays a key role in regulating the structural plasticity of dendritic spines (Stawarski et al., 2014). Additionally, increases in MMP- 9 production have been shown in HIV-related non-human and human studies (Webster and Crowe, 2006), and has been linked with enhanced neurotoxicity in models of neurodegeneration (Jourquin et al., 2003; Mizoguchi et al., 2009). Pretreatment with 100 nM PF3845 significantly reduced levels of MCP-1/CCL2 and MMPs (with MCP-1/CCL2 being reduced below control), while CID partially blocked this effect, suggesting that GPR18 serves an important role in regulating activated microglia (Fig. 4). The finding that Tat PF3845 produces less MCP-1/CCL2 than PF3845 pretreatment alone suggests that PF3845 might exacerbate its effect in the presence of a disease state. As outlined in the present study MCM Tat exposure is associated with neuronal damaging effects, indicated by upregulation of intracellular calcium and decrease in puncta number, thus PF3845 might overcompensate by greatly changing the signaling behavior of glia and driving MCP-1/CCL2 levels below what is seen with baseline or with PF3845 alone. A previous study has demonstrated that WIN55,212 is able to decrease TNF-α levels in brain slices of 7 d old rats to lower levels than those found in controls (Fernandez-Lopez et al., 2006). Further, a compensatory effect of the endocannabinoid system in disease states has been suggested previously by the upregulation of the CB1R and endocannabinoid system activity in experimental models of Parkinson’s disease as well as neuroHIV (Brotchie, 2003; Jacobs et al., 2019).
Considering the heightened levels of these molecules following Tat treatment of our microglial cultures and the resulting neurotoxicity from MCM incubation of neurons, this study lends additional support to both MCP-1/CCL2 and MMP-9 playing an important role, alongside other molecules, in driving HAND related pathology. Likewise, PF3845 could be useful in reducing factors that promote HAND progression.
Microglia have been shown to take on a wide range of morphological features which have been associated with distinct functions. Primary microglia exist mostly in an activated state, making them a good model for neurodegenerative disorders in which this activated morphology has been observed (Timmerman et al., 2018). In the present study, it was determined that the majority of the microglia that make up the cultures used in the present study fall into the unramified/activated morphological state (Karperien et al., 2013; Karperien and Jelinek, 2015). Using a box-counting method for fractal analysis and found that microglia treated with PF3845 took on a distinct profile of DF and Λ compared to untreated glia (Fig. 5). PF3845-treated microglia had significantly higher DF and lower Λ as compared to non-treated cells. As microglia proceed from ramified to unramified states both DF and Λ decrease and increase again as cells “re-ramify” (Karperien and Jelinek, 2015). Thus, based on DF our data indicate that PF3845-mediated changes in our microglia are moving away from a stereotypical activated morphological state. Similarly, another study showed that the effects of another FAAH inhibitor, URB597, decreased production of proinflammatory molecules accompanied by increases in DF and decreases in Λ following ethanol-induced activation of microglia in mouse hippocampus (Rivera et al., 2018). This finding further suggests that the observed decrease in Λ is related to selective FAAH inhibition, with the caveat that the former study focused on ramified and intermediate state microglia. These PF3845-related morphological changes were blocked by CID, providing further evidence of the involvement of GPR18. GPR18-induced changes in microglial morphology have been previously reported, with NAGly incubation enhancing a branched morphology over an ameboid state (McHugh et al., 2014). In contrast, no effects of Tat treatment were noted on DF and Λ, highly likely due to the fact that microglia grown in culture in vitro are more activated than under in vivo conditions and reside in a more amoeboid phenotype/shape (Caldeira et al., 2014; Lai et al., 2013).
FAAH inhibitors represent a promising pharmaceutical treatment for HIV-induced neurotoxicity and HAND progression given decreased FAAH activity leads to heightened levels of AEA and other anti-inflammatory fatty acid amides (Booker et al., 2012). Based on our findings, decreased microglial neurotoxicity likely relies on enhancement of AEA signaling (Fig. 6). Microglia express GPR18, which is stimulated by AEA (McHugh et al., 2012); however, the EC50 is in the low micromolar range. NAGly, proposed endogenous ligand for GPR18, was unlikely involved in the current results as one of its major biosynthesis pathways is FAAH-dependent and FAAH inhibition decreases NAGly levels (Bradshaw et al., 2009). Regardless, NAGly possesses powerful inflammation-resolving properties dependent on GPR18 activity (Burstein et al., 2011), with an EC50 of 44.5 nM (McHugh et al., 2012). Compared to earlier experiments with neuronal cultures (Hermes et al., 2018), our microglial cultures produced roughly 2-fold greater levels of AEA in control conditions. This is similar to earlier reports that described microglia as producing the bulk of endocannabinoids as compared to other cell types in the brain (Walter et al., 2002; Walter et al., 2003). It is important to note that these levels are significantly below the EC50 levels for GPR18 activity described before, suggesting that the observed GPR18-related neuroprotective effect may be dependent on multiple lipids regulated by FAAH. Altogether, these findings further our understanding of the underlying mechanism of FAAH inhibition for the potential treatment of microglial neurotoxicity in conditions like HAND or other neurodegenerative diseases.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Institute on Drug Abuse [(NIDA R01 DA045596 (SF), R21 DA041903 (SF), T32 DA007244 (DJH), R01 DA039942 (AHL)], National Institute of Mental Health [R01 NS108808 (RBM)], National Institute on Aging [R21 AG056924 (RBM)], and the National Center for Research Resources NCRR 1S10OD010366 (NR). Bogna Ignatowska-Jankowska was supported by the fellowship from the Japan Society for Promotion of Science (JSPS). Further, we gratefully acknowledge the assistance of Carlton Anderson with collecting ELISA/multiplex data at CGIBD’s Advanced Analytics Core, UNC Chapel Hill, supported by National Institute of Diabetes and Digestive and Kidney Diseases [P30 DK034987].
Abbreviations
- 2-AG
2-Arachidonoylglycerol
- AEA
anandamide/N-arachidonoylethanolamine
- AM
acetoxymethyl
- ANOVA
analysis of variance
- cART
combined antiretroviral therapy
- CBD
cannabidiol
- CB1R
cannabinoid receptor type 1
- CB2R
cannabinoid receptor type 2
- CID
CID-85469571/GPR18 antagonist
- DF
fractal dimension
- DIV
days in vitro
- FAAH
fatty acid amide hydrolase
- GPCR
G protein coupled receptor
- GPR18
G protein couple receptor 18
- HAND
HIV-associated neurocognitive disorders
- HIV-1
human immunodeficiency virus 1
- KO
knockout
- LPS
lipopolysaccharide
- MCM
microglia-conditioned medium
- MCP-1
monocyte chemoattractant protein 1
- MMP
matrix metalloproteinase
- NAGly
N-Arachidonylglycine
- ROI
region of interest
- SEM
standard error of the mean
- SR141
rimonabant/SR141716/CB1R antagonist
- SR144
SR144528/CB2R antagonist
- Tat
transactivator of transcription
- THC
Δ9-tetrahydrocannabinol
- WT
wildtype
- °C
degrees Celsius
- Λ
lacunarity
Footnotes
Declaration of interest
None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.expneurol.2021.113699.
References
- Ashton JC, Glass M, 2007. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr. Neuropharmacol 5, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haij N, Planès R, Leghmari K, Serrero M, Delobel P, Izopet J, BenMohamed L, Bahraoui E, 2015. HIV-1 tat protein induces production of proinflammatory cytokines by human dendritic cells and monocytes/macrophages through engagement of TLR4-MD2-CD14 complex and activation of NF-κB pathway. PLoS One 10, e0129425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong J-S, 2007. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci 8, 57–69. [DOI] [PubMed] [Google Scholar]
- Booker L, Kinsey SG, Abdullah RA, Blankman JL, Long JZ, Ezzili C, Boger DL, Cravatt BF, Lichtman AH, 2012. The fatty acid amide hydrolase (FAAH) inhibitor PF-3845 acts in the nervous system to reverse LPS-induced tactile allodynia in mice. Br. J. Pharmacol 165, 2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Hu SS-J, Benton VM, Stuart JM, Masuda K, Cravatt BF, O’Dell DK, Walker JM, 2009. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem. 10, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu GC, Brailoiu E, Chang JK, Dun NJ, 2008. Excitatory effects of human immunodeficiency virus 1 Tat on cultured rat cerebral cortical neurons. Neuroscience 151, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotchie JM, 2003. CB1 cannabinoid receptor signalling in Parkinson’s disease. Curr. Opin. Pharmacol 3, 54–61. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, 1999. Microglial–neuronal interactions in synaptic damage and recovery. J. Neurosci. Res 58, 191–201. [DOI] [PubMed] [Google Scholar]
- Burstein SH, McQuain CA, Ross AH, Salmonsen RA, Zurier RE, 2011. Resolution of inflammation by N-arachidonoylglycine. J. Cell. Biochem 112, 3227–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira C, Oliveira AF, Cunha C, Vaz AR, Falcao AS, Fernandes A, Brites D, 2014. Microglia change from a reactive to an age-like phenotype with the time in culture. Front. Cell. Neurosci 8, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K, Pfister SL, Campbell WB, Hillard CJ, 2004. Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol. Pharmacol 65, 999–1007. [DOI] [PubMed] [Google Scholar]
- Carroll A, Brew B, 2017. HIV-associated neurocognitive disorders: recent advances in pathogenesis, biomarkers, and treatment. F1000Res. 6, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang J, Chen C, 2011. Endocannabinoid 2-arachidonoylglycerol protects neurons against β-amyloid insults. Neuroscience 178, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhor V, Le Charpentier T, Lebon S, Ore MV, Celador IL, Josserand J, Degos V, Jacotot E, Hagberg H, Savman K, Mallard C, Gressens P, Fleiss B, 2013. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun 32, 70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholfin JA, Rubenstein JL, 2007. Patterning of frontal cortex subdivisions by Fgf17. Proc. Natl. Acad. Sci. U. S. A 104, 7652–7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ZD, Haney M, 2009. Actions of delta-9-tetrahydrocannabinol in cannabis: relation to use, abuse, dependence. Int. Rev. Psychiatry 21, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Council NR, 2011. Guide for the Care and Use of Laboratory Animals: Eighth Edition. The National Academies Press, Washington, DC. [Google Scholar]
- Cysique LA, Maruff P, Brew BJ, 2004. Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: a combined study of two cohorts. J. Neuro-Oncol 10, 350–357. [DOI] [PubMed] [Google Scholar]
- Dheen ST, Charanjit K, Eng-Ang L, 2007. Microglial activation and its implications in the brain diseases. Curr. Med. Chem 14, 1189–1197. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD, 2005. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflammation 2, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, Hauser KF, 2008. Morphine exacerbates HIV-1 tat-induced cytokine production in astrocytes through convergent effects on [Ca2+]i, NF-κB trafficking and transcription. PLoS One 3, e4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Dever SM, Fitting S, Ahmed T, Hauser KF, 2011. HIV-1 coinfection and morphine coexposure severely dysregulate hepatitis C virus-induced hepatic proinflammatory cytokine release and free radical production: increased pathogenesis coincides with uncoordinated host defenses. J. Virol 85, 11601–11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E, 2007. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat. Rev. Neurosci 8, 33–44. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Dyer G, Calderon TM, Berman JW, 2005. HIV-1 tat protein induces a migratory phenotype in human fetal microglia by a CCL2 (MCP-1)-dependent mechanism: possible role in NeuroAIDS. Glia 49, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lopez D, Martinez-Orgado J, Nunez E, Romero J, Lorenzo P, Moro MA, Lizasoain I, 2006. Characterization of the neuroprotective effect of the cannabinoid agonist WIN-55212 in an in vitro model of hypoxic-ischemic brain damage in newborn rats. Pediatr. Res 60, 169–173. [DOI] [PubMed] [Google Scholar]
- Fisher AG, Feinberg MB, Josephs SF, Harper ME, Marselle LM, Reyes G, Gonda MA, Aldovini A, Debouk C, Gallo RC, Wong-Staal F, 1986. The transactivator gene of HTLV-III is essential for virus replication. Nature 320, 367–371. [DOI] [PubMed] [Google Scholar]
- Fitting S, Knapp PE, Zou S, Marks WD, Bowers MS, Akbarali HI, Hauser KF, 2014. Interactive HIV-1 tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na+ Iinflux, mitochondrial instability, and Ca2+ overload. J. Neurosci 34, 12850–12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi N, Saiyed Z, Thangavel S, Rodriguez J, Rao KVK, Nair MPN, 2009. Differential effects of HIV type 1 clade B and clade C Tat protein on expression of proinflammatory and antiinflammatory cytokines by primary monocytes. AIDS Res. Hum. Retrovir 25, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia-Figueira S, Nording ML, 2015. Validation of a tandem mass spectrometry method using combined extraction of 37 oxylipins and 14 endocannabinoid-related compounds including prostamides from biological matrices. Prostaglandins Other Lipid Mediat. 121, 110–121. [DOI] [PubMed] [Google Scholar]
- Grabiec U, Hohmann T, Ghadban C, Rothgänger C, Wong D, Antonietti A, Groth T, Mackie K, Dehghani F, 2019. Protective effect of N-arachidonoyl glycine-GPR18 signaling after excitotoxical lesion in murine organotypic hippocampal slice cultures. Int. J. Mol. Sci 20, 1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grazia Cascio M, Minassi A, Ligresti A, Appendino G, Burstein S, Di Marzo V, 2004. A structure–activity relationship study on N-arachidonoyl-amino acids as possible endogenous inhibitors of fatty acid amide hydrolase. Biochem. Biophys. Res. Commun 314, 192–196. [DOI] [PubMed] [Google Scholar]
- Green MV, Raybuck JD, Zhang X, Wu MM, Thayer SA, 2019. Scaling synapses in the presence of HIV. Neurochem. Res 44, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhouse SW, Geisser S, 1959. On methods in the analysis of profile data. Psychometrika 24, 95–112. [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY, 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem 260, 3440–3450. [PubMed] [Google Scholar]
- Harkany T, Mackie K, Doherty P, 2008. Wiring and firing neuronal networks: Endocannabinoids take center stage. Curr. Opin. Neurobiol 18, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD, 2001. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem 78, 457–467. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, LeBlanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema- Notestine C, Jernigan TL, Wong J, Grant I, for the C, Groups H, 2011. HIV- associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J. Neuro-Oncol 17, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LJ, Johnson TP, Smith BR, Reoma LB, Santamaria UA, Bachani M, Demarino C, Barclay RA, Snow J, Sacktor N, Mcarthur J, Letendre S, Steiner J, Kashanchi F, Nath A, 2019. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 33, S145–S157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge CM, 2012. Off-target cannabinoid effects mediated by GPR55. Pharmacology 89, 179–187. [DOI] [PubMed] [Google Scholar]
- Hermes DJ, Xu C, Polkis JL, Niphakis MJ, Cravatt BF, Mackie K, Lichtman AH, Ignatowska-Jankowska BM, Fitting S, 2018. Neuroprotective effects of fatty acid amide hydrolase catabolic enzyme inhibition in a HIV-1 Tat model of neuroAIDS. Neuropharmacology 141, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL, 2008. Calcium signaling in dendrites and spines: practical and functional considerations. Neuron 59, 902–913. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC, 2010. Actin in dendritic spines: connecting dynamics to function. J. Cell Biol 189, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Rock RB, 2013. CB2 receptor agonists protect human dopaminergic neurons against damage from HIV-1 gp120. PLoS One 8, e77577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I, 2000. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J. Neuro-Oncol 6, 145–155. [DOI] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Ghosh S, Crowe MS, Kinsey SG, Niphakis MJ, Abdullah RA, Tao Q, ST ON, Walentiny DM, Wiley JL, Cravatt BF, Lichtman AH, 2014. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br. J. Pharmacol 171, 1392–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska B, Wilkerson JL, Mustafa M, Abdullah R, Niphakis M, Wiley JL, Cravatt BF, Lichtman AH, 2015. Selective monoacylglycerol lipase inhibitors: antinociceptive versus cannabimimetic effects in mice. J. Pharmacol. Exp. Ther 353, 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Sahara N, Kanaan NM, Tsukita K, Kondo T, Kutoku Y, Ohsawa Y, Sunada Y, Kawakami K, Hotta A, Yawata S, Watanabe D, Hasegawa M, Trojanowski JQ, Lee VMY, Suhara T, Higuchi M, Inoue H, 2016. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep 6, 34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs IR, Xu C, Hermes DJ, League AF, Xu C, Nath B, Jiang W, Niphakis MJ, Cravatt BF, Mackie K, Mukhopadhyay S, Lichtman AH, Ignatowska- Jankowska BM, Fitting S, 2019. Inhibitory control deficits associated with upregulation of CB1R in the HIV-1 tat transgenic mouse model of hand.J. NeuroImmune Pharmacol. 14, 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Lam L, Sadic E, Fernandez F, Tan J, Giunta B, 2012. HIV-1 tat-induced microglial activation and neuronal damage is inhibited via CD45 modulation: a potential new treatment target for HAND. Am. J. Transl. Res 4, 302–315. [PMC free article] [PubMed] [Google Scholar]
- Jourquin J, Tremblay E, ecanis N, Charton G, Hanessian S, Chollet A-M, Le Diguardher T, Khrestchatisky M, Rivera S, 2003. Neuronal activity-dependent increase of net matrix metalloproteinase activity is associated with MMP-9 neurotoxicity after kainate. Eur. J. Neurosci 18, 1507–1517. [DOI] [PubMed] [Google Scholar]
- Justinova Z, Tanda G, Redhi GH, Goldberg SR, 2003. Self-administration of delta9-tetrahydrocannabinol (THC) by drug naive squirrel monkeys. Psychopharmacology 169, 135–140. [DOI] [PubMed] [Google Scholar]
- Kallendrusch S, Kremzow S, Nowicki M, Grabiec U, Winkelmann R, Benz A, Kraft R, Bechmann I, Dehghani F, Koch M, 2013. The G protein-coupled receptor 55 ligand l-alpha-lysophosphatidylinositol exerts microglia-dependent neuroprotection after excitotoxic lesion. Glia 61, 1822–1831. [DOI] [PubMed] [Google Scholar]
- Karperien AL, Jelinek HF, 2015. Fractal, multifractal, and lacunarity analysis of microglia in tissue engineering. Front. Bioeng. Biotechnol 3, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karperien A, Ahammer H, Jelinek HF, 2013. Quantitating the subtleties of microglial morphology with fractal analysis. Front. Cell. Neurosci 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Waataja JJ, Thayer SA, 2008. Cannabinoids inhibit network-driven synapse loss between hippocampal neurons in culture. J. Pharmacol. Exp. Ther 325, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Buckner CM, Berman JW, 2006. HIV tat and neurotoxicity. Microbes Infect. 8, 1347–1357. [DOI] [PubMed] [Google Scholar]
- Kouzoukas DE, Li G, Takapoo M, Moninger T, Bhalla RC, Pantazis NJ, 2013. Intracellular calcium plays a critical role in the alcohol-mediated death of cerebellar granule neurons. J. Neurochem 124, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP, 1998. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol 154, 276–288. [DOI] [PubMed] [Google Scholar]
- Lai AY, Dibal CD, Armitage GA, Winship IR, Todd KG, 2013. Distinct activation profiles in microglia of different ages: a systematic study in isolated embryonic to aged microglial cultures. Neuroscience 254, 185–195. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K, 2008. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. U. S. A 105, 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Aksenova M, Bertrand SJ, Mactutus CF, Booze R, 2016. Quantification of filamentous actin (F-actin) Puncta in rat cortical neurons. J. Vis. Exp. e53697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Dimen KR, Martin BR, 1995. Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology 119, 282–290. [DOI] [PubMed] [Google Scholar]
- Liu J., Xu P., Collins C., Liu H., Zhang J., Keblesh JP., Xiong H., 2013. HIV-1 Tat protein increases microglial outward K current and resultant neurotoxic activity. PLoS One 8, e64904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longordo F, Feligioni M, Chiaramonte G, Sbaffi PF, Raiteri M, Pittaluga A, 2006. The human immunodeficiency virus-1 protein transactivator of transcription up- regulates N-methyl-D-aspartate receptor function by acting at metabotropic glutamate receptor 1 receptors coexisting on human and rat brain noradrenergic neurones. J. Pharmacol. Exp. Ther 317, 1097–1105. [DOI] [PubMed] [Google Scholar]
- Lu S-M, Tremblay M-È, King IL, Qi J, Reynolds HM, Marker DF, Varrone JJP, Majewska, Dewhurst S, Gelbard HA, 2011. HIV-1 tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One 6, e23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan G, Nadkarni S, 2019. Intracellular calcium stores mediate metaplasticity at hippocampal dendritic spines. J. Physiol 597, 3473–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek N, Popiolek-Barczyk K, Mika J, Przewlocka B, Starowicz K, 2015. Anandamide, acting via CB2 receptors, alleviates LPS-induced neuroinflammation in rat primary microglial cultures. Neural. Plast 2015, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN, 2005. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem 95, 437–445. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt, López-Rodríguez ML, Casanova E, Schütz G, Zieglgänsberger, Di Marzo, Behl C, Lutz B, 2003. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88. [DOI] [PubMed] [Google Scholar]
- Masliah E, Ge N, Morey M, DeTeresa R, Terry RD, Wiley CA, 1992. Cortical dendritic pathology in human immunodeficiency virus encephalitis. Lab. Investig 66, 285–291. [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I, 1997. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann. Neurol 42, 963–972. [DOI] [PubMed] [Google Scholar]
- McHugh D, 2012. GPR18 in microglia: implications for the CNS and endocannabinoid system signalling. Br. J. Pharmacol 167, 1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Page J, Dunn E, Bradshaw HB, 2012. Δ(9)-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br. J. Pharmacol 165, 2414–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Roskowski D, Xie S, Bradshaw H, 2014. Δ9-THC and N-arachidonoyl glycine regulate BV-2 microglial morphology and cytokine release plasticity: implications for signaling at GPR18. Front. Pharmacol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker RB, Poulton W, Clary G, Schriver M, Longo FM, 2016. Novel p75 neurotrophin receptor ligand stabilizes neuronal calcium, preserves mitochondrial movement and protects against HIV associated neuropathogenesis. Exp. Neurol 275 (Part 1), 182–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaluk P, Mikasova L, Groc L, Frischknecht R, Choquet D, Kaczmarek L, 2009. Matrix metalloproteinase-9 controls NMDA receptor surface diffusion through integrin beta1 signaling. J. Neurosci 29, 6007–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi H, Takuma K, Fukuzaki E, Ibi D, Someya E, Akazawa K-H, Alkam T, Tsunekawa H, Mouri A, Noda Y, Nabeshima T, Yamada K, 2009. Matrix metalloprotease-9 inhibition improves amyloid β-mediated cognitive impairment and neurotoxicity in mice. J. Pharmacol. Exp. Ther 331, 14. [DOI] [PubMed] [Google Scholar]
- More SV, Choi D-K, 2015. Promising cannabinoid-based therapies for Parkinson’s disease: motor symptoms to neuroprotection. Mol. Neurodegener 10, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison H, Young K, Qureshi M, Rowe RK, Lifshitz J, 2017. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci. Rep 7, 13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkatti P, Pandey R, Rieder SA, Hegde VL, Nagarkatti M, 2009. Cannabinoids as novel anti-inflammatory drugs. Future Med. Chem 1, 1333–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO, 1999. Transient exposure to HIV-1 tat protein results in cytokine production in macrophages and astrocytes: a hit and run phenomenon. J. Biol. Chem 274, 17098–17102. [DOI] [PubMed] [Google Scholar]
- Nicoli F., Finessi V., Sicurella M., Rizzotto L., Gallerani E., Destro F., Cafaro A., Marconi P., Caputo A., Ensoli B., Gavioli R., 2013. The HIV-1 Tat protein induces the activation of CD8+ T cells and affects in vivo the magnitude and kinetics of antiviral responses. PLoS One 8, e77746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AMM, Bading H, Mauceri D, 2014. Dysfunction of neuronal calcium signaling in aging and disease. Cell Tissue Res. 357, 381–383. [DOI] [PubMed] [Google Scholar]
- Park I-W, Wang J-F, Groopman JE, 2001. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood 97, 352–358. [DOI] [PubMed] [Google Scholar]
- Perry VH, Teeling J, 2013. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol 35, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SW, Barbieri J, Tong N, Polesskaya O, Pudasaini S, Stout A, Lu R, Kiebala M, Maggirwar SB, Gelbard HA, 2010. Human immunodeficiency virus- 1 Tat activates calpain proteases via the ryanodine receptor to enhance surface dopamine transporter levels and increase transporter-specific uptake and Vmax. J. Neurosci 30, 14153–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast MA, Rogers DT, Mulholland PJ, Littleton JM, Wilkins LH Jr., Self RL, Nath A, 2002. Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-D-aspartate receptor. Brain Res. 954, 300–307. [DOI] [PubMed] [Google Scholar]
- Presley C, Abidi A, Suryawanshi S, Mustafa S, Meibohm B, Moore BM, 2015. Preclinical evaluation of SMM-189, a cannabinoid receptor 2-specific inverse agonist. Pharmacol. Res. Perspect 3, e00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce G, Ahmed Z, Hankey DJ, Jackson SJ, Croxford JL, Pocock JM, Ledent C, Petzold A, Thompson AJ, Giovannoni G, Cuzner ML, Baker D, 2003. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 126, 2191–2202. [DOI] [PubMed] [Google Scholar]
- Rao VR, Ruiz AP, Prasad VR, 2014. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res. Ther 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybuck J, Hargus N, Thayer S, 2017. A GluN2B-selective NMDAR antagonist reverses synapse loss and cognitive impairment produced by the HIV-1 protein tat. J. Neurosci 37, 7837–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard SM, Razak K, Ethell IM, 2015. A delicate balance: role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders. Front. Cell. Neurosci 9, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel V, Atzler K, Behrenswerth A, Karcz T, Schoeder C, Hinz S, Kaleta M, Thimm D, Kiec-Kononowicz K, Müller CE, 2014. Bicyclic imidazole-4-one derivatives: a new class of antagonists for the orphan G protein-coupled receptors GPR18 and GPR55. MedChemComm 5, 632–649. [Google Scholar]
- Reyes-Resina I, Navarro G, Aguinaga D, Canela EI, Schoeder CT, Zaluski M, Kiec-Kononowicz K, Saura CA, Muller CE, Franco R, 2018. Molecular and functional interaction between GPR18 and cannabinoid CB2 G-protein-coupled receptors. Relevance in neurodegenerative diseases. Biochem. Pharmacol 157, 169–179. [DOI] [PubMed] [Google Scholar]
- Rivera P, Fernández-Arjona MDM, Silva-Peña D, Blanco E, Vargas A, López-Ávalos MD, Grondona JM, Serrano A, Pavón FJ, Rodríguez de Fonseca F, Suárez J, 2018. Pharmacological blockade of fatty acid amide hydrolase (FAAH) by URB597 improves memory and changes the phenotype of hippocampal microglia despite ethanol exposure. Biochem. Pharmacol 157, 244–257. [DOI] [PubMed] [Google Scholar]
- Roscoe RF Jr., Mactutus CF, Booze RM, 2014. HIV-1 transgenic female rat: synaptodendritic alterations of medium spiny neurons in the nucleus accumbens. J. NeuroImmune Pharmacol 9, 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GR, Lichtman A, Dewey WL, Akbarali HI, 2012. Evidence for the putative cannabinoid receptor (GPR55)-mediated inhibitory effects on intestinal contractility in mice. Pharmacology 90, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba SW, Jauch H, Gargouri B, Keil A, Hurrle T, Volz N, Mohr F, van der Stelt M, Bräse S, Fiebich BL, 2018. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J. Neuroinflammation 15, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sama DM, Norris CM, 2013. Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev 12, 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng WS, Hu S, Hegg CC, Thayer SA, Peterson PK, 2000. Activation of human microglial cells by HIV-1 gp41 and Tat proteins. Clin. Immunol 96, 243–251. [DOI] [PubMed] [Google Scholar]
- Stawarski M, Stefaniuk M, Wlodarczyk J, 2014. Matrix metalloproteinase-9 involvement in the structural plasticity of dendritic spines. Front. Neuroanat 8, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, 2009. Endocannabinoid signaling in microglial cells. Neuropharmacology 56 (Suppl. 1), 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, 2010. Neurotoxicity by microglia: mechanisms and potential therapeutic strategy. Clin. Exp. Neuroimmunol 1, 12–21. [Google Scholar]
- Tchantchou F, Tucker LB, Fu AH, Bluett RJ, McCabe JT, Patel S, Zhang Y, 2014. The fatty acid amide hydrolase inhibitor PF-3845 promotes neuronal survival, attenuates inflammation and improves functional recovery in mice with traumatic brain injury. Neuropharmacology 85, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham CS, Whitaker J, Luo L, Webb M, 2007. Inhibition of microglial fatty acid amide hydrolase modulates LPS stimulated release of inflammatory mediators. FEBS Lett. 581, 2899–2904. [DOI] [PubMed] [Google Scholar]
- Thangaraj A, Periyasamy P, Liao K, Bendi VS, Callen S, Pendyala G, Buch S, 2018. HIV-1 TAT-mediated microglial activation: role of mitochondrial dysfunction and defective mitophagy. Autophagy 14, 1596–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman R, Burm SM, Bajramovic JJ, 2018. An overview of in vitro methods to study microglia. Front. Cell. Neurosci 12, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar M, Keller JN, Chan S, Mattson MP, Marques MA, Crutcher KA, 1999. Truncated apolipoprotein E (ApoE) causes increased intracellular calcium and may mediate ApoE neurotoxicity. J. Neurosci 19, 7100–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma M, Wills Z, Chu CT, 2018. Excitatory dendritic mitochondrial calcium toxicity: implications for Parkinson’s and other neurodegenerative diseases. Front. Neurosci 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Moller T, Stella N, 2002. Astrocytes in culture produce anandamide and other acylethanolamides. J. Biol. Chem 277, 20869–20876. [DOI] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N, 2003. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci 23, 1398–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster NL, Crowe SM, 2006. Matrix metalloproteinases, their production by monocytes and macrophages and their potential role in HIV-related diseases. J. Leukoc. Biol 80, 1052–1066. [DOI] [PubMed] [Google Scholar]
- Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH, 1995. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375, 497–500. [DOI] [PubMed] [Google Scholar]
- Wojda U, Salinska E, Kuznicki J, 2008. Calcium ions in neuronal degeneration. IUBMB Life 60, 575–590. [DOI] [PubMed] [Google Scholar]
- Young K, Morrison H, 2018. Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using ImageJ. J. Vis. Exp 136, 57648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Benson DL, 2001. Stages of synapse development defined by dependence on F-actin. J. Neurosci 21, 5169–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Tang H, Liu J, Dong J, Xiong H, 2011. Chemokine CCL2 modulation of neuronal excitability and synaptic transmission in rat hippocampal slices. J. Neurochem 116, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Tang H, Xiong H, 2016. Chemokine CCL2 enhances NMDA receptor- mediated excitatory postsynaptic current in rat hippocampal slices-a potential mechanism for HIV-1-associated neuropathy? J. NeuroImmune Pharmacol 11, 306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zündorf G, Reiser G, 2011. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 14, 1275–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.