

Abstract
Colorectal cancer (CRC) is an aggressive malignancy with limited options for treatment. Targeting the bromodomain and extra terminal domain (BET) proteins by using BET inhibitors (BETis) could effectively interrupt the interaction with acetylated histones, inhibit genes transcription and have shown a certain effect on CRC inhibition. To improve the efficacy, the inhibitors of Tankyrases, which cause accumulation of AXIN through dePARsylation, in turn facilitate the degradation of β-Catenin and suppress the growth of adenomatous polyposis coli (APC)-mutated CRCs, were tested together with BETi as a combination treatment. We examined the effects of BETi and Tankyrases inhibitor (TNKSi) together on the proliferation, cell cycle and apoptosis of human CRCs cell lines with APC or CTNNB1 mutation, and elucidated the underlying molecular mechanisms affected by the double treatment. The result showed that the TNKSi could sensitize all tested CRC cell lines to BETi, and the synergistic effect was not only seen in cell proliferation inhibition, but also confirmed in decreased colony-forming ability and weaken EdU incorporation compared with monotherapy. Combined treatment resulted in enhanced G1 cell cycle arrest and increased apoptosis. In addition, we found β-Catenin was potentially inhibited by the combination and revealed that both BETi-induced transcriptional inhibition and TNKSi-mediated protein degradation all reduced the β-Catenin accumulation. In all, the synergistic effects suggest that combination of BETi and TNKSi could provide novel treatment opportunities for CRC, but both TNKSi and combination strategy need to be optimized.
Keywords: BET inhibitor, Tankyrases inhibitor, β-Catenin, antitumor activity, combination therapy
Introduction
Epigenetic modifications regulate the occurrence and development of tumors [1]. The bromodomain and extra terminal domain (BET) family, including BRD2, BRD3, BRD4, and BRDT, are epigenetic readers of acetylated histone [2]. They act as transcriptional regulators to identify in the promoter [3] and enhancer [4] regions of oncogenes and promote gene transcriptional expression by specifically recognizing acetylated lysine residues. Targeting the BET family proteins has recently emerged as a promising anti-colorectal cancers (CRCs) approach. The BET inhibitor (BETi) JQ1 could suppress the colon cancer cell proliferation, inhibit WNT activity [5], induce cell cycle arrest [6] and promote apoptosis [7]. However, intriguingly, we found that although there are more than 70 clinical trials undertaken on BETis (ClinicalTrials.gov database), no trial focuses on colon cancer treatment. We proposed that the limited efficacy of BETi monotherapy on CRCs hindered the development of clinical trials. We also noticed that the combinational use of BETi with other molecularly targeted drugs produced very effective therapies. For examples, BETi CPI-0610 in combination with JAK1/2 inhibitor ruxolitinib were generally well-tolerated and provided clinical benefits in myelofibrosis patients, which is now in phase III clinical study [8,9]. Another BETi ZEN-3694 in combination with enzalutamide was very potent in patients with metastatic castration-resistant prostate cancer [10]. Many other combinations were explored, including HDAC inhibitors [11,12], kinase inhibitors [13], HMT inhibitors [14], Bcl-2 inhibitors [15], immunomodulatory drugs [16], proteasome inhibitors [17] and classical chemotherapy agents [18] with BETi in different cancer cells. We also developed one potential BETi HJP-178 (also called as compound 19) as a drug candidate for the treatment of cancers [2]. So, it would be very meaningful and even desirable to find an effective combination with BETi in colon cancer treatment to expand the therapeutic scope of BETis.
Colon cancer is one of the leading causes of cancer-related deaths worldwide [19]. WNT signaling pathway plays an important role in cell proliferation, migration and embryonic development and its deregulation is a major contributor to colorectal carcinogenesis [20]. In approximately 90% of CRCs, WNT pathway is mutated and these mutations are mainly found in the loss-of-function mutation of adenomatous polyposis coli (APC) and β-Catenin gene CTNNB1 [21]. These mutations lead to WNT signaling activation and cytoplasmic accumulation of β-Catenin and its nuclear translocation, which are the central events in canonical WNT pathway [22] and can be found in more than 80% of CRCs [23]. Thus, it is rational targeting WNT/β-Catenin for therapeutic intervention of CRCs. So far, however, this WNT pathway still lacks druggable molecular targets, which hampered the development of therapeutic drugs.
Tankyrases (Tankyrase-1/TNKS1 and Tanky-rase-2/TNKS2, also known as PARP5A/ARTD5 and PARP5B/ARTD6) are members of the poly (ADP-ribose) polymerase (PARP) family with a PARP catalytic domain [24]. Targeting Tankyrases can promote the degradation of β-Catenin and inhibit the WNT/β-Catenin pathway through poly(ADP-ribosyl)ates (PARsylates) of AXIN1/2 [25]. Unfortunately, there is still no TNKSi in clinical studies. Since XAV939 was used as the first TNKSi to start the research on targeting Tankyrases [26], novel TNKSis have been discovered and developed continuously. In 2013, Krauss et al. discovered a novel TNKSi, G007-LK, which showed stronger tumor proliferation inhibition compared with the XAV939 [27]. RK-287107, first published by Seimiya’s lab in 2018, is more sensitive to the inhibition of Tankyrases than G007-LK in cell-free assays, but its advantage in cell activity is not significant [25]. As a classical TNKSi, G007-LK has been mostly used as a positive control and studied in mechanism exploration [28-31], which has been investigated more deeply and extensively than RK-287107. In contrast, relatively limited studies on RK-287107 were reported [25,32,33]. Therefore, G007-LK is a more representative TNKSi. Further, TNKSis were reported to be more sensitive to colon cancer cells with APC mutations [34]. In view of the challenges that BETi and TNKSi face in the clinical treatment of CRCs and the feasibility of targeting Tankyrases to inhibit the WNT pathway, we would like to investigate the effects of the combination treatment of BETi and TNKSi.
We found that the combination of BETi and TNKSi synergistically inhibited growth and proliferation of colon cancer cells with APC mutation (HCT-15 and SW480) or CTNNB1 mutation (HCT-116). Cell cycle arrest and apoptosis were all enhanced by the combination compared with monotherapy. We also found β-Catenin was potentially inhibited by the combination and revealed that both BETi-induced transcriptional inhibition and TNKSi-promoted protein degradation all mediated the β-Catenin accumulation and nuclear translocation. Therefore, an effective combination of BETi and TNKSi could increase the efficacy and expand the antitumor application scope while reducing the dosage of the drug. However, due to the weak activity of single drugs in vitro and in vivo, especially TNKSi, the efficacy of the combination treatment was limited. So the improvement of single drug activity is of great significance to the promotion of combinational application.
Materials and methods
Reagents
Drugs
The BETi OTX-015 (HY-15743), TNKSi G007-LK (HY-12438), RK-287107 (HY-123892), XAV939 (HY-15147), PARPi olaparib (HY-10162), and proteasome inhibitor MG-132 (HY-13259) were purchased from Medchem Express (Shanghai, China). Cycloheximide (CHX) (S7418) was purchased from Selleck (Shanghai, China). The BETi HJP-178 was designed and synthesized as described in previous study [2]. CHX is dissolved in ethanol (10009218) (Sinopharm, Shanghai, China), other compounds used in vitro were dissolved in 100% dimethyl sulfoxide (DMSO) (D2650) (Sigma-Aldrich, Shanghai, China) and stored at -20°C as the stock. In each experiment, dilute the stock solution with normal saline (Sinopharm, Shanghai, China) to the desired concentrations.
Cell lines
HCT-15 and HCT-116 cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). SW480 was obtained from the Cell Bank of the Chinese Academy of Science Type Culture Collection (Shanghai, China). Cells were cultured according to the suppliers’ instructions and authenticated by short tandem repeat (STR) analysis performed by Genesky Biotechnologies (Shanghai, China). Periodic detection of Mycoplasma contamination was performed with the MycAwayTM Plus-Color One-Step Mycoplasma Detection Kit (40612ES25) from YEASEN (Shanghai, China).
Antibodies
Anti-c-Myc (#18583), anti-Rb (#9309), anti-p-Rb (#9307), anti-Caspase 3 (#39662), anti-Cleaved-Caspase-3 (#9661), anti-Cleaved-Caspase-7 (#9491), anti-Cleaved-PARP (#5625), anti-Cleaved-Caspase-8 (#9748), anti-AXIN2 (#2151), anti-Phospho-β-Catenin (Ser675) (#4176), and anti-Cyclin D1 (#2978) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-CDK6 (#177), anti-Tankyrases (#365897), anti-β-Catenin (#59737), anti-RAD51 (#8349) and anti-H3 (#8654) were obtained from Santa Cruz Technology (Dallas, TX). Anti-GAPDH (AF0006) antibody was purchased from Beyotime Biotechnology (Shanghai, China). Secondary HRP-conjugated goat anti-rabbit (#111-035-003) and goat anti-mouse (#115-035-003) antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA).
Methods
Cell proliferation assay
Cell proliferation was detected by sulforhodamine B (SRB; Sigma, St. Louis, MO) assay [35]. In brief, cells were plated and cultured in 96-well plates overnight and then incubated with the indicated compounds for 72 h. Optical density was read with the SpectraMax 190 (Molecular Devices, San Jose, CA) for SRB assay. The IC50s were calculated from at least three independent experiments.
Combination analysis
The Chou-Talalay method for drug combination was used [36]. Cells were treated with the indicated drug combinations simultaneously, and the proliferative inhibition rate (IR) was determined by SRB assay as described above. Combination Index (CI) was analyzed using the CompuSyn software to determine the drug interactions quantitatively. CI<1, CI = 1, and CI>1 represented synergism, additive effect, and antagonism, respectively.
Colony formation assay
According to the different growth rates of three cell lines, HCT-15 and HCT-116 cells were seeded in six-well plates (500 cells/well), and SW480 were seeded in 24-well plates (1000 cells/well) for the formation of colonies. Cells were cultured overnight and treated with desired concentrations of compounds for another 7 days. Concentrations of OTX-015 and HJP-178 were 0.3125 μM in HCT-15 and HCT-116 cells, and 2 μM in SW480 cells. Concentrations of G007-LK were 3.125 μM in HCT-15 and HCT-116 cells, and 10 μM in SW480 cells. After fixing, the cell colonies were stained with SRB and the microscopic images were captured by ChemiDoc MP Imaging System (Bio-rad, Hercules, CA, USA), and then counted the colony numbers in the whole well plate [37].
EdU (5-ethynyl-2’-deoxyuridine) incorporation assay
HCT-15 and HCT-116 cells were seeded at 2×105 cells and SW480 cells was seeded at 3×105 cells per well of 12-well confocal plates. After culturing overnight, cells were treated with compounds for 24 h. The concentrations of HJP-178 and G007-LK were 2 μM in HCT-15 and HCT-116 cells and 10 μM in SW480 cells. Then, cells were incubated with 10 µM EdU for 2 h at 37°C. The fixing and permeating were performed with BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 488 (Beyotime Biotechnology, Shanghai, China) following strictly the manufacturer’s instructions [38]. Finally, cells were incubated and protected from light in Click Additive Solution and stained with DAPI. The fluorescence images of EdU incorporation samples were captured in a florescence microscope (Olympus IX71, Tokyo, Japan).
Real-time quantitative polymerase chain reaction (RT-qPCR)
The effect of different treatments on mRNA levels was detected by standard RT-qPCR. Total RNA was extracted with the HiPure Total RNA Mini Kit (R4111-03) (Magen, Shanghai, China) and transformed into cDNA by reverse transcription with PrimeScript™ RT Master Mix (Perfect Real Time) (RR036A) (TaKaRa; Kusatsu, Shiga, Japan). The resultant cDNA was amplified with specific primers using TB Green®Premix Ex Taq™ kit (Tli RnaseH Plus) (RR420A) (TaKaRa). Each 10-μL reaction system contained 250 ng of total RNA [39]. The primers for human CTNNB1, c-Myc, and GAPDH were as follows: CTNNB1: 5’-GGCGAAGGTGATGGCTTACT-3’ (forward) and 5’-TCCATTTGGCCAGCTTTGGA-3’ (reverse); c-Myc: 5’-CGTCTCCACACATCAGCACAA-3’ (forward) and 5’-TGTTGGCAGCAGGATAGTCCTT-3’ (reverse); and GAPDH: 5’-CCATGGAGAAGGCTGGGG-3’ (forward) and 5’-CAAAGTTGTCATGGATGACC-3’ (reverse). Sequences were from PrimerBank database and synthesized by Sangon Biotech (Shanghai, China). The relative quantitation in gene expression is based on the comparative Cycle Threshold (ΔΔCT), at which cycle number the fluorescence generated within a qRT-PCR amplification reaction crosses the threshold.
Western blotting
Western blotting was performed as previously described [40].
Cell cycle assay
HCT-15 and HCT-116 cells were seeded into six-well plates and incubated overnight. The cells were incubated with either a single drug or a combination for 24 h. Cell sample treatment was the same as described previously [41]. Cell cycle data were obtained by using a FACS Calibur Instrument (BD Biosciences, San Jose, CA) and analyzed with the FlowJo 7.6.1 software.
Annexin V-FITC/PI staining
HCT-15 and SW480 cells were treated with OTX-015, HJP-178 and G007-LK for the indicated concentrations and time. The sample handling procedure is the same as described previously [42]. The percentage of viable cells (Q4: low annexin V-FITC/low PI), early apoptotic cells (Q3: high annexin V-FITC/low PI), and late apoptotic/necrotic cells (Q2: high annexin V-FITC/high PI) was determined by flow cytometer and analyzed by FlowJo.
Caspase-Glo 3/7 assay
The Caspase-Glo® 3/7 Assay Kit (Promega, WI, USA) was used for caspase activity detection [43]. Cells were planted in 96-well plates overnight and incubated with indicated concentrations of compounds for 24 h. Cell samples were treated according to the manufacturer’s instructions. The caspase 3/7 activity was reflected by the luminescent signal which was detected through an EnVision® Multilabel Reader (PerkinElmer, Thermo Fisher Scientific, Shanghai, China).
Preparation of cytoplasmic and nuclear extracts
In order to detect the protein changes in cytoplasm and nucleus, we isolated the nuclear and cytoplasmic proteins by Nuclear and Cytoplasmic Protein Extraction Kit [44] (P0027, Beyotime, Shanghai, China). In brief, 5×105 HCT-15 and SW480 cells were attached to the six-well plate overnight. After 24-h drug treatments, cell precipitate was collected. The nuclear proteins and cytoplasmic proteins were separated according to the manufacturer’s instructions. The protein lysis and loading were performed the same as in Western blotting.
In vivo anticancer activity assays
We established human HCT-15 and HCT-116 xenografts in female BALB/c nude mice (5-6-week-old) as previously described [39,45]. In HCT-15 model, mice were assigned into four groups: 6 mice were treated with 20 mg/kg HJP-178 by i.g. (intragastric) administration once every three days, 6 mice were treated with 10 mg/kg G007-LK by i.p. (intraperitoneal) once a day, 6 mice were treated with HJP-178 and G007-LK combinational administration, and 6 mice were treated with the same vehicle as combinational group. HJP-178 were suspended in 5% dimethylacetamide (271012), 5% solutol HS15 (42966), and 90% distilled water containing 0.5% Methyl Cellulose (SLCC2541) purchased from Sigma-Aldrich (Shanghai, China). G007-LK was suspended in 5% dimethylacetamide (271012), 5% solutol HS15 (42966) purchased from Sigma-Aldrich (Shanghai, China) and 90% distilled water containing 20% hydroxypropyl-β-cyclodextrin (20190920) (Sinopharm, Shanghai, China). Tumor volume and body weight were measured twice a week. After 21-day treatments, the mice were sacrificed. The inhibition rate of tumor volume growth (GI%) was calculated as follows: GI% = [1-(TVt-TV0)/(CVt-CV0)] ×100%, TVt was the tumor volume measured in the treatment groups, TV0 was the tumor volume obtained before administration. CVt and CV0 were corresponding to the volumes of the vehicle group. In HCT-116 model, 6 mice were treated with 40 mg/kg HJP-178 by i.g. administration once every three days, 6 mice received 120 mg/kg OTX-015 in the same way, 6 mice were treated with 25 mg/kg XAV939 by i.p. twice a week, 6 mice were treated with HJP-178 and XAV939 combinational administration, 6 mice for OTX-015 and XAV939 combination group, and 12 were treated with the same vehicle as combinational group. HJP-178 was dissolved as described above, and OTX-015 were suspended in 6% dimethylacetamide (271012), 6% solutol HS15 (42966), and 88% distilled water containing 0.5% Methyl Cellulose (SLCC2541) purchased from Sigma-Aldrich (Shanghai, China). XAV939 was suspended in 5% dimethylacetamide (271012), 5% solutol HS15 (42966) purchased from Sigma-Aldrich (Shanghai, China) and 90% distilled water containing 10% hydroxypropyl-β-cyclodextrin (20190920) (Sinopharm, Shanghai, China). Body weight was measured twice a week. Weigh the tumor after euthanasia. The experimental procedures abided by the ethical guidelines of the Institutional Animal Care and Use Committee (IACUC) in our institute.
Statistical analysis
All data were presented as mean ± standard deviation (SD). Paired Student’s t-test (two tails) was used for statistical difference analysis; P<0.05 was considered statistically significant. All analyses were performed by GraphPad Prism 7, and the photographs were processed with Photoshop CS and Adobe Illustrator CS6.
Results
TNKSi sensitizes colorectal cancer cells to BETi
Based on the limited efficacy of BETi monotherapy, combinational use of TNKSi with BETi was explored. OTX-015 is a classical BETi used in leukemia and lymphoma [46]. However, given the poor monotherapy activity, its phase II development was stopped (NCT02296476). Therefore, effective drug combinations are expected to further expand the therapeutic range of BETis. HJP-178 is a novel BETi independently developed by our lab in comparison with OTX-015 end-to-end. HJP-178 showed more potent antitumor activity at molecular, cellular and animal levels [2]. Therefore, we chose these two BETis for subsequent studies. G007-LK and RK-287107 are two TNKSi, among which G007-LK, as a typical TNKSi, is mostly used as positive control [28-31,47]. Although RK-287107 showed stronger inhibition of Tankyrase activity in cell-free assays, its cell-level activity did not have an apparent advantage [25]. Here we first evaluated the effect of HJP-178, G007-LK, and their combination on the cell viability of three human colorectal cancer cell lines, i.e., APC mutant HCT-15 and SW480 cells, and CTNNB1 mutant HCT-116 cells. We found the combination demonstrated a strong synergistic inhibition effect in all cells (Figure 1A). We next evaluated if this effect can be seen in other drug combinations. We found the proliferative inhibition of HJP-178 was enhanced by another commercially available TNKSi, RK-287107 (Figure 1B). Further, G007-LK also sensitized the cancer cells to OTX-015, another BETi currently undergoing Phase II clinical trial (Figure 1C). The same synergistic sensitization took place in the combination of RK-287107 and OTX-015 (Figure 1D). A compusyn software was used to calculate the combination index (CI). The results showed that BETi and TNKSi displayed apparent synergism (CI<1) in all tested cancer cells. The data indicated that TNKSi can effectively increase the sensitivity of colorectal cancer cells to BETi.
Figure 1.

TNKSi sensitizes colorectal cancer cells to BETi. Cell proliferative inhibition curves in HCT-15, SW480 and HCT-116 with combination treatments of HJP-178 and G007-LK (A), HJP-178 and RK-287107 (B), OTX-015 and G007-LK (C) and OTX-015 and RK-287107 (D) at indicated concentrations for 72 h. The concentration unit of the drugs are μM. The proliferative inhibition rate was determined by SRB assay with monotherapy and combination therapy. The CI value was calculated by Compusyn software with the Chou-Talalay equation and average CI values were shown. CI>1 indicates antagonistic effect, CI = 1 indicates additive effect, and CI<1 indicates a synergistic sensitization effect. The smaller the CI value is, the stronger the synergistic effect is.
Combinations of TNKSi and BETi synergistically inhibit the clone-forming ability and reduce the EdU signals in cancer cells
Based on the results from Figure 1A-D, it is not difficult to see that the synergistic effect of G007-LK combined with different BETi were better than RK-287107 in different cells. Compared with RK-287107 that needs to protect from light (medchemexpress.cn), the stability of G007-LK is better. Therefore, we chose G007-LK for a more in-depth mechanistic exploration. To further determine the synergistic effects of TNKSi and BETi, we performed colony formation assays using three cell lines. The cells were exposed to the combination of the TNKSi G007-LK with the BETi HJP-178 or OTX-015 for 7 days. The results showed that the TNKSi significantly enhanced the inhibition of both BETis against the clone-forming ability of the tested cells (Figure 2A-F). Treatments with HJP-178 and G007-LK alone decreased the colony numbers of HCT-15 cells by 32.53% and 18.02% (Figure 2D), of SW480 cells by 61.49% and 26.95% (Figure 2E), and of HCT-116 cells by 47.04% and 12.62% (Figure 2F), respectively. Comparatively, their combination led to a significantly more decrease in the colony numbers of HCT-15, SW480 and HCT-116 cells by 90.33%, 96.85% and 70.71%, respectively. Similar effects on the colony formation of those cells were also seen when treated with OTX-015, G007-LK and their combination (Figure 2A-F). The findings further strengthen the conclusion that TNKSi and BETi synergistically inhibit tumor cell proliferation. Moreover, HJP-178 is more potent than OTX-015 at the same conditions. Olaparib, an inhibitor of another PARP family member PARP1, has been reported to be synergistic with BETi [48,49]. However, at the same concentration, the TNKSi G007-LK displayed more potent sensitization to BETis against the colony formation of HCT-15 cells than olaparib (Figure S1A).
Figure 2.

TNKSi enhances the anti-proliferative ability of BETi. A-C. Cell proliferation was visualized by colony formation assay in HCT-15, SW480 and HCT-116 cells with combination treatments for seven days. Concentrations of OTX-015 and HJP-178 were 0.3125 μM in HCT-15 and HCT-116 cells, while 2 μM in SW480 cells. Concentrations of G007-LK were 3.125 μM in HCT-15 and HCT-116 cells, and 10 μM in SW480 cells. D-F. Statistical results of counting colony numbers. Data were completed by three independent experiments and expressed as mean ± SD. *P<0.05; **P<0.01; ***P<0.001. G-I. EdU incorporation assay detected green fluorescence intensity after drug treatments for 24 h in HCT-15, SW480, and HCT-116 cells. The concentrations of HJP-178 and G007-LK were 2 μM in HCT-15 and HCT-116 cells and 10 μM in SW480 cells.
Cancer cell proliferation requires new synthesis of DNA. So we conducted the EdU incorporation assay to examine the effects of TNKSi and BETi alone and their combinations on the newly synthesized DNA in cancer cells. In the process of DNA synthesis, thymidine can be replaced by EdU labeled with green fluorescent probes, so that the newly synthesized DNA will show green fluorescent signals. Data showed that the BETi HJP-178 alone reduced the EdU signals and the TNKSi G007-LK alone caused a slight decrease. Their combination apparently synergistically reduced the newly synthesized DNA in all three tested cell lines (Figure 2G-I).
Together, those results consistently indicate the synergism of BETi and TNKSi in suppressing proliferation of cancer cells.
The combination of TNKSi and BETi induces potent G1 cell cycle arrest
Both BETi and TNKSi have been reported to induce G1 arrest [50,51]. We found that the combination of the BETi HJP-178 or OTX-015 with the TNKSi G007-LK caused more potent G1 phase arrest than the corresponding single agents (Figures 3A, 3B, S1B and S1C). The combination of BETi and TNKSi also led to a time-dependent increase in G1 phase arrest (Figure 3C and 3D). Consistently, the combination caused more reduction of G1 phase regulating proteins in HCT-15 cells than the corresponding monotherapy. Though OTX-015, HJP-178 or G007-LK alone lowered the levels of Rb, p-Rb, CDK6 and Cyclin D1 proteins, the combination of either OTX-015 or HJP-178 with G007-LK decreased these proteins more potently (Figures 3E and S1D) and regulated in a time-dependent manner (Figure 3F). The phosphorylation level of Rb protein is positively correlated with the rate of cell proliferation, and the lower the phosphorylation level, the slower the cell proliferation [52]. This result suggests that the enhanced G1 phase arrest might contribute to the synergistic sensitization of BETi and TNKSi.
Figure 3.

The combination of BETi and TNKSi induces more cells arrest at G1 phase. (A) Cell cycle profile of HCT-15 cells after treatment with 2 μM OTX-015, HJP-178, G007-LK, or indicated combinations for 24 h. (B) The statistical results of (A). The cell cycle distribution data were calculated from three independent experiments and expressed as mean ± SD. *P<0.05; **P<0.01; ***P<0.001. (C) Cell cycle profiles of HCT-15 cells were detected at 12 h and 24 h after indicated combination treatments, and statistical analysis was performed in (D). (E and F) Cells were treated as described in (A and C), and the changes of G1 cycle-related proteins were detected using Western blotting. GAPDH was used as the reference control. The relative pixel intensity of representative blot was analyzed by Image J software and vehicle group was normalized as one. All experiments were repeated at least three times.
The TNKSi G007-LK synergistically increases BETi-induced apoptosis in colorectal cancer cells
BETi was reported to induce apoptosis to promote cell death [53]. As expected, either HJP-178 or OTX-015 alone induced apoptosis in colorectal HCT-15 and SW480 cancer cells, and the TNKSi G007-LK significantly potentiated their apoptotic induction while G007-LK alone was poorly active in this aspect (Figures 4A-D, S1E and S1F). Caspase 3/7 are the executors of cell apoptosis and are normally cleaved and activated during apoptosis [54]. Consistently, both HJP-178 and OTX-015 alone caused caspase 3/7 activation in colorectal HCT-15 and SW480 cancer cells, which was further significantly potentiated by the addition of the TNKSi G007-LK (Figure 4E and 4F). Moreover, the combination of G007-LK with HJP-178 or OTX-015 led to more protein cleavage of Caspase 3, Caspase 7, Caspase 8 and PARP than the respective single agents (Figures 4G, 4H and S1G). These data indicate that the TNKSi G007-LK synergistically increases BETi-induced apoptosis.
Figure 4.
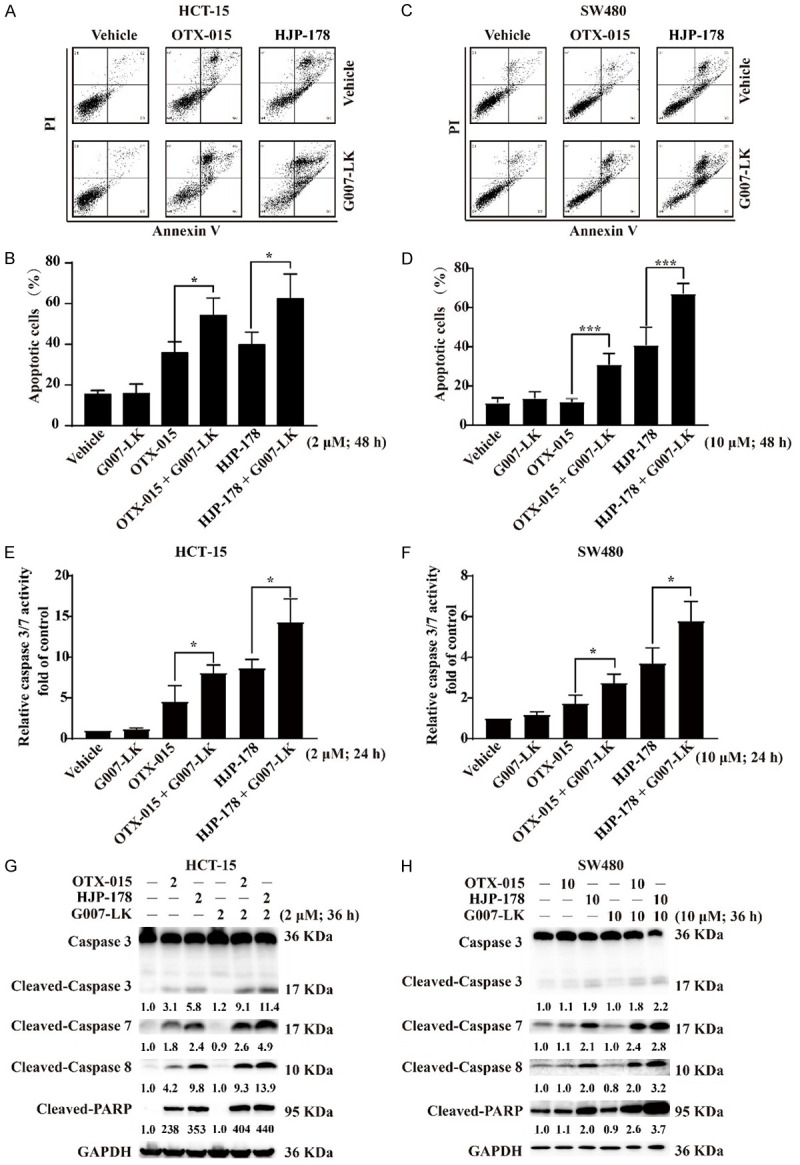
The combination of BETi and TNKSi elicits significant cell apoptosis. (A) Representative images of Annexin V and PI staining of HCT-15 cells treated with 2 μM G007-LK, OTX-015 and HJP-178 for 48 h. The percentage of cells that stained positive for Annexin V/PI is indicated (Q4, viable; Q3, early apoptosis; Q2, late apoptosis). (B) The percentage of apoptosis cells were calculated from three independent experiments and presented as mean ± SD. *P<0.05; **P<0.01; ***P<0.001. (C and D) The same treated way as (A and B) in SW480 cells, but the final concentration of drugs was 10 μM. (E and F) HCT-15 and SW480 cells were treated with the indicated concentrations of drugs for 24 h and detected the activity of caspase 3/7. (G and H) HCT-15 and SW480 cells were treated with the indicated concentrations of drugs for 36 h and collected for apoptosis-related proteins detection using Western blotting. GAPDH was used as the reference control. The relative pixel intensity of representative blot was analyzed by Image J software and the vehicle group without any treatment was normalized as one. In Caspase 3, only Cleaved-Caspase 3 band was quantitatively analyzed. All experiments were repeated at least three times.
The combination of BETi and TNKSi reduces the β-Catenin in transcription and degradation pathways
Since the synergistic sensitization effect of BETi and TNKSi has been verified in different experiments, we further explored the synergistic mechanism. Firstly, we detected the proteins directly regulated in the Tankyrases and BET-related signaling pathways. Β-Catenin is a key transmitter in the typical WNT signaling pathway, and its stability is regulated by destruction complex [55]. Tankyrases could PARsylate AXIN2, and then inhibit the degradation of β-Catenin. Accumulated β-Catenin in the cytoplasm enters the nucleus to regulate the expression of oncogenes and promotes the proliferation of tumor cells [56]. The results showed that BETi had little effect on Tankyrases and AXIN2 expression, but TNKSi G007-LK promoted the expressions of Tankyrases and AXIN2 significantly both in HCT-15 and SW480 cells (Figure 5A and 5B). The change of β-Catenin was slight under the treatment of BETi, and G007-LK induced obvious inhibition in SW480 cells. Different TNKSis RK-287107 and XAV939 showed consistent results in SW480 (Figure S2A). Interestingly, although the expressions of Tankyrases and AXIN2 in the combined group were reduced compared with G007-LK, the decrease in β-Catenin was more significant than any monotherapy treatments. This suggests that there are other regulatory pathways besides the Tankyrases-AXIN2-axis mediated the inhibition of β-Catenin under the combination treatment. Phosphorylation of β-Catenin at S675 could active the transcription of WNT target genes [57], which was also inhibited by the combination treatment. Rad51, a marker of DNA double-strand breaks was down-regulated more evidently by combination treatment, indicating enhanced cell damage induced by BETi and TNKSi simultaneously treatment. We also noticed that, c-Myc, an important target of BET family, was inhibited by BETi and slightly further suppressed in combination with G007-LK (Figure 5A and 5B). The above conclusions were similarly verified in another TNKSi. RK-287107 showed synergistic effects with different BETis OTX-015 and HJP-178 (Figure S2B). Interestingly, the results were consistent with the cellular proliferation inhibition assays (Figure 1), which the combined effect of G007-LK was superior to RK-287107 in the regulation of related proteins (Figure S2C). Therefore, we chose G007-LK as a combinational partner for further research. Meanwhile, consistent with the protein changes, there was no significant difference between single BETi and combination treatments at c-Myc mRNA level (Figure 5C). Because BET family members widely involved in regulating gene transcription, we further tested the mRNA levels of CTNNB1. Although BETi could significantly down-regulate the CTNNB1 level in different cells (Figure 5D), there was no consistent evident changes in the combination group with protein level changes. Consider the above results together (Figure 5A and 5B), we speculated that the synergistic effect of combination was not only due to the transcriptional regulation by BETi. Therefore, we believe that G007-LK may play a role in β-Catenin degradation and contribute to further down-regulation of protein levels in the combination treatment. The result showed that proteasome inhibitor MG-132 could reverse the down-regulation effect of G007-LK on β-Catenin in SW480 and HCT-15 cells (Figure 5E), which confirmed that G007-LK mediate the protein degradation of β-Catenin. Cycloheximide (CHX) is a compound inhibiting mRNA translation [58]. When CHX was added, the newly synthesized protein was inhibited, and the degradation of β-Catenin induced by G007-LK was relatively amplified in SW480. The addition of CHX was more sensitive to HCT-15 cells, which was manifested as a decrease of β-Catenin in the vehicle with CHX. Strikingly, consistent with the results in SW480 cells, the addition of CHX also amplified the down-regulation of β-Catenin in either monotherapy or combination therapy in HCT-15 cells (Figure 5F). In conclusion, we verified that the down-regulation of β-Catenin was the synergistic result of BETi-induced transcriptional inhibition and TNKSi-induced protein degradation.
Figure 5.

The combination of BETi and TNKSi reduces the β-Catenin in transcription and degradation pathways. (A and B) BET and Tankyrases-related protein changes. HCT-15 (A) and SW480 (B) cells were treated with OTX-015 (2 μM and 10 μM), HJP-178 (2 μM and 10 μM), G007-LK (2 μM and 10 μM) or indicated combinations for 24 h. Cells were collected for the protein detections of Tankyrases, AXIN2, β-Catenin, Phospho-β-Catenin (Ser675), c-Myc, RAD51 and GAPDH using Western blotting. GAPDH was used as the reference control. (C and D) Relative mRNA levels of c-Myc and CTNNB1. Cells were treated as described in (A and B). Total RNA was extracted, reverse transcribed to cDNA and amplified by RT-qPCR. Relative mRNA level was calculated from three independent experiments and presented as the mean ± SD. *P<0.05; **P<0.01; ***P<0.001. (E) In the presence and absence of MG-132, the effects of drugs on β-Catenin were detected using Western blotting. HJP-178 and G007-LK were both 10 μM in SW480 and 2 μM in HCT-15 for 24 h. 10 μM MG-132 was treated for 6 h. (F) The treatment condition was consistent with that described in (E), where the concentration of cycloheximide (CHX) was 25 μg/ml for 6 h. GAPDH was used as the reference control. (G and H) β-Catenin expression in nucleus and cytoplasm. Cells were treated as described in (A and B). Nuclear and Cytoplasmic Protein Extraction Kit was used to isolate the nucleus and cytoplasm of HCT-15 (G) and SW480 (H), and the protein expression was detected using Western blotting. GAPDH and H3 were used as internal reference for cytoplasm and nucleus respectively to identify the proteins in nucleus and cytoplasm as completely separated as possible. The relative pixel intensity of representative blot in Western blotting result was analyzed by Image J software and the vehicle group without any treatment was normalized as one. All the experiments were repeated at least three times.
β-Catenin can shuttle between cytoplasm and nucleus. We next detected the distribution of β-Catenin in the nucleus and cytoplasm and found that both BETi and TNKSi could effectively down-regulate β-Catenin, and the inhibition effect was more evident when the two inhibitors were combined (Figure 5G and 5H). Meanwhile, we noticed that the β-Catenin change in the nucleus and cytoplasm together is consistent with the total protein change (Figure 5A and 5B), but the effect of G007-LK on β-Catenin varied in different cells. The inhibition of β-Catenin by G007-LK occurred both in SW480 and HCT-15 cells, but was more significant in SW480 cells (Figure 5A and 5B). Correspondingly, in the cytoplasmic extracts, the reduction effect of G007-LK in SW480 was more obvious (Figure 5H). We believed the changes in any part of the cell extracts are not due to one single action of transcription or degradation, but the results of co-regulation. Briefly in HCT-15 cells, HJP-178-mediated transcriptional repression of β-Catenin is more important, and the β-Catenin reduction caused by G007-LK is more important in SW480 cells.
Based on the above data, it is not difficult for us to conclude that HJP-178 inhibited the synthesis of β-Catenin in the cytoplasm by reducing the transcription of CTNNB1 in the nucleus. G007-LK blocked the entry of β-Catenin into the nucleus by promoting the degradation of β-Catenin in the cytoplasm. The combination treatment of the two inhibitors further down-regulated the expression of β-Catenin, resulting in more effective inhibition of tumor cell proliferation and G1 cell cycle arrest, and more apoptosis induction (Figure 6). This finding provides a preliminary basis for the combinational use of BETi and TNKSi in colorectal cancer treatment.
Figure 6.

Schematic diagram of drug combination mechanism. When the WNT off, a destruction complex consisting of AXIN2, GSK3β, APC, and CK1α degrade β-Catenin through the proteasome system. When the WNT on, Tankyrases PARsylate the AXIN2, which inhibits the degradation of β-Catenin. Accumulated β-Catenin is highly expressed in the cytoplasm and then enters the nucleus through the nuclear pore. β-Catenin can bind to the transcription factor TCF/LEF to induce the cell proliferation and oncogene amplification. TNKSi promotes the degradation of β-Catenin by inhibiting PARsylation. BETi inhibits the transcription process of CTNNB1, and further inhibits CTNNB1 translation and synthesis in cytoplasm. Two inhibitors combine to inhibit the expression of β-Catenin through transcription and degradation pathways, which results in the proliferative inhibition, G1 phase arrest and apoptosis.
Discussion
BETi has been studied for more than 10 years. The weak monotherapy activity and toxicity hinder the development and application of BETi, so combination therapies with BETi have been carried out in clinical research trials in recent years and obtained important results. Activation of WNT signaling pathway is highly common in CRCs. As a candidate target for inhibition of WNT/β-Catenin pathway, Tankyrases are studied. TNKSi can promote the degradation of β-Catenin and reduce the entry of β-Catenin into the nucleus. However, problems such as unsatisfactory single drug activity and poor efficacy in vivo also limit the clinical progress and development of TNKSi. Because BETi also plays a crucial role in WNT pathways by interrupting the transcriptional process of β-Catenin, we consider that these two kinds of inhibitors have certain regulatory effects on the same pathway meanwhile face the similar problem of poor single drug activity. It is meaningful to detect the combination effect to determine the possibility of combination therapy and rescue the clinical failure conditions of BETi and TNKSi simultaneously. Synergistic therapy has shown greater interest in pharmacology and medicine because it achieves greater potency at lower drug doses. Such combination regimens also help to reduce the possible toxicity of the drugs.
In this report, we selected two APC mutant cell lines (HCT-15 and SW480 with mutant in β-Catenin binding domain) and CTNNB1 mutant cell lines (HCT-116 with phosphodegron mutations in β-Catenin) [27] to detect two BETis (OTX-015 and HJP-178) and two TNKSis (G007-LK and RK-287107). The experimental results showed that the combination treatment of multiple drugs all showed synergistic sensitization effect in three cell lines. This indicates that there is indeed a synergistic effect between the two inhibitors and the combination is conducive to further expanding the scope of treatment especially for the insensitive CRCs. Further, three different experimental methods, including cell proliferation assay, colony formation assay, and EdU assay, all verified the obvious synergistic sensitization between BETi and TNKSi. PARP of the same family as Tankyrases, the synergism between BETi and PARPi has been revealed in several literatures successively [59-61]. Meanwhile, the phase I clinical trial of BETi AZD5153 and PARPi olaparib in malignant solid tumors and lymphomas is also progressing, and the synergistic effect of BETi and TNKSi in our study shows an advantage over the combination of BETi and PARPi in the colony formation experiment, which makes the combination of BETi and TNKSi more promising.
The division of tumor cells is extremely active. Cells accelerate the deterioration of the disease by ceaseless proliferation and division [62]. One of the classic mechanisms of BETi is to inhibit the proliferation of tumor cells by inducing G1 phase arrest of tumor cells. The same cycle arrest was induced by G007-LK. Although G1 phase arrest induced by G007-LK was not obvious, when the BETi was added, nearly 70% of the G1 phase arrest occurred in concentration and time dependent manners. This greatly improves the utilization rate of the drug and exerts greater inhibitory activity at a lower dose. The same results were also verified in HCT-116 cells. Continuous cell cycle arrest can trigger the apoptosis. As a pathway of programmed death, the combination of BETi and TNKSi induced more apoptosis of tumor cells than any single drug treatment. Among them, G007-LK hardly showed obvious apoptotic effects, but it could significantly and effectively amplify the pro-apoptotic ability of BETi, activate the activity of caspase 3/7, and regulate the consistent change of caspase proteins. In summary, co-inhibition of BET and Tankyrases induces G1 cycle arrest, and the disorder of cycle progression is consistent with the decrease of new cells labeled with EdU. At the same time, it promoted the death of tumor cells by triggering cell apoptosis. In other words, the combination therapy was demonstrated by inhibiting the proliferation of new tumor cells and promoting the apoptosis of existing tumor cells, ultimately resulting in a reduction of total number of cells detected by the SRB assay and the colony formation assay.
Tankyrases belonging to the family of poly(ADP-ribosyl)ases play an important role in PARsylation by utilizing NAD+ as a substrate in order to generate ADP-ribose polymers [47]. Proteins modified by PARsylation recruit E3 ubiquitin ligase to promote their degradation. By inhibiting PARsylation of Tankyrases, TNKSis block the ubiquitin proteasome pathway, and then upregulate the levels of proteins [26,63]. Moreover, Tankyrases can also autoPARsylate themselves. Therefore, TNKSis block ubiquitination by inhibiting Tankyrases’ PARsylation, which are ultimately manifested as the upregulation of Tankyrases [64]. Since TNKSis are not directly regulated Tankyrases, it is not surprising that the protein levels of Tankyrases and AXIN2 are upregulated by G007-LK. Tankyrases inhibit the degradation of β-Catenin by PARsylating AXIN2 [25]. The accumulated β-Catenin in the cytoplasm is phosphorylated, and the phosphorylation at S675 activates the transcription of WNT target genes [57]. In our study, the expression of Tankyrases and AXIN2 both increased under the treatment of TNKSi, which are due to the autoPARsylation and PARsylation as reported in other test [26]. Although the combination treatment group had slight down-regulation effect, it could still stabilize the protein expression compared with the vehicle and BETi groups. Even SW480 and HCT-15 are both APC mutated cell lines, the codon truncation location mutation both occurred in the region of 20-amino acid repeats which bound to β-Catenin. However, the specific location is different (the mutated site of HCT-15 is 1417 site and SW480 is 1338 site), leading to different sensitivity to TNKSi and regulation of β-Catenin [27]. Different mutation locations may also be the reason causes the different β-Catenin regulation in HCT-15 and SW480 cells. Β-Catenin, as an effector of WNT pathway, it could bind to the transcription factor TCF/LEF in the nucleus to regulate the transcriptional expression of downstream genes. Combined with the results of transcriptional level, we found that different BETi could significantly down-regulate the mRNA level of CTNNB1, but there was no consistent significant difference between the combination and BETi alone. Under MG-132 or CHX treatment, we also found that both G007-LK and the combination showed a promotion effect on β-Catenin degradation, while HJP-178 showed slight change in β-Catenin protein level, except that CHX was sensitive to HCT-15. Therefore, G007-LK played a major role in the protein degradation pathway in the combination group is reasonably presumed. Due to the weak role of BETi in the regulation of degradation and the faint role of G007-LK in the transcription, the protein reduction of β-Catenin of combination in whole cell protein extract was attributed to two pathways: transcription inhibition contributed by BETi and degradation promotion contributed by TNKSi. In addition, we found that the down-regulation of β-Catenin was manifested in the nucleus and cytoplasm, that could be due to two aspects, one is TNKSi-facilitated degradation in the cytoplasm and reduced the accumulation of proteins that will enter the nucleus in excess, the other is inhibitory effect of BETi on the transcriptional regulation of CTNNB1 in the nucleus and inhibit the proteins synthesized by ribosomes in the cytoplasm. These regulations not only reduce β-Catenin entering the nucleus but also inhibit the oncogenes transcription in the nucleus.
In fact, we have done animal studies to test the synergistic effect of HJP-178 and G007-LK in vivo (Figure S2D and S2E). Unfortunately, the result showed that the combination could only slightly inhibit HCT-15 xenograft tumor growth, though G007-LK alone promoted the tumor growth, which was similarly reported in the previous literature [27]. We found that in the solvent formulations used in the literature, G007-LK had poor solubility and insoluble compounds were present. So we improved the solvent formula, and kept the G007-LK completely solved in the new solvent before administration though the undissolved compounds could still occur in a later time. After 21 days of intraperitoneal injection, we found that the tumor volume in the G007-LK group increased gradually with a growth inhibition rate (GI%) of -31.46%, and 5.05% body weight was lost compared with the vehicle group (Figure S2D and S2E). This suggests that certain toxicity has happened at 10 mg/kg G007-LK dosage, and there is no big room for dose optimization. Obviously, our solvent formulation improved the solubility of G007-LK and promoted its absorption and utilization compared with the dose used in the literature [27]. We also found that the similar promoting tumor growth effect of G007-LK was seen in HCT-15 model [27]. The discrepancy between cell and cell xenograft results of G007-LK could be due to the metabolites or complicated microenvironment in vivo. Therefore, optimization of TNKSi is still very important for the antitumor treatment use. In our result, it is hopeful to see the combination treatment with BETi exert enhanced antitumor activity that GI% increased from 7.77% in the HJP-178 group to 12.79% in the combination group. Although G007-LK alone promotes tumor growth, combination therapy still showed an overall suppressive effect on tumor growth, which compared with the GI% of the G007-LK group, P value is 0.053. It is believed that the development of novel TNKSi will lead to positive results in combination therapy. In order to eliminate the chance caused by specific drugs and cell models, we selected the classical TNKSi XAV939 and tried it in the HCT-116 model. However, our data showed that XAV939 itself has not in vivo antitumor activity, and in contrast, it promoted tumor growth again, which were directly reflected in the tumor weight (Figure S2F and S2G). As can be seen from the image (Figure S2H), there was no significant difference between the combination groups and the BETi monotherapy groups. From what have been discussed above, we believe that the optimization of TNKSi may be the key factor to solve the synergistic sensitization effect in vivo at this stage.
In conclusion, our results reveal the synergistic sensitization effect of BETi and TNKSi firstly. Combination therapy of BETi and TNKSi can arrest cells at G1 phase, disrupt the cycle progression of tumor cells and inhibit cell proliferation. It also inhibits cell growth by promoting the apoptosis induction. BETi and TNKSi do not interoperate with each other in the regulation process of β-Catenin, but act on the mRNA level and the protein level of β-Catenin respectively to inhibit its expression. Even though the opposite effect of G007-LK in vivo, this combination therapy still showed signs of synergism and sensitization, and provided a new opportunity for CRCs.
Acknowledgements
We are grateful for financial supports from the Science and Technology Commission of Shanghai Municipality (19ZR1467900 and 20ZR1468100 to Ying-Qing Wang), the Nova Development Program of the Shanghai Institute of Materia Medica, the Chinese Academy of Sciences and the State Key Laboratory of Drug Research.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19:776–800. doi: 10.1038/s41573-020-0077-5. [DOI] [PubMed] [Google Scholar]
- 2.Hu J, Tian CQ, Damaneh MS, Li Y, Cao D, Lv K, Yu T, Meng T, Chen D, Wang X, Chen L, Li J, Song SS, Huan XJ, Qin L, Shen J, Wang YQ, Miao ZH, Xiong B. Structure-based discovery and development of a series of potent and selective bromodomain and extra-terminal protein inhibitors. J Med Chem. 2019;62:8642–8663. doi: 10.1021/acs.jmedchem.9b01094. [DOI] [PubMed] [Google Scholar]
- 3.Xiang Y, Tanaka Y, Patterson B, Hwang SM, Hysolli E, Cakir B, Kim KY, Wang W, Kang YJ, Clement EM, Zhong M, Lee SH, Cho YS, Patra P, Sullivan GJ, Weissman SM, Park IH. Dysregulation of BRD4 function underlies the functional abnormalities of MeCP2 mutant neurons. Mol Cell. 2020;79:84–98. e9. doi: 10.1016/j.molcel.2020.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donati B, Lorenzini E, Ciarrocchi A. BRD4 and cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17:164. doi: 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alghamdi S, Khan I, Beeravolu N, McKee C, Thibodeau B, Wilson G, Chaudhry GR. BET protein inhibitor JQ1 inhibits growth and modulates WNT signaling in mesenchymal stem cells. Stem Cell Res Ther. 2016;7:22. doi: 10.1186/s13287-016-0278-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duan Q, Xiao Y, Zhu L, Liu Z, Mao X, Zhou Z, Liao C, Cai J, Huang F, Liu Z, Zeng J, Xia K, Chang C, Qi J, Chen Z, Huang H, Yang T. BET bromodomain is a novel regulator of TAZ and its activity. Biochim Biophys Acta. 2016;1859:1527–1537. doi: 10.1016/j.bbagrm.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Tian S, Xiong J, Zhou Y, Song H, Liu C. JQ-1 inhibits colon cancer proliferation via suppressing Wnt/β-catenin signaling and miR-21. Chem Res Toxicol. 2018;31:302–307. doi: 10.1021/acs.chemrestox.7b00346. [DOI] [PubMed] [Google Scholar]
- 8.Venugopal S, Mascarenhas J. Current clinical investigations in myelofibrosis. Hematol Oncol Clin North Am. 2021;35:353–373. doi: 10.1016/j.hoc.2020.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Mascarenhas J, Kremyanskaya M, Hoffman R, Bose P, Talpaz M, Harrison CN, Gupta V, Leber B, Sirhan S, Kabir S, Senderowicz A, Shao J, Mertz J, Trojer P, Verstovsek S. MANIFEST, a phase 2 study of CPI-0610, a bromodomain and extraterminal domain inhibitor (BETi), as monotherapy or “add-on” to ruxolitinib, in patients with refractory or intolerant advanced myelofibrosis. Blood. 2019;134:670. [Google Scholar]
- 10.Aggarwal RR, Schweizer MT, Nanus DM, Pantuck AJ, Heath EI, Campeau E, Attwell S, Norek K, Snyder M, Bauman L, Lakhotia S, Feng FY, Small EJ, Abida W, Alumkal JJ. A phase Ib/IIa study of the pan-BET inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2020;26:5338–5347. doi: 10.1158/1078-0432.CCR-20-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhadury J, Nilsson LM, Muralidharan SV, Green LC, Li Z, Gesner EM, Hansen HC, Keller UB, McLure KG, Nilsson JA. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in myc-induced murine lymphoma. Proc Natl Acad Sci U S A. 2014;111:E2721–30. doi: 10.1073/pnas.1406722111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heller G, Schmidt WM, Ziegler B, Holzer S, Müllauer L, Bilban M, Zielinski CC, Drach J, Zöchbauer-Müller S. Genome-wide transcriptional response to 5-aza-2’-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008;68:44–54. doi: 10.1158/0008-5472.CAN-07-2531. [DOI] [PubMed] [Google Scholar]
- 13.Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780–1789. doi: 10.1172/JCI76094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zagni C, Chiacchio U, Rescifina A. Histone methyltransferase inhibitors: novel epigenetic agents for cancer treatment. Curr Med Chem. 2013;20:167–185. doi: 10.2174/092986713804806667. [DOI] [PubMed] [Google Scholar]
- 15.Peirs S, Frismantas V, Matthijssens F, Van Loocke W, Pieters T, Vandamme N, Lintermans B, Dobay MP, Berx G, Poppe B, Goossens S, Bornhauser BC, Bourquin JP, Van Vlierberghe P. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia. 2017;31:2037–2047. doi: 10.1038/leu.2017.10. [DOI] [PubMed] [Google Scholar]
- 16.Siu KT, Ramachandran J, Yee AJ, Eda H, Santo L, Panaroni C, Mertz JA, Sims Iii RJ, Cooper MR, Raje N. Preclinical activity of CPI-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia. 2017;31:1760–1769. doi: 10.1038/leu.2016.355. [DOI] [PubMed] [Google Scholar]
- 17.Teicher BA, Tomaszewski JE. Proteasome inhibitors. Biochem Pharmacol. 2015;96:1–9. doi: 10.1016/j.bcp.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Pollyea DA, Zehnder J, Coutre S, Gotlib JR, Gallegos L, Abdel-Wahab O, Greenberg P, Zhang B, Liedtke M, Berube C, Levine R, Mitchell BS, Medeiros BC. Sequential azacitidine plus lenalidomide combination for elderly patients with untreated acute myeloid leukemia. Haematologica. 2013;98:591–596. doi: 10.3324/haematol.2012.076414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 20.Sebio A, Kahn M, Lenz HJ. The potential of targeting wnt/β-catenin in colon cancer. Expert Opin Ther Targets. 2014;18:611–615. doi: 10.1517/14728222.2014.906580. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Liang B, Song X, Wang H, Evert M, Zhou Y, Calvisi DF, Tang L, Chen X. Loss of apc cooperates with activated oncogenes to induce liver tumor formation in mice. Am J Pathol. 2021;191:930–946. doi: 10.1016/j.ajpath.2021.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katoh M, Katoh M. Molecular genetics and targeted therapy of WNT-related human diseases (review) Int J Mol Med. 2017;40:587–606. doi: 10.3892/ijmm.2017.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wanitsuwan W, Kanngurn S, Boonpipattanapong T, Sangthong R, Sangkhathat S. Overall expression of beta-catenin outperforms its nuclear accumulation in predicting outcomes of colorectal cancers. World J Gastroenterol. 2008;14:6052–6059. doi: 10.3748/wjg.14.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen SH, Yu X. Targeting deparylation selectively suppresses DNA repair-defective and parp inhibitor-resistant malignancies. Sci Adv. 2019;5:eaav4340. doi: 10.1126/sciadv.aav4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizutani A, Yashiroda Y, Muramatsu Y, Yoshida H, Chikada T, Tsumura T, Okue M, Shirai F, Fukami T, Yoshida M, Seimiya H. RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 2018;109:4003–4014. doi: 10.1111/cas.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 27.Lau T, Chan E, Callow M, Waaler J, Boggs J, Blake RA, Magnuson S, Sambrone A, Schutten M, Firestein R, Machon O, Korinek V, Choo E, Diaz D, Merchant M, Polakis P, Holsworth DD, Krauss S, Costa M. A novel tankyrase small-molecule inhibitor suppresses apc mutation-driven colorectal tumor growth. Cancer Res. 2013;73:3132–3144. doi: 10.1158/0008-5472.CAN-12-4562. [DOI] [PubMed] [Google Scholar]
- 28.Katoh M. Multi-layered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/β-catenin signaling activation (review) Int J Mol Med. 2018;42:713–725. doi: 10.3892/ijmm.2018.3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kierulf-Vieira KS, Sandberg CJ, Waaler J, Lund K, Skaga E, Saberniak BM, Panagopoulos I, Brandal P, Krauss S, Langmoen IA, Vik-Mo EO. A small-molecule tankyrase inhibitor reduces glioma stem cell proliferation and sphere formation. Cancers (Basel) 2020;12:1630. doi: 10.3390/cancers12061630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li N, Wang Y, Neri S, Zhen Y, Fong LWR, Qiao Y, Li X, Chen Z, Stephan C, Deng W, Ye R, Jiang W, Zhang S, Yu Y, Hung MC, Chen J, Lin SH. Tankyrase disrupts metabolic homeostasis and promotes tumorigenesis by inhibiting LKB1-AMPK signalling. Nat Commun. 2019;10:4363. doi: 10.1038/s41467-019-12377-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waaler J, Mygland L, Tveita A, Strand MF, Solberg NT, Olsen PA, Aizenshtadt A, Fauskanger M, Lund K, Brinch SA, Lycke M, Dybing E, Nygaard V, Bøe SL, Heintz KM, Hovig E, Hammarström C, Corthay A, Krauss S. Tankyrase inhibition sensitizes melanoma to PD-1 immune checkpoint blockade in syngeneic mouse models. Commun Biol. 2020;3:196. doi: 10.1038/s42003-020-0916-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shirai F, Tsumura T, Yashiroda Y, Yuki H, Niwa H, Sato S, Chikada T, Koda Y, Washizuka K, Yoshimoto N, Abe M, Onuki T, Mazaki Y, Hirama C, Fukami T, Watanabe H, Honma T, Umehara T, Shirouzu M, Okue M, Kano Y, Watanabe T, Kitamura K, Shitara E, Muramatsu Y, Yoshida H, Mizutani A, Seimiya H, Yoshida M, Koyama H. Discovery of novel spiroindoline derivatives as selective tankyrase inhibitors. J Med Chem. 2019;62:3407–3427. doi: 10.1021/acs.jmedchem.8b01888. [DOI] [PubMed] [Google Scholar]
- 33.Shirai F, Mizutani A, Yashiroda Y, Tsumura T, Kano Y, Muramatsu Y, Chikada T, Yuki H, Niwa H, Sato S, Washizuka K, Koda Y, Mazaki Y, Jang MK, Yoshida H, Nagamori A, Okue M, Watanabe T, Kitamura K, Shitara E, Honma T, Umehara T, Shirouzu M, Fukami T, Seimiya H, Yoshida M, Koyama H. Design and discovery of an orally efficacious spiroindolinone-based tankyrase inhibitor for the treatment of colon cancer. J Med Chem. 2020;63:4183–4204. doi: 10.1021/acs.jmedchem.0c00045. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka N, Mashima T, Mizutani A, Sato A, Aoyama A, Gong B, Yoshida H, Muramatsu Y, Nakata K, Matsuura M, Katayama R, Nagayama S, Fujita N, Sugimoto Y, Seimiya H. APC mutations as a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer. Mol Cancer Ther. 2017;16:752–762. doi: 10.1158/1535-7163.MCT-16-0578. [DOI] [PubMed] [Google Scholar]
- 35.Tian CQ, Chen L, Chen HD, Huan XJ, Hu JP, Shen JK, Xiong B, Wang YQ, Miao ZH. Inhibition of the BET family reduces its new target gene IDO1 expression and the production of L-kynurenine. Cell Death Dis. 2019;10:557. doi: 10.1038/s41419-019-1793-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 37.Zhang N, Tian YN, Zhou LN, Li MZ, Chen HD, Song SS, Huan XJ, Bao XB, Zhang A, Miao ZH, He JX. Glycogen synthase kinase 3β inhibition synergizes with PARP inhibitors through the induction of homologous recombination deficiency in colorectal cancer. Cell Death Dis. 2021;12:183. doi: 10.1038/s41419-021-03475-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y, Zhu F, Wang Q, Ding Y, Ying R, Zeng L. Inhibition of EZH2 and EGFR produces a synergistic effect on cell apoptosis by increasing autophagy in gastric cancer cells. Onco Targets Ther. 2018;11:8455–8463. doi: 10.2147/OTT.S186498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Q, Chen DQ, Sun L, Huan XJ, Bao XB, Tian CQ, Hu J, Lv KK, Wang YQ, Xiong B, Miao ZH. Novel bivalent BET inhibitor N2817 exhibits potent anticancer activity and inhibits TAF1. Biochem Pharmacol. 2021;185:114435. doi: 10.1016/j.bcp.2021.114435. [DOI] [PubMed] [Google Scholar]
- 40.Yang B, Wu Q, Huan X, Wang Y, Sun Y, Yang Y, Liu T, Wang X, Chen L, Xiong B, Zhao D, Miao Z, Chen D. Discovery of a series of 1H-pyrrolo[2,3-b]pyridine compounds as potent TNIK inhibitors. Bioorg Med Chem Lett. 2021;33:127749. doi: 10.1016/j.bmcl.2020.127749. [DOI] [PubMed] [Google Scholar]
- 41.Ding C, Tian Q, Li J, Jiao M, Song S, Wang Y, Miao Z, Zhang A. Structural modification of natural product tanshinone I leading to discovery of novel nitrogen-enriched derivatives with enhanced anticancer profile and improved drug-like properties. J Med Chem. 2018;61:760–776. doi: 10.1021/acs.jmedchem.7b01259. [DOI] [PubMed] [Google Scholar]
- 42.Tian QT, Ding CY, Song SS, Wang YQ, Zhang A, Miao ZH. New tanshinone I derivatives S222 and S439 similarly inhibit topoisomerase I/II but reveal different p53-dependency in inducing G2/M arrest and apoptosis. Biochem Pharmacol. 2018;154:255–264. doi: 10.1016/j.bcp.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 43.Li MZ, Meng T, Song SS, Bao XB, Ma LP, Zhang N, Yu T, Zhang YL, Xiong B, Shen JK, Miao ZH, He JX. Discovery of MTR-106 as a highly potent G-quadruplex stabilizer for treating BRCA-deficient cancers. Invest New Drugs. 2021;39:1213–1221. doi: 10.1007/s10637-021-01096-4. [DOI] [PubMed] [Google Scholar]
- 44.Chang X, Chai Z, Zou J, Wang H, Wang Y, Zheng Y, Wu H, Liu C. PADI3 induces cell cycle arrest via the Sirt2/AKT/p21 pathway and acts as a tumor suppressor gene in colon cancer. Cancer Biol Med. 2019;16:729–742. doi: 10.20892/j.issn.2095-3941.2019.0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, Hu P, Li H, Yu XX, Wang XY, Qing YJ, Wang ZY, Wang HZ, Zhu MY, Guo QL, Hui H. LW-213, a newly synthesized flavonoid, induces G2/M phase arrest and apoptosis in chronic myeloid leukemia. Acta Pharmacol Sin. 2020;41:249–259. doi: 10.1038/s41401-019-0270-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Y, Han J, Wang Z, Li X, Sun Y, Hu Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: a systematic study of clinical trials. Front Pharmacol. 2020;11:621093. doi: 10.3389/fphar.2020.621093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamal A, Riyaz S, Srivastava AK, Rahim A. Tankyrase inhibitors as therapeutic targets for cancer. Curr Top Med Chem. 2014;14:1967–1976. doi: 10.2174/1568026614666140929115831. [DOI] [PubMed] [Google Scholar]
- 48.Lui GYL, Shaw R, Schaub FX, Stork IN, Gurley KE, Bridgwater C, Diaz RL, Rosati R, Swan HA, Ince TA, Harding TC, Gadi VK, Goff BA, Kemp CJ, Swisher EM, Grandori C. BET, SRC, and BCL2 family inhibitors are synergistic drug combinations with PARP inhibitors in ovarian cancer. EBioMedicine. 2020;60:102988. doi: 10.1016/j.ebiom.2020.102988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller AL, Fehling SC, Garcia PL, Gamblin TL, Council LN, van Waardenburg R, Yang ES, Bradner JE, Yoon KJ. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine. 2019;44:419–430. doi: 10.1016/j.ebiom.2019.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Damaneh MS, Hu JP, Huan XJ, Song SS, Tian CQ, Chen DQ, Meng T, Chen YL, Shen JK, Xiong B, Miao ZH, Wang YQ. A new BET inhibitor, 171, inhibits tumor growth through cell proliferation inhibition more than apoptosis induction. Invest New Drugs. 2020;38:700–713. doi: 10.1007/s10637-019-00818-z. [DOI] [PubMed] [Google Scholar]
- 51.Solberg NT, Waaler J, Lund K, Mygland L, Olsen PA, Krauss S. Tankyrase inhibition enhances the antiproliferative effect of PI3K and EGFR inhibition, mutually affecting β-catenin and AKT signaling in colorectal cancer. Mol Cancer Res. 2018;16:543–553. doi: 10.1158/1541-7786.MCR-17-0362. [DOI] [PubMed] [Google Scholar]
- 52.Zatulovskiy E, Zhang S, Berenson DF, Topacio BR, Skotheim JM. Cell growth dilutes the cell cycle inhibitor Rb to trigger cell division. Science. 2020;369:466–471. doi: 10.1126/science.aaz6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Letson C, Padron E. Non-canonical transcriptional consequences of BET inhibition in cancer. Pharmacol Res. 2019;150:104508. doi: 10.1016/j.phrs.2019.104508. [DOI] [PubMed] [Google Scholar]
- 54.Ghosh M, Murugadoss S, Janssen L, Cokic S, Mathyssen C, Van Landuyt K, Janssens W, Carpentier S, Godderis L, Hoet P. Distinct autophagy-apoptosis related pathways activated by multi-walled (NM 400) and single-walled carbon nanotubes (NIST-SRM2483) in human bronchial epithelial (16HBE14o-) cells. J Hazard Mater. 2020;387:121691. doi: 10.1016/j.jhazmat.2019.121691. [DOI] [PubMed] [Google Scholar]
- 55.Huang J, Qu Q, Guo Y, Xiang Y, Feng D. Tankyrases/β-catenin signaling pathway as an anti-proliferation and anti-metastatic target in hepatocarcinoma cell lines. J Cancer. 2020;11:432–440. doi: 10.7150/jca.30976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Danek P, Kardosova M, Janeckova L, Karkoulia E, Vanickova K, Fabisik M, Lozano-Asencio C, Benoukraf T, Tirado-Magallanes R, Zhou Q, Burocziova M, Rahmatova S, Pytlik R, Brdicka T, Tenen DG, Korinek V, Alberich-Jorda M. β-Catenin-TCF/LEF signaling promotes steady-state and emergency granulopoiesis via G-CSF receptor upregulation. Blood. 2020;136:2574–2587. doi: 10.1182/blood.2019004664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng Y, Zhou C, Yu XX, Wu C, Jia HL, Gao XM, Yang JM, Wang CQ, Luo Q, Zhu Y, Zhang Y, Wei JW, Sheng YY, Dong QZ, Qin LX. Osteopontin promotes metastasis of intrahepatic cholangiocarcinoma through recruiting MAPK1 and mediating ser675 phosphorylation of β-Catenin. Cell Death Dis. 2018;9:179. doi: 10.1038/s41419-017-0226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thorvaldsen TE, Pedersen NM, Wenzel EM, Stenmark H. Differential roles of AXIN1 and AXIN2 in Tankyrase inhibitor-induced formation of degradasomes and β-Catenin degradation. PLoS One. 2017;12:e0170508. doi: 10.1371/journal.pone.0170508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karakashev S, Zhu H, Yokoyama Y, Zhao B, Fatkhutdinov N, Kossenkov AV, Wilson AJ, Simpkins F, Speicher D, Khabele D, Bitler BG, Zhang R. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep. 2017;21:3398–3405. doi: 10.1016/j.celrep.2017.11.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fiorentino FP, Marchesi I, Schröder C, Schmidt R, Yokota J, Bagella L. BET-inhibitor I-BET762 and PARP-inhibitor talazoparib synergy in small cell lung cancer cells. Int J Mol Sci. 2020;21:9595. doi: 10.3390/ijms21249595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fehling SC, Miller AL, Garcia PL, Vance RB, Yoon KJ. The combination of BET and PARP inhibitors is synergistic in models of cholangiocarcinoma. Cancer Lett. 2020;468:48–58. doi: 10.1016/j.canlet.2019.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin GS. Cell signaling and cancer. Cancer Cell. 2003;4:167–174. doi: 10.1016/s1535-6108(03)00216-2. [DOI] [PubMed] [Google Scholar]
- 63.Callow MG, Tran H, Phu L, Lau T, Lee J, Sandoval WN, Liu PS, Bheddah S, Tao J, Lill JR, Hongo JA, Davis D, Kirkpatrick DS, Polakis P, Costa M. Ubiquitin ligase RNF146 regulates tankyrase and axin to promote Wnt signaling. PLoS One. 2011;6:e22595. doi: 10.1371/journal.pone.0022595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yeh TY, Meyer TN, Schwesinger C, Tsun ZY, Lee RM, Chi NW. Tankyrase recruitment to the lateral membrane in polarized epithelial cells: regulation by cell-cell contact and protein poly(ADP-ribosyl)ation. Biochem J. 2006;399:415–425. doi: 10.1042/BJ20060713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.