

Abstract
Hepatocarcinogenesis is a complex multistep biological process involving genetic and epigenetic alterations that are accompanied by activation of oncoproteins and inactivation of tumor suppressors, which in turn results in Hepatocellular carcinoma (HCC), one of the common tumors with high morbidity and mortality worldwide. The ubiquitin-proteasome system (UPS) is the key to protein degradation and regulation of physiological and pathological processes, and E3 ligases are key enzymes in the UPS that contain a variety of subfamily proteins involved in the regulation of some common signal pathways in HCC. There is growing evidence that many structural or functional dysfunctions of E3 are engaged in the development and progression of HCC. Herein, we review recent research advances in HCC-associated E3 ligases, describe their structure, classification, functional roles, and discuss some mechanisms of the abnormal activation or inactivation of the HCC-associated signal pathway due to the binding of E3 to known substrates. In addition, given the success of proteasome inhibitors in the treatment of malignant cancers, we characterize the current knowledge and future prospects for targeted therapies against aberrant E3 in HCC.
Keywords: E3 ubiquitin ligase, hepatocellular carcinoma, E3-targeting therapy
Introduction
Hepatocellular carcinoma (HCC) accounts for 7% of all cancers worldwide and is the most common primary liver cancer [1]. It is an aggressive tumor with a poor prognosis and is the second leading cause of cancer-related death [2]. The most common cause of HCC is hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, which accounts for more than 90% of HCC cases in developing countries and nearly half of the cases in developed countries [3]. Other risk factors include aflatoxin, alcoholic liver disease, nonalcoholic fatty liver, autoimmune hepatitis, obesity, and diabetes mellitus [3]. Current treatment options, including liver transplantation or surgical resection, can only partially cure patients with early-stage HCC [4]. However, mainly due to the lack of symptoms and effective screening strategies for HCC, 80% of patients are diagnosed with advanced HCC [5]. Due to limited treatment options, broad-spectrum conventional cytotoxic drugs are often used, especially when surgical resection is not possible [6]. Unfortunately, HCC is also highly resistant to conventional chemotherapy using cytotoxic agents [6]. HCC tumorigenesis is a complex multistep biological process involving genetic and epigenetic alterations [7]. These alterations lead to structural activation of oncogenic signal pathways or dysregulation of key pathways that regulate tumor suppressor activity [7]. Several different signal pathways have been identified as responsible for initiating and promoting HCC, such as Wnt/β-catenin pathway, PI3K/AKT/mTOR pathway, RAS/RAF/MEK/ERK pathway, Hippo pathway, Keap1/Nrf2 pathway, HIF-1α pathway, TGF-β/SMAD pathway, Notch pathway and NF-κB pathway [7,8]. These pathways are accomplished by complex, multi-protein and highly precisely regulated molecules that are subject to post-translational modifications such as ubiquitination, phosphorylation and acetylation.
The ubiquitin-proteasome system (UPS) is a common post-translational modification pathway responsible for the degradation of approximately more than 80% of intracellular proteins in normal and pathological states [9]. Ubiquitin, which is highly conserved in eukaryotic cells, is a modified molecule consisting of 76 amino acids that covalently binds and labels target substrates through a cascade reaction of ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2) and ubiquitin-protein ligase (E3) [10]. Subsequently, the modified substrate is recognized by the 26S proteasome complex for ubiquitination-mediated degradation (Figure 1A) [10]. Under physiological conditions, UPS-mediated protein hydrolysis removes misfolded, damaged or excess proteins, which is important for maintaining protein homeostasis [11]. Besides, UPS plays a critical role in the regulation of cell cycle and apoptosis [12-14]. In addition, non-proteasome ubiquitin also exerts an important role in many cellular processes, such as signal transduction, DNA repair, and autophagy (Figure 1A) [15,16]. More importantly, abnormalities in UPS-related proteins have been reported to be closely associated with various human cancers in pathological situations [17].
Figure 1.
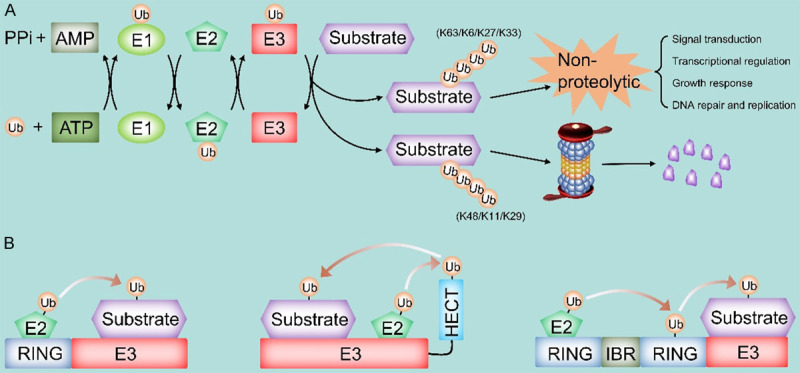
A. Overview of the ubiquitin-proteasome system and the E3 ligase family. Ubiquitin attaches to proteins through a cascade reaction involving E1, E2 and E3 ligases. E3s attaches ubiquitin through specific lysine residues, which determine the fate of the protein after ubiquitin coupling. Among them, K48, K11 and K29 mainly mediate the degradation of substrates via 26s proteasome pathway; while K63, K6, K27 and K33 are primarily involved in the regulation of some cellular processes such as signal transduction, transcriptional regulation, growth response, DNA repair and replication, etc. B. The classification of E3 ligases. Based on its structural and biochemical characteristics, E3s are classified into three major isoforms: the truly interesting novel genes (RING), C-terminus of E6-associated proteins (HECT), and RING homology-in-between-RING (RBR) E3s.
Mechanistically, the E1-E2-E3 catalytic cascade requires three steps: first, ubiquitin is recruited and activated by the E1 enzyme in an ATP-dependent manner, then transferred to the cysteine residue of the E2 enzyme, and then covalently bound to the lysine residue of the target protein via E3 enzyme [9]. The repeated three-step reaction catalyzes the polymerization of ubiquitin to form ubiquitin chains (Figure 1A) [9]. Moreover, the seven acceptor lysines of the ubiquitin molecule (K6, K11, K27, K29, K33, K48 and K63, respectively) can be engaged in sequential conjugation, which in turn leads to different linkages of the ubiquitin chain, thus determining the different fates and functions of the target proteins [18]. Usually, polyubiquitin chains coupled to the K48 site, the K11 site and the K29 site lead to 26S proteasome-mediated degradation of labeled substrates. On the other hand, non-protein-hydrolyzed ubiquitin chains containing K63, K6, K27, and K33, are involved in many key biological processes such as signal transduction, DNA repair, and transcriptional regulation (Figure 1A) [19-21]. Evidently, E3s are particularly important throughout the ubiquitination process as they are responsible for the specific recognition and labeling of substrates [9]. In short, E3s can not only maintain the relative homeostasis of intracellular proteins but also participate in most cellular processes of normal physiological activity. Therefore, dysregulation of E3s would contribute to abnormal activation or inactivation of signal pathways, accumulation of misfolded or dysfunctional proteins, or inadequate assembly of protein complexes [22], thus promoting the development and progression of many types of cancers, including HCC [23]. In recent years, an increasing number of E3 ligases have been identified in HCC, and some of these E3s are oncoproteins, while others function as tumor suppressors. Therefore, a comprehensive study of these multifunctional E3s may provide a new direction for the future treatment of HCC. Herein, we review recent research advances in HCC-associated E3s, describing their structure, classification, functional roles, and discussing some mechanisms of the abnormal activation or inactivation of the HCC-associated signal pathways. In addition, given the success of proteasome inhibitors in the treatment of malignant cancers, we describe current knowledge and future prospects for targeted therapies against aberrant E3s in HCC.
The classification of E3 ligases
To date, more about 650 E3 ligases have been described in humans and classified into three main types based on their structural and biochemical characteristics: homologous to E6-associated protein C-terminus (HECT), really interesting new gene (RING), and RING-in-between-RING (RBR) E3s (Figure 1B) [24]. A detailed classification of E3 ligases is shown in Table 1.
Table 1.
Classification of E3 ligases
| Types | Characteristic domains | Member | Subtypes | Characteristic domains of subtypes | Examples |
|---|---|---|---|---|---|
| RINGs | RING fold structure with/without zinc-binding site (RING/U-box) | >600 | Monomers | Monomeric RING/U-box | c-CBL, TRIM11, E4B |
| Homodimers | Homomeric RING/U-box | Chip, SIAH1 | |||
| Heterodimers | Heteromeric RING | MDM2, HaKai | |||
| Cullin-RING ligases (CRLs) | Cullin scaffold structure | SKP1, RBX1, CRL1/2/3/4/5/7/9, FBPs: FBXL, FBXW, FBXO | |||
| Other Multi-subunit E3s | Multiple subunit structure | APC/C | |||
| HECTs | N-terminal lobe, C-terminal lobe, and a flexible tether between two lobes | About 30 | NEDD4 family | Tryptophan-tryptophan (WW) motifs | NEDD4, ITCH, SMURF1, SMURF2 |
| HERC family | RCC1-like domain | HERC1-2, HERC3-6 | |||
| Other HECTs | Various domain | E6AP | |||
| RBRs | Two RING domains and one-in-between-RING (IBR) domain2 | 14 | Ariadne family | Ariadne domain | HHARI, TRIAD1 |
| Other RBRs | Various domain | PAPKIN, HOIL-1L |
The RING-type subgroup contains the largest number of E3 ligases, up to more than 600 in humans, which can be classified into two categories: typical and atypical. The typical RING structural domain has a RING fold structure with the zinc-binding site, while the atypical one, called the U-box domain, has the same RING fold structure but lacking the zinc-binding site [25]. Remarkably, RING E3s can act as a scaffold to transfer ubiquitin directly from E2 enzyme to the substrate (Figure 1B) [26]. Moreover, both RING and U-box E3 ligases can function as monomers, homodimers, heterodimers, or multiple subunits [24,27]. Cullin-RING ligases (CRLs) are a family of multisubunit RING E3 ligases that can be further divided into Skp1/Cullin 1/F-box protein complex (SCF), Cullin 2-Elongin B/C-VHL or SOCS proteins (CRL2), Cullin 3-BTBs (CRL3), Cullin4-DDB1-DCAFs (CRL4), Cullin5-ElonginB/C-SOCS proteins (CRL5), and the Cullin7/FBXW8 (CRL7) [28,29]. Besides, the anaphase-promoting complex/cyclosome (APC/C) is another key multisubunit E3, consisting of 19 subunits, regulating mitosis and DNA replication [30].
The HECTs are the second largest family of human E3 ligases, consisting of approximately 30 HECT E3 ligases [31]. The HECT domain is located at the C-terminus of these E3 proteins and contains three regions: the N-terminal lobe, the C-terminal lobe, and the flexible chain between these two lobes [32]. The N lobe binds E2 and recognizes specific ligase substrates, the C lobe receives and delivers ubiquitin for the E2 enzyme, and the flexible chain allows for the rotation of both lobes during the enzyme reaction [32]. Usually, HECT E3 ligase catalyzes the ubiquitination of the substrate in two steps: first, the HECT domain interacts with the E2 enzyme and transfers ubiquitin to the C lobe via trans-thioesterification reaction, and then ubiquitin is further transferred to the substrate (Figure 1B) [32]. Based on the N lobe structure, HECT E3s can be further divided into three subfamilies. One is the NEDD4 subfamily, consisting of nine members whose structures contain multiple tryptophan-tryptophan (WW) motifs, such as NEDD4, NEDD4-2 (NEDD4L), ITCH, SMURF1, SMURF2, WWP1, WWP2, etc. The second subfamily is the HECT and RCC1-like structural domain (HERC) subfamily, which consists of six members in humans, an example being HERC1~6. Structurally, HERC proteins can contain single or multiple RCC1-like domains. Finally, the remaining HECT E3s with different structural domains are classified as other HECT E3s like E6AP [31,32].
In recent years, a series of studies have identified another emerging type of E3 ligase, named RBR E3 ligase. To date, 14 RBR E3s have been identified in the human genome [33,34]. RBR E3s contain two RING domains (RING1, RING2) and an in-between-RING (IBR) domain. In general, RING1 is responsible for binding ub-loaded E2, and RING2 catalyzes the trans-thioesterification reaction through cysteine residues and obtains ubiquitin from RING1. In the ubiquitin transfer mechanism, RBR E3s have fusion characteristics of HECT and RING E3s. Specifically, RING1 takes up ubiquitin-loaded E2, transfers the ubiquitin to RING2 via the trans-thioesterification reaction, and subsequently, the RING2 domain transfers the received ubiquitin to the substrate (Figure 1B) [34]. RBR E3s can also be divided into several subfamilies, for instance, the Ariadne family (HHARI, TRIAD1, PARC, ANKIB1) is characterized by the presence of the Ariadne domain, while other subfamily members, such as Parkin, HOIL-1L and HOIP, contains various other domains [35].
E3 ligases: aberrant regulation and their involvement in HCC
Accumulating evidence has shown that E3 ligases perform an important role in HCC by regulating the degradation of tumor promoters or repressors. In addition, some important HCC-related signal pathways and pathological processes, such as Wnt/β-catenin pathway, PI3K/AKT/mTOR pathway, RAS/RAF/MEK/ERK pathway, Hippo pathway, Keap1/Nrf2 pathway, HIF-1α pathway, TGF-β/SMAD pathway, Notch pathway, NF-κB pathway and HBV infection have been rigorously shown to be regulated by multiple E3 ligase targets. In the following sections, we will describe and discuss the links between some important E3 ligases and HCC in different signal pathways and pathological processes (Table 2; Figures 2, 3, 4, 5 and 6).
Table 2.
Summary of abnormal E3s in HCC
| Types | Sub-types | E3s | Substrates in HCC | Degraded or not | Role | Reference |
|---|---|---|---|---|---|---|
| Wnt/β-catenin pathway | ||||||
| RINGs | Monomers | TRIM37 | β-catenin | No | Oncogene | [65] |
| TRIM56 | β-catenin | No | Tumor suppressor | [64] | ||
| Homodimers | SIAH1 | β-catenin | Yes | Tumor suppressor | [58] | |
| Heterodimers | Hakai | Ajuba, E-cadherin | Yes | Oncogene | [59,60] | |
| CRLs | CUL4B | β-catenin | No | Oncogene | [66] | |
| HECTs | ITCH | DVI2, DVI3 | Yes | Tumor suppressor | [52,53] | |
| Mule | β-catenin | Yes | Tumor suppressor | [63] | ||
| PI3K/AKT/mTOR pathway | ||||||
| RINGs | Monomers | TRIM7 | Src | Yes | Tumor suppressor | [109] |
| TRIM11 | AKT | No | Oncogene | [122] | ||
| TRIM31 | TSC1-TSC2 complex | Yes | Oncogene | [117] | ||
| HRD1 | PTEN | Yes | Oncogene | [125] | ||
| Heterodimers | BARD1 | AKT | No | Oncogene | [131] | |
| HECTs | - | NEDD4 | PTEN | Yes | Oncogene | [128] |
| RAS/RAF/MEK/ERK pathway | ||||||
| RINGs | Monomers | c-CBL | EGFR | Yes | Tumor suppressor | [136,137] |
| RNF128 | EGFR | No | Oncogene | [138] | ||
| Hippo pathway | ||||||
| RINGs | Heterodimers | MDM2 | YAP | Yes | Tumor suppressor | [152] |
| CRLs | Fbxw7 | YAP | Yes | Tumor suppressor | [153] | |
| CUL4A | LATS1, YAP | Unknow | Oncogene | [154] | ||
| HECTs | - | NEDD4 | LATS1 | Unknow | Tumor suppressor | [169] |
| Keap1/Nrf2 passway | ||||||
| RINGs | Monomers | TRIM21 | P62 | No | Oncogene | [174,178] |
| TRIM25 | Keap1 | Yes | Oncogene | [175] | ||
| HIF-1α passway | ||||||
| RINGs | Multi-subunit E3s | APCCDC20 | PHD3 | Yes | Oncogene | [194] |
| CRLs | VHL | HIF-1α | Yes | Tumor suppressor | [193] | |
| TGF-β/SMAD passway | ||||||
| RINGs | Monomers | PJA1 | SMAD3 | Yes | Oncogene | [206] |
| NF-κB passway | ||||||
| RBRs | - | Parkin | TRAF2, TRAF6 | Yes | Tumor suppressor | [217] |
| Notch passway | ||||||
| RINGs | CRLs | Fbxw7 | Notch1 | Unknow | Tumor suppressor | [166] |
| HBV infection | ||||||
| RINGs | Monomers | MARCH5 | HBx | Yes | Tumor suppressor | [241] |
| Homodimers | SIAH1 | HBx | Yes | Tumor suppressor | [240] | |
| Heterodimers | HDM2 | HBx | No | Oncogene | [242] | |
| CRLs | CUL4B | HBx | No | Oncogene | [245] | |
| MSL2 | APOBEC3B | Yes | Oncogene | [243] | ||
| DDB1 | Smc5/6 complex | Yes | Oncogene | [244] | ||
| HECTs | - | NEDD4 | HBx | Yes | Tumor suppressor | [238] |
| Other E3s | ||||||
| RINGs | Monomers | ZFP91 | hnRNPA1 | Yes | Tumor suppressor | [278] |
| A20 | PFKL | Yes | Tumor suppressor | [279] | ||
| UHRF1 | MEG3 | Unknow | Oncogene | [280] | ||
| UHRF2 | H3K9ac | Unknow | Oncogene | [281] | ||
| RNF6 | FOXA1 | Yes | Oncogene | [282] | ||
| RNF41 | CACYBP | Yes | Tumor suppressor | [283] | ||
| TRAF6 | HDAC3 | No | Oncogene | [284] | ||
| TRAF7 | KLF4 | Yes | Oncogene | [285] | ||
| TRIM16 | ZEB2 | Unknow | Tumor suppressor | [286] | ||
| TRIM50 | SNAIL | Yes | Tumor suppressor | [287] | ||
| Homodimers | Chip | ERα | Yes | Oncogene | [288] | |
| CRLs | RBX1 | PD-L1 | Yes | Tumor suppressor | [289] | |
| CUL2 | MAF1 | Yes | Oncogene | [290] | ||
| FBXO22 | P21 | Yes | Oncogenev | [291] | ||
| DCAF15 | ZEB1 | Yes | Tumor suppressor | [292] | ||
| SPOP | ZEB2 | Unknow | Tumor suppressor | [293] | ||
| FBXL6 | HSP90AA1 | No | Oncogene | [294] | ||
| SCFβ-TRCP | TRIB2 | Yes | Tumor suppressor | [295] | ||
| SCFskp2 | P21, p27 | Yes | Oncogene | [296] | ||
| HECTs | - | WWP1 | Caspase3, p53 | Unknow | Oncogene | [297] |
Figure 2.
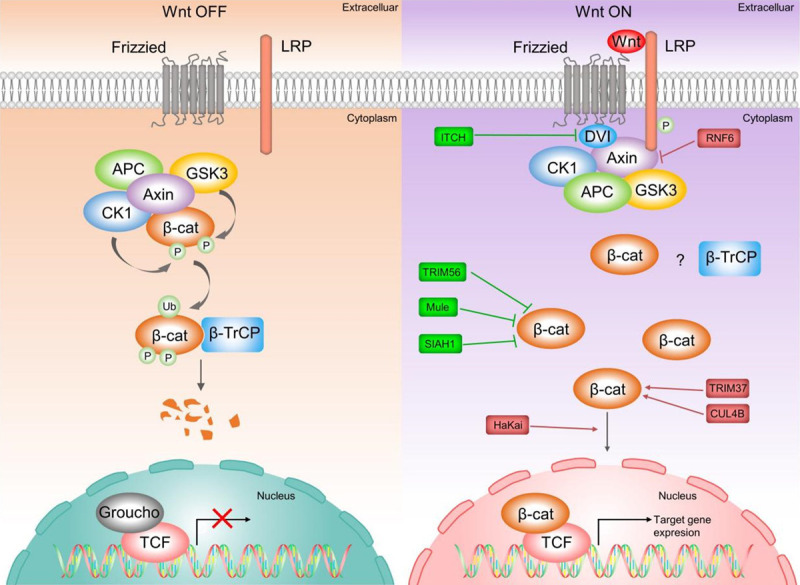
The role of E3 ligases in Wnt/β-catenin passway in HCC. Under normal cellular conditions (Wnt signal off), β-catenin in the cytoplasm undergoes UPS-mediated degradation by interacting with the destructive complex (DC). However, the binding of Wnt to cell surface receptors shuts down the degradation of β-catenin, producing stable β-catenin translocated to the nucleus, which binds to TCF/Lef transcription factors to activate transcription of Wnt target genes. Some E3 ligases are involved in the development of HCC by targeting the Wnt/β-catenin pathway. ITCH promotes ubiquitination and degradation of phosphorylated Dvl2 and Dvl3, thereby inhibiting Wnt signal. Overexpression of SIAH1 inhibits Wnt signal in the form of β-catenin degradation. HaKai causes ubiquitinated degradation of E-calmodulin, leading to nuclear translocation of β-linked proteins and subsequent activation of Wnt signal. Mule can directly degrade β-catenin and suppress Wnt signal. CUL4B represses GSK-3 activity to prevent degradation of β-catenin, which in turn promotes Wnt signal.
Figure 3.
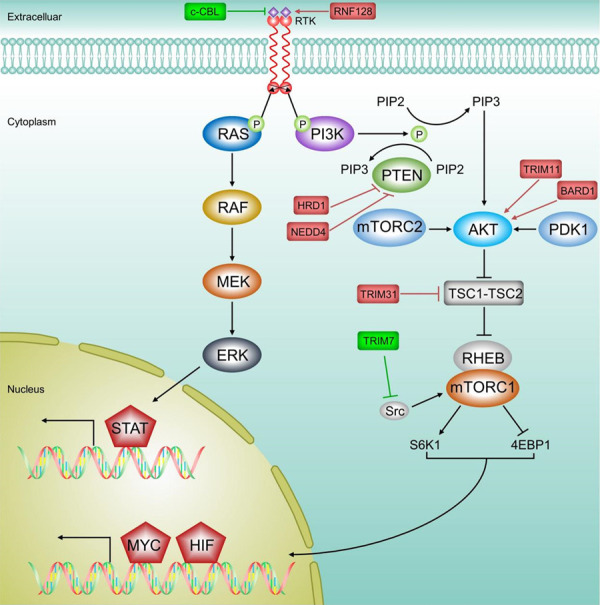
The role of E3 ligases in PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathway in HCC. When RTK binds to their cognate ligands, RTK undergoes autophosphorylation, which generates downstream signals and triggers the activation of several pathways, including the MAPK pathway. PI3K located downstream is subsequently activated following RTK activation. Activated PI3K can gradually activate PIP2 and PIP3, and this signal process can be inhibited by PTEN. Accumulation of PIP3 can trigger activation of AKT by PDK1 and mTORC2. Activated AKT phosphorylates and inhibits the “TSC1-TSC2” complex, which in turn activates mTORC1 and ultimately regulates many oncogenic signal targets. Similar to PI3K signal, upon receiving signals from receptors such as RTKs, RAS is phosphorylated and activated, which in turn activates RAF, and the activated RAF kinase further activates downstream MEK and ERK, ultimately activating multiple transcription factors and other nuclear substrates in the nucleus. Many E3s have been reported to be involved in regulating these two pathways to participate in HCC development. TRIM7 inhibits mTORC1-S6K1 signal by degrading Src. TRIM31 activates AKT-mTORC1 signal by degrading the “TSC1-TSC2” complex. TRIM11 activates PI3K/AKT signal by upregulating PI3K and AKT protein levels. HRD1 promotes PTEN degradation, thereby inhibiting the AKT pathway. BARD1 activates AKT signal by upregulating AKT, mTOR protein expression. c-CBL mediates the degradation of EGFR (a typical transmembrane RTK), thereby inactivating RAS/RAF/MEK/ERK signal. RNF128 activates the RAS/RAF/MEK/ERK pathway in turn by upregulating the expression of EGFR as well as other RAS signal components.
Figure 4.
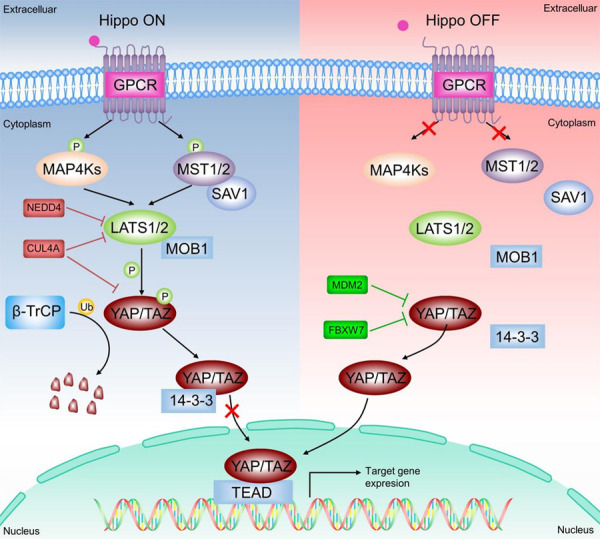
The role of E3 ligases in Hippo pathway in HCC. Under normal cellular conditions (Hippo ON), the binding of extracellular signals to the GPCR triggers the activation of two members of the NDR kinase family, LATS1 and LATS2 (LATS1/2), which are responsible for the phosphorylation of YAP/TAZ and promote its cytoplasmic localization for E3 ligase β-TRCP-mediated ubiquitination and proteasomal degradation, thereby inhibiting YAP/TAZ transcriptional activity. In contrast, in tumor cells (Hippo OFF), inactivation of LATS1/2 results in its inability to phosphorylate YAP/TAZ, which subsequently escapes ubiquitination degradation by β-TRCP and translocates to the nucleus to activate the expression of target genes involved in tumorigenesis and progression. MDM2 mediates cytoplasmic translocation and subsequent degradation of YAP, which in turn inhibits the Hippo pathway. Fbxw7 suppresses Hippo signal by ubiquitinating degradation of YAP. CUL4A ubiquitinates LATS1 and downregulates its protein levels, while repressing phosphorylation of YAP, together leading to inactivation of Hippo signal. NEDD4 represses the Hippo pathway by inactivating LATS1.
Figure 5.

The role of E3 ligases in TGF-β/SMAD, HIF-1α, Notch, Keap1/Nrf2, and NF-κB passway in HCC. Under normal conditions, Nrf2 is targeted for degradation by the Keap1 and remains at low levels. Under oxidative stress, Nrf2 segregates from Keap1 and enters the nucleus to bind to the ARE and activate the transcription of its target gene. In addition, p62 can interact with Keap1 to form protein aggregates and release Nrf2 and enter the nucleus. TRIM21 inhibits Nrf2 release and the Keap1/Nrf2 pathway by ubiquitinating p62. TRIM25 releases Nrf2 and activates the Keap1/Nrf2 pathway by degrading Keap1. Under normoxic conditions, PHD is activated and hydroxylated to modify HIF-1α, which is subsequently degraded by VHL. Under hypoxic conditions, the activity of PHD and VHL was inhibited, thus stabilizing HIF-1α. APCCDC20 mediated the degradation of PHD3 and activated HIF-1α signal. TGF-β binds to TGF-β type I and type II serine/threonine kinase membrane receptors (i.e., TβRI and TβRII) to trigger the formation of heterotetrameric complexes and phosphorylation of SMAD 2/3. Thereafter, phosphorylated SMAD2/3 forms a complex with SMAD4 and translocates to the nucleus, where they are required to interact with other TFs to activate or repress transcription of target genes. PJA1 promotes TGF-β signal through ubiquitination and degradation of SMAD3. Pro-inflammatory cytokines bind to receptors on the cell surface and activate the ligand protein TRAF or receptor-interacting protein RIP, which in turn promotes the activation of IκB kinase β (IKKβ). Subsequently, IκB bound to NF-κB dimers (e.g., p50-RELA, p50-REL) is phosphorylated by activated IKKβ, which induces IκB ubiquitination and proteasome-mediated degradation, ultimately releasing NF-κB dimers into the nucleus and activating transcription of target genes. Parkin inhibits NF-κB pathway through ubiquitination and degradation of TRAF2 and TRAF6. The membrane Notch receptor is activated by the typical ligands DLL1, DLL4, Jagged 1 or Jagged 2. Binding of the ligand to its receptor induces the proteases ADAM10 or ADAM17 to cleave the Notch receptor and produce the Notch extracellular (NEXT) fragment. The NEXT fragment is further cleaved by γ-secretase to form the Notch intracellular structural domain (NICD), which is released from the membrane. Fbxw7 represses Notch signal by downregulating the expression of Notch1 and its downstream molecule NICD.
Figure 6.
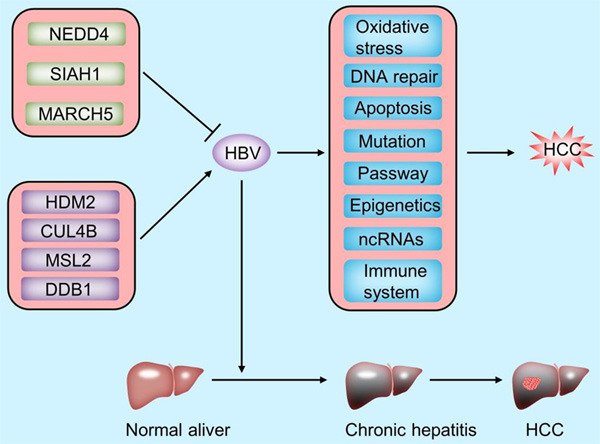
The role of E3 ligases in HBV infection in HCC. HBV DNA integrates into the host genome of normal hepatocytes early in infection, leading to genomic instability and direct insertional mutations of different oncogenes. Sustained expression of HBV regulatory proteins contributes to tumor transformation of hepatocytes. NEDD4, SIAH1 and MARCH5 repress HBV-associated HCC progression by ubiquitinating and degrading HBx. HDM2 stabilizes the structure of HBx by mediating the Neddylation modification of HBx, which in turn promotes HCC development. CUL4B promotes HBV replication and HCC development by inhibiting the ubiquitinated degradation of HBx. MSL2 maintains HBV stability and contributes HCC tumorigenesis by ubiquitination and degradation of APOBEC3B. DDB1 mediates ubiquitination degradation of the Smc5/6 complex, thereby reducing transcriptional repression of HBV by Smc5/6, and ultimately stimulating HBV gene expression and promoting HCC development.
Wnt/β-catenin pathway
As an evolutionarily conservative signal, the Wnt/β-catenin signal pathway serves an essential role in cell fate determination, proliferation, migration, angiogenesis, and organogenesis during embryonic development [36]. Under normal cellular conditions (in the absence of a Wnt signal), β-catenin is a cytoskeletal protein that forms a complex with E-cadherin at the cell membrane to maintain cell-to-cell adhesion and prevent cell migration [37,38]. At the same time, β-catenin in the cytoplasm undergoes UPS-mediated degradation through interaction with a destructive complex (DC), which is composed of scaffolding protein Axin, adenomatous polyposis coli (APC) protein, Ser/Thr kinases glycogen synthase kinase 3 (GSK-3), casein kinase 1 (CK1), and E3-ubiquitin ligase β-TRCP [37]. Besides, protein Disheveled (Dvl) and phosphatase 2A (PP2A) also bind to the complex [38,39]. However, the binding of Wnt to cell surface receptors turns off the degradation of β-catenin, and the stable β-catenin translocates to the nucleus, thereby binding to the TCF/Lef transcription factors to activate transcription of Wnt target genes (such as cyclin D1 and c-Myc) [40,41]. Abnormal accumulation of β-catenin and its targeted gene expression are the main drivers of cell transformation in several human malignant tumors [42-44]. Multiple studies have shown that E3 ligases cause the accumulation of β-catenin and subsequent activation of Wnt signal via mediating the ubiquitination degradation of key molecules on the Wnt/β-catenin pathway, and finally promote tumorigenesis [45-47]. Evidently, E3 ligases are significantly involved in the occurrence and development of HCC by targeting the Wnt/β-catenin pathway (Table 2; Figure 2), including ITCH [48-53], SIAH1 [54-58], Hakai [59,60], Mule [61-63] and other E3 ligases [64-66]. In addition, the substrates targeted by these E3 ligases have also been identified, including Disheveled2 (Dvl2) [52], Dvl3 [53], E-cadherin [59], Ajuba [60] and β-catenin [58,63-66].
ITCH
ITCH, a member of E3 ligases, belonging to the HECT-type E3 subfamily, plays a key role in regulating cell growth and apoptosis [67-69]. During the past 10 years, an increasing number of ITCH’s targets have been implicated in tumorigenesis and chemosensitivity, such as p63, p73, Notch1, and others [70-72], suggesting that ITCH may be significantly associated with tumorigenesis. Data from the International Cancer Genome Consortium (ICGC, https://dcc.icgc.org/genes/ENSG00000078747) database indicate that ITCH has been detected 2.49% of mutations in HBC-associated HCC tissues (10/402). A previous study found that inhibiting ITCH could promote HCC cell colonies formation [48]. Furthermore, another study identified Dvl2 as a downstream substrate of ITCH in HCC [52]. Dvls are the central mediator of Wnt signal and phosphorylated Dvl is required for Wnt signal [73]. ITCH could promote the ubiquitination and degradation of phosphorylated Dvl2 and therefore inhibit Wnt signal [52]. Similarly, Dvl3 has also been reported to be degraded as a downstream substrate of ITCH, thereby repressing the transcriptional activity of β-catenin, and ultimately repressing the cancer stem cells (CSCs) of HCC [53]. Besides, Wu M et al. have demonstrated that micro RNA-411 (miR-411) could bind to the 3’-untranslated region (3’-UTR) of ITCH and inhibit its translation, thus repressing the proliferation of HCC cells [50].
Circular RNAs (CircRNAs) commonly act as miRNA sponges and repress miRNA activities [74]. A recent study found that CircRNA-ITCH (Circ-ITCH) has some miRNA binding sites in the ITCH 3’-UTR [51]. As a miRNA sponge, Circ-ITCH may promote ITCH protein expression by interacting with miR-7 and miR-214 [51]. Moreover, Circ-ITCH down-regulates the protein level of β-catenin and two target genes c-Myc and cyclinD1, thus repressing the cell proliferation and promoting apoptosis in HCC [51]. Clinically, the high expression of Circ-ITCH was associated with favorable survival of HCC [49,50]. Notably, numerous studies have also shown that Circ-ITCH and ITCH could repress the development and progression of many other tumors, such as gastric cancer [75], prostate cancer [76], breast cancer [77], etc., via suppressing the Wnt/β-catenin signal pathway. Together, the above results suggest that ITCH is an inhibitor of the Wnt/β-catenin pathway and functions as a tumor suppressor in HCC.
SIAH1
Seven in absentia homolog 1 (SIAH1), a RING-type E3 ligase, has been implicated in a variety of biological processes, including the cell cycle, programmed cell death and oncogenesis [78]. SIAH1 has been implicated in a variety of human malignancies, including breast cancer [79], gastric cancer [80], and leukemia [81]. Interestingly, SIAH1 appears to have a dual role in HCC, acting as a tumor promoter or a tumor suppressor. Previous studies have shown that SIAH1 expression was changed in HCC tissues, and SIAH1 inactivation contributes to HCC progression [54,55]. Besides, the downregulation of SIAH1 caused an increase in protein stability of Zeb1, a strong EMT-related transcription factor (EMT-TF), which was significantly increased in HCC cells resistant to doxorubicin [56]. However, in another study, nuclear accumulation of the SIAH1 supports numerous pro-tumorigenic cellular processes which is associated with tumor growth and tumor cell dissemination [57]. It may use the transcription factor far-upstream element (FUSE)-binding protein (FBP)-3 (a tumor promoter whose expression level is negatively correlated with the prognosis of HCC patients), at least in part, to increase HCC cell growth, but the molecular mechanism is not fully elucidated [57]. SIAH1 has been shown to be one of the most selective RING-finger E3s in the Wnt/β-catenin signal pathway, mediating ubiquitination and degradation of β-catenin [82]. In one study, overexpression of SIAH1 causes growth arrest and apoptosis of HCC cells in a β-catenin degradation-dependent way [58]. Taken together, SIAH1 has a complicated function in HCC development, which is consistent with the large spectrum of protein substrates that they control. Among them, β-catenin was identified as a new downstream substrate of SIAH1, which will help us better understand the relationship between deregulated β-catenin signal and the tumorigenesis of HCC.
Hakai
Hakai is a Casitas B-lineage lymphoma (Cbl)-like ubiquitin ligase that mediates ubiquitination of E-cadherin and regulates E-cadherin complex endocytosis [83-85]. Hakai-mediated down-regulation of E-cadherin is involved in oncogenic and/or tumor-suppressive signal pathways such as RACK1 and Slit-Robo signal [84,86,87]. The ICGC database shows it with a high mutation rate of 8.93% (59/660) in HCC (virus-associated). Besides, HaKai has been reported to cause ubiquitination degradation of E-cadherin, which led to the nuclear translocation of β-catenin and finally promoted the epithelial-mesenchymal transition (EMT) of HCC [59]. Ajuba belongs to the Ajuba family, and it is required to maintain E-cadherin adhesion [88]. In addition, Hakai degrades Ajuba protein through the Neddylation pathway, which resulted in the loss of E-cadherin, nuclear translocation of β-catenin and activation of Wnt signal, thus promoting the proliferation of HCC [60]. All the evidence points to HaKai may be a potential tumor promoter in HCC and works by targeting the E-cadherin and Wnt/β-catenin pathway. Given there are few relevant studies, further verification is needed.
Mule
Mcl-1 ubiquitin ligase E3 (Mule; also known as HUWE1, ARF-BP1 or HectH9) is a member of HECT E3 ligase family, which is participating in the regulation of many pathophysiological processes, including transcription regulation, protein toxicity stress, DNA replication and injury repair, cell proliferation, differentiation, autophagy and apoptosis [89-92]. Mule is reported to be a tumor suppressor in HCC, which can degrade carcinogenic phosphatidylinositol 3,4,5-trisphosphate-dependent rac exchanger 2 (PREX2) protein through ubiquitination [61]. In addition, Mule expression was inhibited in HCC induced by obesity [62]. Furthermore, a recent study showed that Mule can directly degrade β-catenin and repress β-catenin mediated CSCs in HCC [63]. Interestingly, Mule was able to target β-catenin for degradation to stop abnormal activation of Wnt signal in colorectal cancer [93].
Other E3 ligases
Besides ITCH and Hakai, several other E3 ligases involved in HCC development and progression through regulation of the Wnt/β-catenin pathway have been identified and are described separately because their regulatory mechanisms are not fully understood.
Tripartite motif (TRIM) contains three domains, including an N-terminal RING domain, one or two B-box domains and a C-terminal coiled-coil domain [94,95]. The RING domain confers TRIM as the catalytic function of the E3 ligase, and a single TRIM has been shown to ubiquitinate their interacting partners directly [96]. TRIM56 is an important member of the TRIM family, but the role of TRIM56 in many tumors is not fully understood. In one study, TRIM56 inhibits the proliferation of multiple myeloma cells and induces apoptosis [97]. In another study, TRIM56 can mediate K48 ubiquitin ligation of waveform proteins and promote their degradation, thereby inhibiting ovarian cancer progression [98]. However, in breast cancer, TRIM56 mediates K63 ubiquitination via estrogen receptor-α (ER-α), upregulates ER-α protein stability and promotes cell proliferation [99]. Until recently, Yang Y et al. reported the role and mechanistic studies of TRIM56 in HCC [64]. The results showed that HCC patients expressing low levels of TRIM56 with higher grade clinical stage and poorer survival rate [64]. In addition, overexpression of TRIM56 suppressed the proliferative potential of HCC cells [64]. Mechanistically, TRIM56 can negatively regulate the expression of β-catenin, a key gene in Wnt signal, which in turn inactivates Wnt signal to inhibit the malignant progression of HCC [64]. However, whether TRIM56 regulates β-catenin through its E3 activity is still unclear. In addition, another member of the TRIM family, TRIM37, was significantly up-regulated in HCC tissues and activated Wnt/β-catenin signal and mediated EMT [65]. Collectively, these two studies have not elucidated the molecular mechanism of TRIM in regulating the Wnt/β-catenin passway yet, which needs further investigation.
Cullin-Ring ligases (CRL) 4B, a scaffold protein that assembles CRL4B ubiquitin ligase complex, functions as a tumor promoter in multiple cancers [100,101]. Yuan J et al. showed that CUL4B was up-regulated in HCC tissues, and the development of HCC was accelerated in CUL4B transgenic mice [102]. Moreover, CUL4B could repress the GSK-3 activity to prevent proteasome degradation β-catenin and silence multiple Wnt inhibitors (such as DKK1 and PPP2R2B), thereby promoting hepatocarcinogenesis [66]. However, it is unclear whether CUL4B is involved in the regulation of Wnt signal through its E3 ligase activity.
PI3K/AKT/mTOR pathway
The phosphoinositide 3-kinase/serine/threonine protein kinase AKT/mechanistic target of rapamycin (PI3K/AKT/mTOR) pathway is one of the most common activated pathways in HCC, which is up-regulated in up to 50% (HCC) tumors [103]. Under physiological conditions, this pathway is activated in response to growth factors, cytokines and insulin, regulating key metabolic processes, including glucose metabolism, biosynthesis, and maintenance of redox balance, to support systemic metabolic homeostasis and individual growth and metabolism. In cancer cells, abnormal activation of the PI3K/AKT pathway reprograms cellular metabolism to support the anabolic needs of abnormally grown cells [104]. Mechanistically, PI3K-related downstream signal is subsequently activated after activation of receptor tyrosine kinases (RTK), cytokine receptors, integrins and G protein-coupled receptors (GPCRs). The activated PI3K is recruited to the plasma membrane and phosphorylates phosphatidylinositol 4,5-diphosphate (PIP2), thus producing the second messenger phosphatidylinositol 3,4,5-triphosphate (PIP3), and this signal transduction process can be inhibited by phosphatase and tensin homolog (PTEN). The accumulation of PIP3 then recruits AKT to the plasma membrane, where it is completely activated by phosphorylation of phosphoinositide-dependent protein kinase 1 (PDK1) and mTOR complex2 (mTORC2) protein kinases at T308 and S473, respectively. Activated AKT phosphorylates and inhibits tuberous sclerosis complex1-tuberous sclerosis complex 2 (TSC1-TSC2) complex, and activates mTORC1 via relieving the inhibition of Ras homolog enriched in brain (Rheb) mediated by TSC1-TSC2 complex. Ribosomal protein S6 kinase1 (S6K1) and eukaryotic translation initiation factor 4E binding protein (4EBP1) are typical downstream targets of mTORC1, which are used together with other targets to stimulate mRNA translation of sterol regulatory element binding protein (SREBP), MYC, hypoxia-inducible factor 1α (HIF-1α), and activating transcription factor 4 (ATF4) [104]. Besides, some genetic alterations that lead to abnormal activation of PI3K/AKT/mTOR pathway are important driving factors of tumorigenesis, including activation mutation of p110α catalytic subunit of PI3K, function loss mutation and deletion in PTEN, and amplification and function acquired missense mutation of AKT [105-107], etc. Notably, a series of findings showed that E3 ligases participated in the carcinogenesis of HCC by targeting the AKT pathway (Table 2; Figure 3). Multiple E3 ligases (such as the TRIM7, TRIM11, TRIM31, HRD1, NEDD4, and BARD1) and downstream substrates (such as PTEN, Src, and TSC1-TSC2 complex) have been identified.
TRIM family
TRIM7
The proto-oncogene tyrosine kinase Src, as a non-receptor protein tyrosine kinase, plays a broad role in cellular biological processes, including proliferation, differentiation, survival and metabolism. The activation of Src kinase promotes multiple downstream transduction cascades, including PI3K/AKT, mitogen-activated protein kinase (MAPK), and signal transduction and activator of transcription 3 (STAT3) [108]. Zhu et al. identified Src as the target of tripartite motif (TRIM) 7 ubiquitin ligase [109]. TRIM7 is a new member of the TRIM protein family, and it was originally identified in proteins that interact with glycogen production [110-112]. TRIM7 has an N-terminal RING domain, a B-box domain, a coiled-coil domain and a C-terminal B30.2/SPRY domain, indicating that as a RING-type E3 ligase, it can directly bind to the target for ubiquitination [113]. For example, TRIM7 overexpression enhances the polyubiquitination and degradation of dual-specific phosphatase 6 (DUSP6), which in turn down-regulates the expression of phosphorylated p38 (p-p38) and up-regulates the levels of p53 and p21, ultimately resulting in the occurrence of HCC [114]. What’s more, Zhu L et al. showed that TRIM7 induces K48-linked, RING domain-dependent polyubiquitination of Src protein, which further leads to the suppression of its downstream mTORC1-S6K1 signal and acts as a tumor suppressor in HCC [109].
At present, tyrosine kinase inhibitors (TKIs) such as dasatinib, bosutinib, vandetanib and ponatinib have been approved by the Food and Drug Administration (FDA) as Src inhibitors for drug treatment of malignant diseases, which indicates the great potential of targeted Src in clinical treatment [108]. Therefore, TRIM7, as a new regulator of Src, may provide great therapeutic potential for HCC. At present, there are few studies on TRIM7 in HCC, so it needs further verification and exploration.
TRIM31
TSC1 and TSC2 are upstream inhibitors of the mTORC1 pathway and also well-recognized tumor inhibitors in some cancers including HCC [103,115,116]. In one study, the TSC1-TSC2 complex was identified as a direct downstream target of tripartite motif (TRIM) 31 in HCC [117]. They further proved that TRIM31 directly interacts with the TSC2 complex to mediate K48-linked ubiquitination and degradation of the complex, thereby overactivating the mTORC1 pathway to promote the progression of HCC [117]. Furthermore, the oncogenic effect of TRIM31 has also been confirmed in clinical HCC specimens, cellular models, and animal models [117]. Besides, the same group has proved that TRIM31 can directly target p53 and mediate the degradation of K48-related p53 in a manner that depends on the RING domain, and subsequently promote the anoikis resistance of HCC [118]. In addition, Lv T et al. discovered that miR-29c-3p, as a tumor suppressor, inhibits the malignant progression of HCC by reducing the expression of TRIM31, and the upregulation of TRIM31 can partially abolish the anti-cancer effect of miR-29c-3p in HCC [119]. These findings consistently point out that TRIM31 is a potential tumor suppressor in HCC.
TRIM11
Tripartite motif (TRIM) 11, a member of the TRIM family, has attracted much attention because of its involvement in the development of the central nervous system [120,121]. Zhang Z et al. found that TRIM11 was overexpressed in HCC tissues and cell lines [122], and the down-regulation of TRIM11 inhibited the proliferation, invasion, and EMT of HCC cells in vitro and in vivo. Specifically, down-regulation of TRIM11 reduced the protein levels of phosphorylated PI3K and AKT in HCC cells, thereby inhibiting the activation of the PI3K/AKT signal pathway [122]. In another study, the expression of TRIM11 was significantly increased in HCC tissues, and the overexpression of TRIM11 was closely related to the progression of HCC and the poor survival of patients [123]. Collectively, these results indicate that TRIM11 plays a tumor-promoting effect in HCC, but the specific mechanism is not fully understood.
HRD1
HMG-CoA reductase degradation 1 (HRD1), also known as synoviolin, is an E3 ligase with RING finger domain located on the endoplasmic reticulum (ER) membrane [124]. It is reported to play an important role in the ubiquitination and degradation of unfolded/misfolded major histocompatibility complex I (MHC I) heavy chain proteins [124]. Liu L et al. used proteomics methods to determine that HRD1 is the interacting partner of PTEN [125]. HRD1 can promote PTEN ubiquitination and proteasome degradation, and subsequently inhibit the proliferation of HCC cells [125]. As mentioned previously, PTEN can inhibit the conversion from PIP2 to PIP3, thereby inhibiting the activation of PI3K/AKT signal. Therefore, the ubiquitination and degradation of PTEN mediated by HRD1 may exert anticancer activity by inhibiting PI3K/AKT signal, which needs further confirmation.
NEDD4
Neuronally expressed developmentally downregulated 4 (NEDD4, often known as NEDD4-1) is an E3 ligase containing the HECT domain, which is responsible for the selective ubiquitination of some regulatory proteins involved in transcription and membrane transport [126]. NEDD4-1 was found to be a potential proto-oncogene that negatively regulates PTEN through ubiquitination in a manner similar to that of mouse double minute 2 homolog (MDM2, in human known as HDM2) mediated p53 regulation [127]. Compared with normal hepatocytes and adjacent tissue of HCC, HCC cell lines and human HCC tissues show higher NEDD4 expression level and lower PTEN expression level, and the depletion of NEDD4 inhibits the growth and migration of HCC cells, without affecting the expression level of total AKT, but decreasing the expression of p-AKT (phosphorylated AKT) [128]. Similarly, it was found that depletion of NEDD4 increased the protein level of PTEN and significantly reduced the activity of AKT. Together, NEDD4 may activate the PI3K/AKT pathway by down-regulating the expression of PTEN, and ultimately exerts a carcinogenic effect [129]. However, the molecular mechanism of NEDD4 regulating PTEN and PI3K/AKT pathways is not clear yet, and further research is needed, such as in vivo and in vitro ubiquitination analysis to determine whether NEDD4 regulates PTEN through its E3 ligase function.
BARD1
BRCA1-associated RING domain 1 (BARD1) is an E3 ligase that is required for homologous repair of DNA double-strand breaks [130]. Liao Y et al. discovered that BARD1 was significantly elevated in HCC tumor tissues and the elevation of BARD1 was significantly associated with lower survival rates and advanced stages of HCC patients (34.86 months; 95% confidence interval (CI: 29.23-40.49, P=0.002) [131]. In addition, knockdown of BARD1 inhibits the AKT signal pathway. Specifically, silencing BARD1 represses the signal pathway by reducing the levels of AKT, mTOR, and matrix metalloprotein-9 (MMP-9), and repressing the phosphorylation of AKT (Ser473) and mTOR (Ser2248) [131], but the specific regulation mechanism is quite not clear.
RAS/RAF/MEK/ERK pathway
As the most well-studied signal transduction cascade of the mitogen-activated protein kinase (MAPK) cascade, the RAS/RAF/MEK/ERK pathway regulates basic cell functions like cell proliferation, differentiation, and survival [132]. Besides, it is abnormally activated in more than one-third of human cancers and 90% of skin melanomas, and almost all patients with advanced HCC have reported activation of ERK [133-135]. Similar to PI3K/AKT/mTOR pathway, the RAS/RAF/MEK/ERK pathway is mainly activated by signals generated via membrane-bound receptors such as RTKs, GPCRs, etc. Then GTPase and rat sarcoma (RAS) are activated, which in turn recruits rapidly accelerated fibrosarcoma (RAF) kinase to the plasma membrane for activation. Activated RAF kinase phosphorylates downstream mitogen-activated protein kinase kinase (MEK), which then sequentially phosphorylate Tyr and Thr residues of ERK and drives its nuclear translocation. ERK activates a variety of transcription factors and other nuclear substrates in the nucleus, as well as its cytoplasmic targets [135]. Recently, some studies have found that several E3 ligases are involved in the tumorigenesis and development of HCC by targeting this pathway (Table 2; Figure 3), such as c-CBL [136,137] and RNF128 [138].
c-CBL
c-CBL is a RING domain E3 ligase, consisting of a tyrosine kinase binding (TKB) domain, a RING domain and a C-terminal ubiquitin-related (UBA) domain. The TKB domain binds to the tyrosine phosphorylation target, and the RING domain binds to the E2 binding enzyme [139]. A series of studies have proved that c-CBL promotes the ubiquitination of epidermal growth factor receptor (EGFR) [139,140] and several other RTKs [141] through the recruitment of RING-dependent E2 ubiquitin-conjugating enzyme near the receptor. Specifically, the ligand binds to and initiates receptor autophosphorylation, and then the c-CBL TKB domain is recruited to specific phosphotyrosine site, such as EGFR Tyrosine 1045 (Y1045) [139]. This affinity promotes c-CBL-mediated receptor monoubiquitination and subsequent degradation via the lysosomal pathway, thereby terminating RTK signal transduction [139-141]. Recently, Jiang R et al. discovered that lncRNA-EGFR bound to EGFR and inhibited c-CBL-mediated ubiquitination and subsequent degradation of EGFR, thereby stabilizing the activation of its downstream RAS/RAF/MEK/ERK signal, which ultimately led to regulatory T cells (Tregs) differentiation and HCC development [136]. Similarly, in another study, 14-3-3σ protein can inhibit EGFR-c-CBL binding and subsequent c-CBL-mediated ubiquitination and degradation of EGFR, thereby prolonging the activation of EGFR/ERK signal and causing HCC cells apoptosis resistance [137]. These results suggest that c-CBL exerts as an inhibitor of EGFR to negatively regulate the RAS/RAF/MEK/ERK pathway.
RNF128
RING finger 128 (RNF128), also known as lymphocyte anergy-related gene (GRAIL), is a ring finger protein family E3 ligase that participates in the induction of T cell anergy [142]. The role of RNF128 in tumors is controversial. As reported, RNF128 interacts with p53 and activates the EGFR/MAPK pathway to promote the invasion and migration of esophageal squamous cell carcinoma [143]. However, in melanoma, RNF128 regulates Wnt/β-catenin signal to inhibit EMT and stemness through a mechanism involving CD44 ubiquitination [144]. Besides, down-regulation of RNF128 is associated with poor prognosis of upper urinary tract and bladder cancer [145]. At present, only one study has provided insight into the role of RNF128 in HCC [138]. In vivo and in vitro experiments have found that high levels of RNF128 promote HCC cell proliferation, migration and invasion, inhibit apoptosis, and promote tumor formation. Additionally, the analysis of the clinical data of 171 HCC patients showed that RNF128 is an independent risk factor for HCC recurrence and survival. At the same time, high RNF128 levels are significantly associated with malignant phenotypes, including tumor size, Edmondson-Steiner grade, and TNM staging [138]. Mechanistically, p-EGFR, p-MEK and p-ERK1/2 were significantly up-regulated in cells overexpressing RNF128. In addition, RNF128 can reactivate the EGFR/MEK/ERK pathway that is inhibited by the EGFR antagonist gefitinib [138]. This evidence shows that RNF128 may promote HCC tumorigenesis by activating the EGFR/MEK/ERK pathway, but the specific mechanism still needs further exploration.
Hippo pathway
The Hippo pathway, evolutionarily conserved, plays a central role in organ development, tissue regeneration, wound healing and immune regulation [146,147]. Yes-associated protein (YAP)/transcriptional co-activator with PDZ-binding motif (TAZ) has been identified as the key and main effector of the Hippo pathway, which control gene expression via regulating transcription factors (TFs) represented by the TEA domain transcription factor (TEAD) family, and then promote cell proliferation and survival signals [148]. YAP/TAZ is mainly regulated by phosphorylation, which could promote the cytoplasmic localization of YAP/TAZ to undergo E3 ligase β-TRCP-mediated ubiquitination and proteasome degradation, thus inhibiting the YAP/TAZ transcriptional activity. Two members of the NDR kinase family, LATS1 and LATS2 (hereinafter referred to as LATS1/2) are the major regulators of YAP/TAZ, which phosphorylate YAP/TAZ and cause its destabilization and restricted access to the nucleus [148]. Many upstream signals regulate YAP/TAZ activity, including those signals activated by mechanical force, cell polarity, cell adhesion, mitogen, extracellular matrix (ECM), RTKs, GPCRs, and most of the regulation is relayed by LATS1/2 [148]. However, the dysregulated Hippo pathway and YAP/TAZ-TEAD activity are related to various cancers [149]. Wang H et al. found that the Hippo pathway was inactivated in HCC induced by c-Myc, and the ablation of TAZ completely prevented the development of tumors [150]. In addition, YAP/TAZ is continuously activated to drive the proliferation and dedifferentiation of hepatocytes, thereby promoting the occurrence of hepatobiliary carcinoma in non-alcoholic fatty liver disease [151]. This evidence demonstrates that YAP/TAZ plays a key role in the development of HCC. Most importantly, apart from the physiological ubiquitination modification of YAP (when the Hippo pathway is activated), many E3 ligases have been found to participate in oncogenesis and progression through degrading the core component of the Hippo pathway (Table 2; Figure 4), including MDM2 [152], Fbxw7 [153], CUL4A [154] and NEDD4 [155].
MDM2
As we all know, MDM2 is the major negative regulator of p53, which mediates the ubiquitination modification of p53, and the ubiquitinated p53 is subsequently transferred to the cytoplasm and degraded via the proteasome pathway [156]. Thus, MDM2 can regulate the stability of the p53 signal pathway and act as a tumor promoter in many malignant cancers including lung cancer, breast cancer, HCC, esophagogastric cancer and colorectal cancer [156,157]. As an E3 ligand containing a RING domain, in addition to targeting p53, many downstream substrates of MDM2 have been gradually identified such as p21, insulin-like growth factor 1 receptor (IGF-1R) and retinoblastoma protein (Rb) [158-160]. Recently, one study showed that down-regulation of MDM2 weakened the translocation of YAP from the nucleus to the cytoplasm and increased tumorigenesis in HepG2 cells (a HCC cell line) [152]. The mechanism exploration of the same research showed that MDM2 ubiquitinates YAP in a p53-independent way, and accelerates its cytoplasmic translocation and subsequent degradation [152], suggesting that MDM2 may act as a potential tumor suppressor in a p53-independent manner in HCC, this is contrary to the current mainstream view of MDM2 as a tumor promoter in HCC. Since most of the current research on MDM2 in HCC mainly focuses on the MDM2/p53 axis, it is necessary to continue to search for potential targets and roles of MDM2 in HCC.
Fbxw7
F-box and WD repeat domain-containing 7 (Fbxw7), a F-box protein in the SKP1-CUL1-F-box protein (SCF) E3 ligase complex, which acts as a general tumor suppressor by targeting several well-known oncoproteins for ubiquitination and proteasomal degradation like c-Myc [161], c-Jun [162], mTOR [163], etc. Akhoondi S et al. conducted a comprehensive genetic screening of primary tumors. Their data showed that genetic mutations cause Fbxw7 inactivation in various types of human cancers, and the overall mutation frequency is about 6% [164]. Likewise, Fbxw7 also functions as a tumor suppressor in HCC. Compared with normal tissues, Fbxw7 expression in HCC tissues is reduced [165,166], and it is associated with tumor differentiation, the incidence of portal vein or hepatic vein infiltration, and metastasis [166]. In addition, the expression of Fbxw7 protein in HCC tissues is negatively correlated with its substrate c-Myc and cyclin E [165]. Besides, lower Fbxw7 expression is associated with poor clinicopathological characteristics, including large tumors, high pathological grades, and advanced TNM staging [167]. Accordingly, higher Fbxw7 mRNA expression is associated with better five-year overall survival rate and longer disease-free survival, and Fbxw7 expression can be an independent risk factor for HCC recurrence [167]. However, it is not clear whether Fbxw7 regulates these substrates through its E3 ligase function. Tu K et al. identified YAP as a direct substrate of Fbxw7 in HCC, and Fbxw7 inhibited cell proliferation and induced apoptosis by promoting ubiquitinated degradation of YAP, which in turn inhibited the progression of HCC [153]. The evidence mainly supports Fbxw7 as a tumor suppressor in HCC, but the corresponding mechanistic studies are still poor, especially the identification of its substrates.
Other E3 ligases
As the CUL4B mentioned above, CUL4A is the core component of the CRL4 complex, and its N-terminus binds to the cullin-specific adaptor protein to recruit a large number of substrates. Previous studies have demonstrated that CUL4A directly targets the LATS1 protein and enhances its proteasomal degradation. In another study, the de-inhibition of CRL4DCAF1 inhibited Hippo passway by directly binding and ubiquitinating LATS1/2 in NF2-mutated tumors in the nucleus [168]. Recently, Ni W et al. discovered that CUL4A ubiquitinated LATS1 and down-regulated its protein level and at the same time inhibited YAP phosphorylation at Serine 127 (S127), which in turn leads to the inactivation of Hippo kinase signal and the progression of HCC [154].
As mentioned above, NEDD4 may activate the PI3K/AKT pathway by down-regulating the expression of PTEN, and act as a potential oncogenic protein in HCC [129]. Similarly, it was identified in HCC that LATS1, the main negative regulator of YAP, is the downstream target of NEDD4, which partially exerts its tumor promoter function by inactivating the LATS1 signal in HCC cells [169], but the specific mechanism is not clear. It is worth noting that NEDD4 directly targeted LATS1 and caused its ubiquitination degradation, which in turn increased the transcriptional activity of YAP [155]. This evidence indicates that NEDD4 may directly target the degradation of LATS1 to participate in the occurrence of HCC.
Keap1/Nrf2 passway
The Kelch-like ECH-associated protein 1 (Keap1)/nuclear factor E2-related factor 2 (Nrf2) pathway exerts a major role in regulating cellular redox balance. Under normal conditions, Nrf2 is targeted for degradation by the E3 ligase Kelch-like ECH-related protein 1 (Keap1) and remains at a low level. Under Oxidative stress, it can change the conformation of Keap1 through the oxidation of cysteine residues, thereby separating Keap1 from Nrf2. Then NRF2 enters the nucleus and binds to ARE to activate the transcriptional activity of its target genes, such as NAD(P)H: quinone oxidoreductase 1 (NQO1), heme oxygenase (HO-1), glutamate-half Cystine ligase catalytic subunit (GCLC) and glutamate-cysteine ligase modified subunit (GCLM) [170,171]. In addition to the classic way that Keap1 oxidizes and promotes the failure of Nrf2 proteasome degradation to cause Nrf2 to enter the nucleus, there is also an atypical way that p62 can interact with Keap1 to form protein aggregates and isolate Keap1 to release Nrf2. In response to oxidative stress, p62 dimerizes through the hydrogen bond between Lysine 7 (K7) and Aspartic acid 69 (D69) in its PB1 domain, and is formed by chelating with Keap1 protein aggregates. This Keap1 chelation releases Nrf2, thus promoting nuclear translocation of Nrf2 and transcriptional activation of its targets [172]. Keap1/Nrf2 signals seem to play a contradictory role in cancers. Generally, Nrf2 is considered a tumor suppressor because it has a cytoprotective effect and can resist damage from exogenous substances and oxidative stress. However, some evidence has found high constitutive activation of Nrf2 in many tumors, such as HCC, lung cancer, ovarian cancer, gallbladder cancer, gastric cancer, and breast cancer [173]. Recently, some studies have found that a few E3 ligases play a key role in the tumorigenesis of HCC by targeting molecules related to the Keap1/Nrf2 pathway (Table 2; Figure 5), including TRIM21 [174] and TRIM25 [175].
TRIM21
TRIM21 is an E3 ligase containing a RING finger domain and participates in a variety of biological processes including cellular metabolism, redox homeostasis, immune response, and cancer development [176]. In HCC, while one study suggested that TRIM21 may be a tumor suppressor [177], several comprehensive series of gene expression studies and TCGA datasets have shown the opposite role of TRIM21 (i.e., increased expression is associated with poor HCC development and prognosis) [174]. In addition, TRIM21 promotes diethylnitrosamine (DEN)-induced HCC by inhibiting the p62/Keap1/Nrf2 antioxidant pathway. Specifically, TRIM21 can ubiquitinate p62 and repress p62-mediated Keap1 chelation, thereby repressing the Keap1/Nrf2 pathway to promote DEN-induced oxidative stress [174]. Furthermore, their previous studies have demonstrated that TRIM21 mediates ubiquitination of p62 at K7, which leads to failure of Keap1 chelation and negatively regulates the Keap1/Nrf2 antioxidant pathway [178]. The true role of TRIM21 in HCC is still controversial and further search for downstream substrates and mechanistic studies are necessary.
TRIM25
TRIM25 is also an E3 ligase with the RING domain, which plays an important role in innate immunity and defense against viral infections, control of cell proliferation and cancer cell migration [179]. TRIM25 mainly regulates various physiological functions by regulating the ubiquitination of its target protein. For example, TRIM25 regulates retinoic acid-inducible gene-1 (RIG-1)’s in a K63-linked polyubiquitination, which is essential for host antiviral innate immunity in interferon signal transduction [180]. In addition, recent studies have found that TRIM25 is essential for tumorigenesis of HCC, but its role in HCC is still controversial. In one study, TRIM25 regulates the ubiquitinated degradation of normal hepatocyte metastasis-associated protein (MTA) 1, an essential protein that promotes the progression of HCC metastasis, which in turn inhibits the metastatic ability of HCC cells [181]. However, in another study, TRIM25 promotes the survival and growth of HCC cells by targeting the Keap1-Nrf2 pathway [175]. Mechanistically, TRIM25 directly targets Keap1 through ubiquitination and degradation, which leads to activation of Nrf2, thereby enhancing antioxidant defense and cell survival [175]. Furthermore, Zhang Q, et al. showed that TRIM25 directly ubiquitinates and degrades Fbxw7α, the major E3 ligase of c-Myc, thereby stabilizing c-Myc for HCC development [182]. In addition, high expression of TRIM25 has been shown to occur in a variety of cancers, such as colorectal cancer, lung cancer and breast cancer [183-185]. These results suggest that most of the evidence supports the pro-oncogenic role of TRIM25, but due to the high heterogeneity of HCC, the role of TRIM25 in HCC needs to be further explored.
HIF-1α passway
As the main regulator of oxygen homeostasis, hypoxia-inducible factor-1 (HIF-1) is composed of hypoxia-sensitive HIF-1α subunits and constitutively expressed HIF-1β subunits [186,187]. HIF-1 induces the transcription of more than 100 genes, which are involved in various biological processes, such as angiogenesis, red blood cell production, anaerobic glycolytic metabolism, cell proliferation, survival, inflammation, and cancer metastasis [188]. The activity of HIF-1 is regulated by the stability of HIF-1α, which is hydroxylated by the oxygen-dependent prolyl hydroxylase (PHD) [189]. Under normoxic conditions, PHD is activated by oxygen and cause HIF-1α to be hydroxylated at prolyl residues 402 and 564, which are then recognized by the tumor suppressor protein E3 ligase von Hippel-Lindau (VHL) ubiquitin-labeled, and then HIF-1α rapid degradation through the ubiquitin-proteasome pathway [189,190]. On the contrary, under hypoxia, the activity of PHD and VHL is inhibited by low oxygen and HIF-1α can thus be stabilized [189,190]. Intratumoral hypoxia and low oxygen levels are key features of all solid tumors, especially HCC. Furthermore, hypoxia mainly uses HIF to cause a series of metabolic changes in HCC cells to adapt to hypoxia, and hypoxia creates HCC resistance [191,192]. Given that the HIF-1α pathway is the core hub of HCC cells responding to the hypoxic microenvironment, it is reasonable to participate in HCC tumorigenesis through regulating key molecules in the HIF-1α pathway. Notably, VHL has been reported to exert anticancer effects in HCC through negative regulation of HIF-1α, and intraportal injection of VHL and doxorubicin significantly inhibited HCC growth in mice compared to doxorubicin alone [193]. Besides, another E3 ligase APCCDC20 in HCC that regulates the HIF-1α pathway has recently been discovered (Table 2; Figure 5) [194].
APCCDC20
Anaphase Promoting Complex (APC), a multi-subunit E3 ligase enzyme, has long been thought to be one of the major driving forces governing cell cycle progression [195]. Moreover, the APC complex is associated with the activators CDC20, which determine the APC’s substrate specificity [196]. To carry out its biological functions, CDC20 recruits substrates using a variety of motifs, and it typically binds to substrates with a Destruction-box (D-box) [196]. Furthermore, CDC20 has been linked to the development of HCC, with CDC20 depletion inhibiting cell proliferation and causing mitotic arrest in HCC cells [197], but CDC20’s specific substrates in HCC have yet to be identified. Until recently, Shi M et al. discovered that APCCDC20 is directly upstream of PHD3, and APCCDC20 mediated polyubiquitination and degradation of PHD3 increases the stability and activity of HIF-1 [194]. Specifically, APCCDC20 binds to the D-box motif in the PHD3 protein to exert its ubiquitination effect [194]. Furthermore, it was discovered that CDC20 is overexpressed in HCC in both the TCGA data and clinical HCC samples, and that high CDC20 expression is substantially associated with HCC patient prognosis [194]. Additionally, depletion of endogenous CDC20 also suppresses HCC cell proliferation in vitro and in vivo, indicating that it has a carcinogenic function in HCC [194].
TGF-β/SMAD passway
Transforming growth factor-β (TGF-β) signal is important for tissue homeostasis since it regulates a variety of cellular functions such as proliferation, differentiation, migration, and cell death [198]. Almost all cells, including hepatocytes, can produce and secrete TGF-β [199]. TGF-β binds to TGF-β type I and type II serine/threonine kinase membrane receptors (i.e., TβRI and TβRII) to trigger the formation of heterotetrameric complexes in which constitutive activated TβRII phosphorylates and activates TβRI. After transmembrane transduction of extracellular signals, activated TβRI initiates the typical TGF-β signal pathway through phosphorylation of mothers against decapentaplegic homolog (SMAD) 2 and 3 in its C-terminal serine residues. Thereafter, the phosphorylated SMAD2 and 3 form a complex with the SMAD4 and transfer to the nucleus, where they need to interact with other TFs to activate or inhibit the transcription of target genes [200]. In non-transformed hepatocytes, TGF-β inhibits proliferation [201] and induces apoptosis [202], thus playing a role in suppressing the development of HCC. However, the anti-tumor effect of TGF-β signal can be inactivated in two ways: i. by inactivating the basic components of the pathway; ii by hyperactivating parallel pathways that antagonize TGF-β signal (for example, the synthesis of autocrine substances such as EGFR or PDGF ligands and its receptors) [203,204]. PJA1, a new negative regulator of the TGF-β/SMAD signal pathway has been identified in early research [205], and the latest study showed that PJA1 is involved in HCC tumorigenesis in a TGF-β-dependent manner (Table 2; Figure 5) [206].
PJA1
PJA1 is a RING finger E3 ligase of the Praja family, and a variety of its specific substrates have been identified including SMAD3, homeodomain protein Dlx5, polycomb inhibitory complex 2 protein (PRC2), and zeste homolog 2 enhancement Agent (EZH2) [207-209]. Notably, according to TCGA data, increased PJA1 mRNA expression was associated with a significant decrease in overall survival in HCC patients, implying that PJA1 activity is linked to poor prognosis of HCC patients [210], but the specific mechanism is not clear. Compared with normal liver, the level of PJA1 in HCC is elevated significantly [207]. Furthermore, PJA1 promoted the ubiquitination and degradation of phosphorylated SMAD3, thereby enhancing the TGF-β signal and promoting the development of HCC [206].
NF-κB passway
Nuclear factor kappa-B (NF-κB) is a family of inducible transcription factors expressed in all tissues and cell types. It is activated by a wide range of signals and is necessary for the rapid activation of cellular responses by innate immunity and adaptive immunity [211]. In most cases, the NF-κB-IκB complex is located in the cytoplasm [212]. In the canonical pathway, proinflammatory cytokines such as tumor necrosis factor α (TNFα) and interleukin-1β (IL-1β) bind to the TOLL-like receptor (TLR) and tumor necrosis factor receptor 1 (TNF-R1) on the cell surface to activate the connection protein [TNF receptor-associated factor (TRAF) or receptor-interacting protein (RIP) kinase], which in turn promotes the activation of IκB kinase β (IKKβ). Subsequently, IκB bound to NF-κB dimers (for example, p50-RELA, p50-REL) is phosphorylated by activated IKKβ, thereby inducing ubiquitination and proteasome-mediated degradation of IκBα, and finally releasing NF-κB dimerization enters the nucleus and activates target genes [212]. It is known that NF-κB-induced inflammation can promote HCC tumorigenesis [213,214]. In contrast, the basic NF-κB activity in liver cells prevents HCC by inhibiting liver cell apoptosis [215,216]. In the liver, the combination of hepatocyte apoptosis and compensatory proliferation increases the incidence of carcinogenic mutations, and the anti-apoptotic function of NF-κB can prevent the development of HCC by inhibiting compensatory proliferation [215,216]. At present, a study found that E3 ligase Parkin was involved in the regulation of NF-κB signal transduction and HCC tumorigenesis (Table 2; Figure 5) [217].
Parkin
Parkin is an E3 ligase that belongs to the multi-RING domain family, which was originally found to be associated with autosomal recessive adolescent Parkinson’s syndrome (ARJP) [218]. Accumulating evidence has shown that Parkin serves as a cancer suppressor in the carcinogenesis of multiple tumors, such as HCC, lung cancer, breast cancer, colon cancer, cervical cancer, and so on [219]. A study involving Parkin gene knockout (-/-) transgenic mice model showed that hepatocyte proliferation was promoted in mice with a homozygous deletion of exon 3, while heterozygous (+/-) Parkin siblings proved to have no such effect [220]. Besides, the expression of Parkin in HCC tissues is significantly reduced, and the ectopic overexpression of Parkin in cell lines leads to an overall decline in cell growth and increases the sensitivity to apoptosis [221]. Further mechanism studies have shown that Parkin inhibits the NF-κB pathway through the ubiquitination and degradation of TRAF2 and TRAF6, thereby inducing HCC cell apoptosis [217]. Therefore, the induction of cell apoptosis through this molecular mechanism provides a new UPS-dependent strategy for the treatment of HCC [217].
Notch passway
The Noch pathway controls a variety of processes, including cell differentiation, proliferation, and apoptosis events at various stages of development [222]. Four Notch receptors (Notch1-4) have been identified in humans. Membrane Notch receptors are activated by typical ligands DLL1, DLL4, Jagged 1 or Jagged 2. The binding of the ligand to its receptor induces the protease ADAM10 or ADAM17 to cleave the Notch receptor and produce a Notch extracellular (NEXT) fragment. The NEXT fragment is further cleaved by γ-secretase at the S3 site and forms Notch intracellular domain (NICD) to be released from the membrane. The NICD then translocates into the nucleus, where it is recruited into the Notch transcription complexes (NTCs) containing CBF1 Suppressor of Hairless Lag (CSL), binds to DNA and activates transcription of target genes [223]. The role of Notch signal in HCC is still controversial, and the main reports currently support Notch’s carcinogenic effects. For example, after the constitutive activation of NICD, mice developed HCC [224]. Moreover, aberrant Notch expression and resistance to anticancer therapy are frequently found in human HCC tissues [225-227]. However, Viatour et al. found that in mouse models, the blockade of Notch gene leads to accelerated HCC development, thereby supporting the tumor suppressor effect of Notch signal [228]. The only study to date found that Notch1 acts as a substrate for the E3 ligase Fbxw7 (Table 2; Figure 5) [166].
Fbxw7
As mentioned above, Fbxw7 has been recognized as a universal tumor suppressor in human cancers. It is responsible for the transfer of ubiquitin molecules to substrates, leading to their degradation [229]. Studies have shown that several specific substrates of Fbxw7 include Notch1, c-Myc, etc. [161,230]. In addition, the deletion of Fbxw7 leads to the continued accumulation of Notch1 and its downstream molecule NICD as well as migration and invasion of HCC, suggesting that Fbxw7 may act as a tumor suppressor by negatively regulating the abundance of Notch1 in HCC [166]. However, it is not clear whether Fbxw7 down-regulates the expression of Notch1 through UPS-dependent methods, and further verification is needed.
HBV infection
The most prominent pathogenic factor of HCC is chronic HBV infection, which accounts for up to 54% of HCC cases [231]. HBV DNA integrates into the host genome early in the infection, causing genomic instability and direct insertional mutagenesis of different oncogenes [232]. The neoplastic transformation of liver cells can be accelerated by the persistent expression of HBV regulatory proteins [231]. The HBV X protein (HBx), a multifunctional regulator encoded by the HBV genome, has been shown to be significantly engaged in the malignant transformation process of HCC [233]. For instance, HBx restricts the nucleotide excision repair pathway [234], and prevents apoptosis via the PI3K/AKT and p38/MAPK pathways [235,236]. Furthermore, anti-HBV drugs such as telbivudine, entecavir, and interferon-2b have been shown to inhibit the growth of HBV-related HCC by downregulating the expression of HBx [237]. Increasing evidence shows that Hbx is implicated in the pathophysiology of HBV-related liver diseases, and that HBx downregulation is one of the treatment options for HBV-related HCC [237]. Recently, many studies have demonstrated that various E3 ligases regulate HBV replication and HBx protein expression via ubiquitination modification to exert cancer-promoting or anti-tumor effects in HBV-related HCC (Table 2; Figure 6), such as NEDD4 [238], SIAH1 [239,240], MARCH5 [241], HDM2 [242], MSL2 [243], DDB1 [244], and CUL4B [245].
NEDD4
NEDD4 is overexpressed in a variety of human cancers and can function as either a tumor promoter or suppressor [246]. As described above, NEDD4 has been reported to promote the occurrence of HCC by targeting the PI3K/AKT/mTOR pathways and the Hippo pathway, respectively [128,129,155]. Similarly, other reports have also found the carcinogenic effects of NEDD4 in HCC [127,169]. However, a recent study has provided a different conclusion. Wan T et al. found that the upregulation of NEDD4 in HBV-associated HCC cell lines decreased proliferation, migration, and invasion [238]. Mechanistically, NEDD4-mediated K48-linked ubiquitination of HBx protein inhibits HBV-associated HCC progression [238]. In addition, another previous study also reported the anti-tumor effect of NEDD4 [247]. They showed that NEDD4 induces the degradation of GUCD1, which is upregulated in proliferating hepatocytes [247]. In summary, these findings indicate that the function of NEDD4 in HCC may be context-dependent, and further classification and exploration based on different contexts of HCC (such as virus-related, alcohol-related or adiposity-related, etc.) are expected to reveal the role of NEDD4 in HCC.
SIAH1
As mentioned earlier, SIAH1 plays a tumor-promoting or anti-tumor effect by regulating a variety of downstream substrate proteins [56-58]. Recently, HBx was identified as a novel substrate of SIAH1 that can be specifically recognized and ubiquitinated for degradation by SIAH1, and SIAH1 can inhibit the transcriptional capacity of HBx in vitro and in vivo [239]. Importantly, the same group also showed that SIAH1 mRNA and protein expression were significantly down-regulated in 270 HCC tissue specimens and 9 HCC cell lines [240]. In addition, they further identified three truncated natural HBx variants (HBx-D1, HBx-D2 and HBx-D3) derived from HCC tissue samples, and found that SIAH1 could not reduce the three truncated HBx variants and failed to inhibit their transactivation activity in the heat shock element (HSE) [240]. Moreover, compared with full-length HBx, SIAH1 has a weaker association with three HBx truncations, suggesting that high frequency of HBx truncates in HCC may protect HBx variants from SIAH1 mediated degradation, and the accumulation of HBx variants may improve their ability to promote HCC development [240].
MARCH5
Membrane-associated RING-CH E3 (MARCH) 5/MITOL is a RING-type E3 ligase, which has an important role in mitochondrial dynamics and protein quality control [248]. The study has shown that high expression levels of HBx result in abnormal mitochondrial morphology and function [249]. And mitochondrial damage induced by HBx increases ROS levels and cyclooxygenase-2 gene expression, which in turn leads to liver inflammation and HBV-related HCC [250]. Furthermore, Yoo YS et al. identified the HBx protein as the target of MARCH5 in HCC [241]. Specifically, MARCH5 degrades HBx through the proteasome-dependent pathway, thereby reducing the occurrence of liver inflammation and HBx-mediated HCC [241].
HDM2
Human double minute (HDM) 2 (in mouse known as MDM2) E3 ligase is a major negative regulator of p53. Increased HDM2 expression has been seen in a variety of malignancies, indicating that it has an oncogenic function [251]. Neddylation is a ubiquitin-like modification that regulates a variety of signal pathways, such as apoptosis, DNA damage, and nucleolar stress signals, by controlling the stability and activity of the target protein [252]. In addition to the ubiquitination regulation pathway of p53, HDM2 has been reported to mediate a new type of post-translational modification (Neddylation) of HBx protein [242]. NEDD8, a ubiquitin-like molecule recruited by HDM2, specifically Neddylates HBx at the K91 and K95 sites, thereby preventing the ubiquitination modification of HBx and maintaining the structural stability of HBx [242]. Furthermore, in vivo and in vitro experiments have shown that Neddylation of HBx is beneficial to its functions in promoting cell proliferation and tumor growth [242]. These results indicate that HDM2 exerts a potential carcinogenic effect through Neddylation modification of HBx that is independent from the p53 pathway in HCC.
Other E3 ligases
In addition to the E3 ligase that can directly target HBx, several E3 ligases may play a role in HCC carcinogenesis by regulating HBx in indeterminate or indirect manners. Liu N et al. have demonstrated that HBx-elevated male-specific lethal (MSL)2, an E3 ligase with RING finger domain can maintain the stability of nuclear HBV covalently closed circular DNA (cccDNA) by ubiquitinating and degrading APOBEC3B in HCC cells [243]. It has been reported that APOBEC3B is responsible for the deamination of HBV cccDNA, and overexpression of APOBEC3B can reduce the level of HBV cccDNA in HCC cells infected with HBV particles [253]. In another study, HBx induces DNA-damage binding (DDB) 1 protein E3 ligase targets the structural maintenance of chromosome (Smc)5/6 complex for degradation, thereby reducing the transcriptional inhibition of Smc5/6 and stimulating HBV gene expression [244]. In addition, one latest report showed that CUL4B facilitates HBV replication by interacting with HBx and preventing its ubiquitin-dependent proteasomal degradation [245].
Other E3s
Above we introduced some E3s that regulate important signal pathways and pathological processes involved in the hepatocarcinogenesis. Moreover, many E3s have been identified to be abnormally expressed in HCC or participate in HCC tumorigenesis by targeting other tumor promoters or repressors. Here we will not expand the description further, and summarize them in Table 2.
Targeting E3s for HCC therapy
Accumulating data strongly indicated that deregulation of UPS contributes to cancer progression and that overexpression of certain components of UPS such as E3 ligase is usually associated with poor prognosis. Thus, targeting the activity of UPS components has been considered as a possible cancer treatment strategy. Bortezomib, a selective proteasome inhibitor, is used to treat refractory myeloma and mantle cell lymphoma [254,255]. Its successful application opens up the possibility of regulating the UPS to treat human malignant tumors [254,255]. At the same time, this also suggests that UPS-related regulation may be a new strategy for the treatment of HCC. A previous international multicenter phase II trial evaluated Bortezomib as a monotherapy for patients with unresectable HCC [256]. Despite the lack of a meaningful therapeutic benefit, this trial is nevertheless a ground-breaking clinical trial for UPS-related drug development in HCC. Besides, another study showed that Disulfiram, which is used to treat alcoholism, has been found to have proteasome inhibitory activity as well as specific activity of against zinc fingers and RING-finger ubiquitin ligase [257]. In addition, as a new proteasome inhibitor, the carcinogenic effect of Disulfiram has been confirmed in further preclinical and clinical studies [258]. Thence, Disulfiram may become a powerful anticancer agent in HCC related alcohol or other kinds of HCC.
Generally, inhibitors in the UPS upstream of the proteasome can prevent the degradation of one or several key proteins. Targeting a specific E3 will make it possible to selectively stabilize proteins regulated by related E3, and compared with the inhibition of the proteasome, may in turn lead to reduced toxicity and increased therapeutic index. Considering that many E3s play key roles in HCC tumorigenesis, targeting E3 may become a new approach for HCC treatment. In this regard, the development of inhibitors of some potential tumor promoters in HCC has made certain progress. For example, cis-imidazoline derivatives (nicknamed “Nutlins”) developed by Roche competitively binds to the MDM2-p53 interaction to activate the p53 pathway, leading to cell cycle arrest, cell death, and growth inhibition (Table 3) [259,260]. In addition, inhibitors of E3s such as SKP2, Cdc20 and IAPs have also been used in prostate cancer, breast cancer, and osteosarcoma and other malignant tumors (Table 3) [261-264]. More importantly, a study has demonstrated that the anti-cancer small molecule MLN4924 induces autophagy and apoptosis, and inhibits the growth of HCC cells [265]. Mechanistically, MLN4924 can block the Neddylation modification of cullin and inactivate the function of CRL, leading to the accumulation of CRL substrates, thereby triggering cell cycle arrest, apoptosis and senescence in cancer cells [261]. On the other hand, some E3s are recognized as tumor suppressors in HCC. As well known, VHL inhibits the occurrence and progression of HCC by repressing the expression and activity of HIF and NF-κB [215,266,267]. Wang J et al. have shown that combined intraportal injection of VHL and doxorubicin can significantly inhibit the growth of HCC in mice compared to doxorubicin alone [193]. This makes it possible to use VHL expression vector in combination with doxorubicin for transcatheter arterial chemo-embolization (TACE) [193], which is the standard treatment for unresectable liver cancer [268]. In addition, as mentioned earlier, Fbxw7 can target to degrade some tumor promoters to exert its anti-cancer effect, such as Notch1, c-Myc and so on [186,187]. Several recent studies have shown that Fbxw7 can enhance the sensitivity of liver cancer cells to sorafenib [269,270]. Therefore, the use of E3s analogs, or the use of E3s agonists in combination with other treatment methods, may become a neoadjuvant therapy for HCC within a few years. However, due to frequent mutations of E3s have insufficient specificity to recognize targets, it may cause off-target effects, which may lead to treatment failure. Notably, proteolysis targeting chimeras (PROTACs), a type of multifunctional small molecules that utilize the UPS to degrade proteins of interest (POI) [271]. PROTACs may be preferable to traditional small molecule inhibitors (SMIs) because of their unique mechanism of action, which degrades POI in a substoichiometric manner and allows PROTACs to produce longer and stronger biological effects on the target than SMIs [271]. Furthermore, PROTACs can target “non-drugable” and mutant proteins, and improve the targeting selectivity of downstream tumor promoters [271]. As a result, PROTACs have emerged as a promising technique for the development of novel targeted anti-cancer therapies. In fact, this emerging technology has been applied in various stages of preclinical and clinical development of some hematological malignancies [272-274]. We have reason to believe that it is expected to be used in targeted therapy of HCC in the future.
Table 3.
Summary of inhibitors targeting the E3s in HCC
| Targets | Compound | Chemical nature | Mechanism of action | Clinical stage in HCC | Reference |
|---|---|---|---|---|---|
| IAP | Smac | - | Increasing the apoptosis of XIAP | Proof of principle | [262] |
| LCL161 | Mimetic of Smac | Combining with paclitaxel | Phase I | [263,298,299] | |
| MDM2/p53 | Nutlin-2 | Cis-imidazoline | Targeting the p53-binding site of MDM2 | Proof of principle | [259,260] |
| RITA | - | Preventing the recognition of p53 by MDM2 | Proof of principle | [300] | |
| Spiro-oxindoles | - | Competing MDM2 | Proof of principle | [301] | |
| PXN727/PXN822 | Isoquinolinone | Serving as the MDM2 antagonist | Proof of principle | [302] | |
| TDP521252/TDP | Benzodiazepin | Serving as the MDM2 antagonists | Proof of principle | [302] | |
| -665759 | -edione | ||||
| JNJ26854165 | Tryptamine | Serving as the MDM2 antagonists | Proof of principle | [302] | |
| HLI98 series | - | Serving as the MDM2 antagonists | Proof of principle | [302] | |
| MEL23/MEL24 | Tetrahydro-β-carboline with barbituric acid group | Serving as the MDM2 antagonists | Proof of principle | [302] | |
| SJ172550 | - | Serving as the MDM2 antagonists | Proof of principle | [302] | |
| RO2443/RO5963 | Indolyl hydantoins | Serving as the MDM2 antagonists | Proof of principle | [302] | |
| SCFskp2 | CpdA | - | Interfering with SKP2 ligase function | Proof of principle | [264] |
| C1, C2, C16, C20 | - | Contacting key compound-receptor | Proof of principle | [303] |
Discussion
Tumors are highly heterogeneous diseases and are complex at the genetic and epigenetic levels. In addition to simple mutations and amplifications, post-translational modifications of proteins are now being revealed on a larger scale. In this complex environment, E3 ligases can regulate the activity and function of substrates, including oncogenic and anti-oncogenic proteins, which are critical for tumorigenesis and progression as well as drug treatment sensitivity. In addition to targeting substrates for proteasomal degradation, E3 ligases also regulate the DNA damage response and the localization and activity of key protein kinases in many signal pathways, such as NF-κB, PI3K/AKT, and Wnt/β-catenin [21,59], which makes them a central hub for promoting cancer signatures. The pathogenesis of HCC is complex and its development involves multiple key signal pathways. Apart from their individual independent signal pathways, there are multiple levels of crosstalks between these signal pathways to regulate each other. Therefore, it is not surprising that E3 ligases play different roles in HCC.
The function and significance of E3 ligases in HCC have been well studied, but many questions still remain to be addressed. For example, the main pathways responsible for ectopic expression of E3s are not yet clear. Although the function of most E3s in tumor progression is dependent on E3 activity, whether specific E3s play a proteolytic or non-proteolytic role in HCC remains to be investigated. Furthermore, the substrates and downstream effectors of some E3s in HCC are still largely unknown. One more important question is how E3 ligases recognize their specific substrates, as only a few recognition motifs, called “degron”, have been identified so far [275]. Degron is the target of E3 ligases and directs the destruction of proteins bearing degrons via UPS [275]. Depending on the degron motif, E3 ligases achieve precise targeting and degradation of oncogenic protein, thereby controlling the stability of the protein and its involvement in physiological processes or tumorigenesis [276]. Recent systematic analyses of primary tumors have shown that stable mutations in the oncogenic protein degron and inactivating mutations in E3 that recognize such degron are driving events for tumorigenesis [277]. Specifically, these abnormal mutations disrupt the precise recognition of substrates by E3, resulting in abnormal accumulation of oncogenic protein, ultimately triggering the malignant transformation process in normal cells. Therefore, the design of precision therapies targeting E3 based on degron recognition should be promising. However, only 25 classes of degron motifs have been identified compared to the approximately 600 E3 ligases in the human proteome [275]. Without knowledge of degron motifs, it is difficult to assess whether post-translational modifications regulate the interaction of E3 ligases with degrons and whether there is a direct relationship between E3 ligases and putative substrates. Some of the following measures are expected to solve the above problems: (i) further mining the specific recognition motifs of E3s to substrates; (ii) integration of target E3 structural information, affinity purification-mass spectrometry (AP-MS) and degron motif mining for system-wide identification of downstream substrates in HCC; (iii) identification of the type of E3 modifications to substrates and the major pathways responsible for E3 ectopic expression in HCC.
Given the critical role of E3 ligases in regulating multiple important cellular pathways in HCC, including Wnt/β-catenin, RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, etc., E3 ligases are attractive drug targets for cancer therapy. Compared to proteasome inhibitors that non-selectively blocks protein degradation throughout the system, compounds targeting specific E3 enzymes are expected to more selectively modulate protein levels, thereby increasing specificity with minimal toxicity. Notably, PROTACs have emerged as an important future therapeutic opportunity in the field of E3 ligases. PROTACs induce the proximity of E3 ligases to target proteins (e.g., oncogenic protein) to promote ubiquitination and degradation of the target protein [271]. Central to this technology is the identification of a specific ligase to which it will bind, and this is an area that requires focused attention to accelerate the translation of this technology into the clinical setting. In conclusion, E3 ligases are important tumor regulators and potential therapeutic targets for HCC. The development of novel anticancer drugs targeting specific E3 ligases and E3 ligase-based cancer biomarkers will also be used in the clinic as therapeutic, diagnostic tools or prognostic indicators for the benefit of HCC patients.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Provincial Universities of Zhejiang (No. SJLZ2022004), The Program of Ningbo Medical Research Center for Digestive System Cancer (No. 2019A21003), the Natural Science Foundation of Ningbo (grant nos. 202003N4197, 2021J294 and 2021J065), Zhejiang Medicine and Health Project (No. 2022KY1079), and the K.C.Wong Magna Fund in Ningbo University.
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 4.Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas MB, Abbruzzese JL. Opportunities for targeted therapies in hepatocellular carcinoma. J. Clin. Oncol. 2005;23:8093–8108. doi: 10.1200/JCO.2004.00.1537. [DOI] [PubMed] [Google Scholar]
- 6.Farinati F, Sergio A, Baldan A, Giacomin A, Di Nolfo MA, Del Poggio P, Benvegnu L, Rapaccini G, Zoli M, Borzio F, Giannini EG, Caturelli E, Trevisani F. Early and very early hepatocellular carcinoma: when and how much do staging and choice of treatment really matter? A multi-center study. BMC Cancer. 2009;9:33. doi: 10.1186/1471-2407-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu M, Jiang L, Guan XY. The genetic and epigenetic alterations in human hepatocellular carcinoma: a recent update. Protein Cell. 2014;5:673–691. doi: 10.1007/s13238-014-0065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow AK, Yau SW, Ng L. Novel molecular targets in hepatocellular carcinoma. World J Clin Oncol. 2020;11:589–605. doi: 10.5306/wjco.v11.i8.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 10.Hershko A. Ubiquitin: roles in protein modification and breakdown. Cell. 1983;34:11–12. doi: 10.1016/0092-8674(83)90131-9. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Shaik S, Dai X, Wu Q, Zhou X, Wang Z, Wei W. Targeting the ubiquitin pathway for cancer treatment. Biochim Biophys Acta. 2015;1855:50–60. doi: 10.1016/j.bbcan.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lipford JR, Deshaies RJ. Diverse roles for ubiquitin-dependent proteolysis in transcriptional activation. Nat Cell Biol. 2003;5:845–850. doi: 10.1038/ncb1003-845. [DOI] [PubMed] [Google Scholar]
- 13.Dong LH, Ran ML, Li Z, Peng FZ, Chen B. The role of ubiquitin-proteasome pathway in spermatogenesis. Yi Chuan. 2016;38:791–800. doi: 10.16288/j.yczz.16-120. [DOI] [PubMed] [Google Scholar]
- 14.Chitra S, Nalini G, Rajasekhar G. The ubiquitin proteasome system and efficacy of proteasome inhibitors in diseases. Int J Rheum Dis. 2012;15:249–260. doi: 10.1111/j.1756-185X.2012.01737.x. [DOI] [PubMed] [Google Scholar]
- 15.Brown R, Kaganovich D. Look out autophagy, ubiquilin UPS its game. Cell. 2016;166:797–799. doi: 10.1016/j.cell.2016.07.048. [DOI] [PubMed] [Google Scholar]
- 16.O’Connell BC, Harper JW. Ubiquitin proteasome system (UPS): what can chromatin do for you? Curr Opin Cell Biol. 2007;19:206–214. doi: 10.1016/j.ceb.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8:835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 18.Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett EJ, Harper JW. DNA damage: ubiquitin marks the spot. Nat Struct Mol Biol. 2008;15:20–22. doi: 10.1038/nsmb0108-20. [DOI] [PubMed] [Google Scholar]
- 20.Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 21.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 22.Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Yang LY, Yang ZL, Peng JX, Yang JQ. Elevated expression of autocrine motility factor receptor correlates with overexpression of RhoC and indicates poor prognosis in hepatocellular carcinoma. Dig Dis Sci. 2007;52:770–775. doi: 10.1007/s10620-006-9479-4. [DOI] [PubMed] [Google Scholar]
- 24.Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21:301–307. doi: 10.1038/nsmb.2780. [DOI] [PubMed] [Google Scholar]
- 25.Vander Kooi CW, Ohi MD, Rosenberg JA, Oldham ML, Newcomer ME, Gould KL, Chazin WJ. The Prp19 U-box crystal structure suggests a common dimeric architecture for a class of oligomeric E3 ubiquitin ligases. Biochemistry. 2006;45:121–130. doi: 10.1021/bi051787e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morreale FE, Walden H. Types of ubiquitin ligases. Cell. 2016;165:248–248. e241. doi: 10.1016/j.cell.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Leslie PL, Ke H, Zhang Y. The MDM2 RING domain and central acidic domain play distinct roles in MDM2 protein homodimerization and MDM2-MDMX protein heterodimerization. J Biol Chem. 2015;290:12941–12950. doi: 10.1074/jbc.M115.644435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 2013;32:2307–2320. doi: 10.1038/emboj.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelsall IR, Kristariyanto YA, Knebel A, Wood NT, Kulathu Y, Alpi AF. Coupled monoubiquitylation of the co-E3 ligase DCNL1 by Ariadne-RBR E3 ubiquitin ligases promotes cullin-RING ligase complex remodeling. J Biol Chem. 2019;294:2651–2664. doi: 10.1074/jbc.RA118.005861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kucharski TJ, Minshall PE, Moustafa-Kamal M, Turnell AS, Teodoro JG. Reciprocal regulation between 53BP1 and the anaphase-promoting complex/cyclosome is required for genomic stability during mitotic stress. Cell Rep. 2017;18:1982–1995. doi: 10.1016/j.celrep.2017.01.080. [DOI] [PubMed] [Google Scholar]
- 31.Weber J, Polo S, Maspero E. HECT E3 ligases: a tale with multiple facets. Front Physiol. 2019;10:370. doi: 10.3389/fphys.2019.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lorenz S. Structural mechanisms of HECT-type ubiquitin ligases. Biol Chem. 2018;399:127–145. doi: 10.1515/hsz-2017-0184. [DOI] [PubMed] [Google Scholar]
- 33.Marín I, Lucas JI, Gradilla AC, Ferrús A. Parkin and relatives: the RBR family of ubiquitin ligases. Physiol Genomics. 2004;17:253–263. doi: 10.1152/physiolgenomics.00226.2003. [DOI] [PubMed] [Google Scholar]
- 34.Dove KK, Klevit RE. RING-between-RING E3 ligases: emerging themes amid the variations. J Mol Biol. 2017;429:3363–3375. doi: 10.1016/j.jmb.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smit JJ, Sixma TK. RBR E3-ligases at work. EMBO Rep. 2014;15:142–154. doi: 10.1002/embr.201338166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 37.Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J. 1989;8:1711–1717. doi: 10.1002/j.1460-2075.1989.tb03563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peifer M, McCrea PD, Green KJ, Wieschaus E, Gumbiner BM. The vertebrate adhesive junction proteins beta-catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J Cell Biol. 1992;118:681–691. doi: 10.1083/jcb.118.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stamos JL, Weis WI. The β-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5:a007898. doi: 10.1101/cshperspect.a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamoto H, Hinoi T, Michiue T, Fukui A, Usui H, Janssens V, Van Hoof C, Goris J, Asashima M, Kikuchi A. Inhibition of the Wnt signaling pathway by the PR61 subunit of protein phosphatase 2A. J Biol Chem. 2001;276:26875–26882. doi: 10.1074/jbc.M100443200. [DOI] [PubMed] [Google Scholar]
- 41.Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 42.Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 43.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Xu Z, Li Q, Feng Q, Zheng C, Du Y, Yuan R, Peng X. USP28 facilitates pancreatic cancer progression through activation of Wnt/β-catenin pathway via stabilising FOXM1. Cell Death Dis. 2021;12:887. doi: 10.1038/s41419-021-04163-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flack JE, Mieszczanek J, Novcic N, Bienz M. Wnt-dependent inactivation of the groucho/TLE co-repressor by the HECT E3 ubiquitin ligase Hyd/UBR5. Mol Cell. 2017;67:181–193. e185. doi: 10.1016/j.molcel.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding Y, Zhang Y, Xu C, Tao QH, Chen YG. HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J Biol Chem. 2013;288:8289–8298. doi: 10.1074/jbc.M112.433185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, Myer VE, Finan PM, Porter JA, Huang SM, Cong F. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol. 2011;13:623–629. doi: 10.1038/ncb2222. [DOI] [PubMed] [Google Scholar]
- 48.Xia K, Zhang Y, Cao S, Wu Y, Guo W, Yuan W, Zhang S. miR-411 regulated ITCH expression and promoted cell proliferation in human hepatocellular carcinoma cells. Biomed Pharmacother. 2015;70:158–163. doi: 10.1016/j.biopha.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Guo W, Zhang J, Zhang D, Cao S, Li G, Zhang S, Wang Z, Wen P, Yang H, Shi X, Pan J, Ye H. Polymorphisms and expression pattern of circular RNA circ-ITCH contributes to the carcinogenesis of hepatocellular carcinoma. Oncotarget. 2017;8:48169–48177. doi: 10.18632/oncotarget.18327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu M, Deng X, Zhong Y, Hu L, Zhang X, Liang Y, Li X, Ye X. MafF is regulated via the circ-ITCH/miR-224-5p axis and acts as a tumor suppressor in hepatocellular carcinoma. Oncol Res. 2020;28:299–309. doi: 10.3727/096504020X15796890809840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang B, Zhao J, Huo T, Zhang M, Wu X. Effects of CircRNA-ITCH on proliferation and apoptosis of hepatocellular carcinoma cells through inhibiting Wnt/β-catenin signaling pathway. J Buon. 2020;25:1368–1374. [PubMed] [Google Scholar]
- 52.Wei W, Li M, Wang J, Nie F, Lin L. The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol Cell Biol. 2012;32:3903–3912. doi: 10.1128/MCB.00251-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsui YM, Sze KM, Tung EK, Ho DW, Lee TK, Ng IO. Dishevelled-3 phosphorylation is governed by HIPK2/PP1Cα/ITCH axis and the non-phosphorylated form promotes cancer stemness via LGR5 in hepatocellular carcinoma. Oncotarget. 2017;8:39430–39442. doi: 10.18632/oncotarget.17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsuo K, Satoh S, Okabe H, Nomura A, Maeda T, Yamaoka Y, Ikai I. SIAH1 inactivation correlates with tumor progression in hepatocellular carcinomas. Genes Chromosomes Cancer. 2003;36:283–291. doi: 10.1002/gcc.10170. [DOI] [PubMed] [Google Scholar]
- 55.Jiang X, Shen X. Knockdown of miR-299-5p inhibits the progression of hepatocellular carcinoma by targeting SIAH1. Bull Cancer. 2018;105:873–883. doi: 10.1016/j.bulcan.2018.07.013. [DOI] [PubMed] [Google Scholar]
- 56.Long L, Xiang H, Liu J, Zhang Z, Sun L. ZEB1 mediates doxorubicin (Dox) resistance and mesenchymal characteristics of hepatocarcinoma cells. Exp Mol Pathol. 2019;106:116–122. doi: 10.1016/j.yexmp.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 57.Brauckhoff A, Malz M, Tschaharganeh D, Malek N, Weber A, Riener MO, Soll C, Samarin J, Bissinger M, Schmidt J, Longerich T, Ehemann V, Schirmacher P, Breuhahn K. Nuclear expression of the ubiquitin ligase seven in absentia homolog (SIAH)-1 induces proliferation and migration of liver cancer cells. J Hepatol. 2011;55:1049–1057. doi: 10.1016/j.jhep.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 58.Yoshibayashi H, Okabe H, Satoh S, Hida K, Kawashima K, Hamasu S, Nomura A, Hasegawa S, Ikai I, Sakai Y. SIAH1 causes growth arrest and apoptosis in hepatoma cells through beta-catenin degradation-dependent and -independent mechanisms. Oncol Rep. 2007;17:549–556. [PubMed] [Google Scholar]
- 59.Lu M, Wu J, Hao ZW, Shang YK, Xu J, Nan G, Li X, Chen ZN, Bian H. Basolateral CD147 induces hepatocyte polarity loss by E-cadherin ubiquitination and degradation in hepatocellular carcinoma progress. Hepatology. 2018;68:317–332. doi: 10.1002/hep.29798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu M, Jiang K, Lin G, Liu P, Yan Y, Ye T, Yao G, Barr MP, Liang D, Wang Y, Gong P, Meng S, Piao H. Ajuba inhibits hepatocellular carcinoma cell growth via targeting of β-catenin and YAP signaling and is regulated by E3 ligase Hakai through neddylation. J Exp Clin Cancer Res. 2018;37:165. doi: 10.1186/s13046-018-0806-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li CH, Yen CH, Chen YF, Lee KJ, Fang CC, Zhang X, Lai CC, Huang SF, Lin HK, Arthur Chen YM. Characterization of the GNMT-HectH9-PREX2 tripartite relationship in the pathogenesis of hepatocellular carcinoma. Int J Cancer. 2017;140:2284–2297. doi: 10.1002/ijc.30652. [DOI] [PubMed] [Google Scholar]
- 62.Gruber S, Straub BK, Ackermann PJ, Wunderlich CM, Mauer J, Seeger JM, Büning H, Heukamp L, Kashkar H, Schirmacher P, Brüning JC, Wunderlich FT. Obesity promotes liver carcinogenesis via Mcl-1 stabilization independent of IL-6Rα signaling. Cell Rep. 2013;4:669–680. doi: 10.1016/j.celrep.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 63.Ho NPY, Leung CON, Wong TL, Lau EYT, Lei MML, Mok EHK, Leung HW, Tong M, Ng IOL, Yun JP, Ma S, Lee TKW. The interplay of UBE2T and Mule in regulating Wnt/β-catenin activation to promote hepatocellular carcinoma progression. Cell Death Dis. 2021;12:148. doi: 10.1038/s41419-021-03403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang Y, Mao FF, Guo L, Guo WX. TRIM56 suppresses the malignant development of hepatocellular carcinoma via targeting RBM24 and inactivating the Wnt signaling. Eur Rev Med Pharmacol Sci. 2021;25:722–730. doi: 10.26355/eurrev_202101_24633. [DOI] [PubMed] [Google Scholar]
- 65.Jiang J, Yu C, Chen M, Tian S, Sun C. Over-expression of TRIM37 promotes cell migration and metastasis in hepatocellular carcinoma by activating Wnt/β-catenin signaling. Biochem Biophys Res Commun. 2015;464:1120–1127. doi: 10.1016/j.bbrc.2015.07.089. [DOI] [PubMed] [Google Scholar]
- 66.Yuan J, Han B, Hu H, Qian Y, Liu Z, Wei Z, Liang X, Jiang B, Shao C, Gong Y. CUL4B activates Wnt/β-catenin signalling in hepatocellular carcinoma by repressing Wnt antagonists. J Pathol. 2015;235:784–795. doi: 10.1002/path.4492. [DOI] [PubMed] [Google Scholar]
- 67.Schwarz SE, Rosa JL, Scheffner M. Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J Biol Chem. 1998;273:12148–12154. doi: 10.1074/jbc.273.20.12148. [DOI] [PubMed] [Google Scholar]
- 68.Ho KC, Zhou Z, She YM, Chun A, Cyr TD, Yang X. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability [corrected] . Proc Natl Acad Sci U S A. 2011;108:4870–4875. doi: 10.1073/pnas.1101273108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salah Z, Melino G, Aqeilan RI. Correction: negative regulation of the hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res. 2019;79:3007. doi: 10.1158/0008-5472.CAN-19-1086. [DOI] [PubMed] [Google Scholar]
- 70.Maillard I, Pear WS. Notch and cancer: best to avoid the ups and downs. Cancer Cell. 2003;3:203–205. doi: 10.1016/s1535-6108(03)00052-7. [DOI] [PubMed] [Google Scholar]
- 71.Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, Croce CM, Melino G. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci U S A. 2006;103:12753–12758. doi: 10.1073/pnas.0603449103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, Cesareni G, Melino G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–848. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gao C, Chen YG. Dishevelled: the hub of Wnt signaling. Cell Signal. 2010;22:717–727. doi: 10.1016/j.cellsig.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 74.Zhou R, Wu Y, Wang W, Su W, Liu Y, Wang Y, Fan C, Li X, Li G, Li Y, Xiong W, Zeng Z. Circular RNAs (circRNAs) in cancer. Cancer Lett. 2018;425:134–142. doi: 10.1016/j.canlet.2018.03.035. [DOI] [PubMed] [Google Scholar]
- 75.Peng Y, Wang HH. Cir-ITCH inhibits gastric cancer migration, invasion and proliferation by regulating the Wnt/β-catenin pathway. Sci Rep. 2020;10:17443. doi: 10.1038/s41598-020-74452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li S, Yu C, Zhang Y, Liu J, Jia Y, Sun F, Zhang P, Li J, Guo L, Xiao H, Gao F, Deng X, Cai Z, Cai J. Circular RNA cir-ITCH is a potential therapeutic target for the treatment of castration-resistant prostate cancer. Biomed Res Int. 2020;2020:7586521. doi: 10.1155/2020/7586521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang ST, Liu LB, Li XM, Wang YF, Xie PJ, Li Q, Wang R, Wei Q, Kang YH, Meng R, Feng XH. Circ-ITCH regulates triple-negative breast cancer progression through the Wnt/β-catenin pathway. Neoplasma. 2019;66:232–239. doi: 10.4149/neo_2018_180710N460. [DOI] [PubMed] [Google Scholar]
- 78.Zhang H, Wang J, Ge Y, Ye M, Jin X. Siah1 in cancer and nervous system diseases (review) Oncol Rep. 2022;47:35. doi: 10.3892/or.2021.8246. [DOI] [PubMed] [Google Scholar]
- 79.Wen YY, Yang ZQ, Song M, Li BL, Yao XH, Chen XL, Zhao J, Lu YY, Zhu JJ, Wang EH. The expression of SIAH1 is downregulated and associated with Bim and apoptosis in human breast cancer tissues and cells. Mol Carcinog. 2010;49:440–449. doi: 10.1002/mc.20615. [DOI] [PubMed] [Google Scholar]
- 80.Kim CJ, Cho YG, Park CH, Jeong SW, Nam SW, Kim SY, Lee SH, Yoo NJ, Lee JY, Park WS. Inactivating mutations of the Siah-1 gene in gastric cancer. Oncogene. 2004;23:8591–8596. doi: 10.1038/sj.onc.1208113. [DOI] [PubMed] [Google Scholar]
- 81.Krämer OH, Müller S, Buchwald M, Reichardt S, Heinzel T. Mechanism for ubiquitylation of the leukemia fusion proteins AML1-ETO and PML-RARalpha. FASEB J. 2008;22:1369–1379. doi: 10.1096/fj.06-8050com. [DOI] [PubMed] [Google Scholar]
- 82.Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001;7:915–926. doi: 10.1016/s1097-2765(01)00242-8. [DOI] [PubMed] [Google Scholar]
- 83.Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- 84.Aparicio LA, Valladares M, Blanco M, Alonso G, Figueroa A. Biological influence of Hakai in cancer: a 10-year review. Cancer Metastasis Rev. 2012;31:375–386. doi: 10.1007/s10555-012-9348-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mukherjee M, Chow SY, Yusoff P, Seetharaman J, Ng C, Sinniah S, Koh XW, Asgar NF, Li D, Yim D, Jackson RA, Yew J, Qian J, Iyu A, Lim YP, Zhou X, Sze SK, Guy GR, Sivaraman J. Structure of a novel phosphotyrosine-binding domain in Hakai that targets E-cadherin. EMBO J. 2012;31:1308–1319. doi: 10.1038/emboj.2011.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Swaminathan G, Cartwright CA. Rack1 promotes epithelial cell-cell adhesion by regulating E-cadherin endocytosis. Oncogene. 2012;31:376–389. doi: 10.1038/onc.2011.242. [DOI] [PubMed] [Google Scholar]
- 87.Zhou WJ, Geng ZH, Chi S, Zhang W, Niu XF, Lan SJ, Ma L, Yang X, Wang LJ, Ding YQ, Geng JG. Slit-Robo signaling induces malignant transformation through Hakai-mediated E-cadherin degradation during colorectal epithelial cell carcinogenesis. Cell Res. 2011;21:609–626. doi: 10.1038/cr.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nola S, Daigaku R, Smolarczyk K, Carstens M, Martin-Martin B, Longmore G, Bailly M, Braga VM. Ajuba is required for Rac activation and maintenance of E-cadherin adhesion. J Cell Biol. 2011;195:855–871. doi: 10.1083/jcb.201107162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li L, Martinez SS, Hu W, Liu Z, Tjian R. A specific E3 ligase/deubiquitinase pair modulates TBP protein levels during muscle differentiation. Elife. 2015;4:e08536. doi: 10.7554/eLife.08536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.King B, Boccalatte F, Moran-Crusio K, Wolf E, Wang J, Kayembe C, Lazaris C, Yu X, Aranda-Orgilles B, Lasorella A, Aifantis I. The ubiquitin ligase Huwe1 regulates the maintenance and lymphoid commitment of hematopoietic stem cells. Nat Immunol. 2016;17:1312–1321. doi: 10.1038/ni.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen LJ, Xu WM, Yang M, Wang K, Chen Y, Huang XJ, Ma QH. HUWE1 plays important role in mouse preimplantation embryo development and the dysregulation is associated with poor embryo development in humans. Sci Rep. 2016;6:37928. doi: 10.1038/srep37928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maghames CM, Lobato-Gil S, Perrin A, Trauchessec H, Rodriguez MS, Urbach S, Marin P, Xirodimas DP. NEDDylation promotes nuclear protein aggregation and protects the ubiquitin proteasome system upon proteotoxic stress. Nat Commun. 2018;9:4376. doi: 10.1038/s41467-018-06365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dominguez-Brauer C, Khatun R, Elia AJ, Thu KL, Ramachandran P, Baniasadi SP, Hao Z, Jones LD, Haight J, Sheng Y, Mak TW. E3 ubiquitin ligase Mule targets β-catenin under conditions of hyperactive Wnt signaling. Proc Natl Acad Sci U S A. 2017;114:E1148–E1157. doi: 10.1073/pnas.1621355114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maslov LN, Lishmanov IuB. [Participation of central and peripheral mu- and delta opiate receptors in anti-arrhythmia action of enkephalins] . Biull Eksp Biol Med. 1991;112:124–126. [PubMed] [Google Scholar]
- 95.Venuto S, Merla G. E3 ubiquitin ligase TRIM proteins, cell cycle and mitosis. Cells. 2019;8:510. doi: 10.3390/cells8050510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fletcher AJ, Christensen DE, Nelson C, Tan CP, Schaller T, Lehner PJ, Sundquist WI, Towers GJ. TRIM5α requires Ube2W to anchor Lys63-linked ubiquitin chains and restrict reverse transcription. EMBO J. 2015;34:2078–2095. doi: 10.15252/embj.201490361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen Y, Zhao J, Li D, Hao J, He P, Wang H, Zhang M. TRIM56 suppresses multiple myeloma progression by activating TLR3/TRIF signaling. Yonsei Med J. 2018;59:43–50. doi: 10.3349/ymj.2018.59.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao L, Zhang P, Su XJ, Zhang B. The ubiquitin ligase TRIM56 inhibits ovarian cancer progression by targeting vimentin. J Cell Physiol. 2018;233:2420–2425. doi: 10.1002/jcp.26114. [DOI] [PubMed] [Google Scholar]
- 99.Xue M, Zhang K, Mu K, Xu J, Yang H, Liu Y, Wang B, Wang Z, Li Z, Kong Q, Li X, Wang H, Zhu J, Zhuang T. Regulation of estrogen signaling and breast cancer proliferation by an ubiquitin ligase TRIM56. Oncogenesis. 2019;8:30. doi: 10.1038/s41389-019-0139-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang W, Zhang J, Huo M, Gao J, Yang T, Yin X, Wang P, Leng S, Feng D, Chen Y, Yang Y, Wang Y. CUL4B promotes breast carcinogenesis by coordinating with transcriptional repressor complexes in response to hypoxia signaling pathway. Adv Sci (Weinh) 2021;8:2001515. doi: 10.1002/advs.202001515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leng S, Huang W, Chen Y, Yang Y, Feng D, Liu W, Gao T, Ren Y, Huo M, Zhang J, Yang Y, Wang Y. SIRT1 coordinates with the CRL4B complex to regulate pancreatic cancer stem cells to promote tumorigenesis. Cell Death Differ. 2021;28:3329–3343. doi: 10.1038/s41418-021-00821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yuan J, Jiang B, Zhang A, Qian Y, Tan H, Gao J, Shao C, Gong Y. Accelerated hepatocellular carcinoma development in CUL4B transgenic mice. Oncotarget. 2015;6:15209–15221. doi: 10.18632/oncotarget.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M, Battiston C, Van Laarhoven S, Fiel MI, Di Feo A, Hoshida Y, Yea S, Toffanin S, Ramos A, Martignetti JA, Mazzaferro V, Bruix J, Waxman S, Schwartz M, Meyerson M, Friedman SL, Llovet JM. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–1983. 1983.e1971–1911. doi: 10.1053/j.gastro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88. doi: 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, de Velasco G, Jeong KJ, Akbani R, Hadjipanayis A, Pantazi A, Bristow CA, Lee E, Mahadeshwar HS, Tang J, Zhang J, Yang L, Seth S, Lee S, Ren X, Song X, Sun H, Seidman J, Luquette LJ, Xi R, Chin L, Protopopov A, Westbrook TF, Shelley CS, Choueiri TK, Ittmann M, Van Waes C, Weinstein JN, Liang H, Henske EP, Godwin AK, Park PJ, Kucherlapati R, Scott KL, Mills GB, Kwiatkowski DJ, Creighton CJ. A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell. 2017;31:820–832. e823. doi: 10.1016/j.ccell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, Meyerson M, Beroukhim R. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roskoski R Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res. 2015;94:9–25. doi: 10.1016/j.phrs.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 109.Zhu L, Qin C, Li T, Ma X, Qiu Y, Lin Y, Ma D, Qin Z, Sun C, Shen X, Zhao Y, Han L. The E3 ubiquitin ligase TRIM7 suppressed hepatocellular carcinoma progression by directly targeting Src protein. Cell Death Differ. 2020;27:1819–1831. doi: 10.1038/s41418-019-0464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Skurat AV, Dietrich AD, Zhai L, Roach PJ. GNIP, a novel protein that binds and activates glycogenin, the self-glucosylating initiator of glycogen biosynthesis. J Biol Chem. 2002;277:19331–19338. doi: 10.1074/jbc.M201190200. [DOI] [PubMed] [Google Scholar]
- 111.Zhai L, Dietrich A, Skurat AV, Roach PJ. Structure-function analysis of GNIP, the glycogenin-interacting protein. Arch Biochem Biophys. 2004;421:236–242. doi: 10.1016/j.abb.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 112.Montori-Grau M, Pedreira-Casahuga R, Boyer-Díaz Z, Lassot I, García-Martínez C, Orozco A, Cebrià J, Osorio-Conles O, Chacón MR, Vendrell J, Vázquez-Carrera M, Desagher S, Jiménez-Chillarón JC, Gómez-Foix AM. GNIP1 E3 ubiquitin ligase is a novel player in regulating glycogen metabolism in skeletal muscle. Metabolism. 2018;83:177–187. doi: 10.1016/j.metabol.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 113.Li Y, Wu H, Wu W, Zhuo W, Liu W, Zhang Y, Cheng M, Chen YG, Gao N, Yu H, Wang L, Li W, Yang M. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014;24:762–765. doi: 10.1038/cr.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hu X, Tang Z, Ma S, Yu Y, Chen X, Zang G. Tripartite motif-containing protein 7 regulates hepatocellular carcinoma cell proliferation via the DUSP6/p38 pathway. Biochem Biophys Res Commun. 2019;511:889–895. doi: 10.1016/j.bbrc.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 115.Yamada D, Hoshii T, Tanaka S, Hegazy AM, Kobayashi M, Tadokoro Y, Ohta K, Ueno M, Ali MA, Hirao A. Loss of Tsc1 accelerates malignant gliomagenesis when combined with oncogenic signals. J Biochem. 2014;155:227–233. doi: 10.1093/jb/mvt112. [DOI] [PubMed] [Google Scholar]
- 116.Kang YJ, Lu MK, Guan KL. The TSC1 and TSC2 tumor suppressors are required for proper ER stress response and protect cells from ER stress-induced apoptosis. Cell Death Differ. 2011;18:133–144. doi: 10.1038/cdd.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guo P, Ma X, Zhao W, Huai W, Li T, Qiu Y, Zhang Y, Han L. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1-TSC2 complex. Oncogene. 2018;37:478–488. doi: 10.1038/onc.2017.349. [DOI] [PubMed] [Google Scholar]
- 118.Guo P, Qiu Y, Ma X, Li T, Ma X, Zhu L, Lin Y, Han L. Tripartite motif 31 promotes resistance to anoikis of hepatocarcinoma cells through regulation of p53-AMPK axis. Exp Cell Res. 2018;368:59–66. doi: 10.1016/j.yexcr.2018.04.013. [DOI] [PubMed] [Google Scholar]
- 119.Lv T, Jiang L, Kong L, Yang J. MicroRNA-29c-3p acts as a tumor suppressor gene and inhibits tumor progression in hepatocellular carcinoma by targeting TRIM31. Oncol Rep. 2020;43:953–964. doi: 10.3892/or.2020.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hong SJ, Chae H, Lardaro T, Hong S, Kim KS. Trim11 increases expression of dopamine beta-hydroxylase gene by interacting with Phox2b. Biochem Biophys Res Commun. 2008;368:650–655. doi: 10.1016/j.bbrc.2008.01.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang X, Shi W, Shi H, Lu S, Wang K, Sun C, He J, Jin W, Lv X, Zou H, Shu Y. TRIM11 overexpression promotes proliferation, migration and invasion of lung cancer cells. J Exp Clin Cancer Res. 2016;35:100. doi: 10.1186/s13046-016-0379-y. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 122.Zhang Z, Xu C, Zhang X, Huang L, Zheng C, Chen H, Wang Y, Ju H, Yao Q. TRIM11 upregulation contributes to proliferation, invasion, and EMT of hepatocellular carcinoma cells. Oncol Res. 2017;25:691–699. doi: 10.3727/096504016X14774897404770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen Y, Li L, Qian X, Ge Y, Xu G. High expression of TRIM11 correlates with poor prognosis in patients with hepatocellular carcinoma. Clin Res Hepatol Gastroenterol. 2017;41:190–196. doi: 10.1016/j.clinre.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 124.Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu L, Long H, Wu Y, Li H, Dong L, Zhong JL, Liu Z, Yang X, Dai X, Shi L, Ren M, Lin Z. HRD1-mediated PTEN degradation promotes cell proliferation and hepatocellular carcinoma progression. Cell Signal. 2018;50:90–99. doi: 10.1016/j.cellsig.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 126.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 127.Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang ZJ, Zhu JJ, Yang XY, Biskup E. NEDD4 promotes cell growth and migration via PTEN/PI3K/AKT signaling in hepatocellular carcinoma. Oncol Lett. 2017;14:2649–2656. doi: 10.3892/ol.2017.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hang X, Zhu S, Di H, Wu Z, Chu K, Wang J, Xin H, Yu G, Peng H, Miao X, Xu W. NEDD4 depletion inhibits hepatocellular carcinoma growth via targeting PTEN. Cell Physiol Biochem. 2016;39:768–779. doi: 10.1159/000445667. [DOI] [PubMed] [Google Scholar]
- 130.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 131.Liao Y, Yuan S, Chen X, Zhu P, Li J, Qin L, Liao W. Up-regulation of BRCA1-associated RING domain 1 promotes hepatocellular carcinoma progression by targeting Akt signaling. Sci Rep. 2017;7:7649. doi: 10.1038/s41598-017-07962-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Roskoski R Jr. Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanomas. Pharmacol Res. 2017;117:20–31. doi: 10.1016/j.phrs.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 133.Wortzel I, Seger R. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer. 2011;2:195–209. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A, Ueki T, Hirano T, Yamamoto H, Fujimoto J, Okamoto E, Hayashi N, Hori M. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology. 1998;27:951–958. doi: 10.1002/hep.510270409. [DOI] [PubMed] [Google Scholar]
- 135.Wu PK, Becker A, Park JI. Growth inhibitory signaling of the Raf/MEK/ERK pathway. Int J Mol Sci. 2020;21:5436. doi: 10.3390/ijms21155436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jiang R, Tang J, Chen Y, Deng L, Ji J, Xie Y, Wang K, Jia W, Chu WM, Sun B. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun. 2017;8:15129. doi: 10.1038/ncomms15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Song J, Liu Y, Liu F, Zhang L, Li G, Yuan C, Yu C, Lu X, Liu Q, Chen X, Liang H, Ding Z, Zhang B. The 14-3-3σ protein promotes HCC anoikis resistance by inhibiting EGFR degradation and thereby activating the EGFR-dependent ERK1/2 signaling pathway. Theranostics. 2021;11:996–1015. doi: 10.7150/thno.51646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bai XS, Zhang C, Peng R, Jiang GQ, Jin SJ, Wang Q, Ke AW, Bai DS. RNF128 promotes malignant behaviors via EGFR/MEK/ERK pathway in hepatocellular carcinoma. Onco Targets Ther. 2020;13:10129–10141. doi: 10.2147/OTT.S269606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell. 1999;4:1029–1040. doi: 10.1016/s1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
- 140.Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998;12:3663–3674. doi: 10.1101/gad.12.23.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thien CB, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001;2:294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- 142.Haymaker C, Yang Y, Wang J, Zou Q, Sahoo A, Alekseev A, Singh D, Ritthipichai K, Hailemichael Y, Hoang ON, Qin H, Schluns KS, Wang T, Overwijk WW, Sun SC, Bernatchez C, Kwak LW, Neelapu SS, Nurieva R. Absence of Grail promotes CD8(+) T cell anti-tumour activity. Nat Commun. 2017;8:239. doi: 10.1038/s41467-017-00252-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gao J, Wang Y, Yang J, Zhang W, Meng K, Sun Y, Li Y, He QY. RNF128 promotes invasion and metastasis via the EGFR/MAPK/MMP-2 pathway in esophageal squamous cell carcinoma. Cancers (Basel) 2019;11:840. doi: 10.3390/cancers11060840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wei CY, Zhu MX, Yang YW, Zhang PF, Yang X, Peng R, Gao C, Lu JC, Wang L, Deng XY, Lu NH, Qi FZ, Gu JY. Downregulation of RNF128 activates Wnt/β-catenin signaling to induce cellular EMT and stemness via CD44 and CTTN ubiquitination in melanoma. J Hematol Oncol. 2019;12:21. doi: 10.1186/s13045-019-0711-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee YY, Wang CT, Huang SK, Wu WJ, Huang CN, Li CC, Chan TC, Liang PI, Hsing CH, Li CF. Downregulation of RNF128 predicts progression and poor prognosis in patients with urothelial carcinoma of the upper tract and urinary bladder. J Cancer. 2016;7:2187–2196. doi: 10.7150/jca.16798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1–17. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19:480–494. doi: 10.1038/s41573-020-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang H, Zhang S, Zhang Y, Jia J, Wang J, Liu X, Zhang J, Song X, Ribback S, Cigliano A, Evert M, Liang B, Wu H, Calvisi DF, Zeng Y, Chen X. TAZ is indispensable for c-MYC-induced hepatocarcinogenesis. J Hepatol. 2022;76:123–134. doi: 10.1016/j.jhep.2021.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hyun J, Al Abo M, Dutta RK, Oh SH, Xiang K, Zhou X, Maeso-Díaz R, Caffrey R, Sanyal AJ, Freedman JA, Patierno SR, Moylan CA, Abdelmalek MF, Diehl AM. Dysregulation of the ESRP2-NF2-YAP/TAZ axis promotes hepatobiliary carcinogenesis in non-alcoholic fatty liver disease. J Hepatol. 2021;75:623–633. doi: 10.1016/j.jhep.2021.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jia M, Xiong Y, Li M, Mao Q. Corosolic Acid Inhibits Cancer Progress Through Inactivating YAP in Hepatocellular Carcinoma. Oncol Res. 2020;28:371–383. doi: 10.3727/096504020X15853075736554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tu K, Yang W, Li C, Zheng X, Lu Z, Guo C, Yao Y, Liu Q. Fbxw7 is an independent prognostic marker and induces apoptosis and growth arrest by regulating YAP abundance in hepatocellular carcinoma. Mol Cancer. 2014;13:110. doi: 10.1186/1476-4598-13-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ni W, Zhang Y, Zhan Z, Ye F, Liang Y, Huang J, Chen K, Chen L, Ding Y. A novel lncRNA uc. 134 represses hepatocellular carcinoma progression by inhibiting CUL4A-mediated ubiquitination of LATS1. J Hematol Oncol. 2017;10:91. doi: 10.1186/s13045-017-0449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Salah Z, Cohen S, Itzhaki E, Aqeilan RI. NEDD4 E3 ligase inhibits the activity of the Hippo pathway by targeting LATS1 for degradation. Cell Cycle. 2013;12:3817–3823. doi: 10.4161/cc.26672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bose I, Ghosh B. The p53-MDM2 network: from oscillations to apoptosis. J Biosci. 2007;32:991–997. doi: 10.1007/s12038-007-0103-3. [DOI] [PubMed] [Google Scholar]
- 157.Mendoza M, Mandani G, Momand J. The MDM2 gene family. Biomol Concepts. 2014;5:9–19. doi: 10.1515/bmc-2013-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Girnita L, Girnita A, Larsson O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci U S A. 2003;100:8247–8252. doi: 10.1073/pnas.1431613100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, Allday MJ, Xiao ZX. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 160.Enge M, Bao W, Hedström E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell. 2009;15:171–183. doi: 10.1016/j.ccr.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 161.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374–1378. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- 163.Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P, Nordgren H, Grander D, Reed SI, Widschwendter M, Sangfelt O, Spruck C. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 165.Tu K, Zheng X, Zan X, Han S, Yao Y, Liu Q. Evaluation of Fbxw7 expression and its correlation with the expression of c-Myc, cyclin E and p53 in human hepatocellular carcinoma. Hepatol Res. 2012;42:904–910. doi: 10.1111/j.1872-034X.2012.01005.x. [DOI] [PubMed] [Google Scholar]
- 166.Wang X, Zhang J, Zhou L, Sun W, Zheng ZG, Lu P, Gao Y, Yang XS, Zhang ZC, Tao KS, Dou KF. Fbxw7 regulates hepatocellular carcinoma migration and invasion via Notch1 signaling pathway. Int J Oncol. 2015;47:231–243. doi: 10.3892/ijo.2015.2981. [DOI] [PubMed] [Google Scholar]
- 167.Imura S, Tovuu LO, Utsunomiya T, Morine Y, Ikemoto T, Arakawa Y, Kanamoto M, Iwahashi S, Saito Y, Takasu C, Yamada S, Ishikawa D, Bando Y, Shimada M. Role of Fbxw7 expression in hepatocellular carcinoma and adjacent non-tumor liver tissue. J Gastroenterol Hepatol. 2014;29:1822–1829. doi: 10.1111/jgh.12623. [DOI] [PubMed] [Google Scholar]
- 168.Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, Snuderl M, Ladanyi M, Hanemann CO, Zhou P, Karajannis MA, Giancotti FG. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell. 2014;26:48–60. doi: 10.1016/j.ccr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zheng H, Ke X, Li D, Wang Q, Wang J, Liu X, Deng M, Deng X, Xue Y, Zhu Y, Wang Q. NEDD4 promotes cell growth and motility in hepatocellular carcinoma. Cell Cycle. 2018;17:728–738. doi: 10.1080/15384101.2018.1440879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hayes AJ, Skouras C, Haugk B, Charnley RM. Keap1-Nrf2 signalling in pancreatic cancer. Int J Biochem Cell Biol. 2015;65:288–299. doi: 10.1016/j.biocel.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 171.Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J. 2003;374:337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K, Komatsu M. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang F, Zhang Y, Shen J, Yang B, Dai W, Yan J, Maimouni S, Daguplo HQ, Coppola S, Gao Y, Wang Y, Du Z, Peng K, Liu H, Zhang Q, Tang F, Wang P, Gao S, Wang Y, Ding WX, Guo G, Wang F, Zong WX. The ubiquitin E3 ligase TRIM21 promotes hepatocarcinogenesis by suppressing the p62-Keap1-Nrf2 antioxidant pathway. Cell Mol Gastroenterol Hepatol. 2021;11:1369–1385. doi: 10.1016/j.jcmgh.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu Y, Tao S, Liao L, Li Y, Li H, Li Z, Lin L, Wan X, Yang X, Chen L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat Commun. 2020;11:348. doi: 10.1038/s41467-019-14190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jones EL, Laidlaw SM, Dustin LB. TRIM21/Ro52 - roles in innate immunity and autoimmune disease. Front Immunol. 2021;12:738473. doi: 10.3389/fimmu.2021.738473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ding Q, He D, He K, Zhang Q, Tang M, Dai J, Lv H, Wang X, Xiang G, Yu H. Downregulation of TRIM21 contributes to hepatocellular carcinoma carcinogenesis and indicates poor prognosis of cancers. Tumour Biol. 2015;36:8761–8772. doi: 10.1007/s13277-015-3572-2. [DOI] [PubMed] [Google Scholar]
- 178.Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z, Yang B, Chen JS, Catanzaro JM, Du C, Ding WX, Diaz-Meco MT, Moscat J, Ozato K, Lin RZ, Zong WX. TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell. 2016;62:149–151. doi: 10.1016/j.molcel.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 179.Heikel G, Choudhury NR, Michlewski G. The role of Trim25 in development, disease and RNA metabolism. Biochem Soc Trans. 2016;44:1045–1050. doi: 10.1042/BST20160077. [DOI] [PubMed] [Google Scholar]
- 180.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 181.Li YH, Zhong M, Zang HL, Tian XF. Mechanism of TRIM25 mediated ubiquitination of metastasis associated protein (MTA) 1 in normal liver cells. Exp Cell Res. 2018;371:250–254. doi: 10.1016/j.yexcr.2018.08.018. [DOI] [PubMed] [Google Scholar]
- 182.Zhang Q, Li X, Cui K, Liu C, Wu M, Prochownik EV, Li Y. The MAP3K13-TRIM25-FBXW7α axis affects c-Myc protein stability and tumor development. Cell Death Differ. 2020;27:420–433. doi: 10.1038/s41418-019-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Walsh LA, Alvarez MJ, Sabio EY, Reyngold M, Makarov V, Mukherjee S, Lee KW, Desrichard A, Turcan Ş, Dalin MG, Rajasekhar VK, Chen S, Vahdat LT, Califano A, Chan TA. An integrated systems biology approach identifies TRIM25 as a key determinant of breast cancer metastasis. Cell Rep. 2017;20:1623–1640. doi: 10.1016/j.celrep.2017.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Qin Y, Cui H, Zhang H. Overexpression of TRIM25 in lung cancer regulates tumor cell progression. Technol Cancer Res Treat. 2016;15:707–715. doi: 10.1177/1533034615595903. [DOI] [PubMed] [Google Scholar]
- 185.Sun N, Xue Y, Dai T, Li X, Zheng N. Tripartite motif containing 25 promotes proliferation and invasion of colorectal cancer cells through TGF-β signaling. Biosci Rep. 2017;37:BSR20170805. doi: 10.1042/BSR20170805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 187.Semenza GL. Hypoxia and cancer. Cancer Metastasis Rev. 2007;26:223–224. doi: 10.1007/s10555-007-9058-y. [DOI] [PubMed] [Google Scholar]
- 188.Yee Koh M, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci. 2008;33:526–534. doi: 10.1016/j.tibs.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 189.Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 190.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 191.Chen C, Lou T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget. 2017;8:46691–46703. doi: 10.18632/oncotarget.17358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li JQ, Wu X, Gan L, Yang XL, Miao ZH. Hypoxia induces universal but differential drug resistance and impairs anticancer mechanisms of 5-fluorouracil in hepatoma cells. Acta Pharmacol Sin. 2017;38:1642–1654. doi: 10.1038/aps.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wang J, Ma Y, Jiang H, Zhu H, Liu L, Sun B, Pan S, Krissansen GW, Sun X. Overexpression of von Hippel-Lindau protein synergizes with doxorubicin to suppress hepatocellular carcinoma in mice. J Hepatol. 2011;55:359–368. doi: 10.1016/j.jhep.2010.10.043. [DOI] [PubMed] [Google Scholar]
- 194.Shi M, Dai WQ, Jia RR, Zhang QH, Wei J, Wang YG, Xiang SH, Liu B, Xu L. APC(CDC20)-mediated degradation of PHD3 stabilizes HIF-1a and promotes tumorigenesis in hepatocellular carcinoma. Cancer Lett. 2021;496:144–155. doi: 10.1016/j.canlet.2020.10.011. [DOI] [PubMed] [Google Scholar]
- 195.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wang Z, Wan L, Zhong J, Inuzuka H, Liu P, Sarkar FH, Wei W. Cdc20: a potential novel therapeutic target for cancer treatment. Curr Pharm Des. 2013;19:3210–3214. doi: 10.2174/1381612811319180005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Li J, Gao JZ, Du JL, Huang ZX, Wei LX. Increased CDC20 expression is associated with development and progression of hepatocellular carcinoma. Int J Oncol. 2014;45:1547–1555. doi: 10.3892/ijo.2014.2559. [DOI] [PubMed] [Google Scholar]
- 198.Gonzalez-Sanchez E, Vaquero J, Férnandez-Barrena MG, Lasarte JJ, Avila MA, Sarobe P, Reig M, Calvo M, Fabregat I. The TGF-β pathway: a pharmacological target in hepatocellular carcinoma? Cancers (Basel) 2021;13:3248. doi: 10.3390/cancers13133248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Schon HT, Weiskirchen R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surg Nutr. 2014;3:386–406. doi: 10.3978/j.issn.2304-3881.2014.11.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signal. 2019;12:eaav5183. doi: 10.1126/scisignal.aav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Russell WE, Coffey RJ Jr, Ouellette AJ, Moses HL. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc Natl Acad Sci U S A. 1988;85:5126–5130. doi: 10.1073/pnas.85.14.5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, Schulte-Hermann R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci U S A. 1992;89:5408–5412. doi: 10.1073/pnas.89.12.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gotzmann J, Fischer AN, Zojer M, Mikula M, Proell V, Huber H, Jechlinger M, Waerner T, Weith A, Beug H, Mikulits W. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene. 2006;25:3170–3185. doi: 10.1038/sj.onc.1209083. [DOI] [PubMed] [Google Scholar]
- 204.Caja L, Sancho P, Bertran E, Fabregat I. Dissecting the effect of targeting the epidermal growth factor receptor on TGF-β-induced-apoptosis in human hepatocellular carcinoma cells. J Hepatol. 2011;55:351–358. doi: 10.1016/j.jhep.2010.10.041. [DOI] [PubMed] [Google Scholar]
- 205.Saha T, Vardhini D, Tang Y, Katuri V, Jogunoori W, Volpe EA, Haines D, Sidawy A, Zhou X, Gallicano I, Schlegel R, Mishra B, Mishra L. RING finger-dependent ubiquitination by PRAJA is dependent on TGF-beta and potentially defines the functional status of the tumor suppressor ELF. Oncogene. 2006;25:693–705. doi: 10.1038/sj.onc.1209123. [DOI] [PubMed] [Google Scholar]
- 206.Chen J, Mitra A, Li S, Song S, Nguyen BN, Chen JS, Shin JH, Gough NR, Lin P, Obias V, He AR, Yao Z, Malta TM, Noushmehr H, Latham PS, Su X, Rashid A, Mishra B, Wu RC, Mishra L. Targeting the E3 ubiquitin ligase PJA1 enhances tumor-suppressing TGFβ signaling. Cancer Res. 2020;80:1819–1832. doi: 10.1158/0008-5472.CAN-19-3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Consalvi S, Brancaccio A, Dall’Agnese A, Puri PL, Palacios D. Praja1 E3 ubiquitin ligase promotes skeletal myogenesis through degradation of EZH2 upon p38α activation. Nat Commun. 2017;8:13956. doi: 10.1038/ncomms13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zoabi M, Sadeh R, de Bie P, Marquez VE, Ciechanover A. PRAJA1 is a ubiquitin ligase for the polycomb repressive complex 2 proteins. Biochem Biophys Res Commun. 2011;408:393–398. doi: 10.1016/j.bbrc.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 209.Sasaki A, Masuda Y, Iwai K, Ikeda K, Watanabe K. A RING finger protein Praja1 regulates Dlx5-dependent transcription through its ubiquitin ligase activity for the Dlx/Msx-interacting MAGE/Necdin family protein, Dlxin-1. J Biol Chem. 2002;277:22541–22546. doi: 10.1074/jbc.M109728200. [DOI] [PubMed] [Google Scholar]
- 210.Chen J, A Gingold J. Dysregulated PJA1-TGF-β signaling in cancer stem cell-associated liver cancers. Oncoscience. 2020;7:88–95. doi: 10.18632/oncoscience.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 213.Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, Bremer J, Iezzi G, Graf R, Clavien PA, Thimme R, Blum H, Nedospasov SA, Zatloukal K, Ramzan M, Ciesek S, Pietschmann T, Marche PN, Karin M, Kopf M, Browning JL, Aguzzi A, Heikenwalder M. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16:295–308. doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 215.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 216.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 217.Zhang X, Lin C, Song J, Chen H, Chen X, Ren L, Zhou Z, Pan J, Yang Z, Bao W, Ke X, Yang J, Liang Y, Huang H, Tang D, Jiang L, Liu J. Parkin facilitates proteasome inhibitor-induced apoptosis via suppression of NF-κB activity in hepatocellular carcinoma. Cell Death Dis. 2019;10:719. doi: 10.1038/s41419-019-1881-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 219.Perwez A, Wahabi K, Rizvi MA. Parkin: a targetable linchpin in human malignancies. Biochim Biophys Acta Rev Cancer. 2021;1876:188533. doi: 10.1016/j.bbcan.2021.188533. [DOI] [PubMed] [Google Scholar]
- 220.Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, Kataoka A, Nukina N, Takahashi R, Chiba T. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene. 2008;27:6002–6011. doi: 10.1038/onc.2008.199. [DOI] [PubMed] [Google Scholar]
- 221.Wang F, Denison S, Lai JP, Philips LA, Montoya D, Kock N, Schüle B, Klein C, Shridhar V, Roberts LR, Smith DI. Parkin gene alterations in hepatocellular carcinoma. Genes Chromosomes Cancer. 2004;40:85–96. doi: 10.1002/gcc.20020. [DOI] [PubMed] [Google Scholar]
- 222.Wharton KA, Johansen KM, Xu T, Artavanis-Tsakonas S. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell. 1985;43:567–581. doi: 10.1016/0092-8674(85)90229-6. [DOI] [PubMed] [Google Scholar]
- 223.Majumder S, Crabtree JS, Golde TE, Minter LM, Osborne BA, Miele L. Targeting Notch in oncology: the path forward. Nat Rev Drug Discov. 2021;20:125–144. doi: 10.1038/s41573-020-00091-3. [DOI] [PubMed] [Google Scholar]
- 224.Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, Toffanin S, Rodriguez-Carunchio L, Solé M, Thung S, Stanger BZ, Llovet JM. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143:1660–1669. e1667. doi: 10.1053/j.gastro.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Giovannini C, Gramantieri L, Chieco P, Minguzzi M, Lago F, Pianetti S, Ramazzotti E, Marcu KB, Bolondi L. Selective ablation of Notch3 in HCC enhances doxorubicin’s death promoting effect by a p53 dependent mechanism. J Hepatol. 2009;50:969–979. doi: 10.1016/j.jhep.2008.12.032. [DOI] [PubMed] [Google Scholar]
- 226.Gramantieri L, Giovannini C, Lanzi A, Chieco P, Ravaioli M, Venturi A, Grazi GL, Bolondi L. Aberrant Notch3 and Notch4 expression in human hepatocellular carcinoma. Liver Int. 2007;27:997–1007. doi: 10.1111/j.1478-3231.2007.01544.x. [DOI] [PubMed] [Google Scholar]
- 227.Zhang Q, Lu C, Fang T, Wang Y, Hu W, Qiao J, Liu B, Liu J, Chen N, Li M, Zhu R. Notch3 functions as a regulator of cell self-renewal by interacting with the β-catenin pathway in hepatocellular carcinoma. Oncotarget. 2015;6:3669–3679. doi: 10.18632/oncotarget.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C, Zmoos AF, Mazur PK, Schaffer BE, Ostermeier A, Vogel H, Sylvester KG, Thorgeirsson SS, Grompe M, Sage J. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 2011;208:1963–1976. doi: 10.1084/jem.20110198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wang Z, Inuzuka H, Fukushima H, Wan L, Gao D, Shaik S, Sarkar FH, Wei W. Emerging roles of the FBW7 tumour suppressor in stem cell differentiation. EMBO Rep. 2011;13:36–43. doi: 10.1038/embor.2011.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, Chui I, Deshaies RJ, Kitajewski J. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol Cell Biol. 2001;21:7403–7415. doi: 10.1128/MCB.21.21.7403-7415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ringelhan M, O’Connor T, Protzer U, Heikenwalder M. The direct and indirect roles of HBV in liver cancer: prospective markers for HCC screening and potential therapeutic targets. J Pathol. 2015;235:355–367. doi: 10.1002/path.4434. [DOI] [PubMed] [Google Scholar]
- 232.Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725–12734. doi: 10.1128/JVI.78.23.12725-12734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ali A, Abdel-Hafiz H, Suhail M, Al-Mars A, Zakaria MK, Fatima K, Ahmad S, Azhar E, Chaudhary A, Qadri I. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J Gastroenterol. 2014;20:10238–10248. doi: 10.3748/wjg.v20.i30.10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Qadri I, Fatima K, AbdeL-Hafiz H. Hepatitis B virus X protein impedes the DNA repair via its association with transcription factor, TFIIH. BMC Microbiol. 2011;11:48. doi: 10.1186/1471-2180-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lee YI, Kang-Park S, Do SI, Lee YI. The hepatitis B virus-X protein activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. J Biol Chem. 2001;276:16969–16977. doi: 10.1074/jbc.M011263200. [DOI] [PubMed] [Google Scholar]
- 236.Kuo TC, Chao CC. Hepatitis B virus X protein prevents apoptosis of hepatocellular carcinoma cells by upregulating SATB1 and HURP expression. Biochem Pharmacol. 2010;80:1093–1102. doi: 10.1016/j.bcp.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 237.Zhang S, Gao S, Zhao M, Liu Y, Bu Y, Jiang Q, Zhao Q, Ye L, Zhang X. Anti-HBV drugs suppress the growth of HBV-related hepatoma cells via down-regulation of hepatitis B virus X protein. Cancer Lett. 2017;392:94–104. doi: 10.1016/j.canlet.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 238.Wan T, Lei Z, Tu B, Wang T, Wang J, Huang F. NEDD4 induces K48-linked degradative ubiquitination of hepatitis B virus x protein and inhibits hbv-associated HCC progression. Front Oncol. 2021;11:625169. doi: 10.3389/fonc.2021.625169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhao J, Wang C, Wang J, Yang X, Diao N, Li Q, Wang W, Xian L, Fang Z, Yu L. E3 ubiquitin ligase Siah-1 facilitates poly-ubiquitylation and proteasomal degradation of the hepatitis B viral X protein. FEBS Lett. 2011;585:2943–2950. doi: 10.1016/j.febslet.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 240.Zhao J, Wu J, Cai H, Wang D, Yu L, Zhang WH. E3 ubiquitin ligase Siah-1 is down-regulated and fails to target natural HBx truncates for degradation in hepatocellular carcinoma. J Cancer. 2016;7:418–426. doi: 10.7150/jca.13019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Yoo YS, Park YJ, Lee HS, Oanh NTK, Cho MY, Heo J, Lee ES, Cho H, Park YY, Cho H. Mitochondria ubiquitin ligase, MARCH5 resolves hepatitis B virus X protein aggregates in the liver pathogenesis. Cell Death Dis. 2019;10:938. doi: 10.1038/s41419-019-2175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Liu N, Zhang J, Yang X, Jiao T, Zhao X, Li W, Zhu J, Yang P, Jin J, Peng J, Li Z, Ye X. HDM2 promotes NEDDylation of hepatitis B virus HBx to enhance its stability and function. J Virol. 2017;91:e00340-17. doi: 10.1128/JVI.00340-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gao Y, Feng J, Yang G, Zhang S, Liu Y, Bu Y, Sun M, Zhao M, Chen F, Zhang W, Ye L, Zhang X. Hepatitis B virus X protein-elevated MSL2 modulates hepatitis B virus covalently closed circular DNA by inducing degradation of APOBEC3B to enhance hepatocarcinogenesis. Hepatology. 2017;66:1413–1429. doi: 10.1002/hep.29316. [DOI] [PubMed] [Google Scholar]
- 244.Decorsière A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, Livingston CM, Niu C, Fletcher SP, Hantz O, Strubin M. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531:386–389. doi: 10.1038/nature17170. [DOI] [PubMed] [Google Scholar]
- 245.Shan H, Wang B, Zhang X, Song H, Li X, Zou Y, Jiang B, Hu H, Dou H, Shao C, Gao L, Ma C, Yang X, Liang X, Gong Y. CUL4B facilitates HBV replication by promoting HBx stabilization. Cancer Biol Med. 2021;19:120–131. doi: 10.20892/j.issn.2095-3941.2020.0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wang ZW, Hu X, Ye M, Lin M, Chu M, Shen X. NEDD4 E3 ligase: functions and mechanism in human cancer. Semin Cancer Biol. 2020;67:92–101. doi: 10.1016/j.semcancer.2020.03.006. [DOI] [PubMed] [Google Scholar]
- 247.Bellet MM, Piobbico D, Bartoli D, Castelli M, Pieroni S, Brunacci C, Chiacchiaretta M, Del Sordo R, Fallarino F, Sidoni A, Puccetti P, Romani L, Servillo G, Della Fazia MA. NEDD4 controls the expression of GUCD1, a protein upregulated in proliferating liver cells. Cell Cycle. 2014;13:1902–1911. doi: 10.4161/cc.28760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Nagashima S, Tokuyama T, Yonashiro R, Inatome R, Yanagi S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem. 2014;155:273–279. doi: 10.1093/jb/mvu016. [DOI] [PubMed] [Google Scholar]
- 249.Takada S, Shirakata Y, Kaneniwa N, Koike K. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene. 1999;18:6965–6973. doi: 10.1038/sj.onc.1203188. [DOI] [PubMed] [Google Scholar]
- 250.Lim W, Kwon SH, Cho H, Kim S, Lee S, Ryu WS, Cho H. HBx targeting to mitochondria and ROS generation are necessary but insufficient for HBV-induced cyclooxygenase-2 expression. J Mol Med (Berl) 2010;88:359–369. doi: 10.1007/s00109-009-0563-z. [DOI] [PubMed] [Google Scholar]
- 251.Eischen CM, Lozano G. The Mdm network and its regulation of p53 activities: a rheostat of cancer risk. Hum Mutat. 2014;35:728–737. doi: 10.1002/humu.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16:30–44. doi: 10.1038/nrm3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Hüser N, Durantel D, Liang TJ, Münk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcik-Smith ND, Bhagat R, Lehman MJ, Novick SC, O’Connor OA, Soignet SL. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin. Oncol. 2002;20:4420–4427. doi: 10.1200/JCO.2002.01.133. [DOI] [PubMed] [Google Scholar]
- 255.Kawabata S, Gills JJ, Mercado-Matos JR, Lopiccolo J, Wilson W 3rd, Hollander MC, Dennis PA. Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell Death Dis. 2012;3:e353. doi: 10.1038/cddis.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kim GP, Mahoney MR, Szydlo D, Mok TS, Marshke R, Holen K, Picus J, Boyer M, Pitot HC, Rubin J, Philip PA, Nowak A, Wright JJ, Erlichman C. An international, multicenter phase II trial of bortezomib in patients with hepatocellular carcinoma. Invest New Drugs. 2012;30:387–394. doi: 10.1007/s10637-010-9532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Lövborg H, Oberg F, Rickardson L, Gullbo J, Nygren P, Larsson R. Inhibition of proteasome activity, nuclear factor-KappaB translocation and cell survival by the antialcoholism drug disulfiram. Int J Cancer. 2006;118:1577–1580. doi: 10.1002/ijc.21534. [DOI] [PubMed] [Google Scholar]
- 258.Kona FR, Buac D, M Burger A. Disulfiram, and disulfiram derivatives as novel potential anticancer drugs targeting the ubiquitin-proteasome system in both preclinical and clinical studies. Curr Cancer Drug Targets. 2011;11:338–346. doi: 10.2174/156800911794519798. [DOI] [PubMed] [Google Scholar]
- 259.Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3:419–421. [PubMed] [Google Scholar]
- 260.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 261.Zhang X, Linder S, Bazzaro M. Drug development targeting the ubiquitin-proteasome system (UPS) for the treatment of human cancers. Cancers (Basel) 2020;12:902. doi: 10.3390/cancers12040902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Pei Z, Chu L, Zou W, Zhang Z, Qiu S, Qi R, Gu J, Qian C, Liu X. An oncolytic adenoviral vector of Smac increases antitumor activity of TRAIL against HCC in human cells and in mice. Hepatology. 2004;39:1371–1381. doi: 10.1002/hep.20203. [DOI] [PubMed] [Google Scholar]
- 263.Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, Cohen RB. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014;32:3103–3110. doi: 10.1200/JCO.2013.52.3993. [DOI] [PubMed] [Google Scholar]
- 264.Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, Corral LG, Krenitsky VP, Xu W, Moutouh-de Parseval L, Webb DR, Mercurio F, Nakayama KI, Nakayama K, Orlowski RZ. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood. 2008;111:4690–4699. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, Pan Y, Ding C, Qian J, Wu L, Chu Y, Yi J, Wang X, Sun Y, Jeong LS, Liu J, Jia L. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012;72:3360–3371. doi: 10.1158/0008-5472.CAN-12-0388. [DOI] [PubMed] [Google Scholar]
- 266.Yang H, Minamishima YA, Yan Q, Schlisio S, Ebert BL, Zhang X, Zhang L, Kim WY, Olumi AF, Kaelin WG Jr. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol Cell. 2007;28:15–27. doi: 10.1016/j.molcel.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 268.Llovet JM, Real MI, Montaña X, Planas R, Coll S, Aponte J, Ayuso C, Sala M, Muchart J, Solà R, Rodés J, Bruix J Barcelona Liver Cancer Group. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet. 2002;359:1734–1739. doi: 10.1016/S0140-6736(02)08649-X. [DOI] [PubMed] [Google Scholar]
- 269.Tang X, Yang W, Shu Z, Shen X, Zhang W, Cen C, Cao L, Zhang M, Zheng S, Yu J. MicroRNA-223 promotes hepatocellular carcinoma cell resistance to sorafenib by targeting FBW7. Oncol Rep. 2019;41:1231–1237. doi: 10.3892/or.2018.6908. [DOI] [PubMed] [Google Scholar]
- 270.Zhang Z, Guo M, Li Y, Shen M, Kong D, Shao J, Ding H, Tan S, Chen A, Zhang F, Zheng S. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2020;16:1482–1505. doi: 10.1080/15548627.2019.1687985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, Liu X, Thummuri D, Yuan Y, Wiegand JS, Pei J, Zhang W, Sharma A, McCurdy CR, Kuruvilla VM, Baran N, Ferrando AA, Kim YM, Rogojina A, Houghton PJ, Huang G, Hromas R, Konopleva M, Zheng G, Zhou D. A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25:1938–1947. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zhang C, Han XR, Yang X, Jiang B, Liu J, Xiong Y, Jin J. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK) Eur J Med Chem. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Zhao Q, Ren C, Liu L, Chen J, Shao Y, Sun N, Sun R, Kong Y, Ding X, Zhang X, Xu Y, Yang B, Yin Q, Yang X, Jiang B. Discovery of SIAIS178 as an effective BCR-ABL degrader by recruiting von hippel-lindau (VHL) E3 ubiquitin ligase. J Med Chem. 2019;62:9281–9298. doi: 10.1021/acs.jmedchem.9b01264. [DOI] [PubMed] [Google Scholar]
- 275.Mészáros B, Kumar M, Gibson TJ, Uyar B, Dosztányi Z. Degrons in cancer. Sci Signal. 2017;10:eaak9982. doi: 10.1126/scisignal.aak9982. [DOI] [PubMed] [Google Scholar]
- 276.Abu Ahmad Y, Oknin-Vaisman A, Bitman-Lotan E, Orian A. From the evasion of degradation to ubiquitin-dependent protein stabilization. Cells. 2021;10:2374. doi: 10.3390/cells10092374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Martínez-Jiménez F, Muiños F, López-Arribillaga E, Lopez-Bigas N, Gonzalez-Perez A. Systematic analysis of alterations in the ubiquitin proteolysis system reveals its contribution to driver mutations in cancer. Nat Cancer. 2020;1:122–135. doi: 10.1038/s43018-019-0001-2. [DOI] [PubMed] [Google Scholar]
- 278.Chen D, Wang Y, Lu R, Jiang X, Chen X, Meng N, Chen M, Xie S, Yan GR. E3 ligase ZFP91 inhibits hepatocellular carcinoma metabolism reprogramming by regulating PKM splicing. Theranostics. 2020;10:8558–8572. doi: 10.7150/thno.44873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Feng Y, Zhang Y, Cai Y, Liu R, Lu M, Li T, Fu Y, Guo M, Huang H, Ou Y, Chen Y. A20 targets PFKL and glycolysis to inhibit the progression of hepatocellular carcinoma. Cell Death Dis. 2020;11:89. doi: 10.1038/s41419-020-2278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Zhuo H, Tang J, Lin Z, Jiang R, Zhang X, Ji J, Wang P, Sun B. The aberrant expression of MEG3 regulated by UHRF1 predicts the prognosis of hepatocellular carcinoma. Mol Carcinog. 2016;55:209–219. doi: 10.1002/mc.22270. [DOI] [PubMed] [Google Scholar]
- 281.Zhang T, Zhao L, Zeng S, Bai L, Chen J, Zhang Z, Wang Y, Duan C. UHRF2 decreases H3K9ac expression by interacting with it through the PHD and SRA/YDG domain in HepG2 hepatocellular carcinoma cells. Int J Mol Med. 2017;39:126–134. doi: 10.3892/ijmm.2016.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Cai J, Xiong Q, Jiang X, Zhou S, Liu T. RNF6 facilitates metastasis and radioresistance in hepatocellular carcinoma through ubiquitination of FoxA1. Exp Cell Res. 2019;374:152–161. doi: 10.1016/j.yexcr.2018.11.019. [DOI] [PubMed] [Google Scholar]
- 283.Lian YF, Huang YL, Zhang YJ, Chen DM, Wang JL, Wei H, Bi YH, Jiang ZW, Li P, Chen MS, Huang YH. CACYBP enhances cytoplasmic retention of P27(Kip1) to promote hepatocellular carcinoma progression in the absence of rnf41 mediated degradation. Theranostics. 2019;9:8392–8408. doi: 10.7150/thno.36838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wu H, Yang TY, Li Y, Ye WL, Liu F, He XS, Wang JR, Gan WJ, Li XM, Zhang S, Zhao YY, Li JM. Tumor necrosis factor receptor-associated factor 6 promotes hepatocarcinogenesis by interacting with histone deacetylase 3 to enhance c-Myc gene expression and protein stability. Hepatology. 2020;71:148–163. doi: 10.1002/hep.30801. [DOI] [PubMed] [Google Scholar]
- 285.He H, Wu Z, Li S, Chen K, Wang D, Zou H, Chen H, Li Y, Liu Z, Qu C. TRAF7 enhances ubiquitin-degradation of KLF4 to promote hepatocellular carcinoma progression. Cancer Lett. 2020;469:380–389. doi: 10.1016/j.canlet.2019.11.012. [DOI] [PubMed] [Google Scholar]
- 286.Li L, Dong L, Qu X, Jin S, Lv X, Tan G. Tripartite motif 16 inhibits hepatocellular carcinoma cell migration and invasion. Int J Oncol. 2016;48:1639–1649. doi: 10.3892/ijo.2016.3398. [DOI] [PubMed] [Google Scholar]
- 287.Ma X, Ma X, Qiu Y, Zhu L, Lin Y, You Y, Ma D, Qin Z, Sun C, Zhao Y, Sun Y, Han L. TRIM50 suppressed hepatocarcinoma progression through directly targeting SNAIL for ubiquitous degradation. Cell Death Dis. 2018;9:608. doi: 10.1038/s41419-018-0644-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Wu H, Yao S, Zhang S, Wang JR, Guo PD, Li XM, Gan WJ, Mei L, Gao TM, Li JM. Elevated expression of Erbin destabilizes ERα protein and promotes tumorigenesis in hepatocellular carcinoma. J Hepatol. 2017;66:1193–1204. doi: 10.1016/j.jhep.2017.01.030. [DOI] [PubMed] [Google Scholar]
- 289.Chen Z, Chen Y, Peng L, Wang X, Tang N. 2,5-dimethylcelecoxib improves immune microenvironment of hepatocellular carcinoma by promoting ubiquitination of HBx-induced PD-L1. J Immunother Cancer. 2020;8:e001377. doi: 10.1136/jitc-2020-001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wang X, Rusin A, Walkey CJ, Lin JJ, Johnson DL. The RNA polymerase III repressor MAF1 is regulated by ubiquitin-dependent proteasome degradation and modulates cancer drug resistance and apoptosis. J Biol Chem. 2019;294:19255–19268. doi: 10.1074/jbc.RA119.008849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Zhang L, Chen J, Ning D, Liu Q, Wang C, Zhang Z, Chu L, Yu C, Liang HF, Zhang B, Chen X. FBXO22 promotes the development of hepatocellular carcinoma by regulating the ubiquitination and degradation of p21. J Exp Clin Cancer Res. 2019;38:101. doi: 10.1186/s13046-019-1058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Dong X, Han Y, Zhang E, Wang Y, Zhang P, Wang C, Zhong L, Li Q. Tumor suppressor DCAF15 inhibits epithelial-mesenchymal transition by targeting ZEB1 for proteasomal degradation in hepatocellular carcinoma. Aging (Albany NY) 2021;13:10603–10618. doi: 10.18632/aging.202823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Huang Y, Tan N, Jia D, Jing Y, Wang Q, Li Z, Zhang J, Liu L, Li J, Chen Z, He X. Speckle-type POZ protein is negatively associated with malignancies and inhibits cell proliferation and migration in liver cancer. Tumour Biol. 2015;36:9753–9761. doi: 10.1007/s13277-015-3753-z. [DOI] [PubMed] [Google Scholar]
- 294.Shi W, Feng L, Dong S, Ning Z, Hua Y, Liu L, Chen Z, Meng Z. FBXL6 governs c-MYC to promote hepatocellular carcinoma through ubiquitination and stabilization of HSP90AA1. Cell Commun Signal. 2020;18:100. doi: 10.1186/s12964-020-00604-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Qiao Y, Zhang Y, Wang J. Ubiquitin E3 ligase SCF(β-TRCP) regulates TRIB2 stability in liver cancer cells. Biochem Biophys Res Commun. 2013;441:555–559. doi: 10.1016/j.bbrc.2013.10.123. [DOI] [PubMed] [Google Scholar]
- 296.Du Y, Song W, Chen J, Chen H, Xuan Z, Zhao L, Chen J, Jin C, Zhou M, Tuo B, Zhao Y, Zheng S, Song P. The potassium channel KCa3.1 promotes cell proliferation by activating SKP2 and metastasis through the EMT pathway in hepatocellular carcinoma. Int J Cancer. 2019;145:503–516. doi: 10.1002/ijc.32121. [DOI] [PubMed] [Google Scholar]
- 297.Cheng Q, Cao X, Yuan F, Li G, Tong T. Knockdown of WWP1 inhibits growth and induces apoptosis in hepatoma carcinoma cells through the activation of caspase3 and p53. Biochem Biophys Res Commun. 2014;448:248–254. doi: 10.1016/j.bbrc.2014.04.117. [DOI] [PubMed] [Google Scholar]
- 298.Tian A, Wilson GS, Lie S, Wu G, Hu Z, Hebbard L, Duan W, George J, Qiao L. Synergistic effects of IAP inhibitor LCL161 and paclitaxel on hepatocellular carcinoma cells. Cancer Lett. 2014;351:232–241. doi: 10.1016/j.canlet.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 299.Chen KF, Lin JP, Shiau CW, Tai WT, Liu CY, Yu HC, Chen PJ, Cheng AL. Inhibition of Bcl-2 improves effect of LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem Pharmacol. 2012;84:268–277. doi: 10.1016/j.bcp.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 300.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–1328. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 301.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 302.Patel S, Player MR. Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin Investig Drugs. 2008;17:1865–1882. doi: 10.1517/13543780802493366. [DOI] [PubMed] [Google Scholar]
- 303.Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol. 2012;19:1515–1524. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]