

SUMMARY
Bifurcation of cellular fates, a critical process in development, requires histone 3 lysine 27 methylation (H3K27me3) marks propagated by the polycomb repressive complex 2 (PRC2). However, precise chromatin loci of functional H3K27me3 marks are not yet known. Here, we identify critical PRC2 functional sites at high resolution. We fused a computationally designed protein, EED binder (EB), which competes with EZH2 and thereby inhibits PRC2 function, to dCas9 (EBdCas9) to allow for PRC2 inhibition at a precise locus using gRNA. Targeting EBdCas9 to four different genes (TBX18, p16, CDX2, and GATA3) results in precise H3K27me3 and EZH2 reduction, gene activation, and functional outcomes in the cell cycle (p16) or trophoblast transdifferentiation (CDX2 and GATA3). In the case of TBX18, we identify a PRC2-controlled, functional TATA box >500 bp upstream of the TBX18 transcription start site (TSS) using EBdCas9. Deletion of this TATA box eliminates EBdCas9-dependent TATA binding protein (TBP) recruitment and transcriptional activation. EBdCas9 technology may provide a broadly applicable tool for epigenomic control of gene regulation.
Graphical Abstract
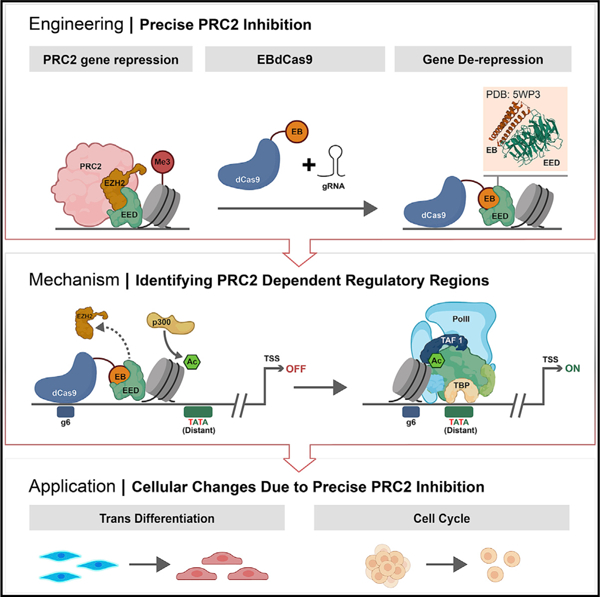
In brief
Levy et al. fused a computationally designed protein, EED binder (EB), which competes with EZH2 and thereby inhibits PRC2 function, to dCas9 (EBdCas9). EBdCas9 represses PRC2 action in precise loci, remodels epigenomic marks, exposes transcriptional elements, and induces transdifferentiation.
INTRODUCTION
Chromatin modification mediated by the polycomb repressive complex 2 (PRC2) plays a critical role in developmental transitions. However, the specific genetic loci at which PRC2 function is critical for determining cell fate has remained unknown, as there has been no way to target PRC2 inhibition to specific chromosomal locations. A central question in epigenomics and developmental biology is the role of PRC2-dependent specific histone 3 lysine 27 methylation (H3K27me3) marks in cell fate decisions (Margueron and Reinberg, 2011). While broad upstream regions of developmental genes are decorated with H3K27me3 marks, it is not known which, if any, single nucleosomes require H3K27me3 marks for gene repression and cell-fate determination.
The two main complexes involved in polycomb-based repression are PRC1 and PRC2. PRC1 catalyzes monoubiquitylation of histone H2A Lys 119 (H2AK119ub), while PRC2 catalyzes the mono-, di-, and trimethylation of histone H3 Lys 27 (H3K27me1/me2/me3) (Lee et al., 2019). Recent structural analysis has revealed the mechanism of PRC1/PRC2 spatiotemporal regulation and H3K27me3 mark spreading (Banaszynski et al., 2013; Laugesen et al., 2016). Non-canonical PRC1 containing RYBP ubiquitinates H2K119 to recruit accessory proteins JARID2 and MTF2 (Coleman and Struhl, 2017; Laprell et al., 2017; Yu et al., 2019). JARID2 recruits PRC2 and contributes to the allosteric activation of PRC2 (Lee et al., 2018). PRC2 containing EED and EZH2 facilitates H3K27me3 marks by reading (EED) and writing/catalyzing (EZH2) new marks. H3K27me3 marks are then read by canonical PRC1 containing CBX, resulting in chromatin compaction, H2AK119ub, and gene repression (Brockdorff, 2017; Holoch and Margueron, 2017; Francis et al., 2009; Eskeland et al., 2010; Illingworth et al., 2015; Pengelly et al., 2015; Oksuz et al., 2018). During developmental transitions, the chromatin undergoes extensive epigenetic remodeling at the promoter and enhancer regions, where H3K27me3 marks are added or removed to produce the required decrease or increase in gene expression (Hawkins et al., 2010; Battle et al., 2019). Importantly, only the repressive marks, but not the activating histone tail marks, are instrumental for histone mark inheritance during development (Escobar et al., 2019). Recent studies suggest that proximal regulatory marks governing gene expression are associated with histone modification domains within 2.5 kb of known transcriptional start sites (Woo Jun Shim et al., 2019). However, it is not known if any specific H3K27me3-marked nucleosomes are critical for control of gene expression or if the entire broad 2.5 kb region is required for gene repression. This issue has been challenging to address as existing genetic and chemical methods eliminate all H3K27me3 marks without precision.
Recent studies that fuse catalytically dead Cas9 (dCas9) to proteins that promote or remove histone methyl groups provide tools for manipulating the epigenome. For example, the repressor KRAB recruits methyltransferases to increase H3K9me3 repression levels (Pengue and Lania, 1996; Groner et al., 2010), and its fusion to dCas9 results in 50%–99% gene repression in various cell lines, primary cultures, and pluripotent stem cells (Gao et al., 2013; Kearns et al., 2015; Shechner et al., 2015; THAKORE et al., 2015; Amabile et al., 2016; Pradeepa et al., 2016). Fusion of dCas9 to the transcriptional activators VP16, VP64, and VP65 promotes chromatin de-condensation, accumulation of H3K27ac and H3K4me3 marks, RNA polymerase II (RNA Pol II) recruitment, and transcriptional activation (Chavez et al., 2016; Gilbert et al., 2014). Likewise, gene expression was found to be regulated following the recruitment of histone acetyltransferase dCas9-p300 (Chen et al., 2019; Hilton et al., 2015). Similarly, fusion of dCas9 to the histone demethylase LSD1/KDM1A results in H3K4 and H3K9 methyl group removal and affects embryonic stem cell (ESC) self-renewal, differentiation, cancer cell proliferation, and development (Kearns et al., 2015; Adamo et al., 2011; Goodman and Smolik, 2000).
Despite the considerable array of tools available to modify the epigenome, currently there is no method to inhibit PRC2 function at a particular genomic locus and precisely at a single nucleosome in order to determine which specific H3K27me3 marks result in transcriptional repression. We have generated a computer-designed protein that binds EED (EED binder [EB]) and competes with EZH2 (Moody et al., 2017). By fusing EB to dCas9, we enable probing H3K27me3 function at precise gene loci. Here, we show that EBdCas9/gRNA is able to affect the PRC2 complex, remodel the histone marks of specific target sites, upregulate genes of interest, and promote epigenomic memory. We reveal precise loci for PRC2 requirement for transcription repression; in the TBX18 gene, PRC2 activity normally represses the distal TATA box region. Using EBdCas9, we show that specific H3K27me3 marks are required for inner cell mass (ICM) versus trophectoderm (TE) cell-fate decision (Saha et al., 2013; Xiao et al., 2020) and proliferation control of the lethal pediatric CNS tumor diffuse midline glioma (DMG). These data suggest that EBdCas9 technology is broadly applicable for the regulation of histone marks at a single locus to control gene expression in different cell types.
RESULTS
EBdCas9/gRNA activates TBX18 transcription
The catalytic and substrate recognition functions of PRC2, mediated by the SET domain containing EZH2 subunit and the trimethyl lysinbinding EED subunit, respectively, are coupled by binding of the N-terminal helix of EZH2 to an extended groove on EED (Kim et al., 2013). Disrupting PRC2 function can be achieved by chemical drugs or knocking out key PRC2 components (Kong et al., 2014; Knutson et al., 2013). We previously generated and characterized a computationally designed protein (EB) that binds to the EZH2 binding site on EED by two orders of magnitude higher affinity than EZH2 (Moody et al., 2017). As a control, we created an EB negative control (NC) where two amino acid mutations, F47E and I54E on the EED binding interface, abolish binding to EED (Moody et al., 2017). EB, but not NC, with subnanomolar affinity, forms tight complexes with EED, reduces EZH2 and JARID2 global levels, and exhibits a significant genome-wide reduction of H3K27me3 repressive marks in promoter regions (Moody et al., 2017).
To target EB to a specific chromatin locus, we fused EB to dCas9 (EBdCas9). This enables disruption of PRC2 function in specific regions within H3K27me3 domains to determine which, if any, control gene expression (Figure 1A). We fused EB to dCas9 with a 30 aa residue 6 × 5 (SGGGG) linker under the control of the AAVS1-TREG inducible promoter of the dCas9-NLS-2A-mCherry plasmid (Mandegar et al., 2016) (Figure 1B). Similarly, we fused the NC control to dCas9 to distinguish between dCas9 non-specific effects on chromatin and EB-specific effects on PRC2. The EB-linker-dCas9-NLS-2A-mCherry (EBdCas9) and NC-linker-dCas9-NLS-2A-mCherry (NCdCas9) constructs were transfected to induced pluripotent stem cells (iPSCs) (WTC) using TALENS to enforce recombinant homology at the AAVS1 site on chromosome 19. Following antibiotic selection, EBdCas9 and NCdCas9 mCherry expression were doxycycline (Dox) inducible, and stem cell morphology was maintained with no changes in EZH2 and H3K27me3 levels (Figures 1C, 1D, and S1A–S1D; Table S1) or gene expression in general (Figures S1E and S1F). Expression of EB alone has more global effects; the difference may reflect the 50-fold lower expression of EBdCas9 (Figure S1G) or the steric hindrance of the EB-EED interaction by the fused dCas9 when not bound to DNA (Cofsky Joshua et al., 2021) (colocalization of EED and EBdCas9 increases 2.7 × in the presence of +gRNA versus –gRNA [Figures S1H and S1I]).
Figure 1. EBdCas9 upregulates TBX18 expression in precise loci.
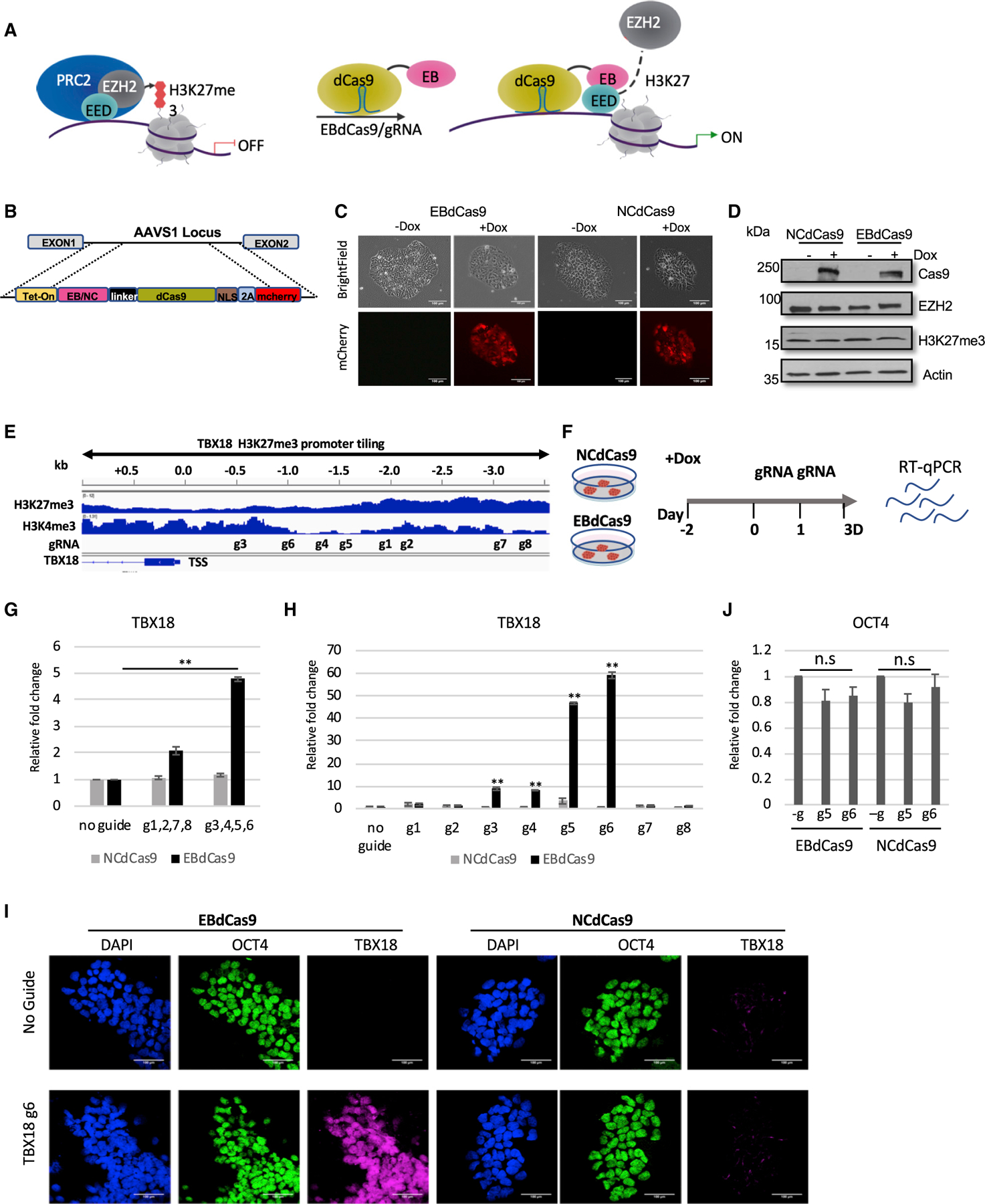
(A) Model of EBdCas9 precise elimination of PRC2 activity in targeted loci.
(B) EBdCas9-mCherry and NCdCas9-mCherry construct under Tet-On operator.
(C) Generation of stable EBdCas9 or NCdCas9 transgenic iPSC (WTC) lines after 3 days of induction of (2 µg/ml) doxycycline (Dox).
(D) Immunoblot analysis of EBdCas9 and NCdCas9 whole-cell lysate after 3 days of Dox induction.
(E) Integrative genomic viewer of TBX18 H3K27me3 and H3K4me3 promoter tiling.
(F) Timeline of EBdCas9 or NCdCas9 induction and gRNA transfection.
(G and H) qRT-PCR analysis of TBX18 expression of EBdCas9 and NCdCas9 normalized to β-actin and calculated as the relative fold change compared with no guide (induced with Dox) of each respected cell line (G) after TBX18 gRNAs cocktail transfection; g1, g2, g7, and g8 or g3, g4, g5, and g6 (H) after individual TBX18 gRNA (1–8) transfection (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(I) Immunofluorescent imaging of EBdCas9 WTC and NCdCas9 WTC for either no guide or after transfection with TBX18 gRNA 6 (TBX18 g6). Blue, DAPI; green, OCT4; purple, TBX18; scale bar, 100 µm.
(J) qRT-PCR analysis of OCT4 after individual transfection of TBX18 gRNA g5 and g6.
We analyzed EBdCas9 or NCdCas9 in genes that were responsive to previous EB treatment (Moody et al., 2017). TBX18 is a growth-promoting transcription factor required for embryonic development and the conversion of myocytes into sinoatrial cells (Kapoor et al., 2013; Wiese et al., 2009). TBX18 was upregulated with reduced H3K27me3 marks after EB expression (Figures 1E and S2A) (Moody et al., 2017). In iPSCs, the TBX18 promoter is bivalent—the upstream region is simultaneously decorated with H3K27me3 repressive marks and H3K4me3 active marks. We tiled the TBX18 upstream promoter region, based on UCSC Genome Browser (GRCh36/hg36) Assembly, with guides specific to the region using CRISPRscan (Moreno-mateos et al., 2015). Guides were used with EBdcas9 and NCdCas9 to screen for loci sensitive for PRC2 (Figure 1E). EBdCas9 was induced at day —2 using Dox and transfected with in-vitro-synthesized gRNA on days 0 and 1, and the cells were collected on day 3 (Figure 1F).
To identify upstream regions that are more sensitive to EB action, we transfected the cells with a combination of guides. While guides 1.9–3.5 kb upstream from transcription start sites (TSSs) (guide 1 [g1], g2, g7, and g8) show no noticeable effects in TBX18 transcription, guides 0–1.9 kb upstream of TSSs (g3, g4, g5, and g6) showed 5-fold TBX18 upregulation compared with no guide treatment or with the NCdCas9 control (Figure 1G). Induction of EBdCas9 with individual TBX18 gRNAs increased TBX18 transcript levels by 10-fold (gRNAs 3 and 4) and 50–60 fold (gRNAs 5 and 6) compared with EBdCas9 with no guide or with g1,2,7,8 and NCdCas9 or KRABdCas9 (Figures 1H and S2B). TBX18 protein was overexpressed with EBdCas9/g6 but not with NCdCas9/g6 (Figure 1I). gRNAs 3–6 (−0.5 to −1.5 kb) localized to chromatin domains where H3K4me3 marks are low and H3K27me3 marks and EZH2 are enriched (Figures 1E and S2C). To ensure tiled gRNAs have access to targeted TBX18 DNA, we localized EBdCas9 after transfection with TBX18 gRNAs (1–8) using Cas9 antibody (Ab) for chromatin immunoprecipitation (ChIP)-qPCR (Figure S2D). To test the specificity of EBdCas9, we monitored POU5F1 (OCT4) gene expression and observed no significant changes in OCT4 in cells treated with gRNAs targeting the TBX18 promoter (Figure 1J); hence, TBX18 upregulation is not a secondary effect of cellular differentiation. Assay for transposase-accessible chromatin sequencing (ATAC-seq) analysis of EBdCas9/g6- or NCdCas9/g6-transfected cells resulted in similar open chromatin regions. Since no significant changes in chromatin accessibility were detected, we conclude that transcript upregulation is dependent on local changes in the PRC2 complex (Figure S2A) and that the process is PRC2-dependent since EBdCas9, but not NCdCas9, activates TBX18 gene expression at the precise loci.
EBdCas9/gRNA remodels epigenomic marks and memory in TBX18 locus
To dissect the mechanism of EBdCas9 action at precise genomic loci, we used ChIP-qPCR (PIXUL-Matrix-ChIP [Bomsztyk et al., 2019]). As noted above, EBdCas9 or NCdCas9 expression was induced in iPSCs (WTC) using Dox followed by TBX18 gRNA6 (g6) transfections on days 0 and 1 and analysis on day 3 by qRT-PCR and ChIP-qPCR (Figure 2A). As expected, while both constructs showed similar dCas9 expression, only EBdCas9/g6 showed a significant increase of TBX18 transcription (Figure 2B). Using the ChIP-qPCR method, we quantified the abundance of EB/NCdCas9 (Cas9), histone 3 with K27me3 marks (H3K27me3), and EZH2 proteins at the TBX18 g6 region by generating and quantifying the 150 bp amplicon flanking the g6 chromatin region. ChIP-qPCR using a Cas9 Ab confirmed that both EBdCas9 and NCdCas9 are recruited to the g6 locus (Figure 2C). However, only EBdCas9 results in a reduction of EZH2 (Figure 2C), most likely by directly blocking EZH2 binding to EED (Moody et al., 2017). This resulted in a reduction of H3K27me3 marks in EBdCas9/g6 at the g6 locus (Figure 2C). In contrast, while the ChIP-qPCR assay of EBdCas9/g7 confirmed recruitment of Cas9 and reductions of EZH2 and H3K27me3 marks at the g7 locus, TBX18 transcription was not affected (Figures 1H, 2D–2E, and S3A). Thus, while EBdCas9 is able to disrupt EED-EZH2 interactions at many loci, transcription is only affected at certain locations.
Figure 2. EBdCas9 precisely eliminates EZH2 from the PRC2 complex, remodels epigenomic marks, and actuates epigenomic memory on TBX18 locus.
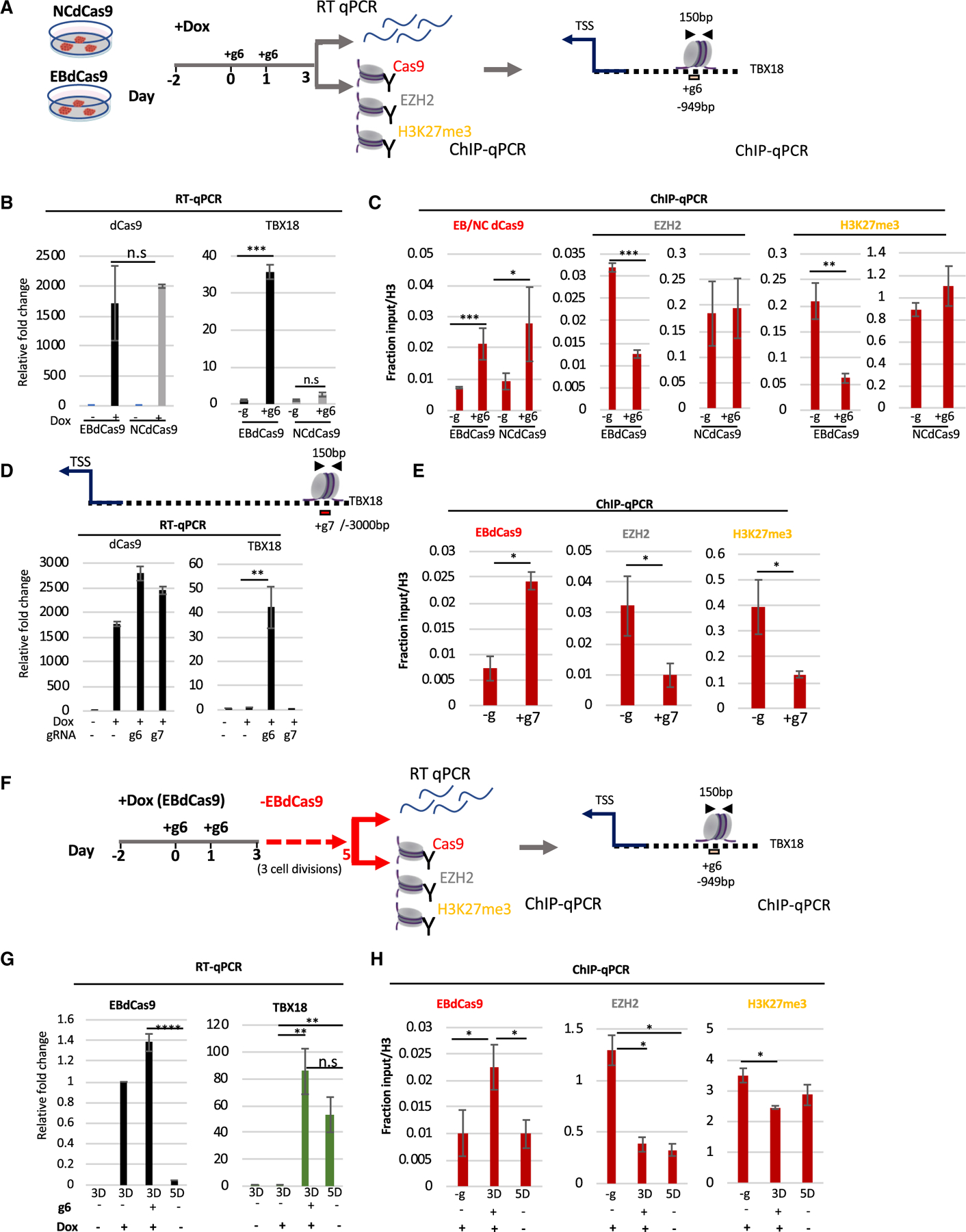
(A) EBdCas9 and NCdCas9 timeline for Dox induction, gRNA transfection, and analysis: qRT-PCR or ChIP-qPCR using the antibodies Cas9, EZH2, and H3K27me3 and analyzing TBX18 g6 DNA region ~1.0 kb upstream of the TSS (150 bp).
(B) qRT-PCR of dCas9 or TBX18 relative fold change after 3 days of Dox induction (dCas9) or 3 days of TBX18 g6 RNA transfection (TBX18) normalized to β-actin and compared with no guide (Dox induced for TBX18) of each respective cell line.
(C) ChIP-qPCR of induced (+Dox) EBdCas9 and NCdCas9 after 3 days of transfection with TBX18 g6 RNA (+g6) or no transfection (–g). Normalized as fraction of input/H3. Antibodies for ChIP are listed above the graphs, and the genomic region analyzed by qPCR includes TBX18 g6 locus (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(D) qRT-PCR of dCas9 and TBX18 relative fold change after 3 days of Dox induction and TBX18 g6 RNA (+g6) or g7 RNA (+g7) transfection normalized to β-Actin and compared with no guide (Dox induced) (–g) exactly as described in (A).
(E) ChIP-qPCR of induced (+Dox) EBdCas9 after 3 days of transfection with TBX18 g7 RNA (+g7) or no transfection (–g). Normalized as fractioned of input/H3 and analyzed exactly as in (C).
(F) EBdCas9 timeline for measuring memory. EBdCas9 induction and TBX18 g6 RNA transfection exactly as in (A). On day 3, EBdCas9 media was replaced with no Dox (–EBdCas9) for 2 days and analyzed on day 5 for qRT-PCR and ChIP-qPCR.
(G) qRT-PCR analysis of EBdCas9 and TBX18 for 3 or 5 days while inducing with Dox (+) or not (–) and in the presence of TBX18 g6 RNA (g6) (+) or not (–).
(H) ChIP-qPCR of no guide (–g), 3- or 5-day EBdCas9 either induced with Dox (+) or not (–), or transfected with TBX18 g6 RNA (+) or not (–). Normalized to fractioned Input/H3 and analyzed exactly as in (C).
Recent data have shown that PRC2-dependent marks are faithfully inherited during cell division (Escobar et al., 2019). To learn whether EBdCas9-dependent reduction of EZH2 occupancy and H3K27me3 marks are inherited from the cell from which they descended (epigenomic memory), we repeated the assay as before but allowed the cells to grow for an additional 2 days (3 cell cycles; Figure S3B) after eliminating EBdCas9/gRNA expression (Figure 2F). qRT-PCR shows that the EBdCas9 transcript is upregulated at day 3 post-transfection but is not detected 2 days after elimination of induction (day 5) (Figure 2G). In contrast, TBX18 transcription increased 80-fold at day 3 and 50-fold at day 5 (Figure 2G). To test if TBX18 transcription 2 days after EBdCas9 elimination correlated with epigenomic memory, we performed PIXUL-ChIP for the EZH2 protein and H3K27me3 marks at the g6 region at 3 and 5 days. ChIP-qPCR showed a depletion of both H3K27me3 and EZH2 at day 3, as expected. Importantly, this reduction of EZH2 and H3K27me3 persisted until day 5 (Figure 2H), showing an epigenomic inheritance of PRC2 elimination in the g6 region. These data reveal that EBdCas9 not only remodels the epigenome but also leads to inheritance of the changes.
Spreading of EBdCas9 induced epigenomic changes
To learn whether (1) EBdCas9/TBX18g6 epigenomic changes are limited to the g6 chromatin locus and (2) the spread chromatin changes are inherited upon cell division, we tiled the TBX18 genomic area with primer sets between the TSS and g6 and upstream of g6. As expected, at 3, but not at 5, days, EBdCas9 (Cas9) is localized to the g6 region (Figure 3A). At neither time point was EBdCas9 observed in any other loci upstream of the TBX18 TSS. However, the EZH2 protein was significantly downregulated not only at g6 but also in loci between the TSS and g6 both at day 3 and 5 time points (Figure 3B). Since EBdCas9 was specifically localized to the g6 region at day 3, but not at day 5, EZH2 reduction in these regions suggest that PRC2 disruption by EBdCas9 at g6 at day 3 (1) spreads toward the TSS and (2) is inherited during cell division. Accordingly, disruption of the PRC2 complex also resulted in an ongoing reduction of H3K27me3 marks between the TSS and g6 (Figure 3C). The PRC2 component, JARID2, showed reduced binding to chromatin between the TSS and g6 at the day 3 time point (Figure 3D). Since JARID2 is critical for PRC2 homing and the stimulation of EZH2 catalytic activity, its reduction in this region is a further indication of reduced PRC2 activity (Kasinath et al., 2018). EED, on the other hand, binds to EBdCas9 and remains at g6 at day 3; however, it is depleted between the TSS and g6 (Figure 3D). Thus, PRC2 disruption and consequent reduction of H3K27me3 histone marks spread from g6 toward the TSS, and these changes persist after multiple cell divisions.
Figure 3. EBdCas9 causes epigenomic spreading and recruits activation marks.
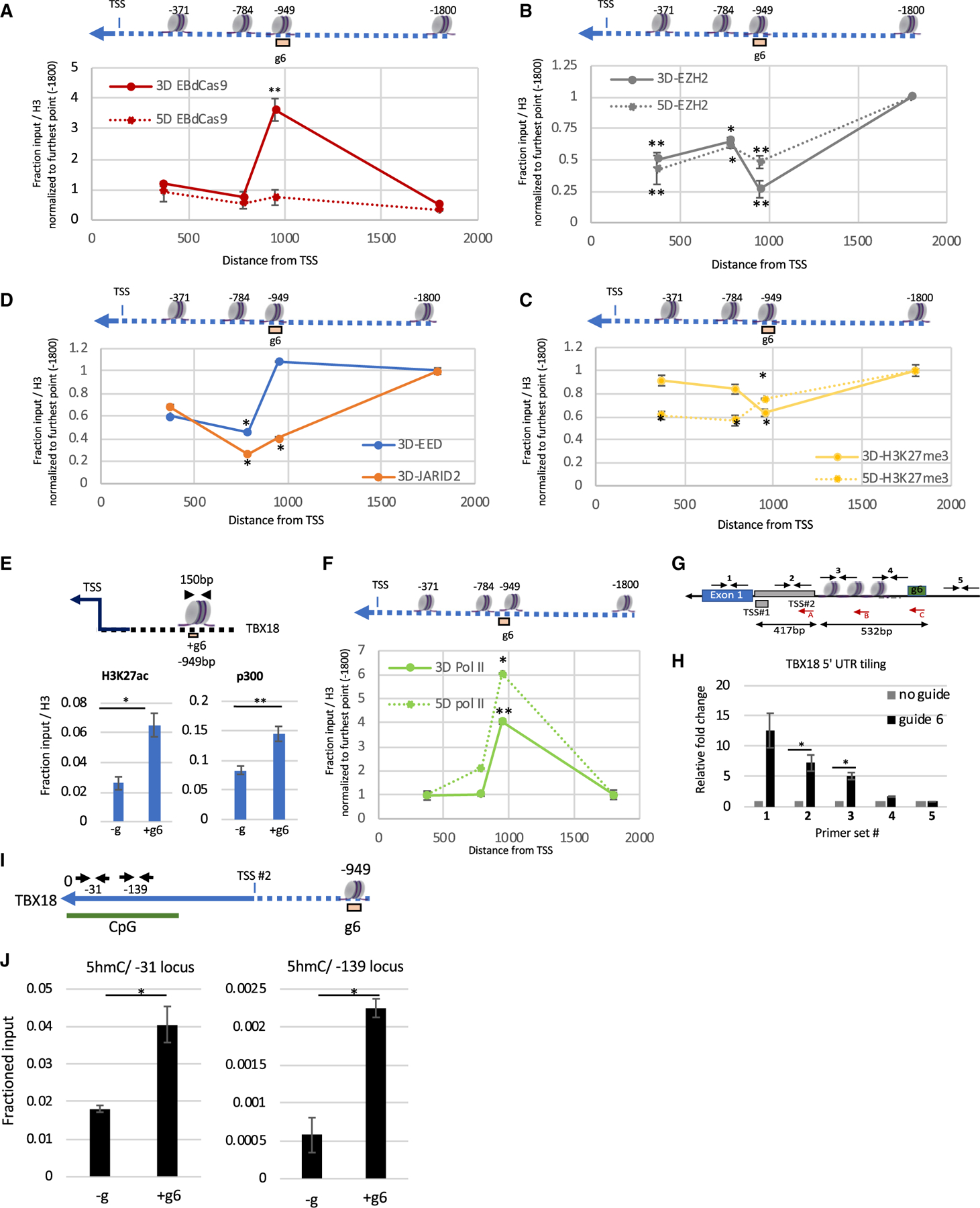
(A–D) Tiling of EBdCas9/+g6 ChIP (A) Cas9, (B) EZH2, (C)H3K27me3, (D) EED, and JARID 2 on TBX18 genomic loci (bp) relative to the TSS (listed above nucleosome) using qPCR; solid red lines are day 3 and dashed red lines are day 5. Each point is the relative fold change TBX18 g6RNA (+g6) versus no guide (–g), normalized to the respective fraction of input/H3 and compared with the relative fold change using −1,800 bp primer set as control (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(E) ChIP-qPCR of 3-days-induced EBdCas9 and TBX18 g6 RNA (+g6) transfected or not (–g) using H3K27ac and p300 and normalized as a fraction of input/H3. Genomic region analyzed by qPCR is the TBX18 g6 locus and is denoted above (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(F) Tiling of EBdCas9/+g6 ChIP (RNA Pol II CTD) on TBX18 genomic loci (bp) relative to the TSS (listed above nucleosome) using qPCR exactly as in (A); solid green lines are day 3 and dashed green lines are day 5 (as described in A–D). Analyzed exactly as in (A).
(G) Illustration of TBX18 TSS peak position relative to the g6 region in bp.
(A–C) Red arrows denote the reversed primer used for cDNA reverse transcription. Internal black arrows with Arabic numbers denote the PCR amplicon generated following cDNA production: (C) corresponds to primer set #4; (B) corresponds to #3; and (A) corresponds to primer sets #2, #1, and #5.
(H) RNA expression of TBX18 5′UTR region using qRT-PCR (reverse primer in red) after 3 days of transfection with TBX18 g6 RNA (g6) or no transfection (no guide). Regions amplified correspond to #1–#5 and are indicated with black arrows. Normalized to β-actin, and the relative fold change is compared with no guide (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(I) Illustration of TBX18 CpG locus using UCSC Genome Browser (GRCh36/hg36) Assembly. ‘‘0’’ denotes first Met relative to the g6 region in bp. Small black arrows annotate the loci for qPCR amplification in (J).
(J) MedIP-qPCR of induced (+Dox) EBdCas9 after 3 days of transfection with TBX18 g6 RNA (+g6) or no transfection (–g) using 5 hydroxymethylcytosine (5hmC) Abs. Normalized to fractioned input (n = 2, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
To test if the depletion of PRC2 results in epigenomic modifications indicative of transcription activation, we performed ChIP-qPCR using antibodies against H3K27ac, p300, RNA Pol II C-terminal domain (CTD), and RNA Pol II CTD Ser5P (indicative of RNA Pol II pause [Boehm et al., 2003]). We observed recruitment of RNA Pol II, histone acetyltransferase (p300), and acetylation marks (H3K27ac) at the TBX18 g6 locus (Figures 3E and S3C), indicative of active transcription. RNA Pol II was not only highly enriched in the TBX18 g6 locus at the day 3 time point but also at the day 5 time point, 2 days after the EBdCas9 was eliminated (Figure 3F). Since RNA Pol II is recruited to the g6 region, we tested if TBX18 nascent RNA is transcribed in that region. We observed increased nascent RNA amplification in EBdCas9 cells transfected with TBX18g6 compared with no guide using qRT-PCR (Figures 3G and 3H; upregulation on exon 1 but not on the 2.0 kb upstream region). There are two transcription start sites for TBX18 (UCSC Genome Browser GRCh37/hg19); EBdCas9/g6 activates transcription in the TSS#2 region (532 bp downstream of g6; Figures 3G and 3H), which is the dominant start site based on cap analysis gene expression (CAGE)-seq data (Figures S3D and S3E).
To test if DNA methyl marks are affected by EBdCas9/TBX18g6, we performed Matrix-MeDIP (Yu et al., 2011) using 5 methylcytosine (5mC) and 5 hydroxymethylcytosine (5hmC) antibodies (Figure 3I). While EBdCas9/TBX18g6 did not alter methylation (5mC) compared with no guide, it increased hydroxymethylation (5hmC; hydroxymethylations is actively involved in DNA demethylation and transcription activation [Santiago et al., 2014]) (Figure 3J). Tiling regions outside of the CpG island on the TBX18 promoter did not show significant 5hmC changes (Figures S3F–S3H).
EBdCas9 reveals distal TATA box function
Since targeting the TBX18g6 site led to transcription activation, epigenetic remodeling, and epigenomic memory, we analyzed the chromatin region for transcription factor binding sites, core promoter sequence elements, and G-quadruplex patterns (Figures S4A–S4C). Quadruplex forming G-rich sequences (QGRSs) (Kikin et al., 2006) are structure motifs linked to increased transcriptional activity (Hansel-hertsch et al., 2016). In the 1 kb upstream from the TSS, we identified a combination of TATA box and mammalian initiator factor binding sites (INRs) near the TBX18 g6 locus (Figures 4A and S4C; Table S2; TATA box 21 bp and INR 48–57 bp from g6). We tested if this distal TATA box is required for TBX18 transcription by precisely deleting 40 bp surrounding it using CRISPR-Cas9-homology-directed repair (HDR) (Figure 4B). Two independent, pluripotent EBdCas9 cell lines with homozygous TATA box deletions were generated, TATA D #1 and TATA ∆ #2 (see STAR Methods for details) (Figure 4B) and analyzed for TBX18 transcription. In contrast to the significant increase of TBX18 transcription in wild-type EBdCas9/g6 (WT), TATA ∆ #1 and TATA ∆ #2 EBdCas9 lines failed to upregulate TBX18 transcription (Figure 4C). Similar levels of EBdCas9 mRNA expression after Dox induction were observed in all cell lines (Figure 4C). To test if other transcriptional elements may be responsible for TBX18 expression, we transfected WT or TATA ∆ #1 with TBX18g5 (900 bp upstream of g6). Only WT was able to upregulate TBX18 (Figures S4D and S4E). TATA ∆ #1 also reduced activation by the EZH2 chemical inhibitor EPZ-6438 (Knutson et al., 2013) (Figures S4F and S4G). We conclude that the distal, upstream, PRC2-regulated TATA box is essential for TBX18 gene expression.
Figure 4. EBdCas9 reveals functional distal TATA box.
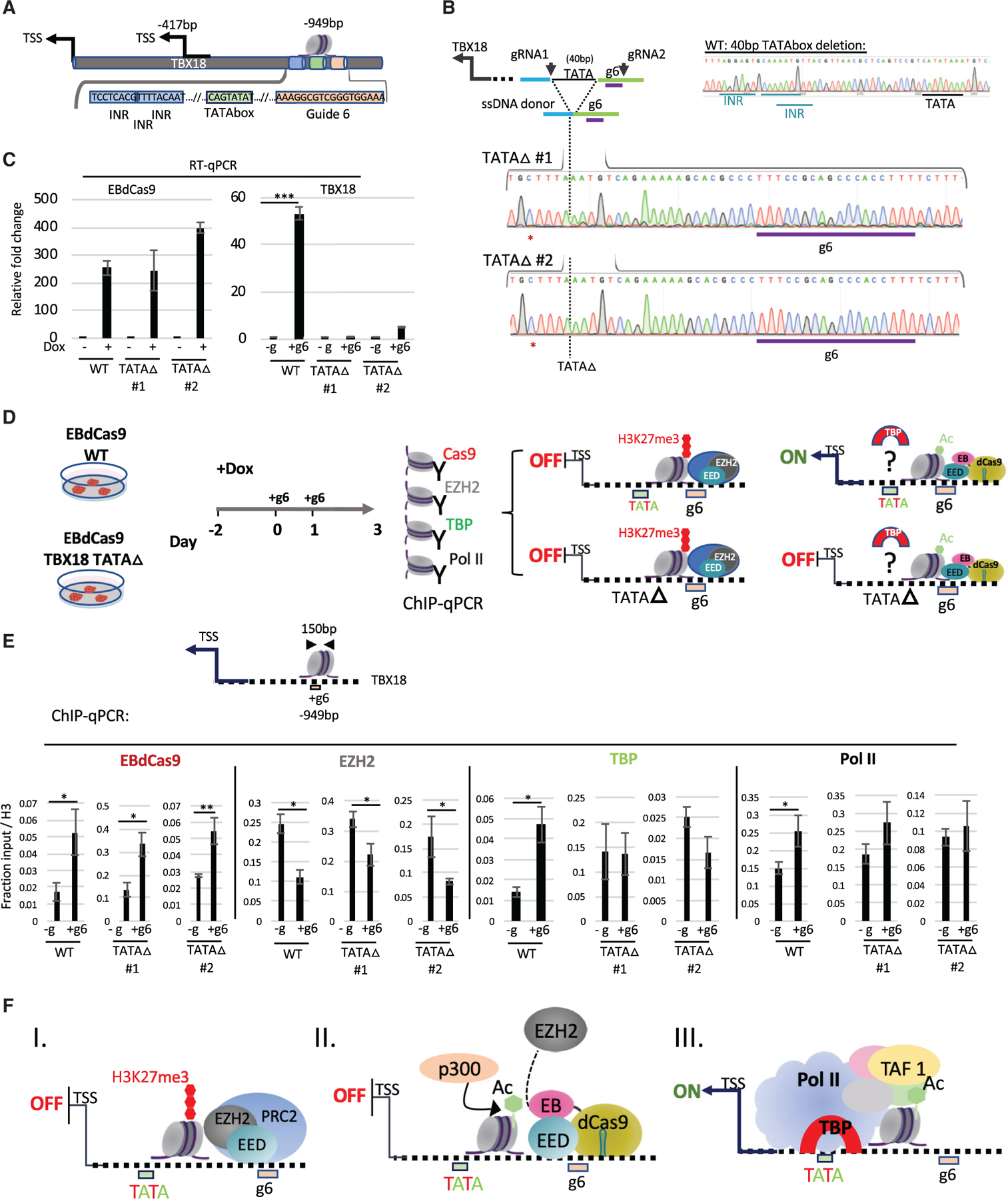
(A) TATA box and initiator (INR) representation with respect to g6 locus using the Element Navigation Tool for detection of core promoter elements.
(B) Generation of TATA box deletion (TATA ∆) clones TATA ∆ #1 and TATA ∆ #2 using CRISPR-Cas9. gRNA1 and −2 excise an 88 bp fragment, including the g6 region. The g6 region was reconstructed using a 168 bp single-stranded DNA (ssDNA) donor. Depicted here is a deletion of 40 bp including the TATA box and 3 initiator elements (INR). Genomic DNA of TATA ∆ #1 or TATA ∆ #2 displaying homozygous clones of 40 bp TATA box deletion using Sanger sequencing. PAM-site-blocking mutation is denoted with a red asterisk.
(C) qRT-PCR of EBdCas9 and TBX18 relative fold change after 3 days of Dox induction and TBX18 g6 RNA transfection normalized to β-actin and compared with no guide for each respective cell line (WT: EBdCas9, TATA ∆ #1, TATA ∆ #2).
(D) EBdCas9 WT and EBdCas9 TATA ∆ #1 and TATA ∆ #2 timeline for Dox induction, gRNA transfection, and ChIP-qPCR analysis using the antibodies Cas9, EZH2, TATA binding protein (TBP), and RNA Pol II and analyzing for TBX18 g6 DNA region. Right panel depicts 4 possible model scenarios for TATA box and TATA ∆ with and without EBdCas9.
(E) ChIP-qPCR of induced (+Dox) EBdCas9 (WT) and EBdCas9 TATA ∆ #1 and TATA ∆ #2 3 days following transfection with TBX18 g6 RNA (+g6) or no transfection (–g). Normalized to fraction input/H3. Antibodies that were used for ChIP are listed above the graphs, and the genomic region analyzed by qPCR includes the TBX18 g6 locus (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(F) Model describing EBdCas9/TBX18 mode of operation: (I) The TBX18 distal TATA box function is repressed by PRC2. (II) EBdCas9 derepresses the TBX18 TATA box region. (III) TBP, TBP associated factors (TAFs), and RNA Pol II are recruited to the TBX18 TATA box region to allow TBX18 transcription.
To explore why deletion of the distal TATA box affects TBX18 gene expression, we examined recruitment of TATA binding protein (TBP), and RNA Pol II (Wasylyk et al., 1980), at the g6 region after EBdCas9/g6 treatment (Figure 4D). We found that TBP was recruited to the g6 region, showing that the TATA box 21 bp away from the g6 region and 900 bp away from TSS1 can bind TBP after local PRC2 inhibition (Figures 4D and 4E). EBdCas9 (Cas9) and RNA Pol II were also recruited to the g6 site, and the EZH2 protein was reduced, as observed earlier (Figures 2C, 3F, and S3C). However, when the TATA box region was deleted (TATA D #1 and TATA ∆ #2 EBdCas9), TBP and RNA Pol II were not recruited (Figure 4E). Deletion of the TATA box did not alter EBdCas9 recruitment to, and EZH2 depletion at, the g6 region (Figure 4E). Thus, the TATA box >500 bp away from TBX18 TSS can recruit TBP when PRC2 is inhibited at the locus (model in Figure 4F).
EBdCas9 activates CDKN2A (p16) by epigenomic remodeling in iPSCs and DMG tumor cells
We studied EBdCas9/g action on PRC2 function at a second gene locus, CDKN2A (p16). p16 is a critical regulator of cell division and a tumor suppressor that inhibits cyclin-D-dependent protein kinase activity and the G1-S transition (Piunti et al., 2017; Mohammad et al., 2017; Cordero et al., 2017; Chen et al., 2020). In rapidly dividing cells, such as in DMG, p16 is repressed due to hypermethylation at the promoter area (Cordero et al., 2017). We searched for potential nucleation sites in the p16 upstream region responsive to PRC2 reduction by EBdCas9. We tiled the promoter area and gene body of p16 with eight gRNAs ranging from 0.3 to 2.3 kb upstream of the TSS and 0.2–0.7 kb downstream of the TSS (Figure 5A) and assessed DNA accessibility using the human ESC (hESC) Elf iCas9 cell line (Ferreccio et al., 2018) (Figure S5A). WTC EBdCas9 or NCdCas9 were induced prior to transient transfection of the gRNAs followed by cell harvest at day 3 and p16 transcript analysis (Figure 5B). EBdCas9 activated p16 transcript expression on 6 out of the 8 gRNAs tested, but none activated transcription in the control NCdCas9-expressing line (Figure 5C). As observed with TBX18 tiling, gRNAs that are within 0.5–1.5 kb of the TSS showed the highest p16 transcript activation. g1, g2, g3, g4, g6, and g7 induced transcription of p16 by 20- to 80-fold compared with no guide (–g) or NCdCas9 (Figures 5A–5C); g5 and g8 did not increase transcription significantly, even though EBdCas9/g had access to the DNA in these regions. TATA box and mammalian initiator factor binding sites in the p16 −424 and —750 bp promoter regions, upstream from the TSS (Figure 5D), are in close proximity to g1. p16 was also upregulated at the protein level by using immunofluorescence analysis (Figure 5E).
Figure 5. EBdCas9 upregulates CDKN2A (p16) expression and compromises cell viability.
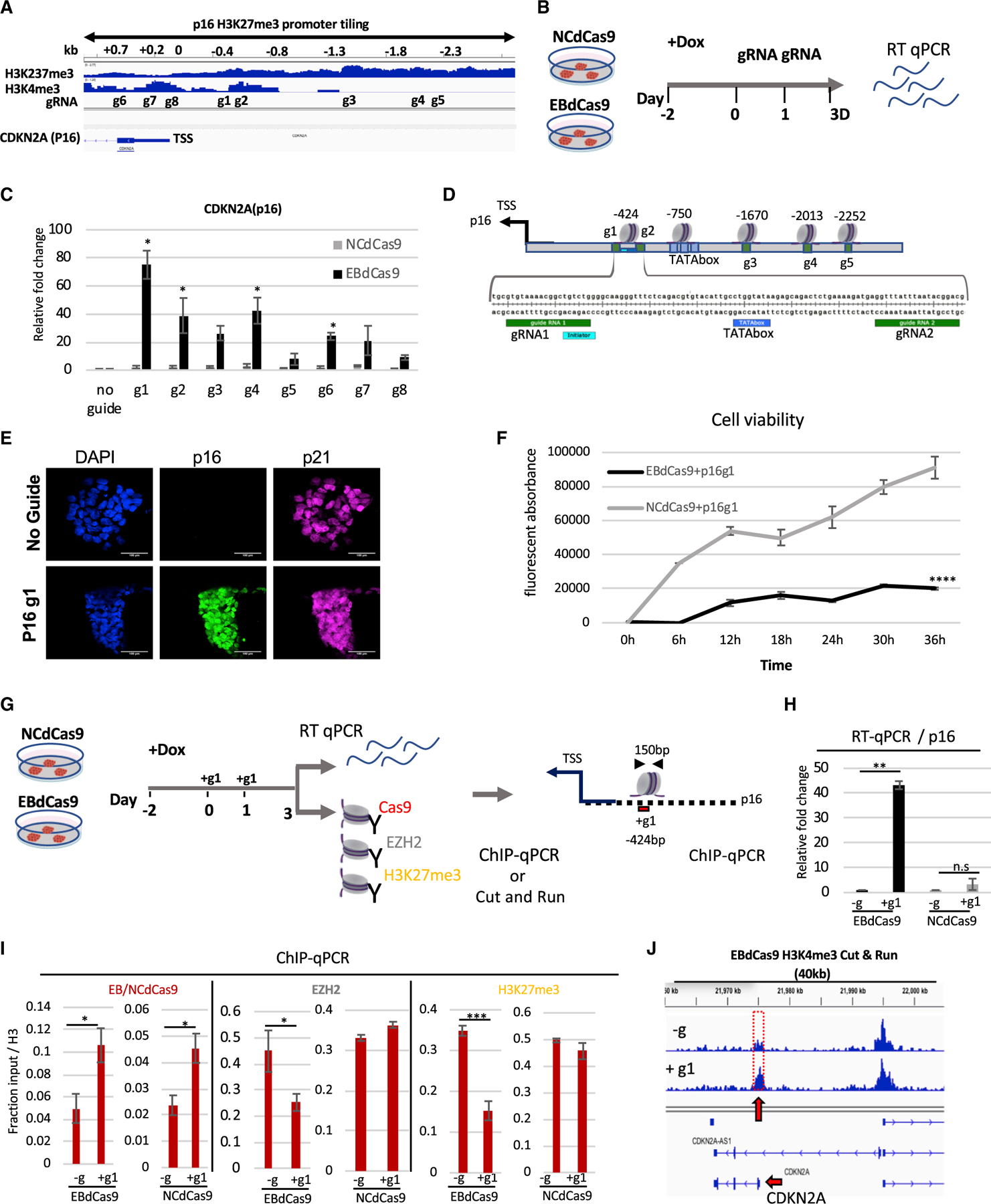
(A) Integrative genomic viewer of CDKN2A H3K27me3 and H3K4me3 promoter tiling.
(B) Timeline of EBdCas9 or NCdCas9 induction and gRNA transfection.
(C) qRT-PCR analysis of EBdCas9 or NCdCas9 after 3 days of transfection with single gRNA (g1–g8) normalized to β-actin and relative fold change compared with no guide (–g).
(D) TATA boxes/initiator representation with respect to surrounding guide loci using the Element Navigation Tool for detection of core promoter elements.
(E) Immunofluorescent imaging of EBdCas9 after 3 days of transfection with single p16 gRNA 1 (+g1) or no transfection (–g). Blue, DAPI; green, p16; far red, p21; scale bar, 100 µm.
(F) Cell viability of EBdCas9 and NCdCas9 transfected with p16 g1 (+g1) compared with no guide (–g). Time points were analyzed every 6 h using Alamar Blue (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(G) EBdCas9 timeline for measuring epigenetic remodeling.
(H) qRT-PCR of p16 relative fold change of EBdCas9 and NCdCas9 after 3 days of Dox induction and p16 g1 transfection. Samples were normalized to β-actin and compared with no guide (–g) of each respective cell line.
(I) ChIP-qPCR of induced (+Dox) EBdCas9 and NCdCas9 after 3 days of transfection with p16 g1 RNA (+g1) or no transfection (–g). Antibodies used for ChIP are listed above (Cas9, EZH2, H3K27me3). Normalized to fraction input/H3 (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(J) H3K4me3 marks increase after EBdCas9/p16 g1 (+g1) induction. CUT&RUN analysis of induced (+Dox) EBdCas9 after 3 days of transfection with p16 g1 RNA (+g1) or no transfection (–g) for H3K4me3. H3K4me3 tracks are normalized to immunoglobulin G (IgG) and displayed on the Integrated Genome Viewer (IGV); 40 kb region of the CDKN2A (p16) gene dispalyed in the integrated genome viewer.
Since activation of p16 expression results in cell-cycle arrest in gliomas (Cordero et al., 2017), we tested the effect of p16 upregulation in iPSCs. Transfection of WTC EBdCas9 with g1(p16) resulted in a 2-fold cell number and colony size reduction compared with no-guide controls (–g) on day 3 (Figures S5B and S5C). Consistent with this, EBdCas9/g1(p16) reduced cell viability compared with no guide (–g) or NCdCas9 using a resazurin fluorescent (Alamar Blue) detection reagent (Figure 5F). Transfection of g1 resulted in 40-fold transcriptional upregulation of the p16 gene with EBdCas9 but not the NC (Figures 5G and 5H). Similarly, EBdCas9/g1, but not NCdCas9/g1, reduced EZH2 protein and H3K27me3 marks at the g1 site (the two proteins were recruited equally to the locus) (Figure 5I) and increased H3K4me3 marks (Figures 5J [Skene and Henikoff, 2017] and S5D). H3K27ac marks also increased on EBdCas9/g1 compared with -g samples (Figure S5E).
DMG is an aggressive form of brain cancer that develops in glial cells in the pons and primarily affects children (Mohammad et al., 2017; Stafford et al., 2018). A lysine-to-methionine mutation in the histone H3.3 gene (H3.3K27M) defines DMG tumors (Mohammad et al., 2017; Stafford et al., 2018). Genome-wide studies of H3K27me3 marks in DMG cells have revealed that while a global loss of H3K27me3 is observed due to a H3.3K27M mutation, surprisingly, a sharp increase in H3K27 me3 repressive marks is observed at certain genes (presumably due to other H3 variants than H3.3 being methylated) (Stafford et al., 2018), including the tumor suppressor CDKN2A/p16/INK4a (Mohammad et al., 2017). Previous work has shown that overexpression of p16 reduces DMG cell growth and viability (Cordero et al., 2017). We were able to transiently transfect both EBdCas9 and NCdCas9 into DMG cells with the p16-targeting guide g1 (Figure S6A), but only the former reduced cell viability and cell replication (by 50%; Figures S6B and S6C) and upregulated p16 gene expression (Figure S6D). As in iPSCs, there was a reduction of EZH2 and H3K27me3, and an upregulation of H3K4me3, for EBdCas9 but not for NCdCas9 (Figure S6E). Thus, EBdCas9 can function in different cell types, and targeting EBdCas9 to specific loci produces similar epigenomic and transcriptional effects in different cell types.
H3K27me3 remodeling induces trophoblast fate
The placental (trophectoderm [TE]) versus embryonic (inner cell mass [ICM]) cellular fate decision (the first in animal development) is dependent on PRC2 activity (Saha et al., 2013). While overexpression of H3K27me3 is associated with ICM lineage, genome-wide depletion of H3K27me3 marks is associated with TE lineage (Saha et al., 2013; Banaszynski et al., 2013; Liu et al., 2016; Yang et al., 2017a, 2017b; Yan et al., 2013) (Figure 6A). We first tested if ubiquitously expressed EB (Moody et al., 2017) can induce trophoblast fate (Figure 6A). Recently, two groups have generated culture conditions that enabled the establishment of extended (EPS) cells from either cleavage state of mouse embryos or hESCs (Yang et al., 2017a, 2017b). The EPS cell stage has a developmental potency to generate both embryonic (ICM) and extraembryonic placental tissue (TE cell lineage) (Yang et al., 2017a, 2017b). Moreover, EPS epigenetic analysis validated the enrichment of bivalent, H3K27me3, and H3K4me3 marks in developmental genes (Yang et al., 2017a, 2017b). We reprogrammed WTC EB-FLAG and WTC NC-FLAG (Moody et al., 2017) iPSC lines to the EPS stage using a hLIF, chir99021, (S)-(+)-dimethindene maleate (DiM), and minocycline hydrochloride (MiH) (LCDM) reprogramming cocktail (Yang et al., 2017b). Once established, we validated the EPS cell stage by colony dome-shaped morphology, single-cell colony forming efficiency, expression of pluripotency markers, and TE lineage differentiation capacity in trophoblast differentiation (TX) media containing tumor growth factor beta 1 (TGF-β1), fibroblast growth factor 4 (FGF4), and heparin (Kubaczka et al., 2014) (Figures 6B and S7A). EB-FLAG-but not NC-FLAG-expressing cells lost EPS colony morphology and upregulated GATA3 and TBX3 expression at day 4, suggesting accelerated differentiation (Figures 6B–6D). We further showed that while confocal imaging confirmed the tight, dome-shaped morphology, the expression of nuclear stem cell transcription factor OCT4, and the absence of GATA3 expression for both EPS (EB-FLAG) and EPS(NC-FLAG) at day 0 (Figure 6E), TE differentiation (4 days) with EB-FLAG, but not NC-FLAG, resulted in a dramatic reduction of the OCT4 expression, loss of colony morphology, and upregulation of GATA3 (Figure 6E). Furthermore, immunoblot analysis revealed that 4 days of TE differentiation with EB-FLAG reduced H3K27me3 and EZH2, while GATA3 was upregulated Figure S7.
Figure 6. Trophoblast trans-differentiation using EBdCas9.
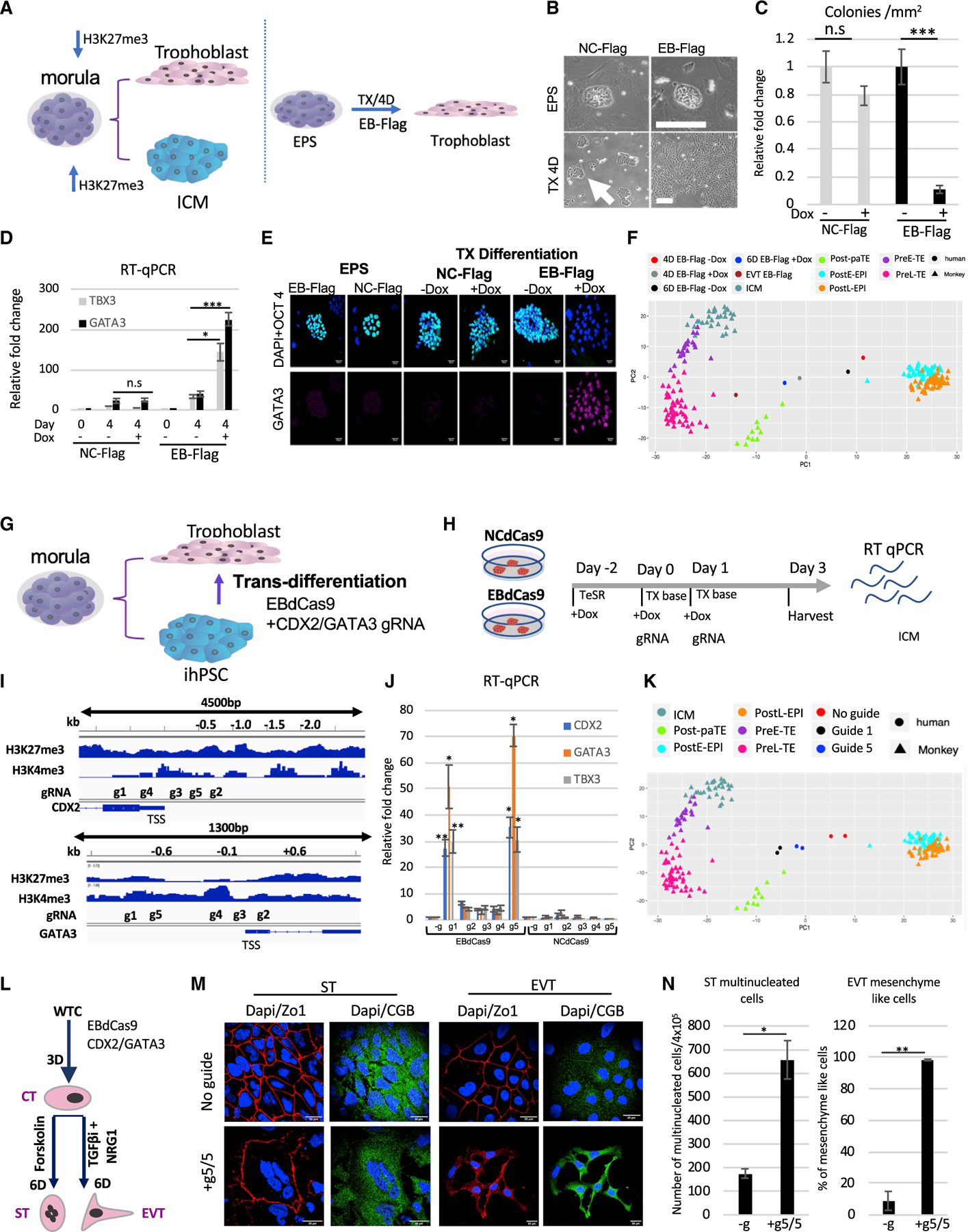
(A) Morula bifurcation to trophoblast and ICM is H3K27me3 dependent (left panel). EPS differentiation using EB-FLAG and TX media (Kubaczka et al., 2014) (right panel).
(B) WTC EB-FLAG or WTC NC-FLAG as described previously (Moody et al., 2017) were reprogramed to EPS for 2 weeks (3 passages). Bright field of colony morphology of EPS EB-FLAG or NC-FLAG induced with Dox on MEFs for 4 days or plated on matrigel in TX media (TGF-b1, FGF4, and heparin) for 4 days.
(C) Colony morphology count following 4 days in TX media EB-FLAG compared with NC-FLAG (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(D) qRT-PCR of EB-FLAG and NC-FLAG either in EPS stage or 4 days in trophoblast differentiation (TX media) with (+) or without (–) Dox. Normalized to b-actin and relative to EPS EB-FLAG stage (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
(E) Immunofluorescence of EPS EB-FLAG or EPS NC-FLAG on MEF/LCDM media or differentiation using matrigel/TX media with (+) or without Dox (–). DAPI, blue; WGA, red; OCT4, green; GATA3, far red. Scale bar, 50 µm.
(F) PCA of EPS samples compared to monkey single-cell RNA-seq (Nakamura et al., 2017). EB-FLAG EPS cells were differentiating in TX media with or without Dox for 4 days or 6 days or passaged 3 times as extravillous cytotrophoblasts (EVTs). Cell types in the monkey single-cell data include the following: Post-paTE, post-implantation parietal trophectoderm; PreL-TE, pre-implantation late TE; PreE-TE, pre-implantation early TE; ICM, inner cell mass; Pre-EPI, pre-implantation epiblast; PostE-EPI, post-implantation early epiblast; PostL-EPI, post-implantation late epiblast.
(G) Model of WTC EBdCas9 trans-differentiation to trophoblasts using CDX2 and GATA3 gRNA cocktail.
(H) Timeline of EBdCas9 or NCdCas9 induction and gRNA transfection.
(I) Tiling of CDX2 and GATA3 promoter and gene body gRNA.
(J) qRT-PCR analysis of CDX2 and GATA3 of co-transfected gRNAs in the presence of EBdCas9 or NCdCas9 induction (+Dox). g1–g5 correspond to g1/g1, g2/g2, g3/g3, g4/g4, and g5/g5 CDX2/GATA3 co-transfection. Normalized to β-actin and compared with no guide (–g) (n = 3, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test)
(K) PCA of day 3 WTC EBdCas9 co-transfected with g5/g5 CDX2/GATA3 and g1/g1 CDX2/GATA3 or not transfected (–g) bulk RNA-seq compared with monkey single-cell RNA-seq (Nakamura et al., 2017). Cell types are exactly as in (F).
(L) Timeline of EVT and syncytio trophoblast (ST) differentiation after EBdCas9:CDX2/GATA3 g5,g5 transfection (cytotrophoblast [CT]-like stage) and the factors used.
(M) Immunofluorescence of EBdCas9 3 days post-CDX2/GATA3 g5,g5 RNA transfection compared with no guide and further 6-day differentiation to either EVT using differentiation factors (see STAR Methods). DAPI, blue; ZO-1, red; chorionic gonadotropin beta (CGB), green. Scale bar, 30 mm.
(N) Quantification of ST-(multinucleation) and EVT-(mesenchyme-like morphology) positive cell count. Area of total count is 18 mm2 (n = 2, SEM; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test).
To investigate these fate changes in more detail, we analyzed gene expression utilizing RNA sequencing (RNA-seq). We projected gene expression data for TE-differentiated EPS(EB-FLAG) cells (4 days, 6 days, and differentiated extravillus cytotrophoblast [EVT] [Okae et al., 2018]) onto principal-component analysis (PCA) plots of single-cell transcriptomes of early cynomolgus monkeys (Nakamura et al., 2017) and found acceleration of TE differentiation in EB-FLAG-expressing cells (Figures 6F; Table S3); the projection is based on 773 highly variable genes (standard deviation >2) in the monkey single-cell dataset). TE-differentiated EPS EB-FLAG cells that were induced with Dox during differentiation migrate from post-implantation early or late epiblast (PostE-EPI or PostL-EPI), respectively, to post-implantation partial TE (Post-paTE) and pre-implantation late TE (PreL-TE) compared with no-Dox EB-FLAG-differentiated cells. The EB-FLAG samples are distant from ICM on the TE differentiation lineage, consistent with EB-FLAG accelerating TE differentiation. EVT EB-FLAG cells were first induced to express EB for 4 days and thereafter passaged 3 times (in trophoblast stem cell [TSC]-conditioned media) (Okae et al., 2018) without Dox (and therefore without further EB expression) and were found to be closest to PreL-TE. Thus, elimination of H3K27me3 marks by induction of the EB-FLAG protein dramatically accelerates TE lineage differentiation.
Targeted PRC2 inhibition at CDX2 and GATA3 loci induces trophoblast trans-differentiation
During mouse blastocyst formation, the relative levels of EED and KDM6B determine PRC2 complex recruitment and incorporation of H3K27me3 marks at the chromatin domains of target genes (Figure 6A). The TE-lineage-specific transcription factors CDX2 and GATA3 show PRC2-dependent repression in the ICM (Saha et al., 2013; Xiao et al., 2020). However, it is not known if PRC2 activity in the promoter regions of these two genes is sufficient to induce the bifurcation between TE and ICM lineages. We tested if ICM-like cells, such as iPSCs, which have already passed the bifurcation point, are able to trans-differentiate to TE if CDX2 and GATA3 H3K27me3 epigenetic marks are reduced (Figure 6G). To do so, we targeted EBdCas9/gRNA to the gene-upstream regions of TE transcription factors CDX2 and GATA3. After WTC(EBdCas9) induction and gRNA transfection (+Dox, 2 days, TeSR), the media was changed to TX-base media (+Dox) without factors (no TGF-β1, FGF4, and heparin) (Figure 6H). This created a permissive environment for TE differentiation, in which EBdCas9/gRNAs are the sole drivers for potential trans-differentiation. CDX2 and GATA3 were tiled across the promoter and gene body region with 5 different guides (Figure 6I). Since these two transcription factors are critical players in TE differentiation in mouse (Saha et al., 2013), we screened for the PRC2-responsive regions by co-transfecting guides covering upstream regions of CDX2 and GATA3 genes (g1/g1, g2/g2, …, g5/g5). WTC EBdCas9, but not NCdCas9, with gRNA pairs g1 and g5 resulted in a significant gene activation (20- to 80-fold increase) for CDX2 and GATA3, as well as the TE marker TBX3, and a reduction of EZH2 and H3K27me3 at targeted loci (Figures 6J and S7C–S7F; the other gRNA pairs had little effect).
We analyzed the global transcriptomes of EBdCas9 g1/g1 or g5/g5 CDX2 and GATA3 samples by RNA-seq. PCA showed that the g1/g1 and the g5/g5 samples were more similar to TE samples than the no-guide controls (migration from PostE-EPI/PostL-EPI toward Post-paTE and PreL-TE state) (Figure 6K; Table S4). Furthermore, PCA projection of developmental genes indicated similarity to TE fate (Figure S7G; Krendl et al., 2017) in g1/g1 and g5/g5-treated WTC EBdCas9 cells but not in WTC EBdCas9 with no gRNA. After 3 additional days of differentiation using TGF-βi and Neuregulin (Okae et al., 2018), the guide-treated cells produced chorionic gonadotropin beta (CGB) consistent with the EVT fate, while in the presence of Forskolin, they produced mesenchyme-like cells with multinucleation morphology, consistent with the syncytiotrophoblast (ST) fate (Figures 6L–6N and S7H). Thus, targeting EBdCas9 to precise CDX2 and GATA3 loci results in trans-differentiation of iPSCs toward TE fate. To explore the effects on normal differentiation, we reprogrammed WTC EBdCas9 to the EPS state and transfected with the g1/g1 and g5/g5 guides, which resulted in an increase in GATA3 gene expression of 100- to 500-fold (Figures S7I–S7K; CDX2 was less responsive) and loss of the pluripotency marker Oct4. Thus, inhibition of PRC2 activity at specific sites in only two genes can induce the bifurcation between TE and ICM fates.
DISCUSSION
Control of epigenomic regulation holds immense therapeutic promise for human disease without manipulation of endogenous gene sequences. Here, we develop a system for the targeted inhibition of PRC2 that can identify the precise nucleosomes that require H3K27me3 marks for transcription repression. We identified a distal TATA box that is regulated by PRC2 repression and required for transcript activation. We show that this PRC2-inhibiting tool, EBdCas9, allows the study of epigenomic memory of PRC2 at specific loci. Furthermore, we show the general applicability of EBdCas9 by identifying the regions where PRC2 disruption induces transcription of bivalent genes (TBX18, p16, CDX2, and GATA3). In total, we have targeted 26 specific sites in promoter regions upstream of four different genes and observed significant PRC2-dependent transcriptional derepression in 8 loci. NCdCas9, which differs from EB by two amino acids required for EED binding, does not result in transcriptional changes. This suggests that EBdCas9 functions by locally inhibiting PRC2 activity. When targeting PRC2 in the promoter regions of known TE transcription factors CDX2 and GATA3, EBdCas9 application identified specific loci in which PRC2 elimination is sufficient to upregulate the gene transcription and therefore induce TE cell-fate commitment. Furthermore, EBdCas9/g-based epigenetic control of p16 can result in reduced cell growth in DMG tumor cells.
dCas9 is the most promising method for targeted delivery of agents to chromatin. However, previous fusions of dCas9 to enzymes can result in off-target activity outside of the targeted locus. Existing histone modifying enzyme fusions are continuously active, potentially introducing or removing marks at off-target locations independent of the endogenous epigenetic modification machinery. In contrast, the computationally designed protein EB fused to dCas9 (EBdCas9) with appropriate gRNA presented here is able to precisely inhibit PRC2 at specific genetic loci, enabling (1) target and inhibition of PRC2 at a single nucleosome level; (2) reduced H3K27me3 at precise, targeted loci; (3) induced targeted transcription; (4) mediation of neighborhood spreading of remodeled epigenomics; (5) generation of epigenomic memory; (6) discovery of functional distal TATA box and TBP >500 bp of the TSS; (7) change of cell functionality; (8) transdifferentiation of one cell fate to another; and (9) reduction of tumor cells. Recently, several groups have activated targeted genes using synergistic activation mediator (SAM) such as dCas9-VP64, sgRNA, and MS2/p65/HSF1(Konermann et al., 2015; Liao et al., 2017; Joung et al., 2017; Weltner et al., 2018) by recruitment of transcription factors to the promoter region. Our approach of targeted PRC2 inhibition utilizing EBdCas9/gRNA allows organic expression of targeted genes as the cell makes holistic decisions for transcriptional activation. EBdCas9 function is specific to PRC2-repressed genes.
One of the fundamental findings of this study is that most broad H3K27me3 marks on promoter regions appear indiscriminate. Using EBdCas9/gRNA, we identified key PRC2-dependent single nucleosomes on promoter regions that control gene expression. In eukaryotes, genes are classified as TATA-containing and TATA-less in their promoter regions (Basehoar et al., 2004). The TATA-containing core promoter, in a range of −150 to −1 relative to the TSS, constitute ~17% of the total promoters in yeast, ~10% in worms, ~14% in fruit flies, ~10% in zebrafish, and ~3% in human and mouse, making 97% of core promoters TATA-less (Yella and Bansal, 2017). If the −500 to +500 region spanning the TSS is considered, the number of predicted TATA-containing promoter sequences grows to 14% (Bansal et al., 2014). While TATA-containing promoters have a TATA sequence at region −30 to −1 relative to the TSS region, TATA-less promoters have higher guanine-cytosine (GC) content in the TSS −150 to —1 region (Yella and Bansal, 2017; Yang et al., 2007). In addition, human TATA-less promoters have different structural motifs such as G-quadruplex (GNGNGNG) and CpG islands that are enriched at approximately ~42% at TATA-less promoter regions (Yella and Bansal, 2017). In the scenario of a distal TATA box, we hypothesize that a G-quadruplex structure contributes to active transcription as nucleosome-depleted regions in euchromatin associate with H3K4me3, RNA Pol II, and transcription factor recruitment (Hansel-hertsch et al., 2016). Here, we identify single nucleosomes on key promoter regions that are transcriptionally responsive to EBdCas9, suggesting that these regions are normally repressed due to PRC2 activity. In the case of TBX18, we show that PRC2-dependent sites co-localize with the distal TATA box and initiator binding elements. Furthermore, we show that the TATA box region is required for EBdCas9-based gene upregulation since TBP is recruited to the TATA box site after PRC2 derepression by EBdCas9. CRISPR-Cas9-based deletion of the TATA box region eliminates TBP recruitment, which subsequently results in transcript inactivation. This suggests that remote TATA boxes are regulated by biological signals culminating in PRC2 activity. We now show that the EBdCas9 biological tool can reveal PRC2-dependent, masked regulatory elements on promoter regions through EBdCas9/gRNA multiplex promoter screens.
The combination of controlled epigenomic gain- and loss-of-function manipulations are the most desirable for elastic gene-expression-based epigenomic memory. In the future, EBdCas9 expression under tissue-specific promoters or by transfection of ribonucleoproteins (RNPs) will allow us to explore the in vivo applicability of this technology. The adaptive and efficient targeted PRC2 inhibition by EBdCas9 identifies functional H3K27me3 marks and mediates gene activation, which holds promise both as an epigenetic tool in biomedical research and as an approach for treating a wide range of human diseases, including cancer.
Limitations of the study
In this study, we show that EBdCas9 inhibits local H3K27me3 and EZH2 levels. Since EBdCas9/gRNA targets 2 loci in each cell, local co-immunoprecipitation experiments of EED are technically limiting. Additionally, the gRNAs designed in this study were selected using CRISPR-Cas9 tools that are optimized for PAM sites recognition only. This limits the study to only those sites that are in close proximity to PAM sites.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact. Hannele Ruohola-Baker (hannele@uw.edu)
Materials availability
All unique constructs and stable cell lines generated in this study are available from the lead contact with a completed Uniform Biological Materials Transfer Agreement.
Data and code availability
RNA sequencing, ChIP sequencing, Cut and Run sequencing and ATAC sequencing data have been deposited at NIH GEO and are publicly available as of the date of publication. Accession numbers are listed in the key and resources table. Bulk RNAseq and DEseq values are found in Data S1. Original western blot images, RT-qPCR Ct, ChIP-qPCR Ct, and plate reader values are available in Data S2. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at NIH GEO and is publicly available as of date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-OCT4 | Santa Cruz | Cat#sc-5279; RRID: AB_628051 |
| Anti-TBX18 | Santa Cruz | Cat#sc-514486 |
| Anti-mCherry | Abcam | Cat#ab183628 |
| Anti-EZH2 | cell signaling | Cat#5246s |
| Anti-H3K27me3 | active motif | Cat#39155 |
| Anti-EED | EMD Millipore | Cat#09–774 |
| Anti-JARID | cell signaling | Cat#13594s |
| Anti-H3K27ac | Active motif | 39133 |
| Anti-p300 | Santa Cruz | sc-584 |
| Anti-Pol II CTD | santa cruz | sc-47701 |
| Anti-Pol II CTD PS5 | covance | MMS-134R-200 |
| Anti-p16 | thermo fisher Scientific | MA517054 |
| Anti-p21 | cell signaling | Cat#2946 |
| Anti-H3K4me3 | abcam | ab272143 |
| Anti-H3 | abcam | AB1791 |
| Anti-GATA3 | cell signaling | Cat#5852 |
| Anti-CGB | Invitrogen | Cat#PA558598 |
| Anti-HLA | Novus Biologicals Inc | Cat#NBP1–43123 |
| Anti-Actin | Cell Signaling | Cat#4970S |
| Anti-Flag | Sigma-Aldrich | Cat#F7425 |
| Anti-Cas9 | cell signaling | Ab #14697 |
| Anti 5mC | Active Motif | AB_2793653 |
| Anti 5hmC | Active Motif | AB_10013602 |
|
Bacterial and virus strains | ||
| 10-BETA Competent Cells | New England BioLabs | C3019H |
| DH5-alpha Competent Cells | New England BioLabs | C2987I |
|
Chemicals, peptides, and recombinant proteins | ||
| Matrigel | Corning | 356231 |
| Doxycycline | Millipore-Sigma | D9891–10G |
| Neurobasal | Thermo Fisher Scientific | 21103–049 |
| N2 supplement | Thermo Fisher Scientific | 17502–048 |
| B27 supplement | Thermo Fisher Scientific | 12587–010 |
| knockout serum replacement | Thermo Fisher Scientific | A3181502 |
| hLIF | SPEED Biosystems, LLC | YSP1249 |
| GS3Ki | Selleckchem | S7566 (2017), S2924 (2014), 72054 (2016) |
| Rocki Y27632 | R & D Systems (R&D) | 1254 |
| (S)-(+)-Dimethindene maleate Mino | santa Cruz Biotechnology Inc | sc-361329 |
| IWR | Selleckchem | S7086 |
| DMEM/F12 w/o HEPES or L-Glu | Invitrogen (Gibco/BRL Life Tech) | 21331–020 |
| l-ascorbic acid 2-phosphate Mg | sigma | A8960–5G |
| Insulin-Transferrin-Selenium | Fisher Scientific | 41400045 |
| NaHCO3 | sigma | S7277 |
| FGF4 | Life technologies | PHG0263 |
| TGF-beta1 | peprotech | 100–21 |
| Heparin | Stem Cell Technologies | 7980 |
| ITS-X supplement | Thermo fisher | 51500056 |
| NRG1 | Cell Signaling | 5218SC |
| A83–01 | Miltenyi Biotec Inc. | 130–105-333 |
| Forskolin | StemCell Technologies | 72112 |
| Alamar Blue | Thermo fisher Scientific | A50100 |
| Click iT EdU | ThermoFisher Scientific | C10337 |
|
Critical commercial assays | ||
| qiagen PCR purification | Quiagen | 28104 |
| qiagen gel purification | Quiagen | 28706 |
| mini-prep | Quiagen | 27104 |
| maxi-prep | Quiagen | 12662 |
| T7 maxi invitro transcription | invitrogen | AM1314 |
| Iscript | biorad | 1708891 |
| RT | Applied Biosytems | 205111 |
| RNAiMax | invitrogen | 13778030 |
| spCas9 | sigma | CAS9PROT |
| Cell Line NucleofectorTM Kit V | lonza | VCA-1003 |
| Quick Extract | Lucigen | QE09050 |
| ExoSAP-IT | Thermofisher | 78200.200.UL |
| GoTaq® DNA Polymerase | promega | M3001 |
| pGEM®-T Easy Vector Systems | promega | A1360 |
| Q5® High-Fidelity DNA Polymerase | NEB | M0491S |
|
Deposited data | ||
| RNA-seq data | GEO | GSE155022 |
| ChIP-seq data | GEO | GSE195552 |
| Cut and Run data | GEO | GSE195553 |
| ATAC-seq data | GEO | GSE195699 |
| DEseq and bulk RNAseq | This study | Data S1 |
|
Experimental models: Cell lines | ||
| WTC-11 | Coriell Institute | GM25256 |
| Elf1-iCas9 | Ferreccio et al. | RRID:CVCL_VR49 |
| IR-MEF | ISCRM hESC CORE | N/A |
| DIPG | gift from M Monje at Stanford University | N/A |
| WTC-11 EBdCas9 | This study | N/A |
| WTC-11 NCdCas9 | This study | N/A |
| WTC-11 EBdCas9 TBX18 TATAdeletion clone 11 |
This study | N/A |
| WTC-11 EBdCas9 TBX18 TATAdeletion clone 23 |
This study | N/A |
| Elf1 EBdCas9 | This study | N/A |
| Elf1 NCdCas9 | This study | N/A |
|
Oligonucleotides | ||
| See Table S5 for oligonucleotide information | N/A | |
|
Recombinant DNA | ||
| KRAB-dCas9 | Concklin Lab | Mandegar et al., 2016 |
| EB-Flag | Our Lab | Moody et al., 2017 |
| NC-Flag | Our Lab | Moody et al., 2017 |
| EBdCas9 | This paper | N/A |
| NCdCas9 | This paper | N/A |
|
Software and algorithms | ||
| Element | Element | http://lifefaculty.biu.ac.il/gershon-tamar/index.php/element-description/element |
| Fantom | FANTOM 5 | http://fantom.gsc.riken.jp/5/ |
| R | The R Foundation | https://www-r-project-org.offcampus.lib.washington.edu/ |
| ImageJ | NIH | https://imagej-nih-gov.offcampus.lib.washington.edu/ij/ |
| Microsoft office | Microsoft | https://products.office.com |
| Snapgene | Snapgene | https://www.snapgene.com |
| Transcription factor prediction Tool | http://www.ifti.org/cgi-bin/ifti/Tfsitescan.pl | |
| QGRS Mapper Tool | http://bioinformatics.ramapo.edu/QGRS/analyze.php | |
| GEO DOI | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE195555 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
hiPSC line WTC #11, previously derived in the Conklin laboratory (Kreitzer et al., 2013), WTC line EB-Flag and NC-Flag, previously derived in our lab (Moody et al., 2017), Elf iCas9, previously derived in our lab (Ferreccio et al., 2018) and DMG (DIPG), a gift provided by M Monje at Stanford University were used in this study.
METHOD DETAILS
hiPSC and hESC Cell culture
The hiPSC line WTC #11, previously derived in the Conklin laboratory (Kreitzer et al., 2013), was cultured on Matrigel (1:30) growth factor-reduced basement membrane matrix (Corning) in mTeSR media (StemCell Technologies). Cells were passaged using versene once reaching 70% confluency and plated at 1:6 density. For EPS conditions, cells were grown as previously described(Yang et al., 2017b). Briefly, WTC cells were reprogrammed in base medium containing 100 mL DMEM/F12, 100 mL Neurobasal, 1 mL N2 supplement, 2 mL B27 supplement, 1% GlutaMAX, 1% NEAA, 0.1 mM β-mercaptoethanol, penicillin-streptomycin and 5% KSR, and freshly supplemented with 10 ng/mL hLIF, GSK3i (1 µM), ROCKi (2 µM), (S)-(+)-Dimethindene maleate (2 µM; Tocris), Minocycline hydrochloride (2 µM; Santa Cruz Biotechnology) and IWR-endo-1 (0.5–1 µM; Selleckchem). Cells were adapted to EPS conditions for at least 3 passages before analysis. EPS cells were pushed toward differentiation using TX media (Kubaczka et al., 2014): TX medium formulation was DMEM/F12 without HEPES and L-glutamine (Life Technologies), 64 mg/l L-ascorbic acid-2-phosphate magnesium, 14 mg/L sodium selenite, 19.4 mg/L insulin, 543 mg/L NaHCO3, 10.7 mg/L holo-transferrin (all Sigma-Aldrich), 25 ng/mL human recombinant FGF4 (Reliatech), 2 ng/mL human recombinant TGF-β1 (PeproTech), 1mg/ml heparin (Sigma-Aldrich), 2 mM L-glutamine, 1% penicillin, and streptomycin (all PAN-biotech). Medium was prepared without growth factors (TX-growth factors) and stored at 4°C. To prepare complete TX, the growth factors: FGF4, heparin, and TGF-β1 were added prior to use. Medium was changed every other day. All cells were cultured at 37°C in 5% CO2. For Elf1 iCas9 conditions, cells were treated as described before(Ferreccio et al., 2018) but adapted to TeSR culture media and single cell passaging: the cells were grown on Matrigel (1:30) growth factor-reduced basement membrane matrix (Corning) in mTeSR media (StemCell Technologies) and passaged using versene once reaching 70% confluency and plated at 1:6 density.
EBdCas9 and NCdCas9 plasmid construction
we used the AAVS1 TREG KRAB-dCas9 plasmid previously derived in the Conklin laboratory (Kreitzer et al., 2013), eliminated KRAB using PacI and AgeI restriction cut and then constructed and religated the EEDbinder-linker-dCas9-NLS-mCherry (EBdCas9) or EEDbinder Negative Control-linker-dCas9-NLS-mCherry (NCdCas9) to the cut plasmid, screened colonies and verified the sequence by Sanger sequencing (SI Table S5).
Insertion of inducible EBdCas9 and NCdCas9 into AAVS1 site of WTC cells
1×106 cells of WTC p42 were transfected with 5mg AAVS1-TALEN R plasmid (Addgene #59026), 5mg AAVS1-TALEN L plasmid (Addgene #59025), and 5mg donor plasmid (AAVS1 TREG EBdCas9 or AAVS1 TREG NCdCas9) using the Amaxa Lonza Human stem cell Kit #2. The cells were then plated with 5µM of Rock inhibitor (ROCKi) onto 10cm with fresh media. Three days following the nucleofection, the cells were selected for neomycin resistance with Genetecin (50mg/ml) for four days. Clones that survived after selection were expanded as a pool. The clones were plated onto matrigel with or without Doxycycline (2mg/ml) and RNA was extracted in order to analyze the level of Cas9 expression by qPCR. Insertion of EBdCas9 or NCdCas9 into the AAVS1 site was confirmed by cellular genomic isolation, PCR amplification and Sanger sequencing.
Trophoblast differentiation using EB-Flag/NC-Flag EPS cells
50k-100k EPS Cells grown on matrigel (1:30) coated plate in TX base medium (DMEM/F12 w/o HEPES or L-Glu (21331–020), 64mg/ml ascorbic acid phosphate Mg, 14ug/l sodium selenite, 19.4mg/L insulin, 10.7 mg/L holo-transferrin, 543mg/L NaHCO3, 1% Pen Strep and, 2mM Glutamax) and TX factors (25ng/ml FGF4, 2ng/ml TGF-β1 1 mg/mL Heparin) TX media was changed every other day. 2µg/ml Doxycycline was added to the medium on day of differentiation.
Trophoblast trans-differentiation using EBdCas9/NCdCas9 WTC cells
100k WTC cells were plated on matrigel plates (1:30) in the presence TeSR and Doxycycline (2mg/ml) for 48h. Prior to gRNA transfections the media was changed to TX media as described in derivation and maintenance of murine trophoblast stem cells under defined conditions(Kubaczka et al., 2014) (without factors). gRNAs were transfected using RNAimax for 2 consecutive days and were either harvest for RT-qPCR, ChIP-qPCR at day 3 or continued for further EVT/ST differentiation using the below media conditions for EVT or ST, without EB or NC induction.
EVT/ST differentiation
3d EBdCas9 TX cells were grown to 80% confluency in TX medium (Kubaczka et al., 2014) and dissociated with 0.05% Trypsin-EDTA for 5 min at 37° and harvested as described in derivation of human trophoblast stem cells(Okae et al., 2018). Briefly, trypsinization was stopped using EVT/ST base medium (DMEM/F12 0.1 mM 2 mercaptoethanol, 0.5% Penicillin-Streptomycin, 0.3% BSA, 1% ITS-X supplement, 2.5 mM Y27632 (Rock i), 4% KnockOut Serum Replacement) and cells were plated on matrigel (1:30) 0.75×105 density per well and cultured in 2 mL of EVT/ST medium with either EVT differentiation factors (100 ng/mL NRG1,7.5 mM TGF-βi A83–01) or ST differentiation factor (2mM forskolin). Cell were cultured for additional 6 days, with media changes every other day. On day 9 cells were fixed and permeabilized for immunofluorescent staining and imaging.
Guide RNA design, synthesis and transfection
The gRNAs targeting TBX18, p16, CDX2 and GATA3 genes were designed using the CRISPRscan web prediction tools (Moreno-mateos et al., 2015) and ordered as T7-gRNA primers. The T7-gRNA forward primer and the reverse scaffold primer were used in primer extension reaction to synthesized a double stranded DNA fragment by using Q5 High Fidelity-based PCR (New England Biolabs) followed by PCR purification (Qiagen). The 120 bp dsDNA served as a template for IVT (MAXIscript T7 kit, applied Biosystems). The RNA was then purified using Pellet Paint Co-Precipitant (Novagen). For transfection, 1×10^5 WTC EBdCas9 or 1×10^5 NCdCas9 cells were seeded at day 0, and treated with Dox (2µg/ml) for 2 days before and during transfection. On day 2 cells were transfected with gRNAs using Lipofectamine RNAiMAX (Life Technologies). gRNA was added at a 40 nM final concentration when added alone or 20nM in co-gRNA transfection. A second transfection was performed after 24 h. All no guide controls were induced by Dox and harvested with study groups. Two days after the last gRNA transfection, cells were harvest for either DNA, RNA, protein, ChIP-qPCR, or Cut and Run analysis (SI Table S5). N = 1 for gRNA transfection. p value was calculated based on 3 independent transfections.
Elf iCas9 transfection
Elf iCas9 cell line(Ferreccio et al., 2018) was induced with Dox for 2 days prior to gRNA (SI Table S5) transient transfection. individual gRNAs were transfected transiently (800ng/ml) and cell were collected for genomic DNA isolation, amplification of designated regions and indel validation using Sanger sequencing.
DNA extraction and sequencing
Genomic DNA was collected using DNAzol reagent (Invitrogen) according to manufacturer’s instructions and quantified using Nanodrop ND-1000. Genomic regions flanking the AAVS1 were PCR amplified with the designed primers, purified by PCR Purification Kit (Invitrogen) and sent to Genewiz for sequencing.
RNA extraction and RT-qPCR analysis
RNA was extracted using Trizol (Life Technologies) according to manufacturer’s instructions. RNA samples were treated with Turbo DNase (ThermoFischer) and quantified using Nanodrop ND-1000. Reverse transcription was performed using iScript (BioRad). 10 ng of cDNA was used to perform qRT-PCR using SYBR Green, with suitable primers (SI Table S5) on an Applied Biosystems 7300 real-time PCR system with PCR conditions as stage 1 50°C for 2mins, stage 2 as 95°C for 10mis, 95°C for 15sec, 60°C for 1min(40 Cycles). β-actin was used as an endogenous control), related to Data S2.
Protein extraction and western blot analysis
Cells were lysed directly on the plate with lysis buffer containing 20mM Tris-HCl pH 7.5, 150mM NaCl, 15% Glycerol, 1% Triton X-100, 1M ß-Glycerolphosphate, 0.5M NaF, 0.1M Sodium Pyrophosphate, Orthovanadate, PMSF and 2.3% SDS. 25 U of Benzonase Nuclease (EMD Chemicals, Gibbstown, NJ) was added to the lysis buffer right before use. Proteins were quantified by Bradford assay (Bio-rad), using BSA as Standard using the EnWallac Vision. The protein samples were combined with the 4x Laemli sample buffer (900 µL of sample buffer and 100 µL β-Mercaptoethanol), heated (95°C, 5mins) and run on SDS-PAGE (protean TGX pre-casted gradient gel, 4%–20%, Bio-rad) and transferred to the Nitro-Cellulose membrane (Bio-Rad) by semi-dry transfer (Bio-Rad). Membrane was blocked for 1hr with 5% milk, and incubated in the primary antibodies overnight in 4°C. The antibodies used for Western blot were beta-Actin (Sigma A5441, 1:2000), Cas9 (Cell Signaling 1:1000), Oct-4 (Santa Cruz sc-5279, 1:1000, Novus Biologicals NB110–90606, 1:500), H3K27me3 (Active Motive 39,155 1:1000), EZH2 (Cell Signaling D2C9, 1:1000), GATA3 (Cell signaling D13C9,1:1000), Flag (sigma F1804, 1:1000) The membranes were then incubated with secondary antibodies (1:10,000, goat anti-rabbit or goat anti-mouse IgG HRP conjugate (Bio-Rad) for 1hr and the detection was performed using the immobilon-luminol reagent assay (EMP Millipore) (SI Table S6).
Immunostaining and confocal imaging
Cells were fixed in 4% paraformaldehyde for 15 min, washed with PBS (3 × 5min), and blocked in the presence of 0.1% Triton X-100 and 2% BSA for 1h at room temp. Cells were then incubated in primary antibody overnight, washed with PBS (3 × 5min), incubated with the secondary antibody in 2% BSA for 1hr, washed (4 × 10mins, adding 1mg/ml DAPI at the second wash), mounted using Vectashield (Vector Labs) and imaged using a Leica TCS-SPE confocal microscope at 10x,40x or 63x. Antibodies use in immunostaining were: TBX18 (Santa Cruz sc-514486, 1:200), Oct 4 (Novus Biologicals, NB11–1:150), p16 (Invitrogen MA5–14260, 1:250), p21 (Cell Signaling 2947S, 1:200), GATA3 (Cell signaling D13C9, 1:250), CGB (DAKO GA508; 1:200). Slides were rinsed with PBS-T and incubated with secondary antibodies: DAPI (0.02 µg/mL, Molecular Probes), goat anti-rabbit 488, and goat anti-mouse 647 (1:500, Molecular Probes) for 2 h at room temperature in blocking buffer. (EBdCas9 or NCdCas9 were visualized using endogenous mCherry expression) (SI Table S6).
Cell harvesting cross-linking and PIXUL sonication
PIXUL 96-well plate sonicator (Matchstick Technologies, Inc, Kirkland, WA and Active Motif, Carlsbad, CA) was used to fragment chromatin. All steps began from 1 × 35 mm plate exactly as described in Guide RNA design, synthesis and transfection above. Cells were harvested using Versene (Thermo Fisher), followed by 3 min centrifugation (1.2 xg). Pelleted cells were washed with 13 PBS. Cells were cross-linked by adding 500ul 1% formaldehyde in PBS for 20min in RT. Supernatant was removed and 500ul of PBS/glycine (125mM) was added for 5 min at RT for quenching. Supernatant was removed and the cells were washed with 500ul of PBS. PBS was removed and samples were stored in −80°C. For shearing, cells were resuspended in 100ul chromatin sheering buffer (Active Motif) and transferred into wells of 96 well plate, sealed with PCR film (MiniAMP Optical Adhesive Film) and placed in PIXUL for shearing. PIXUL parameters were as follows: Cycles = 50; PRF = 1kHz; Burst = 20 for 6min x 4. Once sheered the cells were chromatin immunoprecipitated using matrix-ChIP. N = 1 for gRNA transfection and ChIP. p value was calculated based on 3 independent transfections.
PIXUL-matrix-ChIP-qPCR (PIXUL-ChIP) analysis
PIXUL-ChIP was performed as previously described (Flanagin et al., 2008; Bomsztyk et al., 2019). Briefly, UV treated 96-well polypropylene microplates were incubated with protein A on a low-speed shaker at room temperature overnight. The next day, the wells of the 96-well plate-coated with protein A were blocked with blocking buffer containing 5% BSA, sheared salmon sperm DNA (10µg/µL final) in immunoprecipitation buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 5 mM EDTA, NP-40 (0.5% vol/vol), Triton X-100 (1.0% vol/vol)) on a shaker at room temperature for 60 min. Simultaneously, sheered chromatin samples (see above),blocking buffer and antibodies were added to a another polypropylene 96-well microplate (untreated) and incubated in ultrasonic bath for 60 min at 4°C. The blocking buffer was aspirated from the protein A-coated plate, and the chromatin + antibody mix from the untreated plate was transferred to the protein A-coated wells and incubated in the ultrasonic bath for 60 min at 4°C. The wells were then washed 3 times with immunoprecipitation buffer followed by 3 washes with TE buffer. Finally, elution buffer containing 25 mM Tris base, 1 mM EDTA (pH10) with proteinase K 200 µg/mL was added to the wells, nutated for 30 s at 1400 rpms and incubated for 45 min at 55°C and then 10 min at 95°C. The 96-well plates were then briefly agitated and centrifuged for 3 min at ~ 500 g at 4°C and were used for PCR. The antibodies (SI Table S6) used for Matrix ChIP were: mCherry (Abcam ab167453), H3K27me3 (Active motif 39,155), EZH2 (Cell Signaling D2C9), H3K27ac (Active motif 39,133), p300(Santa Cruz sc-48343), Pol II 4H8 (Santa Cruz, sc-47701), Pol II Ser 5P+ (Santa Cruz), H3K4me3(Active motif 39,159), Cas9 (Active motif 61,757). Matrix ChIP experiments were performed in triplicate followed by qPCR in 4–8 replicates. ChIP-qPCR analysis and calculation are described below.
ChIP-qPCR calculations
All PCR reactions were run in quadruplicated using syber green using primer sets found in (SI Table S5). PCR calibration curves were generated for each primer set from a dilution series of total human genomic DNA. For each qPCR run the primer efficiency curve was fit to cycle threshold (CT) versus log (genomic DNA concentration) using an r-squared best fit. DNA concentration values for each ChIP and input chromatin DNA sample were calculated from their respective average Ct values as previously described (Flanagin et al., 2008; Bomsztyk et al., 2019). Final results are expressed as fraction of input DNA normalized to H3 antibody.
PIXUL-matrix-ChIP-seq
Cells were dissociated using versene, washed once in cold PBS and counted. Approximately 6×10^6 cells were fixed and chromatin was PIXUL sonicated as described above. H3K27me3 (Active Motif Ab) Matrix-ChIP’ed DNA was purified using phenol-chloroform extraction and ChIP-seq libraries were prepared (Next Gen DNA Library Kit, Active Motif, Cat# 53,216, 53,264). Libraries were sequenced on NextSeq2000 in single end run and approximately 45 million raw reads were obtained for each sample. For analysis: reads were normalized via RPKM and aggregate read signal for all 22K genes was calculated in a 5k window centered at the TSS. Significance by first calculating average signal in 5k window for all TSS′, then comparing means across conditions via t test.
Cut and Run analysis
Cut and Run method and analysis was performed as previously described(Skene and Henikoff, 2017). 1million WTC EBdCas9 cells gRNA transfected or not were harvested by centrifugation (600 g, 3 min in a swinging bucket rotor) and washed in ice-cold phosphate-buffered saline (PBS). Nuclei were isolated by hypotonic lysis in 1 mL NE1 (20 mM HEPES-KOH pH 7.9; 10 mM KCl; 1 mM MgCl2; 0.1% Triton X-100; 20% Glycerol) for 5 min on ice followed by centrifugation as above. Nuclei were briefly washed in 1.5 mL Buffer 1 (20 mM HEPES pH 7.5; 150 mM NaCl; 2 mM EDTA; 0.5 mM Spermidine; 0.1% BSA) and then washed in 1.5 mL Buffer 2 (20 mM HEPES pH 7.5; 150 mM NaCl; 0.5 mM Spermidine; 0.1% BSA). Nuclei were resuspended in 500 µL Buffer 2 and 10 µL antibody was added and incubated at 4°C for 2 h. Nuclei were washed 3 x in 1 mL Buffer 2 to remove unbound antibody. Nuclei were resupended in 300 µL Buffer 2 and 5 µL pA-MN added and incubated at 4°C for 1 h. Nuclei were washed 3 x in 0.5 mL Buffer 2 to remove unbound pA-MN. Tubes were placed in a metal block in ice-water and quickly mixed with 100 mM CaCl2 to a final concentration of 2 mM. The reaction was quenched by the addition of EDTA and EGTA to a final concentration of 10 mM and 20 mM respectively and 1 ng of mononucleosome-sized DNA fragments from Drosophila DNA added as a spike-in. Cleaved fragments were liberated into the supernatant by incubating the nuclei at 4°C for 1 h, and nuclei were pelleted by centrifugation as above. DNA fragments were extracted from the supernatant and used for the construction of sequencing libraries. We have also adapted this protocol for use with magnetic beads (Skene and Henikoff, 2017).
Matrix-MeDIP-qPCR
Single-stranded DNA (ssDNA) was isolated form iPSC (WTC) EBdCas9 +Dox cells either after 3 day treatment with TBX18 gRNA (+g6) or untreated (-g). DNA was sonicated for 6 min in PIXUL followed Matrix-MeDIP-qPCR protocol as described above and in (Flanagin et al., 2008; Bomsztyk et al., 2019; Yu et al., 2011). Note that cells were not fixed or quenched for MeDIP (SI Table S5).
ATAC-seq data analysis
Protocol used for ATAC-seq is described in (Corces et al., 2017). Briefly. cells were dissociated using versene, washed once in cold PBS and counted. Approximately 500,000 cells were resuspended to final concentration of ATAC resuspension buffer (10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2) in the presence of 0.1% NP40, 0.1% Tween 20, and 0.01% Digitonin as previously describe in (Corces et al., 2017). Nuclei were pellet at 500 RCF for 10 min at 4°C and supernatant was aspirated. Cells were then resuspended to final concertation of transposition mix (Tris-HCl 20mM, MgCl210mM, 20% Dimethyl Formamide, transposase 100nM, 1% digitonin, 10% Tween 20) and incubated at 37°C for 30 min in a thermomixer with 1000 RPM mixing. The reaction was cleaned up using Zymo DNA Clean and Concentrator-5 Kit (cat# D4014). The eluted DNA was amplified using NEBNext 2x MasterMix. KAPA Library Quantification kit (cat# KK4854) was used for final concentration determination.
RNA-seq data analysis
RNA-seq samples were aligned to the custom human reference genome hg38 with EBdCas9sequence inserted using Hisat2 (version 2.1.0) (Kim et al., 2019). Gene-level read counts were quantified using subread(v2.0.0) (Liao et al., 2014) using custom Ensembl GRCh38 gene annotations with EBdCas9 added. Custom reference genome and annotation files with EBdCas9 were generated as follows: 1) Genome sequence (GRCh38.p13) was downloaded from the GENECODE browser (07–08-2021)the transcript-level annotation was downloaded from the GENECODE browser (07–08-2021). 2) The EBdCas9 sequence and annotation were added at the end of the reference genome and annotation. Filtered estimated gene count matrices generated for each RNA-seq sample were normalized (median of ratios) with the R package DESeq2(v1.32.0) (Love et al., 2014) and subjected to differential expression analysis with significance cutoff of an actual fold-change of at least 2 (Log2 FC > 1) with a false discovery rate (FDR) cutoff of 5% (adjusted p value < 0.05) was used to determine whether a gene was differentially expressed. Volcano plots were generated to display the list of DEG for each RNA-seq dataset using the R package EnhancedVolcano (v1.10.0) (Blighe et al., 2018).
For trophoblast transdifferentiation: RNA-seq samples were aligned to hg19 using Tophat (version 2.0.13) (Trapnell et al., 2009). Gene-level read counts were quantified using htseq-count using Ensembl GRCh37 gene annotations. Processed single cell RNA-seq data from Nakamura et al. (Nakamura et al., 2017) were used. Only genes expressed above 10 Reads Per Million in 3 or more samples were kept. t-SNE was performed with the Rtsne package, using genes with the top 20% variance across samples. Cluster labels from Nakamura et al. were used. A Principle Component Analysis (PCA) was performed using all of the cynomolgus monkey samples from Nakamura et al. (Nakamura et al., 2017) using R software. Genes used in the analysis were restricted to defined homologs expressed at non-zero Transcripts Per Million (TPM) in human in vitro cell lines, and in the preprocessed mouse and cynomolgus monkey single cell samples from Nakamura et al. RNA-seq data from human cell lines were corrected for batch effects using ComBat (Johnson et al., 2007). Human bulk RNA-seq samples were projected onto the PCA coordinate via matrix multiplication. Human, cynomolgus monkey and mouse RNA-seq data were separately centered and scaled within each species before PCA and projection was performed. These data can be found in the Data S1.
Cell count
Cells were lifted using Versene and resuspended in media for cell count using hemocytometer. 100ul of cell suspension were applied to hemocytometer using 10× objective. 4 most outward corners were counted, averaged, multiplied by 104, and multiplied by the dilution factor to get the final number of cells.
Cell viability
Cell viability assay was measured by AlamarBlue (thermofisher scientific, DAL1025). Cells were induced and transfected with gRNA as discussed above. 6h after last transfection, 10× of viability reagent was added to media. Time ‘‘0’’ was measured 4h after. Fluo-rescence measurements were read on PerkinElmer Envision plate reader after transferring 50ul of media from each condition to 96 well microplate reader.
Cell proliferation and FACS analysis
Cell proliferation assay was measured using Click-iT EdU Cell Proliferation Kit (ThermoFisher Scientific, C10337). Approximately 0.5M DMG cells were transfected with either EBdCas9/p16 + g1, NCdCas9/p16 + g1, or untreated (-g) on day 0 in the presence of Dox (0.5ug/ml) following EdU incorporation (10uM) on day 1. On day 3 DMG were collected using accutase, washed twice with PBS and resuspended with 4% PFA. Subsequently, a Click-iT reaction was percoemed as directed by the manufacture’s protocol. Finally, cells were washed twice with PBS, resuspended in 500ul of PBS-5% FBS, analyzed by FACS and FlowJo software.
CRISPR-based deletion of TBX18 TATA box region
For WTC EBdCas9 TBX18 TATA box deletion (40 bp deletion), we used CRISPR/Cas9 to delete an 88bp region by using 2 gRNAs flanking TATA box 5’ AND-3′ ends. Since gRNA 2 overlaps with TBX18 g6 locus we reconstructed guide6 region using a ssDNA donor (168bp, IDT) that lacks the 40bp region around the TATA box (TATATGAC) but contains intact guide6 region and mutated PAM region. Two guides RNAs (Synthego) that are flanking TATA box region were designed using Benchling (Table S5). Briefly, 0.5µM of spCas9 (sigma) were mixed with 1µM of gRNAs and incubated at RT for 15min. 1×106 EBdCas9 cells were transfected with the above RNP complex and 100µM of ssDNA (HDR) (Table S5) using AMAXA electroporation kit (LONZA) and plated in TeSR media (supplemented with 5µM ROCKi). Several EBdCas9 colonies were picked, followed by chromosomal DNA isolation (Quick Extract/Lucigen), PCR amplification (Table S5) and PCR purification (ExoSAP-IT/Thermofisher). PCR products were sequenced to identify potential 40bp TATA box region deletions and g6 reconstruction. To produce a homozygous deletion of TBX18 TATA box and guide 6 reconstruction a second hit of CRISPR/Cas9 was introduced using the same exact 2 guide RNAs and ssDNA as describe above. Genomic DNA sequencing analysis of selected clones confirmed homozygous deletion of 40bp of TATA box region on TBX18. To validate that the lines analyzed were homozygous TATA box deletion and guide6 reconstruction, we ligated PCR amplicons to pGEM-T (Promega) vector system and confirmed that all picked clones encompassed TATA box deletion and guide6 reconstruction using Sanger sequencing (Table S5).
ElemeNT Navigation Tool
TATA box prediction and other detection of core promoter elements were assayed using ElemeNT Navigation Tool(Sloutskin et al., 2015). Each analyzed sequence included 1000bp region. https://www.juven-gershonlab.org/resources/element/run/
Fantom5 cage tool
Functional annotation of the mammalian genome was used to evaluate TBX18 transcription start site (TSS) against human RNAseq datasets. https://fantom.gsc.riken.jp/5/sstar/EntrezGene:9096.
QGRS mapper tool
Distribution of putative Quadruplex forming G-Rich Sequences (QGRS) in nucleotide sequences was predicted using QGRS mapper too. http://bioinformatics.ramapo.edu/QGRS/analyze.php.
Transcription factor prediction tool
Prediction of transcription factor site scan on promoter region was analyzed using http://www.ifti.org/cgi-bin/ifti/Tfsitescan.pl.
SIM and image processing
Three-color, 3D-SIM images were acquired with a commercial OMX-SR system (GE Healthcare). Toptica diode lasers with excitation at 405nm, 488nm, and 640nm were used. Emission was collected on three separate PCO.edge sCMOS cameras using an Olympus 60 × 1.42NA PlanApochromat oil immersion lens. 512 × 512 images (pixel size 6.5 µm) were captured with no binning. Acquisition was controlled with AcquireSR Acquisition control software. Z-stacks were collected with a step size of 125 nm and 15 images per optical slice (3 angles and 5 phases) using immersion oil with a refractive index 1.516, volumes varied between 2.0 and 3.5 µm per cell. Images were reconstructed using Wiener filter settings of 0.005 for the 405 nm (nuclei) channel and 0.001 for all other channels and optical transfer functions (OTFs) measured specifically for each channel with SoftWoRx 7.0.0 (GE Healthcare). Images from different color channels were registered using parameters generated from a gold grid registration slide (GE Healthcare). SIM data, including raw and reconstructed images, were subjected to SIMcheck FIJI Plugin (Ball et al., 2015) to ensure that image quality was within acceptable standards.
Cell images were analyzed using a combination of custom ImageJ/Fiji and python scripts. Images channels were separated followed by nuclear segmentation via Gaussian blur (sigma = 30) to even out illumination and auto thresholding using the Huang method (Liang-kai and Wang, 1995) and filling binary holes to create the final mask. This mask was then applied to each channel containing proteins of interest. The masked protein images were converted to 8-bit and background corrected (rolling ball radius = 2) followed by using the find maxima function in Fiji (prominence = 50) and measuring the mean intensity and location of each spot. A custom python script was used to determine puncta colocalization using a separation threshold distance of 0.25 µm as the cutoff. Colocalized puncta number and percentage relative to total puncta were recorded.
EZH2 chemical inhibitory drugs
Cells were plated on 6 well plates at day 0 and were treated with EPZ-6438 (Selleckchem, stock dilution 10mM) working solution of 1mM to final concentration of 5µM or untreated (DMSO) or treated on day 1. On day 3 the cells were harvested for RNA isolation and purification using Trizol. EPZ-6438, is a selective EZH2 inhibitor that is FDA approved for B-cell and follicular Lymphomas (NCT01897571, Epizyme). EPZ-6438 (EPZ) competes with the substrate S-adenosylmethionine (SAM) (Knutson et al., 2013).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical methods for each experiment can be found in the Figure legends as well as in the method details section. p values and individual data points for the main figures can be found in the Data S2.
Supplementary Material
Highlights.
EBdCas9 inhibits PRC2 function in precise genomic locations
EBdCas9 upregulates TBX18, p16, CDX2, and GATA3 by derepression
EBdCas9 identifies PRC2-controlled, active TATA box >500 bp upstream of TBX18 TSS
EBdCas9 is sufficient to induce transdifferentiation and repress cancer cell cycle
ACKNOWLEDGMENTS
We thank members of the H.R.-B. laboratory for helpful discussions, inspiration, and advice throughout this work. We thank Christopher Cavanaugh, Jennifer Hesson, Mary C. Regier, Pushpa Pushpa, Stuart Harper, and James Moody for help and advice throughout this work. We thank Jim Olsson at the Fred Hutchinson Cancer Center and M. Biery at the Seattle Children Research Institute for help with the DMG (DIPG-XVIIB) cell line, which was developed and generously provided by M. Monje at Stanford University. S.L. was supported by the WRF Postdoctoral Fellowship and the ISCRM Fellows program. A.P. was supported by the ISCRM Fellows Program. This work is supported in part by grants from the Brotman Baty Institute (BBI) for Precision Medicine (Catalytic Collaborations Award) for N.V., J.S., D.B., and H.R.-B. and National Institutes of Health grants R01GM097372, R01GM97372-03S1, R01GM083867, 1P01GM081619, DOD PR203328 W81XWH-21-1-0006, and AHA 19IPLOI34760143 to H.R.-B. and R42HG010855 and U01CA246503 to K.B.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110457.
DECLARATION OF INTERESTS
S.L., H.R.-B., and D.B. are co-inventors on US patent application no. 17/434,832. S.L. is a founder and stockholder at Histone Therapeutics Corp., a company that aims to develop inventions described in this manuscript. K.B. is a co-founder, board member, and equity holder of Matchstick Technologies, Inc., and the developer and maker of the PIXUL instrument.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
REFERENCES
- Adamo A, Sese B, Boue S, Castano J, Paramonov I, Barrero MJ, and Izpisua Belmonte JC (2011). LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat. Cell Biol 13, 652–659. [DOI] [PubMed] [Google Scholar]
- Amabile A, Migliara A, Capasso P, Biffi M, Cittaro D, Naldini L, and Lombardo A (2016). Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing. Cell 167, 219–232 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball G, Demmerle J, Kaufmann R, Davis I, Dobbie IM, and Schermelleh L (2015). SIMcheck: a Toolbox for Successful super-resolution structured illumination microscopy. Sci. Rep 5, 15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Wen D, Dewell S, Whitcomb SJ, Lin M, Diaz N, Elsasser SJ, Chapgier A, Goldberg AD, Canaani E, et al. (2013). Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell 155, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal M, Kumar A, and Yella VR (2014). Role of DNA sequence based structural features of promoters in transcription initiation and gene expression. Curr. Opin. Struct. Biol 25, 77–85. [DOI] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, and Pugh BF (2004). Identification and distinct regulation of yeast TATA box-containing genes. Cell 116, 699–709. [DOI] [PubMed] [Google Scholar]
- Battle SL, Doni Jayavelu N, Azad RN, Hesson J, Ahmed FN, Overbey EG, Zoller JA, Mathieu J, Ruohola-Baker H, Ware CB, and Hawkins RD (2019). Enhancer chromatin and 3D genome architecture changes from naive to primed human embryonic stem cell states. Stem Cell Rep 12, 1129–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blighe K, Rana S, and Lewis M (2018). EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling https://bioconductor.org/packages/release/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html.
- Boehm AK, Saunders A, Werner J, and Lis JT (2003). Transcription factor and polymerase recruitment, modification, and movement on dhsp70 in vivo in the minutes following heat shock. Mol. Cell Biol 23, 7628–7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsztyk K, Mar D, Wang Y, Denisenko O, Ware C, Frazar CD, Blattler A, Maxwell AD, Macconaghy BE, and Matula TJ (2019). PIXUL-ChIP: integrated high-throughput sample preparation and analytical platform for epigenetic studies. Nucleic Acids Res 47, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N (2017). Polycomb complexes in X chromosome inactivation. Philos. Trans. R. Soc. Lond. B Biol. Sci 372, 20170021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D, Haque SJ, Cecchi RJ, Kowal EJK, Buchthal J, et al. (2016). Comparison of Cas9 activators in multiple species. Nat. Methods 13, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lin Z, Barrett L, Dai L, and Qin Z (2020). Identification of new therapeutic targets and natural compounds against diffuse intrinsic pontine glioma (DIPG). Bioorg. Chem 99, 103847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Lin YT, Gallegos DA, Hazlett MF, Gomez-Schiavon M, Yang MG, Kalmeta B, Zhou AS, Holtzman L, Gersbach CA, et al. (2019). Enhancer histone acetylation Modulates transcriptional bursting dynamics of neuronal activity-inducible genes. Cell Rep 26, 1174–1188 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cofsky Joshua C, Soczek KM, Knott Gavin J, Eva N, and Doudna Jennifer A (2021). CRISPR-Cas9 bends and twists DNA to read its sequence Preprint at. bioRxiv 10.1101/2021.09.06.459219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RT, and Struhl G (2017). Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356, eaai8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Arm-strong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero FJ, Huang Z, Grenier C, He X, Hu G, Mclendon RE, Murphy SK, Hashizume R, and Becher OJ (2017). Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol. Cancer Res 15, 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar TM, Oksuz O, Saldana-Meyer R, Descostes N, Bonasio R, and Reinberg D (2019). Active and repressed chromatin domains exhibit distinct nucleosome Segregation during DNA replication. Cell 179, 953–963 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D, Gilbert N, Fan Y, Skoultchi AI, Wutz A, and Bickmore WA (2010). Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 38, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreccio A, Mathieu J, Detraux D, Somasundaram L, Cavanaugh C, Sopher B, Fischer K, Bello T, Hussein AM, Levy S, et al. (2018). Inducible CRISPR genome editing platform in naive human embryonic stem cells reveals JARID2 function in self-renewal. Cell Cycle 17, 535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagin S, Nelson JD, Castner DG, Denisenko O, and Bomsztyk K (2008). Microplate-based chromatin immunoprecipitation method, Matrix ChIP: a platform to study signaling of complex genomic events. Nucleic Acids Res 36, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NJ, Follmer NE, Simon MD, Aghia G, and Butler JD (2009). Polycomb proteins remain bound to chromatin and DNA during DNA replication in vitro. Cell 137, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Dong R, Du J, Ma P, Wang S, and Fan Z (2013). Depletion of histone demethylase KDM2A inhibited cell proliferation of stem cells from apical papilla by derepression of p15INK4B and p27Kip1. Mol. Cell Biochem 379, 115–122. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RH, and Smolik S (2000). CBP/p300 in cell growth, transformation, and development. Genes Dev 14, 1553–1577. [PubMed] [Google Scholar]
- Groner AC, Meylan S, Ciuffi A, Zangger N, Ambrosini G, Denervaud N, Bucher P, and Trono D (2010). KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. Plos Genet 6, e1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel-hertsch R, Beraldi D, Lensing SV, Marsico G, Zyner K, Parry A, Di Antonio M, Pike J, Kimura H, Narita M, et al. (2016). G-quadruplex structures mark human regulatory chromatin. Nat. Genet 48, 1267–1272. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. (2010). Distinct epigenomic land-scapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D’ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, and Gersbach CA (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, and Margueron R (2017). Mechanisms regulating Prc2 recruitment and enzymatic activity. Trends Biochem. Sci 42, 531–542. [DOI] [PubMed] [Google Scholar]
- Illingworth RS, Moffat M, Mann AR, Read D, Hunter CJ, Pradeepa MM, Adams IR, and Bickmore WA (2015). The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev 29, 1897–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, and Zhang F (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor N, Liang W, Marban E, and Cho HC (2013). Direct conversion of quiescent cardiomyocytes to pacemaker cells by expression of Tbx18. Nat. Biotechnol 31, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinath V, Faini M, Poepsel S, Reif D, Feng XA, Stjepanovic G, Aeber-sold R, and Nogales E (2018). Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 359, 940–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, and Maehr R (2015). Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 12, 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikin O, D’antonio L, and Bagga PS (2006). QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res 34, W676–W682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, and Orkin SH (2013). Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol 9, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, et al. (2013). Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. U S A 110, 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Chen L, Jiao L, Jiang X, Lian F, Lu J, Zhu K, Du D, Liu J, Ding H, et al. (2014). Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J. Med. Chem 57, 9512–9521. [DOI] [PubMed] [Google Scholar]
- Kreitzer FR, Salomonis N, Sheehan A, Huang M, Park JS, Spindler MJ, Lizarraga P, Weiss WA, So PL, and Conklin BR (2013). A robust method to derive functional neural crest cells from human pluripotent stem cells. Am. J. Stem Cells 2, 119–131. [PMC free article] [PubMed] [Google Scholar]
- Krendl C, Shaposhnikov D, Rishko V, Ori C, Ziegenhain C, Sass S, Simon L, Muller NS, Straub T, Brooks KE, et al. (2017). GATA2/3-TFAP2A/C transcription factor network couples human pluripotent stem cell differentiation to trophectoderm with repression of pluripotency. Proc. Natl. Acad. Sci. U S A 114, E9579–E9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubaczka C, Senner C, Arauzo-bravo MJ, Sharma N, Kuckenberg P, Becker A, Zimmer A, Brustle O, Peitz M, Hemberger M, and Schorle H (2014). Derivation and maintenance of murine trophoblast stem cells under defined conditions. Stem Cell Rep 2, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprell F, Finkl K, and Muller J (2017). Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356, 85–88. [DOI] [PubMed] [Google Scholar]
- Laugesen A, Hojfeldt JW, and Helin K (2016). Role of the polycomb repressive complex 2 (PRC2) in transcriptional regulation and cancer. Cold Spring Harb Perspect. Med 6, a026575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Yu JR, Granat J, Saldana-meyer R, Andrade J, Leroy G, Jin Y, Lund P, Stafford JM, Garcia BA, et al. (2019). Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev 33, 1428–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Yu JR, Kumar S, Jin Y, Leroy G, Bhanu N, Kaneko S, Garcia BA, Hamilton AD, and Reinberg D (2018). Allosteric activation dictates PRC2 activity independent of its recruitment to chromatin. Mol. Cell 70, 422–434 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang-kai H, and Wang M-J (1995). Image thresholding by minimizing the measures of fuzziness. Pattern Recognition 28, 41–51. [Google Scholar]
- Liao HK, Hatanaka F, Araoka T, Reddy P, Wu MZ, Sui Y, Yamauchi T, Sakurai M, O’keefe DD, Nunez-delicado E, et al. (2017). In vivo target gene activation via CRISPR/Cas9-Mediated trans-epigenetic Modulation. Cell 171, 1495–1507 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang C, Liu W, Li J, Li C, Kou X, Chen J, Zhao Y, Gao H, Wang H, et al. (2016). Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537, 558–562. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et al. (2016). CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell 18, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, and Reinberg D (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med 23, 483–492. [DOI] [PubMed] [Google Scholar]
- Moody JD, Levy S, Mathieu J, Xing Y, Kim W, Dong C, Tempel W, Robitaille AM, Dang LT, Ferreccio A, et al. (2017). First critical repressive H3K27me3 marks in embryonic stem cells identified using designed protein inhibitor. Proc. Natl. Acad. Sci. U S A 114, 10125–10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, and Giraldez AJ (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yabuta Y, Okamoto I, Sasaki K, Iwatani C, Tsuchiya H, and Saitou M (2017). Single-cell transcriptome of early embryos and cultured embryonic stem cells of cynomolgus monkeys. Sci. Data 4, 170067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okae H, Toh H, Sato T, Hiura H, Takahashi S, Shirane K, Kabayama Y, Suyama M, Sasaki H, and Arima T (2018). Derivation of human trophoblast stem cells. Cell Stem Cell 22, 50–63 e6. [DOI] [PubMed] [Google Scholar]
- Oksuz O, Narendra V, Lee CH, Descostes N, Leroy G, Raviram R, Blumenberg L, Karch K, Rocha PP, Garcia BA, et al. (2018). Capturing the onset of PRC2-mediated repressive domain formation. Mol. Cell 70, 1149–1162 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly AR, Kalb R, Finkl K, and Muller J (2015). Transcriptional repression by PRC1 in the absence of H2A monoubiquitylation. Genes Dev 29, 1487–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengue G, and Lania L (1996). Kruppel-associated box-mediated repression of RNA polymerase II promoters is influenced by the arrangement of basal promoter elements. Proc. Natl. Acad. Sci. U S A 93, 1015–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, et al. (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med 23, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeepa MM, Grimes GR, Kumar Y, Olley G, Taylor GC, Schneider R, and Bickmore WA (2016). Histone H3 globular domain acetylation identifies a new class of enhancers. Nat. Genet 48, 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha B, Home P, Ray S, Larson M, Paul A, Rajendran G, Behr B, and Paul S (2013). EED and KDM6B coordinate the first mammalian cell lineage commitment to ensure embryo implantation. Mol. Cell Biol 33, 2691–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago M, Antunes C, Guedes M, Sousa N, and Marques CJ (2014). TET enzymes and DNA hydroxymethylation in neural development and function - how critical are they? Genomics 104, 334–340. [DOI] [PubMed] [Google Scholar]
- Shechner DM, Hacisuleyman E, Younger ST, and Rinn JL (2015). Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat. Methods 12, 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloutskin A, Danino YM, Orenstein Y, Zehavi Y, Doniger T, Shamir R, and Juven-gershon T (2015). ElemeNT: a computational tool for detecting core promoter elements. Transcription 6, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-meyer R, Yu JR, Leroy G, Oksuz O, Chapman JR, Suarez F, et al. (2018). Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv 4, eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, D’ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, and Gersbach CA (2015). Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasylyk B, Derbyshire R, Guy A, Molko D, Roget A, Teoule R, and Chambon P (1980). Specific in vitro transcription of conalbumin gene is drastically decreased by single-point mutation in T-A-T-A box homology sequence. Proc. Natl. Acad. Sci. U S A 77, 7024–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weltner J, Balboa D, Katayama S, Bespalov M, Krjutskov K, Jouhilahti EM, Trokovic R, Kere J, and Otonkoski T (2018). Human pluripotent reprogramming with CRISPR activators. Nat. Commun 9, 2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Grieskamp T, Airik R, Mommersteeg MT, Gardiwal A, De Gier-De Vries C, Schuster-Gossler K, Moorman AF, Kispert A, and Christoffels VM (2009). Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ. Res 104, 388–397. [DOI] [PubMed] [Google Scholar]
- Woo Jun Shim ES, Xu J, Vitrinel B, Alexanian M, Andreoletti G, Shen S, Balderson B, Peng G, Jing N, Sun Y, et al. (2019). Comparative analysis of diverse cell states establishes an epigenetic basis for inferring regulatory genes governing cell identity Preprint at. bioRxiv 10.1101/635516. [DOI] [Google Scholar]
- Xiao L, Ma L, Wang Z, Yu Y, Lye SJ, Shan Y, and Wei Y (2020). Deciphering a distinct regulatory network of TEAD4, CDX2 and GATA3 in humans for trophoblast transition from embryonic stem cells. Biochim. Biophys. Acta Mol. Cell Res 1867, 118736. [DOI] [PubMed] [Google Scholar]
- Yan L, Yang M, Guo H, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng X, Yan J, et al. (2013). Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol 20, 1131–1139. [DOI] [PubMed] [Google Scholar]
- Yang C, Bolotin E, Jiang T, Sladek FM, and Martinez E (2007). Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 389, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ryan DJ, Wang W, Tsang JC, Lan G, Masaki H, Gao X, Antunes L, Yu Y, Zhu Z, et al. (2017a). Establishment of mouse expanded potential stem cells. Nature 550, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu B, Xu J, Wang J, Wu J, Shi C, Xu Y, Dong J, Wang C, Lai W, et al. (2017b). Derivation of pluripotent stem cells with in vivo embryonic and extraembryonic potency. Cell 169, 243–257 e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yella VR, and Bansal M (2017). DNA structural features of eukaryotic TATA-containing and TATA-less promoters. FEBS Open Bio 7, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Feng Q, Ruan Y, Komers R, Kiviat N, and Bomsztyk K (2011). Microplate-based platform for combined chromatin and DNA methylation immunoprecipitation assays. BMC Mol. Biol. 12, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JR, Lee CH, Oksuz O, Stafford JM, and Reinberg D (2019). PRC2 is high maintenance. Genes Dev 33, 903–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing, ChIP sequencing, Cut and Run sequencing and ATAC sequencing data have been deposited at NIH GEO and are publicly available as of the date of publication. Accession numbers are listed in the key and resources table. Bulk RNAseq and DEseq values are found in Data S1. Original western blot images, RT-qPCR Ct, ChIP-qPCR Ct, and plate reader values are available in Data S2. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at NIH GEO and is publicly available as of date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-OCT4 | Santa Cruz | Cat#sc-5279; RRID: AB_628051 |
| Anti-TBX18 | Santa Cruz | Cat#sc-514486 |
| Anti-mCherry | Abcam | Cat#ab183628 |
| Anti-EZH2 | cell signaling | Cat#5246s |
| Anti-H3K27me3 | active motif | Cat#39155 |
| Anti-EED | EMD Millipore | Cat#09–774 |
| Anti-JARID | cell signaling | Cat#13594s |
| Anti-H3K27ac | Active motif | 39133 |
| Anti-p300 | Santa Cruz | sc-584 |
| Anti-Pol II CTD | santa cruz | sc-47701 |
| Anti-Pol II CTD PS5 | covance | MMS-134R-200 |
| Anti-p16 | thermo fisher Scientific | MA517054 |
| Anti-p21 | cell signaling | Cat#2946 |
| Anti-H3K4me3 | abcam | ab272143 |
| Anti-H3 | abcam | AB1791 |
| Anti-GATA3 | cell signaling | Cat#5852 |
| Anti-CGB | Invitrogen | Cat#PA558598 |
| Anti-HLA | Novus Biologicals Inc | Cat#NBP1–43123 |
| Anti-Actin | Cell Signaling | Cat#4970S |
| Anti-Flag | Sigma-Aldrich | Cat#F7425 |
| Anti-Cas9 | cell signaling | Ab #14697 |
| Anti 5mC | Active Motif | AB_2793653 |
| Anti 5hmC | Active Motif | AB_10013602 |
|
Bacterial and virus strains | ||
| 10-BETA Competent Cells | New England BioLabs | C3019H |
| DH5-alpha Competent Cells | New England BioLabs | C2987I |
|
Chemicals, peptides, and recombinant proteins | ||
| Matrigel | Corning | 356231 |
| Doxycycline | Millipore-Sigma | D9891–10G |
| Neurobasal | Thermo Fisher Scientific | 21103–049 |
| N2 supplement | Thermo Fisher Scientific | 17502–048 |
| B27 supplement | Thermo Fisher Scientific | 12587–010 |
| knockout serum replacement | Thermo Fisher Scientific | A3181502 |
| hLIF | SPEED Biosystems, LLC | YSP1249 |
| GS3Ki | Selleckchem | S7566 (2017), S2924 (2014), 72054 (2016) |
| Rocki Y27632 | R & D Systems (R&D) | 1254 |
| (S)-(+)-Dimethindene maleate Mino | santa Cruz Biotechnology Inc | sc-361329 |
| IWR | Selleckchem | S7086 |
| DMEM/F12 w/o HEPES or L-Glu | Invitrogen (Gibco/BRL Life Tech) | 21331–020 |
| l-ascorbic acid 2-phosphate Mg | sigma | A8960–5G |
| Insulin-Transferrin-Selenium | Fisher Scientific | 41400045 |
| NaHCO3 | sigma | S7277 |
| FGF4 | Life technologies | PHG0263 |
| TGF-beta1 | peprotech | 100–21 |
| Heparin | Stem Cell Technologies | 7980 |
| ITS-X supplement | Thermo fisher | 51500056 |
| NRG1 | Cell Signaling | 5218SC |
| A83–01 | Miltenyi Biotec Inc. | 130–105-333 |
| Forskolin | StemCell Technologies | 72112 |
| Alamar Blue | Thermo fisher Scientific | A50100 |
| Click iT EdU | ThermoFisher Scientific | C10337 |
|
Critical commercial assays | ||
| qiagen PCR purification | Quiagen | 28104 |
| qiagen gel purification | Quiagen | 28706 |
| mini-prep | Quiagen | 27104 |
| maxi-prep | Quiagen | 12662 |
| T7 maxi invitro transcription | invitrogen | AM1314 |
| Iscript | biorad | 1708891 |
| RT | Applied Biosytems | 205111 |
| RNAiMax | invitrogen | 13778030 |
| spCas9 | sigma | CAS9PROT |
| Cell Line NucleofectorTM Kit V | lonza | VCA-1003 |
| Quick Extract | Lucigen | QE09050 |
| ExoSAP-IT | Thermofisher | 78200.200.UL |
| GoTaq® DNA Polymerase | promega | M3001 |
| pGEM®-T Easy Vector Systems | promega | A1360 |
| Q5® High-Fidelity DNA Polymerase | NEB | M0491S |
|
Deposited data | ||
| RNA-seq data | GEO | GSE155022 |
| ChIP-seq data | GEO | GSE195552 |
| Cut and Run data | GEO | GSE195553 |
| ATAC-seq data | GEO | GSE195699 |
| DEseq and bulk RNAseq | This study | Data S1 |
|
Experimental models: Cell lines | ||
| WTC-11 | Coriell Institute | GM25256 |
| Elf1-iCas9 | Ferreccio et al. | RRID:CVCL_VR49 |
| IR-MEF | ISCRM hESC CORE | N/A |
| DIPG | gift from M Monje at Stanford University | N/A |
| WTC-11 EBdCas9 | This study | N/A |
| WTC-11 NCdCas9 | This study | N/A |
| WTC-11 EBdCas9 TBX18 TATAdeletion clone 11 |
This study | N/A |
| WTC-11 EBdCas9 TBX18 TATAdeletion clone 23 |
This study | N/A |
| Elf1 EBdCas9 | This study | N/A |
| Elf1 NCdCas9 | This study | N/A |
|
Oligonucleotides | ||
| See Table S5 for oligonucleotide information | N/A | |
|
Recombinant DNA | ||
| KRAB-dCas9 | Concklin Lab | Mandegar et al., 2016 |
| EB-Flag | Our Lab | Moody et al., 2017 |
| NC-Flag | Our Lab | Moody et al., 2017 |
| EBdCas9 | This paper | N/A |
| NCdCas9 | This paper | N/A |
|
Software and algorithms | ||
| Element | Element | http://lifefaculty.biu.ac.il/gershon-tamar/index.php/element-description/element |
| Fantom | FANTOM 5 | http://fantom.gsc.riken.jp/5/ |
| R | The R Foundation | https://www-r-project-org.offcampus.lib.washington.edu/ |
| ImageJ | NIH | https://imagej-nih-gov.offcampus.lib.washington.edu/ij/ |
| Microsoft office | Microsoft | https://products.office.com |
| Snapgene | Snapgene | https://www.snapgene.com |
| Transcription factor prediction Tool | http://www.ifti.org/cgi-bin/ifti/Tfsitescan.pl | |
| QGRS Mapper Tool | http://bioinformatics.ramapo.edu/QGRS/analyze.php | |
| GEO DOI | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE195555 |