

Abstract
2-Aryl-2-fluoroacetonitriles have garnered increasing interest as versatile building blocks in asymmetric synthesis. However, the configurational stability of these organofluorines is poorly understood and analytical methods that can be used to differentiate between their enantiomers remain underdeveloped. In this study, baseline HPLC enantioseparation of ten 2-aryl-2-fluoroacetonitriles was achieved by screening frequently used chiral stationary phases. While Chiralcel OD, Chiralpak AD and Chiralpak AS proved to be most broadly useful, preparative separation of the enantiomers of 2-(2-naphthyl)-2-fluoroacetonitrile was possible on Chiralcel OJ. This enabled racemization studies at various temperatures and in the presence of organic bases which showed that this compound is configurationally stable under neutral conditions upon heating to 130 oC for 6 hours but undergoes complete racemization within 10 hours in the presence of stoichiometric amounts of a guanidine base at room temperature. The racemization is likely to proceed via formation of an achiral keteniminate intermediate and obeys reversible first order reaction kinetics with a half-life time of 87.7 minutes in ethanolic hexanes at 23.2 oC. Racemization is significantly slower and occurs with a half-life time of 23.1 hours at 22.4 °C when the guanidine is replaced with a weaker amidine base.
Keywords: Chiral nitriles, organofluorines, enantiomer separation, high performance liquid chromatography, chiral stationary phase, configurational stability, enantioconversion
Graphical Abstract
Introduction
The incorporation of fluorine atoms into biologically active compounds has become a widely recognized strategy to improve physicochemical properties and therapeutic activities of promising drug candidates or even marketed pharmaceuticals.1,2 As a result, small fluorinated chiral molecules are of considerable interest to synthetic and medicinal chemists who use them as versatile building blocks and precursors of important pharmacophores.3–4,5 To address this demand, a structurally diverse pool of commercially available or easily prepared chiral compounds exhibiting a carbon-fluorine bond among other functionalities has been introduced over the past few years. However, the unique stereochemical properties of organofluorines are often poorly understood and analytical tools that can be used to differentiate between their enantiomers remain elusive. The synthetic utility of 2-aryl-2-fluoroacetonitriles in asymmetric catalysis and stereodivergent synthesis, for example, was only recently discovered and little is known about the chemistry of chiral fluoronitriles.6–7,8 The increasing interest in this class of chiral organofluorines demands methods that allow separation of enantiomers and insights into their susceptibility to racemization which may assist in the discovery of dynamic kinetic resolution protocols and new asymmetric transformations.
Based on our longstanding interest in fluorinated compounds,7,9–10,11,12,13,14,15,16,17,18,19,20 we decided to screen a variety of chiral stationary phases (CSPs) to separate the enantiomers of the 2-aryl-2-fluoroacetonitriles 1-10 by liquid chromatography, Figure 1. It was expected that the development of HPLC conditions that allow baseline separation of 1-10 would not be a simple task and most likely be achieved with some of the most frequently used CSPs originally introduced by Pirkle and Okamoto who without a doubt have been the most influential inventors of broadly useful chiral HPLC columns.21–22,23,24,25,26,27 We therefore chose to test the brush-type Whelk-O 1, Chirex 3001 and 3014 CSPs as well as the polysaccharide derived Chiralcel OD, OJ and OA and Chiralpak AD and AS. We now wish to disclose the result of our chiral HPLC investigation which demonstrates that baseline enantioseparation of compounds 1-10 is possible. The column screening revealed that Okamoto’s cellulose and amylose derived CSPs, in particular Chiralcel OD, Chiralpak AD and Chiralpak AS, are superior choices when the task is to separate 2-aryl-2-fluoroacetonitriles into enantiomers. However, there is not a single column that is generally successful and our study shows that the screening of several CSPs as well as mobiles phase optimization are both necessary. The exceptional performance of Chiralcel OJ allowed preparative enantioseparation of 2-(2-naphthyl)-2-fluoroacetonitrile, 8, and racemization studies that were conducted by external heating of enantiopure samples in a closed vessel or in the presence of base and subsequent chiral HPLC analysis of the change in the enantiomeric composition.
Figure 1.
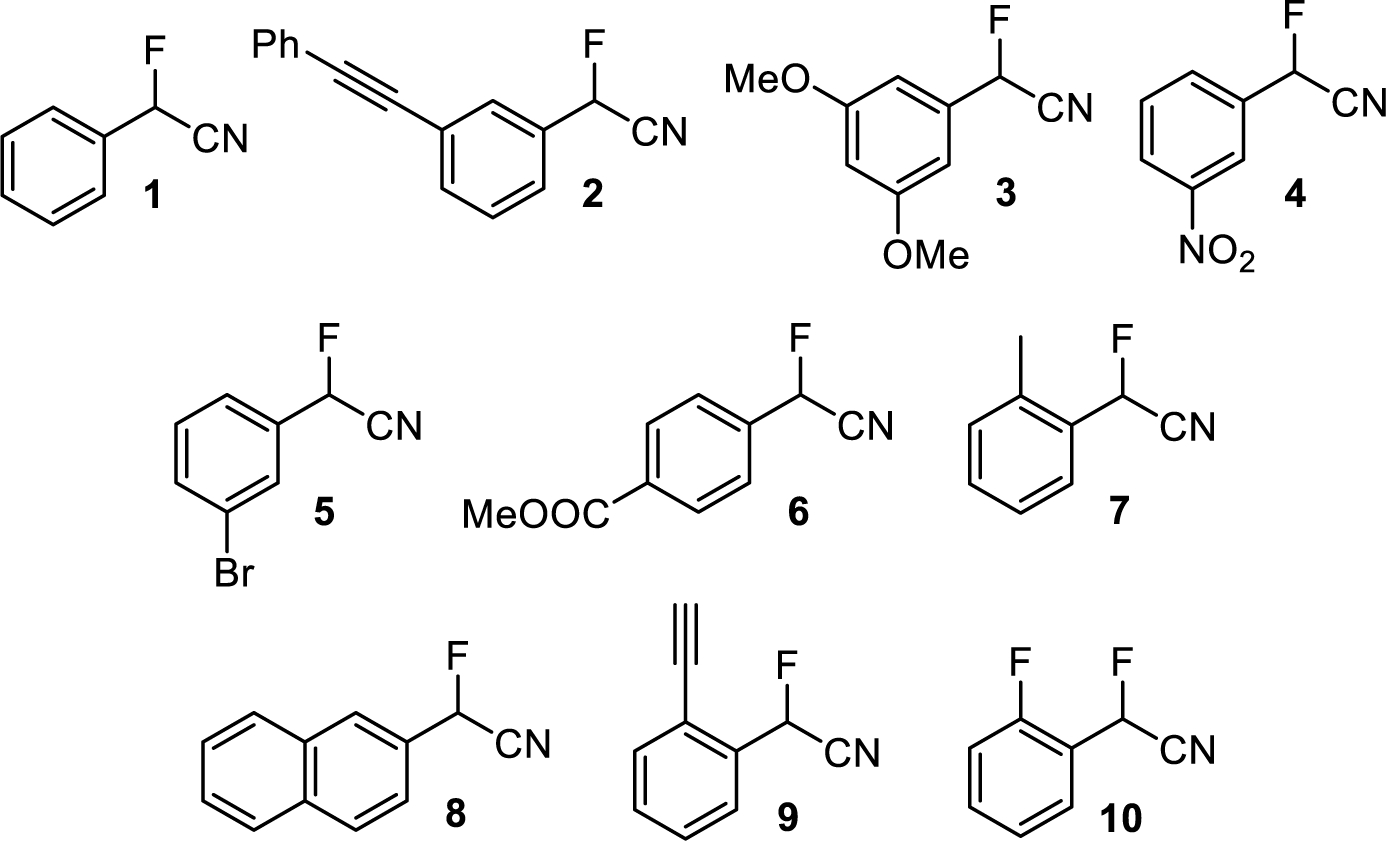
Structures of 2-aryl-2-fluoroacetonitriles investigated.
Materials and Methods
Chiral HPLC
HPLC enantioseparations were performed at room temperature with a flow rate of 1.0 mL/min using approximately 1 mg/mL solutions of the analytes dissolved in hexanes:ethanol mixtures. The chromatograms were recorded at 205, 214, 230, 254 and 280 nm with a diode array detector. The polarity of the mobile phase was adjusted to optimize peak separation and retention times. Only hexanes:ethanol mixtures were used as mobile phase for HPLC studies with Chiralcel and Chiralpak columns. The brush-type columns were tested with hexanes:ethanol and hexanes:dichloromethane mobile phases. The 2-aryl-2-fluoroacetonitriles 1-10 used in this study were available in sufficient amounts for analytical purposes from a previous study and the CSPs are commercially available.7 The Chiralcel OD, OJ and the Chiralpak AD and AS columns used were coated on 10 μm particles. The enantioselectivity, α, column efficiency, N, and resolution, R, were determined according to equations 1–3. The column efficiency values were averaged for both peaks. A solution of 1,3,5-tri-tert-butylbenzene dissolved in hexanes:EtOH (95:5 v/v, 1.0 mg/mL) was used to determine t0 for all columns with hexanes:EtOH (95:5 v/v) as mobile phase.
| Eq 1 |
| Eq 2 |
| Eq 3 |
where t0 is the void, tR2 and tR1 are the retention times of the enantiomer peaks, W0.5 is the peak width at half height.
Synthesis of 2-(2-Naphthyl)-2-fluoroacetonitrile
For preparative enantioseparation and racemization studies, compound 8 was prepared from aldehyde 11 by cyanide addition and subsequent deoxyfluorination of 12 with DAST (diethylaminosulfur trifluoride) following a previously reported protocol, Figure 2.7,28,29 Flash chromatographic purification gave 8 as a pale yellow powder in 50% overall yield. 1H NMR (400 MHz, CDCl3) δ 8.02 (s, J = 2.6 Hz, 1H), 7.97 – 7.85 (m, 3H), 7.64 – 7.52 (m, 3H), 6.20 (d, J = 47.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 134.3, 132.7, 129.6, 128.5, 128.5 (JC-F = 20.5 Hz), 128.0, 127.9, 127.2, 123.6, 115.4 (d, JC-F = 33.7 Hz), 80.4 (d, JC-F = 181.4 Hz). 19F NMR (376 MHz, CDCl3) δ −166.86 (d, J = 47.1 Hz).
Figure 2.
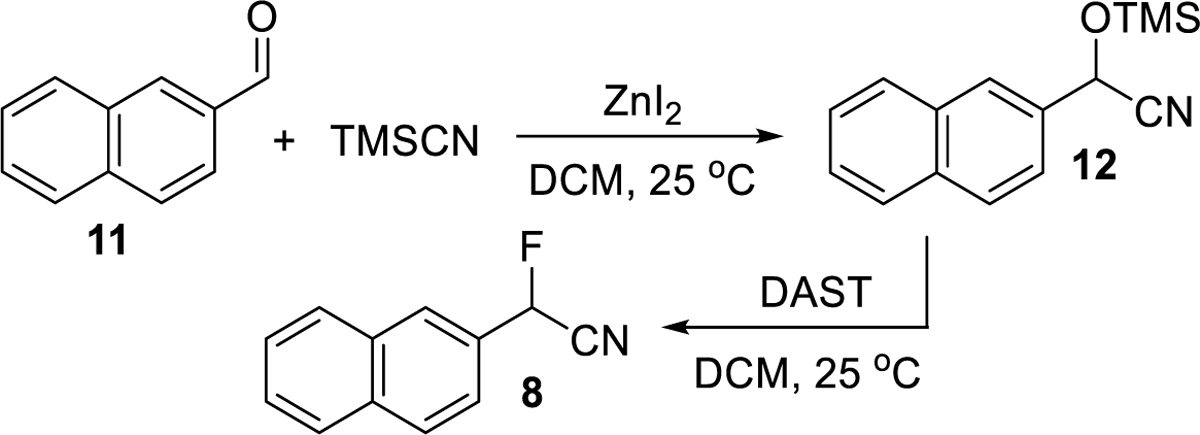
Synthesis of 8.
Racemization Study
The enantiomers of 2-(2-naphthyl)-2-fluoroacetonitrile were isolated by chiral HPLC on Chiralcel OJ using a mobile phase of 90:10 hexanes:ethanol, a flow rate of 1 mL/min, and a sample containing racemic 8 at a concentration of 5.40 mM in 95:5 hexanes:ethanol. The enantiopurity of the collected fractions was verified with the same chiral HPLC method. The solutions containing the preparatively separated enantiomers in hexanes:ethanol solution (36.0 μM to 0.21 mM, 90:10, v/v) were directly applied in racemization experiments at varying temperatures and in the presence of base additives. A closed glass vessel designed for high pressure experiments and equipped with a Teflon seal was used to avoid solvent and base loss during heating. The racemization course of heated samples was monitored with reaction aliquots cooled to room temperature without further dilution prior to injection on the chiral HPLC column using the method described above. The racemization of enantiopure samples of 2-(2-naphthyl)-2-fluoroacetonitrile in the presence of one equivalent of Barton’s base (BTMG, 2-tert-butyl-1,1,3,3-tetramethylguanidine) and DBU (1,8-diazabicyclo[5.4. 0]undec-7-ene), respectively, in hexanes:EtOH (95:5 v/v) at room temperature was monitored by analyzing 50 μL aliquots at different time intervals and the half-life time was calculated according to reversible first order kinetics described in Equation 4. To assure accurate and reproducible %ee measurements, a linearity study of the UV signals at 205, 214, 230, 254 and 280 nm and blank injections to eliminate the chance of sample carryovers from previous injections were conducted, see Supporting Information for details.
| Eq 4 |
Results and Discussion
Chiral HPLC Development
As mentioned above, it was expected that HPLC enantioseparation of 1-10 will require the screening of a variety of chiral stationary phases and subsequent mobile phase optimization. Arylfluoronitriles do not form strong hydrogen bonds which often play a major role in the formation of thermodynamically distinct, transient diastereomeric adducts, in particular when the Whelk-O 1 and similar brush-type CSPs are used. We therefore anticipated that polysaccharide CSPs would be more suitable for this class of compounds. The structures of the eight CSPs (Whelk-O 1, Chirex 3001 and 3014, Chiralcel OD, OJ and OA, Chiralpak AD and AS) selected for this study are shown in Figure 3. All columns are frequently used in numerous laboratories and have proven quite successful for the separation of the enantiomers of a large variety of chiral compounds. To evaluate the usefulness of these CSPs for the separation of the enantiomers of 1-10 we chose to use hexanes:ethanol mixtures as the mobile phase. The polarity of the mobile phase was adjusted to allow sufficient analyte retention on the column.
Figure 3.
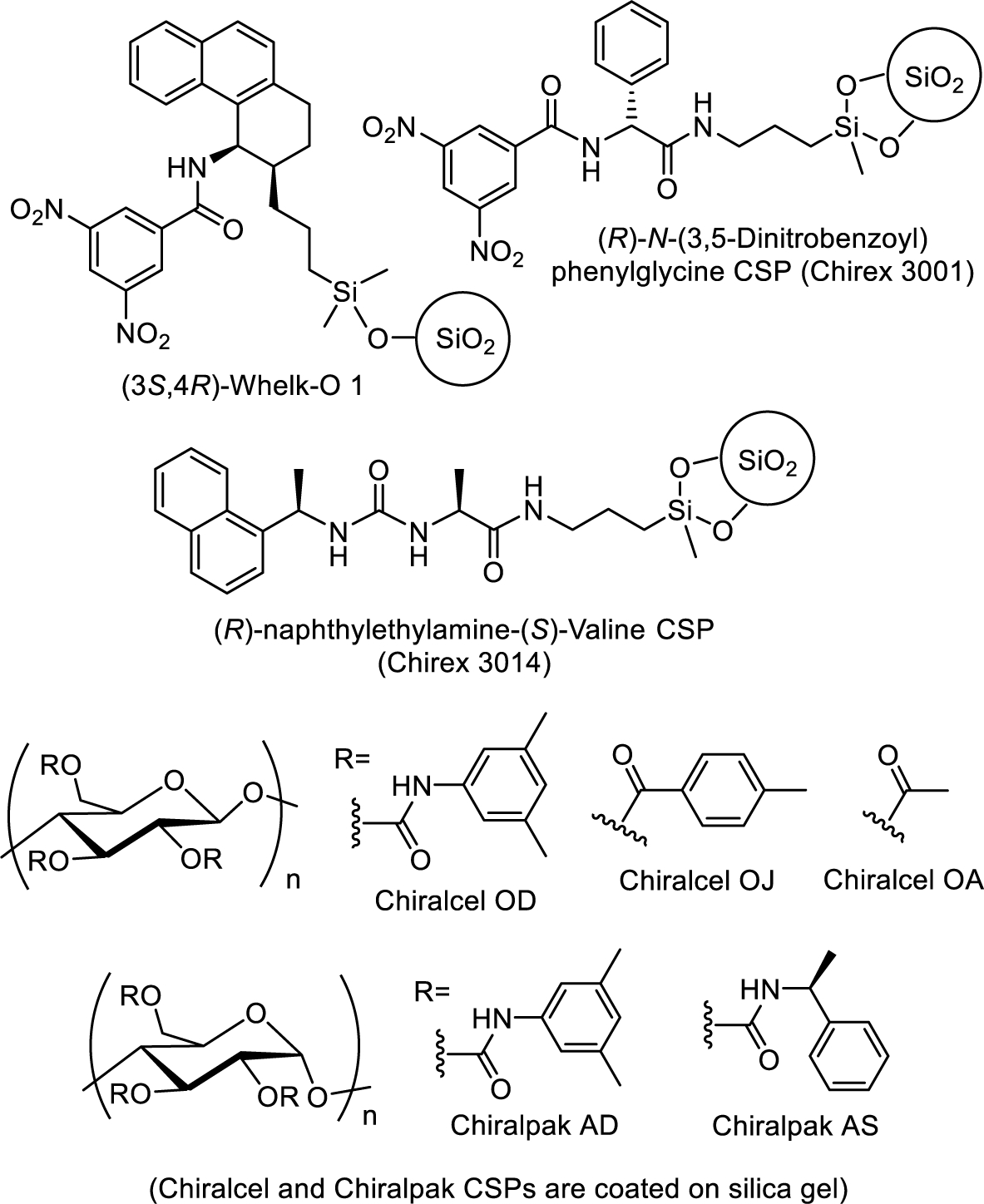
Structures of chiral stationary phases tested.
We found that Chiralcel OD allows full separation of the enantiomers of the nitriles 3, 5, 6 and 8 whereas only partial resolutions were achieved with 9 and 10. Despite the considerable structural similarity, there was no sign of enantioselectivity of this CSP for 1, 4 and 7. These results emphasized the potential of polysaccharide phases for HPLC enantioseparation of the 2-aryl-2-fluoroacetonitriles 1-10 but also indicated that satisfactory resolution of all ten compounds would require a trial-and-error screening process. We therefore continued with Chiralcel OJ which successfully separated the enantiomers of 2 and 8 but only partial separations were possible for 5, 7 and 10. We then discovered that the most broadly applicable CSPs are the amylose derivatives Chiralpak AD and AS. The nitriles 2, 3, 4, 6, 7 and 10 were well resolved on Chiralpak AD and partial separations were observed for 5, 8 and 9. The somewhat complementary performance of Chiralcel OD and Chiralpak AD was encouraging and we were delighted to see that all compounds except 2 can be fully resolved into the enantiomers on the Chiralpak AS column. By contrast, Chiralcel OA was significantly less useful and we observed baseline separation only in the case of 8 while 3 and 6 were partially separated into enantiomers.
Screening of the Whelk-O 1 and Chirex columns showed no sign of resolution when hexanes:ethanol mixtures were used as the mobile phase. To favor hydrogen bonding interactions between 1-10 and the three brush-type CSPs we also investigated hexanes:dichloromethane mobile phase compositions. This uncovered, however, only a partial separation of 8 on the Whelk-O 1 column. Selected chromatograms showing baseline enantioseparations of analytes 4, 6 and 10 are shown in Figure 4 and all separations including calculated enantioselectivity, α, column efficiency, N, and resolution, R, values can be found in the Supporting Information. Although the screening of eight CSPs did not yield a single column that can separate all ten analytes into enantiomers, we found that this is possible with a combination of Chiralcel and Chiralpak columns. The utility of the CSPs tested is illustrated with an enantioseparation heat map provided in Table 1.
Figure 4.
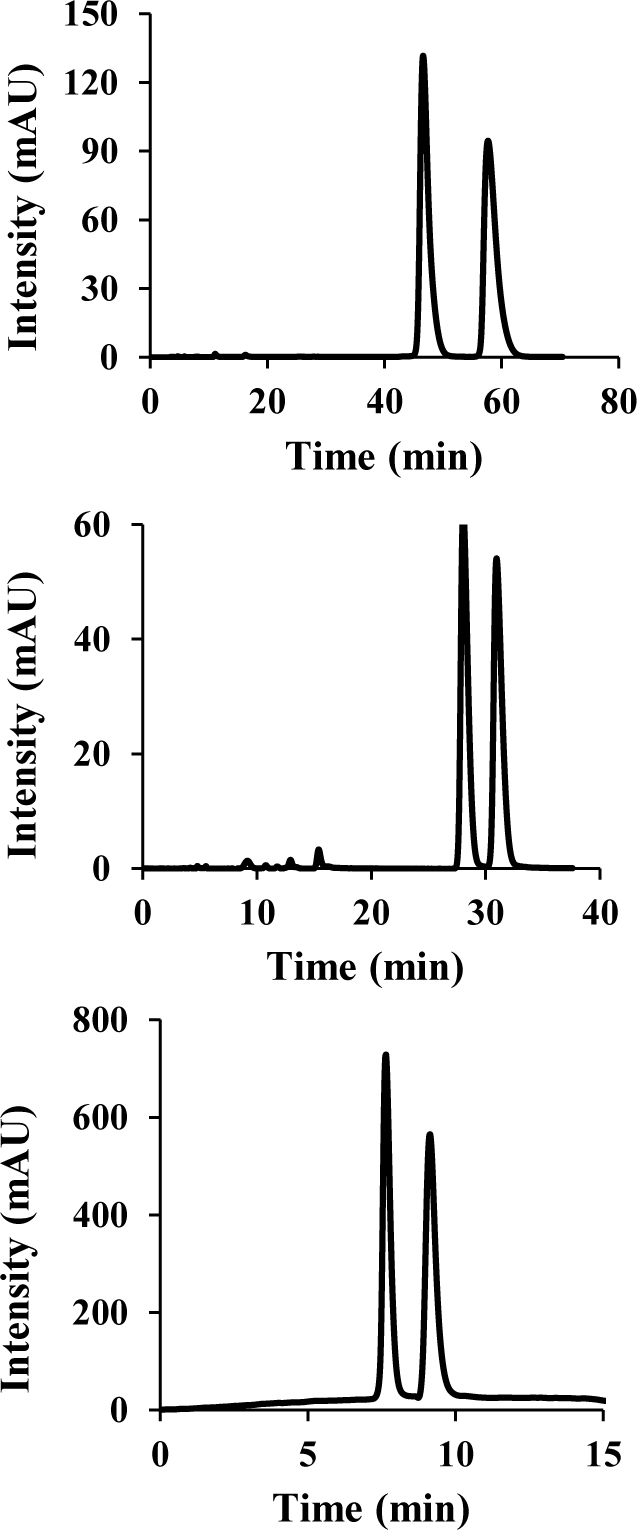
Representative HPLC chromatograms. Top: Resolution of 4 on Chiralpak AS (α = 1.26, N = 4440, Rs =3.48). Middle: Enantioseparation of 6 on Chiralpak AD (α = 1.12, N = 9050, Rs = 2.33). Bottom: Separation of 10 on Chiralpak AS (α = 1.35, N = 4560, Rs = 3.00). See SI for details.
Table 1.
Enantioseparation heat map.
| Column | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Chiralcel OD | A | A | A | A | A, B | A, B | |||||
| Chiralcel OJ | C | E | C | E | E | ||||||
| Chiralpak AD | A | A | A, C | A, B | A, C | A, B | A | A | A | ||
| Chiralpak AS | A, B | A | A, C | A | A | A | A | A | A, B | ||
| Chiralcel OA | D | D | D | F | |||||||
| Whelk-O 1 | G | ||||||||||
| Chirex 3001 | |||||||||||
| Chirex 3014 | |||||||||||
Green: Baseline or almost complete enantioseparation; Yellow: Partial separation, Red: No separation. Mobile phase compositions: A = 99:1 Hex:EtOH; B = 99.5:0.5 Hex:EtOH; C = 95:5 Hex:EtOH; D = 98:2 Hex:EtOH; E = 90:10 Hex:EtOH; F = 80:20 Hex:EtOH; G = 90:10 Hex:CH2Cl2.
Racemization study with compound 8
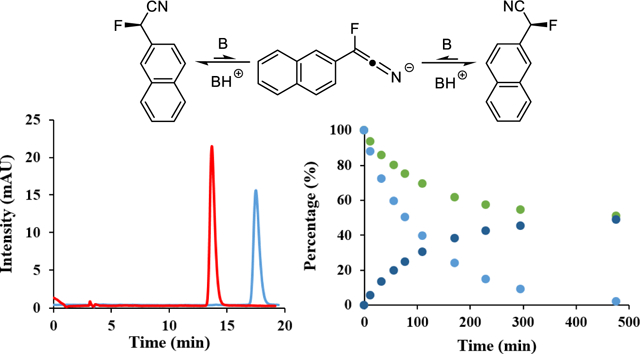
The excellent separation of the enantiomers of 2-(2-naphthyl)-2-fluoroacetonitrile, 8, on Chiralcel OJ allowed preparative isolation of enantiopure samples that were applied in racemization studies, Figure 5. We found that 2-fluoro-2-arylacetonitriles are configurationally stable under neutral conditions even at high temperatures. In the absence of base, we observed no sign of racemization with an enantiopure sample of 8 that was dissolved in hexanes:ethanol (95:5 v/v) and heated to 130 oC in a closed vessel for 6 hours. The addition of an amine base did not result in enantioconversion at room or ambient temperatures but slow racemization toward approximately 80 %ee occurred after heating a sample containing diisopropylethylamine (5% v/v) to 100 oC for 19 hours. We finally resorted to using a stronger base and discovered that complete racemization of 8 can be achieved in the presence of 0.1% (v/v) of Barton’s base at room temperature in just 10 minutes. These results are consistent with a racemization course via an achiral keteniminate intermediate as shown in Figure 5. Having established that guanidines cause enantioconversion under mild conditions we were able to set up a racemization experiment with stoichiometric amounts of Barton’s base. A solution of enantiopure 8 and BTMG (each 0.20 mM, hexanes:ethanol 95:5, v/v) was stirred at 23.2 oC and small aliquots were analyzed over time with our chiral HPLC method. We observed an exponential decline in the sample %ee which proved to obey reversible first order reaction kinetics. The racemization half-life under these conditions was determined as 87.7 minutes. When we used a stoichiometric amount of the weaker base DBU under otherwise similar conditions (0.14 mM in hexanes:ethanol (95:5), 22.4 °C) the racemization was significantly slower and we determined a half-life time of 23.1 hours.
Figure 5.
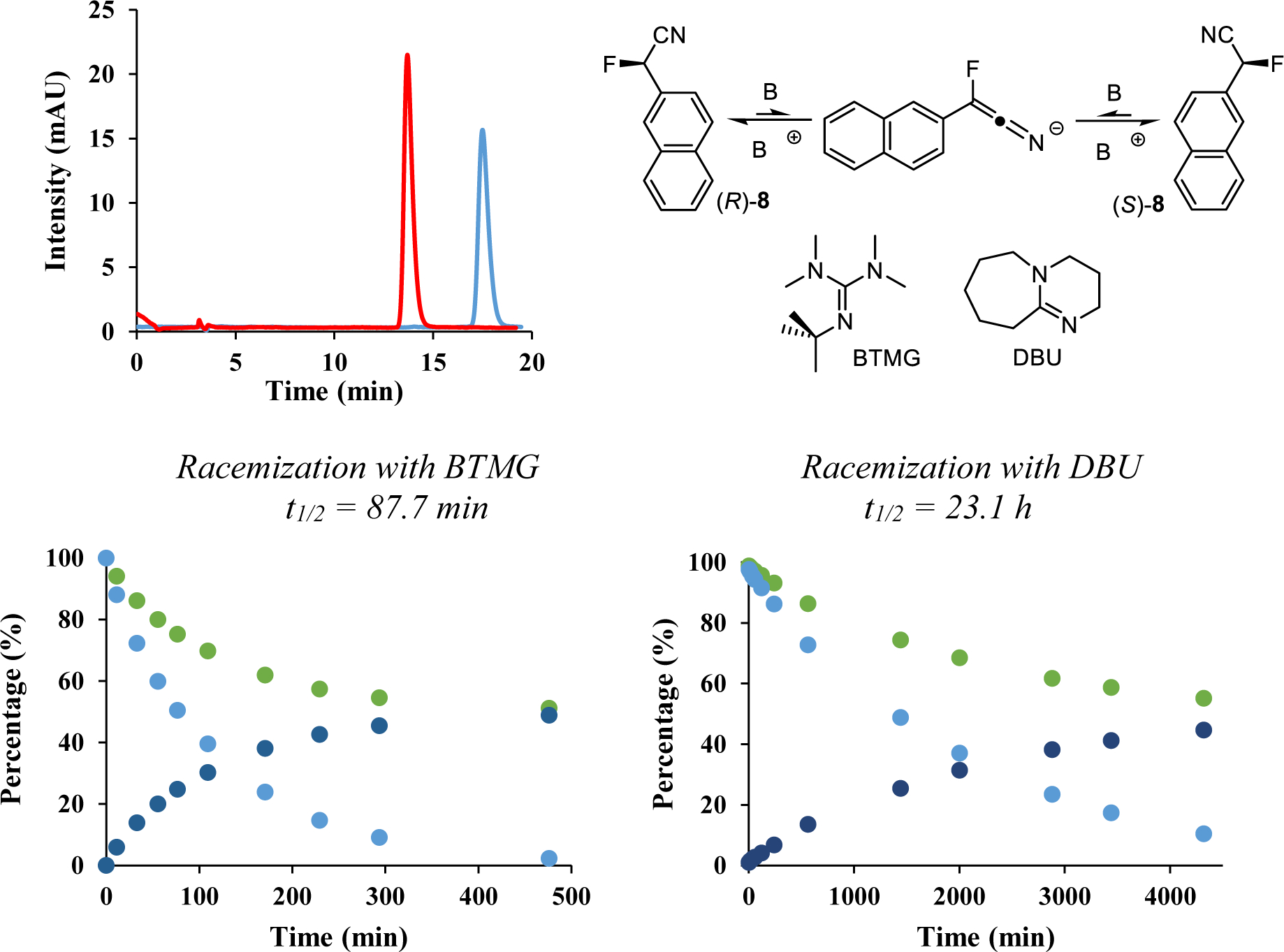
Racemization study. Top: Overlaid HPLC chromatograms of the isolated enantiomers of 8 and base promoted racemization mechanism. Bottom: Change in the enantiomeric composition of 8 in the presence equimolar amounts of BTMG and DBU (enantiomer percentages are shown in green and dark blue, %ee in light blue). See SI for details.
Conclusion
In summary, baseline HPLC enantioseparation of ten 2-aryl-2-fluoroacetonitriles was achieved by screening eight frequently used polysaccharide and brush-type chiral stationary phases. Okamoto’s cellulose and amylose derived columns, in particular Chiralcel OD, Chiralpak AD and Chiralpak AS, were found to be superior choices for this task. Preparative separation of the enantiomers of 2-(2-naphthyl)-2-fluoroacetonitrile on Chiralcel OJ enabled racemization studies that were performed at various temperatures and with amine, amidine or guanidine bases. It was found that this compound is configurationally stable under neutral conditions even upon heating to 130 oC but it racemizes within several hours in the presence of stoichiometric amounts of a guanidine base at room temperature. The enantioconversion obeys reversible first order reaction kinetics and the racemization half-life time was determined as 87.7 minutes using a 0.20 mM solution of enantiopure 2-(2-naphthyl)-2-fluoroacetonitrile and BTMG in hexanes:ethanol (95:5 v/v) at 23.2 oC. Racemization was found to be significantly slower in the presence of one equivalent of DBU (0.14 mM in hexanes:ethanol (95:5), 22.4 °C) and a half-life time of 23.1 hours was obtained under these conditions.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the U.S. National Institutes of Health (GM106260) and the U.S. National Science Foundation (CHE1764135).
Footnotes
Supporting Information
Additional supporting information including HPLC chromatograms may be found in the online version of this article at the publisher’s website.
REFERENCES AND NOTES
- 1.Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Acena JL, Soloshonok VA, Izawa K, Liu H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016;116: 422–518. [DOI] [PubMed] [Google Scholar]
- 2.Smith BR, Eastman CM, Njardarson JT. Beyond C, H, O, and N Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem. 2014;57:9764–9773. [DOI] [PubMed] [Google Scholar]
- 3.Liang T, Neumann CN, Ritter T. Introduction of Fluorine-containing Functional Groups. Angew. Chem. Int. Ed. 2013;52:8214–8264. [DOI] [PubMed] [Google Scholar]
- 4.Yang X, Wu T, Phipps RJ, Toste FD. Advances in Catalytic Enantioselective Fluorination, Mono‑, Di‑, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015;115:826–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu Y, Han J, Wang J, Shibata N, Sodeoka M, Soloshonok VA, Coelho JAS, Toste FD. Modern Approaches for Asymmetric Construction of Carbon–Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018;118:3887–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balaji PV, Brewitz L, Kumagai N, Shibasaki M. Achiral Trisubstituted Thioureas as Secondary Ligands to CuI Catalysts: Direct Catalytic Asymmetric Addition of α-Fluoronitriles to Imines. Angew. Chem. Int. Ed. 2019;58:2644–2648. [DOI] [PubMed] [Google Scholar]
- 7.Ding R, De los Santos ZA, Wolf C. Catalytic Asymmetric Mannich Reaction of α-Fluoronitriles with Ketimines: Enantioselective and Diastereodivergent Construction of Vicinal Tetrasubstituted Stereocenters. ACS Catal. 2019;9:2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balaji PV, Li Z, Saito A, Kumagai N, Shibasaki M. Direct Catalytic Asymmetric Addition of a-Fluoronitriles to Aldehydes. Chem. Eur. J. 2020;26:15524–15527. [DOI] [PubMed] [Google Scholar]
- 9.Xu H, Wolf C. Synthesis of Chiral Tertiary Trifluoromethyl Alcohols by Asymmetric Nitroaldol Reaction with a Cu(II)-Bisoxazolidine Catalyst. Chem. Commun. 2010;46:8026–8028. [DOI] [PubMed] [Google Scholar]
- 10.Wolf C, Zhang P. Asymmetric Friedel-Crafts Reaction of Indoles with Ethyl Trifluoropyruvate Using a Copper(I)-Bisoxazolidine Catalyst. Adv. Synth. Catal. 2011;353:760–766. [Google Scholar]
- 11.Zhang P, Wolf C. Catalytic Enantioselective Difluoroalkylation of Aldehydes. Angew. Chem. Int. Ed. 2013;52:7869–7873. [DOI] [PubMed] [Google Scholar]
- 12.Balaraman K, Moskowitz M, Liu Y, Wolf C. Detrifluoroacetylative Generation of Halogenated Enolates: Practical Access to Perhalogenated Ketones and Alkenes. Synthesis 2016;2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding R, Wolf C. Catalytic Insertion of Aldehydes into Dihalonitroacetophenones via Sequential Bond Scission-Aldol Reaction-Acyl Transfer. Chem. Commun. 2016;52:3576–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cook AM, Wolf C. Efficient Access to Multifunctional Trifluoromethyl Alcohols through Base-Free Catalytic Asymmetric C−C Bond Formation with Terminal Ynamides. Angew. Chem. Int. Ed. 2016;55:2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding R, Bakhshi PR, Wolf C. Organocatalytic Insertion of Isatins into Aryl Difluoronitromethyl Ketones. J. Org. Chem. 2017;82:1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balaraman K, Wolf C. Catalytic Enantioselective and Diastereoselective Allylic Alkylation with Fluoroenolates: Efficient Access to C3-Fluorinated and All-Carbon Quaternary Oxindoles. Angew. Chem. Int. Ed. 2017;56:1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balaraman K, Ding R, Wolf C. Stereoselective Synthesis of 3,3’-Bisindolines by Organocatalytic Michael Additions of Fluorooxindole Enolates to Isatylidene Malononitriles in Aqueous Solution. Adv. Synth. Catal. 2017;359:4165–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding R, Wolf C. Organocatalytic Asymmetric Synthesis of α-Oxetanyl and α-Azetidinyl Tertiary Alkyl Fluorides and Chlorides. Org. Lett. 2018;20:892–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moskowitz M, Balaraman K, Wolf C. Organocatalytic Stereoselective Synthesis of Fluorinated 3,3’-Linked Bisoxindoles. J. Org. Chem. 2018;83:1661–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balaraman K, Moskowitz M, Wolf C. Organocatalytic Decarboxylative Cyanomethylation of Difluoromethyl and Trifluoromethyl Ketones. Adv. Synth. Catal. 2018;360:4705–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pirkle WH, Welch CJ, Lamm B. Design, Synthesis, and Evaluation of an Improved Enantioselective Naproxen Selector. J. Org. Chem. 1992;57:3854–3860. [Google Scholar]
- 22.Okamoto Y, Kaida Y. Resolution by High-performance Liquid Chromatography Using Polysaccharide Carbamates and Benzoates as Chiral Stationary Phases. J. Chromatogr. A. 1994;666:403–419. [Google Scholar]
- 23.Okamoto Y, Yashima E. Polysaccharide Derivatives for Chromatographic Separation of Enantiomers. Angew. Chem., Int. Ed. 1998;37:1021–1043. [DOI] [PubMed] [Google Scholar]
- 24.Wolf C Dynamic Stereochemistry of Chiral Compounds, RSC Publishing, Cambridge, 2008, pp. 136–179. [Google Scholar]
- 25.Wolf C, Pirkle WH. Synthesis and Evaluation of a Copolymeric Chiral Stationary Phase. J. Chromatogr. A 1998;799:177–184. [Google Scholar]
- 26.Welch CJ. Evolution of Chiral Stationary Phase Design in the Pirkle Laboratories. J. Chromatogr. A 1994;666:3–26. [Google Scholar]
- 27.Okamoto Y, Ikai T. Chiral HPLC for Efficient Resolution of Enantiomers. Chem. Soc. Rev. 2008;37:2593–2608. [DOI] [PubMed] [Google Scholar]
- 28.LeTourneau ME, McCarthy JR. A Novel Synthesis of α-Fluoroacetonitriles. Application to A Convenient Preparation of 2-Fluoro-2-phenethylamines. Tetrahedron Lett. 1984;46:5227–5230. [Google Scholar]
- 29.Venkatachalam TK, Uckun FM. Synthesis of β-Fluorophenethyl Halopyridyl Thiourea Compounds as Non-nucleoside Inhibitors of HIV-1 Reverse Transcriptase. Synth. Commun. 2004;13:2463–2472. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.