

Abstract
Despite of the improvement in gastric cancer (GC) therapies patients still suffer from cancer recurrence and metastasis. Recently, the high ratio of these events combined with increased chemoresistance has been related to the asymptomatic Helicobacter pylori (Hp) infections. The limited efficiency of GC treatment strategies is also increasingly attributed to the activity of tumor stroma with the key role of cancer-associated fibroblasts (CAFs). In order to investigate the influence of Hp infection within stromal gastric tissue on cancer initiation and progression, we have exposed normal gastric epithelial cells to long-term influence of Hp-activated gastric fibroblast secretome. We have referred obtained results to this secretome influence on cancer cell lines. The invasive properties of cells were checked by time-lapse video microscopy and basement membrane assays. The expression of invasion-related factors was checked by RT-PCR, Western Blot, immunofluorescence and Elisa. Hp-activated gastric fibroblast secretome induced EMT type 3-related shifts of RGM1 cell phenotype; in particular it augmented their motility, cytoskeletal plasticity and invasiveness. These effects were accompanied by Snail1/Twist activation, the up-regulation of cytokeratin19/FAP/TNC/Integrin-β1 and MMPs, and by the induction of cMethigh/pEGFRhigh phenotype. Mechanistic studies suggest that this microevolution next to TGFβ relies also on c-Met/EGFR signaling interplay and engages HGF-Integrin-Ras-dependent Twist activation leading to MMP and TNC upregulation with subsequent positive auto- and paracrine feedback loops intensifying this process. Similar shifts were detected in cancer cells exposed to this secretome. Collectively, we show that the secretome of Hp-infected fibroblasts induces reprogramming/microevolution of epithelial and cancer cells towards type 3 EMT-related invasive phenotype in a manner reciprocally reliant next to TGFβ on cMet/Integrin-β1/p-EGFR-dependent axis. Apparently, the phenotypical plasticity of Hp-activated fibroblast reprogrammed gastric epithelial cells determines their susceptibility to the pro-invasive signaling, which results in re-organization of gastric niches and provides the cues for GC promotion/progression.
Keywords: Helicobacter pylori-activated fibroblasts, gastric cancer, cancer associated fibroblasts, metastasis, epithelial-mesenchymal transition, mesenchymal-epithelial transition, epithelial-myofibroblast transition
Introduction
Gastric cancer (GC) remains the fourth most common cause of cancer-related death in the world with 90% of all stomach tumors being malignant [1]. Despite the improvement in therapies patients still suffer from GC recurrence and metastasis [2] due to the high invasiveness, rapid proliferation and the activity of anti-apoptotic systems in tumor cells [3]. Helicobacter pylori (Hp)-induced GC development is a complex cascade of events, characterized mainly by the interplay between Hp infection, host and environmental factors. The particular emphasis has been placed on direct interactions of Hp with the epithelium. However, despite that Hp colonizes mainly gastric epithelium; it may also penetrate the mucus layer reaching pits of gastric glands. The presence of Hp was found in the lamina propria and the correlation between asymptomatic Hp infections and subsequent GC development has been reported [3-8]. Additionally, this bug has recently been correlated with stromal tumor development [6]. Parallely, the limited efficiency of GC treatment strategies is increasingly attributed to the activity of tumor stroma with the key role of cancer-associated fibroblasts (CAFs) [9]. It’s well known, that CAFs are responsible for the constitution of tumor microenvironment [10-12] through mediating cancer-related inflammation [13,14], re-arrangement of extracellular matrix (ECM) proteins [14,15] and epithelial-mesenchymal transition (EMT) type 3 initiation in epithelial cells [9-14,16]. EMT is a biological process in which polarized epithelial cells lose the adherence and tight cell-cell junctions, enhance their migratory capacity, and become resistant to apoptosis. EMT type 1 and 2 are essential for numerous developmental as well as regenerative processes like wound healing, while EMT type 3 initiation in epithelial cell compartment is a prerequisite for tumor promotion and progression [9-14].
The depth of invasion and the regional lymph node activation represent two critical survival-influencing factors in GC [9-14], with the initiation of metastatic cascade remaining one of the most enigmatic aspects of this disease [17]. An essential steps in metastasis are: EMT type 3 progression, which enables tumor cells to acquire invasive properties [17] and its reversal counterpart MET (mesenchymal-epithelial transition) responsible for establishing secondary tumors. Both processes demand precise regulation of master EMT-TFs, such as Twist and Snail. Snail initiates EMT leading to the induction of motile, mesenchymal phenotype [18,19], while Twist is involved in tumor invasion and metastasis [18,20-26]. Despite increasing knowledge in this area, a better understanding of EMT-TF function and regulation of expression is required, particularly in Hp-induced GC.
Recently, we have reported that oncogenic Hp strain (cagA+vacA+s1m1) activates fibroblasts towards cells bearing the features of CAFs [27,28]. Such Hp-activated fibroblasts were capable of EMT process initiation in normal gastric epithelium in short-term (up to 96 hrs) cultures [19,29], while in long-term cultures (one month) they switched normal gastric epithelium microevolution towards cancer stem cell-related differentiation program [28]. Herein, we have attempted to assess further consequences of the prolonged exposition of gastric epithelial cells to Hp-AGF secretome. In particular, (i) we have concentrated on the hallmarks of the permanent phenotypic reprogramming of normal gastric epithelial cells towards EMT type 3 related invasive phenotype (ii) and we have attributed these reprogramming events to the activation of Hp-AGF triggered signaling pathways in gastric epithelial cells. Finally we have focused on the versatility of Hp-AGF short-term evoked reprogramming as well as on the assessment how this influence will affect already developed gastric/colon cancer cells and to what extent these mechanisms can participate in neoplasm.
Materials and methods
Experimental design
The Hp strain expressing CagA and VacA cytotoxins (43504 Hp cagA+vacA+(s1/m1)) was purchased from American Type Culture Collection [19,27-30]. Stock cultures were maintained and cultured as previously described [19,27-30]. Gastric samples were harvested from 8-week-old Spraque-Dowley rats for fibroblasts isolation and subsequent activation by Hp as described [19,27-29]. RGM1 cells (RCB0876, Riken Cell Bank, Ibaraki, Japan) were cultured in 5 ml of neat rat gastric Hp-activated fibroblast (Hp-AGF) supernatant for 96 hrs on 6-well plates then trypsinized and resuspended in serum-free DMEM with antibiotics. The cells were seeded on the upper side of transwell inserts containing native microporous membranes (pore diameter: 8 μm) at a density of 1×104 and allowed to transmigrate towards the Hp-AGF supernatant for 24 hrs. RGM1 cells, which successfully transmigrated through the cell culture inserts (s.t.EMT+RGM1 cells) were then cultured in the supernatant from Hp-AGFs for at least 30 days long-term RGM1 cells originating from EMT-positive short-term RGM1 cells (l.t.EMT+RGM1 cells) in 25 cm2 flasks. The neat Hp-AGF supernatant was added in an amount of 4 ml and changed every 3 days. Simultaneously, RGM1 cells cultured for 96 hrs in GF supernatant (abbreviated as s.t.EMT-RGM1 cells) were cultured in normal rat gastric fibroblast (GF) supernatant for at least 30 days long-term RGM1 cells originating from short-term EMT negative RGM1 cells (l.t.EMT-RGM1 cells). RGM1 cells were cultured in DMEM+10% FBS (RGM1) for the identical period of time. During all experiments, the amounts of the media as well as the procedures applied to the cells were standardized [28] (Figure 11A).
Figure 11.
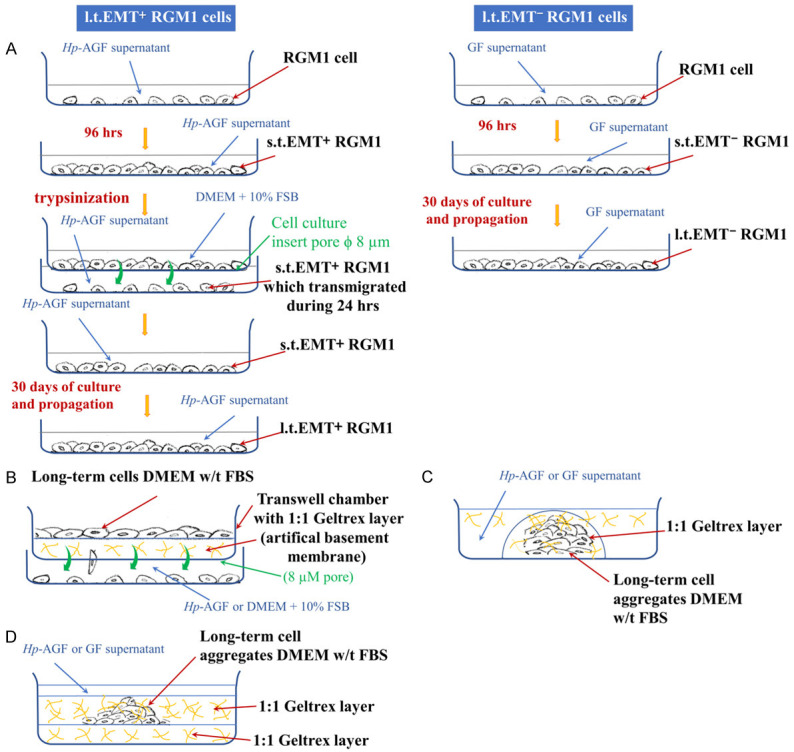
Flowchart of the experimental procedures.
Human fibroblasts were isolated from the biopsies of patients without systemic inflammatory and autoimmunologic diseases and Hp infection, qualified to laparoscopic, sleeve gastrectomy. The cells were isolated and cultured as previously described for rat fibroblasts [19,27-30] and then activated by Hp for 120 hrs according to previusly established protocol [19,27-30] in F12/DMEM+10% FBS. For the experiments with AGS cells (ATCC CRL-1739, Manassas, VA, USA), the supernatant from Hp-activated human gastric fibroblasts (Hp-hAGF) and from normal human gastric fibroblasts (hGF) was collected as described previously [19,28].
HaCaT (CLS 300493 GmbH, Heidelberg, Germany) and HT29 (ATCC HTB38, Manassas, VA, USA) cells were cultured in: DMEM+10% FBS (control), hGF and in Hp-hAGF supernatants for 24 and 96 hrs [19,28] and then used for analysis. AGS cells were cultured in F12/DMEM+10% FBS (control), hGF and in Hp-hAGF supernatants [19,28] for 24 and 96 hrs and then used for analysis.
Immunofluorescence
Image acquisition was performed with a Leica DMI6000B microscope equipped with the total internal reflection fluorescence (TIRF) and Nomarski interference contrast (DIC) modules. LAS-AF deconvolution software was used for image processing. Actin distribution was analyzed in formaldehyde (3.7%)-fixed, Triton X-100 (0.1%) permeabilized cells. Nuclear translocation of Twist was analyzed in aceton: metanol-fixed, Triton X-100 (0.1%) permeabilized cells. Specimens were labeled with the TRITC conjugated phalloidin (A2547, Sigma-Aldrich, Saint Louis, MO, USA) to stain F-actin and with anti-Twist primary antibody (ab50581, Abcam, Cambridge, UK). Then the secondary, mouse anti-rabbit IgG (F4890, Sigma-Aldrich, Saint Louis, MO, USA) antibody was used and counterstained with Hoechst 33258 (No.B2883, Sigma-Aldrich, Saint Louis, MO, USA).
Spheroid/aggregate production-gravity forced aggregation
Tripsinized cells were suspended in DMEM and in the case of AGS cells in F12/DMEM. Hanging drops (0.5×104 cells/aggregate in 40 µl of DMEM) were formed on the lid of a 60 mm tissue culture dish, inverted onto the PBS filled bottom chamber and incubated (5% CO2, 37°C) until cell aggregates formed (5 hrs).
Cell 3D expansion potential, Geltrex invasion chambers and analysis of MMP activity
The invasion of l.t.EMT+RGM1 and l.t.EMT-RGM1 cells towards Hp-AGF supernatant and DMEM+10% FBS was investigated using invasion chambers. The chambers consisted of cell culture inserts with 8 μm pore membrane (Corning, NY, USA), and thin layer of Geltrex BM (GeltrexTMLDEV-Free, Gibco, Dublin, Ireland). The cells (5.0×104 cells/ml) were re-suspended in a serum-free DMEM and placed on the layer of Geltrex in invasion chambers. DMEM+10% FBS and Hp-AGF supernatants were added to the outer chamber. The cells were incubated under humidified conditions (37°C, 5% CO2) for 48 hrs. The invasive cells found on the surface of the outer chamber were counted under the microscope (Nikon TMS) (Figure 11B). The invasion of HT29 and AGS cells towards culture media (DMEM+10% FBS and F12/DMEM+10% FBS respectively), hGF supernatant, and Hp-hAGF supernatant has been investigated for 24 hrs identically to the invasion of l.t.EMT+RGM1 and l.t.EMT-RGM1 cells. For AGS cells additionally the bottom side of the invasion chamber membrane was stained with bisbenzimide (Hoechst 33342, Sigma-Aldrich) to visualize transmigrated cells attached to the membrane. Three independent experiments were performed for each condition.
For Geltrex MMP activity assay, the l.t.EMT+RGM1, l.t.EMT-RGM1 cell aggregates and groups were suspended in 1:1 GeltrexTMLDEV-Free/DMEM (FBS free) and the drops of 40 µl were placed on 6-well plates, incubated under humidified conditions (37°C, 5% CO2) and carefully monitored to prevent Geltrex drying. Then the Geltrex/cells drops were flooded with Hp-AGF and GF supernatants (Figure 11C). For HT29 and AGS cells, the analogous technique has been applied. The Geltrex/cell drops were flooded with culture medium, hGF and Hp-hAGF supernatants. To visualize l.t.EMT+RGM1 and l.t.EMT-RGM1 cell expansion potential, the technique based on cell migration in 1:1 solidified Geltrex/DMEM (FBS free) cell sandwiches in 6 well plates was developed. The cell aggregates were placed on 1:1 solidified layer of GeltrexTMLDEV-Free/DMEM (FBS free) in 6 well plates. After the aggregates attached to the Geltrex surface, they were covered with the next layer of GeltrexTMLDEV-Free/DMEM (FBS free) (1:1). The solidification was monitored to prevent cell and Geltrex drying (Figure 11D).
Assessment of aggregate dynamics
To compare the dynamics of l.t.EMT+RGM1 and l.t.EMT-RGM1 cell groups in 3D ECM matrices, we have developed a new method, based on solution of reversed Mahalonobis problem [31] enabling direct computation of standard deviations (σ X,σ Y), centroid position (µ X, µ Y) and correlation coefficient r (according to original method designed by dr Janusz Opila).
Image acquisition and time-lapse video microscopy
Image acquisition and cell movement recording was performed with a Leica DMI6000B time-lapse system. Deconvolution software (Leica Microsystems) was used for image processing. The trajectories were constructed from a sequence of cell centroid positions recorded at 300 s time intervals using a dry ×20, NA-0.75 objective. Total length of cell trajectory (µm), velocity of cell movement (speed; total length of cell trajectory/time of recording; µm/min) and total length of cell displacement (i.e. the distance from the starting point directly to the cell’s final position; µm) were quantified with the Hiro program [19,32].
RT-PCR technique
Tripsinized cells were seeded at 8×106 cells/well and cultured for 96 hrs. The PCR was carried out, using 1 µg cDNA and Promega PCR reagent. Specific primers for rat and human Snail, Twist, Fap, MMP2, MMP3, MMP9, HGF, c-Met, EGFR, TNC, Oct4, Sox-2, c-Myc and 18S RNA (RNA integrity verification) were used (Sigma-Aldrich, Saint Louis, MO, USA). Primer sequences and annealing temperatures are listed in the Table 1. PCR products were separated by electrophoresis in 2% agarose gel containing 0.5 µg/mL ethidium bromide and then visualized under UV light. Location of predicted PCR product was confirmed by using O’GeneRuler 50 bp DNA ladder (Thermo Fisher Scientific, Waltham, MA, USA) and O’GeneRuler TM Low Range DNA Ladder (Thermo Fisher Scientific, Waltham, MA, USA) as standard marker.
Table 1.
Rat and human oligonucleotide primers for detection of mRNA by RT-PCR, annealing temperature and size of PCR products employed in experimental protocol
| Gene | Primer sequence | Size of PCR product (bp) | Annealing temp. (°C) |
|---|---|---|---|
| Rat 18S | Forward 5’-GTTGGTTTTGATCTGATAAATGC-3’ | 143 | 60 |
| Reverse 5’-CATTAAATCAGTTATGGTTCCTTTG-3’ | |||
| Rat Fap | Forward 5’-AGCCATATGGGGATGGTCCT-3’ | 154 | 60 |
| Reverse 5’-TGTTGGGAGGCCCATGAATC-3’ | |||
| Rat MMP9 | Forward 5-AGGTGCCTCGGATGGTTATCG-3’ | 156 | 63,3 |
| Reverse 5’-TGCTTGCCCAGGAAGACGAA-3’ | |||
| Rat MMP3 | Forward 5’-CTCTCCCAAGATGATGTAGATGGTATTC-3’ | 124 | 59 |
| Reverse 5’-AGCTACACATTGGTAAGGTCTCAG-3’ | |||
| Rat MMP2 | Forward 5’-CAGGGAATGAGTACTGGGTCTATT-3’ | 126 | 60 |
| Reverse 5’-ACTCCAGTTAAAGGCAGCATCTAC-3’ | |||
| Rat Twist | Forward 5’-GCCGGAGACCTAGATGTCATT-3’ | 185 | 60 |
| Reverse 5’-GGCCTG TCTCGCTTT CTCTT-3’ | |||
| Rat Snail | Forward 5’-CTGGGCGCT CTGAAGATGCA-3’ | 250 | 60 |
| Reverse 5’-GGAGCAGCCAGACTCTTGGTGT-3’ | |||
| Rat HGF | Forward 5’-TCTTGGTGTCAT TGTTCCTG-3’ | 157 | 60 |
| Reverse 5’-CCATGGATGCTTCAAATACA-3’ | |||
| Rat cMet | Forward 5’-TCCAGCTGTTGCAGGGAAG-3’ | 66 | 60 |
| Reverse 5’-GGCGTGCCAACATCGC-3’ | |||
| Rat EGFR | Forward 5’-GTAGCATTTATGGAGAGTG-3’ | 454 | 64 |
| Reverse 5’- GAGAGGAGAACTGCCAGAA-3’ | |||
| Rat TNC | Forward 5’-AGGCCACTGAGTACG AAATT-3’ | 360 | 55 |
| Reverse 5’-GACCATCGAGAGGCTGTGATT-3’ | |||
| Human Snail | Forward 5’-GGCAATTTAACAATGTCTGAAAAGG-3’ | 105 | 60 |
| Reverse 5’- GAATAGTTCTGGGAGACACATCG-3’ | |||
| Human Twist | Forward 5’-CTCAAGAGGTCGTGCCAATC-3’ | 119 | 60 |
| Reverse 5’-CCCAGTATTTTTATTTCTAAAGGTGTT-3’ | |||
| Human FAP | Forward 5’-AGCCATATGGGGATGGTCCT-3’ | 154 | 60 |
| Reverse 5’-TGTTGGGAGGCCCATGAATC-3’ | |||
| Human MMP2 | Forward 5’-AGATCTTCTTCTTCAAGGACCGGTT-3’ | 225 | 67,2 |
| Reverse 5’-GGCTGGTCAGTGGCTTGGGGTA-3’ | |||
| Human MMP3 | Forward 5’-AGGCTGTATGAAGGAGAGGCTGAT-3’ | 246 | 56,6 |
| Reverse 5’-AGTGTTGGCTGAGTGAAAGAGACC-3’ | |||
| Human MMP9 | Forward 5’-GCCACTACTGTGCCTTTGAGTC-3’ | 125 | 60 |
| Reverse 5’-CCCTCAGAGAATCGCCAGTACT-3’ | |||
| Human cMet | Forward 5’-TGCACAGTTGGTCCTGCCATGA-3’ | 142 | 60 |
| Reverse 5’-CAGCCATAGGACCGTATTTCGG-3’ | |||
| Human EGFr | Forward 5’-AACACCCTGGTCTGGAAGTACG-3’ | 106 | 60 |
| Reverse 5’-TCGTTGGACAGCCTTCAAGACC-3’ | |||
| Human Oct4 | Forward 5’-AACCTGGAGTTTGTGCCAGGGTTT-3’ | 123 | 60 |
| Reverse 5’-TGAACTTCACCTTCCCTCCAACCA-3’ | |||
| Human Sox2 | Forward 5’-AGAACCCCAAGATGCACAAC-3’ | 204 | 48 |
| Reverse 5’-CTCCGGGAAGCGTGTACTTA-3’ | |||
| Human c-Myc | Forward 5’-CTGCTTGAATGGACAGGATGTA-3’ | 320 | 60 |
| Reverse 5’-CTCCACTCACCAGCACAACTAC-3’ | |||
| Human 18S | Forward 5’-TAGTAGCGACGGGCGGTGTG-3’ | 230 | 60 |
| Reverse 5’-CAGCCACCCCAGATTGAGCA-3’ | |||
| Human αSMA | Forward 5’-ATCACCAACTGGGACGACAT-3’ | 175 | 60 |
| Reverse 5’-CATACATGGCTGGGACATTG-3’ | |||
| Human E-Cadherin | Forward 5’-AACGAGGGCATTCTGAAAACA-3’ | 75 | 60 |
| Reverse 5’-CACTGTCACGTGCAGAATGTACT-3’ | |||
| Human N-Cadherin | Forward 5’-ATGAAGAAGGTGGAGGAGA-3’ | 152 | 60 |
| Reverse 5’-AGATCGGACCGGATACT-3’ | |||
| Human Integrin-β1 | Forward 5’-GGATTCTCCAGAAGGTGGTTTCG-3’ | 143 | 60 |
| Reverse 5’-TGCCACCAAGTTTCCCATCTCC-3’ | |||
| Human TGFβ | Forward 5’-CTCGCCAGAGTGGTTATCTT-3’ | 123 | 60 |
| Reverse 5’-AGTGTGTTATCCCTGCTGTCA-3’ | |||
| Human EGF | Forward 5’-TGCCAGCTGCACAAATACAGAGGG-3’ | 167 | 58 |
| Reverse 5’-CATCGTGGGACAGGGGACATTCA-3’ |
Western blot
The cells were harvested with 0.25% trypsin and the proteins were then extracted with the Subcellular Protein Fractionation Kit (Thermo Fisher Scientific, Waltham, MA, USA). Total protein concentration was determined by nanodrop measurement. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (NuPAGE, Invitrogen, Carlsbad, CA, USA) and transferred to nitrocellulose membranes (iBlot System Invitrogen, Carlsbad, CA, USA). Membranes were washed with Tris-buffered saline containing 0.05% Tween-20 (TBST) and blocked with 3% BSA. Next, the membranes were incubated overnight with primary antibodies at 4°C and washed with TBST. Then, the membranes were incubated in the presence of HRP-conjugated secondary antibodies for 1 hr at RT.
The primary antibodies used: anti-GAPDH (D16H11 Cell Signaling), anti-Snail (ab180714, Abcam, Cambridge, UK), anti-Twist (ab50887 Abcam, Cambridge, UK), anti-Cytokeratine19 (GTX112666 Genetex, Irvine, CA, USA), anti-TNC (GTX17605 Genetex, Irvine, CA, USA), anti-β1-integrin (GTX128839 Genetex, Irvine, CA, USA), anti-c-Met (25869-1-AP ProteinTech, Manchester, UK) and anti-p-EGFR (2231S, Cell Signaling, Danvers, MA, USA) and anti-EGFR (ab52894, Abcam, Cambridge, UK). The HRP-conjugated secondary antibodies used: goat antirabbit IgG (ab97051 Abcam, Cambridge, UK) and goat anti-mouse IgG (ab205719, Abcam, Cambridge, UK). Immunoreactive bands were visualized and quantified by chemiluminescence imaging densitometry Image Studio Lite software (Li-Cor).
Influence of EGFR kinase activity receptor inhibition on AGS cells
AGS cells were cultured in Hp-hAGF supernatant as described in the section Experimental design with the addition of inhibitor of EGF receptor kinase activity A46 (0.1 mM/l) [33,34].
Determination of TGFβ1 release
Cells were placed on the 6 well plates in DMEM+10% FBS (0.3×106 cells per well) and allowed to release HGF for 48 hrs. Then, the supernatants were collected and the concentration of HGF was measured by ELISA (Biorbyt, Cambridge, UK, orb180665) according to manufacturer protocol.
Statistical analyses
Statistical analysis of the data was performed with the use of Excel Software. Each variable was expressed as the mean (± S.E.M.). Statistical significance of difference was determined using analysis of variance (one-way ANOVA) test (Statistica Software). Further statistical analysis for post hoc comparisons was carried out with Newman-Keuls test. Differences were considered statistically at P<0.05.
Results
Migrative properties of long-term RGM1 cells
The motility of l.t.EMT+RGM1, l.t.EMT-RGM1 and RGM1 in DMEM+10% FBS, Hp-AGF and GF supernatants have been checked to identify migrative properties of each cell type as well as to examine the influence of different environmental niches on cell motility parameters. Hp-AGF supernatant significantly enhanced migration of l.t.EMT+RGM1 and l.t.EMT-RGM1 cells (Figure 1A-C). Even though higher directionality of l.t.EMT-RGM1 cells in Hp-AGF, movement was observed (measured as the ratio of cell displacement over distance; 0.64 compared to 0.44 for l.t.EMT+RGM1; Figure 1F), their distance/speed of movement was comparable and significantly higher than of control RGM1 cells (Figure 1A). Similarly, in GF supernatant the values for distance/speed and directionality of cell movement of l.t.EMT+RGM1 and l.t.EMT-RGM1 cells were significantly higher than these of RGM1 cells (Figure 1A, 1B, 1D).Nevertheless these values were much lower than in Hp-AGF secretome (Figure 1A, 1B, 1D). DMEM+10% FBS again enhanced both, the distance/speed and displacement of l.t.EMT+RGM1 and l.t.EMT-RGM1 cells (Figure 1A, 1B, 1E). L.t.EMT+RGM1 and l.t.EMT-RGM1 cells were motile in all media which indicates general reprogramming of l.t.EMT+RGM1 and l.t.EMT-RGM1 cells towards motile phenotype regardless of the medium (Figure 1A-E). However l.t.EMT-RGM1 cells were characterized with higher persistence of movement (Figure 1F) accompanied with fibroblastoid morphology (Figure 1G), while l.t.EMT+RGM1 cells presented medium-dependent heterogeneity of movement starting from more settle epithelioid-like cells (Figure 1H) to polarized more motile cells showing intermediate morphology/movement (both mesenchymal and epithelial) (Figure 1I). This heterogeneity was particularly prominent in DMEM+10% FBS medium, which confirms our previous observations [28] on phenotypical, niche-dependent plasticity of l.t.EMT+RGM1 cells. Our data also underlie pro-metastatic, migration-promoting properties of Hp-AGF supernatant favoring excessive migration of normal and pro-cancerogenic cells and anti-metastatic migration-limiting properties of GF supernatant (Figure 1A, 1B, 1D).
Figure 1.

Motility of RGM1, l.t.EMT-RGM1 and l.t.EMT+RGM1 cells in Hp-AGF supernatant, GF supernatant and DMEM+10% FBS. Total length of cell trajectory (µm), velocity of cell movement (speed; total length of cell trajectory/time of recording; µm/min) and total length of cell displacement (i.e. the distance from the starting point directly to the cell’s final position; µm) were quantified with the Hiro program. Column charts show migration parameters at the population level (registered for 8 h; N=50): distance (A) and displacement (B). Cell trajectories are presented as circular diagrams (axis scale in µm) drawn with the initial point of each trajectory placed at the origin of the plot. Circular diagrams represent migration parameters at the single-cell level of all three cell types in: Hp-AGF supernatant (C), in GF supernatant (D) and in DMEM+10% FBS (E). Distance over displacement ratio for each cell type in respective media (F). For constant directionality the parameter equals 1, for random movement the parameter tends to zero. The movement strategies of l.t.EMT-RGM1 cells (G) and l.t.EMT+RGM1 cells (H, I).
Hp-AGF induces phenotypical heterogeneity and plasticity of actin cytoskeleton organization in l.t.EMT+RGM1 cells
Since cytoskeleton is regarded as the primary force-generating machinery in the cell, being particularly important for cell migration [35-37], we have focused on the long-term effect of Hp-AGF and GF secretomes on actin organization in epithelial cells. Control RGM1 cells established colonies with typical actin organization [19] characteristic for non-motile cells (Figure 2A). L.t.EMT-RGM1 cells (Figure 2B) were characterized by flat lamellipodia-broad zones of branched F-actin at their leading fronts (Figure 2C, white arrow), which has been known to provide pushing forces for cell migration and its persistence [35,38]. L.t.EMT-RGM1 cells were also characterized with the presence of abundant stress fibers characteristic for fibroblasts (Figure 2B, 2C, blue arrows). In migrating cells, stress fibers generate contractile forces applied to focal adhesions [35,39] which serve for cell motility and ECM remodeling. These results indicate that prolonged GF secretome exposure induces epithelial reprogramming prompting their epithelial-fibroblast transition (EFT). In turn, l.t.EMT+RGM1 cells were characterized by enhanced formation of filopodia (Figure 2D, 2E white arrows), believed to guide cell locomotion during normal tissue morphogenesis or cancer metastasis [35,40], which was accompanied by broad diversity of actin cytoskeleton organization. It ranged from the sparse cells possessing vestigial stress fibers, through the cells with an elongated morphology and diffusible actin distribution devoid of stress fibers, to cells with epithelial-like morphology and typical actin cytoskeleton (Figure 2D). The limited presence of lamellipodia and stress fibers reflects lower directionality of l.t.EMT+RGM1 cell movement (Figures 1F and 2E), while diversity of actin organization indicates high degree of phenotypic plasticity (Figures 1H, 1I, 2D, 2E) correlating with overall increase of l.t.EMT+RGM1 cell motility. Additionally, we have observed tumor-like nuclear localization of actin [41,42] (Figure 2E, yellow arrows) exclusively in l.t.EMT+RGM1 cells (Figure 2A-E). These observations confirm different reprogramming scenarios induced by long-term influence of GF and Hp-AGF secretomes.
Figure 2.

Hp-AGF induces phenotypical diversity and plasticity of actin cytoskeleton organization in l.t.EMT+RGM1 cells associated with nuclear actin localization. The composition of Nomarski contrast and immunofluorescence micrographs of actin cytoskeleton in RGM1 cells characterized by the thin bundles of actin filaments in the perinuclear zone and thick cortical bundles of actin filaments at cellular peripheries, which formed a tangential system at the periphery of epithelial islets (A); in l.t.EMT-RGM1 cells showing their reprogramming towards fibroblast-like phenotype: blue arrows point stress fibers (B), white arrow points lamellipodium (C) and in l.t.EMT+RGM1 cells characterized by enhanced formation of filopodia and broad diversity of actin cytoskeleton organization (D). Nuclear localization of actin in l.t.EMT+RGM1 cells (respectively: actin, chromatin and composite); white arrows indicate filopodia, yellow arrows indicate nuclear actin (E). Red: F-actin and blue: Hoechst.
Long-term exposition of l.t.EMT+RGM1 cells to Hp-AGF secretome induces their shift towards invasive potential
The pro-migratory phenotype and cytoskeleton plasticity pushed us to check l.t.EMT+RGM1 cell behavior in 3D BM. We have focused on the responsiveness of l.t.EMT+RGM1 and l.t.EMT-RGM1 to the strongest motility inducers: Hp-AGF and DMEM+10% FBS gradients. Geltrex invasion assay showed highly accelerated movement of l.t.EMT+RGM1 towards DMEM+10% FBS, additionally intensified towards Hp-AGF supernatant (Figure 3A). Only a few l.t.EMT-RGM1 cells managed to pass through the Geltrex layer exclusively towards Hp-AGF supernatant (Figure 3A). RGM1 cells failed to pass the BM layer (Figure 3A). These results confirm differential abilities of l.t.EMT+RGM1 and l.t.EMT-RGM1 cells to move across BM.
Figure 3.

Hp-AGF secretome prompts invasive properties of long-term RGM1 cells. Geltrex invasion assay of RGM1, l.t.EMT-RGM1 and l.t.EMT+RGM1 cells towards Hp-AGF supernatant and towards DMEM+10% FBS showing enhanced invasiveness of l.t.EMT+RGM1 cells (A). RT-PCR analysis of the expression of 18S mRNA and of FAP and MMP 2, 3 and 9 mRNA expression in l.t.EMT-RGM1, l.t.EMT+RGM and original RGM1 cells and the ratio of selected genes over 18S mRNA showing their strong upregulation in l.t.EMT+RGM1 cells (B). Phase contrast microscopy of l.t.EMT-RGM1 and l.t.EMT+RGM1 Geltrex metalloproteinase activity assay (1:1) showing the ability of l.t.EMT+RGM1 to degrade basement membrane components (C). Results are mean ± SEM of four to six independent experimental repeats. Asterisk (*) indicates a significant change (P<0.05) as compared to the control RGM1 value. Hash (#) indicates a significant change (P<0.05) as compared to l.t.EMT-RGM1 value.
To substantiate this notion we have checked the expression of MMPs, responsible for ECM degradation [43-46]. L.t.EMT+RGM1 cells showed strong up-regulation of mRNAs encoding MMP2, MMP3 and MMP9 accompanied by the appearance of FAP mRNA (Figure 3B) characteristic for CAFs and tumor cells [47,48]. L.t.EMT-RGM1 cells were characterized with up-regulation of MMP9 mRNA expression and vestigial MMP3 and MMP2 mRNA expressions (Figure 3B). These results led us to visualize the ability of l.t.EMT+RGM1 and l.t.EMT-RGM1 cells to degrade BM. Geltrex MMP activity assay showed dense central concentration of l.t.EMT-RGM1 cells, whereas the profound dispersion of l.t.EMT+RGM1 cells combined with complete destruction of artificial BM matrix within 48 hrs (Figure 3C). These results underline pro-invasive long-term influence of Hp-AGF secretome as compared to GF secretome.
Hp-AGF secretome induces increased dynamics of long-term RGM1 cell groups and aggregates in 3D BM
Next, we have checked the behavior of l.t.EMT+RGM1 cells in 3D BM. 8 hrs after seeding, RGM1 cells were organized in aggregates, while l.t.EMT-RGM1 and particularly, l.t.EMT+RGM1 cells stayed largely scattered due to E-cadherin down-regulation [25]. The already existing RGM1 cell aggregates significantly reduced their area within next 8 hrs (Figure 4A, 4D). At the same time l.t.EMT-RGM1 (Figure 4B, 4E) and l.t.EMT+RGM1 (Figure 4C, 4F) cell groups started to cluster in the aggregates. The cell group area reduction and the rate of group area relative change were significantly higher for l.t.EMT+RGM1 cells (Figure 4F) and the lowest for RGM1 cells (Figure 4D). The overall migration speed of l.t.EMT-RGM1 and l.t.EMT+RGM1 cells were comparable (0.12 µm/s vs. 0.13 µm/s) (Figure 4E, 4F) while overall migration speed of RGM1 cells was more than three times lower (0.04 µm/s) (Figure 4D). The translation spotted was approximately linear in all cases, i.e. no random movement was observed. No change of sign was spotted, i.e. general orientation of the aggregate movement was not changing (Figure 4D-F). Given dynamics of cell behavior during first 8 hrs of observation, we have then checked the organization of l.t.EMT+RGM1 (Figure 4G) and l.t.EMT-RGM1 cells (Figure 4H) after 24 and 48 hrs. After 24 hrs both cell types were arranged in dense, round aggregates, however while l.t.EMT-RGM1 cells kept compacting cell aggregate areas up to 48 hrs (Figure 4H), l.t.EMT+RGM1 cells started to actively migrate from the aggregates (Figure 4G). Although the total area of l.t.EMT+RGM1 cell groups increased, the area of densely aggregated cells decreased due to cell migration (Figure 4H). Apparently l.t.EMT+RGM1 cells exhibited prominent two stage behavior first compacting into densely packed aggregates minimizing their area (Figure 4C, 4F), then invasively migrating from the aggregates towards Hp-AGF secretome (Figure 4D). The underlying mechanism of such two-step behavior requires following in-depth study. The obtained results confirm that GF ensures invasive restrictions.
Figure 4.

Dynamics of cell groups/aggregates in 3D Geltrex BM. Nomarski contrast of initial and final distribution of cell groups in 3D Geltrex BM flooded with appropriate culture medium: RGM1 cells in DMEM+10% FBS (A), l.t.EMT-RGM1 cells in GF supernatant (B) and l.t.EMT+RGM1 in Hp-AGF supernatant (C). Upper and lower pictures show the initial and final distribution of cell groups (8 hrs after seeding and 16 hrs after seeding) with visible computed ellipses (magenta) coinciding with error ellipse (white). Cell group dynamics was registered with Leica DMI6000B time-lapse system for 8 hrs. The computed parameters of relevant aggregates e.g. standard deviations (σX, σY), centroid positions (µX, µY) and correlation coefficient r for given cell type are shown in the tables: RGM1 cells in DMEM+10% FBS (D), l.t.EMT-RGM1 in GF supernatant (E) and l.t.EMT+RGM1 cells in Hp-AGF secretome (F). The changing values of correlation coefficient reflect the aggregate shape changes and standard deviations changes reflect the area changes. Decreasing value of correlation coefficient points to striving round shape adaptation (evolution of linearly elongated aggregates into more circularly shaped ones) and decreasing values of standard deviations point to the overall area decrease. Nomarski contrast of l.t.EMT+RGM1 (G) and l.t.EMT-RGM1 (H) depicting aggregated cells in 3D Geltrex basement membrane 24 hrs after seeding and 48 hrs after seeding and showing invasive properties of l.t.EMT+RGM1 cells. Computed ellipses (magenta) and computed parameters of relevant aggregates: standard deviations (σX, σY), centroid positions (µX, µY) and correlation coefficient r. The aggregate area was calculated according to the formula: S=9πσXσY. Cell group dynamics was registered with Leica DMI6000B.
Invasive potential of l.t.EMT+RGM1 cells is related to the function of Twist
Obtained results prompted us to analyze Snail and Twist expression in l.t.EMT+RGM1 and l.t.EMT-RGM1 cells. Considering the invasive potential of l.t.EMT+RGM1 cells, we’ve analyzed Snail and Twist expression in their population. Both l.t.EMT+RGM1 and l.t.EMT-RGM1 cells displayed significant Snail and Twist transcriptional up-regulation (Figure 5A). The increase of Snail mRNA expression led to the induction of Snail protein expression in both l.t.EMT-RGM1 and l.t.EMT+RGM1 cells, however, in l.t.EMT+RGM1 cells it was significantly higher (Figure 5B). At the same time increased expression of Twist mRNA resulted in Twist protein expression only in l.t.EMT+RGM1 cells (Figure 5B). To further confirm Twist expression in l.t.EMT+RGM1, we have applied immunofluorescence which is more sensitive than Western Blot technique. We have not only confirmed the exclusive Twist expression in l.t.EMT+RGM1 cells but also its translocation into the nucleus (Figure 5C). Thus, we conclude that long-term administration of Hp-AGF secretome induces both, Snail and functional Twist protein expression in normal gastric epithelial cells, while GF secretome induces only Snail protein expression, implicating Twist in pro-invasive behavior of l.t.EMT+RGM1 cells. Additionally the pro-cancerogenic, elastic phenotype of l.t.EMT+RGM1 cells was confirmed by Cytokeratin19, which increasing expression is characteristic for reprogramming of normal tissues to premalignant lesions and finally to adenocarcinoma in situ [48]. On the contrary, GF secretome downregulated Cytokeratin19 which together with Snail expression in l.t.EMT-RGM1 cells clarifies their reprogramming and differentiation towards myofibroblasts (EmyoT) which strengthen our previous results [28].
Figure 5.

L.t.EMT+RGM1 cell pro-invasive and pro-metastatic abilities are related to Twist protein expression. RT-PCR analysis of the expression of 18S mRNA and of Snail and Twist mRNAs expression in l.t.EMT-RGM1 cells, l.t.EMT+RGM1 cells and the original RGM1 cells and the ratio of selected genes over 18S mRNA (A). Western Blot analysis of Snail Twist and Cytokeratin19 expression in total cellular proteins isolated from l.t.EMT-RGM1 cells, l.t.EMT+RGM1 cells and original RGM1 cells and the semi-quantitative densitometry analysis of the ratio of selected proteins over GAPDH showing the upregulation of Snail, Twist and Cytokeratin19 in l.t.EMT+RGM1 cells. 10 µg of total cellular proteins were loaded per each lane for Snail and Cytokeratin 19 and 30 µg of Twist (B). The composite of Nomarski contrast and immunofluorescence of chromatin and Twist protein in RGM1 cells, l.t.EMT-RGM1 and l.t.EMT-RGM1 cells showing the appearance of Twist and additionally its translocation into the nucleus (sequentially: composite and Twist). Green: Twist and blue: chromatine (Hoechst 33258) (C). Results are mean ± SEM of four to six independent experimental repeats. Asterisk (*) indicates a significant change (P<0.05) as compared to the control RGM1 value. Hash (#) indicates a significant change (P<0.05) as compared to l.t.EMT-RGM1 value.
Invasive potential of l.t.EMT+RGM1 cells is associated with HGF/TenascinC/β1-integrin pathway
To look into the mechanistic insight of the pathways governing motility and invasion of l.t.EMT+RGM1 cells, we have checked the level of c-Met, the activator of signaling pathways responsible for motility and cell scattering. Both l.t.EMT+RGM1 and l.t.EMT-RGM1 cells were characterized by elevated level of c-Met mRNA as well as c-Met protein expression (Figure 6A, 6C). Additionally l.t.EMT+RGM1 cells gained the ability to produce and release HGF (Figure 6A, 6B). These observations together with highly increased HGF concentration in Hp-AGF secretome [27] implicates, that despite of c-Met up-regulation in l.t.EMT+RGM1 cells, the increased amount of HGF ligands in Hp-AGF and l.t.EMT+RGM1 cell secretome provides l.t.EMT+RGM1 cells with higher para- and autocrine stimulation. Additionally, l.t.EMT+RGM1 cells increased expression of functional c-Met partner, Integrin-β1 [49] which was only slightly up-regulated in l.t.EMT-RGM1 (Figure 6C). Importantly, Hp-AGFs were characterized with upregulation of TenascinC (TNC) [29], the HGF responsive factor involved in cancer cell scattering [50-52], β1-integrin clustering and activation [53,54]. We have found that also l.t.EMT+RGM1 cells have started to express small, hardly detectable amounts of TNC (Figure 6A, 6C). L.t.EMT+RGM1 cells showed also increased expression of EGFR and its activation (Figure 6A, 6C), which is frequently over expressed in many types of cancers including GC. These findings are consistent with the findings that among other signaling molecules, also EGFR activation leads to E-cadherin repression through STAT3 induced Twist activation and thereby, promote EMT [55].
Figure 6.

Pro-migratory and pro-metastatic potential of l.t.EMT+RGM1 cells is associated with the activation of HGF/TNC/Integrin-β1 pathway. RT-PCR analysis of the adhesion and motility related mRNA expression (18S, cMet, HGF, EGFR and TNC) in l.t.EMT-RGM1, l.t.EMT+RGM1 and original RGM1 cells and the ratio of selected genes over 18S mRNA (A). Elisa analysis of HGF content in the secretome. The cells were transferred to DMEM+10% FBS and allowed for 48 hrs secretion. The analysis showed that only l.t.EMT+RGM1 cells were able to secrete HGF (B). Western Blot analysis of c-Met, p-EGFR, integrin-β1 and TNC protein expression in total cellular proteins isolated from l.t.EMT-RGM1, l.t.EMT+RGM1 and the original RGM1 cells and the semi-quantitative densitometry analysis of the ratio of selected proteins over GAPDH showing the upregulation of c-Met, p-EGFR, Integrin-β1 and TNC proteins in l.t.EMT+RGM1 cells. 10 µg and in the case of EGFR and TNC 25 µg of total cellular proteins were loaded per each lane (C). Results are mean ± SEM of four to six independent experimental repeats. Asterisk (*) indicates a significant change (P<0.05) as compared to the control RGM1 value. Hash (#) indicates a significant change (P<0.05) as compared to l.t.EMT-RGM1 value.
The short-term influence of Hp-hAGF secretome on EMT induction in human skin keratinocytes
To verify if Hp-activated fibroblast secretome-induced reprogramming is not one cell-line specific and to what extend Hp-AGF short-term evoked reprogramming can be universal mechanism eliciting EMT in other cell types of epithelial origin, we have checked the morphology and motility of normal skin keratinocytes HaCaT after 96 hrs of culture in Hp-activated human gastric fibroblast (Hp-hAGF) supernatant. In control conditions (DMEM+10% FBS), HaCaT cells adopted predominantly polygonal, epithelioid morphology, which was characterized by the lack of polarization. The cells showed the tendency to form islets in the subconfluent culture (Figure 7A) and were characterised with very slow, collective cell movement (Figure 7C). The morphology of HaCaT cells cultured in the supernatant from hGFs was similar to this, estimated for the cells cultured in control medium (Figure 7A). The cells in Hp-hAGF supernatant were also characterized with collective cell movement, accompanied by the formation of multicellular leading fronts (Figure 7A-C). The front cells moved ahead of follower cells, providing the guidance for the migrating group, which is characteristic for wound healing and morphogenesis [58]. Only a few cells were characterized by single, but typical epithelial cell movement (Figure 7A).
Figure 7.

Hp-hAGFs elicit EMT in short-term cultures of HaCaT cells. Nomarski contrast of the phenotype of HaCaT cells placed in DMEM+10% FBS (control), hGF supernatant and Hp-hAGF supernatant (A). The trajectories of cell movement in respective supernatants and control medium presented as circular diagrams (axis scale in µm) drawn with the initial point of each trajectory placed at the origin of the plot (B). Corresponding motility parameters: total length of cell trajectory (µm), velocity of cell movement (speed; total length of cell trajectory/time of recording; µm/min) and total length of cell displacement (i.e. the distance from the starting point directly to the cell’s final position; µm) were quantified with the Hiro program. Column charts show migration parameters at the population level (registered for 8 h; N=50): distance and displacement (C). RT-PCR analysis of the expression of mRNA for 18S, Snail, Twist and EMT markers in HaCaT cells cultured in DMEM+10% FBS (control), hGF supernatant and Hp-hAGF supernatant and the ratio of selected genes over 18S mRNA. Results are mean ± SEM of four independent experimental repeats. Asterisk (*) indicates a significant change (P<0.05) as compared to the control value. Hash (#) indicates a significant change (P<0.05) as compared to the value for cells cultured in hGF secretome (D).
In turn, Hp-hAGF secretome induced scattering of HaCaT cells, with the appearance of few bigger and more elongated cells with the tendency to pro-fibroblastic movement. The cells failed to form islets and the collective migration was replaced by single cell movement (Figure 7A). Both, hGF and Hp-hAGF secretomes enhanced motility of HaCaT cells, which were almost non-motile in culture medium (Figure 7B, 7C). All motility parameters were the highest in hGF secretome, particularly the displacement (Figure 7C) which pointed to ordering exerted by normal fibroblasts during physiological wound healing. Nevertheless, only Hp-hAGF secretome induced pro-EMT phenotypical shift and cell scattering of HaCaT cells (Figure 7A, 7B).
To further confirm the influence of Hp-hAGF secretome on HaCaT cells, we have checked the expression of EMT-TFs and EMT markers. Both, hGF and Hp-hAGF secretome induced Snail mRNA upregulation, however only Hp-hAGF secretome elicited Twist mRNA expression (Figure 7D). Twist appearance was accompanied by increased expression of mRNA for EMT markers: α-SMA and N-cadherin and characteristic decrease of E-cadherin mRNA expression (Figure 7D) confirming EMT commitment evoked by Hp-hAGF. The secretome from hGF slightly decreased E-cadherin mRNA expression, nevertheless failed to significantly change N-cadherin and α-SMA mRNA expression (Figure 7D). The obtained results were uniform with the results obtained by our team for short-term EMT induction in RGM1 cells, which strongly points to the versatility of EMT-inducing capacity of Hp-activated fibroblasts.
The influence of Hp-hAGF secretome induces EMT related changes of motility mode in cancer cells
To assess how Hp-hAGF will affect already developed gastric/colon cancer cells we have checked the morphology and motility of human colon adenocarcinoma HT29 and human gastric adenocarcinoma AGS after 24 hrs of culture in Hp-hAGF supernatant.
Neither hGF, nor Hp-hAGF secretome induced the change of HT-29 cell morphology and motility in applied 2D culture conditions (culture dish surface) and the cells stayed non-motile (Figure 8A). Then, we investigated morphology and motility parameters of AGS cells. The hGF secretome didn’t induce any changes in cell morphology as compared to control conditions (Figure 8A). On the contrary, in Hp-hAGF secretome, some AGS cells changed their morphology from epithelioid towards strongly elongated, fibroblastoid shape (Figure 8B) with the appearance of fibroblastoid motility mode (Figure 8E). Additionally, the distance rate and the speed of displacement of epithelioid type of movement were only slightly lower in Hp-hAGF secretome comparing to control and hGF secretome (Figure 8D). In non-gradient conditions, the AGS cells in Hp-hAGF secretome were also characterized with more random movement (Figure 8C, 8D). Previously, we have shown EMT type 3 commitment of RGM1 cells to be dependent on TGFβ signaling [19,28]. It’s now well recognized that EMT engages discrete interplay between different growth factor and cytokine signaling pathways. Thus, focusing on GC, to specify the influence evoked by Hp-hAGF on progression of this type of cancer, we have attempted to determine if EGFR signaling can participate in this mechanism. Though, the addition of A46 (inhibitor of EGF receptor kinase activity) to AGS cells cultured in Hp-hAGF secretome, didn’t significantly influence motility parameters of epithelioid cells (Figure 8C, 8D), it resulted in disappearance of AGS fibroblastoid motility mode (Figure 8E). Collectively, the above results confirm EMT-inducing and migration promoting activity of Hp-hAGF secretome in normal and cancer motile cells in non-gradient 2D conditions. The EMT-inducing, but not migration-promoting influence of Hp-hAGF on motile human gastric adenocarcinoma AGS was at least partially dependent on EGFR signaling in short-term conditions. These findings are consistent with the findings that among other signaling molecules, also EGFR activation leads to E-cadherin repression through STAT3 induced Twist activation and thereby, promote EMT [55]. Nevertheless, one should still remember, that EGFR downstream signaling cascades can shift to be activated via EGFR-independent mechanisms [56-59].
Figure 8.

Hp-hAGF secretome induces EMT related changes of motility mode in cancer cells. Nomarski contrast of the phenotype of HT29 cells placed in DMEM+10% FBS (control), hGF supernatant and Hp-hAGF supernatant (A) and of AGS cells placed in DMEM/F12HAM+10% FBS, hGF supernatant, Hp-hAGF supernatant and Hp-hAGF supernatant with the addition of EGFR inhibitor tyrphostin A46 (0.1 mM) (B). Cell trajectories of epithelioid AGS cells in DMEM/F12HAM+10% FBS, hGF supernatant, Hp-hAGF supernatant and Hp-hAGF supernatant with the addition of A46 (0.1 mM) (C) and corresponding motility parameters (D). Cell trajectories of fibroblastoid AGS cells in DMEM/F12HAM+10% FBS, hGF supernatant, Hp-hAGF supernatant and Hp-hAGF supernatant with the addition of A46 (0.1 mM) and corresponding motility parameters (E). Cell trajectories are presented as circular diagrams (axis scale in µm) drawn with the initial point of each trajectory placed at the origin of the plot. Total length of cell trajectory (µm), velocity of cell movement (speed; total length of cell trajectory/time of recording; µm/min) and total length of cell displacement (i.e. the distance from the starting point directly to the cell final position; µm) were quantified with the Hiro program. Column charts show migration parameters at the population level (registered for 8 h; N=50): distance and displacement.
The influence of Hp-hAGF secretome induces Twist upregulation accompanied with EMT type 3-related pro-pluripotent changes in cancer cells
Next, we have focused on the correlation between EMT-TF expression and pro-pluripotent plasticity of cancer cells upon the influence of Hp-hAGF secretome. We have checked Snail and Twist gene upregulation in cancer HT29 and AGS cell lines. Consistently, Hp-hAGF secretome induced increased expression of Snail mRNA in both cell lines as well as Twist gene upregulation in HT29 and Twist mRNA expression appearance in AGS cells (Figure 9A). Accordingly to its EMT-inducing capacity, Hp-hAGF secretome elicited increased expression of mRNA for pluripotency markers [28]: c-Myc, Oct4 and Sox-2 confirming pro-pluripotent activity of Hp-activated fibroblast secretome. At the same time hGF secretome only slightly increased mRNA expression for Twist, c-Myc and Oct4 mRNA in HT29 cells. In AGS cells hGF secretome didn’t elicit neither Snail transcriptional upregulation nor Twist appearance, additionally decreasing the expression of c-Myc and Sox-2 mRNA (Figure 9A, 9B). The appearance of Twist expression in AGS cells was dependent on EGFR signaling at least within 96 hrs of experimental procedure and disappeared after the addition of A46, which also diminished Snail, c-Myc, Oct4 and Sox-2 gene upregulation (Figure 9A, 9B), which is consistent with the supportive role of EGFR in EMT and stemness [60,61]. Above results confirm that Hp-AGF secretome induces both, Snail and Twist gene upregulation in normal cells and predominantly Twist upregulation in cancer cells. At the same time, GF secretome induces Snail gene upregulation and doesn’t influence Twist. In cancer cells, Twist mRNA upregulation was combined with EMT with EMT type 3-connected propluripotent markers upregulation. The appearance of Twist gene activity and associated pluripotency markers gene upregulation in AGS cells, depended on EGFR signaling in our experimental conditions. These results confirm our previous observations on plasticity inducing properties of Hp-activated-fibroblast secretome and the opposite role of GF secretome [28].
Figure 9.
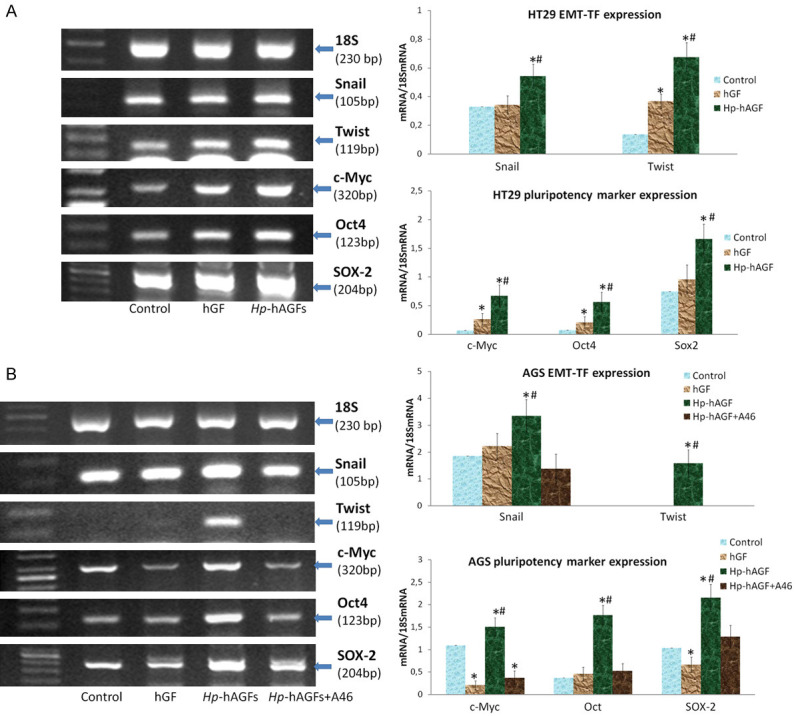
Hp-hAGF secretome prompts Twist related EMT type 3 pro-pluripotent changes in cancer cells. RT-PCR analysis of the expression of mRNA for 18S, Snail, Twist and pluripotency-related Yamanaka factors: c-Myc, Oct4 and Sox-2 in HT29 (A) and AGS (B) cells cultured for 96 hrs in control medium, hGF and Hp-hAGF supernatants and the ratio of selected genes over 18S mRNA. Additionally AGS cells were cultured in Hp-hAGF secretome with the addition of A46 (0.1 mM). The analysis shows Snail mRNA expression increase and strong Twist transcriptional upregulation. Results are mean ± SEM of four independent experimental repeats. Asterisk (*) indicates a significant change (P<0.05) as compared to the control value. Hash (#) indicates a significant change (P<0.05) as compared to the value for cells cultured in hGF secretome.
Increased invasiveness in adenocarcinoma cells engages the interplay of cMet/HGF/β1-integrin/TNC and EGFR signaling
To verify further influence of Hp-hAGF on cancer cells, we have focused on their invasiveness. We have also checked EGFR dependence of the mechanism leading to changes in invasiveness of AGS cells. We have first checked the impact of Hp-hAGF secretome gradient on HT29 and AGS cells. Surprisingly, 3D gradient conditions evoked chemotactic invasive response of 2D non-motile HT29 cells, accompanied with their morphology change. Both HT29 and AGS cells showed highly accelerated movement towards Hp-hAGF secretome already after 24 hrs, as compared to control medium and hGF supernatant (Figure 10A, 10B). We have also observed decrease of invasive properties of AGS cells towards Hp-hAGF secretome after EGFR signaling inhibition (Figure 10A, 10B). Additionally, we have observed that Hp-hAGF secretome induced motile, elongated phenotype in 3D gradient conditions also in 2D non-motile HT29 cells (Figure 10C), which underlie not only promotion of BM degradation ability induced by Hp-hAGFs, but also high chemotactic properties of Hp-hAGF secretome (Figure 10A). These results again confirm pro-invasive influence of Hp-hAGF secretome as compared to both, hGF and control medium.
Figure 10.

Hp-hAGF secretome induces increased invasiveness of cancer cells. This phenomenon in adenocarcinoma cells is related to cMet/HGF/Integrin-β1/TNC and EGFR signaling. Geltrex invasion assay of HT29 and AGS cells towards culture medium, hGF and Hp-hAGF supernatants and in the case of AGS cells towards Hp-hAGF secretome with the addition of tyrphostin A46 (0.1 mM) showing enhanced invasiveness towards Hp-hAGF supernatant and the decrease after A46 (A). Fluorescent microscopy of transmigrated AGS cells attached to the bottom side of the geltrex/transwell membrane stained with bisbenzimide (Hoechst 33342) (B). The phase contrast microscopy showing HT29 and AGS cells that transmigrated through invasion chambers towards Hp-hAGF supernatant. Three independent experiments were performed for each condition (C). RT-PCR analysis of the expression of 18S, Fap, MMP 2, 3, 9 and mRNA in HT29 (D) and AGS (E) cells cultured in culture medium, hGF, Hp-hAGF supernatants and in the case of AGS cells in Hp-hAGF secretome with addition of tyrphostin A46 (0.1 mM) and the ratio of selected genes over 18S mRNA showing their strong upregulation in Hp-hAGF secretome. Phase contrast microscopy of Geltrex metalloproteinase activity assay (1:1) of HT29 (F) and AGS (G) flooded with culture medium, hGF, Hp-hAGF supernatants and in the case of AGS cells with Hp-hAGF secretome with the addition of A46 (0.1 mM), showing enhanced ability of cells flooded with Hp-hAGF supernatant to cross basement membrane components already after 24 hrs. RT-PCR analysis of the mRNA expression for 18S, cMet, HGF, EGFR and Integrin-β1 in AGS cells cultured in control medium, hGF, Hp-hAGF and in Hp-hAGF secretome with the addition of A46 (0.1 mM) and the ratio of selected genes over 18S mRNA (H). Results are mean ± SEM of four independent experimental repeats. Asterisk (*) indicates a significant change (P<0.05) as compared to the control cell value. Hash (#) indicates a significant change (P<0.05) as compared to the value of cells grown in GF supernatant.
To specify this notion, we have checked the expression of MMPs. Both, HT29 and AGS cells showed strong up-regulation of mRNAs encoding MMP2, MMP3 and MMP9 (Figure 10D, 10E) accompanied by the upregulation of FAP mRNA (Figure 10D, 10E). On the contrary, hGF secretome strongly decreased the expression of MMP9 only slightly increasing MMP2 in HT29 cells. In AGS cells hGF secretome induced decreased expression of MMPs tested, as compared to control values. The upregulated expression of MMPs as well as FAP expression appearance in AGS cells disappeared after EGFR signaling inhibition with A46 (Figure 10E). Next, we have visualized the ability of HT29 and AGS cells to move across and degrade BM. The Geltrex assay showed that both cell types actively migrated towards Geltrex/Hp-hAGF secretome interface and actively crossed it already after 24 hrs (Figure 10F, 10G). Consistently, the acceleration of invasive properties in AGS cells was again leveled by EGFR inhibition, however it’s worth noting that these properties weren’t entirely abolished as seen in Figure 10A, 10E, 10G. These results underline general, pro-invasive properties of Hp-AGF secretome exerted on cancer cells at least partially by EGFR signaling activation.
Further verifying the mechanism of EMT type 3-relared invasive expansion exerted by Hp-activated fibroblasts, we have confirmed the upregulation of c-Met, EGFR and Integrin-β1 mRNA combined with strong upregulation of EGF mRNA in AGS cells (Figure 10H). We have found that above increases in mRNA expression were dependent on EGFR signaling at least within 96 hrs of stimulation (Figure 10H). The decrease, but not complete disappearance of mRNA level for EGFR points to its partial autoregulation. The obtained results in the combination with Hp-induced upregulation of TNC in Hp-activated fibroblasts [29] strengthen our notion that the process of proinvasive cell stimulation evoked by Hp-activated fibroblasts engages the interplay of cMet/HGF/Integrin-β1/TNC and EGFR signaling. We have additionally found some upregulation of TGFβ mRNA in Hp-hAGF stimulated AGS cells (Figure 10H).
Discussion
The development of cancer is a multistep process in which cells gradually increase capacity for proliferation and survival, progressively becoming malignant through a series of alterations [62]. Recently, we have shown that long-term presence of Hp-AGF secretome switched microevolution of normal gastric epithelium towards EMT type 3 cancer stem cell-related program, while GF secretome induced differentiation related EmyoT in these cells [28]. Type 3 EMT occurs in neoplastic cells that have previously undergone genetic and epigenetic changes, specifically in genes that favor clonal outgrowth and the development of localized tumors [63]. EMT type 3 is observed in these cells as a part of dedifferentiation program conferring cancer cells with the traits of stemness [64]. Beside neoplastic growth, this type of transition has been also associated with cancer progression and metastasis. Nowadays, cancer metastasis is considered as an adaptive process requiring increased motility and the pluripotency-related cell phenotypical plasticity acquired through EMT type 3 and its reversed counterpart MET.
Guided by these premises, we have decided herein to check if Hp-AGFs are not only responsible for the induction of pro-pluripotent phenotype in normal gastric epithelial cells [28] but can also elicit their permanent reprogramming towards EMT-type 3 related invasive phenotype.
The analysis of obtained results presented herein showed that Hp-AGF secretome was characterized with general migration-promoting properties favoring excessive migration of normal and pro-cancerogenic cells. Parallely GF secretome exerted strong, migration-limiting effect.
We have also observed that both l.t.EMT+RGM1 and l.t.EMT-RGM1 cells undergone reprogramming towards motile phenotype. However, while l.t.EMT-RGM1 cells were characterized with higher persistence of movement and permanent fibroblastoid morphology, l.t.EMT+RGM1 cells presented phenotypical, niche-dependent heterogeneity of motility mode with the presence of intermediate, both mesenchymal and epithelial types of movement. These results stay in agreement with our previous observations [28] on phenotypical, niche-dependent plasticity of l.t.EMT+RGM1 cells. Above observations were reflected in highly differential actin distribution in l.t.EMT+RGM1 and l.t.EMT-RGM1 cells. L.t.EMT-RGM1 cells were characterized with the presence of abundant stress fibers characteristic for fibroblasts, which together with our previous results [28] showing α-SMA incorporation into their stress fibers, confirm permanent EmyoT of epithelial cells under prolonged GF secretome exposure. Such changes are the hallmarks of EMT type 2 ultimately leading to fibrosis. In turn, l.t.EMT+RGM1 cells showed heterogeneous, diffused F-actin distribution characteristic for transition between epithelial and mesenchymal state, accompanied with enhanced formation of filopodia, believed to guide cell locomotion during normal tissue morphogenesis or cancer metastasis [35,40]. Such dynamic transition between epithelial and mesenchymal actin organization is known to facilitate EMT type 3/MET switch [65,66]. EMT type 3/MET transition enables cell dissemination under diverse microenvironmental conditions during the process of metastasis and then formation of secondary tumors to some extent returning back to epithelial characteristic [67-71]. Our observations on pro-pluripotent, pro-cancerogenic motile characteristic of l.t.EMT+RGM1 cells were strengthened by nuclear accumulation of actin in l.t.EMT+RGM1 cells. Actin presence in the nucleus has been linked to DNA transcription and repair. In the normal epithelium, extracellular cues maintain low level of nuclear actin, while tumorigenic microenvironment promotes relocation of actin into the nucleus. This event leads to further cell transcriptome reprogramming towards tumor-like phenotype [41,42]. Additionally the pro-cancerogenic, elastic phenotype of l.t.EMT+RGM1 cells was confirmed by Cytokeratin19, which increasing expression is characteristic for reprogramming of normal tissues to premalignant lesions and finally to adenocarcinoma in situ [48].
We have previously shown that comparing to GF secretome, Hp-AGF secretome is enriched in TGFβ and HGF [27]. Accordingly, it has been proposed, that while the presence of TGFβ itself triggers EMT type 2, the parallel administration of TGFβ and HGF/c-Met is a potent EMT type 3 inducer leading to dynamic actin reorganization in epithelial cells [72-75]. Additionally, we have previously shown, that although TGFβ acted as EMT inducer in short-term gastric epithelial cultures [19], strongly reduced, but still sustained activity of TGFβRII in long-term cultures (l.t.EMT+RGM1) was a necessary condition enabling EMT type 3 related proliferation arrest escape and pro-pluripotent phenotype of epithelial cells [27]. Accordingly, Kubiczkova and coworkers [76] have shown complementary activation of HGF receptor cMet in tissues where TGFβRII had been suppressed. This fact implicates paracrine dependence between TGFβ and HGF signaling in regulation of carcinoma development [71,76] which seems to be also true for Hp-AGF-reprogrammed gastric epithelial cells, as shown in this study.
HGF/c-Met pathway has been shown to be essential for growth, survival and invasiveness of GC [77]. It stimulates multiple downstream pathways, including ERK/MAPK [78], which may collectively trigger signaling cascades resulting in the loss of cell-cell contacts, scattering, motility, angiogenesis, apoptosis escape and the ability to proliferate and settle down in a new environment [79-83].
Indeed, next to TGFβ, also HGF has been shown to trigger activation of ones of the main EMT type 3 related transcription factors (EMT-TFs): Snail and Twist [71,80,81]. Snail has been shown to repress epithelial and to enhance mesenchymal gene expression leading to induction of motile phenotype [84-86], i.e. by actin dynamic related Cdc42 activation [70,73-75]. Additionally, Snail was proposed to act as a key regulator of RhoA/α-SMA axis [85] leading to α-SMA incorporation into stress fibers. Concomitantly, in our present studies the overexpression of Snail in l.t.EMT-RGM1 and l.t.EMT+RGM1 cells was correlated with their accelerated motility. It also correlated with increased α-SMA content as reported by our team previously [28]. However, although α-SMA expression was much higher in l.t.EMT+RGM1 cells, it was not incorporated into stress fibers like in the case of l.t.EMT-RGM1 [28]. The observed dispersion of α-SMA in l.t.EMT+RGM1 suggests the existence of mechanism uncoupling Snail/RhoA/α-SMA machinery under the influence of Hp-AGF resulting in more plastic, motile phenotype.
According to the observation showing induction of Twist expression under increased HGF signaling [81-83,87-89] we have observed exclusive upregulation of Twist expression (Snail+Twist+l.t.EMT+RGM1 versus Snail+Twist-l.t.EMT-RGM1) followed by its nuclear translocation in l.t.EMT+RGM1 cells. Twist expression has been shown to promote invadopodia formation with upregulation and increased activation of MMPs. As the consequence, cells acquire ability to degrade tight barriers, such as the BM surrounding epithelial organs and to invade surrounding tissues [18,89-94]. Moreover, through degradation of proteins and ECM components, MMP expression participates in the formation of tumor microenvironment, characterized e.g. by altered matrix structural tension and release of ECM-bound signaling molecules (cytokines and growth factors). Such changed outside-in signaling resulted in the acceleration of cancer cell activation [88-92]. Both, MMP2 and MMP9 which are responsible for digestion of type IV collagen, constituting the main BM component [43-45], have been linked to invasive growth and cancer-related inflammation [44,94-97]. In turn, high expression of MMP3 has been shown to contribute to the late stage of GC [98] and to negatively correlate with GC differentiation [99,100]. Consistently with these findings, Snail+Twist+l.t.EMT+RGM1 cells were characterized with upregulated MMP2, MMP3 and MMP9 mRNA expression, revealing strong correlation with increased ability of these cells to degrade and migrate through BM.
Long-term guidance exerted by Hp-AGF secretome resulted also in Fap gene expression in Snail+Twist+l.t.EMT+RGM1 cells, which is a characteristic feature of CAFs [101] and metastastatic tumor cells [47,102,103]. It has been shown that MMP2, MMP9 and Fap localizing to invadopodia facilitate the process of cell invasion [104,105]. MMP2 and MMP9 were also shown to be responsible for HGF release from the ECM and their proteolytic processing to active form [106-108] which in turn coincided with c-Met overexpression [109]. While cMet receptor is expressed by epithelial cells, HGF production has been restricted to cells of mesenchymal origin [107], CAFs and cancer cells [110].
Accordingly, we have found previously, that prolonged administration of Hp-AGF secretome resulted in the induction of HGF release by overall pro-pluripotent Snail+Twist+Fap+Cytokeratin19+LGR5+l.t.EMT+RGM1 cell population [LGR5 determined in 28]. Herein, we have shown that these cells were also characterized with increased expression of c-Met enabling next to strengthened paracrine, also autocrine c-Met activation. All above findings implicate accelerated HGF/c-Met signaling in cytoskeletal plasticity, increased motility and invasiveness of l.t.EMT+RGM1 cells, triggered by increased content of HGF in both Hp-AGF [27] and l.t.EMT+RGM1 secretomes and by increased expression of c-Met on the surface of l.t.EMT+RGM1 cells.
Additionally, our results underline the notion of EMT type 3 conferring stem cell-like properties consistent with the migratory CSC concept [64]. In the light of these findings, we postulate that long-term Hp-AGF secretome-reprogrammed epithelial cells may constitute the source of both, CAFs and cancer cells, possibly switching from pro-invasive cancerous phenotype to CAF characteristic. Nevertheless, this notion demands further studies. Simultaneously, long-term administration of GF secretome failed to induce such properties in Snail+Twist-Fap-Cytokeratin19-LGR5-l.t.EMT-RGM1 cell population [LGR5 determined in 28]. Spix and coworkers postulated that HGF promotes motility and tumor progression in part by EGFR activation [72]. Elevated level of EGFR is frequently observed in different forms of cancer and often correlates with cancer progression and poor prognosis also in Hp-induced GC [111]. Indeed, it has been stated, that activation of c-Met, can result in proteolytic cleavage of EGF ligand precursors located in the cell membrane, which can then activate EGFR (the triple membrane-passing signaling mechanism) [112,113]. This interaction seems to be reciprocal, as increased c-Met protein level after increased EGFR activation has also been reported [114]. Moreover, it has been shown, that TGFβ over released by activated fibroblasts [114], including Hp-activated fibroblasts [19] can lead to production of several mitogenic growth factors including EGF [114]. In turn, EGFR has been proposed to potentiate TGFβ induction of a subset of invasion-associated genes, along with transcriptional regulation of HBEGF, a heparin-binding EGFR ligand [114]. This phenomenon could give additional explanation of decreased expression of TGβRII active form in l.t.EMT+RGM1 cells [28], which on one hand allowed the escape from TGFβ-induced arrest of proliferation and on the other hand still sustained cell pro-pluripotent [28] and invasive properties presented herein. The sustained activation of TGFβ elicited also by autologous TGFβ release [28], increased expression of c-Met, EGFR and activated form of EGFR point to the role of these reciprocal interactions in Hp-AGF induced reprogramming of epithelial cells. Thus, we conclude that cMet, TGFβ and EGFR interplay participate in the process of cell reprogramming evoked by Hp-AGF secretome at least at some stage of malignant transformation. Additionally, the cross-talk between growth factor receptors (GFR) and integrins has been well established i.e. in metastatic diffusion of tumor cells [115]. In vitro, mesenchymal fibroblasts and cancer cells migrate along 2D or through 3D ECM using integrins composed of β1, β3, β5 and β6 subunits [116,117] with the abundance of β1 subunits in the contact with collagen type IV [108,115,118]. C-Met activation leads to activation of Ras/Raf/MEK/ERK cascade which is known to promote proliferation and invasion of tumor cells [119-121] by increased expression of integrin-α6β1 [122]. Apart from the increased expression of integrins- β1, the physical and functional interactions between c-Met and Integrins-β1 where shown to drive the process of metastases [49] e.g. by MMP modulation [123-125]. The Integrin-β1-FAK-JNK signaling pathway has been shown to up-regulate the expression of both, MMP2 and MMP9 [126,127]. All this observations are again consistent with our previous results showing MEK1 upregulation in l.t.EMT+RGM1 cells [28] and present findings showing an increased expression of Integrin-β1 in these cells which points to the interplay between c-Met/Integrin-β1/MMP pathways. We have also previously shown that Hp infection induced TNC upregulation in gastric fibroblasts [29] and herein we have observed that Hp-AGF secretome induced expression of small amounts of TNC in l.t.EMT+RGM1 cells. It is known, that TNC is an Integrin-β1 ligand and is expressed at sites of epithelial-mesenchymal interactions during chronic inflammation and cancer [52,128]. TNC is transcriptionally upregulated mainly in CAFs, however it may also be upregulated in cancer cells. Importantly, TNC was found to promote migration through shifts from the stable focal adhesion with stress fibers to intermediate adherence [129] which are favorable for cell migration and spreading [50,51]. In GC, Twist-associated TNC expression has been linked to metastasis and poor prognosis [50,52,128,130]. Additionally, De Weaver group identified HGF and TGFβ as potent inducers of TNC expression [131]. In turn, cell contact with TNC was shown to induce expression of MMPs e.g. MMP2 [11,132] which subsequently cleaved TNC, creating adhesive sites for cell adhesion receptors. This event further led to clustering and activation of Integrins-β1 resulting in increased cancer cell spreading [53,54,128]. It is also worth noting, that TNC is susceptible to proteolytic degradation also by MMP3 in vitro [133,134].
Therefore, we conclude that long-term influence of Hp-AGF secretome induces reprogramming of normal epithelial cells towards pro-pluripotent, cancerogenic and invasive phenotype which can be extrapolated to EMT type 3-linked plasticity and accelerated invasiveness in vivo. We hypothesize, that this microevolution next to TGFβ relies also on c-Met/EGFR signaling interplay and involves the HGF-Integrin-Ras-dependent Twist activation leading to MMP and TNC upregulation with subsequent positive auto- and paracrine feedback loops intensifying this process.
To assess versatility of Hp-induced activation within fibroblast populations and subsequent impact of such activated fibroblasts on epithelial cells, we have applied short-term cultures of normal skin keratinocytes (HaCaT) in the secretome from Hp-hAGFs. The obtained results showed that the effect of human fibroblast activation is similar to that of rat fibroblast activation. Hp-hAGFs induced EMT type 3 related changes in HaCaT cells. These changes were similarly to RGM1 cells manifested by fibroblastoid morphology, transcriptional upregulation of EMT markers, EMT-TFs: Snail and Twist as well as for pluripotency markers: Oct, Sox-2 and c-Myc. We have also observed typical E- to N-cadherin mRNA switch. These findings indicate that EMT type 3 related mechanisms evoked in RGM1 cells are not restricted to one given cell line and even to the origin of epithelium.
Thus we have further attempted to assess how Hp-activated gastric fibroblast secretome may affect already developed gastric/colon cancer cells and to what extent the mechanisms engaged in RGM1 reprogramming towards l.t.EMT+RGM1 cells can participate in the neoplasm. Our results on short-term cultures of human colon adenocarcinoma HT29 and human gastric adenocarcinoma cells AGS in Hp-hAGF secretome, revealed the appearance of fibroblastoid motility mode in AGS cells. The EMT-inducing, but not migration-promoting influence of Hp-hAGF on motile human gastric adenocarcinoma AGS was at least partially dependent on EGFR signaling in these conditions. This observation is consistent with the findings that among other signaling molecules, also EGFR activation leads to E-cadherin repression through STAT3 induced Twist activation and thereby, promote EMT [53]. To further verify the influence of Hp-hAGF on cancer cells, we have focused on their invasiveness. We have discovered that similarly as for RGM1 cells, Hp-hAGS secretome evoked chemotactic response and increased invasiveness of both HT29 and AGS cells within 24 hrs of stimulation. The increased invasiveness was again accompanied by strong upregulation of mRNAs encoding MMP2, MMP3 and MMP9 and by FAP mRNA appearance. Importantly, we have observed the decrease of invasive properties of AGS cells towards Hp-hAGF secretome after EGFR signaling pathway inhibition. Decreased invasiveness was followed by the reduction of MMP and disappearance of Fap expression. These results underline general, pro-invasive properties of Hp-AGF secretome exerted on cancer cells at least partially by EGFR signaling activation.
To look deeper into Hp-hAGF elicited changes, we have checked EMT-TF and pluripotency marker expression. Hp-hAGF secretome induced some increase in Snail mRNA expression in both, HT29 and AGS cells as well as strong Twist transcriptional upregulation in HT29 and Twist mRNA appearance in AGS cells. Accordingly to its EMT type 3 inducing capacity, Hp-hAGF secretome elicited an increase in expression of mRNA for pluripotency markers [28]: c-Myc, Oct4 and Sox-2 in these cell types, confirming general pro-pluripotent activity of Hp-activated fibroblast secretome.
Above results also indicate that Hp-AGF secretome induces predominantly Twist upregulation in cancer cells, followed by pluripotency marker upregulation. The appearance of Twist gene activity and associated pluripotency markers gene upregulation in AGS cells, depended on EGFR signaling in our experimental conditions. These results confirm our previous observations on cell phenotypical plasticity inducing properties of Hp-AGF secretome and the opposite role of GF secretome and are consistent with the supportive role of EGFR in EMT and stemness [60,61].
To get insight into the mechanism of Hp-hAGF induced, EMT type 3-related invasive expansion of gastric cancer cells exerted by Hp-hAGFs, we have checked the expression of factors involved in cancerogenic and invasive reprogramming of RGM1 also in AGS cells. We have confirmed the upregulation of c-Met, EGFR and Integrin-β1 mRNA expression combined with strong upregulation of EGF and increased expression of TGFβ mRNAs in these cells. Again, the upregulation depended on EGFR signaling at least within 96 hrs of stimulation. The decrease, but not complete disappearance of mRNA level for EGFR after EGFR signaling inhibition suggests its partial autoregulation. These findings indicate that Hp-activated fibroblasts involve similar mechanisms to reprogram epithelial cells and already existing cancer cells.
EGFR signaling dependent upregulation of c-Met, EGFR and TGFβ mRNA in gastric adenocarcinoma cells indicate the cross-talk between these pathways also under Hp-hAGF induced stimulation at least at some stage of malignant transformation. Accordingly, the abolishment of Hp-hAGF secretome-induced c-Met transcriptional upregulation triggered by EGFR signaling inhibition was accompanied by decreased expression of MMP and Integrin-β1 mRNA. The obtained results in the combination with Hp-induced upregulation of TNC in Hp-activated fibroblasts [29] strengthen our notion that the process of proinvasive cancer cell stimulation evoked by Hp-hAGFs next to the cross-talk between growth factors and their receptors engages also cMet/HGF/Integrin-β1/TNC signaling.
Therefore we conclude that long-term influence of Hp-infected fibroblast secretome induces reprogramming of normal epithelial cells towards pro-pluripotent, cancerogenic and invasive phenotype which can be extrapolated to EMT type 3-linked plasticity and accelerated invasiveness of cancer cells in vivo. It also potentiates pro-pluripotent and invasive phenotype of already existing cancer cells. We hypothesize, that this microevolution next to TGFβ relies also on c-Met/EGFR signaling interplay at least at some stage of invasive growth and engages HGF/Integrin/Ras-dependent Twist activation leading to MMP and TNC upregulation with subsequent positive auto- and paracrine feedback loops intensifying this process. However, one should still remember, that EGFR downstream signaling cascades can shift to be activated via EGFR-independent mechanisms [56-59].
Thus, it would be beneficial to investigate how inhibition of each of these pathways affects the activation of others in this complex interplay. It would be also important to check how these interrelations change with the time of stimulation.
We also postulate that long-term influence of Hp-activated fibroblast secretome induces phenotypical plasticity of normal epithelial cells which can serve as the source of both cancer cells and CAFs, being able to switch between phenotypes. This hypothesis needs to be further elucidated with precise identification of eventual phenotypical subpopulations within l.t.EMT+RGM1 cells. Concomitantly, GF secretome reprograms epithelial cells towards pro-fibrotic, EMyoT non-invasive phenotype reflecting microenvironmental induction of EMT type 2. The influence exerted by Hp-infected fibroblast secretome reflects in some simplification the guidance of cancerous niche in the process of carcinogenesis and the development of invasive traits in cancer cells and can give the hint to the correlation between initially asymptomatic Hp (cagA+, vacA+) and further GC incidence. It also provides a convenient start-up model for studying more complex interactions between gastric mucosal epithelium and its microenvironment contributing to GC development.
Acknowledgements
This article was supported by The National Science; the National Science, Centre, Grant/Award Number: 2018/02/X/NZ3/00276 and statutory grant (N41/DBS/000576) from Jagiellonian University Medical College.
Disclosure of conflict of interest
None.
References
- 1.Dicken BJ, Bigam DL, Cass C, Mackey JR, Joy AA, Hamilton SM. Gastric adenocarcinoma review and considerations for future directions. Ann Surg. 2005;241:27–39. doi: 10.1097/01.sla.0000149300.28588.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen JQ, Kong YY, Weng S, Dong C, Zhu L, Yang Z, Zhong J, Yuan Y. Outcomes of surgery for gastric cancer with distant metastases: a retrospective study from the SEER database. Oncotarget. 2017;8:4342–4351. doi: 10.18632/oncotarget.14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apicella M, Corso S, Giordano S. Targeted therapies for gastric cancer: failures and hopes from clinical trials. Oncotarget. 2017;8:57654–57669. doi: 10.18632/oncotarget.14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, Ricci V, Solcia E. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology. 2007;132:1009–1023. doi: 10.1053/j.gastro.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 5.Konturek PC, Konturek SJ, Brzozowski T. Helicobacter pylori infection in gastric cancerogenesis. J Physiol Pharmacol. 2009;60:3–21. [PubMed] [Google Scholar]
- 6.Kagihara J, Matsuda B, Young KL, Li X, Lao X, Deshpande GA, Omata F, Burnett T, Lynch CF, Hernandez BY, Kuwada SK. Novel association between Helicobacter pylori infection and gastrointestinal stromal tumors (GIST) in a multi-ethnic population. Gastrointestinal Stromal Tumor. 2020:3. [Google Scholar]
- 7.Rokkas T, Rokka A, Portincasa P. A systematic review and meta-analysis of the role of H. pylori eradication in preventing gastric cancer. Ann Gastroenterol. 2017;30:414–423. doi: 10.20524/aog.2017.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bae SE, Choi KD, Choe J, Kim SO, Na HK, Choi JY, Ahn JY, Jung KW, Lee JH, Kim DH, Chang HS, Song HJ, Lee GH, Jung HY. The effect of eradication of H. pylori on GC prevention in healthy asymptomatic populations. Helicobacter. 2018;23:e12464. doi: 10.1111/hel.12464. [DOI] [PubMed] [Google Scholar]
- 9.Ham IH, Lee D, Hur H. Role of cancer-associated fibroblast in gastric cancer progression and resistance to treatments. J Oncol. 2019;2019:6270784. doi: 10.1155/2019/6270784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paraiso KH, Smalley KS. Fibroblast-mediated drug resistance in cancer. Biochem Pharmacol. 2013;85:1033–41. doi: 10.1016/j.bcp.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 11.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi H, Sakai R. Direct interaction between carcinoma cells and cancer associated fibroblasts for the regulation of cancer invasion. Cancers. 2015;7:2054–62. doi: 10.3390/cancers7040876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim H, Moon A. Inflammatory fibroblasts in cancer. Arch Pharm Res. 2016;39:1021–31. doi: 10.1007/s12272-016-0787-8. [DOI] [PubMed] [Google Scholar]
- 14.Monteran L, Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol. 2019;10:1835. doi: 10.3389/fimmu.2019.01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzet SE, Gaggioli C. Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res. 2016;365:607–19. doi: 10.1007/s00441-016-2467-x. [DOI] [PubMed] [Google Scholar]
- 16.Wu CW, Hsieh MC, Lo SS, Lui WY, P’eng FK. Results of curative gastrectomy for carcinoma of the distal third of the stomach. J Am Coll Surg. 1996;183:201–207. [PubMed] [Google Scholar]
- 17.Wu CW, Hsieh MC, Lo SS, Tsay SH, Li AF, Lui WY, P’eng FK. Prognostic indicators for survival after curative resection for patients with carcinoma of the stomach. Dig Dis Sci. 1997;42:1265–1269. doi: 10.1023/a:1018814426278. [DOI] [PubMed] [Google Scholar]
- 18.Mousami T, Khan MA, Fu J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. 2014;35:7335–42. doi: 10.1007/s13277-014-2163-y. [DOI] [PubMed] [Google Scholar]
- 19.Krzysiek-Maczka G, Wrobel T, Targosz A, Szczyrk U, Strzalka M, Ptak-Belowska A, Czyz J, Brzozowski T. Helicobacter pylori-activated gastric fibroblasts induce epithelial-mesenchymal transition of gastric epithelial cells in vitro in a TGF-β-dependent manner. Helicobacter. 2019;24:e12653. doi: 10.1111/hel.12653. [DOI] [PubMed] [Google Scholar]
- 20.Pereda MP, Ledda MF, Goldberg V, Chervín A, Carrizo G, Molina H, Müller A, Renner U, Podhajcer O, Arzt E, Stalla GK. High levels of matrix metalloproteinases regulate proliferation and hormone secretion in pituitary cells. J Clin Endocrinol Metab. 2000;85:263–269. doi: 10.1210/jcem.85.1.6248. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Patron C, Zouki C, Whittal R, Chan JS, Davidge ST, Filep JG. Matrix metalloproteinases regulate neutrophil- endothelial cell adhesion through generation of endothelin-1. FASEB J. 2001;15:2230–2240. doi: 10.1096/fj.01-0178com. [DOI] [PubMed] [Google Scholar]
- 22.Giannelli G, Falk-Marzillier J, Schiraldi R, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–228. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- 23.Kim JH, Kim JH, Cho CS, Jun HO, Kim DH, Yu YS, Kim KW. Differential roles of matrix metalloproteinase-9 and-2, depending on proliferation or differentiation of retinoblastoma cells. Investig Ophthalmol Vis Sci. 2010;51:1783–1788. doi: 10.1167/iovs.09-3990. [DOI] [PubMed] [Google Scholar]
- 24.Augustin S, Berard M, Kellaf S, Peyri N, Fauvel-Lafève F, Legrand C, He L, Crépin M. Matrix metalloproteinases are involved in both type I (apoptosis) and type II (autophagy) cell death induced by sodium phenylacetate in MDA-MB-231 breast tumour cells. Anticancer Res. 2009;29:1335–1344. [PubMed] [Google Scholar]
- 25.Isaacson KJ, Subrahmanyam NB, Ghandehari H. Matrix-metalloproteinases as targets for controlled delivery in cancer: an analysis of upregulation and expression. J Control Release. 2017;259:62–75. doi: 10.1016/j.jconrel.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan KT, Cortesio CL, Huttenlocher A. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J Cell Biol. 2009;185:357–370. doi: 10.1083/jcb.200809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krzysiek-Maczka G, Targosz A, Ptak-Belowska A, Korbut E, Szczyrk U, Strzalka M, Brzozowski T. Molecular alterations in fibroblasts exposed to Helicobacter pylori: a missing link in bacterial inflammation progressing into gastric carcinogenesis? J Physiol Pharmacol. 2013;64:77–87. [PubMed] [Google Scholar]
- 28.Krzysiek-Maczka G, Targosz A, Szczyrk U, Wrobel T, Strzalka M, Brzozowski T, Czyz J, Ptak-Belowska A. Long-term Helicobacter pylori infection switches gastric epithelium reprogramming towards cancer stem cell-related differentiation program in Hp-activated gastric fibroblast-TGFβ dependent manner. Microorganisms. 2020;8:1519. doi: 10.3390/microorganisms8101519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krzysiek-Maczka G, Targosz A, Szczyrk U, Strzałka M, Sliwowski Z, Brzozowski T, Czyz J, Ptak-Belowska A. Role of Helicobacter pylori infection in cancer-associated fibroblast-induced epithelial-mesenchymal transition in vitro. Helicobacter. 2018;23:e12538. doi: 10.1111/hel.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krzysiek-Maczka G, Targosz A, Szczyrk U, Strzalka M, Brzozowski T, Ptak-Belowska A. Involvement of epithelial-mesenchymal transition-inducing transcription factors in the mechanism of Helicobacter pylori-induced fibroblasts activation. J Physiol Pharmacol. 2019;70 doi: 10.26402/jpp.2019.5.08. [DOI] [PubMed] [Google Scholar]
- 31.Gustafson DE, Kessel WC. Fuzzy clustering with a fuzzy covariance matrix. IEEE Conference on Decision and Control including the 17th Symposium on Adaptive Processes; 1978. pp. 761–766. [Google Scholar]
- 32.Madeja Z, Master A, Michalik M, Sroka J. Contact-mediated acceleration of migration of melanoma B16 cells depends on extracellular calcium ions. Folia Biol (Krakow) 2001;49:113–24. [PubMed] [Google Scholar]
- 33.Majka J, Wierdak M, Szlachcic A, Magierowski M, Targosz A, Urbanczyk K, Krzysiek-Maczka G, Ptak-Belowska A, Bakalarz D, Magierowska K, Chmura A, Brzozowski T. Interaction of epidermal growth factor with COX-2 products and peroxisome proliferator-activated receptor-system in experimental rat Barrett’s esophagus. Am J Physiol Gastrointest Liver Physiol. 2020;318:G375–G389. doi: 10.1152/ajpgi.00410.2018. [DOI] [PubMed] [Google Scholar]
- 34.Lee K, Yun ST, Yun CO, Ahn BY, Jo EC. S100A2 promoter-driven conditionally replicative adenovirus targets non-small-cell lung carcinoma. Gene Ther. 2012;19:967–77. doi: 10.1038/gt.2011.168. [DOI] [PubMed] [Google Scholar]
- 35.Svitkina T. The actin cytoskeleton and actin-based motility. Cold Spring Harb Perspect Biol. 2018;10:a018267. doi: 10.1101/cshperspect.a018267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 37.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 38.Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JL, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–21. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- 39.Wolfenson H, Bershadsky A, Henis YI, Geiger B. Actomyosin-generated tension controls the molecular kinetics of focal adhesions. J Cell Sci. 2011;124:1425–32. doi: 10.1242/jcs.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Bio. 2008;19:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 41.Fiore APZP, Spencer VA, Mori H, Carvalho HF, Bissell MJ, Bruni-Cardoso A. Laminin-111 and the level of nuclear actin regulate epithelial quiescence via exportin-6. Cell Rep. 2017;19:2102–2115. doi: 10.1016/j.celrep.2017.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelpsch DJ, Tootle TL. Nuclear actin: from discovery to function. Anat Rec (Hoboken) 2018;301:1999–2013. doi: 10.1002/ar.23959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sobue T, Hakeda Y, Kobayashi Y, Hayakawa H, Yamashita K, Aoki T, Kumegawa M, Noguchi T, Hayakawa T. Tissue inhibitor of metalloproteinases 1 and 2 directly stimulate the bone-resorbing activity of isolated mature osteoclasts. J Bone Miner Res. 2001;16:2205–2214. doi: 10.1359/jbmr.2001.16.12.2205. [DOI] [PubMed] [Google Scholar]
- 44.Emara M, Cheung PY, Grabowski K, Sawicki G, Wozniak M. Serum levels of matrix metalloproteinase-2 and -9 and conventional tumor markers (CEA and CA 19-9) in patients with colorectal and gastric cancers. Clin Chem Lab Med. 2009;47:993–1000. doi: 10.1515/CCLM.2009.212. [DOI] [PubMed] [Google Scholar]
- 45.Bibak F, Ahmadi S, Khateri Z, Ahmadi A, Yari K. The role of matrix metalloproteinase-2 expression in gastric cancer susceptibility: a systematic review. Int J Cancer Manag. 2019;12:e94185. [Google Scholar]
- 46.Xu Y, Zhang J, Liu X, Huo P, Zhang Y, Chen H, Tian Q, Zhang N. MMP-2-responsive gelatin nanoparticles for synergistic tumor therapy. Pharm Dev Technol. 2019;24:1002–1013. doi: 10.1080/10837450.2019.1621899. [DOI] [PubMed] [Google Scholar]
- 47.Liu F, Qi L, Liu B, Liu J, Zhang H, Che D, Cao JY, Shen J, Geng JX, Bi Y, Ye LG, Pan B, Yu Y. Fibroblast activation protein overexpression and clinical implications in solid tumors: a meta-analysis. PLoS One. 2015;10:e0116683. doi: 10.1371/journal.pone.0116683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jain R, Fischer S, Serra S, Chetty R. The use of Cytokeratin 19 (CK19) immunohistochemistry in lesions of the pancreas, gastrointestinal tract, and liver. Appl Immunohistochem Mol Morphol. 2010;18:9–15. doi: 10.1097/PAI.0b013e3181ad36ea. [DOI] [PubMed] [Google Scholar]
- 49.Jahangiri A, Nguyen A, Chandra A, Sidorov MK, Yagnik G, Rick J, Han SW, Chen W, Flanigan PM, Schneidman-Duhovny D, Mascharak S, De Lay M, Imber B, Park CC, Matsumoto K, Lu K, Bergers G, Sali A, Weiss WA, Aghi MK. Cross-activating c-Met/β1 integrin complex drives metastasis and invasive resistance in cancer. PNAS. 2017;114:E8685–E8694. doi: 10.1073/pnas.1701821114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida T, Akatsuka T, Imanaka-Yoshida K. Tenascin-C and integrins in cancer. Cell Adh Migr. 2015;9:96–104. doi: 10.1080/19336918.2015.1008332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katoh D, Nagaharu K, Shimojo N, Hanamura N, Yamashita M, Kozuka Y, Imanaka-Yoshida K, Yoshida T. Binding of αvβ1 and αvβ6 integrins to tenascin-C induces epithelial-mesenchymal transition-like change of breast cancer cells. Oncogenesis. 2013;2:e65. doi: 10.1038/oncsis.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orend G, Chiquet-Ehrismann R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006;244:143–163. doi: 10.1016/j.canlet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 53.Watanabe K, Takahashi H, Habu Y, Kamiya-Kubushiro N, Kamiya S, Nakamura H, Yajima H, Ishii T, Katayama T, Miyazaki K, Fukai F. Interaction with heparin and matrix metalloproteinase 2 cleavage expose a cryptic anti-adhesive site of fibronectin. Biochemistry. 2000;39:7138–7144. doi: 10.1021/bi992670r. [DOI] [PubMed] [Google Scholar]
- 54.Saito Y, Imazeki H, Miura S, Yoshimura T, Okutsu H, Harada Y, Ohwaki T, Nagao O, Kamiya S, Hayashi R, Kodama H, Handa H, Yoshida T, Fukai F. A peptide derived from tenascin-C induces beta1 integrin activation through syndecan-4. J Biol Chem. 2007;282:34929–34937. doi: 10.1074/jbc.M705608200. [DOI] [PubMed] [Google Scholar]
- 55.Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100:8933–8. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yarar D, Lahdenranta J, Kubasek W, Nielsen UB, MacBeath G. Heregulin-ErbB3-driven tumor growth persists in PI3 Kinase mutant cancer cells. Mol Cancer Ther. 2015;14:2072–80. doi: 10.1158/1535-7163.MCT-15-0075. [DOI] [PubMed] [Google Scholar]
- 58.Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68:5878–87. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 59.Liu Q, Yu S, Zhao W, Qin S, Chu Q, Wu K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol Cancer. 2018;17:53. doi: 10.1186/s12943-018-0793-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han W, Lo HW. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012;318:124–134. doi: 10.1016/j.canlet.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lv XX, Zheng XY, Yu JJ, Ma HR, Hua C, Gao RT. EGFR enhances the stemness and progression of oral cancer through inhibiting autophagic degradation of SOX2. Cancer Med. 2020;9:1131–1140. doi: 10.1002/cam4.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buder T, Deutsch A, Klink B, Voss-Böhme A. Patterns of tumor progression predict small and tissue-specific tumor-originating niches. Front Oncol. 2019;8:668. doi: 10.3389/fonc.2018.00668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng X, Dai F, Feng L, Zou H, Feng L, Xu M. Communication between epithelial mesenchymal plasticity and cancer stem cells: new insights into cancer progression. Front Oncol. 2021;11:617597. doi: 10.3389/fonc.2021.617597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–52. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. Br J Pharmacol. 2014;171:5507–23. doi: 10.1111/bph.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188:11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sahai E. Illuminating the metastatic process. Nat Rev Cancer. 2007;7:737–49. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 69.Sanz-Moreno V, Marshall CJ. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr Opin Cell Biol. 2010;22:690–6. doi: 10.1016/j.ceb.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 70.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 71.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 72.Spix JK, Chay EY, Block ER, Klarlund JK. Hepatocyte growth factor induces epithelial cell motility through transactivation of the epidermal growth factor receptor. Exp Cell Res. 2007;313:3319–25. doi: 10.1016/j.yexcr.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Díaz-López A, Moreno-Bueno G, Cano A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag Res. 2014;6:205–16. doi: 10.2147/CMAR.S38156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev. 2003;120:1351–83. doi: 10.1016/j.mod.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 75.Izdebska M, Zielińska W, Grzanka D, Gagat M. The role of actin dynamics and actin-binding proteins expression in epithelial-to-mesenchymal transition and its association with cancer progression and evaluation of possible therapeutic targets. Biomed Res Int. 2018;2018:4578373. doi: 10.1155/2018/4578373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S. TGF-β-an excellent servant but a bad master. J Transl Med. 2012;10:183. doi: 10.1186/1479-5876-10-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bradley CA, Salto-Tellez M, Laurent-Puig P, Bardelli A, Rolfo C, Tabernero J, Khawaja HA, Lawler M, Johnston PG, Van Schaeybroeck S MErCuRIC consortium. Targeting c-MET in gastrointestinal tumours: rationale, opportunities and challenges. Nat Rev Clin Oncol. 2017;14:562–576. doi: 10.1038/nrclinonc.2017.40. [DOI] [PubMed] [Google Scholar]
- 78.Slattery ML, Lundgreen A, Wolff RK. Dietary influence on MAPK-signaling pathways and risk of colon and rectal cancer. Nutr Cancer. 2013;65:729–738. doi: 10.1080/01635581.2013.795599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tahir AA, Sani NF, Murad NA, Makpol S, Ngah WZ, Yusof YA. Combined ginger extract & Gelam honey modulate Ras/ERK and PI3K/AKT pathway genes in colon cancer HT29 cells. Nutr J. 2015;14:31. doi: 10.1186/s12937-015-0015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ye J, Talaiti A, Ma Y, Zhang Q, Ma L, Zheng H. Allergies and risk of colorectal cancer: a systematic review and meta-analysis of observational studies. Oncotarget. 2017;8:14646–14654. doi: 10.18632/oncotarget.14599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 82.Mai A, Muharram G, Barrow-McGee R, Baghirov H, Rantala J, Kermorgant S, Ivaska J. Distinct c-Met activation mechanisms induce cell rounding or invasion through pathways involving integrins, RhoA and HIP1. J Cell Sci. 2014;127:1938–1952. doi: 10.1242/jcs.140657. [DOI] [PubMed] [Google Scholar]
- 83.Zhou G, Yang J, Song P. Correlation of ERK/MAPK signaling pathway with proliferation and apoptosis of colon cancer cells. Oncol Lett. 2019;17:2266–2270. doi: 10.3892/ol.2018.9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park KJ, Choi HJ, Roh MS, Kwon HC, Kim C. Intensity of tumor budding and its prognostic implications in invasive colon carcinoma. Dis Colon Rectum. 2005;48:1597–1602. doi: 10.1007/s10350-005-0060-6. [DOI] [PubMed] [Google Scholar]
- 85.Karamitopoulou E, Zlobec I, Born D, Kondi-Pafiti A, Lykoudis P, Mellou A, Gennatas K, Gloor B, Lugli A. Tumour budding is a strong and independent prognostic factor in pancreatic cancer. Eur J Cancer. 2013;49:1032–1039. doi: 10.1016/j.ejca.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 86.Morris HT, Machesky LM. Actin cytoskeletal control during epithelial to mesenchymal transition: focus on the pancreas and intestinal tract. Brit J Cancer. 2005;112:613–620. doi: 10.1038/bjc.2014.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Puisieux A, Valsesia-Wittmann S, Ansieau S. A twist for survival and cancer progression. Brit J Cancer. 2006;94:13–7. doi: 10.1038/sj.bjc.6602876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ansieau S, Morel AP, Hinkal G, Bastid J, Puisieux A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene. 2010;29:3173–3184. doi: 10.1038/onc.2010.92. [DOI] [PubMed] [Google Scholar]
- 89.Zhao Z, Rahman MA, Chen ZG, Shin DM. Multiple biological functions of Twist1 in various cancers. Oncotarget. 2017;8:20380–20393. doi: 10.18632/oncotarget.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372–386. doi: 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Romero S, Musleh M, Bustamante M, Stambuk J, Pisano R, Lanzarini E, Chiong H, Rojas J, Castro VG, Jara L, Berger Z, Gonzalez-Hormazabal P. Polymorphisms in TWIST1 and ZEB1 are associated with prognosis of gastric cancer patients. Anticancer Res. 2018;38:3871–3877. doi: 10.21873/anticanres.12671. [DOI] [PubMed] [Google Scholar]
- 92.Zhao XL, Sun T, Che N, Sun D, Zhao N, Dong XY. Promotion of hepatocellular carcinoma metastasis through matrix metalloproteinase activation by epithelial-mesenchymal transition. J Cell Mol Med. 2011;15:691–700. doi: 10.1111/j.1582-4934.2010.01052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jena MK, Janjanam J. Role of extracellular matrix in breast cancer development: a brief update. F1000Res. 2018;7:274. doi: 10.12688/f1000research.14133.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koyama S. Significance of cell-surface expression of matrix metalloproteinases and their inhibitors on gastric epithelium and infiltrating mucosal lymphocytes in progression of Helicobacter pylori-associated gastritis. Scand J Gastroenterol. 2004;39:1046–1053. doi: 10.1080/00365520410003245. [DOI] [PubMed] [Google Scholar]
- 95.Koyama S. Enhanced cell surface expression of matrix metalloproteinases and their inhibitors, and tumor-induced host response in progression of human gastric carcinoma. Dig Dis Sci. 2004;49:1621–1630. doi: 10.1023/b:ddas.0000043375.35611.dd. [DOI] [PubMed] [Google Scholar]
- 96.Rautelin HI, Oksanen AM, Veijola LI, Sipponen PI, Tervahartiala TI, Sorsa TA, Lauhio A. Enhanced systemic matrix metalloproteinase response in Helicobacter pylori gastritis. Ann Med. 2009;41:208–215. doi: 10.1080/07853890802482452. [DOI] [PubMed] [Google Scholar]
- 97.Bergin PJ, Anders E, Sicheng W, Johnsson E, Andersson J, Lönroth H, Michetti P, Pan-Hammarström Q, Quiding-Järbrink M. Increased production of matrix metalloproteinases in Helicobacter pylori-associated human gastritis. Helicobacter. 2004;9:201–210. doi: 10.1111/j.1083-4389.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- 98.Nie K, Shi L, Wen Y, Pan J, Li P, Zheng Z, Liu F. Identification of hub genes correlated with the pathogenesis and prognosis of gastric cancer via bioinformatics methods. Minerva Med. 2020;111:213–225. doi: 10.23736/S0026-4806.19.06166-4. [DOI] [PubMed] [Google Scholar]
- 99.Xu J, Changyong E, Yao Y, Ren S, Wang G, Jin H. Matrix metalloproteinase expression and molecular interaction network analysis in gastric cancer. Oncol Lett. 2016;12:2403–2408. doi: 10.3892/ol.2016.5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yeh YC, Sheu BS, Cheng HC, Wang YL, Yang HB, Wu JJ. Elevated serum matrix metalloproteinase-3 and -7 in H. pylori-related gastric cancer can be biomarkers correlating with a poor survival. Dig Dis Sci. 2010;55:1649–1657. doi: 10.1007/s10620-009-0926-x. [DOI] [PubMed] [Google Scholar]
- 101.Wang J, Zhang G, Wang J, Wang L, Huang X, Cheng Y. The role of cancer-associated fibroblasts in esophageal cancer. J Transl Med. 2016;14:30. doi: 10.1186/s12967-016-0788-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Puré E, Blomberg R. Pro-tumorigenic roles of fibroblast activation protein in cancer: back to the basics. Oncogene. 2018;37:4343–4357. doi: 10.1038/s41388-018-0275-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Siveke JT. Fibroblast-activating protein: targeting the roots of the tumor microenvironment. J Nuclear Med. 2018;5:1412–1414. doi: 10.2967/jnumed.118.214361. [DOI] [PubMed] [Google Scholar]
- 104.Chen WT. Proteases associated with invadopodia, and their role in degradation of extracellular matrix. Enzyme Prot. 1996;49:59–71. doi: 10.1159/000468616. [DOI] [PubMed] [Google Scholar]
- 105.Eckert MA, Yang J. Targeting invadopodia to block breast cancer metastasis. Oncotarget. 2011;2:562–568. doi: 10.18632/oncotarget.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mohammed FF, Pennington CJ, Kassiri Z, Rubin JS, Soloway PD, Ruther U, Edwards DR, Khokha R. Metalloproteinase inhibitor TIMP-1 affects hepatocyte cell cycle via HGF activation in murine liver regeneration. Hepatology. 2005;41:857–867. doi: 10.1002/hep.20618. [DOI] [PubMed] [Google Scholar]
- 107.Catizone A, Ricci G, Caruso M, Galdieri M, Corano Scheri K, Di Paolo V, Canipari R. HGF modulates actin cytoskeleton remodeling and contraction in testicular myoid cells. Biomedicines. 2015;3:89–109. doi: 10.3390/biomedicines3010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Blandin AF, Renner G, Lehmann M, Lelong-Rebel I, Martin S, Dontenwill M. β1 integrins as therapeutic targets to disrupt hallmarks of cancer. Front Pharmacol. 2015;6:279. doi: 10.3389/fphar.2015.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mai A, Muharram G, Barrow-McGee R, Baghirov H, Rantala J, Kermorgant S, Ivaska J. Distinct c-Met activation mechanisms induce cell rounding or invasion through pathways involving integrins, RhoA and HIP1. J Cell Sci. 2014;127:1938–52. doi: 10.1242/jcs.140657. [DOI] [PubMed] [Google Scholar]
- 110.Owusu BY, Galemmo R, Janetka J, Klampfer L. Hepatocyte growth factor, a key tumor-promoting factor in the tumor microenvironment. Cancers (Basel) 2017;9:35. doi: 10.3390/cancers9040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Talukdar S, Emdad L, Das SK, Fisher PB. EGFR: an essential receptor tyrosine kinase-regulator of cancer stem cells. Adv Cancer Res. 2020;147:161–188. doi: 10.1016/bs.acr.2020.04.003. [DOI] [PubMed] [Google Scholar]
- 112.Fischer OM, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203–8. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 113.Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291:C1–C10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- 114.Dulak AM, Gubish CT, Stabile LP, Henry C, Siegfried JM. HGF-independent potentiation of EGFR action by c-Met. Oncogene. 2011;30:3625–3635. doi: 10.1038/onc.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–68. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 116.Even-Ram S, Yamada KM. Cell migration in 3D matrix. Curr Opin Cell Biol. 2005;17:524–532. doi: 10.1016/j.ceb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 117.Maaser K, Wolf K, Klein CE, Niggemann B, Zänker KS, Bröcker EB, Friedl P. Functional hierarchy of simultaneously expressed adhesion receptors: integrin alpha2beta1 but not CD44 mediates MV3 melanoma cell migration and matrix reorganization within three-dimensional hyaluronan-containing collagen matrices. Mol Biol Cell. 1999;10:3067–79. doi: 10.1091/mbc.10.10.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schaffner F, Ray AM, Dontenwill M. Integrin alpha5beta1, the fibronectin receptor, as a pertinent therapeutic target in solid tumors. Cancers. 2013;5:27–47. doi: 10.3390/cancers5010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Honma N, Genda T, Matsuda Y, Yamagiwa S, Takamura M, Ichida T, Aoyagi Y. MEK/ERK signaling is a critical mediator for integrin-induced cell scattering in highly metastatic hepatocellular carcinoma cells. Lab Invest. 2006;86:687–96. doi: 10.1038/labinvest.3700427. [DOI] [PubMed] [Google Scholar]
- 120.Enayat S, Ceyhan MŞ, Başaran AA, Gürsel M, Banerjee S. Anticarcinogenic effects of the ethanolic extract of Salix aegyptiaca in colon cancer cells: involvement of Akt/PKB and MAPK pathways. Nutr Cancer. 2013;65:1045–1058. doi: 10.1080/01635581.2013.850966. [DOI] [PubMed] [Google Scholar]
- 121.Zhou G, Yang J, Song P. Correlation of ERK/MAPK signaling pathway with proliferation and apoptosis of colon cancer cells. Oncol Lett. 2019;17:2266–2270. doi: 10.3892/ol.2018.9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen JC, Chen YJ, Lin CY, Fong YC, Hsu CJ, Tsai CH, Su JL, Tang CH. Amphiregulin enhances alpha6beta1 integrin expression and cell motility in human chondrosarcoma cells through Ras/Raf/MEK/ERK/AP-1 pathway. Oncotarget. 2015;6:11434–11446. doi: 10.18632/oncotarget.3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.White DE, Muller WJ. Multifaceted roles of integrins in breast cancer metastasis. J Mammary Gland Biol. 2007;12:135–142. doi: 10.1007/s10911-007-9045-5. [DOI] [PubMed] [Google Scholar]
- 124.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 125.Huttenlocher A, Horwitz AR. Integrins in cell migration. Csh Perspect Biol. 2011;3:a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yue J, Zhang K, Chen J. Role of integrins in regulating proteases to mediate extracellular matrix remodeling. Cancer Microenviron. 2012;5:275–83. doi: 10.1007/s12307-012-0101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hauck CR, Hsia DA, Puente XS, Cheresh DA, Schlaepfer DD. FRNK blocks v-Src-stimulated invasion and experimental metastases without effects on cell motility or growth. EMBO J. 2002;21:6289–6302. doi: 10.1093/emboj/cdf631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Midwood KS, Orend G. The role of tenascin-C in tissue injury and tumorigenesis. J Cell Commun Signal. 2009;3:287–310. doi: 10.1007/s12079-009-0075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest. 2001;107:785–90. doi: 10.1172/JCI12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Qi W, Yang Z, Li H, Cui Y, Xuan Y. The role of Tenascin-C and Twist1 in gastric cancer: cancer progression and prognosis. APMIS. 2019;127:64–71. doi: 10.1111/apm.12919. [DOI] [PubMed] [Google Scholar]
- 131.De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, Mareel M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004;18:1016–8. doi: 10.1096/fj.03-1110fje. [DOI] [PubMed] [Google Scholar]
- 132.Tremble P, Chiquet-Ehrismann R, Werb Z. The extracellular matrix ligands fibronectin and tenascin collaborate in regulating collagenase gene expression in fibroblasts. Mol Biol Cell. 1994;5:439–453. doi: 10.1091/mbc.5.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Giblin SP, Midwood KS. Tenascin-C: form versus function. Cell Adh Migr. 2015;9:48–82. doi: 10.4161/19336918.2014.987587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lowy CM, Oskarsson T. Tenascin C in metastasis: a view from the invasive front. Cell Adh Migr. 2015;9:112–124. doi: 10.1080/19336918.2015.1008331. [DOI] [PMC free article] [PubMed] [Google Scholar]