

Abstract
The iPSC Neurodegenerative Disease Initiative (iNDI) is the largest-ever iPSC genome engineering project. iNDI will model more than 100 mutations associated with Alzheimer’s disease and related dementias (ADRD) in isogenic iPSC lines. Resulting cell lines and phenotypic datasets will be broadly shared.
ADRD research in an aging population
As the global population ages, the number of people suffering from Alzheimer’s disease and related dementias (ADRD) will grow. Heroic efforts have been undertaken by the research community to develop disease-modifying therapies for ADRD. Unfortunately, these endeavors have yielded multiple failed clinical trials, leaving patients with few treatment options. Our limited understanding of the underlying mechanisms of disease initiation and progression continue to hamper development of effective therapeutics.
Fortunately, convergent breakthroughs in several fields have set the stage for a renaissance in ADRD research. First, human genetic studies have identified the majority of gene mutations associated with familial ADRD, and genome-wide association studies (GWASs) have identified many variants that contribute to overall lifetime risk of sporadic ADRD. Second, human induced pluripotent stem cell (iPSC) disease models have emerged and can now be genetically engineered to harbor or correct disease-associated gene mutations (Kwart et al., 2019). Finally, development of powerful new “omic” and computational tools enable genome-wide characterization of how mutations alter proximate biological processes. These advances collectively provide an opportunity to discover the fundamental mechanisms of ADRD and identify new therapeutic targets. However, progress has been hindered by a lack of high-quality, standardized iPSC models and foundational datasets describing biological consequences of disease mutations. Here, we introduce the iPSC Neurodegenerative Disease Initiative (iNDI), a large-scale project that leverages new technologies to develop an ADRD iPSC repository and data resource for the research community.
Advancements and challenges to modeling ADRD with iPSCs
Traditional cellular models of ADRD have revealed important disease-relevant biology but suffer from several limitations (Slanzi et al., 2020). Commonly used cancer cell lines are subject to karyotypic changes and genomic drift, patient fibroblasts are non-neuronal, and cultured rodent neurons may not reflect human-specific pathobiology, particularly for non-coding variants that are poorly conserved across species. In the past decade, laboratories worldwide have adopted iPSCs to examine human biology and genomics in an endogenous context. iPSCs can be derived from human patients, are easily grown at scale, and can be differentiated into neurons and glia (Abud et al., 2017; Wang et al., 2017). Combined with discoveries of numerous gene mutations associated with disease and advancements in CRISPR-Cas9 gene-editing technologies, it is now possible to model and study the effects of specific disease mutations in relevant cell types.
Despite their potential, multiple barriers currently hinder widespread use of iPSCs. iPSCs from different donors have unique genetic backgrounds and varied differentiation potentials. Traditionally, separate iPSC lines are derived from patients with either sporadic or familial disease and age/sex-matched controls. Each independent line is then differentiated into CNS cell types for phenotypic evaluations. Determining whether a biological effect is driven by the disease state of the cells or other genomic differences between the iPSC lines is therefore challenging. Culturing multiple independent patient and control lines in parallel may mitigate this concern, but substantially increases the cost and effort of even straightforward experiments, and can be unfeasible for complex experimental workflows. For studies of familial ADRD, the use of mutation-corrected “isogenic” control lines has become de rigueur, created from patient-derived mutant iPSC lines via CRISPR-Cas9 gene editing (Xiong et al., 2016). Theoretically, these paired iPSC lines circumvent inter-individual variability and enable comparison of multiple mutations in one genetic background. In practice, off-target mutations and chromosomal abnormalities can occur during the editing and expansion of engineered lines and are sometimes missed due to a lack of standardized post-editing quality control assays (Kwart et al., 2019). Consequentially, whether lines truly vary only at one base pair across the genome is often ambiguous. Furthermore, rare familial mutations are difficult to study with patient-derived lines, as these lines often either do not exist, cannot be readily shared, or are of uncertain quality. An alternative approach involves the generation of familial mutation lines from well-characterized reference lines, which can be readily engineered at or below the cost of generating iPSC lines from patient-derived cells. Given the caveats of non-isogenic controls, it is likely that isogenic genome-edited lines will become standard tools in ADRD research.
Large-scale efforts to understand ADRD
Although iPSC-related techniques are robust, the process of deriving, engineering, and characterizing novel iPSC lines remains burdensome. To address this challenge, consortia have emerged to generate and characterize iPSCs from large numbers of patients. These efforts harness economies of scale in iPSC work and sometimes generate accompanying phenotypic datasets, including transcriptomic/proteomic/epigenomic analyses of differentiated disease-relevant cells. For example, Answer ALS aims to understand amyotrophic lateral sclerosis (ALS) disease mechanisms by integrating detailed patient clinical features with multi-omic datasets from iPSC-derived motor neurons (Rothstein et al., 2020). Answer ALS plans to overcome the limitations of iPSC donor variability by analyzing data from motor neurons differentiated from more than 1,000 new patient and control iPSC lines. This type of study is particularily suited to the study of sporadic ALS, which is not associated with highly penetrant single mutations and thus cannot be modeled with isogenic iPSCs. Once iPSCs and data are generated, they will be openly shared, critical for promoting discovery by the community as a whole. At the time of this publication, Answer ALS has made whole-genome sequencing, RNA sequencing, and epigenomic data available from ~150 iPSC lines (Rothstein et al., 2020; https://data.answerals.org/).
Smaller efforts have been undertaken to study familial ADRD. One group recently generated an isogenic series of AD mutations in a single iPSC background (Kwart et al., 2019). Transcriptomic analysis of neurons derived from these iPSCs implicated endocytic pathways as a converging disease pathway, consistent with known biology of this disease. A second study modeled AD mutations in isogenic iPSC-derived microglia and pointed to APOE as a common player in familial AD (Konttinen et al., 2019). Such cell-type-specific analysis in isogenic series has the potential to uncover new therapeutic targets for familial disease. Comparisons of isogenic iPSCs on a single parental background may additionally uncover relationships across multiple disease-associated genes.
Because transferring iPSC lines between labs is often hindered by consenting and tech transfer barriers, there have been several attempts to create central resources to facilitate wider sharing. The NINDS Human Cell and Data Repository (NHCDR) is an iPSC banking and engineering effort to create iPSC lines from several adult-onset neurodegenerative diseases. Despite expansion of this resource in recent years, the catalog of iPSC lines remains relatively limited, reflecting the challenges both of recruiting rare disease patients and in applying genome-editing technology efficiently. To date, NHCDR has generated 150 iPSC lines, of which 12 lines have matched isogenic controls. International efforts to generate isogenic iPSCs include the Human Induced Pluripotent Stem Cell initiative for rare disorders (HipSci) (Streeter et al., 2017), the Tau Consortium to model tauopathies (Karch et al., 2019), and extensions of the Parkinson’s Progression Markers initiative (Marek et al., 2011). Each of these initiatives focuses on a relatively limited range of disorders.
What is iNDI?
Here, we present the iPSC Neurodegenerative Disease Initiative (iNDI). iNDI is a collaborative effort, anchored within the new NIH Intramural Center for Alzheimer’s and Related Dementias, to generate a large series of isogenic iPSC lines and related foundational datasets for the research community. Cross-cutting by design, iNDI will model disease-causing mutations from multiple disorders: AD, frontotemporal dementia and amyotrophic lateral sclerosis (FTD/ALS), dementia with Lewy bodies/Parkinson’s disease dementia (DLB/PDD), and other adult-onset neurodegenerative disorders with dementia as a component. Unlike companion initiatives that focus on patient-derived lines, iNDI will engineer a large number of mutations into deeply characterized parental lines derived from unaffected individuals. Because mutant lines will share common parental backgrounds, this approach will also allow for cross-analysis of different ADRD-associated genes and mutations. iNDI comprises two phases (Figure 1). Phase 1 will focus on iPSC line generation and phase 2 on multi-dimensional phenotypic characterization and analysis.
Figure 1. Overview of iNDI.
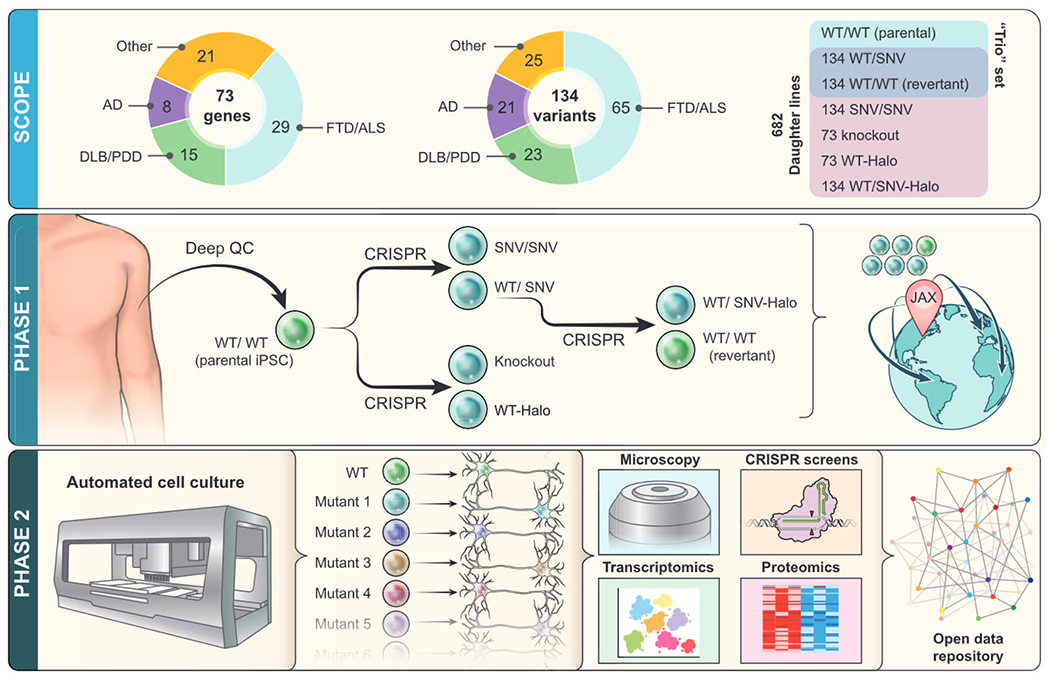
iNDI is a large iPSC genome-engineering initiative, with more than 600 engineered daughter lines to be generated from each parental line. Companion lines will include revertants, Halo-tagged knockins, and knockouts. Phase 1: iNDI will genetically engineer isogenic iPSC lines harboring many ADRD variants, knockouts, and endogenous tags across several deeply characterized parental lines from unaffected individuals. Following stringent quality control assays, each line will be available for distribution through The Jackson Laboratory (JAX). Phase 2: using high-throughput robotic platforms, iNDI will differentiate mutation harboring iPSC lines into CNS-relevant cell types, such as neurons and microglia, followed by a series of phenotypic assays. These include transcriptomics, proteomics, imaging, and genetic interaction screens, yielding a rich multi-dimensional picture of how mutations disrupt cellular function. Open access datasets will enable the community to analyze these data in new ways.
Phase 1
The goal of iNDI phase 1 is to generate high-quality isogenic ADRD iPSC lines for the research community. Partnering with multiple external organizations with proven genome engineering track records (The Jackson Laboratory [JAX], Thermo Fisher Scientific, and Synthego), iNDI will generate cell lines for 134 variants across 73 ADRD genes. Genes and variants will be selected through automated identification from online databases, augmented with additional alleles nominated by expert geneticists. Starting from a panel of iPSCs from multiple healthy donors, each candidate iPSC line will be subcloned and characterized for growth rate, morphology, chromosome stability, and differentiation potential and undergo both short- and long-read whole-genome sequencing. Following selection and expansion of a lead parental line, all mutations and endogenous tags will be engineered in early passage cells.
A core set of lines will be generated for each variant: homozygous mutation, heterozygous mutation, and a revertant line in which the mutation is edited back to the wild-type allele. For autosomal dominant disease, we will revert heterozygous lines, while for recessive mutations, we will engineer homozygous lines back to wild type. However, both homozygous and heterozygous lines will be available to study allelic dosage effects. The use of revertant lines may allow discrimination of phenotypes caused by the mutation of interest from those due to off-target consequences of editing and clonal expansion. These isogenic “trio” sets of the parental line, mutant, and revertant will be available for distribution at early stages of this project. Knockout lines will also be available to probe normal function, and we will engineer endogenous Halo-tagged wild-type and mutant forms of each of the selected genes to study how mutations affect protein localization and protein-protein interaction networks. Final engineered cell lines will be validated by quality control and distributed openly to interested researchers by JAX via a simplified electronic material transfer agreement, along with all related quality control data (Figure 1). Ultimately, we plan to generate a complete set of mutant lines from one male and one female parental line and partial sets of mutant lines for the most highly requested mutation lines from diverse racial and ethnic backgrounds. Generation of numerous additional isogenic mutant lines that fall outside of the scope of ADRD research is also planned through ongoing partnerships between the NIH, Aligning Science Across Parkinson’s (ASAP), and through a Chan Zuckerberg Initiative grant to JAX.
Phase 2
The goal of iNDI phase 2 is to generate foundational datasets that define proximate effects of ADRD mutations in disease-relevant CNS cells. Each cell line will be differentiated into relevant cell types, including both neurons and microglia, and will be characterized through multiple assays described below. Given the number of edited iPSC lines to characterize, iNDI will make use of high-throughput robotics to automate iPSC culture and differentiation. Analysis of genes across the ADRD spectrum will undoubtedly provide novel insights into proximal perturbations and gene-gene relationships.
The characterization of iNDI lines will fall into several categories (Figure 1). (1) Transcriptomics and proteomics: we will identify effects of ADRD mutations on cellular functions through transcriptomic and proteomic profiling of iPSC-derived neurons and other relevant cell types. (2) Protein interaction networks: using affinity purification of Halo-tagged lines in iPSC-derived neurons followed by mass spectrometry, we will map protein interaction networks and how these networks are changed by disease mutations. (3) Microscopy: we will employ automated longitudinal imaging and immunostaining approaches to assess morphology, survival, and pathology of mutant cell lines. We will also evaluate protein (mis) localization throughout the course of differentiation to mature cellular states. (4) Synthetic lethality screens: to examine how other genes may interact genetically with ADRD genes, we will perform genetic interaction synthetic lethality screens in differentiated cells. Whole-genome CRISPR inhibition lentiviral libraries will be delivered to each mutant cell line and survival will be assessed to determine whether other genes may exacerbate or buffer phenotypes caused by mutations. All data generated during the course of iNDI, including quality control and downstream phenotypic outcomes, will be made freely available on the interactive iNDI online data portal. Researchers can visit the portal to easily request cell lines and visualize and download data generated by iNDI.
Summary
iNDI directly addresses the need for readily available, high-quality isogenic iPSCs to model neurodegenerative diseases. The aim of iNDI is to be an open and easily accessible resource for researchers to help promote novel discoveries in the biology of ADRD without assumptions about the relationships between different diagnostic labels. iNDI will yield hundreds of engineered cell lines and rich multi-omic datasets characterizing effects of more than 100 familial ADRD mutations, openly shared through an interactive iNDI data portal. Validated cell lines can be requested on a rolling basis along with accompanying quality control datasets. Data and results from phase 2 will be uploaded to the iNDI portal as they become available. Because all data are made public, integration and cross-analysis of datasets are readily possible and encouraged. These types of integrative analyses are likely to lead to interesting and actionable discoveries that no one approach would be able to learn in isolation, providing the best chance at truly understanding the basis of ADRD and promising treatment possibilities.
ACKNOWLEDGMENTS
We thank Erika Lara Flores, Andy Qi, Luke Reilly, Mike Nalls, Lirong Peng, Faraz Faghri, Jizhong Zou, Kailyn Anderson, Julia Stadler, Caroline Pantazis, Bruce Conklin, the Chan Zuckerberg Initiative, and the Allen Institute for their helpful input on this project and manuscript. This work was supported in part by the Intramural Research Programs of the NIA and NINDS, NIH, Bethesda.
REFERENCES
- Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, et al. (2017). iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 94, 278–293.e9, 10.1016/j.neuron.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Kao AW, Karydas A, Onanuga K, Martinez R, Argouarch A, Wang C, Huang C, Sohn PD, Bowles KR, et al. ; Tau Consortium Stem Cell Group (2019). A Comprehensive Resource for Induced Pluripotent Stem Cells from Patients with Primary Tauopathies. Stem Cell Reports 13, 939–955, 10.1016/j.stemcr.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konttinen H, Cabral-da-Silva MEC, Ohtonen S, Wojciechowski S, Shakirzyanova A, Caligola S, Giugno R, Ishchenko Y, Hernández D, Fazaludeen MF, et al. (2019). PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human iPSC-Derived Microglia. Stem Cell Reports 13, 669–683, 10.1016/j.sterner.2019.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, Fak J, Olsen O, Darnell RB, and Tessier-Lavigne M (2019). A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron 104, 256–270.e5, 10.1016/j.neuron.2019.07.010. [DOI] [PubMed] [Google Scholar]
- Marek K, Jennings D, Lasch S, Siderowf A, Tanner C, Simuni T, Coffey C, Kieburtz K, Flagg E, Chowdhury S, et al. ; Parkinson Progression Marker Initiative (2011). The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol 95, 629–635, 10.1016/j.pneurobio.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein J, Berry J, Svendsen C, Thompson L, Finkbeiner S, Van Eyk J, Fraenkel E, Cudkowicz M, Maragakis N, Sareen D, et al. (2020). Answer ALS: A Large-Scale Resource for Sporadic and Familial ALS Combining Clinical Data with Multi-Omics Data from Induced Pluripotent Cell Lines. Research Square, 10.21203/rs.3.rs-96858/v1. [DOI] [Google Scholar]
- Slanzi A, Iannoto G, Rossi B, Zenaro E, and Constantin G (2020). In vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol 8, 328, 10.3389/fcell.2020.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeter I, Harrison PW, Faulconbridge A, Flicek P, Parkinson H, and Clarke L; The HipSci Consortium (2017). The human-induced pluripotent stem cell initiative-data resources for cellular genetics. Nucleic Acids Res. 45 (D1), D691–D697, 10.1093/nar/gkw928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ward ME, Chen R, Liu K, Tracy TE, Chen X, Xie M, Sohn PD, Ludwig C, Meyer-Franke A, et al. (2017). Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Reports 9, 1221–1233, 10.1016/j.stemcr.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Chen M, Lim WA, Zhao D, and Qi LS (2016). CRISPR/Cas9 for Human Genome Engineering and Disease Research. Annu. Rev. Genomics Hum. Genet 17, 131–154, 10.1146/annurev-genom-083115-022258. [DOI] [PMC free article] [PubMed] [Google Scholar]