

Abstract
Liquid biopsy test has a better uptake for colorectal cancer (CRC) screening. However, suboptimal detection of early-staged colorectal neoplasia (CRN) limits its application. Here, we established an early-staged CRN blood test using error-corrected sequencing by comparing clonal hematopoiesis (CH) of 63 CRN patients and that of 32 controls. We identified 1,446 variants and classified the uniqueness in CRN patients. There was no significance difference in the amount of variant between CRNs and controls, but the uniqueness of variants with defective DNA mismatch repair-related mutational signature was addressed from peripheral blood in early-staged CRN patients. By machine learning approach, the early-staged CRNs was discriminated from controls with an AUC of 0.959 and an accuracy of 0.937 (95% CI, 0.863 to 0.968). The CRN predictive model was further validated by additional 20 CRNs and 10 controls and showed the accuracy, sensitivity, specificity, positive prediction value (PPV) and negative prediction value (NPV) of 0.933 (95% CI: 0.779 to 0.992), 0.95, 0.90, 0.95 and 0.90, respectively. In summary, we develop a CH-based liquid biopsy test with machine learning approach, which not only increase screening uptake but also improve the detection rate of early-staged CRN.
Keywords: Colorectal neoplasia, clonal hematopoiesis, early diagnosis, machine learning
Introduction
Colorectal cancer (CRC) draws attentions because it is the third most common cancer and the second leading cause of cancer-related deaths worldwide [1]. In Taiwan, the incidence of CRC was 41.84 cases per 100,000 residents in 2018 and was the most common cancer than lung cancer and breast cancer. CRC has high mortality in advanced stages but it is preventable by removal of precancerous lesions (advanced adenoma, AA) or detection of CRC in early stage [2]. Colonoscopy remains the gold standard among the various screening modalities and reduces the mortality rates by 67% [3,4]. However, it remains debated whether colonoscopy qualified as first-line screening modality because of its inconvenience, invasiveness, and cost, which limit the adherence of general population to the screening program. Currently, fecal immunochemical test (FIT) and fecal DNA test are the main approaches for non-invasive CRC screening. Although FIT contributes towards a reduction in CRC mortality, low specificity and sensitivity hinder its clinical utility for identifying early-staged CRN [5-7]. Therefore, there is an overwhelming preference for liquid biopsy versus stool-based screening, highlighting the need and potential merit for developing highly robust liquid biopsy tests for identifying early-staged CRN. However, sub-optimal liquid biopsy detection remains an unmet need for CRN screening in current clinical practice.
Clonal hematopoiesis (CH) is referred to the clonal expansions of mutated hematopoietic cells and is common in aging human [8-11]. In the past decade, emerging data have demonstrated that CH in the peripheral blood may imprecate the microenvironment in disease [12-17]. Cancer patients have higher rate of CH and this may be associated with exposure to environmental mutagens, radiation or chemotherapy [18,19]. The DNMT3A, TET2, ASXL1 and JAK2 are canonical CH-related genes and common mutated in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) [20,21]. Recent genetic studies have shown that CH is common in individuals without hematological malignancies and is associated with the cardiovascular disease and stroke by promoting inflammation of blood vessel walls [22-24]. Furthermore, CH is also reported to be relevant with solid cancer and approximately 30% of patients in solid cancer harbor CH mutations [25]. Currently, CH is thought to serve as predictors of progression to hematologic malignancy and to have the potential role of precision oncology in treatment [26,27]. For example, mutations in DNA damage response genes (TP53, PPM1D, CHEK20) were clonal selected with exposure to cancer therapies (radiotherapy, platinum and topoisomerase II inhibitors) and the presence of those selected cancer predisposition mutations may increase the risk of therapy-related myeloid neoplasms (tMNs) development in patient of solid cancer [28]. Additionally, CH in patients with cancers has fundamental differences comparing to healthy individuals [29]. However, the mechanisms of CH selection and malignant transformation are still not fully understood.
With increasing use and improvement of next-generation sequencing (NGS) technique, recent studies have shown the applications of “error-corrected sequencing” (ECS) in CH researches. “Error suppression and mutation call” of ECS are designed as the “unique molecular index” (UMI), which improves the detection sensitivity of achieving variant allele frequency (VAF) ≥0.0001 [30]. Genetic analysis of human blood samples using ECS has proven that low allelic variants (defined as VAF lower than 0.02) have found in almost everyone over the age of 50 [31,32]. In note, most of those low allelic variants are below the detection limit of standard whole exome sequencing (WES). On the other hand, the higher sensitivity achieved by the advanced NGS technologies results in more variants identified. The huge number of variants increases the difficulty of using traditional analysis methods to process the ever-increasing CH mutations. Currently, machine learning is wildly applied in complex genetic analysis due to its ability to identify the multivariate statistical properties that distinguish two different groups of data [33]. For example, MetaNet, a computational framework, is developed by analyzing clinical and pan-cancer DNA sequencing data for assessment of metastatic risk by using a machine learning approach [34]. Features learned from the metastatic tumors enable MetaNet to identify patients with primary cancers at high risk of metastasis before the onset of symptoms.
In this study, we hypothesize that the precancerous environments provide pressures to lead the evolutional trajectory of CH and these CRN-related CH variants may be used as detection markers for early CRN screening in liquid biopsy. Thus, we enrolled coloscopy-informed early-staged CRN patients and healthy controls to identify CH through ECS technique. Unique spectrum of CH predictors was discovered and a CH-based liquid biopsy test was established through machine-learning approach.
Materials and methods
Patients
This study protocol was approved by the Institutional Review Board of the National Taiwan University Hospital (No. 201712033RIN). The informed consent was signed by the patients. Respective colorectal neoplasm patients and healthy controls were retrieved from Health Management Center at National Taiwan University Hospital. Eligible patients were at least 40 years of age and classified based on the histological examination and coloscopy assay. Early-stage CRN was defined as AA and stage I cancer.
Blood collection and DNA extraction
Whole blood was collected in K2EDTA tube and processed from 2-6 hours after blood drawn. Peripheral blood DNA was extracted from PBMC using QIAamp Blood DNA Mini kit (Qiagen, Hilden Germany) according to the manufacturer’s instructions. Additional fragmentation was performed from 100 ng peripheral blood DNA using KAPA Freg kit (KAPA Biosystems, Wilmington, MA) under the condition of 37°C incubation for 30 minutes.
Library preparation and next-generation sequencing
Sequencing libraries were prepared according to the manufacturer’s instructions of AVENIO Expanded kit (Roche sequencing solution, Pleasanton, CA) with 30 ng of fragmented peripheral blood DNA. The library profile was analyzed with the Agilent 2200 Bioanalyzer (Agilent Technologies, Palo Alto, CA) and quantified using QuBit dsDNA HS Assay kit (ThermoFisher, Waltham, MA). For a single sequencing run, the 8 or 16-multiplexed library was created by pooling the libraries and sequenced on a lane of Illumina HiSeq 4000 flow cell or NovaSeq 6000 S1 flow cell with 2×150-bp paired-end reads. Raw sequencing was analyzed using AVENIO ctDNA analysis software (version 1.1.0) with the setting of “Default-Expanded-Panel-Workflow”.
Statistical analysis
Statistical analysis was performed in R (version 3.6.1). Two-sample T test was used for comparisons of the VAF distributions. Wilcoxon rank-sum test was performed to test the probability of difference in VAFs observed between the CRN patients and controls. P-value <0.05 was considered statistically significant.
Mutational signature analysis
The contribution of known mutation processes to the single nucleotide variations was measured by “MiutationalPatterns” package using the COSMIC signature set v2 [35]. Non-negative matrix factorization (NMF) package was used to determine the minimal components with may explain maximum variance among CRN group or control group in training cohort. Cosine correlation similarity was in using the “MutationalPatterns” package to measure the closeness of mutational signatures. Visualizations of variants in different groups were carried out in using the R package “ggplot2”.
Data pre-process and model-training method
A learning framework with pre-processing filtering was used for the establishment of CRN predictive model. The classification framework of model-training used the R package “caret”. The discovery cohort was randomly split into training set and testing set in the ratio 70:30 and the CRN classifier was developed by partial least square regression with repeated 10-fold cross-validation in 1,000 times. Accuracy of the CRN classifier was assessed in the testing set and another independent validation cohort, composed of 20 CRNs and 10 controls. In addition, random sampling of the discovery cohort using 1,000 bootstraps was used to assess the performance of prediction model and estimate the confidence interval of accuracy in using the BCa bootstrap method by R package “boot”.
Results
Patients’ characteristics
We retrieved respective cohorts of colorectal neoplasm patients and healthy controls from Health Management Center at National Taiwan University Hospital. Eligible patients were at least 40 years of age and classified based on the histological examination and coloscopy assay. Early-staged CRN was defined as AA and stage I cancer. Under the selection criteria, 63 early-staged CRN patients (CRNs), including 18 stage I CRC patients and 45 AA patients, and 32 healthy normal controls (controls) were enrolled in this study as the discovery cohort. The positive rate of FIT test in the CRNs was 55.6%. All the controls were negative in FIT test and confirmed by coloscopy assay. Conditional probabilities for the effect of age and sex on screening were tested by Student’s t and Fisher’s exact tests, resulting in P-values of 0.155 and 0.096, respectively. Next, additional 20 CRN patients, including 16 AA patients and 4 stage I CRC patients, and 10 healthy normal controls were enrolled and served as the validation cohort (Table 1).
Table 1.
Demographic and clinical information
| Discovery cohort | Validation cohort | |||
|---|---|---|---|---|
|
|
|
|||
| CRNs, n=63 | Controls, n=32 | CRNs, n=20 | Controls, n=10 | |
| Male, n (%) | 30 (47.6) | 21 (65.6) | 7 (35.0) | 5 (50.0) |
| Age, years (SD) | 64.6 (9.6) | 61.9 (6.4) | 65.6 (9.9) | 61.6 (5.6) |
| Location, n (%) | ||||
| Proximal | 32 (50.8) | - | 10 (50.0) | - |
| Distal | 10 (15.9) | - | 6 (30.0) | - |
| Rectum | 21 (33.3) | - | 4 (20.0) | - |
| Tumor size, mm (SD) | 3.4 (1.7) | - | 3.1 (1.3) | - |
| Histology, n (%) | ||||
| Advanced adenoma | - | |||
| Tubular | 11 (17.5) | - | 5 (25.0) | |
| Tubulovillous | 31 (49.2) | - | 11 (55.0) | - |
| Villous | 3 (4.7) | - | 0 | - |
| Stage I cancer | 18 (28.6) | - | 4 (20.0) | - |
| FIT, n (%) | ||||
| Positive | 35 (55.6) | 0 | 10 (50.0) | 0 |
| Negative | 28 (44.4) | 32 (100) | 10 (50.0) | 10 (100) |
FIT: fecal immunochemical test.
Reshaping the mutational profiles of CH in peripheral blood by error-corrected sequencing
As a step towards developing a non-invasive method for early-stage CRN screening, we characterized mutations including CH-related variants in peripheral blood mononuclear cells (PBMCs) using the ECS approach (Figure 1A). Initially, 63 early-staged CRN patients and 32 healthy individuals were enrolled as the discovery cohort for the ECS analysis. We implemented a quality control to ensure that the unique depth of each individual is greater than 3,000× before further analysis of the variants (Figure 1B). As the result, 1,446 variants were identified in the discovery cohort (Figure 1C). There were 1,171 variants identified in CRNs and 753 of 1,171 (64.3%) variants were unique and not shared with controls. On the other hand, 693 variants were identified in controls and 275 of 693 (39.7%) variants were unique and not shared with CRNs. The average number of detected variants in CRNs and controls were 128.2 (92-167) and 127.2 (101-166), respectively. Somatic mutations with VAF at least 0.02 are the traditional definition of clonal hematopoiesis indeterminate potential (CHIP) in clinic. With the specification, there were 7 (11.11%) somatic variants with VAF greater than 0.02 found in 63 CRNs and 3 (9.38%) were found in 32 controls (Table 2). Not surprisingly, the low allelic variants with VAF below 0.02 were detected in all samples (Figure 2A). The average proportions of low allelic variants were 39.05% (12.63%-53.33%) in CRNs and 37.07% (8.6%-54.3%) in controls (Figure 2B). Notably, the mean proportions of those low allelic variants with VAF lower than 0.001 were 15.88% (3.16%-27.5%) and 16.1% (3.31%-26.53%) in CRNs and controls, respectively (Figure 2C). However, there was no statistical significance in the difference of the variant quantities between CRNs and controls.
Figure 1.

The mutational spectrum reshaped by ECS in CRNs and controls. A. The experimental workflow. B. Metrics of Error-corrected Sequencing. C. The mutational spectrum. The level of VAF is addressed from dark blue to red as low to high. The clinicopathological features are noted for each individual. FIT: fecal immunochemical test; AA: advanced adenoma.
Table 2.
Somatic mutations with VAF greater than 0.02
| Sample group | Age | Gender | Gene | Variant | VAF (Deplex) |
|---|---|---|---|---|---|
| CRN patient | 65.8 | M | BRAF | Gly446val | 0.027 |
| CRN patient | 56.3 | F | DDR2 | Pro496Ser | 0.044 |
| CRN patient | 59.3 | M | MSH2 | Arg382His | 0.021 |
| CRN patient | 72.4 | M | ROS1 | Arg1942Trp | 0.024 |
| CRN patient | 49.4 | F | FGFR2 | Arg422Cys | 0.024 |
| CRN patient | 62 | F | AR | Leu57Gln | 0.025 |
| CRN patient | 76.4 | M | CSF1R | Trp159Ter | 0.050 |
| Control group | 62 | M | PMS2 | His479Gln | 0.027 |
| Control group | 69 | M | ROS1 | Val332Ala | 0.040 |
| Control group | 64 | M | FLT1 | Ser1279Asn | 0.031 |
| GNA11 | Arg114Gln | 0.026 |
Figure 2.

Genetic profiling of variants in CRN patients and risk-matched controls. (A) Distribution of VAF in each individual was addressed in CRN patients and risk-matched control groups. The VAFs of PBMCs were determined by error corrected sequencing with greater than 3,000× unique-depth. (B) The prevalence of VAF <0.02 in CRNs and controls. (C) The prevalence of VAF <0.001 in CRNs and controls. P-values were calculated using two-sample T test with Welch’s correction in (B and C).
Mutational signature analysis reveals the influence of genetic architecture in defective DNA mismatch repair
To understand the influence of identified somatic variants, we performed the mutational signature analysis in both groups. Initially, we identified 845 somatic variants observed in the CRNs and 450 somatic variants observed in the controls followed by performing the 96 mutational profiles analysis for each sample. By using the algorithms of non-negative matrix factorization (NMF), three mutational profiles were identified from CRNs (Figure 3) and four mutational profiles were identified from controls (Figure 4). To investigate the potential contribution of mutagens in those profiles, cosine similarity analysis was used to identify the similarity between those profiles and mutational signatures with the activity of 30 specific mutational processes investigated by COSMIC mutational signature v2 in human cancer (Figures 3A, 4A). Next, we compared the difference between those groups and found the Signature 20 was contributed uniquely from CRNs. The proposed etiology of Signature 20 is associated with defective DNA mismatch repair, which is commonly reveled in sporadic CRCs [36,37]. Furthermore, we found that Signature 1 did not contribute to both groups in this study, which may indicate that age-related variants are not common in this target panel and may not contribute to the construction of predictive models for CRN detection.
Figure 3.

Three enriched mutational profiles in CRNs. A. Trinucleotide motif frequency plot and enriched mutational signature for three mutational profiles in CRNs. On the x axis, the 16 possible trinucleotide contexts are repeated once for each of the six substitution types. B. Relative contribution. Proportional distribution of each enriched mutational profile was addressed. C. Absolute contribution. The number of mutations was addressed for each enriched mutational profile.
Figure 4.

Four enriched mutational profiles in controls. A. Trinucleotide motif frequency plot and enriched mutational signature for four mutational profiles in controls. B. Relative contribution. Proportional distribution of each enriched mutational profile was addressed. C. Absolute contribution. The number of mutations was addressed for each enriched mutational profile.
Construction of CH-based predictive model for early-stage CRN identification
In this case-control study, those variants were scattered and it was difficult to deal with those statistical extremes through traditional analysis methods. Therefore, we implemented machine learning approach to discriminate early-stage CRN patients from risk-matched controls (Figure 5A). Initially, the pre-processing filtering was performed using the 1,446 variants from the discovery cohort with following filters: (1) removed the germline mutations; (2) removed the common shared variants present in CRN patients and risk-matched controls with a Wilcoxon rank sum test P-value >0.1; (3) removed the unique variants (only presented in CRNs or controls) with less than two cases in the training cohort; (4) rescued the filtered unique variants which have been reported with the pathogenicity significantly. Under the filter criteria, 88 resulting variants were used as the variables for model training (Figure 5B). Next, we split the discovery cohort to training set and testing set by 70:30 resampling and trained our predictive model using partial least squares regression with repeated 10-fold cross-validation in 1,000 times. The AUC of predictive model was 0.959 (Figure 5C). Confidence interval for accuracy of training-model was generated by 1,000 bootstraps from discovery cohort, resulting in 0.937 (95% CI, 0.863 to 0.968) (Figure 5D).
Figure 5.

Prediction of CRNs using machine learning algorithms. A. Study framework. B. The diagram of pre-processing filtering. C. The analysis method of machine-learning was based on a 10-fold cross-validation framework. D. Confidence interval for accuracy of training-model was generated by 1,000 bootstraps from the discovery cohort.
Validation of CH-based CRN predictive model
An independent cohort, which included 20 early-staged CRN patients and 10 healthy controls was used to validate the CH-based CRN prediction model. As the result shown in Table 3, our prediction model could correctly discriminate those 20 CRNs from 10 controls in an accuracy, sensitivity, specificity, positive prediction value (PPV) and negative prediction value (NPV) of 0.933 (95% CI: 0.779 to 0.992), 0.95, 0.90, 0.95 and 0.90, respectively. It is worth noting that, in the same validation cohort, the sensitivity in FIT test of the AA group and the stage I cancer group were 48.9% and 72.2%, respectively (Table 3). That is, by using the CH-based CRN predictive model, it may improve the ability of early CRN detection compared to the traditional FIT test and provide the potential in clinical practice.
Table 3.
Predictive performance in validation cohort
| Predictor | Reference | |
|---|---|---|
|
| ||
| CRNs | Controls | |
| CRNs | 19 | 1 |
| Controls | 1 | 9 |
Accuracy (95% CI): 0.9333 (0.7793, 0.9918). P-value [Acc>NIR]: 0.00065. Kappa: 0.85. Sensitivity: 95.0%. Specificity: 90.0%.
Characteristics of variants in CH-based CRN predictive model
The 88 resulting variants were retained from the 1,446 variants under the screening criteria and used as variables for model training. Among these 88 variants, 71 were unique in the CRNs, 8 were unique in controls and 9 were overlaid in both groups (Figure 6A). On the other hand, these 88 variants were located into 42 genes, and 27 of 42 were unique in CRNs (Figure 6B). To characterize the CRN unique variants, we implemented the mutational signature analysis to analyze these 71 variants. As the result in Figure 6C, 9 mutational signatures were identified, including Signature 3 with BRCA1/BRCA2 mutation; Signature 10 with POLE mutations; Signature 15, 20 with defective DNA mismatch repair; Signature 21 with microsatellite unstable tumors; Signature 22 with exposure of aristolochic acid and Signature 12, 28, 30 with unknown etiologies.
Figure 6.
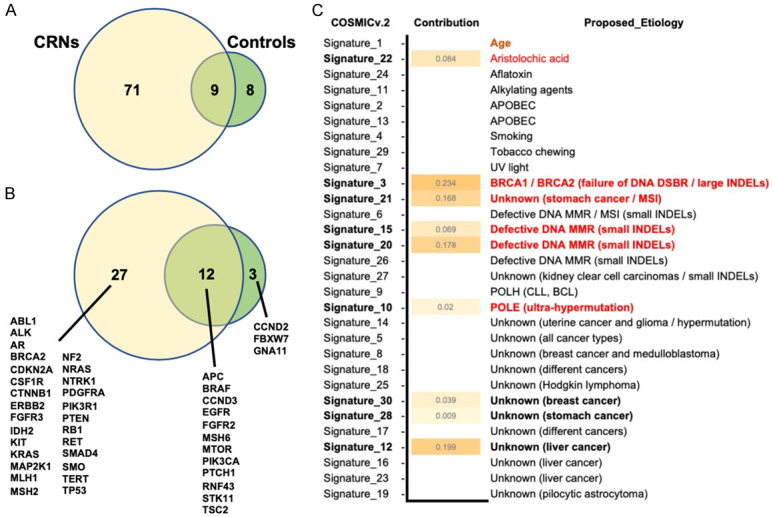
Overview of resulting variants and genes in CRN predictive model. A. Overlap of the resulting variants in CRNs and controls. B. Overlap the genes in CRNs and controls. C. The COSMIC mutation signature v.2 analysis was shown with the level of contribution in 71 CRN-specific resulting variants.
Discussion
CH is common in the elderly. Due to the lifestyle factors and the exposure of environmental carcinogens, the accumulation of mutations in malignant tumors increases. In this study, we applied the ECS technique with higher sensitivity to detect low allelic variants and reshaped the CH landscape in early-stage CRN patients. It was not a surprise that high rate of low allelic variants was measured in almost all samples when using the ECS technique for variant profiling. Most of those low allelic variants do not directly act as driver mutations for malignant transformation but the clonal evolution of CH may reflect the condition of stress or the status of precancerous environment [38]. Previous report also indicated that most of those low allelic variants might remain stable at low levels for many years in healthy individuals [39]. However, as far as we know, there is limited understanding about the mechanisms underpinning the evolutionary trajectory of CH. Indeed, most studies identified those low allelic variants in white blood cells and suggested those variants as “background” or “contaminate” in cell-free DNA (cfDNA) analysis [40,41].
What sets us apart in this study is that we reshaped the genetic variation profile of each individual and determined the uniqueness of the variation distribution from this case-control study. It is worth noting that these unique variants are scattered in the cohort, which increases the difficulty of using traditional analysis methods to deal with those statistical extremes. Nevertheless, we proved the potential of machine learning methods in improving statistical analysis and established a non-invasive CRN predictive model with a performance of 0.959 in AUC. When applying this method to panel design, it is critical to reduce the risk of model overfitting. Otherwise, model overfitting may lead to overly optimistic results. Therefore, at least one independent cohort is essential for model validation. On the other hand, sufficient unique sequencing depth (coverage of more than 3,000× as shown in this study) is required to ensure the accuracy and sensitivity of detection.
In the pathogenicity of view, some pathogenic mutations in low-allele fraction were selected by this machine-learning approach. For example, BRAF G466V was reported in 0.06% of all CRC patients in AACR Project GENIE [42] and 2 of 63 (3.17%) CRNs in our training cohort were altered in this mutation with VAF 0.0269 and 0.0003. On the contrary, most variants of those low allelic variants are not canonical pathogenic variants but still selected by the machine-learning method for the CRN predictive model.
To test the causality, the mutational signature analysis was performed in this study. As the result from 845 somatic variants observed in the CRNs, Signature 20, as the etiology as defective DNA mismatch repair, was identified. Furthermore, 9 mutational signatures were contributed from the 71 CRN-specific resulting variants of CRN predictive model. Among those 9 mutational signatures, 5 mutational signatures were associated with BRCA1/BRCA2 mutations, ultra-hypermutation, defective DNA mismatch repairs and microsatellite-instability. Interestingly, the characteristics of these 5 mutational signatures are related to the effect of DNA damages. This observation indicated that the ML approach could effectively enrich CRN-associated variants, improve the accuracy of early cancer screening and characterize the possible causality of CRN. For example, CRC tumors with MSI have defective in DNA repair enzymes and are infiltrated by a large number of lymphocytes [43]. In metastatic colon cancer, the test of MSI/dMMR and the process of POLE mutation signature are used either in guiding the therapeutic decision and predicting the survival outcome [44,45], or in studying the colorectal tumorigenesis [46-48]. In addition, metastatic CRCs with MSI were found to have a better response to immune checkpoint inhibitors [49]. Although there is not enough information from our current data to figure out the consequence of the mutation process, but we would like to demonstrate the potential of mutational signature analysis in studying the tumorigenesis.
Additionally, the Signature 22 as “exposures to aristolochic acid” provides the contribution at the level of 0.084 in this study but not found in the COSMIC CRC database. It makes sense because the exposures of aristolochic acid are a critical issue in Taiwan and almost one third Taiwanese were caught in the exposures of aristolochic acid [50]. Further studies with the comparison in different populations may provide the evidence to study the relationship between aristolochic acid exposure and higher incidence of CRC in Taiwan. We do not exclude the possibility that the prediction accuracy of our CRN predictive model might be affected by ethnicity and environmental agents. Whether this machine learning-based CRN detection is suitable for Caucasian population remains to further investigation. Lastly, as current knowledge as we know, the CH is correlated with several cancers. However, it is not currently available to access the ECS-based CH profiles from public database and to investigate the similarity of CH mutations between different types of cancers. For this reason, it is limited to accessing whether the model is capable of distinguishing early-staged CRNs from other types of cancers.
In conclusion, we establish a liquid biopsy-based non-invasive early-stage CRN detection by improved NGS technique and machine learning algorithm for decoding the information from CH in CRN patients. The use of this CH-based blood test is still limited in clinical practice. Although further prospective studies with larger sample size would be needed to clarify the clinical effectiveness, the present study has demonstrated the potential of CH for early cancer diagnosis and helps to decode the aberration of CH for imprecating the microenvironment in disease.
Acknowledgements
We acknowledge the technical support from Pharmacogenomics Laboratory of National Core Facility for Biopharmaceuticals (NCFB), the NGS and Microarray Core Facility of NTU Centers of Genomic and Precision Medicine. This work was supported by grants from Pharmacogenomics Laboratory of NCFB and Center of Precision Medicine’ from The Featured Areas Research Center Program and by the Ministry of Science and Technology (MOST108-2319-B-002-001, MOST-109-2634-F-002-043).
Disclosure of conflict of interest
None.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Zauber AG, Winawer SJ, O’Brien MJ, Lansdorp-Vogelaar I, van Ballegooijen M, Hankey BF, Shi W, Bond JH, Schapiro M, Panish JF, Stewart ET, Waye JD. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N Engl J Med. 2012;366:687–696. doi: 10.1056/NEJMoa1100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh H, Nugent Z, Demers AA, Kliewer EV, Mahmud SM, Bernstein CN. The reduction in colorectal cancer mortality after colonoscopy varies by site of the cancer. Gastroenterology. 2010;139:1128–1137. doi: 10.1053/j.gastro.2010.06.052. [DOI] [PubMed] [Google Scholar]
- 4.Kahi CJ, Imperiale TF, Juliar BE, Rex DK. Effect of screening colonoscopy on colorectal cancer incidence and mortality. Clin Gastroenterol Hepatol. 2009;7:770–775. doi: 10.1016/j.cgh.2008.12.030. quiz 711. [DOI] [PubMed] [Google Scholar]
- 5.Knudsen AB, Zauber AG, Rutter CM, Naber SK, Doria-Rose VP, Pabiniak C, Johanson C, Fischer SE, Lansdorp-Vogelaar I, Kuntz KM. Estimation of benefits, burden, and harms of colorectal cancer screening strategies: modeling study for the US Preventive Services Task Force. JAMA. 2016;315:2595–2609. doi: 10.1001/jama.2016.6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ladabaum U, Mannalithara A. Comparative effectiveness and cost effectiveness of a multitarget Stool DNA test to screen for colorectal neoplasia. Gastroenterology. 2016;151:427–439. e426. doi: 10.1053/j.gastro.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, Ahlquist DA, Berger BM. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med. 2014;370:1287–1297. doi: 10.1056/NEJMoa1311194. [DOI] [PubMed] [Google Scholar]
- 8.Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, Fitzgerald RC, Handford PA, Campbell PJ, Saeb-Parsy K, Jones PH. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911–917. doi: 10.1126/science.aau3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, Ozenberger BA, Welch JS, Link DC, Walter MJ, Mardis ER, Dipersio JF, Chen F, Wilson RK, Ley TJ, Ding L. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Challen GA, Goodell MA. Clonal hematopoiesis: mechanisms driving dominance of stem cell clones. Blood. 2020;136:1590–1598. doi: 10.1182/blood.2020006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen KZ, Kazi R, Porter CC, Qu CK. Germline mutations: many roles in leukemogenesis. Curr Opin Hematol. 2020;27:288–293. doi: 10.1097/MOH.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.SanMiguel JM, Young K, Trowbridge JJ. Hand in hand: intrinsic and extrinsic drivers of aging and clonal hematopoiesis. Exp Hematol. 2020;91:1–9. doi: 10.1016/j.exphem.2020.09.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Severson EA, Riedlinger GM, Connelly CF, Vergilio JA, Goldfinger M, Ramkissoon S, Frampton GM, Ross JS, Fratella-Calabrese A, Gay L, Ali S, Miller V, Elvin J, Hadigol M, Hirshfield KM, Rodriguez-Rodriguez L, Ganesan S, Khiabanian H. Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood. 2018;131:2501–2505. doi: 10.1182/blood-2018-03-840629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018;2:3404–3410. doi: 10.1182/bloodadvances.2018020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swierczek SI, Agarwal N, Nussenzveig RH, Rothstein G, Wilson A, Artz A, Prchal JT. Hematopoiesis is not clonal in healthy elderly women. Blood. 2008;112:3186–3193. doi: 10.1182/blood-2008-03-143925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, Balasis ME, Mesa TE, Sallman DA, Lancet JE, Komrokji RS, List AF, McLeod HL, Alsina M, Baz R, Shain KH, Rollison DE, Padron E. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18:112–121. doi: 10.1016/S1470-2045(16)30627-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ptashkin RN, Mandelker DL, Coombs CC, Bolton K, Yelskaya Z, Hyman DM, Solit DB, Baselga J, Arcila ME, Ladanyi M, Zhang L, Levine RL, Berger MF, Zehir A. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol. 2018;4:1589–1593. doi: 10.1001/jamaoncol.2018.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M, Höglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF, Sklar P, Grönberg H, Hultman CM, McCarroll SA. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, Campbell PJ. Universal patterns of selection in cancer and somatic tissues. Cell. 2018;173:1823. doi: 10.1016/j.cell.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coombs CC, Gillis NK, Tan X, Berg JS, Ball M, Balasis ME, Montgomery ND, Bolton KL, Parker JS, Mesa TE, Yoder SJ, Hayward MC, Patel NM, Richards KL, Walko CM, Knepper TC, Soper JT, Weiss J, Grilley-Olson JE, Kim WY, Earp HS 3rd, Levine RL, Papaemmanuil E, Zehir A, Hayes DN, Padron E. Identification of clonal hematopoiesis mutations in solid tumor patients undergoing unpaired next-generation sequencing assays. Clin Cancer Res. 2018;24:5918–5924. doi: 10.1158/1078-0432.CCR-18-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, Schmid T, Brüne B, Wagner S, Serve H, Hoffmann J, Seeger F, Dimmeler S, Zeiher AM, Rieger MA. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4:25–33. doi: 10.1001/jamacardio.2018.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, Duyvesteyn K, Haidari S, van Hoeck A, Onstenk W, Roepman P, Voda M, Bloemendal HJ, Tjan-Heijnen VCG, van Herpen CML, Labots M, Witteveen PO, Smit EF, Sleijfer S, Voest EE, Cuppen E. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575:210–216. doi: 10.1038/s41586-019-1689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, Samuel M, Ritchie EK, Guzman ML, Ballman KV, Roboz GJ, Hassane DC. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018;24:1015–1023. doi: 10.1038/s41591-018-0081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, Barda N, Zuzarte PC, Heisler L, Sundaravadanam Y, Luben R, Hayat S, Wang TT, Zhao Z, Cirlan I, Pugh TJ, Soave D, Ng K, Latimer C, Hardy C, Raine K, Jones D, Hoult D, Britten A, McPherson JD, Johansson M, Mbabaali F, Eagles J, Miller JK, Pasternack D, Timms L, Krzyzanowski P, Awadalla P, Costa R, Segal E, Bratman SV, Beer P, Behjati S, Martincorena I, Wang JCY, Bowles KM, Quirós JR, Karakatsani A, La Vecchia C, Trichopoulou A, Salamanca-Fernández E, Huerta JM, Barricarte A, Travis RC, Tumino R, Masala G, Boeing H, Panico S, Kaaks R, Krämer A, Sieri S, Riboli E, Vineis P, Foll M, McKay J, Polidoro S, Sala N, Khaw KT, Vermeulen R, Campbell PJ, Papaemmanuil E, Minden MD, Tanay A, Balicer RD, Wareham NJ, Gerstung M, Dick JE, Brennan P, Vassiliou GS, Shlush LI. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559:400–404. doi: 10.1038/s41586-018-0317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, Patel M, Berthon A, Syed A, Yabe M, Coombs CC, Caltabellotta NM, Walsh M, Offit K, Stadler Z, Mandelker D, Schulman J, Patel A, Philip J, Bernard E, Gundem G, Ossa JEA, Levine M, Martinez JSM, Farnoud N, Glodzik D, Li S, Robson ME, Lee C, Pharoah PDP, Stopsack KH, Spitzer B, Mantha S, Fagin J, Boucai L, Gibson CJ, Ebert BL, Young AL, Druley T, Takahashi K, Gillis N, Ball M, Padron E, Hyman DM, Baselga J, Norton L, Gardos S, Klimek VM, Scher H, Bajorin D, Paraiso E, Benayed R, Arcila ME, Ladanyi M, Solit DB, Berger MF, Tallman M, Garcia-Closas M, Chatterjee N, Diaz LA Jr, Levine RL, Morton LM, Zehir A, Papaemmanuil E. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52:1219–1226. doi: 10.1038/s41588-020-00710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, Hyman DM, Solit DB, Robson ME, Baselga J, Arcila ME, Ladanyi M, Tallman MS, Levine RL, Berger MF. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21:374–382. e374. doi: 10.1016/j.stem.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong WH, Bhatt S, Trinkaus K, Pusic I, Elliott K, Mahajan N, Wan F, Switzer GE, Confer DL, DiPersio J, Pulsipher MA, Shah NN, Sees J, Bystry A, Blundell JR, Shaw BE, Druley TE. Engraftment of rare, pathogenic donor hematopoietic mutations in unrelated hematopoietic stem cell transplantation. Sci Transl Med. 2020;12:eaax6249. doi: 10.1126/scitranslmed.aax6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crowgey EL, Mahajan N, Wong WH, Gopalakrishnapillai A, Barwe SP, Kolb EA, Druley TE. Error-corrected sequencing strategies enable comprehensive detection of leukemic mutations relevant for diagnosis and minimal residual disease monitoring. BMC Medical Genomics. 2020;13:32. doi: 10.1186/s12920-020-0671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abbosh C, Swanton C, Birkbak NJ. Clonal haematopoiesis: a source of biological noise in cell-free DNA analyses. Ann Oncol. 2019;30:358–359. doi: 10.1093/annonc/mdy552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Yang J, Wang J. Leverage large-scale biological networks to decipher the genetic basis of human diseases using machine learning. Methods Mol Biol. 2021;2190:229–248. doi: 10.1007/978-1-0716-0826-5_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang B, Mu Q, Qiu F, Li X, Xu W, Yu J, Fu W, Cao Y, Wang J. Machine learning of genomic features in organotropic metastases stratifies progression risk of primary tumors. Nat Commun. 2021;12:6692. doi: 10.1038/s41467-021-27017-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Díaz-Gay M, Vila-Casadesús M, Franch-Expósito S, Hernández-Illán E, Lozano JJ, Castellví-Bel S. Mutational signatures in cancer (MuSiCa): a web application to implement mutational signatures analysis in cancer samples. BMC Bioinformatics. 2018;19:224. doi: 10.1186/s12859-018-2234-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carethers JM, Jung BH. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology. 2015;149:1177–1190. e1173. doi: 10.1053/j.gastro.2015.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stadler ZK. Diagnosis and management of DNA mismatch repair-deficient colorectal cancer. Hematol Oncol Clin North Am. 2015;29:29–41. doi: 10.1016/j.hoc.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Gao T, Ptashkin R, Bolton KL, Sirenko M, Fong C, Spitzer B, Menghrajani K, Ossa JEA, Zhou Y, Bernard E, Levine M, Martinez JSM, Zhang Y, Franch-Expósito S, Patel M, Braunstein LZ, Kelly D, Yabe M, Benayed R, Caltabellotta NM, Philip J, Paraiso E, Mantha S, Solit DB, Diaz LA Jr, Berger MF, Klimek V, Levine RL, Zehir A, Devlin SM, Papaemmanuil E. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nat Commun. 2021;12:338. doi: 10.1038/s41467-020-20565-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, Abida W, Juluru K, De Bruijn I, Hou C, Venn O, Lim R, Anand A, Maddala T, Gnerre S, Vijaya Satya R, Liu Q, Shen L, Eattock N, Yue J, Blocker AW, Lee M, Sehnert A, Xu H, Hall MP, Santiago-Zayas A, Novotny WF, Isbell JM, Rusch VW, Plitas G, Heerdt AS, Ladanyi M, Hyman DM, Jones DR, Morrow M, Riely GJ, Scher HI, Rudin CM, Robson ME, Diaz LA Jr, Solit DB, Aravanis AM, Reis-Filho JS. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25:1928–1937. doi: 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan HT, Nagayama S, Chin YM, Otaki M, Hayashi R, Kiyotani K, Fukunaga Y, Ueno M, Nakamura Y, Low SK. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol Oncol. 2020;14:1719–1730. doi: 10.1002/1878-0261.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.AACR Project GENIE Consortium. AACR Project GENIE: powering precision medicine through an International Consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dolcetti R, Viel A, Doglioni C, Russo A, Guidoboni M, Capozzi E, Vecchiato N, Macrì E, Fornasarig M, Boiocchi M. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathol. 1999;154:1805–1813. doi: 10.1016/S0002-9440(10)65436-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinicrope FA, Shi Q, Smyrk TC, Thibodeau SN, Dienstmann R, Guinney J, Bot BM, Tejpar S, Delorenzi M, Goldberg RM, Mahoney M, Sargent DJ, Alberts SR. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99. doi: 10.1053/j.gastro.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, Sinicrope FA, Rosty C, Buchanan DD, Potter JD, Newcomb PA. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77–87. e72. doi: 10.1053/j.gastro.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Temko D, Van Gool IC, Rayner E, Glaire M, Makino S, Brown M, Chegwidden L, Palles C, Depreeuw J, Beggs A, Stathopoulou C, Mason J, Baker AM, Williams M, Cerundolo V, Rei M, Taylor JC, Schuh A, Ahmed A, Amant F, Lambrechts D, Smit VT, Bosse T, Graham TA, Church DN, Tomlinson I. Somatic POLE exonuclease domain mutations are early events in sporadic endometrial and colorectal carcinogenesis, determining driver mutational landscape, clonal neoantigen burden and immune response. J Pathol. 2018;245:283–296. doi: 10.1002/path.5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castellsagué E, Li R, Aligue R, González S, Sanz J, Martin E, Velasco À, Capellá G, Stewart CJR, Vidal A, Majewski J, Rivera B, Polak P, Matias-Guiu X, Brunet J, Foulkes WD. Novel POLE pathogenic germline variant in a family with multiple primary tumors results in distinct mutational signatures. Hum Mutat. 2019;40:36–41. doi: 10.1002/humu.23676. [DOI] [PubMed] [Google Scholar]
- 48.Sun J, Wang C, Zhang Y, Xu L, Fang W, Zhu Y, Zheng Y, Chen X, Xie X, Hu X, Hu W, Zheng J, Li P, Yu J, Mei Z, Cai X, Wang B, Hu Z, Shu Y, Shen H, Gu Y. Genomic signatures reveal DNA damage response deficiency in colorectal cancer brain metastases. Nat Commun. 2019;10:3190. doi: 10.1038/s41467-019-10987-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ng AWT, Poon SL, Huang MN, Lim JQ, Boot A, Yu W, Suzuki Y, Thangaraju S, Ng CCY, Tan P, Pang ST, Huang HY, Yu MC, Lee PH, Hsieh SY, Chang AY, Teh BT, Rozen SG. Aristolochic acids and their derivatives are widely implicated in liver cancers in Taiwan and throughout Asia. Sci Transl Med. 2017;9:eaan6446. doi: 10.1126/scitranslmed.aan6446. [DOI] [PubMed] [Google Scholar]