

Abstract
2,7-Diazapyrenes are promising azaaromatic scaffolds with a unique structural geometry and supramolecular properties. This core moiety and its derivatives with some N-methyl cations like N-methyl-2,7,-diazapyrenium, and N,N′-dimethyl-2,7-diazapyrenium attract special attention due to their challenging photophysical properties, especially in the context of interactions with DNA and some of its mononucleotides. This review focuses on the analysis of the main synthetic approaches to 2,7-diazapyrene and its functional derivatives employing various strategies under different reaction conditions. The opportunities of applications of 2,7-diazapyrenes, including their remarkable photophysical and supramolecular properties, DNA-bindings, in sensors, molecular electronics, supramolecular systems, and related areas are also highlighted.
This review discusses the synthetic routes for 2,7-diazapyrene derivatives and highlights the opportunities for application of 2,7-diazapyrenes, including their remarkable photophysical and supramolecular properties, in sensors and related areas.
Introduction
Among many polyaromatic hydrocarbons (PAHs) pyrenes are probably the most explored ones due to their unique photophysical/optical properties and a broad range of their applications has been found in various fields from single molecule-based chemosensors,1–5 including those for gases6–8 to fluorescent materials for bioimaging applications,9–11 stimuli (temperature, pH, pressure) responsive molecular sensors12–14 and advanced functional materials.15–19 Modification of pyrene at 1-, 3-, 6- and 8-positions makes the maximum contributions to the HOMO–LUMO levels of pyrene and significantly influences the S1 ← S0 transitions.16,20–23 The 2,7-positions of pyrenes (Fig. 1) are located on the nodal planes of the HOMO and LUMO, and in general have no influence on HOMO–LUMO levels. However, according to several reports,16,23 the modification of 2- and 7-positions in pyrene can reverse the order of HOMO/HOMO−1 and/or LUMO/LUMO+1 and, at the same time, adjust the band gap. In general, donor moieties in these positions destabilize the HOMO−1 and the acceptor moieties stabilize the LUMO+1 of the pyrene core.23 2,7-Diazapyrene (DAP) as well as N-alkyl (alkyl = Me, allyl or benzyl) cations like N-alkyl-2,7,-diazapyrenium (ADAP+), and N,N′-dialkyl-2,7-diazapyrenium (DADAP2+) can be considered as examples of pyrenes with acceptor moieties introduced in 2,7-positions of pyrene core. Due to the intriguing electronic properties 2,7-azapyrenes attract special attention, especially in context of interactions with various charged and neutral guest molecules or in construction of supramolecular assemblies.
Fig. 1. (a) Atom numbering system in pyrene with principle Cartesian coordinate system used for pyrene; (b) the four frontier orbitals of pyrene; (c) optical transitions of pyrene. Reproduced be the permission of ref. 23 Copyrights © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.

Owing to broad applications of 2,7-diazapyrenes in different fields, synthesis of these scaffolds is a challenging task to the chemists. So far, the synthetic approaches towards 2,7-diazapyrenes are not very well explored. In this short review, we have depicted and discussed the most common synthetic routes for 2,7-diazapyrene and its derivatives. In addition, we have highlighted the most important opportunities on applications of 2,7-diazapyrenes, based on their intriguing photophysical and supramolecular properties, in sensorics, molecular electronics, supramolecular assembling, and some related areas.
Electronic structure and HOMO–LUMO-distribution for 2,7-diazapyrenes vs. pyrene
Unlike parent pyrene 2,7-diazapyrenes have unique architecture with N-atoms located in 2,7-positions of pyrene core which influence dramatically on the electronic structures of the azapyrene π-system and also provides some attractive functionalities, such as imine–amine redox conversion and ability to coordinate metal cations or ability to form proton or quaternary alkyl salts. Thus, 2,7-diazapyrenes are able to form mono- and dications (Fig. 2). According to the reports24,25 the character of the singly occupied MO in one-electron reduced compounds can be effected by removal of electron density from the potentially coordinating pyridyl nitrogen centres.
Fig. 2. Pyrene vs. 2,7-diazapyrenes.

The dication form of N,N′-dimetylated 2,7-diazapyrene and 2,7-dimethyl-2,7-dihydrobenzo[lmn][3,8]phenanthrolinium salt is able to participate in imine–amine interconversion to provide molecules with switchable aromaticity/antiaromaticity by means of two-electron reduction/oxidation to form dihydro-2,7-diazapyrene or transform back to the 2,7-diazapyrenium dication.25,26 According to the literature, the two-electron-reduced forms of 2,7-diazapyrene exhibit distinct antiaromatic character owing to their 16π electron conjugation. And two-electron reduction of the conventional N,N′-dimethyl-4,4′-bipyridyl dication, methyl viologen (MV2+) did not result in antiaromaticity due to the lack of a cyclic conjugation circuit. In addition, the authors24 observed a quite low oxidation potential (−1.34 V) for 1,3,6,8-tetraarylated dihydro-2,7-diazapyrene as a result of combination of electron-rich N-heterocyclic system of 2,7-dimetylated dihydro-2,7-diazapyrene and the intrinsic destabilization of the HOMO energy level derived from its 16π antiaromatic nature.
To study the influence of the nature of substituents at the 2,7-positions of (aza)pyrene core on the HOMO–LUMO distributions we carried out the DFT calculations of 2,7-diazapyrene, its mono- and dicationic forms, as well as dihydro-2,7-diazapyrenes compare to pyrene. The results are presented in Table 1 and the details are presented in ESI.†
Calculated energy of HOMO/LUMO and energy gaps for pyrene and 2,7-diazapyrenesa.
| Compound | HOMO, eV | LUMO, eV | ΔE, eV |
|---|---|---|---|

|

|

|
3.8445b |

|

|

|
3.8658 |

|

|

|
3.5648 |
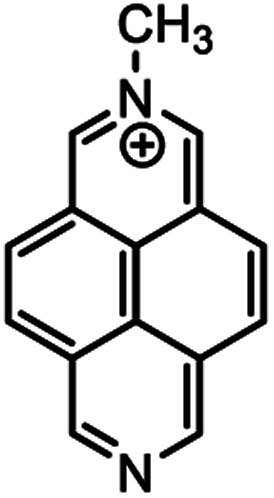
|

|

|
3.6016 |

|

|

|
3.5324 |
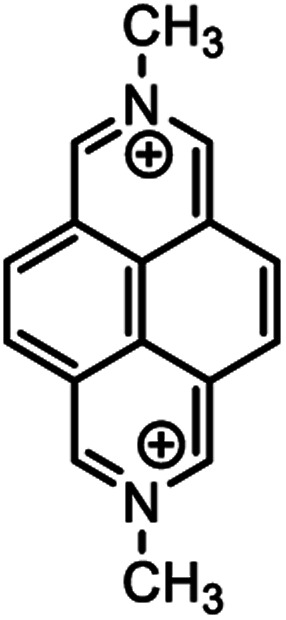
|
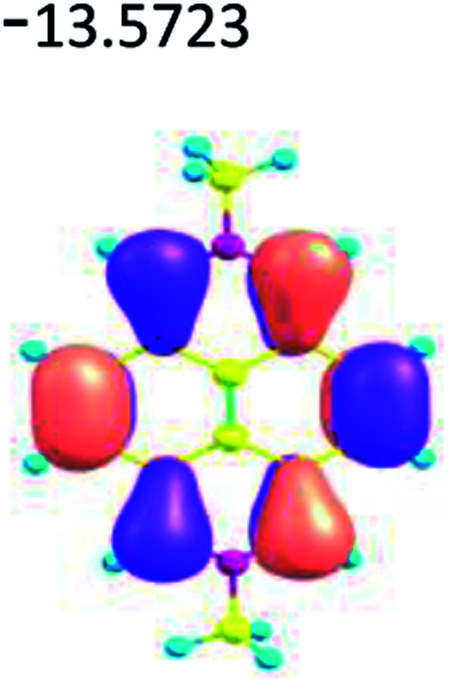
|

|
3.6042 |

|

|

|
1.0514 |

|

|

|
1.1159 |
According to the results of the calculations, the introduction of nitrogen atoms at the 2 and 7 positions of pyrene in case of 2,7-diqazapyre has almost no influence on both distribution of HOMO–LUMO and on the band gap. And the introduction of quaternary nitrogen atoms into the 2,7-positions of pyrene core reduces the band gap and LUMO energy levels (−6.17 to −10.43 eV) of resulted diazapyrenes, while LUMO orbitals are located on 2,7-positions of 2,7-diazapyrenes unlike pyrene. It is worthy to mention that, for n-type organic semiconductors the LUMO energy levels must be lower than −4.0 eV to protect the material against oxidation by ambient O2 and H2O.32,33 In case of dihydro-2,7-diazapyrenes the band gap could be reduced to values as low as 1.05–1.11 eV, and this is a characteristic feature of antiaromatic compounds, which are expected to find applications as key components of field-effect transistors as well as other organic electronic devices. Some common synthetic approached to dihydro-2,7-diazapyrenes will be discussed below.
Common synthetic approaches towards 2,7-diazapyrene and its derivatives
According to a literature, four main synthetic approaches can be highlighted to construct of 2,7-diazapyrene core 1 (Scheme 1).
Scheme 1. Four main routes to construct of 2,7-diazapyrene derivatives.

The route 1 is the most common to construct the 2,7-diazapyrene core 1 since 1,4-dihydronaphthalene-1,4,5,8-tetracarboxylic acid 2 or its cyclic anhydride 3 are the mostly commercially available. While routes 2–4 so far were reported only by few examples. Indeed, the synthesis of 1 from 1,4,5,8-substituted naphthalene 5, [2.2](3,5)pyridinophane 6 and 2,3-dihydro-1H-benzo[de]isoquinoline 7 is a non-trivial task and requires harsh conditions in multystep synthesis.
Several approaches to construct naphthalenetetracarboxylic diimide 4 as a main precursor of 2,7-diazapyrene are reported starting from 1,5-dichloro-8 (ref. 34) or 1,4-dihalogen-substituted 9 (ref. 35 and 36) naphthalenes via the ipso-cyanation reaction and the following cyclocondensation (Scheme 2).
Scheme 2. Synthesis of compound 4via cyclocondensation. Reagents and conditions: (i), KCN, DMF; (ii), cyclization.

Route 1
In 1973, Hünig and co-workers first developed a convenient method for the synthesis of 2,7-diazapyrene 1, as well as N,N′-dimethyl-2,7-diazapyrenium salt (DMDAP2+) 10.37 In a typical experimental procedure, 1,4,5,8-naphthalenetetracarboxylic acid 11a was converted into its tetrakis(N-methylammonium) salt 11b by evaporation in aqueous methylamine solution, which was transformed into bis(N-methyl)imide 12 by heating in N-methylpyrrolidone. In the next step, obtained imide 12 was reduced to diamine, namely, 2,7-dimethyl-1,2,3,6,7,8-hexahydrobenzo[lmn][3,8]phenanthroline 13 in presence of LiAlH4 and aluminum chloride in THF. After that treating with NBS or Hg(ii) acetate converted the tertiary amine to N,N′-dimethyl-2,7-diazapyrenium salt 10. Finally, the expected 2,7-diazapyrene 1 was obtained from that salt by heating with potassium iodide in triethylene glycol.
The authors also developed another straightforward approach by using selenium as an oxidant. Thus, if 2,7-dimethyl-1,2,3,6,7,8-hexahydrobenzo[lmn][3,8]phenanthroline 13 was heated at 265 °C in presence of selenium, the 2,7-diazapyrene 1 was formed directly. Later, various researchers followed this same procedure to synthesize this moiety by modifying few conditions (Scheme 3).
Scheme 3. Synthetic route towards 2,7-diazapyrene & N,N′-dimethyl-2,7-diazapyrenium salt. Reagents and conditions: (i), MeNH2 (aq.), heating; (ii), LiAlH4, AlCl3, THF; (iii), Se, 265 °C, 4 h; (iv), (1). NBS, (2). HBF4; (v), Hg(OAc)2, HBF4; (vi), (CH3)3O+BF4−; (vii), CH3I, DMSO; (viii), KI, triethylenglycol, 260 °C.

Several examples of the synthesis of 2,7-diazapyrene core 1 from the commercially available precursor, 1,4,5,8-naphthalenetetracarboxylic anhydride 3, were presented in the literature.38–40 For example, Stang et al.38 proposed a modified Hünig procedure37 for the synthesis 2,7-daiazapyrene by converting 3 to diimide 14a (R = Me), which was reduced to 15a by LiAlH4/AlCl3 in THF and aromatized in the presence of Pd/C to afford 2,7-diazapyrene 1. Sotiriou-Leventis and co-workers40 developed an alternative and convenient approach via very fast, low-priced, and high yielded synthesis of 2,7-diazapyrene. In their work, compound 1 was synthesized in three steps with high yields starting from commercially available 1,4,5,8-napthalene tetracarboxylic dianhydride 3 which first reacts with concentrated ammonium hydroxide solution to give 1,4,5,8-napthalenetetracarboxylic diimide 14b (R = H). The latter compound was reduced with borane in refluxing THF to give 1,2,3,6,7,8-hexahydro-2,7-diazapyrene 15b (R = H) which was oxidized with activated manganese dioxide in refluxing benzene giving 2,7-diazapyrene 1 (Scheme 4).
Scheme 4. Synthesis of 2,7-diazapyrene from 1,4,5,8-naphthalenetetracarboxylic anhydride. Reagents and conditions: (R = Me) (i) 40% aq. MeNH2, reflux, 97%; (ii) LiAlH4/AlCl3, THF, heating, 16 h, 73%; (iii) 10% Pd/C, 300 °C, 1 h, 45%; (R = H) (i) (a) NH4OH, r.t., 96%; (ii) BH3, THF, reflux; (b) HCl; (c) K2CO3; 77%; (iii) MnO2, benzene, reflux, 71%.

An alternative simple procedure of the synthesis of various N,N′-bis(2-propenyl)-2,7-diazapyrenium salts 16a–d in good yields was reported (Scheme 5).41 The first step involves the preparation of N,N′-bis(2-propenyl)-1,4,5,8-naphthalenetetra-carboxylic diimide 18 by the reaction of 1,4,5,8-naphthalenetetracarboxylic dianhydride 3 with allylamine 17 in water at room temperature for 6 h which is a simplified modification of the Hünig procedure.37 The second step involves AlCl3-catalyzed reduction of 18 with LiAlH4 to yield 1,3,6,8-tetrahydro-N,N′-bis(2-propenyl)-2,7-diazapyrene 19. The last step involves the oxidation of 19 by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in anhydrous MeCN at room temperature followed by the addition of concentrated acid to obtain the corresponding diazapyrenium salt 16a–d.
Scheme 5. Synthesis of N,N′-bis(2-propenyl)-2,7-diazapyrenium quaternary salts. Reagents and conditions: (i), H2O; (ii), LiAlH4, AlC3, THF; (iii), DDQ, CH3CN, (iv), HBr; (v), HCl; (vi), HClO4.

Routes 2–4
In 1968, unsubstituted 2,7-diazapyrene 1 was synthesized from pyridinophane42via the method of rather theoretical interest, which involved the oxidative cyclocondensation of [2.2](3,5)pyridinophane 6 with Pd/C upon heating (Scheme 6).
Scheme 6. Synthesis of 2,7-diazapyrene via oxidative cyclocondensation. Reagents and conditions: (i), Pd/C, 290 °C, 3 h.

In 2008, Aksenov et al. developed a convenient method for the synthesis of 2,7-diazapyrene 1 by heating dihydrophenalene 20 (namely, 2,3-dihydro-1H-benzo[de]isoquinoline) with 1,3,5-triazine 21 in polyphosphoric acid (PPA) affording 55% yield (Scheme 7).43
Scheme 7. Synthesis of 2,7-diazapyrene in presence of PPA. Reagents and conditions: 1,3,5-triazine, PPA, 100–140 °C, 3 h.

Kamata and Wasada44 reported synthesis of 2,7-diazapyrene 1 from 1,4,5,8-tetrakis(bromomethyl)naphthalene 21 according to the Scheme 8, involving the cyclization, carboxylation, hydrolysis and oxidative aromatization. The disadvantage of the method is the low availability of starting substrates.
Scheme 8. Synthesis of 2,7-diazapyrene from 1,4,5,8-tetrakis(bromomethyl)naphthalene. Reagents and conditions: (i), PhCH2NH2, K2CO3; (ii), ClCO2Et, THF, 55 °C; (iii), KOH, H2O-1,4-dioxane = 1 : 1; (iv), MnO2, Benzene, reflux.

Other synthetic approaches
Synthetic modification of 2,7-azapyrenes core via C–C-coupling reactions
As a common way to introduce extra functionalities to 2,7-diazapyrene core can be used the organometallic coupling procedures.
In 2018, Miyake et al. developed a novel protocol for the synthesis of various 1,3,6,8-tetrasubstituted 2,7-diazapyrenes 23, 24a–d using a combination of a reductive aromatization of naphthalene diimide 14a and Ni-catalysed Suzuki–Miyaura cross-coupling reactions. In addition, the authors also synthesized the alkylated 2,7-diazapyrene derivative 25 in presence of RANEY® nickel (Scheme 9).45
Scheme 9. Synthesis of 2,7-diazapyrene derivatives. Reagents and conditions: (i), Zn dust (16 eq.), Piv2O (16 eq.), 120 °C, 24 h; (ii), ArB(OH)2 (16 eq.), Ni(COD)2 (40 eq.), PCy3 (80% mol.), K3PO4 (16 eq.), toluene, 60 °C, 18 h; (iii), RANEY® Ni (excess), toluene, 60 °C, 5 h.

Later, the same group provided another approach to afford DAP derivatives bearing various alkyl substituents at 1,3,6,8 positions 27a–c.46 Being reminiscent to the method outlined above, the new approach was based on 1,3,6,8-tetramethoxy-2,7-diazapyrene 26 as a key intermediate. It was then subject to reductive aromatization, followed by alkylation of the latter through Ni-catalyzed cross-coupling with alkyl Grignard reagents (Scheme 10).
Scheme 10. Synthesis of 1,3,6,8-tetrasubstituted 2,7-diazapyrene derivatives bearing alkyl or methoxy groups. Reagents and conditions: (i), Zn dust (16 equiv.), MeOTf (6 eq.), 1,4-dioxane, 60 °C, 24 h.; (ii), AlkMgI (6 eq.), Ni(COD)2 (40% mol.), dcype (40% mol.), toluene, 80 °C, 24 h.

Very recently Sundermeyer and co-authors47 reported facile reductive O-triflylation and O-silylation of naphthalene diimide 14a. The resulting highly reactive 1,3,6,8-tetratriflato-2,7-diazapyrene was served as a versatile platform for further functionalizations via Sonogashira cross coupling or nucleophilic aromatic substitution by piperidine. Thus, the treatment of naphthalene diimide 14a with either Zn or potassium graphite (KC8) with the following addition of SiR3Cl or Tf2O afforded tetra-O-silylated 2,7-diazapyrenes 28a–c in 30–71% yields or tetratriflate derivative of 2,7-diazapyrene 29 in 75% yield (Scheme 11). The latter one was subject to react with acetylenes via Sonogashira cross-coupling reaction to afford tetraalkynyl 2,7-diazapyrenes 30a–b in 25–27% yields or, by ipso-amination reaction with piperidine, tetraamino-substituted 2,7-diazapyrene 31 in 65% yield. It is worthy to mention that quite similar approach was reported earlier for 2,9-diazaperylens.48 In addition, tetra-O-TMS 2,7-diazapyrenes were obtained via O-Li salts, generated in situ.49
Scheme 11. Synthesis of 2,7-diazapyrene derivatives. Reagents and conditions: (i), Zn, (8 equiv.), TMSCl (8 equiv.), 1,4-dioxane, r.t. 6 h, 68%; (ii), KC8 (4.4 equiv.), TIPSCl (5 equiv.), THF, reflux, 16 h, 30%; (iii), Zn, (8 equiv.), TBDMSCl (8 equiv.), imidazole (8 equiv.) 1,4-dioxane, 80 °C, 3 h, 71%; (iv), (1) KC8 (4.4 equiv.), 80 °C, 3 h, (2) Tf2O (4.4 equiv.), −50 °C, 3 h, DME, 76%; v, piperidine (exc.), DMSO, 90 °C, 1 h; (b) trimethylsilyl acetylene (6 equiv.), CuI (20 mol%), [Pd(dppf)Cl2] (10 mol%), THF/NEt3, 65 °C, 16 h; (vi), p-tert-butylphenyl acetylene (6 equiv.), NMe4I, CuI (20 mol%), [Pd(dppf)Cl2] (10 mol%), 1,4-dioxane/NEt3, 85 °C, 16 h.

Two-electron reduction of N,N′-dimethyl-2,7-diazapyrenium dications as an approach to 16π antiaromatic N,N′-dimethyl-2,7-dihydrodiazapyrenes
In 1996, Wardör and Kaim observed the formatin of N,N′-dimethyl-2,7-dihydrodiazapyrene (MDAP0) 32via its absorption spectrum after the two-electron reduction of MDAP2+33, however without its isolation and detailed characterization (Scheme 12).50
Scheme 12. Dicationic and neutral forms of N,N′-dimethyl-2,7-diazapyrene (MDAP).

In 2021, for the first time, 1,3,6,8-tetraaryl-N,N′-dimethyl-2,7-dihydrodiazapyrene derivatives were synthesized and fully characterized as examples of highly electron-rich 16π antiaromatic systems.24 Thus, treatment of diazapyrenes 34a and 24a with 6 equiv. of methyl triflate (MeOTf) in 1,2-dichloroethane at 80 °C for 6 h afforded dications 35a2+ and 35b2+ in 50% and 81% yield (Scheme 13a). The reaction of 35a2+ and 35b2+ with 3 equiv. of bis(pentamethylcyclopentadienyl)cobalt(ii) (CoCp*2) as reducing agent in THF at room temperature for 3 h afforded the corresponding two-electron reduced forms 36a0 and 36b0 in 52% and 78% yield (Scheme 13b). The X-ray diffraction analysis of 36a0 revealed a quinoidal structure. The 1H NMR chemical shift of the proton on the diazapyrene core of 36b0 was shifted upfield relative to that of the corresponding aromatic diazapyrene 24a. Theoretical calculations clearly supported the presence of an anticlockwise paratropic ring current.
Scheme 13. (a) Synthesis of N,N′-dimethyl-2,7-diazapyrenium dications. (b) Reduction reaction of N,N′-dimethyl-2,7-diazapyrenium dications. Reagents and conditions: (i), MeOTf (6 eq.), 1,2-dichloroethane, 80 °C, 6 h; (ii), CoCp*2 (3 equiv.), THF, r.t., 3 h, glove box.

Photophysical properties of 2,7-diazapyrenes
Changing the molecular architecture of the pyrene core by doping the carbon framework with heteroatoms, for instance, pyridine-type nitrogen, proved to be an effective strategy for tuning the electrochemical and optical properties of the resulted hetero-PAHs.31 The 2,7-diazapyrene derivatives, as nitrogen-doped analogues of pyrene, should have possess highly efficient optoelectronic properties. However, studies of the parent 2,7-diazapyrene 1 remain limited due to the lack of effective pathways for the synthesis of 2,7-diazapyrene derivatives.45
Aryl-substituted diazapyrene fluorophores 24a–d, 25, obtained by Miyake and co-workers, showed intense fluorescence both in solution of CH2Cl2 (Φf = 0.13–0.47) and in the solid state (Φf = 0.01–0.41).45 The introduction of electron-donating aryl groups to the azapyrene's periphery resulted in a significant bathochromic shift of the emission bands due to more efficient π-conjugation. The fluorescence lifetime of azapyrenes, measured in a diluted solution (1.0 × 10−6 M) at 425 nm, averaged 8 ns. Fig. 3a and b shows the electronic absorption spectra of 24a, 24d and 25 in CH2Cl2 and emission spectra of 25 in CH2Cl2. A significant change in the photophysical properties of 25 was caused by the use of protic solvents. Experiments were carried out on the protonation of trifluoroacetic acid 25 in CH2Cl2, which led to the appearance of a new absorption band at 427 nm and the emission spectrum was bathochromically shifted (Fig. 3b).
Fig. 3. (a) Absorption spectra of 24a (red), 24d (black) and 25 (blue) in CH2Cl2. (b) Absorption (black solid line: in CH2Cl2; red solid line: in 2,2,2-trifluoroethanol) and fluorescence (black dashed line: in CH2Cl2; red dashed line: in 2,2,2-trifluoroethanol) spectra (λex = 360 nm) of 25. Reproduced be the permission of ref. 45 Copyrights © 2018 Royal Society of Chemistry.

The introduction of peripheral aryl groups also exhibits a bathochromic shift of emission bands: the colours of emission were changed dramatically based on effective π-conjugation of the aryl groups (Fig. 4b and c). A new emission band at ∼500 nm was observed with increasing concentration of 25 (Fig. 4a, d and c). The lifetime measured in dilute solution (1.0 × 10−6 M) at 425 nm was 7.6 ns, while it was 19.4 ns when measured at 500 nm in a concentrated solution (1.0 × 10−2 M). Based on these results, the new emission at 500 nm was assigned to the excimer emission.
Fig. 4. (a) Fluorescence spectra of 25 in methylcyclohexane at various concentrations (normalized at 425 nm). (b) Photo of cuvette containing 24a in CH2Cl2 (4.9 × 10−5 M) under illumination (λex = 365 nm). (c) Photo of cuvette containing 24d in CH2Cl2 (6.9 × 10–5 M) under illumination (λex = 365 nm). Photos of cuvettes containing 25 in methylcyclohexane under illumination (λex = 365 nm) at (d) 1.0 × 10−6 M and (e) 1.0 × 10−2 M. Reproduced be the permission of ref. 45 Copyrights © 2018 Royal Society of Chemistry.

In addition, the electrochemical properties of diazapyrene 25 and 1,3,6,8-tetrapentylpyrene were examined by cyclic voltammetry (CV). Diazapyrene 25 exhibited one reversible reduction potential (−2.56 V), while a reduction peak was not observed for 1,3,6,8-tetrapentylpyrene up to −3.0 V, supporting the notion that the LUMO level of 25 is lower than that in 1,3,6,8-tetrapentylpyrene. The observed value of reduction potential for 2,7-diazapyrene 25 is close to the LUMO value obtained for unsubstituted 2,7-diazapyrene by using DFT calculations at the B3LYP/6-311G* level of theory (Table 1). The experimentally confirmed low LUMO level motivated the authors to evaluate the electron mobility of 25 in the hope to find a new n-type semiconducting material.45
In 2021, the same Miyake group comprehensively carried out photophysical studies of 16π antiaromatic 1,3,6,8-tetraaryl-N,N′-dimethyl-2,7-dihydrodiazapyrene 36b0 in comparison with N,N′-dimethyl-2,7-diazapyrenium dication 35b2+ and 1,3,6,8-tetraaryl-N,N′-dimethyl-2,7-diazapyrene 24a.24 The absorption spectrum in THF of 35b2+ showed a bathochromic shift relative to spectrum 24a and a vibrational structure. However, the longest wavelength maxima of the absorption spectrum in THF (up to 600 nm), as well as a weak and broad absorption band up to 1200 nm, were observed for dihydrodiazapyrene 36b0 in UV/Vis-NIR region (Fig. 5).
Fig. 5. UV/Vis-NIR absorption spectra of 24a (black), 35b2+ (blue), and 36b0 (red) in THF. Reproduced be the permission of ref. 24 Copyrights © 2021 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.

In addition, 2,7-diazapyrene 35b2+ exhibits an emission with a larger quantum yield (61%) than 24a (38%). The authors suggested that these changes in the optical properties resulted from not only the electric modulation due to the quaternization of the nitrogen atoms but also from the suppression of the rotational dynamics of the peripheral aryl groups due to the steric repulsion between methyl groups on the nitrogen atoms and the aryl groups.24 According to the results of cyclic voltammetry studies, 35b2+ showed a particularly low oxidizing potential (Ered1 = −1.01 and Ered2 = −1.34 V). These values were drastically shifted to higher potential compared to those of 24a (Ered1 = −2.14 and Ered2 = −2.53 V), which indicated that imparting a dicationic nature to the diazapyrenes efficiently increased their electron affinity.24 According to time-dependent DFT calculations at the B3LYP/6-31G(d) level of theory for the compound 24a the calculated LUMO value was −2.09 eV, which is somewhat close to the one (−2.66 eV) estimated based on results on CV (ELUMO = − (4.80 + Ered1) eV). The lowest energy band of 36b0 was assigned to the forbidden HOMO–LUMO transition. The HOMO–LUMO energy gap was 1.87 eV, which is significantly less than that of aromatic 2,7-diazapyrene (data article24 and Table 1), and is a characteristic feature of antiaromatic compounds.
Absorption and emission spectra of 1,3,6,8-tetratriflato-2,7-diazapyrene 29, 1,3,6,8-tetraalkynyl-2,7-diazapyrene derivatives 30a,b, 1,3,6,8-tetraamino-2,7-diazapyrene 31 and tert-butyl-dimethylsilyl ether 28c as analogues of Miyake's tetraaryl-2,7-diazapyrene derivatives 24a–d, 25 were recorded in DCM (Fig. 6).47
Fig. 6. (a) UV-Vis spectra of 28c, 29, 30a, 30b, 31 recorded in DCM (c ≈ 10−5 M); (b) photoluminescence (PL) spectra of 28c, 29, 30a, 30b, recorded in DCM (c ≈ 10−7 M, λex. = 350 nm); (c) photographs of cuvettes containing, 28c, 29, 30a, 30b under UV-light (λ = 366 nm); (d) Kohn–Sham molecular orbitals of 30a (def2-TZVPP/PBE level of theory). Reproduced be the permission of ref. 47 Copyrights © 2021 Royal Society of Chemistry.

Long-wavelength of both absorption (from 435 to 480 nm) and emission (up to 500 nm), high extinction coefficients of the order of 4 × 105 M−1 cm−1 for tetraalkynyl-2,7-diazapyrene derivatives 30a,b, (Fig. 6a–c) as well as the high value of the fluorescent quantum yield for 30b (33%) (Fig. 6b) were comparable to spectroscopic data of Miyake's derivatives 24a–d, 25. This shows that the use of alkyne substituents at 1,3,6,8-positions is an effective way to tune the optical properties by conjugation to the coplanar π-system. This is supported by DFT-calculated Kohn–Sham frontier molecular orbitals of 30a (Fig. 6d). This is different from Miyake's derivatives displaying four aryl substituents preferentially in a nonconjugated conformation with respect to the central plane. Therefore, the aryl substituents do only contribute minorly to the HOMO and LUMO, resulting in larger HOMO–LUMO energy gaps and more hypsochromically shifted UV-Vis and photoluminescence maxima.47 According to the CV data tetraalkyne 30a, tetratriflate 29 and, remarkably, tetraamine 31 showed comparably high first reduction potentials (Ered1 = −1.13 − 1.59 eV), but also low first oxidation potentials (Eox1 = 0.14–1.00 eV), which corresponds to the ELUMO = −3.57 − 3.31 eV and EHOMO = −4.94 − 5.80 eV (EHOMO/LUMO = − (4.80 + Eox1/red1) eV). According to the authors, these 2,7-azapyrenes can be classified as electron mediators, both electron donors and acceptors. Both alkyne and amine functionalization leads to smaller optical HOMO–LUMO energy gaps than silyl ethers 28a–c and also small electrochemical gaps due to energetically low first oxidation (electrochemical HOMO) and high first reduction (electrochemical LUMO) waves. Azapyrenes 30a and 30b show a higher first oxidation potential in comparison to silyl ethers 28a–c. This is characteristic of the energetic stabilization of the LUMO of alkynes 30a and 30b in comparison to silyl ethers. It is worthy to mention that according to time-dependent DFT calculations at the def2-TZVPP/PBE level of theory for the compounds 28, 29a, 30a-b, 31 their LUMO energy level values of −3.95 eV (28), −1.96 eV (29a), −3.39 eV (30a), −3.24 eV (30b), −1.87 eV (31) and HOMO energy level values of −6.29 eV (28), −4.22 eV (29a), −5.15 eV (30a), −4.72 eV (30b), −3.73 eV (31) were of some difference from the electrochemical HOMO and LUMO values, which suggests an overestimation of HOMO and LUMO levels by DFT.
Structural and electronic tuning via complexation of α-hydroxylated tetraaryl-2,7-diazapyrenes with boron Lewis acids was reported very recently.51 Tetracoordinate diazapyrene boron complexes 39a-b as separable anti- and syn-isomers were obtained via complexation of 1,3,6,8-tetra(2-hydroxyphenyl)-2,7-diazapyrene 38 with boron precursors (Scheme 14).
Scheme 14. Synthesis of 2,7-diazapyrene boron complexes 39a-b. (a) 2-Methoxyphenylboronic acid (16 equiv.), K3PO4 (16 equiv.), Ni(cod)2 (40 mol%), PCy3 (80 mol%), toluene, 80 °C, 12 h, 30%; (b) pyridine hydrochloride (400 equiv.), 200 °C, 5 h, 84%. Method A: (i) BBr3 (400 equiv.), CH2Cl2, 0 °C to r.t., 3 h, 58% (39a), 7% (39b); (ii) PhMgBr (6 equiv.), toluene, 0 °C to r.t., 18 h. Method B: PhB(OH)2 (8 equiv.), trimethylamine (20 equiv.), benzene, 90 °C, 12 h, 10% (39a), 68% (39b).

The reaction of 1,3,6,8-tetrapivaloxy-2,7-diazapyrene 23 with 2-methoxyphenylboronic acid in the presence of catalytic amounts of Ni(cod)2 (cod: 1,5-cyclooctadiene) and tricyclohexylphosphine (PCy3) was carried out to obtain a 2,7-diazapyrene 37 in 30% yield. The reaction of demethylation of compound 37 to 1,3,6,8-tetra (2-hydroxyphenyl)-2,7-diazapyrene 38 using pyridine hydrochloride was performed. The reaction of 38 with boron tribromide followed by the addition of PhMgBr gave the corresponding boron complex as a mixture of two stereoisomers 39a-b, which were separated by column chromatography in 58% and 7% yields (method A in Scheme 14). In the case of the reaction of 38 with 8 equiv. phenylboronic acid gave 39a (anti-) and 39b (syn-) in 10% and 68% yields respectively in the presence of triethylamine (method B in Scheme 14).
The broad absorption bands of 39a (anti-) and 39b (syn-) from 400 to 600 nm were bathochromically shifted relative to 1,3,6,8-tetra(4-tert-butylphenyl)-2,7-diazapyrene 24a as comparison compound in CH2Cl2 due to the boron complexation. Both diazapyrene boron complexes 39a-b were emissive, of which quantum yields were 25% and 41%, respectively (Fig. 7).
Fig. 7. (a) UV-Vis absorption spectra of 39a (anti-) (red line), 39b (syn-) (blue line) and 24a (black line) in CH2Cl2. (b) Emission spectra of 39a (anti-) (red line), 39b (syn-) (blue line) (λex = 450 nm) and 24a (λex = 400 nm). Reproduced be the permission of ref. 51 Copyrights © 2021 Royal Society of Chemistry.

Supramolecular charge-transfer (CT) complex 40 between bis(18-crown-6)stilbene (E) (π-donor) and 2,7-bis(3-ammoniopropyl)benzo[lmn][3,8]phenanthrolinediium tetraperchlorate (π-acceptor) was obtained as dark-colored fine-crystalline powders by slow precipitation from acetonitrile solutions of equimolar mixtures of components.52 CT complex 40 has been tested for use as a fluorescent sensor for alkaline-earth metal ions, in particular for Ca2+ cations. Complex 40 is non-fluorescent in nature, but its solution exhibits very weak fluorescence due to the presence of diazapyrene salt molecules 41, the percentage of which at C = 1.0 × 10−5 M is <1%. When Ca(ClO4)2 was added, the percentage of 41 increases due to the binding of Ca2+ ions to crown ether fragments of stilbene 42, which leads to an increase in fluorescence intensity (fluorescence quantum yield 41 in MeCN was 31%) (Scheme 15).
Scheme 15. Reaction of Ca2+ cations with Complex 40.

The inclusion complexation behaviors of the tetrasulfonated 1,5-dinaphtho-38-crown 434− with N,N′-dimethyl-2,7-diazapyrenium salt 44 (DMDAP2+) and N,N′-dibenzyl-2,7-diazapyrenium salt 45 (DBDAP2+) were studied systematically by Liu et al.53 This 2,7-diazapyrenium dications 44-45 were chosen to study the cooperative π-stacking interactions in 434− with the final goal to enhance the binding ability of flexible and non preorganized macrocyclic compounds. Furthermore, in order to demonstrate the roles of π-conjugate and substituent effect clearly, the diquaternary salts of 4,4′-bipyridine 46 (BV2+) and 1,10-phenanthroline 47 (DP2+) were chosen as the reference compounds (Fig. 8).53
Fig. 8. Structures of host (43) and guest (44–47) azaheterocyclic molecules.

According to the absorption spectra of 430 nm for 434−, 44 (DMDAP2+) or 45 (DBDAP2+), there was no absorption, while the complexes of 44⊂434− or 45⊂434− showed the appearance of a broad band from 440 to 500 nm. (Fig. 9b), which confirms the significant charge-transfer (CT) interaction between electron-donor naphthalenesulfone fragments and electron-withdrawing 2,7-diazapyrenium dications (DAP2+) units. The donor–acceptor interaction in these supramolecular complexes could also be readily distinguished by the characteristic color changing of the solution. The dicationic substrates alone were colorless or pale yellow, but instantly turn to brilliant orange in the presence of macrocycle 434−(inset photos in Fig. 9).
Fig. 9. UV-Vis absorption spectra of (a) the spectral sum of the individual components 43 and DMDAP2+ and (b) complex DMDAP2+⊂43 in water at 20 °C. Inset: visible color changes of DP2+, BV2+, DMDAP2+, and DBDAP2+ (from left to right) in the absence and presence of 43. Reproduced be the permission of ref. 53 Copyrights © 2014 American Chemical Society.

According to the authors, a more enhanced association constant Ka value up to 108 M−1 was observed for the complex 44 (DMDAP2+)⊂43 (Fig. 10). Different from previous results, the electrostatic attraction is replaced by the π-stacking interaction as the most important driving force to preorganize the host–guest superstructures. These obtained results can greatly overcome the obstacles of binding affinity in the flexible and large-sized crown ether and then lead to a multicomponent assembly of MV2+·DMDAP2+⊂43 involving two different guest molecules in the solid state.
Fig. 10. An extraordinarily strong association constant (Ka) up to 108 M−1 order of magnitude in water for the complex 44(DMDAP2+)⊂43. Reproduced be the permission of ref. 53 Copyrights © 2014 American Chemical Society.

Applications of 2,7-diazapyrene derivatives
In this section, we succinctly spotlight some current prospective directions of the utilization of 2,7-diazapyrenes in applied science to further emphasize the significance of DAP derivatives.
For instance, the intercalation of 2,7-diazapyrenes with DNA54–56 and some of its mononucleotides57 were reported. Upon interaction of 2,7-diazapyrenes with nucleic acids form stacked (“intercalation”) complexes, which for the methylated derivatives exhibit new absorption features assigned as charge-transfer (CT) transitions.58
It was observed that DMDAP2+ can function as a redox dependent receptor for aromatic carboxylates; in addition, due to water solubility, that dication could be potentially useful in electrochemical sensors for nonactivated aromatics in water.59 On the other hand, N-(3-trimethoxysilylpropyl)-2,7-diazapyrenium bromide60 can covalently bond to the framework of silica aerogels,61 and the derived luminescent material has shown sensitivity to oxygen. The corresponding dicationic salt, namely, N,N′-bis(3-trimethoxysilylpropyl)-2,7-diazapyrenium dibromide, having two possible points of attachment onto a sol–gel framework, would be expected to retain an alignment imposed by an externally applied force field during gelation.62–65
DNA binding
Lehn et al.57 described that the DMDAP2+ cations 48a-c, 49 were an attractive molecular subunits for designing artificial receptor molecules66 capable of binding anionic or neutral molecular substrates, in particular flat molecules via a stacking type of interaction as present also in intercalation processes. For this study, the authors synthesized DAP as well as various DAP salts as shown in Scheme 16. The authors first showcased the ability of DMDAP2+48a–c, 49 and bis-DAP4+50 to bind with DNA via intercalation.56 Due to the distinctive binding and photophysical properties of these intercalators, the latter can enable visible-light-induced photocleavage of DNA strands through photooxidation at the binding site. Later, a number of different works were devoted in investigating the mechanism of the related intercalations.54,59,67
Scheme 16. Synthetic routes towards various DMDAP2+ mono- and dications and bis-DAP4+ tetracations.

Obviously, such interactions between DAP derivatives and nucleic acids can be responsible for producing a multitude of potentially advantageous biological activities, including antiproliferative and antitumour ones, as well as antibacterial, antiviral and antiprotozoal effects. All these activities were discussed in detail in the recent review by Zhirov et al.68
Construction of metallacycles and metal–organic frameworks (MOFs)
Beyond binding with nucleic acids, DAP derivatives are considered as attractive scaffolds to bind with transition metals,69–75 as well as metalloids (such as boron51). Various supramolecular assemblies originated upon complexation of these azaaromatic ligands with palladium,70–73 platinum,70–72 copper,74 silver,69,74 zinc75 cations have been reported over past three decades. Depending on their structure, these frameworks can serve for different purposes. For example, DAP-based chiral MOF thin films [Zn(Cam)2DAP]n (Cam = camphoric acid) were shown75 to have strong circularly polarized luminescence (CPL) properties. Materials exhibiting such property are of great interest in terms of their potential applications in the fields of optical displays, information transmission/storage, photoelectric devices, and chiral sensors. Meanwhile, the rectangular metallacycles self-assembled from a 2,7-diazapyrenium-based ligand and square-planar Pd(ii) or Pt(ii) complexes were found71 to function as receptors for polycyclic aromatic hydrocarbons (PAHs), e.g., pyrene, phenanthrene, triphenylene, and benzo[a]pyrene. In particular, Quintela et al. demonstrated71 the potential of metallacycle 51 (Fig. 11) to be applied for the selective extraction of PAHs from an organic media to an aqueous phase. The above-mentioned example also refers to another rich area of exploiting DAP derivatives, which is discussed below.
Fig. 11. Structure of metallacyclic receptor for PAHs.

Stang's group performed interaction of a variety of bidentate ligands (bipyridine, diazapyrene, dicyanobenzene, and dicyanobiphenyl) with square-planar cis-bis(phosphine)Pt and bis(triflate)Pd complexes resulting in molecular squares in high yields via self-assembly.38
In another work, the one dimensional silver(i) co-ordination polymers one of which included a 2,7-diazapyrene ligand with formula {[Ag(2,7-diazapyrene)]BF4·MeCN} were obtained. Complex representing linear, coplanar ribbons of ligands has been synthesized for studying the influence of anions and intermolecular aromatic interactions on the orientation.69
Host–guest chemistry: construction of molecular machines
For over two decades, DAP derivatives are extensively exploited for the construction of various host–guest supramolecular systems. Specifically, diazapyrene scaffolds were shown to be employed as both the guest units and the parts of the host architectures. For example, a new synthesized photoresponsive cryptand 52 as new efficient photocontrolled host–guest recognition motif was carried out and investigated by Huang et al.76 It was found that cryptand 52 exhibit an ON-OFF binding ability to 2,7-diazapyrenium salts 53a-b as a cis-isomer only, which has been well demonstrated by various characterization techniques (Scheme 17).
Scheme 17. Complexation of cryptand 47 with DAP derivatives 48a-b.

Later, complexation of DAP-derived guests of diverse structures (including dendronised ones77,78) was also explored with regard to other hosts, such as molecular clips,77 pillar[7]arenes,79 and cucurbiturils.78,80 In particular, the liposome-encapsulated pair of cucurbit[8]uril (CB8) host and N,N′-dimethyl-2,7-diazapyrenium (DMDAP) guest was utilized for monitoring of membrane transport of peptides in real time via a label-free fluorescence-based method.80 Also, the ternary complexation of CB8 host, DMDAP guest, and certain chiral analytes was employed for the real-time monitoring of chemical reactions.81
Furthermore, 2,7-diazapyrene derivatives are successfully used for the assembly of pseudorotaxane-type molecular machines,82–85 which can be driven, for instance, by action of light82 or by chemical means.85 Herein, DAP cores may not only be found in the structures of molecular guests,82,85 but also be embedded into the macrocyclic host frameworks.83,84 The latter was showcased by Mencarelli et al., who synthesized DAP-based [2]pseudorotaxanes 55 comprising tetrathiafulvalene guests (Fig. 12).84 It was also exemplified by García and Peinador et al., who achieved DAP/bipyridinium-based hermaphroditic host–guest aggregates, pseudo[1]rotaxanes 56 (Fig. 13).83
Fig. 12. Structure of DAP-based [2]pseudorotaxanes 50 comprising tetrathiafulvalene guests.

Fig. 13. Schematic representation of DAP-containing pseudo[1]rotaxanes 51.

Besides, the latter research team exploited DAP scaffolds in the design of catenane-type molecular machines.72,86,87 For the construction of such supramolecules, they applied both stepwise86 and self-assembly72,87 protocols, involving metallacycles related to that depicted in Fig. 11. As a result, both [2]catenanes86,87 and [3]catenanes72 were afforded. Meanwhile, Stoddart et al. reported88 the assembly of two DAP-containing [2]catenanes (57 and 58, Fig. 14), both of which were found to be persistent air-stable radicals. These structures are expected to serve as prototypes in the development of high-density data memories.
Fig. 14. Structures of DAP-containing air-stable radical catenanes 57, 58.

In 2013, the Stoddart's group reported the synthesis of two [2]catenane-containing struts that were composed of a tetracationic cyclophane (TC4+) encircling a 1,5-dioxynaphthalene (DNP)-based crown ether bearing two terphenylene arms.89 The TC4+ rings comprise either two bipyridinium (BIPY2+) units or a BIPY2+ and a diazapyrenium (DAP2+) unit. To work on this, the authors synthesized the DAP2+ according to the previously reported method as mentioned earlier.69
It is noteworthy that some of the works discussed above80,81 intertwine also with another scope of DAP derivatives application, namely the sensing ability of the latter. In the following subsection, we mention a few more examples related to this topic.
Construction of sensors
Prominent photophysical properties of DAP derivatives afford one to utilize them as fluorescent probes for sensing various analytes. For instance, Raymo et al. showed that 2,7-diazapyrenium films on glass and silica surfaces could be responsive to sub-millimolar concentrations of dopamine.90 This fact is associated with the decrease of emission intensity of DAP-derived cationic motifs upon interaction with the neurotransmitter mentioned (Fig. 15).
Fig. 15. Fluorescent diazapyrenium films and their response to dopamine. Reproduced be the permission of ref. 90 Copyrights © 2005 American Chemical Society.

Later, Chang et al. disclosed that the fluorescence of DMDAP cation could be almost completely quenched upon intercalation with aptamers of aptamer-modified gold nanoparticles (Apt-AuNPs).91 The elaborated DMDAP/Apt-AuNP system was demonstrated to bind in homogenous solution with platelet-derived growth factors (PDGFs) and their receptors, both being the breast cancer markers. Upon binding of DMDAP/Apt-AuNP with PDGFs, high turn-on fluorescence signal increasing was observed, governing the sensitivity for the detection of cancer markers (platelet-derived growth factors (PDGFs) and their receptors) (Fig. 16).
Fig. 16. Schematic representations of PDGF nanosensors that operate based on modulation of the FRET between DMDAP and Apt-AuNPs. Reproduced be the permission of ref. 91 Copyrights © 2007 American Chemical Society.

Furthermore, Balkus Jr. et al. developed the novel 2,7-diazapyrene-based periodic mesoporous organosilica (PMO) materials emerged as effective optical sensors for nitrated explosive taggants because of quenching of the PMO emission.92
Finally, Coskun et al. showcased the capability of porous cationic polymers incorporating DAP moieties to detect and capture aliphatic amines, uptake capacities being up to 31 wt% for primary amines from CO2 scrubbing systems.93
In summary, the application range of DAP derivatives appears to be very broad, which is mostly based on remarkable photophysical and binding properties of 2,7-diazapyrene unit. Of course, these features are expected to be the cornerstone for the further developments as well. Obviously, new relevant research focused on employing DAP derivatives in various fields of science won't be long in coming.
Conclusions
2,7-Diazopyrenes are promising azaaromatic scaffolds with a unique structural geometry and supramolecular properties. Due to the presence of pyridine-type nitrogen atoms in 2,7-positions of the pyrene core these heterocyles exhibit both pyrene- and pyridine-like supramolecular properties. Thus, similar to pyrene and other PAHs 2,7-diazapyrenes exhibit an excimer emission in concentrated solutions, while the presence of pyridine-type nitrogen atoms provides an intensive acid-responsive changes in the absorption and fluorescence spectra, which might be useful for the designing pH-responsive excimer probes based on 2,7-diazapyrenes. The electronegativity of the nitrogen atoms lowers the LUMO level of diazapyrene core and provides higher electron mobility in some 2,7-diazapyrenes compare to pyrene.32,94–97 Moreover, quaternization of nitrogen atom(s) dramatically reduces LUMO level of 2,7-diazapyrene-based mono- and dications as well as band gap. Additionally, in dication form 2,7-diazapyre was subject to two electron reduction to form 16π antiaromatic 2,7-dihydrodiazapyrenes,24,48 possessing unprecedentedly low band gap and a quite low oxidation potential (−1.34 V). All the above suggests the possible application of 2,7-diazapyrenes as promising n-type organic semiconductors for OFETs or similar devices.
In addition to electronic properties the nature of substituents dramatically affected the packing structures of tetra-substituted 2,7-diazapyrenes in the solid state. Compare to tetrasubstituted pyrenes the replacement of the C–H moieties with nitrogen atoms at the 2 and 7 positions in 2,7-diazapyrene reduces the steric repulsion between the central core and the peripheral aryl groups.98 As a result, tetraaryl and tetramethyl-substituted 2,7-diazapyrene exhibited more distinct excimer emission. The nature of the substituents in 2,7-diazapyrenes also modulates both the intermolecular interactions in solution and packing structures in the solid state and, compare to other tetraalkyl/tetramethoxy-substituted 2,7-diazapyrene, tetramethyl-substituted 2,7-diazapyrene was found to possess both stronger excimer emission and high electron mobility than other due to the overlaps of low LUMOs because of less steric hindrances.46
2,7-Diazapyrenes were found to be promising candidates for materials science and molecular recognition. Thus, 2,7-diazapyrene-based chiral MOF thin films were shown75 to have strong circularly polarized luminescence (CPL) properties, and 2,7-diazapyrenium-based ligand and square-planar Pd(ii) or Pt(ii) complexes were found71 to recognize polycyclic aromatic hydrocarbons (PAHs). Some, 2,7-diazapyrene-derived guests to form inclusion complexes with cucurbit[n]urils (n = 7, 8)78,80,99 and photoresponsive inclusion complexes with azobenzene-bridged cryptands were also reported.76 Finally, 2,7-diazapyrenes were successfully used for the assembly of pseudorotaxane-type molecular machines,82–85 which can be photo-82 or chemically-activated.85
In medicinal applications, 2,7- as well as other azapyrenes were successfully used to recognize nucleic acids and bind to specific areas of DNA, major or minor grooves, or integrate between DNA bases arranged in specific nucleotide sequences.68
Dispirit unique electronic and supramolecular properties the photophysical studies of 2,7-diazapyrenes so far reported only in very few publications, which might be due to the limited synthetic approaches towards these azaheterocycles.
So far, the most common methods involve reductive aromatization of naphthalene diimides, obtained either from naphthalene-1,4,5,8-tetracarboxylic acid or its anhydride,37,39–41,69 with the following transition metal (TM)-catalyzed coupling reactions with aryl-boronic acids,45 Grignard reagents46 or acetylenes.47 An alternative approach to non-substituted 2,7-diazapyrene involves the reaction 2,3-dihydro-1H-benzo[de]isoquinoline with 1,3,5-triazine in polyphosphoric acid.43 So far, no examples on TM-catalyzed/TM-free direct C–H-functionalization in 2,7-diazapyrenes were reported, and one might expect a gaining interest in this area. As a consequence, this review enables one to stimulate research in this fascinating area of organic synthesis and supramolecular chemistry. So, we believe that this updated account of developments will be of much utility in organic synthesis, the pharmaceutical industry, and material science.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the Russian Science Foundations (Grants ## 20-73-10205 and 21-13-00304) and Council on Grants of the President of the Russian Federation (grant no. NSh-2700.2020.3). DFT calculation for this work were performed using “Uran” supercomputer of IMM UB RAS.
Biographies
Biography
Anindita Mukherjee.

Anindita Mukherjee graduated from the University of Burdwan, India in 2014. She received her master's degree at Ural Federal University, Russia in 2017. Currently she is focusing on her doctoral research with Prof. G. V. Zyryanov, Ural Federal University, Russia. Her research interest lies in developing new methodologies to synthesize heterocyclic compounds exhibiting biological activity and/or luminescent properties for photochemistry, new analytical methods, chemosensors, molecular recognition, supramolecular chemistry, etc.
Biography
Alexey A. Akulov.

Alexey A. Akulov obtained his MSc degree in Chemistry at Ural Federal University in 2020. In 2016, he joined the Department of Organic & Biomolecular Chemistry at the same university, where he is currently working as a research engineer. His scientific activities are focused on the C–H functionalization methodology development with regard to azaheterocyclic compounds and, in particular, on the elaboration of both novel transition metal-catalyzed and metal-free cross-dehydrogenative coupling approaches.
Biography
Sougata Santra.

Sougata Santra obtained his PhD in 2014 from Visva-Bharati (A Central University of India) under the supervision of Prof. Adinath Majee. Since January 2015, he is working as a post-doctoral researcher in Ural Federal University, Russian Federation under the supervision of Prof. Grigory V. Zyryanov. In July 2015, he joined as a Senior Scientist in the Department of Organic and Biomolecular Chemistry, Chemical Engineering Institute; Ural Federal University, Yekaterinburg, Russian Federation. His research interest is to develop new methodologies to synthesize heterocyclic compounds along the lines of green chemistry. Currently he is working on syntheses of (hetero)macrocycles, their functionalization and applications in materials chemistry.
Biography
Mikhail V. Varaksin.

Mikhail V. Varaksin, Head of Chemical Engineering Institute, graduated from Ural State Technical University in 2008. In 2010, he earned his PhD in Organic Chemistry. In 2011, he joined the Department of Organic & Biomolecular Chemistry at Ural Federal University as an Associate Professor. His current research interests include the development of novel pot, atom, step economy approaches based on methodology of C–H bond functionalization in both (hetero)aromatic and non-aromatic substrates in design of novel heterocyclic derivatives of nitroxide radicals, calixarenes, carboranes, and also advanced materials for molecular electronics.
Biography
Grigory A. Kim.

Grigory A. Kim is a junior researcher in the laboratory of perspective organic materials in Institute of Organic Synthesis UB RAS. His scientific interest is heterocycle compounds, UV-Vis and fluorescent spectroscopy and quantum calculations in organic chemistry.
Biography
Dmitry S. Kopchuk.

Dmitry S. Kopchuk earned his PhD in Organic Chemistry in 2010. In 2010, he joined as a Researcher in the Postovsky Institute of Organic Synthesis of Ural Branch of the RAS. In 2019, he earned the Doctor of Science degree in Organic Chemistry. His current scientific interests include the development of new synthetic approaches to (hetero)aromatic compounds, including new bi- and terpyridine ligands, substituted 1,2,4-triazines, chemosensors for metal cations, and the detection of explosives.
Biography
Olga S. Taniya.

Olga S. Taniya earned her PhD (Organic chemistry) in 2018 from the Ural Federal University. Her scientific research interests include the synthesis and studying of the photophysical properties of poly(aza)aromatic chemosensors/fluorophores, supramolecular chemistry, molecular recognition, etc.
Biography
Grigory V. Zyryanov.

Grigory V. Zyryanov earned his PhD in Organic Chemistry in 2000. In 2001–2003 he joined the research group of Prof. Dmitry M. Rudkevich (UT Arlington, Arlington, TX) as a post-doctoral fellow. In 2004, he joined the Prof. l Anzenbacher Jr. group. In 2010 he moved to Russia and joined the Department of Organic and Biomolecular Chemistry at the Ural Federal University. In 2011, he joined as a Researcher in the Postovsky Institute of Organic Synthesis of Ural Branch of the RAS. In 2012, he earned the Doctor of Science degree in Organic Chemistry. In 2018, he became a Professor of the RAS. His current research interests include the development of novel approaches to new (hetero)aromatic compounds exhibiting biological activity and/or luminescent properties; green chemistry; coordination chemistry, photochemistry, new analytical methods, chemosensors, molecular recognition and supramolecular chemistry.
Biography
Oleg. N. Chupakhin.

Oleg N. Chupakhin is a full member of the Russian Academy of Sciences, Professor and ex-Head of the Department of Organic & Biomolecular Chemistry at Ural Federal University, and Chief Researcher at Postovsky Institute of Organic Synthesis of Ural Branch of the RAS. Prof. Chupakhin graduated with honors from Ural Polytechnical Institute in 1957. He earned his PhD in Organic Chemistry in 1962 and Doctor of Science degree in 1976. Current research interests: new methodologies in organic synthesis, structural analysis of organic compounds, mechanisms of organic reactions, heterocyclic chemistry, green chemistry, industrial chemistry, environmental chemistry, medicinal chemistry, chemosensors, molecular recognition and supramolecular chemistry.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00260d
Notes and references
- Khasanov A. F. Kopchuk D. S. Kovalev I. S. Taniya O. S. Giri K. Slepukhin P. A. Santra S. Rahman M. Majee A. Charushin V. N. Chupakhin O. N. New J. Chem. 2017;41:2309–2320. [Google Scholar]
- Sriyab S. Jorn-Iat K. Prompinit P. Wolschann P. Hannongbua S. Suramitr S. J. Lumin. 2018;203:492–499. [Google Scholar]
- Ruiu A. Vonlanthen M. Morales-Espinoza E. G. Rojas-Montoya S. M. González-Méndez I. Rivera E. J. Mol. Struct. 2019;1196:1–7. [Google Scholar]
- Gowri A. Khamrang T. Velusamy M. Kathiresan M. Kumar M. D. Jaccob M. Kathiravan A. Sensors and Actuators Reports. 2020;2:100007. [Google Scholar]
- Kowser Z. Rayhan U. Akther T. Redshaw C. Yamato T. Mater. Chem. Front. 2021;5:2173–2200. [Google Scholar]
- Fujiwara Y. Amao Y. Sens. Actuators, B. 2003;89:58–61. [Google Scholar]
- Nagel C. C., Bentsen J. G., Yafuso M., Katritzky A. R., Dektar J. L. and Kipke C. A., US Pat., US5498549, 1996
- Lee E. D. Werner T. C. Seitz W. R. Anal. Chem. 1987;59:279–283. [Google Scholar]
- Chao J. Wang H. Zhang Y. Yin C. Huo F. Sun J. Zhao M. New J. Chem. 2018;42:3322–3333. [Google Scholar]
- Saravanan A. Shyamsivappan S. Suresh T. Subashini G. Kadirvelu K. Bhuvanesh N. Nandhakumar R. Mohan P. S. Talanta. 2019;198:249–256. doi: 10.1016/j.talanta.2019.01.114. [DOI] [PubMed] [Google Scholar]
- Srinivasan V. Jhonsi M. A. Dhenadhayalan N. Lin K. C. Ananth D. A. Sivasudha T. Narayanaswamy R. Kathiravan A. Spectrochim. Acta, Part A. 2019;221:117150. doi: 10.1016/j.saa.2019.117150. [DOI] [PubMed] [Google Scholar]
- Birks I. H. M. J. B. Lumb M. D. Proc. R. Soc. London, Ser. A. 1964;280:289–297. [Google Scholar]
- Templer R. H. Castle S. J. Curran A. R. Rumbles G. Klug D. R. Faraday Discuss. 1999;111:41–53. doi: 10.1039/a806472e. [DOI] [PubMed] [Google Scholar]
- Pokhrel M. R. Bossmann S. H. J. Phys. Chem. B. 2000;104:2215–2223. [Google Scholar]
- Wang T. Zhang N. Ge Y. Wang C. Hang Z. Zhang Z. Macromol. Chem. Phys. 2020;221:1900463. [Google Scholar]
- Zhang Y. Tan L. Shi J. Ji L. New J. Chem. 2021;45:14869–14878. [Google Scholar]
- Figueira-Duarte T. M. Müllen K. Chem. Rev. 2011;111:7260–7314. doi: 10.1021/cr100428a. [DOI] [PubMed] [Google Scholar]
- Jia W. L. McCormick T. De Liu Q. Fukutani H. Motala M. Wang R. Y. Tao Y. Wang S. J. Mater. Chem. 2004;14:3344–3350. [Google Scholar]
- Kalyanasundaram K. Thomas J. K. J. Am. Chem. Soc. 1977;99:2039–2044. [Google Scholar]
- Crawford A. G. Dwyer A. D. Liu Z. Steffen A. Beeby A. Pålsson L. O. Tozer D. J. Marder T. B. J. Am. Chem. Soc. 2011;133:13349–13362. doi: 10.1021/ja2006862. [DOI] [PubMed] [Google Scholar]
- Casas-Solvas J. M. Howgego J. D. Davis A. P. Org. Biomol. Chem. 2014;12:212–232. doi: 10.1039/c3ob41993b. [DOI] [PubMed] [Google Scholar]
- Feng X. Hu J.-Y. Redshaw C. Yamato T. Chem.–Eur. J. 2016;22:11898–11916. doi: 10.1002/chem.201600465. [DOI] [PubMed] [Google Scholar]
- Merz J. Fink J. Friedrich A. Krummenacher I. Al Mamari H. H. Lorenzen S. Haehnel M. Eichhorn A. Moos M. Holzapfel M. Braunschweig H. Lambert C. Steffen A. Ji L. Marder T. B. Chem.–Eur. J. 2017;23:13164–13180. doi: 10.1002/chem.201702594. [DOI] [PubMed] [Google Scholar]
- Nakazato T. Takekoshi H. Sakurai T. Shinokubo H. Miyake Y. Angew. Chem., Int. Ed. 2021;60:13877–13881. doi: 10.1002/anie.202103667. [DOI] [PubMed] [Google Scholar]
- Waldhör E. Zulu M. M. Zalis S. Kaim W. J. Chem. Soc., Perkin Trans. 2. 1996;6:1197–1204. [Google Scholar]
- Nakazato T. Takekoshi H. Sakurai T. Shinokubo H. Miyake Y. Angew. Chem. 2021;133:13996–14000. doi: 10.1002/anie.202103667. [DOI] [PubMed] [Google Scholar]
- Frisch M. J. Pople J. A. Binkley J. S. J. Chem. Phys. 1984;80:3265–3269. [Google Scholar]
- Clark T. Chandrasekhar J. Spitznagel G. W. Schleyer P. V. R. J. Comput. Chem. 1983;4:294–301. [Google Scholar]
- McLean A. D. Chandler G. S. J. Chem. Phys. 1980;72:5639–5648. [Google Scholar]
- Krishnan R. Binkley J. S. Seeger R. Pople J. A. J. Chem. Phys. 1980;72:650–654. [Google Scholar]
- Taniya O. S. Khasanov A. F. Varaksin M. V. Starnovskaya E. S. Krinochkin A. P. Savchuk M. I. Kopchuk D. S. Kovalev I. S. Kim G. A. Nosova E. V. Zyryanov G. V. Chupakhin O. N. New J. Chem. 2021;45:20955–20971. [Google Scholar]
- Usta H. Facchetti A. Marks T. J. Acc. Chem. Res. 2011;44:501–510. doi: 10.1021/ar200006r. [DOI] [PubMed] [Google Scholar]
- Okamoto T. Kumagai S. Fukuzaki E. Ishii H. Watanabe G. Niitsu N. Annaka T. Yamagishi M. Tani Y. Sugiura H. Watanabe T. Watanabe S. Takeya J. Sci. Adv. 2020;6:427–469. doi: 10.1126/sciadv.aaz0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzlein G. and Vollmann H., Ger. Pat., DE552760, 1930
- Stoddart J. F., Nalluri S. K. M. and Liu Z., PCT Pat., WO2017161095, 2017
- Kranzlein G. and Vollmann H., US Pat., US1920406, 1933
- Hünig S. Groβ J. Lier E. F. Quast H. Justus Liebigs Ann. Chem. 1973;1973:339–358. [Google Scholar]
- Stang P. J. Cao D. H. Saito S. Arif A. M. J. Am. Chem. Soc. 1995;117:6273–6283. [Google Scholar]
- Sehnal P. Holý P. Tichý M. Závada J. Císařová I. Collect. Czech. Chem. Commun. 2002;67:1236–1246. [Google Scholar]
- Sotiriou-Leventis C. Mao Z. J. Heterocycl. Chem. 2000;37:1665–1667. [Google Scholar]
- Sotiriou-Leventis C. Mao Z. Rawashdeh A. M. M. J. Org. Chem. 2000;65:6017–6023. doi: 10.1021/jo0004557. [DOI] [PubMed] [Google Scholar]
- Philip S. M. R. and Keehn M., Cyclophanes, Elsevier, 1983 [Google Scholar]
- Aksenov A. V. Borovlev I. V. Aksenova I. V. Pisarenko S. V. Kovalev D. A. Tetrahedron Lett. 2008;49:707–709. [Google Scholar]
- Kamata T. and Wasada N., Jpn. Pat., 11322747, 1999
- Nakazato T. Kamatsuka T. Inoue J. Sakurai T. Seki S. Shinokubo H. Miyake Y. Chem. Commun. 2018;54:5177–5180. doi: 10.1039/c8cc01937a. [DOI] [PubMed] [Google Scholar]
- Nakazato T. Matsuda W. Sakurai T. Seki S. Shinokubo H. Miyake Y. Chem. Lett. 2020;49:465–468. [Google Scholar]
- Werner S. Vollgraff T. Fan Q. Bania K. Gottfried J. M. Sundermeyer J. Org. Chem. Front. 2021;8:5013–5023. [Google Scholar]
- Sundermeyer J., Baal E. and Werner S., PCT Pat., WO2019229134A1, 2019
- Sachdev H., Eur. Pat., EP2390253A1, 2011
- Waldhör E. Zulu M. M. Zalis S. Kaim W. J. Chem. Soc., Perkin Trans. 2. 1996;6:1197–1204. [Google Scholar]
- Nakazato T. Shinokubo H. Miyake Y. Chem. Commun. 2021;57:327–330. doi: 10.1039/d0cc07804b. [DOI] [PubMed] [Google Scholar]
- Vedernikov A. I. Ushakov E. N. Efremova A. A. Kuz’mina L. G. Moiseeva A. A. Lobova N. A. Churakov A. V. Strelenko Y. A. Alfimov M. V. Howard J. A. K. Gromov S. P. J. Org. Chem. 2011;76:6768–6779. doi: 10.1021/jo201172w. [DOI] [PubMed] [Google Scholar]
- Zhang Y. M. Wang Z. Chen L. Bin Song H. Liu Y. J. Phys. Chem. B. 2014;118:2433–2441. doi: 10.1021/jp4110008. [DOI] [PubMed] [Google Scholar]
- Brun A. M. Harriman A. J. Am. Chem. Soc. 1991;113:8153–8159. [Google Scholar]
- Brun A. M. Harriman A. J. Am. Chem. Soc. 1992;114:3656–3660. [Google Scholar]
- Blacker A. J. Jazwinski J. Lehn J.-M. Wilhelm F. X. J. Chem. Soc., Chem. Commun. 1986:1035. [Google Scholar]
- Blacker A. J. Jazwinski J. Lehn J.-M. Helv. Chim. Acta. 1987;70:1–12. [Google Scholar]
- Becker H. C. Nordén B. J. Am. Chem. Soc. 1997;119:5798–5803. [Google Scholar]
- Lilienthal N. D. Enlow M. A. Othman L. Smith E. A. F. Smith D. K. J. Electroanal. Chem. 1996;414:107–114. [Google Scholar]
- Leventis N. Elder I. A. Rolison D. R. Anderson M. L. Merzbacher C. I. Chem. Mater. 1999;11:2837–2845. [Google Scholar]
- Hüsing N. Schubert U. Angew. Chem. 1998;110:22–47. doi: 10.1002/(SICI)1521-3773(19980202)37:1/2<22::AID-ANIE22>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Kalluri S. Shi Y. Steier W. H. Yang Z. Xu C. Wu B. Dalton L. R. Appl. Phys. Lett. 1994;65:2651–2653. [Google Scholar]
- Sung P. H. Hsu T. F. Polymer. 1998;39:1453–1459. [Google Scholar]
- Sung P. H. Hsu T. F. Ding Y. H. Wu A. Y. Chem. Mater. 1998;10:1642–1646. [Google Scholar]
- Yang Z. Chengzeng X. Wu B. Dalton L. R. Kalluri S. Steier W. H. Shi Y. Bechtel J. H. Chem. Mater. 1994;6:1899–1901. [Google Scholar]
- Lehn J. M. Pure Appl. Chem. 1978;50:871–892. [Google Scholar]
- Terenzi A. Ducani C. Blanco V. Zerzankova L. Westendorf A. F. Peinador C. Quintela J. M. Bednarski P. J. Barone G. Hannon M. J. Chem.–Eur. J. 2012;18:10983–10990. doi: 10.1002/chem.201201519. [DOI] [PubMed] [Google Scholar]
- Zhirov A. M. Kovalev D. A. Ulshina D. V. Pisarenko S. V. Demidov O. P. Borovlev I. V. Chem. Heterocycl. Compd. 2020;56:674–693. doi: 10.1007/s10593-020-02717-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake A. J. Baum G. Champness N. R. Chung S. S. M. Cooke P. A. Fenske D. Khlobystov A. N. Lemenovskii D. A. Li W. S. Schröder M. J. Chem. Soc., Dalton Trans. 2000:4285–4291. [Google Scholar]
- Stang P. J. Olenyuk B. Fan J. Arif A. M. Organometallics. 1996;15:904–908. [Google Scholar]
- Blanco V. García M. D. Terenzi A. Pía E. Fernández-Mato A. Peinador C. Quintela J. M. Chem.–Eur. J. 2010;16:12373–12380. doi: 10.1002/chem.201002051. [DOI] [PubMed] [Google Scholar]
- Blanco V. García M. D. Peinador C. Quintela J. M. Chem. Sci. 2011;2:2407–2416. [Google Scholar]
- Rama T. Alvariño C. Domarco O. Platas-Iglesias C. Blanco V. García M. D. Peinador C. Quintela J. M. Inorg. Chem. 2016;55:2290–2298. doi: 10.1021/acs.inorgchem.5b02650. [DOI] [PubMed] [Google Scholar]
- Blake A. J. Champness N. R. Khlobystov A. N. Lemenovskii D. A. Li W. S. Schröder M. Chem. Commun. 1997:1339–1340. [Google Scholar]
- Chen S. M. Chang L. M. Yang X. K. Luo T. Xu H. Gu Z. G. Zhang J. ACS Appl. Mater. Interfaces. 2019;11:31421–31426. doi: 10.1021/acsami.9b11872. [DOI] [PubMed] [Google Scholar]
- Liu M. Yan X. Hu M. Chen X. Zhang M. Zheng B. Hu X. Shao S. Huang F. Org. Lett. 2010;12:2558–2561. doi: 10.1021/ol100770j. [DOI] [PubMed] [Google Scholar]
- Bergamini G. Molloy J. K. Fermi A. Ceroni P. Klärner F. G. Hahn U. New J. Chem. 2012;36:354–359. [Google Scholar]
- Molloy J. K. Bergamini G. Baroncini M. Hahn U. Ceroni P. New J. Chem. 2018;42:16193–16199. [Google Scholar]
- Li Z. Yang J. Huang F. Chin. J. Chem. 2018;36:59–62. [Google Scholar]
- Barba-Bon A. Pan Y. C. Biedermann F. Guo D. S. Nau W. M. Hennig A. J. Am. Chem. Soc. 2019;141:20137–20145. doi: 10.1021/jacs.9b09563. [DOI] [PubMed] [Google Scholar]
- Biedermann F. Nau W. M. Angew. Chem., Int. Ed. 2014;53:5694–5699. doi: 10.1002/anie.201400718. [DOI] [PubMed] [Google Scholar]
- Ashton P. R. Ballardini R. Balzani V. Constable E. C. Credi A. Kocian O. Langford S. J. Preece J. A. Prodi L. Schofield E. R. Spencer N. Stoddart J. F. Wenger S. Chem.–Eur. J. 1998;4:2413–2422. [Google Scholar]
- Rama T. Blanco-Gómez A. Neira I. Domarco O. García M. D. Quintela J. M. Peinador C. Chem.–Eur. J. 2017;23:16743–16747. doi: 10.1002/chem.201704540. [DOI] [PubMed] [Google Scholar]
- Bruschini M. Ercolani G. Gallina S. Mencarelli P. J. Org. Chem. 2018;83:11446–11449. doi: 10.1021/acs.joc.8b01786. [DOI] [PubMed] [Google Scholar]
- Yan X. Wu X. Wei P. Zhang M. Huang F. Chem. Commun. 2012;48:8201–8203. doi: 10.1039/c2cc33783e. [DOI] [PubMed] [Google Scholar]
- López-Vidal E. M. García M. D. Peinador C. Quintela J. M. Chem.–Eur. J. 2015;21:2259–2267. doi: 10.1002/chem.201405297. [DOI] [PubMed] [Google Scholar]
- Rama T. Lõpez-Vidal E. M. García M. D. Peinador C. Quintela J. M. Chem.–Eur. J. 2015;21:9482–9487. doi: 10.1002/chem.201501087. [DOI] [PubMed] [Google Scholar]
- Sun J. Liu Z. Liu W. G. Wu Y. Wang Y. Barnes J. C. Hermann K. R. Goddard W. A. Wasielewski M. R. Stoddart J. F. J. Am. Chem. Soc. 2017;139:12704–12709. doi: 10.1021/jacs.7b06857. [DOI] [PubMed] [Google Scholar]
- Cao D. Juríček M. Brown Z. J. Sue A. C. H. Liu Z. Lei J. Blackburn A. K. Grunder S. Sarjeant A. A. Coskun A. Wang C. Farha O. K. Hupp J. T. Stoddart J. F. Chem.–Eur. J. 2013;19:8457–8465. doi: 10.1002/chem.201300762. [DOI] [PubMed] [Google Scholar]
- Cejas M. A. Raymo F. M. Langmuir. 2005;21:5795–5802. doi: 10.1021/la0502793. [DOI] [PubMed] [Google Scholar]
- Huang C. C. Chiu S. H. Huang Y. F. Chang H. T. Anal. Chem. 2007;79:4798–4804. doi: 10.1021/ac0707075. [DOI] [PubMed] [Google Scholar]
- Balkus K. J. Pisklak T. J. Hundt G. Sibert J. Zhang Y. Microporous Mesoporous Mater. 2008;112:1–13. [Google Scholar]
- Kim K. Buyukcakir O. Coskun A. RSC Adv. 2016;6:77406–77409. [Google Scholar]
- Anthony J. E. Facchetti A. Heeney M. Marder S. R. Zhan X. Adv. Mater. 2010;22:3876–3892. doi: 10.1002/adma.200903628. [DOI] [PubMed] [Google Scholar]
- Zhao X. Zhan X. Chem. Soc. Rev. 2011;40:3728–3743. doi: 10.1039/c0cs00194e. [DOI] [PubMed] [Google Scholar]
- Zhan X. Facchetti A. Barlow S. Marks T. J. Ratner M. A. Wasielewski M. R. Marder S. R. Adv. Mater. 2011;23:268–284. doi: 10.1002/adma.201001402. [DOI] [PubMed] [Google Scholar]
- Zhou K. Dong H. li Zhang H. Hu W. Phys. Chem. Chem. Phys. 2014;16:22448–22457. doi: 10.1039/c4cp01700e. [DOI] [PubMed] [Google Scholar]
- El-Assaad T. H. Auer M. Castañeda R. Hallal K. M. Jradi F. M. Mosca L. Khnayzer R. S. Patra D. Timofeeva T. V. Brédas J. L. List-Kratochvil E. J. W. Wex B. Kaafarani B. R. J. Mater. Chem. C. 2016;4:3041–3058. [Google Scholar]
- Miskolczy Z. Megyesi M. Biczók L. Prabodh A. Biedermann F. Chem.–Eur. J. 2020;26:7433–7441. doi: 10.1002/chem.201905633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.