

Abstract
Recreating features of the native extracellular matrix (ECM) with engineered biomaterials has become a valuable tool to probe the influence of ECM properties on cellular functions (e.g., differentiation) and towards the engineering of tissues. We developed a protocol to visualize and quantify the spatiotemporal evolution of newly synthesized and deposited matrix by cells that are either cultured atop (two-dimensional, 2D) or embedded within (three-dimensional, 3D) biomaterial systems (e.g., hydrogels, fibrous matrices). This technique relies on the incorporation of a non-canonical amino acid (azidohomoalanine, AHA) into proteins and proteoglycans as they are synthesized. Deposited nascent ECM components are then visualized with fluorescent cyclooctynes via copper-free cycloaddition or modified with cleavable biotin probes for identification. Here, we describe the preparation of hyaluronic acid (HA) hydrogels through ultraviolet or visible light induced crosslinking for 2D and 3D cell culture, as well as the fluorescent labelling of nascent ECM deposited by cells during culture. We also provide protocols for secondary immunofluorescence of specific ECM components and ImageJ-based ECM quantification methods. HA polymer synthesis takes 2 weeks to complete and hydrogel formation for 2D or 3D cell culture is performed in 2–3 h. Lastly, we detail the identification of nascent proteins, including enrichment, preparation and analysis with mass spectrometry, which can be completed in 10 days.
Keywords: metabolic labelling, hydrogels, cell culture, tissue engineering, extracellular matrix, proteomics
INTRODUCTION
The extracellular matrix (ECM) is a network of macromolecules, including proteins and sugars, which constitute the cellular microenvironment and present numerous chemical and physical cues to regulate cell behavior during tissue development, homeostasis and repair. Cells interact with and constantly remodel their surrounding ECM and thus, play an active role in shaping their own microenvironment1. Any disruption to these control mechanisms may lead to disease, such as tissue fibrosis, which is characterized by aberrant remodelling and excessive accumulation of ECM components2. Beyond regulating cell behaviour, ECM organization and composition also gives rise to tissue properties and function, such as the mechanical strength provided by the assembly of various fibrous proteins or directional properties through ECM anisotropy3.
Our understanding of cell-ECM interactions has been limited due to a lack of tools to probe this interface either in vivo where imaging is challenging or in vitro with standard culture systems (e.g., tissue culture plates) that are non-physiological. Biomaterials have emerged as a valuable tool to recreate certain aspects of the ECM, allowing researchers to explore the influence of ECM properties (e.g., mechanics, structure) on cellular functions. As one example, hydrogels (water-swollen polymer networks) are of particular interest since they can be engineered with specific biophysical or biochemical cues that mimic those of a cell’s extracellular environment4. Hydrogels can be engineered such that cells can be either seeded atop or encapsulated within for two- and three-dimensional (2D, 3D) cell culture, respectively5. The potential of these ECM-mimetic hydrogels has been leveraged for numerous applications, including studies in tissue engineering, disease modeling and mechanobiology. As another biomaterial example, fibrous materials have been investigated to better recapitulate the fibrous structure of the ECM6. Fibrous biomaterials have been fabricated through techniques such as electrospinning, phase separation, or self-assembly and can be processed with anisotropy to mimic the organization of many tissues7. Like hydrogels, numerous fibrous biomaterials have been investigated in applications of tissue engineering and mechanobiology.
While these biomaterial platforms can be used to model properties of the ECM, cells will also deposit their own local matrix over time, which may influence the cell-biomaterial interface. Cell-deposited matrix may indeed be desirable, for applications such as tissue engineering where the biomaterial is only a temporary matrix during the evolution of the neo-tissue. For applications such as this, it is important to evaluate the spatio-temporal deposition and composition of the ECM to better understand and guide cell behaviour. However, most assays towards the visualization and quantification of ECM dynamics either lack spatiotemporal resolution (e.g., biochemical assays, immunofluorescence) or are inherently complicated by the use of radio-labelled isotopes (autoradiography). In addition, most of these assays provide limited information regarding the structure and organization of the secreted ECM. Thus, new tools are needed to evaluate the properties and composition of cell-produced ECM.
To address this, metabolic labelling techniques using a cell’s endogenous synthesis and translation machinery have been employed. In particular, metabolic labelling utilizes chemical analogs (e.g., functional non-canonical amino acids and other building blocks) that can be incorporated into newly synthesized biomolecules (e.g., proteins) that present reactive handles for the labelling or purification of these biomolecules8. As an example, amino acid analogs that carry bio-orthogonal functional groups can selectively be detected by fluorescent tags9 or ligated to an affinity tag for enrichment and identification10,11. Following initial demonstration in bacteria12, the capability to track proteome-wide changes found wide applications in mammalian cell cultures to probe intracellular protein localization13, degradation14 and turnover15. Recent developments have further increased spatial and cell-specific control over the labelling of nascent proteins. For example, non-canonical amino acids have been engineered to only incorporate upon direct light exposure using photocaging16 or into cells expressing a mutant enzyme for protein biosynthesis17,18, allowing for cell-selective labelling. The potential of these systems has further been leveraged in vivo to discern protein expression of transplanted cells within host tissue19 or for diagnosis and biomarker discovery20. However, these techniques often require advanced chemistries or genetic engineering of cells or organisms, which may be a limiting factor for many laboratories. In addition, many such studies have focused on intracellular proteins, with fewer focusing on the role of cell-produced and secreted ECM in controlling cell function in engineered culture systems and towards tissue repair. Thus, we have been adapting metabolic labelling to probe the dynamics of extracellular proteins in vitro and in vivo21–26. This nascent ECM labelling technique can be readily adapted to investigate a range of cell types and with varied engineered biomaterial platforms.
Development of the protocol
We developed a protocol to visualize and identify the spatiotemporal evolution of newly synthesized and deposited matrix components by cells cultured in combination with biomaterials. This technique relies on the reactive non-canonical amino acids azidohomoalanine (AHA) or homopropargylglycine (HPG) which, when added to culture media, are incorporated into proteins as they are synthesized (Fig. 1).
Figure 1. Overview of nascent matrix labelling approach.
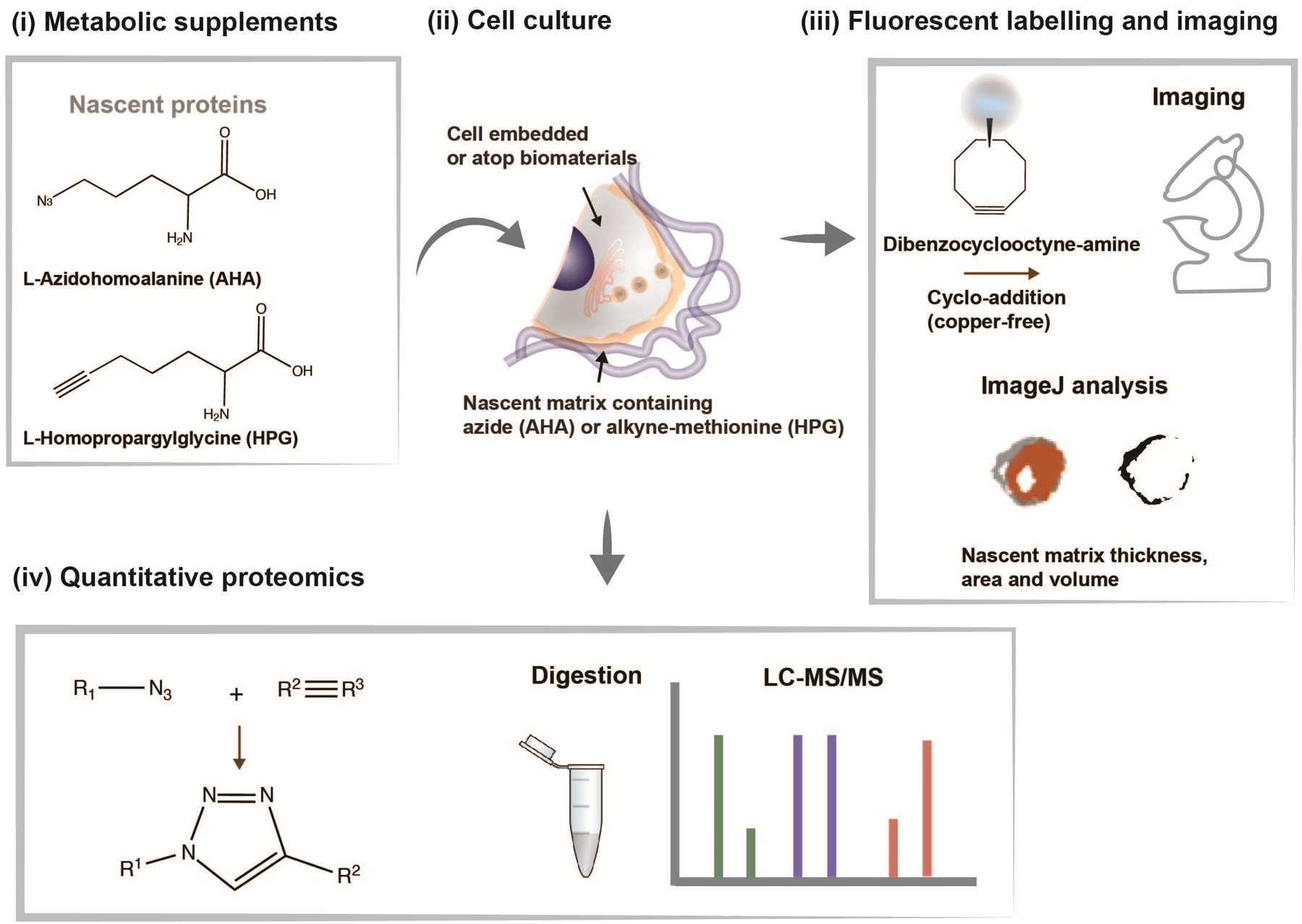
(i) Chemical structures of analogs of the amino acid methionine (L-azidohomoalanine (AHA) and L-homopropargylglycine (HPG)) to label nascent proteins. (ii) Schematic of cell interacting with a biomaterial and cultured in methionine-free media supplemented with AHA or HPG to label nascent proteins. (iii) Incorporated methionine analogs can be fluorescently labelled using click-chemistry (copper-free cycloaddition) using a fluorophore-conjugated cyclooctyne (e.g., dibenzocyclooctyne-amine (DBCO)), imaged and nascent matrix properties (thickness, area, volume) quantified using ImageJ. (iv) Incorporated methionine-analogs can be quantified using liquid chromatography-mass spectrometry (LC-MS/MS) upon release from the biomaterial.
The incorporated azide/alkyne functional groups can then be reacted specifically with fluorophores using copper-catalyzed azide-alkyne click chemistry23,25. While the advantage of this labelling strategy is the availability of commercially available kits (e.g., Click-iT™ HPG Alexa Fluor™ Protein Synthesis Assay Kit, Invitrogen), the copper-catalyzed reaction is cytotoxic, preventing the use on live cells. Building upon previous advances in biocompatible click reaction chemistry, we further advanced the labelling strategy by using copper-free cycloaddition, which retains labelled proteins that are extracellular22. The labelling technique has a wide range of applications, as it addresses the challenge of distinguishing between existing and newly secreted matrix components. We have applied this technique primarily to address questions related to mechanosensing within engineered biomaterial systems21,22 and to measure the dynamics of the cell-hydrogel interface26. In addition, pulse-labelling studies - the supplementation of analogs for only a certain time-period - offer an additional strategy to assess temporal aspects of nascent matrix deposition and remodeling23.
For analysis of nascent matrix properties, we used confocal fluorescence microscopy for quantitative measurements of local matrix dimensions for which immunofluorescence staining may be further performed to evaluate co-localization with specific ECM proteins (Fig. 1). In addition, we developed a method to isolate the labelled matrix proteins using a cleavable biotin-alkyne linker, which allows for identification and quantification of secreted proteins via mass spectrometry (Fig. 1)24. Thus, using bio-orthogonal amino acid analogs, we have adapted a metabolic labelling strategy that provides a global representation of all secreted matrix components, enabling visualization and comprehensive analysis of nascent ECM.
Applications
The labelling of newly secreted matrix components has a wide range of applications as it enables visualization and quantitative analysis of the spatial and temporal changes of newly secreted matrix in response to various chemo-mechanical cues, and can be performed on a single-cell basis. Monitoring the deposition and accumulation of secreted matrix has direct implications in the context of tissue engineered constructs such as for the replacement of degenerated or injured cartilage tissue27,28. In cartilage tissue engineering, cells (e.g., chondrocytes) are commonly encapsulated within hydrogels that recreate aspects of cartilage ECM and direct the distribution of secreted matrix29,30. Towards the quantification of nascent matrix dynamics, we measured the spatiotemporal evolution of nascent matrix secreted by chondrocytes when encapsulated within hydrogels (Fig. 2a). In our recent study, we cultured cells in covalently crosslinked hyaluronic acid (HA) hydrogels that are non-degradable and monitored the distribution of nascent proteins over a time course of seven days (Fig. 2b)26. With time, the nascent matrix components successively extended from the cell, indicating accumulation and distribution throughout the hydrogel (Fig. 2c). In addition, co-encapsulation of fluorescent beads into the hydrogel revealed that the emerging matrix physically displaces the hydrogel from the cell body (Fig. 2d)26. These findings indicate that the nascent matrix increasingly masks the presentation of engineered cues, an approach previously not accessible due to the limited number of ECM components that can be labelled simultaneously with traditional immunofluorescence. We further investigated how this nascent matrix is modulated by the hydrogel microenvironment23, including with the use of physically crosslinked agarose hydrogels where biophysical properties can be altered (Fig. 2e). Encapsulation of chondrocytes into softer, loosely crosslinked gels (1% polymer (wt/vol)) resulted in a readily distributed fibrous protein network, while encapsulation in a more crosslinked gel (2%, 3% (wt/vol)) resulted in a less permissive environment and the nascent proteins were located more adjacent to the cell body (Fig. 2f).
Figure 2. Application of metabolic labelling to monitor nascent protein accumulation at the cell-hydrogel interface in 3D hydrogels.

a Schematic illustrating the encapsulation of chondrocytes in synthetic hydrogels and accumulation of nascent matrix at the cell-hydrogel interface during culture in chondrogenic media. b Representative images (scale bars 10 μm) of nascent protein (AHA, grey) accumulation by chondrocytes (cell membrane marker, red) encapsulated in covalently crosslinked, non-degradable hyaluronic acid hydrogels and cultured up to 7 days in chondrogenic media supplemented with the methionine-analog AHA. c Intensity profile plots of nascent protein staining (median, shaded areas represent 95% confidence interval, n = 40 cells) at various times during culture. d Representative images (magnifications on right) of accumulated nascent proteins (white) of chondrocytes co-embedded with fluorescent beads (orange) and cultured for 7 days (scale bars 10 μm). e Representative images (scale bar 10 μm) of nascent protein accumulation (HPG) by chondrocytes cultured in agarose hydrogels of varied concentrations for 9 days. f Intensity profile plots of nascent protein staining (median, shaded areas represent 95% confidence interval, n = 20 cells). b,d adapted with permission from Loebel et al.26, Wiley), e,f adapted with permission from McLeod & Mauck23, Nature Research).
Beyond the monitoring of ECM accumulation for tissue engineering, nascent protein labelling provides a means to probe how cells perceive and respond to mechanical signals from their extracellular environment, including varying biomaterial moduli and architecture31. To demonstrate the influence of hydrogel stiffness on cell behavior and ECM deposition, we labelled nascent proteins secreted by mesenchymal stromal cells (MSCs) cultured atop hydrogels with different elastic moduli (5 and 15 kPa Young’s modulus, Fig. 3a,b). The deposition of matrix around the cell body increased for MSCs on stiff substrates when compared to softer hydrogels (Fig. 3c). We extended this approach for in vitro models of fibrotic tissue remodeling, which is associated with extensive matrix deposition and disruption of ECM organization in vivo32. In our recent study, we visualized nascent matrix deposited by MSCs seeded atop aligned and non-aligned electrospun scaffolds to mimic healthy and injured connective tissues (Fig. 3d, e)21. As a result, nascent matrix deposition increased for MSCs cultured on non-aligned fibers, indicating an increased fibrotic response to an injury-mimetic microenvironment (Fig. 3f). With these studies, our metabolic labelling was important to visualize the ECM architecture and organization, particularly as the specific composition was unknown, making traditional labelling and imaging approaches difficult. Furthermore, these studies support previous reports on the interplay of the role of substrate stiffness and other environmental factors, including dimensionality and architecture on cell function and matrix deposition5,33.
Figure 3. Application of nascent protein labelling to assess cell mechanobiologic response atop 2D biomaterials.
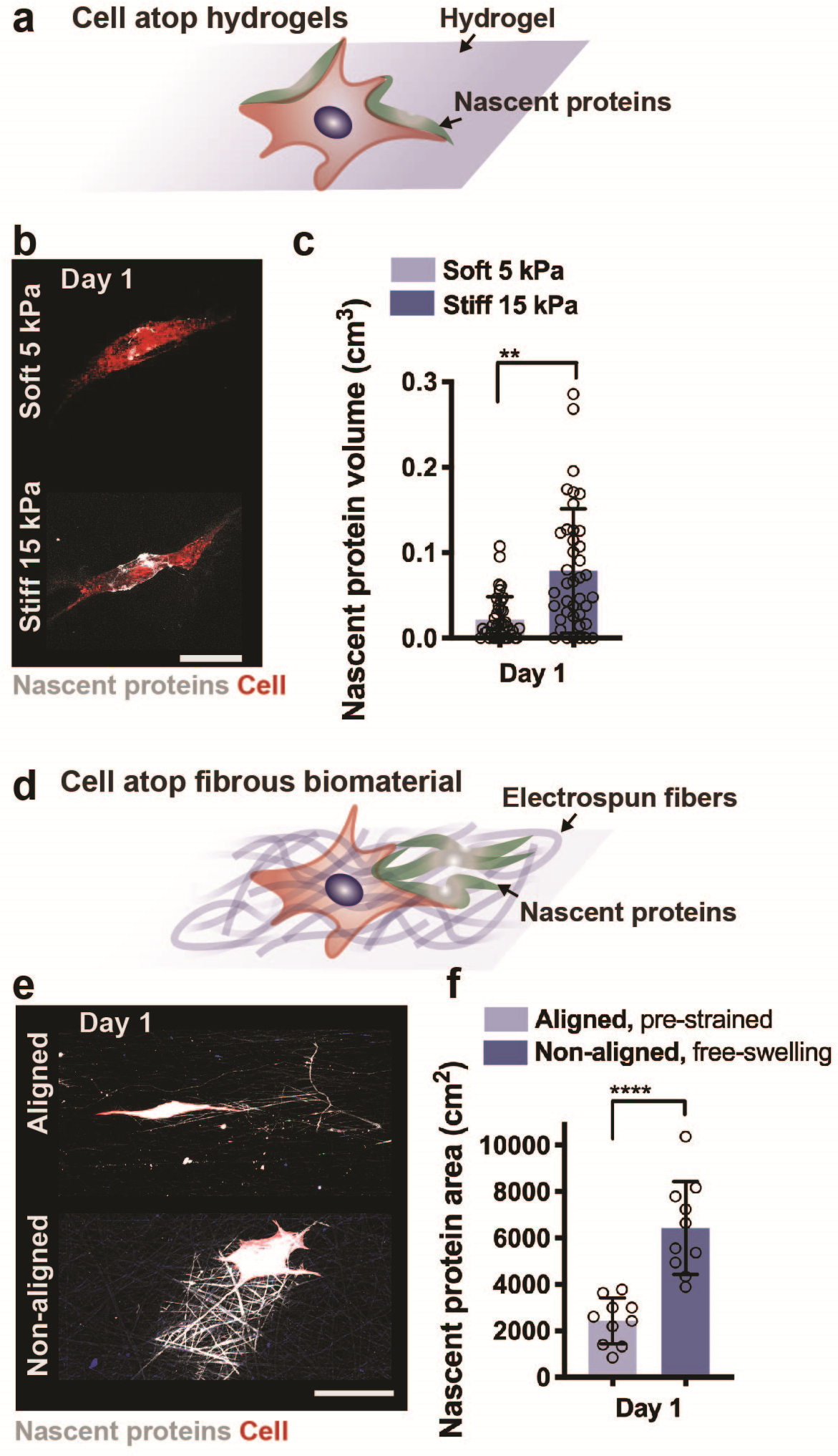
a Schematic illustrating the culture of a cell atop a synthetic hydrogel and accumulation of nascent proteins adjacent to the cell body. b Representative images (scale bar 50 μm) of nascent protein (AHA, grey) deposition by human mesenchymal stromal cells (hMSCs, cell membrane marker, red) seeded on non-degradable soft (5 kPa Young’s modulus) and stiff (15 kPa Young’s modulus) hyaluronic acid hydrogels and cultured for 1 day (24 hours) in growth media supplemented with the methionine-analog AHA. c Quantification of nascent protein volume deposited by hMSCs within 1 day of culture (n = 25 cells, mean ± SD, **p ≤ 0.01 by two-tailed Student’s t-test). d Schematic illustrating the culture of a cell atop a fibrous biomaterial and deposition of nascent proteins. e Representative images (scale bar 50 μm) of nascent protein (AHA, grey) deposition by bovine mesenchymal stromal cells (bMSCs, cell membrane marker, red) seeded on electrospun fibronectin-coated polycaprolactone fibers (aligned, pre-strained or non-aligned, free-swelling) and cultured for 1 day (24 hours) in growth media supplemented with the methionine-analog AHA. f Quantification of nascent protein area deposited by bMSCs within 1 day of culture (n = 10 cells, mean ± SD, ****p ≤ 0.0001 by two-tailed Student’s t-test). e,f adapted with permission from Bonnevie et al.21, Nature Research).
We further applied the metabolic labelling technique to probe questions related to mechanosensing in 3D22. Specifically, we used nascent protein labelling to address questions related to the mechanosensing of MSCs when embedded within hydrogels that accommodate cell spreading through either protease-dependent degradation or protease-independent remodelling (Fig. 4a). In both hydrogel types, cells deposited matrix rapidly within 24 hours, which expanded with extended culture times. In proteolytically degradable hydrogels, we treated embedded MSCs with monoclonal antibodies to block cellular adhesion to secreted fibronectin or collagen, which decreased their ability to spread (Fig. 4b,c). In addition, within dynamic viscoelastic hydrogels, we found that disruption of the secretion or remodelling of these nascent proteins also inhibits spreading and consequently reduces downstream signalling such as osteogenic differentiation (Fig. 4d,e)22. Furthermore, by increasing the dissociation rate of these dynamic crosslinks, our recent studies indicated rapid cell spreading that is regulated by cellular interactions with their nascent proteins (Fig. 4f,g)34. These studies indicate the importance of adhesion to and remodelling of nascent matrix within 3D cellular microenvironments, beyond only the presentation of the biomaterial. Again, the metabolic labelling approach was needed to provide a global representation of the dynamic evolution of proteins, with findings that may have been missed if only select proteins were labelled.
Figure 4. Application of nascent protein labelling to probe mechanosensing and spreading in 3D hydrogels.
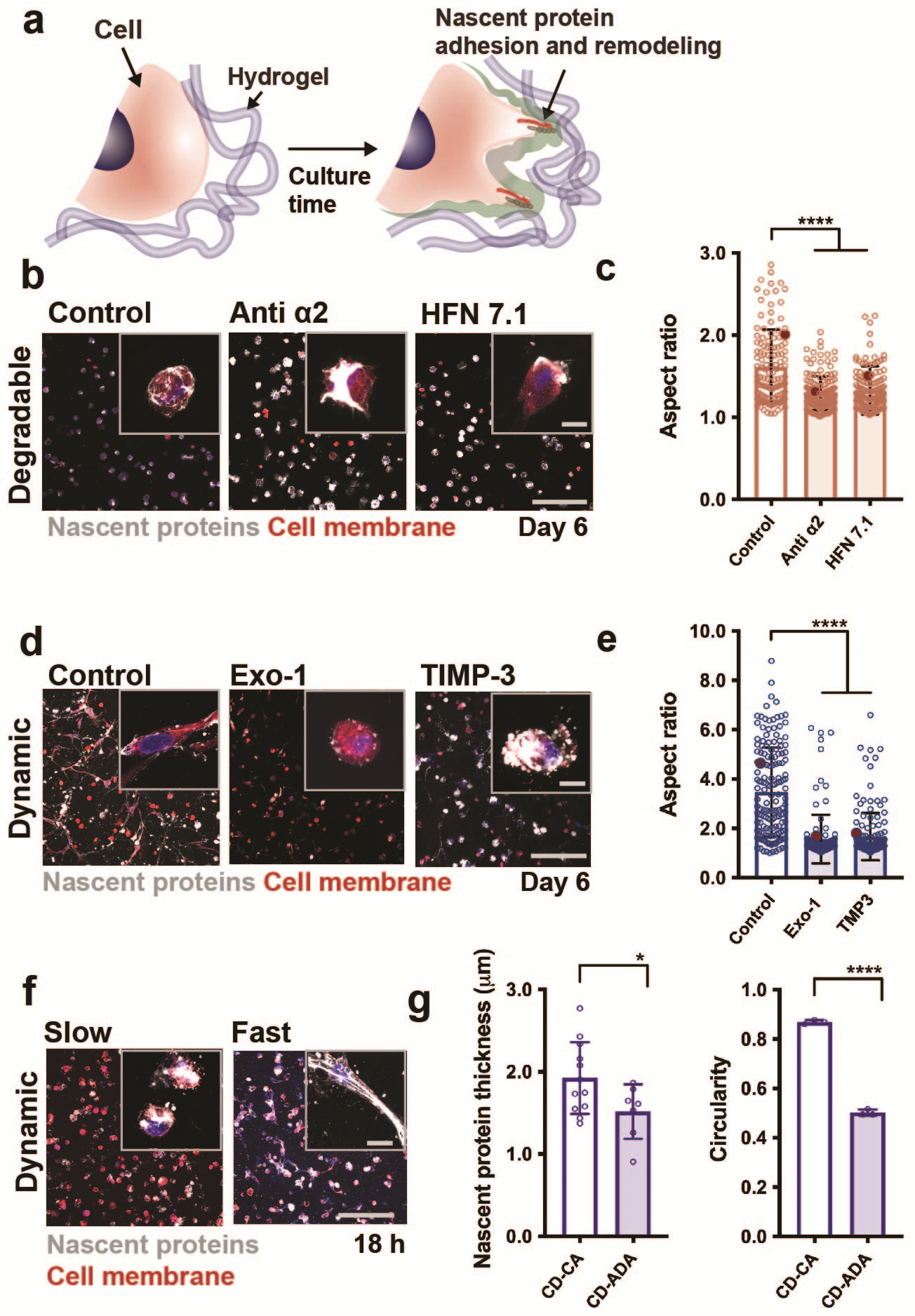
a Schematic illustrating the culture of a cell within a synthetic hydrogel and adhesion to and remodeling of nascent proteins that enables cell spreading. b Representative images (scale bar 200 μm (main image) and 20 μm (inset)) of nascent protein deposition (AHA, grey) by hMSCs embedded in enzymatically degradable hydrogels and cultured for 6 days without treatment (control) or when treated with monoclonal antibodies against integrin alpha2 (anti-α2) or human fibronectin (HFN7.1). c Quantification of cell aspect ratios of hMSCs embedded in enzymatically degradable hydrogels at day 6 (n = 133 cells (control), n = 155 cells (anti-α2) n = 152 cells (HFN7.1), mean ± SD, ****p ≤ 0.0001 by one-way ANOVA with Bonferroni post hoc). d Representative images (scale bar 200 μm (main image) and 20 μm (inset)) of nascent protein deposition (AHA, grey) by hMSCs in dynamically crosslinked hydrogels (double-networks with guest-host and covalent bonds) and cultured for 6 days without treatment (control) or when treated with an inhibitor of exocytosis (Exo-1) or a recombinant tissue inhibitor of metalloproteinase 3 (TIMP-3). e Quantification of cell aspect ratios of hMSCs embedded in dynamically crosslinked hydrogels at day 6 (n = 161 cells (control), n = 122 cells (Exo-1) n = 150 cells (TIMP-3), mean ± SD, ****p ≤ 0.0001 by one-way ANOVA with Bonferroni post hoc). f Representative images (magnifications inserted) of nascent proteins (white and fibronectin (red) secreted by hMSCs encapsulated in dynamic hydrogels with crosslinks based on slow or fast dissociation rate constants and cultured for 18 hours (scale bars 200 μm, insert 20 μm, mean ± SD, *p ≤ 0.05, ****p ≤ 0.0001 by two-tailed Student’s t-test). b,c,d,e,f adapted with permission from Loebel et al.22 and Yang et al.34, Nature Research).
While metabolic labelling provides information on the spatio-temporal properties of nascent matrix in engineered culture systems, metabolic supplements are also being used to map protein synthesis and turnover during tissue development in vitro and in vivo (Fig. 5a). For the identification of newly secreted proteins, we developed a strategy for isolating AHA-labelled proteins from engineered cellular environments and developing tissues (Fig. 5b)24,25. This method uses a cleavable biotin-alkyne linker and NeutrAvidin beads to selectively enrich for AHA-labelled proteins prior to identification by liquid chromatography-tandem mass spectrometry (LC-MS/MS). We applied this approach to identify extracellular matrix proteins secreted by chondrocytes cultured within the 3D hydrogels in vitro and resolved significant upregulation of chondrogenic markers (Fig. 5c). In vivo, we have used this strategy to compare the newly synthesized proteome. Data from two embryonic (E) time points (E12.5, E15.5) showed significant differences that are characteristic for these developmental time frames (Fig. 5d). These studies demonstrate the potential use of AHA-labelled protein enrichment to measure protein dynamics during tissue formation in vitro and in vivo. Alternative methods have been developed to monitor protein dynamics, such as stable isotope labeling (SILAC); however, selective enrichment of AHA-labelled proteins using cleavable biotin-alkyne linker enables detection of lower abundant proteins because unlabeled proteins are removed during enrichment, whereas with SILAC all proteins are analyzed by LC-MS/MS. Furthermore, combining this with the spatio-temporal information obtained through fluorescent labeling provides insight into the dynamics of cell-matrix interactions such as cell-material dynamics in 3D hydrogel studies22.
Figure 5. Enrichment of AHA-labelled proteins within engineered hydrogels in vitro and from murine embryos in vivo.
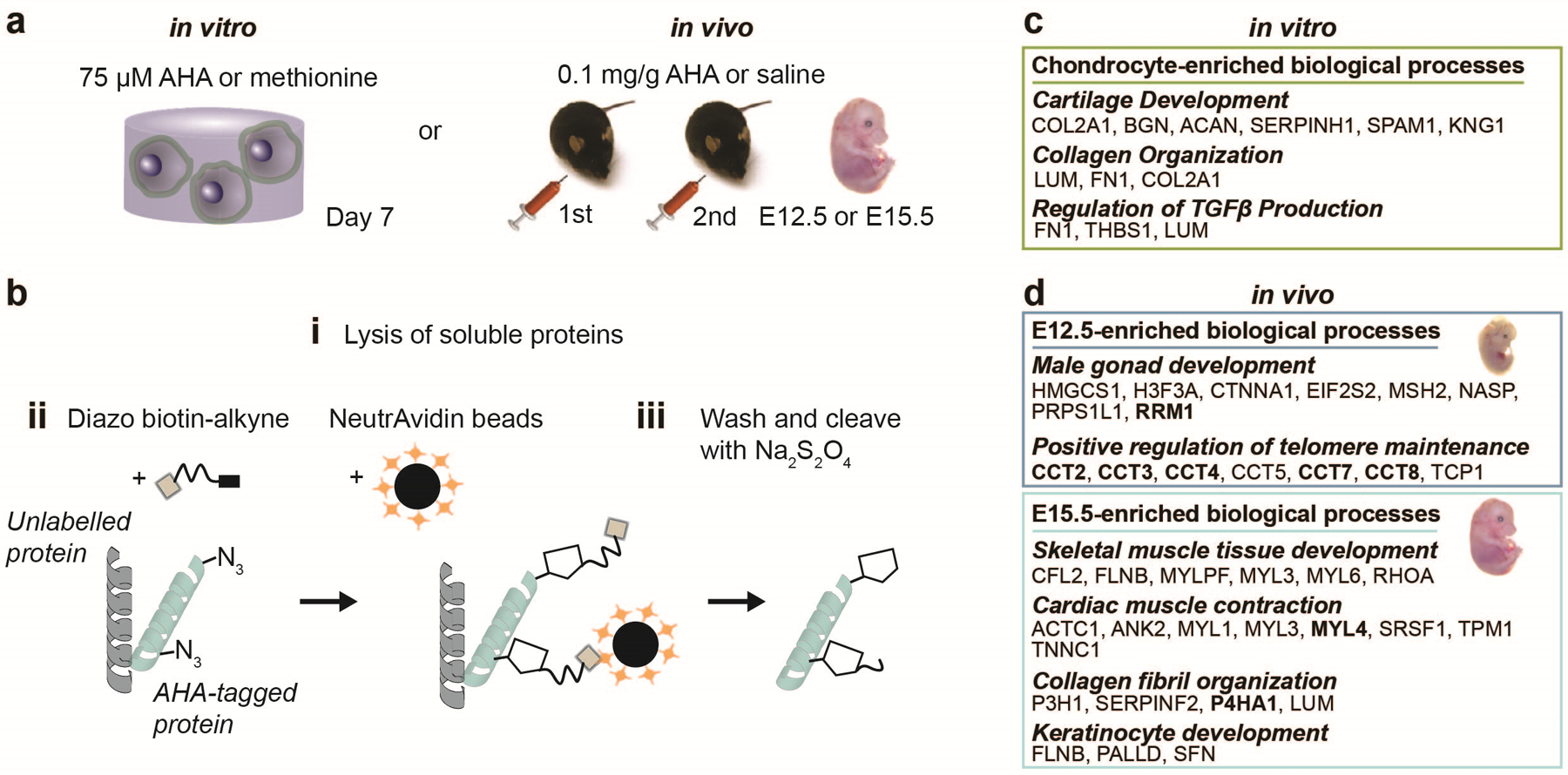
a Workflow for enrichment of AHA-labelled proteins using a diazo biotin alkyne linker. AHA is added to methionine-free media to identify secreted proteins during in vitro culture of cells embedded within 3D hydrogels. For in vivo studies, AHA is injected into time-mated dams at 0.1 mg/g, once a day at E12.5 and E13.5, whereas 10 μL/g saline is injected into control animals. b Cells or E15.5 embryos are harvested, and lysates of soluble proteins (i) are reacted with the linker and isolated using neutralizing streptavidin beads (ii). Unlabelled proteins are removed through washing and AHA-labelled proteins are released using sodium dithionite (Na2S2O4) (iii). c Select biological process terms identified by conducting gene ontology analyses on proteins exclusive to and >2-fold enriched and p < 0.05 between AHA and methionine controls, calculated using a two-tailed Student’s t-test. d Select biological process terms identified by conducting gene ontology analyses on proteins exclusive to and >2-fold enriched compared with saline for individual time points. d adapted with permission from Saleh et al.24, Elsevier).
Limitations
Here, we describe a strategy for visualizing and identifying newly secreted proteins by cells cultured within or atop biomaterials. One limitation of the nascent protein labelling is the restriction to methionine-containing proteins. However, the majority of ECM proteins contain methionine and our previous study indicated that both AHA and HPG incorporate into the ECM of various tissues including lungs, hearts, brains and kidneys25. An important exception is elastin - a protein that contains a very low content of methionine - and may preclude investigations into elastin-rich tissues and organs. While alternative non-canonical amino acids exist such as through the incorporation of tyrosine analogs (e.g., 4-Azido-L-phenylalanine35, p-Acetylphenylalanine36), these amino acids are not readily incorporated and require transgenic lines that express a mutant aminoacyl tRNA synthetase capable of charging the tyrosine analog. An additional limitation of these azide-modified biomolecules is the incorporation into all methionine-containing ECM components synthesized by cells. Thus, we implemented secondary labelling of specific components (e.g., collagens, glycosaminoglycans) through immunofluorescence staining as a tool to assess and distinguish their role in the organization and timing of ECM assembly22,23,26. Furthermore, this metabolic labelling approach serves as a tool to visualize the nascent matrix and as such may not directly provide mechanistic insight into the role of newly secreted proteins in cellular behaviours. Here, we and others have applied additional techniques such as labelling of specific ECM/material components21,22,26,37 or mechanical analysis of the secreted matrix26,38,39, which can add additional mechanistic insight to the question being asked. Given that metabolic labelling visualizes all newly secreted proteins, we have also developed techniques to identify labelled proteins using mass spectrometry24,25, which further expands the utility and understanding of these cellular microenvironments. A limitation of using AHA enrichment for LC-MS/MS analysis is the nonspecific labeling of proteins with the cleavable biotin alkyne linker. However, we addressed this limitation by adding ascorbate as a catalyst of the copper-mediated click reaction and aminoguanidine to scavenge ascorbate byproducts. These modifications substantially reduce nonspecific protein labeling24 and have further been optimized for 3D cell culture studies (Supplementary Fig. 4). Finally, while this protocol allows identification of newly secreted proteins, measuring protein concentrations and turnover rates may be limited by the generally poor solubility of ECM proteins, especially when compared to other cellular compartments. Therefore, to increase protein solubility, we have optimized the protocol to solubilize ECM proteins in high molar urea followed by sonication to mechanically disrupt non-covalent protein-protein interactions24,40.
Experimental design
Hydrogel formation (Steps 1–27).
In this protocol, the synthesis of norbornene-modified hyaluronic acid (NorHA) polymers to form NorHA hydrogels is described. NorHA hydrogels are an example of a hydrogel that has been used in the culture of cells, either with cells seeded atop (Fig. 2b,c) or encapsulated within (Fig. 3b,c)22,33. We have extensive experience with the use of norbornene-modified hydrogels and modification is based on previously described protocols41; however, chemical modifications may be applied to other polymers such as polyethylene glycol42,43 or gelatin44. Hydrogels are then formed through the photo-crosslinking of aqueous NorHA polymer solutions containing crosslinkers and photoinitiators with either ultraviolet (UV) or visible light (depending on the photoinitiator used). The initial mechanical properties can be tuned by varying the concentrations of NorHA polymer and crosslinker (e.g., dithiothreitol, dithiol peptide crosslinkers)22,41. If desirable, the mechanical properties can be measured through bulk (e.g., rheology, dynamic mechanical analysis) or local measurements such as nanoindentation45,46.
Cell seeding and encapsulation (Steps 28–37).
For cell culture, we describe the fabrication of NorHA hydrogels for 2D cell seeding and 3D cell encapsulation. In general, the crosslink density and polymer concentration depend on the application; however, we typically use hydrogels with an initial Young’s modulus of at least 2 kPa to ensure that constructs are stable during culture. For 2D cell culture, we describe the thiolation of glass coverslips to promote the adherence of thin NorHA hydrogel films (ca. 100 μm) prior to cell seeding. This strategy can be adapted to other chemistries for covalently attaching hydrogels to coverslips, where the treatment depends on the type of crosslinking (e.g. methacrylated47 or gelatin-coated45 coverslips) (Fig. 6a). Further, we illustrate the preparation of NorHA hydrogels with cells embedded for 3D cell culture experiments. A cell suspension is mixed with the NorHA precursor solution and then crosslinked between two untreated coverslips separated by spacers (ca. 300 μm thick) with light (Fig. 6b). Hydrogels of 5 mm × 5 mm are then cut from the hydrogel and cultured separately. We recommend this strategy for studies involving Live/Dead staining, fluorescent imaging and analysis.
Figure 6. Hydrogel preparation for 2D and 3D cell culture studies.
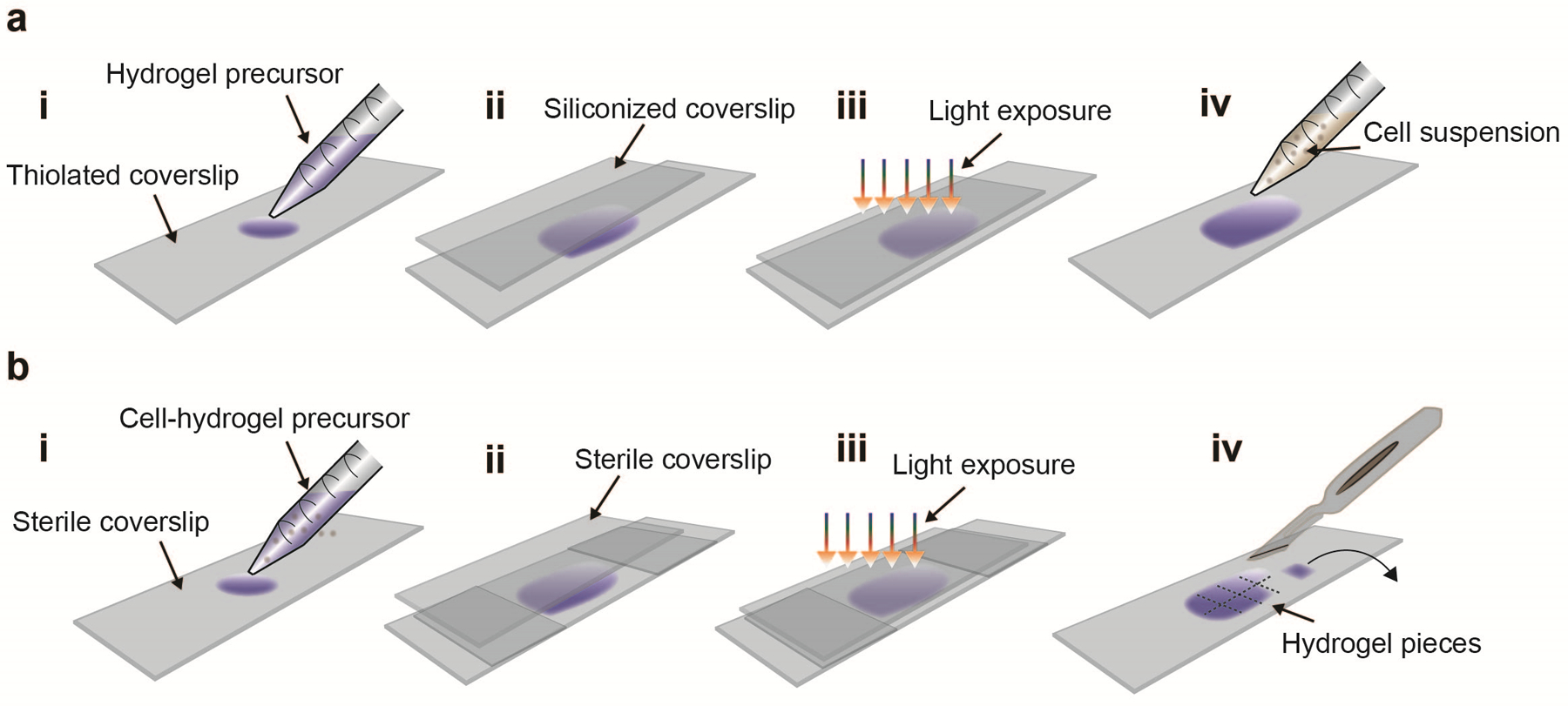
a For 2D cell culture, first add the hydrogel precursor solution onto a thiolated glass coverslip (i). Next, place a Sigmacote-treated coverslip on top and gently press to allow the hydrogel precursor to spread out (ii). For crosslinking, expose the coverslips to ultraviolet or visible light, depending on the photoinitiator used (iii). Gently remove the coverslip on top, sterilize the hydrogel with a germicidal lamp, wash with sterile PBS and add the cell suspension in culture media to the hydrogel surface for cell seeding (iv). b For 3D cell culture, first mix the cell suspension with the hydrogel precursor and add onto an untreated sterile coverslip (i). Next, place a second coverslip on top with two spacers (e.g. small coverslips) and gently press (ii). Expose to ultraviolet or visible light (depending on the photoinitiator used) under sterile conditions (iii). Remove the coverslip on top, cut the hydrogel film into ca. 5 mm × 5 mm pieces and transfer the cell-laden hydrogels into culture media (iv).
Nascent matrix labelling and image analysis (Step 38–49).
Labelling media is prepared using methionine-free Dulbecco’s Modified Eagle Medium (DMEM), which is supplemented with a methionine analog (AHA) and additives (e.g., serum, growth factors) according to its application. We recommend culture conditions be customized for each cell type and time of culture to ensure high cell viability. For short culture periods (up to 7 days), we typically substitute methionine entirely with the analog AHA (100 μM, Supplementary Fig. 1); however, for studies that include sensitive cell types (e.g., primary cells, progenitor cells) or extensive culture, various ratios of L-methionine and analog may be desired. The concentration is based upon cell viability and culture period. The fluorescent labelling of incorporated proteins is then performed using a copper-free click-reaction with a fluorophore-conjugated cyclooctyne (DBCO, Fig. 1). In addition, towards optimizing the method for particular cell types, we recommend including labeling periods ranging between one and a few days to ensure matrix synthesis and deposition. This bio-orthogonal chemistry can be performed on living cells, which prevents staining of intracellular proteins when compared to the labelling of cells after fixation (Supplementary Fig. 2). We further describe membrane labelling techniques to visualize the cell boundaries and additional immunofluorescence for evaluation of intracellular and extracellular markers, such as specific ECM components. We use confocal microscopy with high resolution to acquire z-stack images of single cells within or atop hydrogels. To quantify nascent matrix properties, we describe area, volume and local thickness measurements using the image processing program, ImageJ (Supplementary Fig. 3, Fig. 7). The analysis of local thickness is based on ‘BoneJ’, an open source ImageJ plugin48 and can be applied for analysis of a wide variety of different ECM components.
Figure 7. Quantification of nascent matrix thickness in ImageJ.
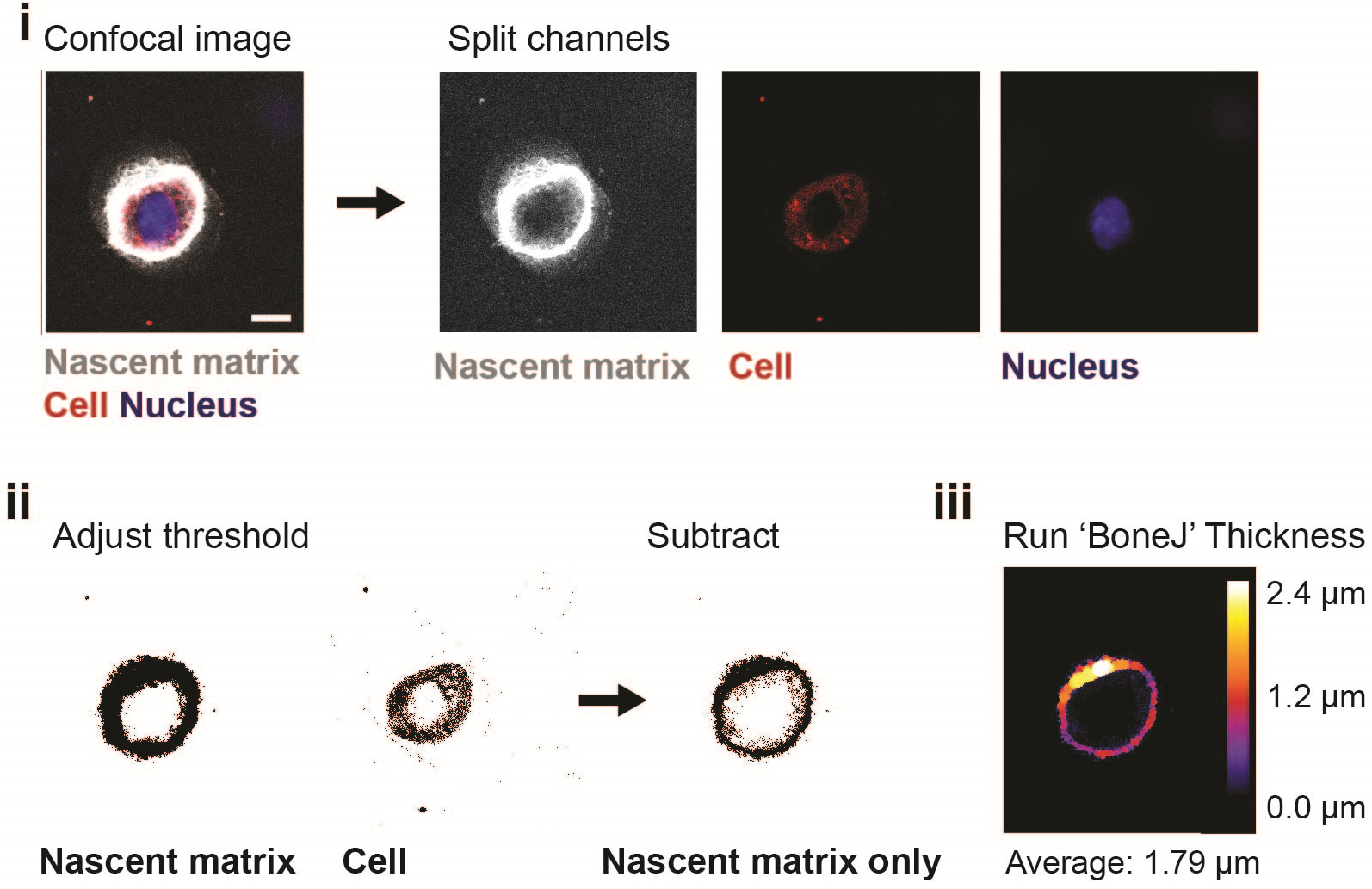
Acquire a confocal image of the nascent matrix, cell membrane and nucleus, and split the channels into single images using ImageJ (scale bar 10 μm). (i). To obtain an image of the nascent matrix only, adjust the threshold for each channel with ‘Otsu thresholding’ and subtract the ‘cell’ image from the ‘nascent matrix’ image (ii). Use the ImageJ plugin ‘BoneJ’ to measure the average nascent matrix thickness (iii).
Protein enrichment and mass spectrometry (Step 50–100).
For identifying proteins secreted by cells cultured within 3D NorHA hydrogels, we describe an approach to solubilize intra- and extracellular proteins in concentrated urea to denature intra- and extracellular proteins, which can then be selectively enriched for nascent proteins (Fig. 5c, Supplementary Fig. 4). Hydrogel constructs are treated with hyaluronidase (to digest the hydrogel), chondroitinase ABC, high molar urea and sonication (to disrupt strong, non-covalent protein-protein interactions). The AHA-containing nascent proteins are then modified with a cleavable diazo biotin alkyne linker and isolated using streptavidin beads (NeutrAvidin). We further describe the preparation and analysis of these AHA-containing proteins using liquid chromatography-tandem mass spectrometry. This procedure can be readily adapted for culture atop 2D biomaterials or with other types of hydrogel systems by selecting appropriate material digestion protocols.
REAGENTS
!CAUTION When using the chemicals described in this protocol, always wear suitable personal protective equipment, including lab coat, gloves, safety goggles, and where indicated face shield, respirator and thermo-insulated gloves. Follow the institutional and governmental safety guidelines and refer to the appropriate materials safety data sheets. All synthesis steps should be performed within a chemical fume hood and cell experiments in a class II biological safety hood.
Preparation of hydrogels
Sodium hyaluronic acid, 74 kDa (HA, Lifecore, cat. no. HA-60K; store under nitrogen at −20 °C). CRITICAL Other molecular weights of sodium hyaluronic acid can be used for this protocol. However, this affects the viscosity and properties of resulting hydrogels.
Sodium hydroxide (NaOH, Millipore Sigma, cat. no. S8045 store at RT). !CAUTION Sodium hydroxide can cause skin corrosion and serious eye damage. Avoid any direct contact.
Dowex resin (Sigma Aldrich, cat. no. 217506, store at RT). !CAUTION Dowex is acutely toxic and causes skin and eye irritations and presents organ toxicity after single exposure. Wear a respirator.
Tetrabutylammonium hydroxide solution 40% (w/v) in water (TBA-OH, Millipore Sigma cat. no. 178780, store at RT). !CAUTION Tetrabutylammonium hydroxide can cause skin corrosion and serious eye damage. Avoid any direct contact and wear a face shield and respirator.
5-Norbornene-2-carboxylic acid (TCI America, cat. no. N0667, store at RT). !CAUTION 5-norbornene-2-carboxylic acid causes skin corrosion and serious eye irritation. Wash hands and face thoroughly after handling and wear protective gloves, eye protection.
4-(Dimethylamino)pyridine (DMAP, Millipore Sigma cat. no. 107700, store at RT). !CAUTION DMAP is acutely toxic and causes respiratory, skin and eye irritation. Wear protective gloves/protective clothing/eye protection and face shield and use only in a chemical hood.
Ditert-butyl dicarbonate (Boc2O, Millipore Sigma cat. no. 205249, store at 4 °C, container tightly closed). !CAUTION Boc2O is flammable liquid and vapor, and causes skin and respiratory irritation, serious eye damage and is fatal if inhaled. Wear protective gloves, eye protection/face shield and respiratory protection.
Acetone (Fisher Scientific, cat. no. A18–20, store at RT). !CAUTION Acetone is flammable, may cause irritation of respiratory tract and vapors may cause dizziness. Keep away from heat and hot surfaces.
Liquid nitrogen. !CAUTION Contact with liquid nitrogen may cause serious freezing injuries. When handling liquid nitrogen, always wear thermo-insulated gloves, a full-face shield and laboratory coat.
Sodium chloride (Millipore Sigma cat. no. S9888, store at RT).
Deuterium oxide (D2O, Millipore Sigma cat. no. 151882, store at RT in a desiccator).
2-Hydroxyethyl agarose (Millipore Sigma cat. no. A4018, store at RT).
1,4-Dithiothreitol (DTT, Millipore Sigma cat. no. D0632, store under nitrogen at 4 °C). !CAUTION DTT is acutely toxic and harmful if swallowed. Dispose container to an approved waste disposal plant.
RGD peptide (GCGYGRGDSPG, Genscript customized, store at −20 °C).
Matrix metalloproteinase (MMP)-degradable crosslinker (GCNSVPMSMRGGSNCG, Genscript customized, store at −20 °C).
2-Hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959, Millipore Sigma cat. no. 410896, store at RT).
Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, Colorado Photopolymer Solutions cat. no. TPO-Li, store at RT). !CAUTION LAP may cause allergic skin reaction. Wear protective gloves.
(3-Mercaptopropyl)trimethoxysilane (Millipore Sigma cat. no. 175617). !CAUTION (3-mercaptopropyl)trimethoxysilane may cause an allergic skin reaction and is toxic to aquatic life. Wear protective gloves.
Sodium hydroxide pellets (NaOH, Fisher Scientific, cat. no. S318100). !CAUTION NaOH may be corrosive to metals and causes severe skin burns and eye damage. Wear protective gloves, protective clothing, eye protection and face shield. Use in a chemical hood.
Toluene (Fisher Scientific, cat. no. S25611). !CAUTION Toluene is highly flammable and causes skin irritation, dizziness and may cause damage to organs through prolonged exposure. Wear protective gloves, protective clothing, eye protection and face shield. Use in a chemical hood.
Hexylamine (Millipore Sigma cat. no. 219703) !CAUTION Hexylamine is flammable, toxic if swallowed or in contact with skin, can cause severe skin burns and eye damage. Wear protective gloves, protective clothing, eye protection and face shield. Use in a chemical hood.
Nascent protein labelling
Dulbecco’s Modified Eagle Medium glutamine-, methionine- and cystine-free high-glucose (DMEM, Thermo Fisher Scientific, cat. no. 21013024, store at 4°C).
L-cystine (Millipore Sigma cat. no. C7602, store at RT).
GlutaMAX™ Supplement (Thermo Fisher Scientific, cat. no. 35050061, store at 4°C).
Sodium pyruvate (Thermo Fisher Scientific, cat. no. 11360070, store at 4°C).
Ascorbate 2-phosphate (Millipore Sigma cat. no. A8960, store at RT).
L-methionine (Millipore Sigma cat. no. M5308, store at RT).
L-azidohomoalanine (AHA, Click Chemistry Tools cat. no. 1066, store at −20°C).
Dimethyl sulfoxide (DMSO, sterile, Millipore Sigma, cat. no. D2438, store at RT). !CAUTION Wear protective gloves, protective eyewear and full-face respirator.
Hydrochloric acid 12M (HCl, Fisher Scientific, cat. no. S25358, store at RT). !CAUTION HCl causes serious eye damage, skin burns and may cause respiratory irritation.
Bovine serum albumin (BSA, Millipore Sigma cat. no. A7906, store at 4°C).
Dibenzocyclooctyne-amine-488 (DBCO-488, Click Chemistry Tools cat. no. 1278–1, store at −20°C).
CellMask Orange Plasma membrane stain (Thermo Fisher Scientific cat. no. C10045, store at −20°C).
Paraformaldehyde solution (PFA, Fisher Scientific, cat. no. AAJ19943K2, store at 4°C). !CAUTION PFA causes eye burns and severe eye damage and may cause allergic skin reaction. Wear personal protective equipment and face protection and use only in chemical hood.
ProLong Gold Antifade Mountant (Thermo Fisher Scientific cat. no. P36930, store at RT, protect from light).
Cell seeding and encapsulation
Human mesenchymal stromal cells (MSCs, e.g. Lonza cat. no. PT-2501, store in liquid nitrogen). !CAUTION These cells should be routinely checked to ensure that they are authentic and not contaminated with mycoplasma.
Chondrocytes (CH, e.g., isolated from bovine chondrocytes from juvenile bovine knees (Research 87, Boylston MA)26,49 or Promocell cat. no. C-12710 (human), store in liquid nitrogen). !CAUTION These cells should be routinely checked to ensure that they are authentic and not contaminated with mycoplasma.
Minimum Essential Media (α-MEM, Thermo Fisher Scientific, cat. no. 11095080, store at 4 °C).
Fetal bovine serum (FBS, Thermo Fisher Scientific cat. no. 1437028, store at −20 °C).
Penicillin/streptomycin, 10.000 U/ml (Life Technologies cat. no. 15140–122, store at −20 °C).
Gentamycin (Life Technologies cat. no. 15710–064, store at 4 °C).
Phosphate-Buffered Saline (Life Technologies cat. no. 10010023, store at 4 °C).
Trypsin (Life Technologies, cat. no. 15090–046, store at 4 °C).
Live/Dead Viability/Cytotoxicity Kit for mammalian cells (e.g. Thermo Fisher Scientific, cat. no. L3224, store at −20 °C).
Immunofluorescence
Triton-X100 (Sigma Aldrich, cat. no. T8787, store at RT). !CAUTION Triton-X100 can cause skin irritation and serious eye damage. Wear protective gloves, eye protection and face shield.
Sucrose (Fisher Scientific, cat. no. S25590, store at RT).
Dulbecco’s Phosphate-Buffered Saline (PBS, Thermo Fisher Scientific cat. no. 14040141, store at 4 °C).
Anti-fibronectin (Milllipore Sigma, cat. no. F6140, store at −20 °C).
Anti-collagen type I (Abcam, cat. no. ab138492, store at −20 °C).
Anti-collagen type IV (ThermoFisher, cat. no. MA1–22148, store at −20 °C).
Alexa Fluor-647 IgG H&L (Abcam cat. no. 150143, store at −20 °C).
Alexa Fluor-594 IgG H&L (Abcam cat. no. 150080, store at −20 °C).
Rhodamine-conjugated phalloidin (Invitrogen R415, store at −20 °C). !CAUTION Phalloidin is acutely toxic when inhaled and fatal if swallowed or in contact with skin. Wear protective gloves, eye protection and face shield.
Hoechst 33342 (ThermoFisher, cat. no. 62249, store at 4 °C). !CAUTION Hoechst can cause serious eye damage and eye irritation. Wear protective gloves and eye and face protection.
Nail polish (e.g. Electron Microscopy Sciences, cat. no. 72180). !CAUTION Nail polish causes skin and serious eye irritation and may cause drowsiness or dizziness. Wear protective gloves, eye protection and face shield.
Sigmacote (Sigma Aldrich, cat. no. SL2, store at 4 °C). !CAUTION Sigmacote causes skin and serious eye irritation and may cause drowsiness or dizziness. Wear protective gloves, face and eye protection.
Proteomic-based analyses
Water (high-performance liquid chromatography (HPLC)-grade; Fisher Scientific, cat. no. W5SK-1, store at RT) ▲CRITICAL Unless otherwise specified, HPLC-grade water is used for reagent preparation.
1× Phosphate-Buffered Saline (1xPBS, Gibco, cat. no. 10010023, store at RT).
Hyaluronidase from bovine testes (Milllipore Sigma, cat. no. H3506, store at −20 °C).
Chondroitinase ABC from Proteus vulgaris (Milllipore Sigma, cat. no. C3667, store at −20 °C).
Sodium acetate (Milllipore Sigma, cat. no. S8750, store at RT).
Dimethyl sulfoxide (DMSO, sterile, Millipore Sigma, cat. no. D2438, store at RT). !CAUTION Wear protective gloves, protective eyewear and full-face respirator.
Tris-hydroxypropyltriazolylmethylamine (THPTA, Click Chemistry Tools, cat. no. 1010–100, store at 4 °C).
Diazo Biotin Alkyne (Click Chemistry Tools, cat. no. 1042–5, store at −20 °C).
Copper (II) sulfate pentahydrate (Alfa Aesar, cat. no. AA14178A3, store at RT). !CAUTION Copper sulfate is toxic to aquatic life. Dispose in an appropriate waste container. Wear dust mask type N95, protective gloves and face shield.
Sodium ascorbate (Milllipore Sigma, cat. no. A7631, store at RT).
Aminoguanidine hydrochloride (Milllipore Sigma, cat. no. 396494, store at RT).
NeutrAvidin agarose resin (Thermo Fisher Scientific, cat. no. 29200, store at 4 °C).
Sodium dithionite (Na2S2O4, Milllipore Sigma, cat. no. 157953, store at RT). !CAUTION Na2S2O4 can cause serious eye irritation. Wear protective gloves, protective eyewear and full-face respirator.
20% (wt/vol) sodium dodecyl sulfate (SDS, Bio-Rad, cat. no. 1610418, store at RT). !CAUTION SDS causes skin and serious eye irritation. Wear protective gloves, protective eyewear and face protection.
Sodium hydroxide solution (10 M) (NaOH, Milllipore Sigma, cat. no. 72068, store at RT). !CAUTION NaOH is corrosive and causes severe skin and eye damage. Wear protective gloves, face shield, protective eyewear and full-face respirator.
Acetone (HPLC-grade, Milllipore Sigma, cat. no. 650501, store at RT). !CAUTION Acetone is flammable, may cause irritation of respiratory tract and vapors may cause dizziness. Keep away from heat and hot surfaces.
Urea (Proteomics grade, VWR, cat. no. 97061916, store at RT).
Ammonium bicarbonate (Milllipore Sigma, cat. no. 09830, store at RT). !CAUTION Wear dust mask type N95, protective gloves and protective eyewear.
1,4-Dithiothreitol (DTT, Millipore Sigma cat. no. D0632, store under nitrogen at 4 °C). !CAUTION DTT is acutely toxic and harmful if swallowed. Dispose container to an approved waste disposal plant.
Iodoacetamide (IAA, VWR, cat. no. 97064926, store at 4 °C). !CAUTION IAA causes severe skin burns and eye damage and may cause allergy or breathing difficulties if inhaled. Wear protective gloves, protective eyewear and face protection.
Endoproteinase LysC (New England Biolabs, cat. no. P8109S, store at −20 °C).
Trypsin (Mass spectrometry (MS) grade, Thermo Fisher Scientific, cat. no. 90057, store at −20 °C).
Acetonitrile (ACN, HPLC-grade, Fisher Scientific, cat. no. A998–1, store at RT). !CAUTION ACN is a highly flammable liquid and vapor and causes serious eye irritation. Wear protective gloves and protective eyewear and use only in a chemical hood.
Trifluoroacetic acid (TFA, VWR, cat. no. 89399844, store at RT). !CAUTION TFA can cause severe skin burns and eye damage, and is harmful upon inhalation. Wear protective gloves and protective eyewear and use only in a chemical hood.
Formic acid (FA, Fisher Scientific, cat. no. A117–50, store at RT). !CAUTION FA is acute toxic when inhaled, causes severe skin burns and eye damage. Wear protective gloves and protective eyewear and use only in a chemical hood.
Pierce Detergent Removal Spin Columns (Thermo Fisher Scientific, cat. no. 87777, store at 4 °C).
C18 MicroSpin columns (The Nest Group, Inc, cat. no. SS18V, store at RT).
4× Laemmli protein sample buffer (Bio-Rad, cat. no. 1610747, store at RT).
β-Mercaptoethanol (Sigma, cat. no. M6250, store at 4 °C). !CAUTION β-Mercaptoethanol is toxic. Wear respiratory protection, protective gloves and use only in a chemical hood.
Mini-PROTEAN TGX Precast Protein Gels 4–20% (Bio-Rad, cat. no. 4561094, store at 4 °C).
Tans-Blot Turbo Mini 0.2 PVDF Transfer Packs (Bio-Rad, cat. no. 1704156, store at 4 °C).
Pierce Protein-Free (tris-buffered saline) Blocking Buffer (Thermo Fisher Scientific, cat. no. 37570, store at 4 °C).
Tris base (Millipore Sigma, cat. no. 10708976001, store at RT).
Tween 20 (Millipore Sigma, cat. no. P1379, store at RT).
IRDye 680 Streptavidin (LICOR, cat. no. 926–68079, store at 4 °C).
EQUIPMENT
Preparation of NorHA hydrogels
Heated stir plate (e.g., RCT Basic, IKA cat. no. 0003810001).
1-neck round-bottom flasks, assorted sizes, with a magnetic stir bar.
Rubber septa (Sigma Aldrich cat. no. Z553980).
pH meter (e.g. Fisher Scientific™ accumet ™ cat. no. AB150BCERT).
Cannula 20 gauge, (e.g. Chemglass cat. no. A-0519–08).
Glass pipettes with dropper bulbs.
Beaker with a minimum of 3 L volume.
Separatory funnel (e.g. Chemglass cat. no. CG-1742–04).
Filter No. 1 (e.g. Millipore Sigma cat. no. WHA1004110).
Dialysis tubing (6000–8000 MW cut-off regenerated cellulose, e.g. SpectrumLabs Spectra/Por 1 cat. no. 132670) and tubing closures (e.g. SpectrumLabs cat. no. 142734).
Schlenk line with nitrogen line.
Lyophilizer (e.g.Thermo Fisher Scientific Labconco™ 7387020).
Nuclear magnetic resonance spectrometer (NMR, e.g. Bruker DMX360).
Hydrogel preparation, cell encapsulation
Tissue culture hood (e.g. Thermo Fisher Scientific 1300 Series Class II, Type A2 Biological Safety Cabinet).
CO2 cell culture incubator set to 5% CO2 and 37 °C (e.g. Thermo Fisher Scientific Heracell™ VIOS).
Microwave
Epifluorescence microscope (e.g. Olympus BX51).
Confocal microscope (e.g. Leica TCS SP5).
Hemocytometer (e.g. Hausser Scientific cat. no. 0630010).
Water bath set to 37 °C.
Sterile, individually wrapped 0.5 mL syringes 1/2cc with attached needle 27Gx1/2” (e.g. BD cat. no. 305620).
Filter 0.22 μm (e.g. EMD Millipore cat. no. SLGP033RS).
Sterile 48- well plates for 3D cell culture (e.g. Corning Costar Untreated, Fisher Scientific, cat. no. 0720086).
Sterile 4- well rectangular dishes for glass slides (e.g. Rectangular Tray untreated, Thomas Scientific cat. no. 267061).
Coverslips 22 × 22 mm #1.5 (e.g. TedPella, cat. no. 260148).
Coverslips 22 × 50 mm #1.5 (e.g. TedPella, cat. no. 260154).
Coverslip Mini-Rack (e.g. ThermoFisher Scientific, cat. no. C14784).
Scalpel handle (e.g. Fisher Scientific, cat. no. 12000164).
Scalpel blade (e.g. No. 22 Fisher Scientific cat. no. 22079697).
UV-lamp (e.g. EXFO OmniCure Series 2000 with a 320–390 filter).
Visible light-lamp (e.g. EXFO OmniCure Series 2000 with a 400–500 filter).
Proteomic-based analyses
Thermomixer R (e.g. Eppendorf, cat. no. 05400205).
Rotisserie-Rotator for conical and microcentrifuge tubes (e.g. Fisher Scientific cat. no. 22505007).
Sonifier S-450A (Branson Ultrasonics, cat. no. 22–309783). !CAUTION The sonicator should be placed in a sound-proof box or a hearing protector should be worn while sonicating.
CentriVap vacuum concentrator (e.g. Labconoco, cat. no. 7970010).
LC system (Dionex UltiMate 3000 RSLCnano) coupled to a high resolution tandem mass spectrometer (e.g. Thermo Scientific Q Exactive HF Hybrid Quadrupole-Orbitrap or Orbitrap Fusion Lumos Tribrid).
Database search engine such as MaxQuant Andromeda (Max Planck Institute of Biochemistry)50, Mascot (Matrix Science)51 or Sequest (Thermo Fisher Scientific)52.
Trans-Blot Turbo Transfer System (e.g. Bio-Rad, cat. no. 1704150).
Azure c600 imaging system (e.g. Azure Biosystems).
Other laboratory equipment
Sterile 1.5-, 2-, 15- and 50-mL conical (micro)centrifuge tubes
Glass beaker, 200–500 L
Spatula
Forceps
Micropipettes and tips
Disposable Pasteur glass pipettes
Pasteur pipette rubber bulbs
Sterile syringes and needles
Fridge set to 4 °C.
Freezer set to −20 °C.
Deep freezer set −80 °C.
Standard laboratory bench-top refrigerated centrifuge for 50 mL conical tubes (e.g. Thermo Fisher Scientific cat. no. 75004521).
Microcentrifuge (e.g. Eppendorf® Refrigerated Microcentrifuge, model 5417R).
Vortex Mixer (e.g. Fisher Scientific cat. no. 02–215-414) and tube holder (e.g. Fisher Scientific cat. no 11–676-363).
REAGENT SETUP
Solutions for nascent protein labelling
Prepare 50 mM stock solution of AHA by adding 25 mg to 2.768 mL MiliQ water, vortex until fully dissolved. The AHA stock solution can be stored for 6 months at −20 °C. Prepare L-methionine stock solution with 10 mg L-methionine dissolved in 200 μL 1M HCl, vortex until fully dissolved. The L-methionine stock solution can be stored for 1 month at 4 °C. Prepare L-cystine stock solution with 10 mg L-cystine dissolved in 200 μL 1M HCl, vortex until fully. The L-cystine stock solution can be stored for 1 month at 4 °C. Prepare ascorbic acid stock solution with 50 mg ascorbic acid dissolved in 1 mL MilliQ water, vortex until fully dissolved. ▲CRITICAL The ascorbic acid stock solution can be stored for 6 months at −20 °C. Avoid storage at RT for longer than 2 hours as it will reduce the ability to catalyze reactions. ▲CRITICAL Sterile filter water-based stock solutions through a 0.22 μm filter into sterile tubes before storage.
Staining solution for DBCO labelling
Prepare 5 mM stock solution of DBCO-488 by adding 187 μl DMSO per 1 mg DBCO-488 and vortex until fully dissolved. This DBCO-488 stock solution can be stored for 6 months at −20 °C. Prepare 2% BSA solution in PBS with 20 mg BSA dissolved in 2 mL PBS and vortex until fully dissolved (washing buffer). To prepare a 30 μM DBCO-488 staining buffer add 12 μL of DBCO-488 stock buffer to washing buffer. ▲CRITICAL The staining solution should be prepared fresh at the time of the assay.
Polymer stock solutions for hydrogel formation
Prepare 130 mM stock solution of DTT crosslinker by dissolving 20 mg in 1 mL PBS and vortex until fully dissolved. Prepare 53 mM MMP-degradable peptide crosslinker by dissolving 85 mg in 100 μL MiliQ water and vortex until fully dissolved. The DTT and peptide crosslinker solution can be stored for 1 month at −20 °C. ▲CRITICAL Avoid freeze-thawing as it induces formation of spontaneous dithiols and reduces crosslinking efficiency. Prepare 50 mM stock solution of thiolated RGD by dissolving 20 mg in 290 mL and vortex until fully dissolved. Given the desired weight fraction (wt) and total gel volume (VNorHA), determine the total mass of NorHA polymer (mNorHA). Based on the modification of HA with norbornene (%NorHA), the molecular weight of HA disaccharide repeat unit (MWHA) and the conjugated norbornene groups (MWNor) determine the molecular weight of the modified HA dissacharide repeat unit (MWNorHA). Along with the molar concentration of NorHA ([NorHA]) and the effective molar concentration of norbornene functional groups ([Nor]), determine the concentration of dithiol crosslinker ([Crosslinker]) for complete consumption (i.e., XCrosslinker = 1]) or fractional consumption (i.e., XCrosslinker < 1) and the required mass of crosslinker (mCrosslinker) accordingly.
Determine the mass of crosslinker with the following equations:
Prepare 0.5% stock solution of photoinitiator LAP (for UV and visible blue light) or Irgacure 2959 (for UV light) and vortex until fully dissolved. Photoinitiators can be stored for at least 1 month when protected from light at 4 °C. Photoinitiator concentrations of 0.05% have been successfully used in our laboratory. To prepare the polymer precursor solution, dissolve NorHA polymer in PBS at the desired concentration and place on a shaker for 1 hour to ensure complete dissolution. Add the photoinitiator, thiolated RGD and crosslinker, vortex for 10 sec and protect from light. ▲CRITICAL Polymer stock solutions should be prepared fresh the day of each experiment.
Thiolation solution
Add 10.5 mL (3-mercaptopropyl)trimethoxysilane and 3.5 mL hexylamine to 70 mL toluene. ▲CRITICAL Thiolation solution should be prepared fresh the day of each treatment. !CAUTION Use a glass beaker to prevent hexylamine-induced melting of plastic ware.
Human MSCs
Expand cells in α-MEM, 10% FBS, 1% penicillin/streptomycin. This media can be stored for 1 week at 4 °C.
Serum-free DMEM
Count cells in DMEM, 1% penicillin/streptomycin. This media can be stored for 4 weeks at 4 °C.
Solution for Live/Dead staining
Add 0.5 μl calcein AM (2 μM) and 2 μl (4 μM) ethidium homodimer-1 to 1 mL serum-free DMEM and allow it to warm to 37 °C. Protect vial from light. ▲CRITICAL Live/Dead solution should be prepared fresh at the time of the assay. Prepare at least 300 μl per sample, adjusting the volume according to the number of samples to be assayed.
Permeabilization buffer
Add 1.4 g sucrose and 260 μL Triton-X100 to 13 mL PBS, vortex until fully dissolved. The buffer can be stored for at least 1 week at 4 °C.
Hyaluronidase solution
Add 0.5 mg in 1 ml PBS, mix gently until fully dissolved. ▲CRITICAL Hyaluronidase solution should be prepared fresh for each experiment.
Chondroitinase ABC solution
Prepare a 0.1 Unit/μL enzyme solution by reconstituting the lyophilized powder in 0.1 M Tris-HCl, pH 8 containing 0.03 M sodium acetate. This solution can be stored for at least 6 months at − 80°C. ▲CRITICAL Avoid repeated freeze-thaw cycles as it may reduce the activity of the enzyme.
Diazo biotin alkyne solution
Dissolve 5 mg in 628.5 μL DMSO to obtain a stock solution of 10 mM. This solution can be stored in 50–100 μL aliquots for 1 year at −20°C.
THPTA ligand solution Dissolve 43.5 mg in 1 mL HPLC-grade water to obtain a stock solution of 100 mM. This solution can be stored for 3 months at −20°C.
Copper sulfate solution
Dissolve 12.48 mg in 1 mL HPLC-grade water to obtain a stock solution of 50 mM. ▲CRITICAL Copper sulfate solution should be prepared fresh for each use.
Aminoguanidine solution
Dissolve 22.11 mg in 2 mL PBS, pH 7.4. to obtain a stock solution of 100 mM. ▲CRITICAL Aminoguanidine solution should be prepared fresh for each use.
Sodium ascorbate solution
Dissolve 79.2 mg in 1 mL HPLC-grade water to obtain a stock solution of 400 mM. ▲CRITICAL Aminoguanidine solution should be prepared fresh for each use.
IAA solution for click reaction
Dissolve 92.5 mg in 1 mL in HPLC-grade water to obtain a stock solution of 0.5 M. ▲CRITICAL The IAA solution should be prepared fresh for each use and protected from light.
NeutrAvidin wash buffer 1
Prepare NeutrAvidin wash buffer 1 by dissolving 12 g urea (4 M) and 250 μL 20% SDS (0.1% wt/vol) in 50 mL PBS (pH 7.4). ▲CRITICAL NeutrAvidin wash buffer 1 should be prepared fresh for each use.
NeutrAvidin wash buffer 2
Prepare NeutrAvidin wash buffer 2 by adding 250 μL 20% SDS (0.1% wt/vol) to 50 mL PBS (pH 7.4). The NeutrAvidin wash buffer 2 can be stored for 3 – 4 months at RT.
NeutrAvidin elution buffer
Prepare NeutrAvidin elution buffer by dissolving 87 mg Na2S2O4 (50 mM) and 50 μL 20% SDS (0.1% wt/vol) in 10 mL PBS (pH 7.4). Adjust the pH to 7–7.3 by adding 10 M NaOH solution. ▲CRITICAL The NeutrAvidin elution buffer should be prepared immediately before use and protected from light. A pH between 7–7.3 is required for optimal cleavage of the diazo biotin alkyne linker. The presence of SDS in the buffer is critical for efficient elution of labelled proteins.
Ammonium bicarbonate buffer
Dissolve 395 mg in 100 mL HPLC-grade water to obtain a concentration of 50 mM. The ammonium bicarbonate buffer can be stored for up to 1 month at RT.
8 M urea buffer
Dissolve 4.8 g urea in 10 mL ammonium bicarbonate buffer (50 mM). ▲CRITICAL The urea buffer should be prepared fresh for each use
Urea/DTT buffer
Dissolve 1.54 mg DTT in 1 mL urea buffer (8 M) to obtain a concentration of 10 mM. ▲CRITICAL The urea/DTT buffer should be prepared fresh for each use.
IAA solution for MS
Dissolve 92.5 mg IAA in 1 mL 50 mM ammonium bicarbonate buffer to obtain a concentration of 0.5 M. ▲CRITICAL The IAA solution should be prepared fresh for each use and protected from light.
LysC solution
Reconstitute 20 μg of lyophilized Endoproteinase LysC powder in 40 μL HPLC-grade water. The LysC solution can be stored in aliquots for 6 months at −80°C. ▲CRITICAL Avoid repeated freeze-thaw cycles as it may reduce the activity of the enzyme.
Trypsin solution
Reconstitute 20 μg of lyophilized powder in 20 μL 50 mM acetic acid. The trypsin solution can be stored in aliquots for 6 months at −80°C. ▲CRITICAL Avoid repeated freeze-thaw cycles as it may reduce the activity of the enzyme.
C18 column equilibration buffer
Prepare 0.1% (vol/vol) TFA in HPLC-grade water. The equilibration buffer can be stored for 1 month at RT.
C18 column elution buffer
Prepare 80% (vol/vol) ACN and 0.1% (vol/vol) FA in HPLC-grade water. The elution buffer can be stored for 1 month at RT.
MS loading buffer
Prepare 3% (vol/vol) ACN and 0.1% (vol/vol) FA in HPLC-grade water. The loading buffer can be stored for 1 month at RT.
1 × TBST buffer
Dissolve 6.05 g Tris base (50 mM) and 8.76 g NaCl in 800 mL MilliQ water (150 mM). Add 0.5 mL Tween 20 (0.05% (vol/vol)) and adjust the pH to 7.6 with 1 N HCl. Bring the final volume up to 1 L with MilliQ water.
PROCEDURE
Preparation of NorHA, part 1: synthesis of tetrabutylammonium salts of HA (HA-TBA)53 ●TIMING 2h, 3 days lyophilization
-
1
Dissolve 2.0 g HA in 100 mL deionized water in a 250 mL one-neck round-bottom flask while stirring with a magnetic stir bar.
-
2
Add 6.0 g Dowex 50Wx8 to the stirring HA solution for 30 min at RT.
▲CRITICAL the reaction is scaled by adjusting the amount of HA while maintaining a 2.0% w/v solution and a 300% w/w ratio of Dowex/HA.
!CAUTION Hydrogen form Dowex is a strong acid. Avoid any direct contact, always wear appropriate personal protective equipment including gloves, safety goggles, and laboratory coat.
-
3
Collect the HA from the resin via vacuum filtration using a No.1 filter paper.
-
4
Prepare 1:1 (20%) and 1:5 (8%) dilutions of tetrabutylammonium hydroxide (TBA-OH, 40%) against deionized water.
-
5
Titrate HA solution by 40% TBA-OH to pH 5, by 20% TBA-OH to pH 7, and by 8% TBA-OH to a pH of 7.02–7.05.
▲CRITICAL After filtration, set aside 5 mL HA solution in case the solution is over-titrated during addition of TBA-OH. Slowly add TBA-OH as the pH of the solution can change rapidly near neutral pH.
-
6
Transfer HA-solution to 50 mL tubes and freeze at −80 °C overnight.
■ PAUSE POINT Samples can be stored at −80 °C for at least 1 month.
-
7
Lyophilize the sample until the synthesized polymer is fully dry (3–4 days). Cover the tubes with gauze and seal with rubber bands before lyophilization. Ensure that samples do not thaw when transferred to the lyophilizer. Once dry, store polymers under nitrogen at −20 °C to avoid absorption of water.
-
8
Confirm product and degree of modification using 1H NMR and D2O as a solvent.
■ PAUSE POINT Lyophilized HA-TBA samples can be stored under nitrogen for at least one year at −20 °C.
Preparation of NorHA, part 2: Synthesis of NorHA41 ●TIMING 24 h, 11 days dialysis
-
9
Allow HA-TBA to warm up to RT.
▲CRITICAL Keep HA-TBA in a desiccator, as it is hydroscopic and will adsorb water from the atmosphere.
-
10
Equip a dry one-neck round bottom flask with a magnetic stir bar and stopper.
▲CRITICAL It is important to perform this reaction with exclusion of moisture as the presence of water reduces the coupling efficiency of the reaction.
-
11
Add 2 g (2.74 mmol, 1 equiv.) HA-TBA, 0.49 g (3.52 mmol, 1.3 equiv.) 4-(dimethylamino)pyridine (DMAP), 1.12 g (0.99 mL, 8.1 mmol, 3 equiv.) 5-norbornene-2-carboxylic acid to the flask. Purge with nitrogen before adding 100 mL anhydrous DMSO (2% wt/vol) via cannulation. Stir at 350 rpm until fully dissolved.
!CAUTION 5-norbornene-2-carboxylic acid has a strong odor. Avoid any direct contact, always wear appropriate personal protective equipment including gloves, safety goggles, and laboratory coat.
▲CRITICAL Secure the stopper with a copper wire to prevent the stopper from becoming dislodged due to slight pressure when heated during the reaction.
▲CRITICAL It is important to perform this reaction with exclusion of moisture as the presence of water reduces the coupling efficiency of the reaction.
-
12
Melt di-tert-butyl dicarbonate (BOC2O) in a 37 °C water bath and add 0.25 mL (0.36 equiv.) to the reaction with a plastic syringe. Purge the reaction vessel with nitrogen and stir for 20 h at 45 °C in an oil bath.
▲CRITICAL The degree of modification can be controlled by adjusting the amount of BOC2O; modifications of 10–40% have been achieved in our laboratory (quantity used to afford the desired modification: 10%, 0.12 equiv.; 15%, 0.18 equiv.; 30%, 0.36 equiv.; 40%, 0.48 equiv.).
-
13
Quench reaction with 10 mL cold deionized water and transfer to dialysis tubing.
-
14
Dialyze for 5 days at RT and change water twice daily. During the first three days add sodium chloride (NaCl) (1.5 g per liter dialysate) to force dialysis of the TBA salt.
▲CRITICAL STEP Precipitation of excess norbornene is expected and can be removed by either precipitation (option A) or filtration of the product followed by extensive dialysis (option B).
(A) Precipitation
Add 0.95 g NaCl to the reaction solution (0.75 g/100 mL).
Prepare 1.25 L ice-cold acetone (1 L per 100 mL) and stir at 500 rpm.
Add the NorHA dropwise with a separation funnel.
Stop stirring and allow the precipitate to settle (approximately 10 min).
Decant excess acetone.
Partition remaining acetone and precipitated product into 50 mL conical tubes.
Centrifuge for 2 min at 3200 g and decant acetone.
Re-dissolve product in 100 mL deionized water by vortexing, and return the product to dialysis for 5 days. Change water twice daily.
(B) Filtration
Remove the precipitate from the solution by vacuum filtration using a No. 1 filter paper.
Return the filtrate to dialysis for an additional 7 days. Change water twice daily.
-
15
Freeze at −80° and lyophilize (see step 7).
-
16
Confirm product and degree of modification using 1H NMR and D2O as a solvent.
?TROUBLESHOOTING
■ PAUSE POINT Lyophilized NorHA samples can be stored under nitrogen for at least one year at −20 °C.
Hydrogel fabrication: thiolation of coverslips ●TIMING 3 h
-
17
Prepare thiolated coverslips by immersing them in 10M NaOH in a 10 cm2 dish for 20 min at RT, flip after 10 min.
-
18
Place coverslips in racks, wash 2× 2 min with MilliQ water and allow to air-dry in chemical hood.
-
19
Incubate coverslips for 1 h in thiolation solution (see Reagent setup).
!CAUTION Use a glass beaker to prevent hexylamine-induced melting of plastic ware.
-
20
Wash with 70 mL toluene.
-
21
Place coverslips on aluminum foil tray and bake for 1h at 100 °C.
!CAUTION Contact with heated tray may cause serious skin burns. Always wear heat resistant gloves.
■ PAUSE POINT Thiolated coverslips can be stored under nitrogen at −20 °C for 1 week.
Hydrogel fabrication: Photopolymerization of NorHA hydrogels ●TIMING 1 h
-
22
Prepare stock solution of polymer by dissolving NorHA in PBS at the desired concentration (e.g. 4.0% (wt/vol): 4 mg of NorHA in 83.8 μL PBS, see Reagent setup).
-
23
Prepare hydrophobic glass coverslips by immersing them in Sigmacote for 10 seconds.
-
24
Allow the slides to air-dry in a chemical hood.
-
25
Add crosslinker, thiolated RGD and photoinitiator to hydrogel precursor solution (e.g. 1.97 mM DTT (1.5 μL), 2 mM RGD (4 μL) and 10 μL LAP, see Reagent setup) and vortex briefly. Add 100 μL hydrogel solution onto a thiolated glass coverslip and place a hydrophobic coverslip on the top of the spacers (Fig. 6a).
-
26
Expose to UV (3 mW/cm2) or blue light (10 mW/cm2) for 3 min.
-
27
Gently detach the top coverslip with forceps. Allow the hydrogels to swell in PBS overnight at 37 °C.
?TROUBLESHOOTING
■ PAUSE POINT Hydrogels can be stored in PBS for 1 week at 4 °C.
Cell seeding: preparation of NorHA hydrogel components ●TIMING 2 h
▲CRITICAL For cell culture applications, work in a class II biological safety hood and use sterile instruments. Clean all working surfaces with 70% (vol/vol) ethanol before use.
-
28
Sterilize untreated, thiolated (steps 17–21) and hydrophobic coverslips (steps 23–24) by placing them beneath a germicidal ultraviolet lamp for 60 min in a class II biological safety cabinet.
-
29
Sterilize the desired amount of dry NorHA polymer by placing the polymer beneath a germicidal ultraviolet lamp for 60 min in a class II biological safety cabinet. Dissolve in sterile PBS and place the polymer solutions on a shaker for 1 hour to ensure complete dissolution of the polymer. Add photoinitiator and crosslinker at the desired concentration and vortex for 10 sec (see Reagent Setup).
▲CRITICAL 10% of the final volume should be reserved for resuspending the cell pellet, to yield the desired final concentration.
Cell seeding: preparation of cells ●TIMING 1–7 d for cell expansion, 1 h for cell preparation
-
30
Expand human MSCs or any other cell type (e.g. fibroblasts, chondrocytes) in tissue culture dishes (e.g. 150 cm2) using MSC expansion media (see Reagent Setup).
-
31
Rinse cells with 15 mL sterile PBS, add 5 mL sterile trypsin-EDTA and incubate for 5 min in a cell culture incubator at 37 °C.
-
32
Add 15 mL of sterile MSC expansion media and pellet the cells by centrifuging at 300g for 3min at RT.
-
33
Resuspend the cell pellet in 1 mL serum-free DMEM and count using a hemocytometer.
Cell seeding: 2D and 3D cell culture ●TIMING 1–2 h
-
34
Culture cells atop hydrogels for 2D cell culture (option A) or encapsulate within the hydrogels for 3D culture (option B).
(A) 2D cell culture
Prepare hydrogels attached to thiolated coverslips (see steps 22-27, Fig. 6a).
Add AHA media with desired concentrations (see Reagent Setup, Table 1) and incubate for 30 min in a cell culture incubator at 37 °C.
Add cells at desired number (e.g. 1000 cells/cm2) and incubate in a cell culture incubator at 37 °C.
Table 1.
Recipe for preparing AHA media (5 mL per condition, values listed in μL)
| AHA concentration | 25% | 50% | 75% | 100% |
|---|---|---|---|---|
|
| ||||
| DMEM | 4335 | 4340 | 4338 | 4335 |
| AHA | 2.5 | 5.0 | 7.5 | 10 |
| L-Methionine | 1.125 | 0.75 | 0.375 | 0 |
| L-Cystine | 4.83 | 4.83 | 4.83 | 4.83 |
| Glutamax | 50 | 50 | 50 | 50 |
| Ascorbic acid | 5 | 5 | 5 | 5 |
| FBS | 500 | 500 | 500 | 500 |
| Penicillin/Streptomycin | 50 | 50 | 50 | 50 |
| Sodium Pyruvate | 45 | 45 | 45 | 45 |
?TROUBLESHOOTING
▲CRITICAL Gently shake cell culture well plates to evenly distribute the cells on the hydrogel.
(B) 3D cell culture
Resuspend desired number of cells in 10% (e.g. 10 μL PBS) of the final hydrogel volume (e.g., 100 μL) and add to the hydrogel precursor solution (see step 29). Mix gently with a pipette to ensure homogenous cell dispersion.
Pipet cell-hydrogel suspension onto an untreated coverslip. Place three 22 × 22 mm coverslips as spacers on both ends of the slide and place another untreated coverslip on the top of the spacers (Fig. 6b).
Expose to UV (3 mW/cm2) or blue light (10 mW/cm2) for 3 min.
Gently detach the top coverslip with forceps and cut hydrogel film into ca. 5 mm × 5 mm pieces.
Incubate individually in 48-well plates with AHA media in a cell culture incubator at 37 °C (see Reagent Setup, Table 1).
▲CRITICAL Concentration of AHA for 2D and 3D cell culture depend on the cell and culture time (Supplementary Fig. 1).
Cell seeding: assessment of cell viability within 3D hydrogels ●TIMING 2 h
▲CRITICAL To visualize and quantify the viability of encapsulated cells, perform fluorescent staining of live samples protected from light and take images of the hydrogels using confocal laser scanning microscopy (CLSM).
-
35
Remove media from the wells and add 200 μL Live/Dead staining solution per sample in a 24 well-plate (see Reagent Setup).
-
36
Incubate for 30 min in a cell culture incubator at 37 °C. Aspirate the staining solution and replace with 0.5 mL serum-free DMEM.
-
37
Image the samples with a CLSM with a 10/20× objective. Quantify cell viability by counting cells that stain positive for live and dead stains using ImageJ or CellProfiler.
?TROUBLESHOOTING
Nascent matrix labelling
▲CRITICAL Perform DBCO labelling prior to fixation to avoid labelling of intracellular proteins.
-
38
To stain for nascent matrix, prepare washing and DBCO staining buffers (see Reagent Setup).
-
39
Remove media from the wells and add 300 μL washing buffer. Incubate for 2 min at RT.
-
40
Remove washing buffer and add 300 μL DBCO staining buffer (see Reagent Setup).
-
41
Incubate for 35 min in a cell culture incubator at 37 °C.
▲CRITICAL Use small pipette tips (e.g. 200 μL) during washes to prevent damage of hydrogels.
-
42
Wash samples 3× 2 min with the washing buffer.
-
43
Remove the washing buffer and fix them with 300 μL 4% PFA for 30 min at RT.
-
44
Wash 3× 2 min with PBS.
-
45
Add 300 μL of 0.1% CellMask in PBS and incubate for 30 min at RT.
-
46
Wash 3× 2 min with PBS.
?TROUBLESHOOTING
■ PAUSE POINT Hydrogels can be stored for ca. 1 week at 4 °C. Protect samples from light.
Immunofluorescence of specific ECM components
-
47
To analyze immunofluorescence of 2D cell culture samples follow option A, for 3D cell culture follow option B.
(A) Immunofluorescence of cells cultured atop hydrogels (2D)
Add 300 μL of permeabilization buffer (see Reagent Setup) and incubate for 4 h at 4 °C.
-
Wash the samples 3× 10 min with PBS.
▲CRITICAL Skip these permeabilization steps (i-ii) if analysing components of the ECM or surface proteins only.
Add 300 μL of blocking buffer (2% BSA) and incubate for 1 h at RT.
Incubate the sample with primary antibody dissolved at the desired concentration in 300 μL blocking buffer for 24 h at 4 °C.
Wash the samples 3× 10 min with PBS.
-
Incubate the sample with secondary antibody dissolved at the desired concentration (e.g. 1:200) in 300 μL blocking buffer for 2 h at RT.
▲CRITICAL Blocking buffer should be prepared freshly each day.
Wash the samples 3× 10 min with PBS.
Incubate in 2 μM Hoechst in PBS for 30 min at RT.
Was 2× 10 min with PBS.
■ PAUSE POINT Hydrogels can be stored for at least 1 month at 4 °C. Protect samples from light.
(B) Immunofluorescence of embedded cells (3D)
Add 1.5 mL of permeabilization buffer (see Reagent Setup) and incubate for 30 min at 4 °C.
-
Wash the samples 3× 2 min with PBS.
■CRITICAL Skip these permeabilization steps (i-ii) if analysing components of the ECM or surface proteins only.
Incubate in blocking buffer (2% BSA) for 1 h at RT.
Incubate the sample with primary antibody (e.g., anti-collagen type I, anti-collagen type IV) dissolved at the desired concentration in 1.5 mL blocking buffer for 24 h at 4 °C.
Wash the samples 3× 2 min with PBS.
-
Incubate the sample with secondary antibody (e.g., Alexa Fluor-594 IgG H&L, Alexa Fluor-647 IgG H&L) dissolved at the desired concentration (e.g. 1:200) in 1.5 mL blocking buffer for 1 h at RT.
▲CRITICAL Blocking buffer should be prepared freshly each day.
Wash the samples 3× 10 min with PBS.
Incubate in 2 μM Hoechst in PBS for 30 min at RT.
Was 2× 10 min with PBS.
Mount the sample with ProLong Gold Antifade Mountant.
Immediately seal around the coverslip edges using nail polish.
■ PAUSE POINT Hydrogels can be stored for several months at −20 °C. Protect samples from light.
Image quantification of nascent matrix properties
-
48
Acquire confocal images of single cells including the nascent matrix, cell membrane and nucleus.
▲CRITICAL A resolution of ≤ 0.3 μm/pixel is recommended to capture differences in nascent matrix properties.
?TROUBLESHOOTING
-
49
For analysis of nascent matrix local thickness, follow option A, for nascent matrix area follow option B and for nascent matrix volume option C.
(A) Nascent matrix local thickness (Fig. 7)
(B) Nascent matrix volume (Supplementary Fig. 3a)
Transform z-stack into 3D objects and split the channels into single 3D images using ImageJ.
Subtract the ‘cell’ image from the ‘nascent matrix’ image.
Adjust the threshold for each channel with ‘Otsu thresholding’.
Subtract the ‘cell’ image from the ‘nascent matrix’ image.
Use the ImageJ ‘3D object counter’ function to measure the nascent matrix volume.
(C) Nascent matrix area (Supplementary Fig. 3b)
Split the channels into single z-stack images using ImageJ Subtract the ‘cell’ image from the ‘nascent matrix’ image.
Adjust the threshold for each channel with ‘Otsu thresholding’.
Subtract the ‘cell’ image from the ‘nascent matrix’ image and generate a z-stack max projection.
Use the ImageJ ‘Analyze particles’ function to measure the nascent matrix area of the max projected ‘nascent matrix only’ image.
?TROUBLESHOOTING
Analysis of nascent proteins, part 1: sample preparation for AHA enrichment ●TIMING 3 d
-
50
Snap-freeze the cell-laden hydrogels for 2 min in liquid nitrogen (see Step 34B).
▲CRITICAL STEP Measure the hydrogel mass prior to snap-freezing.
■ PAUSE POINT Hydrogel samples can be stored at −80 °C for at least 1 month.
▲CRITICAL The volumes of reagents are based on 50 mg average wet weight of hydrogel. For larger amounts, scale the volume of reagents proportionally.
-
51
Thaw AHA-labelled and unlabelled (control) hydrogel samples. Add 500 μL of 0.5 mg/mL hyaluronidase solution to each sample (see Reagent Setup). Incubate overnight at 37 °C in a thermal mixer with constant agitation at 900rpm. Shorter incubation times may be used if complete degradation of the hydrogel is observed.
-
52
Add 2 μL of chondroitinase ABC (see Reagent Setup). Incubate overnight at 900rpm at 37 °C in a thermal mixer.
-
53
Allow the samples to cool down to RT. Add 100% ice-cold acetone to the samples at a 4:1 ratio (vol/vol), vortex briefly and incubate for at least 6 h at −20 °C.
▲CRITICAL Samples can be incubated for up to 24 h at −20 °C.
-
54
Centrifuge at 21,000g for 20 min at 4 °C. Discard the supernatant. Vacuum-dry the pellets for 15 min at RT using CentriVap. Alternatively, air-dry the pellets for 30 – 45 min at RT.
▲CRITICAL Do not over dry the pellet to avoid difficulties with resuspension.
■ PAUSE POINT Protein pellets can be stored for up to 2 months at −80 °C.
-
55
Resuspend the pellets in 500 μL of 8 M urea (see Reagent Setup). Sonicate for 10 s on ice with a 50% duty cycle and power output 3, followed by 1 min incubation on ice. Repeat the sonication step two times.
▲CRITICAL STEP Keep the samples on ice during sonication to avoid excessive heating.
-
56
Centrifuge the samples at 16,000g for 20 min at RT. Transfer the supernatant into new tubes.
-
57
Measure the protein concentration using Pierce Coomassie (Bradford) Protein Assay Kit (follow the instructions provided by the kit).
-
58
Adjust all samples to the same protein concentration using 8 M urea (see Reagent Setup).
Analysis of nascent proteins, part 2: click chemistry reaction ●TIMING 1 d
■CRITICAL For optimum results, we recommend adding 0.6–0.8 mg protein in a total click reaction volume of 800 μL.
-
59
Take the appropriate sample volume (up to 464 μL) to obtain the recommended protein amount. Add 8 M Urea (see Reagent Setup) to obtain a final volume of 464 μL if a smaller volume is taken.
-
60
Add 40 μL of 0.5 M IAA (final 39.7 mM, see Reagent Setup). Rotate end-over-end for 30 min at RT protected from light.
-
61
Add the click reagents in the following order, vortex the tube for 10 s after adding each reagent: 4 μL of 10 mM diazo biotin alkyne (final 50 μM), 80 μL of 100 mM THPTA (final 10 mM), 32 μL of 50 mM copper sulfate (final 2 mM), 160 μL of 100 mM aminoguanidine (final 20 mM) and 20 μL of 400 mM sodium ascorbate (final 10 mM) (See Reagent Setup).
-
62
Incubate for 2 h at RT with end-over-end rotation.
?TROUBLESHOOTING
-
63
Add 100% ice-cold acetone to the samples at a 4:1 ratio (vol/vol), vortex briefly and incubate overnight at −20°C.
-
64
Centrifuge at 21,000g for 20 min at 4°C. Discard the supernatant. Vacuum-dry the pellets for 10 – 15 min at RT using CentriVap. Alternatively, air-dry the pellets for 30 – 45 min at RT.
▲CRITICAL Do not over dry the pellet or it may be hard to resuspend.
■ PAUSE POINT Protein pellets can be stored for up to 2 months at −80 °C.
Analysis of nascent proteins, part 3: NeutrAvidin enrichment ●TIMING 3.5 h
-
65
Transfer a suitable volume of NeutrAvidin resin into a microcentrifuge tube. We typically use 100 μL settled resin for each sample of 0.6–0.8 mg starting protein amount. The resin is provided as a 50% slurry, i.e., 200 μL slurry is equivalent to ~100 μL of settled resin.
▲CRITICAL STEP Keep the starting protein amount and resin volume the same for biological replicates.
-
66
Wash the resin with 1 mL PBS (pH 7.4). Spin down for 2 min at 1,200g at RT. Discard the supernatant. Repeat this washing step two times.
-
67
Resuspend the pellets from Step 64 in 530 μL of 4.5 M urea in PBS. Centrifuge at 18,000g for 20 min at RT to remove any insoluble particles. Transfer the supernatant into a new tube.
▲CRITICAL Save 30 μL for western blot analysis (Steps 71–76, click reaction, Supplementary Fig. 4).
-
68
Transfer the supernatant to the washed resin. Incubate for 1.5 h at RT with end-over-end rotation.
-
69
Spin down at 1,200g for 2 min.
▲CRITICAL Save 30 μL of the supernatant for western blot analysis (Steps 71–76, unbound fraction, Supplementary Fig. 4).
-
70
Wash the resin with 1 mL of NeutrAvidin wash buffer 1 for 10 min at RT with end-over-end rotation (see Reagent Setup). Spin down at 1,200g for 2 min at RT and discard the supernatant. Repeat this washing step three more times.
-
71
Confirm the enrichment via Western Blotting. Mix each of the 30 μL samples collected in Steps 67 and 69 with 10 μL 4× Laemmli sample buffer containing 5% (vol/vol) β-Mercaptoethanol. Boil the samples at 95°C for 5 min in a thermal mixer.
-
72
Assemble the blotting sandwich with the Trans-Blot Turbo Transfer Packs and transfer the proteins onto the PVDF membrane using the Trans-Blot Turbo Transfer System. Note that a standard wet transfer system can also be used.
-
73
Incubate the membrane in Pierce Protein-Free (TBS) Blocking Buffer for 1 h at RT.
-
74
Incubate the membrane with IRDye 680 Streptavidin diluted 1:3000 (vol/vol) in 1:1 (vol/vol) TBST buffer and Protein-Free TBS Blocking Buffer for 2 h at RT or overnight at 4 °C.
-
75
Wash the membrane three times with TBST buffer for 5 min (see Reagent Setup).
-
76
Image the membrane with infrared imaging system (e.g. Azure c600 imaging system).
Analysis of nascent proteins, part 4: protein elution ●TIMING 1 d
-
77
Add 400 μL of NeutrAvidin elution buffer (see Reagent Setup). Incubate for 45 min at RT protected from light with end-over-end rotation.
▲CRITICAL STEP The elution buffer should be protected from light and prepared immediately before use. For efficient cleavage of the diazo biotin alkyne linker, the buffer pH should range between 7–7.3 (see Reagent Setup).
?TROUBLESHOOTING
-
78
Spin down at 1,200g for 2 min at RT and transfer the supernatant into a new tube. The supernatant represents “Elution 1”. Repeat Steps 77 and 78 two times and combine the three elutions.
-
79
Add 100% ice-cold acetone to the eluted proteins at a 4:1 ratio (vol/vol), vortex briefly and incubate overnight at −20 °C.
-
80
Centrifuge at 21,000g for 20 min at 4 °C. Discard the supernatant. Vacuum-dry the pellets for 10 – 15 min at RT using CentriVap. Alternatively, air-dry the pellets for 30 – 45 min at RT.
▲CRITICAL Do not over dry the pellet or it may be hard to resuspend.
■ PAUSE POINT Protein pellets can be stored for up to 2 months at −80 °C.
Analysis of nascent proteins, part 5: sample preparation for mass spectrometry ●TIMING 2 d
▲CRITICAL The volumes of reagents are based on 0.6–0.8 mg starting protein amount. For larger amounts, scale the volume of reagents proportionally.
-
81
Resuspend the pellets in 50 μL of Urea/DTT buffer (see Reagent Setup). Incubate for 1 h at 37 °C in a thermal mixer with constant agitation at 1000rpm.
-
82
Cool down the samples to RT. Add 2.5 μL of 0.5 M IAA (final is 25 mM, see Reagent Setup). Incubate for 30 min at RT and protected from light.
-
83
Add 2 μL LysC (see Reagent Setup) and incubate for 3 h at 37 °C and 1000rpm in a thermal mixer.
-
84
Dilute the samples to 2 M Urea by adding 150 μL of 50 mM ammonium bicarbonate buffer (see Reagent Setup). Add 3 μL trypsin (see Reagent Setup) and incubate overnight at 37 °C and 1000rpm in a thermal mixer.
▲CRITICAL STEP Urea concentration should be ≤ 2 M before adding trypsin to avoid enzyme denaturation.
-
85
Acidify the samples to deactivate trypsin by adding TFA to a final concentration of 1% (vol/vol).
-
86
Centrifuge the samples at 16,000g for 20 min at RT. Transfer supernatants into new tubes.
■ PAUSE POINT Peptide samples can be stored for 3 – 4 months at −80°C.
-
87
Remove the residual SDS from the peptide samples using Pierce Detergent Removal Spin Columns (follow the instructions provided by the manufacturer).
-
88
Place the C18 column in a 2 mL microcentrifuge tube and add 200 μL 100% ACN. Centrifuge at 110g for 1 min at RT.
-
89
Add 100 μL of the equilibration buffer to the column (see Reagent Setup). Centrifuge at 110g for 1 min at RT. Repeat this step.
-
90
Add the peptide sample into the column. Centrifuge at 110g for 1 min at RT.
-
91
Wash the column with 100 μL of the equilibration buffer (See Reagent Setup). Centrifuge at 110g for 1 min at RT. Repeat this step.
-
92
Place the column in a new 1.5 mL microcentrifuge tube. Elute peptides by adding 50 μL of the elution buffer (see Reagent Setup). Centrifuge at 110g for 1 min. Repeat this step two times.
-
93
Vacuum-dry the eluted peptides for at least 3 h at 45 °C in a CentriVap.
■ PAUSE POINT Peptide pellets can be stored for 3 – 4 months at −80 °C.
-
94
Resuspend the peptide pellets in 10 μL of the MS loading buffer (see Reagent Setup).
Analysis of nascent proteins, part 6: LC-MS/MS analysis ●TIMING 3 h per sample
-
95
The method of LC-MS/MS analysis depends on the liquid chromatography and mass spectrometer system in use. A high-resolution tandem mass spectrometer such as Thermo Scientific Q Exactive HF Hybrid Quadrupole-Orbitrap or Orbitrap Fusion Lumos Tribrid should be used. We perform our analysis in the positive ion mode using 2 h LC gradient. MS data are acquired in a data-dependent mode, monitoring the Top 20 precursor ions.
Analysis of nascent proteins, part 7: MS Data analysis ●TIMING ≥ 4 d, according to the number of samples and the computing power
-
96
Search the raw MS data against a protein sequence database using MaxQuant or other search engines such as Mascot or Sequest (can be customized to the sample). Include cysteine carbamidomethylation (+57.021) as a fixed modification, and AHA substitution for methionine (−4.986) and cleaved diazo biotin alkyne-tagged AHA substitution for methionine (+257.14) as variable modifications. In addition, consider including other variable modifications such as methionine oxidation (+15.995), N-terminal acetylation (+42.011), asparagine deamidation (+0.984)54, the conversion of glutamine to pyro-glutamic acid (−17.027), or ECM-rich hydroxylysine (+15.995) and, hydroxyproline (+15.995) and 55,56. Note that the number of modifications greatly influences the search time and can lead to false discoveries.
-
97
Exclude proteins that are identified by less than two unique and razor peptide, contaminants, reverse hits and proteins identified only by a modification site.
-
98
Classify the newly synthesized proteins according to their cellular compartment and filter to ECM proteins. We developed a set of classification to classify proteins as cytosolic, nuclear, membrane, cytoskeletal or ECM (matrisome-related proteins)24 based on the Gene Ontology (GO) Consortium57 and the Matrisome Project55.
-
99
Log2-transform protein intensities and calculate the fold change of proteins identified in AHA-labelled and control samples by subtracting the intensity of each protein in the AHA-labelled samples from the intensity in the corresponding control samples.
-
100
We define the “newly synthesized proteins” as those proteins exclusive to AHA-labelled samples or have a fold change > 2 and p ≤ 0.05 in AHA-labelled compared to control samples, in at least two of the biological replicates.
?TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
|
| |||
| 16 | Low degree of norbornene modification | Residual water quenches the anhydrous reaction (steps 11,12). | Ensure that the glassware is completely dry and purged with nitrogen before starting the reaction |
| Preweigh HA-TBA before storage under nitrogen (Step 7) to avoid long exposure to the atmosphere before starting NorHA synthesis (Step 9) | |||
| Sufficiently purge by dry nitrogen before the reaction | |||
| 27 | Gels are very soft or form only with high crosslinker concentration | Low content of reactive thiols within the crosslinker results in insufficient crosslink formation | Use freshly prepared crosslinker solutions (see Reagent Setup) Freeze-thaw aliquots only once to avoid spontaneous dithiol formation before hydrogel formation |
| Low degree of norbornene modification | Synthesize NorHA with higher norbornene modifications by increasing Boc2O concentrations (Step 16) | ||
| 34 A | Hydrogels detach during culture | Thiolation of coverslips is not homogeneous | Use freshly prepared thiolation solution (see Reagent Setup) |
| Sufficiently purge the container with coverslips by dry nitrogen before storage (Step 21) | |||
| 37 | Low cell viability | Absence or low activity of growth factors and additives | Use freshly prepared cell culture media and components (Step 34) |
| Cells do not tolerate AHA concentrations (see Supplementary Fig. 1 as an example) | Reduce the concentration of AHA (Step 34, Table 1) | ||
| 46 | Intracellular staining | Cells do not tolerate AHA concentrations | Reduce the concentration of AHA (Step 34, Table 1) |
| 48 | High background staining | Cells do not tolerate DBCO labelling Unspecific binding of DBCO to hydrogel | Reduce the concentration of DBCO (See Reagent Setup) Shake sample during all washing steps to allow excess DBCO to be removed washing (Steps 42, 47) |
| Low fluorescence intensities | Fluorophores are photobleached | Lower the laser power during image acquisition. Adjust microscopy settings to avoid photobleaching58 | |
| 49 | Spotted threshold images | Cell and nascent matrix boundaries are not homogenous | Image with high resolution and adequate exposure times to ensure high quality images |
| 62 | Inefficient click reaction | Low level of AHA is incorporated or concentration of AHA-labelled proteins is low | Increase the concentration and/or time of AHA supplementation |
| The quality of reagents is poor | Use freshly prepared copper sulfate, sodium ascorbate and aminoguanidine (see Reagent Setup) | ||
| 77 | Low levels of eluted proteins | The cleavage reaction of the diazo biotin is incomplete | Use freshly prepared elution buffer and ensure a pH of 7 – 7.3 (see Reagent Setup) |
| Perform elution steps protected from light (Step 77) | |||
| The SDS concentration in the elution buffer is low or absent | Ensure that the elution buffer contains at least 0.1% SDS to ensure efficient elution of labelled proteins (see Reagent Setup) | ||
| 100 | Small number of proteins are exclusively identified or > 2 fold-enriched in AHA-labelled compared to control samples | Low level of AHA is incorporated or concentration of AHA-labelled proteins is low Nonspecific labelling during click reaction | Increase the concentration and/or time of AHA supplementation (Step 34) Ensure that samples are alkylated with IAA before the click reaction (Step 60, see Reagent Setup) |
| Ensure that aminoguanidine is added to the click reaction (Step 61) | |||
| Use western blot analysis to optimize the concentration of the alkyne linker to determine the best signal-to-noise ratio (Supplementary Figure 4, Steps 71–76) | |||
| Measure peptide concentrations to optimize MS sample preparation (e.g., Pierce™ Quantitative Fluorometric Peptide Assay, Supplementary Figure 5, Steps 81–94) | |||
| The number of eluted proteins is low | Use freshly prepared elution buffer and ensure a pH of 7 – 7.3 (see Reagent Setup) | ||
| Perform elution steps protected from light (Step 77) Ensure that the elution buffer contains at least 0.1% SDS to ensure efficient elution of labelled proteins (see Reagent Setup) | |||
| Measure protein concentrations to optimize protein cleavage conditions (e.g., Pierce™ Coomassie (Bradford) Protein Assay, Supplementary Figure 5, Steps 65–80) | |||
| The incubation with NeutrAvidin resin is too long | Ensure that the incubation time is no longer that 1.5 h (Steps 68) | ||
| Nonspecific binding to NeutrAvidin resin | Decrease the resin volume (Supplementary Figure 4, Step 65) | ||
| Increase the number of washes (Steps 66, 70) | |||
| Increase the urea concentration in wash buffer 1 to 5 M and/or SDS to 1% (see Reagent Setup) | |||
| Protein identification is too low | Match between runs feature to increase identifications (Step 97) | ||
●TIMING
NorHA synthesis, 12 days
Hydrogel fabrication 4 hours
Cell seeding, material preparation, 2 hours; seeding, 0.5 hours
Cell encapsulation, material preparation, 2 hours; encapsulation, 1 hours; Live-Dead assay, 2 hours
Nascent matrix labelling 3 hours
Immunofluorescence, 2 days
Image quantification, 1–3 hours
AHA-labelled protein enrichment and mass spectrometry sample preparation, 3 days; click chemistry reaction, 2.5 hours; protein precipitation, 18 hours; NeutrAvidin enrichment: 3.5 hours; protein elution, 2.5 hours; protein precipitation, 18 hours; sample preparation for mass spectrometry, 2 days; LC-MS/MS analysis, 3 hours per sample; MS data analysis, 4 days
ANTICIPATED RESULTS
Analytical data
Tetrabutylammonium salt of HA.
The recovered product is a spongy white solid. 1H NMR (D2O) HA-TBA δ =0.9–1.04 (t, 3H), 1.33–1.47 (m, 2H), 1.62–1.76 (m, 2H), 2.1 (s, 3H), 3.13–3.3 (m, 2H), 3.30–4.20 (10 H).
Norbornene HA.
The recovered product is a spongy white solid with an approximate yield of 23%. 1H NMR (D2O) based on integration of the vinyl protons (δ = 5.8−6.4, 2H) relative to the HA backbone (δ = 3.20−4.20, 10H). For troubleshooting, see entries for Step 16 in Table 2.
Nascent matrix visualization and quantification.
The nascent matrix thickness depends on the culture technique (i.e. 2D versus 3D culture) and the time. In 3D culture a thickness of 0.5–2 μm is anticipated (Fig. 7).
Analysis of nascent proteins.
The content of proteins depends on the cell type, culture conditions (e.g., differentiation media) and the time. For chondrocytes embedded in 3D NorHA hydrogels and cultured in chondrogenic media (DMEM supplemented with insulin-transferrin-sodium selenite, transforming growth factor beta 3 (TGFβ3)), About 35% of the total protein intensity is anticipated to be comprised of ECM proteins involved in various processes of ECM organization, adhesion and development. At least 3 biological replicates are required for identification and analysis of newly synthesized proteins (Supplementary table 1).
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful for financial support from the National Institutes of Health (R01 AR077362, R01 AR056624, R01 AR071359, K99 HL151670), as well as the Center for Engineering MechanoBiology through the National Science Foundation’s STC Program (CMMI: 15–48571). The authors thank Claire M. McLeod, Edward D. Bonnevie, and Jonathan H. Galarraga for helpful discussions.
Footnotes
Reporting Summary: Further information on research design is available in the Nature Research Reporting Summary linked to this article.
DATA AVAILABILITY
All the data generated or analyzed during this study are included within this article or references cited and its Supplementary Information. Proteomics data are available in the MassIVE repository (accession number: MSV000087857, password: chondrocyte). Additional information is available from the corresponding author on request.
References
- 1.Gjorevski N & Nelson CM Bidirectional extracellular matrix signaling during tissue morphogenesis. Cytokine Growth Factor Rev 20, 459–465, doi: 10.1016/j.cytogfr.2009.10.013 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrera J, Henke CA & Bitterman PB Extracellular matrix as a driver of progressive fibrosis. J Clin Invest 128, 45–53, doi: 10.1172/JCI93557 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frantz C, Stewart KM & Weaver VM The extracellular matrix at a glance. Journal of Cell Science 123, 4195–4200, doi: 10.1242/jcs.023820 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffith LG & Swartz MA Capturing complex 3D tissue physiology in vitro. Nature Reviews Molecular Cell Biology 7, 211–224, doi: 10.1038/nrm1858 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Caliari SR & Burdick JA A practical guide to hydrogels for cell culture. Nat Methods 13, 405–414, doi: 10.1038/nmeth.3839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prince E & Kumacheva E Design and applications of man-made biomimetic fibrillar hydrogels. Nature Reviews Materials 4, 99–115, doi: 10.1038/s41578-018-0077-9 (2019). [DOI] [Google Scholar]
- 7.Ma PX Biomimetic materials for tissue engineering. Advanced Drug Delivery Reviews 60, 184–198, doi: 10.1016/j.addr.2007.08.041 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saleh AM, Wilding KM, Calve S, Bundy BC & Kinzer-Ursem TL Non-canonical amino acid labeling in proteomics and biotechnology. Journal of Biological Engineering 13, 43, doi: 10.1186/s13036-019-0166-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dieterich DC et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nature Neuroscience 13, 897–905, doi: 10.1038/nn.2580 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dieterich DC, Link AJ, Graumann J, Tirrell DA & Schuman EM Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proceedings of the National Academy of Sciences 103, 9482–9487, doi: 10.1073/pnas.0601637103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nessen MA et al. Selective Enrichment of Azide-Containing Peptides from Complex Mixtures. Journal of Proteome Research 8, 3702–3711, doi: 10.1021/pr900257z (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Hest JCM, Kiick KL & Tirrell DA Efficient Incorporation of Unsaturated Methionine Analogues into Proteins in Vivo. Journal of the American Chemical Society 122, 1282–1288, doi: 10.1021/ja992749j (2000). [DOI] [Google Scholar]
- 13.Beatty KE et al. Fluorescence Visualization of Newly Synthesized Proteins in Mammalian Cells. Angewandte Chemie International Edition 45, 7364–7367, doi: 10.1002/anie.200602114 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Wang J, Ng S, Lin Q & Shen H-M Development of a novel method for quantification of autophagic protein degradation by AHA labeling. Autophagy 10, 901–912, doi: 10.4161/auto.28267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang MM, Tsou LK, Charron G, Raghavan AS & Hang HC Tandem fluorescence imaging of dynamic S-acylation and protein turnover. Proceedings of the National Academy of Sciences 107, 8627–8632, doi: 10.1073/pnas.0912306107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adelmund SM, Ruskowitz ER, Farahani PE, Wolfe JV & DeForest CA Light-Activated Proteomic Labeling via Photocaged Bioorthogonal Non-Canonical Amino Acids. ACS Chemical Biology 13, 573–577, doi: 10.1021/acschembio.7b01023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvarez-Castelao B et al. Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nature Biotechnology 35, 1196–1201, doi: 10.1038/nbt.4016 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Alvarez-Castelao B, Schanzenbächer CT, Langer JD & Schuman EM Cell-type-specific metabolic labeling, detection and identification of nascent proteomes in vivo. Nature Protocols 14, 556–575, doi: 10.1038/s41596-018-0106-6 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Liu Y et al. Application of bio-orthogonal proteome labeling to cell transplantation and heterochronic parabiosis. Nature Communications 8, 643, doi: 10.1038/s41467-017-00698-y (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadlowski C et al. Graphene-based biosensor for on-chip detection of bio-orthogonally labeled proteins to identify the circulating biomarkers of aging during heterochronic parabiosis. Lab on a Chip 18, 3230–3238, doi: 10.1039/C8LC00446C (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonnevie ED et al. Aberrant mechanosensing in injured intervertebral discs as a result of boundary-constraint disruption and residual-strain loss. Nat Biomed Eng 3, 998–1008, doi: 10.1038/s41551-019-0458-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loebel C, Mauck RL & Burdick JA Local nascent protein deposition and remodelling guide mesenchymal stromal cell mechanosensing and fate in three-dimensional hydrogels. Nat Mater 18, 883–891, doi: 10.1038/s41563-019-0307-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLeod CM & Mauck RL High fidelity visualization of cell-to-cell variation and temporal dynamics in nascent extracellular matrix formation. Sci Rep 6, 38852, doi: 10.1038/srep38852 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saleh AM, Jacobson KR, Kinzer-Ursem TL & Calve S Dynamics of Non-Canonical Amino Acid-Labeled Intra- and Extracellular Proteins in the Developing Mouse. Cell Mol Bioeng 12, 495–509, doi: 10.1007/s12195-019-00592-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calve S, Witten AJ, Ocken AR & Kinzer-Ursem TL Incorporation of non-canonical amino acids into the developing murine proteome. Sci Rep 6, 32377, doi: 10.1038/srep32377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loebel C et al. Metabolic Labeling to Probe the Spatiotemporal Accumulation of Matrix at the Chondrocyte–Hydrogel Interface. Advanced Functional Materials n/a, 1909802, doi: 10.1002/adfm.201909802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan AR & Hung CT Concise Review: Mesenchymal Stem Cells for Functional Cartilage Tissue Engineering: Taking Cues from Chondrocyte-Based Constructs. STEM CELLS Translational Medicine 6, 1295–1303, doi: 10.1002/sctm.16-0271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen FH, Rousche KT & Tuan RS Technology Insight: adult stem cells in cartilage regeneration and tissue engineering. Nature Clinical Practice Rheumatology 2, 373–382, doi: 10.1038/ncprheum0216 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Vega SL, Kwon MY & Burdick JA Recent advances in hydrogels for cartilage tissue engineering. Eur Cell Mater 33, 59–75, doi: 10.22203/eCM.v033a05 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eslahi N, Abdorahim M & Simchi A Smart Polymeric Hydrogels for Cartilage Tissue Engineering: A Review on the Chemistry and Biological Functions. Biomacromolecules 17, 3441–3463, doi: 10.1021/acs.biomac.6b01235 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Huebsch N Translational mechanobiology: Designing synthetic hydrogel matrices for improved in vitro models and cell-based therapies. Acta Biomaterialia 94, 97–111, doi: 10.1016/j.actbio.2019.05.055 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C & Brown RA Myofibroblasts and mechano-regulation of connective tissue remodelling. Nature Reviews Molecular Cell Biology 3, 349–363, doi: 10.1038/nrm809 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Caliari SR, Vega SL, Kwon M, Soulas EM & Burdick JA Dimensionality and spreading influence MSC YAP/TAZ signaling in hydrogel environments. Biomaterials 103, 314–323, doi: 10.1016/j.biomaterials.2016.06.061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang B et al. Enhanced mechanosensing of cells in synthetic 3D matrix with controlled biophysical dynamics. Nat Commun 12, 3514, doi: 10.1038/s41467-021-23120-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuet KP et al. Cell-specific proteomic analysis in Caenorhabditis elegans. Proc Natl Acad Sci U S A 112, 2705–2710, doi: 10.1073/pnas.1421567112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Datta D, Wang P, Carrico IS, Mayo SL & Tirrell DA A designed phenylalanyl-tRNA synthetase variant allows efficient in vivo incorporation of aryl ketone functionality into proteins. J Am Chem Soc 124, 5652–5653, doi: 10.1021/ja0177096 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Dunham C, Havlioglu N, Chamberlain A, Lake S & Meyer G Adipose stem cells exhibit mechanical memory and reduce fibrotic contracture in a rat elbow injury model. The FASEB Journal 34, 12976–12990, doi: 10.1096/fj.202001274R (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jowett GM et al. ILC1 drive intestinal epithelial and matrix remodelling. Nature Materials 20, 250–259, doi: 10.1038/s41563-020-0783-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norman MDA, Ferreira SA, Jowett GM, Bozec L & Gentleman E Measuring the elastic modulus of soft culture surfaces and three-dimensional hydrogels using atomic force microscopy. Nature Protocols 16, 2418–2449, doi: 10.1038/s41596-021-00495-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naba A et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 11, M111.014647, doi: 10.1074/mcp.M111.014647 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gramlich WM, Kim IL & Burdick JA Synthesis and orthogonal photopatterning of hyaluronic acid hydrogels with thiol-norbornene chemistry. Biomaterials 34, 9803–9811, doi: 10.1016/j.biomaterials.2013.08.089 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shih H & Lin C-C Cross-Linking and Degradation of Step-Growth Hydrogels Formed by Thiol–Ene Photoclick Chemistry. Biomacromolecules 13, 2003–2012, doi: 10.1021/bm300752j (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aimetti AA, Machen AJ & Anseth KS Poly(ethylene glycol) hydrogels formed by thiol-ene photopolymerization for enzyme-responsive protein delivery. Biomaterials 30, 6048–6054, doi: 10.1016/j.biomaterials.2009.07.043 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mũnoz Z, Shih H & Lin C-C Gelatin hydrogels formed by orthogonal thiol–norbornene photochemistry for cell encapsulation. Biomaterials Science 2, 1063–1072, doi: 10.1039/C4BM00070F (2014). [DOI] [PubMed] [Google Scholar]
- 45.Loebel C et al. Cross-Linking Chemistry of Tyramine-Modified Hyaluronan Hydrogels Alters Mesenchymal Stem Cell Early Attachment and Behavior. Biomacromolecules 18, 855–864, doi: 10.1021/acs.biomac.6b01740 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Vega SL et al. Combinatorial hydrogels with biochemical gradients for screening 3D cellular microenvironments. Nat Commun 9, 614, doi: 10.1038/s41467-018-03021-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cosgrove BD et al. N-cadherin adhesive interactions modulate matrix mechanosensing and fate commitment of mesenchymal stem cells. Nat Mater 15, 1297–1306, doi: 10.1038/nmat4725 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doube M et al. BoneJ: Free and extensible bone image analysis in ImageJ. Bone 47, 1076–1079, doi: 10.1016/j.bone.2010.08.023 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mauck RL, Yuan X & Tuan RS Chondrogenic differentiation and functional maturation of bovine mesenchymal stem cells in long-term agarose culture. Osteoarthritis Cartilage 14, 179–189, doi: 10.1016/j.joca.2005.09.002 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Cox J & Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372, doi: 10.1038/nbt.1511 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Perkins DN, Pappin DJ, Creasy DM & Cottrell JS Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567, doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2 (1999). [DOI] [PubMed] [Google Scholar]
- 52.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989, doi: 10.1016/1044-0305(94)80016-2 (1994). [DOI] [PubMed] [Google Scholar]
- 53.Loebel C, Rodell CB, Chen MH & Burdick JA Shear-thinning and self-healing hydrogels as injectable therapeutics and for 3D-printing. Nat Protoc 12, 1521–1541, doi: 10.1038/nprot.2017.053 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Catterall JB et al. Protein modification by deamidation indicates variations in joint extracellular matrix turnover. J Biol Chem 287, 4640–4651, doi: 10.1074/jbc.M111.249649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naba A et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 11, M111 014647, doi: 10.1074/mcp.M111.014647 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naba A et al. Characterization of the Extracellular Matrix of Normal and Diseased Tissues Using Proteomics. J Proteome Res 16, 3083–3091, doi: 10.1021/acs.jproteome.7b00191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29, doi: 10.1038/75556 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jonkman J, Brown CM, Wright GD, Anderson KI & North AJ Tutorial: guidance for quantitative confocal microscopy. Nature Protocols, doi: 10.1038/s41596-020-0313-9 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data generated or analyzed during this study are included within this article or references cited and its Supplementary Information. Proteomics data are available in the MassIVE repository (accession number: MSV000087857, password: chondrocyte). Additional information is available from the corresponding author on request.