

Abstract
Introduction
Alzheimer's disease (AD) is characterized by neurotoxic immuno‐inflammation concomitant with cytotoxic oligomerization of amyloid beta (Aβ) and tau, culminating in concurrent, interdependent immunopathic and proteopathic pathogeneses.
Methods
We performed a comprehensive series of in silico, in vitro, and in vivo studies explicitly evaluating the atomistic–molecular mechanisms of cytokine‐mediated and Aβ‐mediated neurotoxicities in AD. Next, 471 new chemical entities were designed and synthesized to probe the pathways identified by these molecular mechanism studies and to provide prototypic starting points in the development of small‐molecule therapeutics for AD.
Results
In response to various stimuli (e.g., infection, trauma, ischemia, air pollution, depression), Aβ is released as an early responder immunopeptide triggering an innate immunity cascade in which Aβ exhibits both immunomodulatory and antimicrobial properties (whether bacteria are present, or not), resulting in a misdirected attack upon “self” neurons, arising from analogous electronegative surface topologies between neurons and bacteria, and rendering them similarly susceptible to membrane‐penetrating attack by antimicrobial peptides (AMPs) such as Aβ. After this self‐attack, the resulting necrotic (but not apoptotic) neuronal breakdown products diffuse to adjacent neurons eliciting further release of Aβ, leading to a chronic self‐perpetuating autoimmune cycle. AD thus emerges as a brain‐centric autoimmune disorder of innate immunity. Based upon the hypothesis that autoimmune processes are susceptible to endogenous regulatory processes, a subsequent comprehensive screening program of 1137 small molecules normally present in human brain identified tryptophan metabolism as a regulator of brain innate immunity and a source of potential endogenous anti‐AD molecules capable of chemical modification into multi‐site therapeutic modulators targeting AD's complex immunopathic–proteopathic pathogenesis.
Discussion
Conceptualizing AD as an autoimmune disease, identifying endogenous regulators of this autoimmunity, and designing small molecule drug‐like analogues of these endogenous regulators represents a novel therapeutic approach for AD.
Keywords: Alzheimer's disease, amyloid beta, antimicrobial peptide, arginine, autoimmune, cytokine, tryptophan
1. INTRODUCTION
As new potentially explanatory biochemical mechanisms for Alzheimer's disease (AD) emerge, they are often regarded as mutually exclusive and in competition—a situation resulting in pronouncements that the amyloid hypothesis, plagued by numerous clinical trial failures, is dead and needs to be replaced. 1 However, a variety of data (including in vitro neurotoxicity studies, Down's syndrome studies, and other genetic linkage analyses) compellingly link amyloid beta (Aβ) to the pathogenesis of AD. 2 , 3 Accordingly, rather than categorically rejecting the role of Aβ, the need for a new widely encompassing conceptualization of AD that unifies seemingly divergent theories into a single harmonized explanation emerges as an effective strategy. Incorporating protein misfolding mechanisms into a broader‐based immunopathic model of AD could attain such a goal—a goal which can be achieved by repositioning Aβ as an immunopeptide. 4
Herein, based on an extensive series of in silico, in vitro, and in vivo studies, a new mechanistic model of AD as a brain‐centric autoimmune disorder of the innate immune system is presented: in response to various initiating stimuli (e.g., infection, trauma, ischemia), Aβ is released as an early responder immunopeptide triggering an innate immunity cascade in which Aβ exhibits both immunomodulatory and antimicrobial properties (whether bacteria are present or not), resulting in a misdirected attack upon “self” neurons, arising from analogous electronegative surface topologies between neurons and bacteria, and rendering them similarly susceptible to membrane penetration by antimicrobial peptides (AMPs) such as Aβ. After this self‐attack, the resulting necrotic neuronal breakdown products diffuse to adjacent neurons eliciting further release of Aβ, leading to a chronic self‐perpetuating autoimmune cycle. A subsequent comprehensive screening program of small molecules endogenous to the human brain identified tryptophan metabolism as a biochemical regulator of innate immunity and thus a source of potential endogenous anti‐AD molecules capable of chemical modification into multi‐site modulators targeting AD's complex immunopathic–proteopathic pathogeneses.
2. MATERIALS AND METHODS
The in silico, in vitro, and in vivo methods used in this study are described in detail in Appendix 1 in supporting information. Briefly, compounds were prepared by standard synthetic routes, purified by high‐performance liquid chromatography and recrystallization, and characterized by proton nuclear magnetic resonance and mass spectrometry. Compounds were then evaluated via multiple in vitro assays including: inhibition of biotinylated Aβ1‐42 oligomerization and Aβ1‐40 fibrillization, inhibition of tau aggregation, prevention of conformational change in Aβ1‐40 from α‐helical to β‐sheet in circular dichroism (CD) studies, prevention of Aβ1‐40 toxicity against SH‐SY5Y neuroblastoma cells, indoleamine‐2,3‐dioxygenase (IDO‐1) enzymatic inhibition studies, and inhibition of pro‐inflammatory cytokine release from microglial cells. In vivo studies used radial arm and Morris water maze studies in APP/PS1 transgenic mice.
RESEARCH IN CONTEXT
Systematic Review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts and presentations. While the amyloid hypothesis has been the subject of extensive study, the immunological basis of Alzheimer's disease (AD) is still evolving and attempts to combine the two have not been systematically studied. Relevant studies are appropriately cited.
Interpretation: Our findings lead to an integrated mechanistic explanation of AD. This explanation is consistent with AD as a brain‐centric proteopathic‐triggered autoimmune disorder of innate immunity. In response to stimulating events (e.g., microbes, trauma, ischemia, pollution, depression), amyloid beta (Aβ) is produced as an early molecular component of the innate immunity cascade; however, Aβ’s immunomodulatory/antimicrobial duality results in a misdirected cytotoxic attack upon “self” neurons, culminating in progressive neurodegeneration.
Future Directions: The article proposes a framework for the generation of new experiments and the development of novel therapeutics. Examples include: (a) the role of tryptophan metabolism as a key regulator of innate immunity the in brain, (b) developing tryptophan metabolites or analogs thereof as disease‐modifying therapeutics for AD, and (c) further exploration of Aβ as an immunopeptide.
3. RESULTS
3.1. In silico studies of Aβ–membrane interactions
A molecular mechanistic understanding of AD necessitates dynamically modeling AD's pathogenesis at an explicit atomistic–molecular level (Figure 1). To fundamentally explore the initial stages of Aβ1‐42 neuronal engagement, we started with simple simulations of Aβ adjacent to model membrane lipids without proteins present (Figure S1 in supporting information). In silico simulations, using dipalmitoylphosphatidylcholine bilayers and dipalmitoylphosphatidylethanolamine monolayers, demonstrated pH‐dependent membrane attachment of Aβ via its hydrophilic N‐terminal (Aβ5‐16), electrostatically anchored by its polycationic H+H+QK+ domain (Aβ13‐16; Figures 2A‐2C). Inserting polyanionic glycosaminoglycans (GAGs; e.g., polysulfated heparin fragments) or gangliosides (GM1; monosialotetrahexosyl‐ganglioside) into the membrane improved Aβ’s electrostatic binding. Focused quantum mechanics calculations showed that the inclusion of metal cations (Cu2+) further strengthens the HHQK–GAG interaction, acting as a chelating bridge between “soft” histidine side‐chain nitrogen atoms and “hard” GAG sulphate anions—enabled by vacant d orbitals in the Cu2+ d 9 inner orbital complex; other ions (Na+, Ni2+) did not demonstrate stabilization properties.
FIGURE 1.
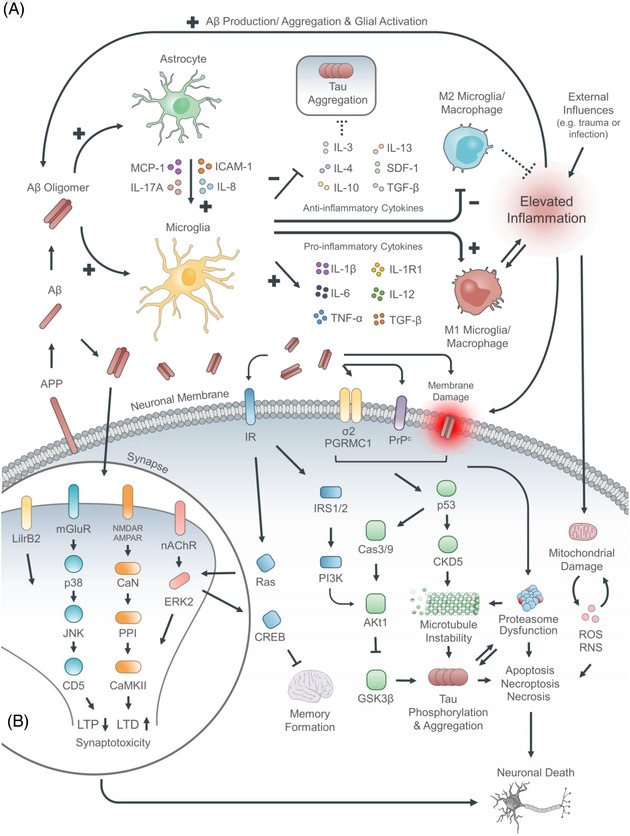
Combined proteopathic–immunopathic conception of AD. (A) Summary of cellular and protein elements implicated in AD pathogenesis. (B) Summary of synaptotoxic pathologies and signaling in AD. Conceptualizing Aβ as an immunopeptide enables a unifying harmonization of the multiple postulated pathogeneses for AD. Aβ oligomers are directly neurotoxic damaging the neuronal membrane and eliciting excitotoxicity via neurotransmitter receptors such at the NMDA receptor. Concomitantly, Aβ oligomers are directly immunotoxic interacting with microglia and astrocytes (with enhanced microglia‐astrocyte cross‐talk mediated in part by MCP‐1, ICAM‐1), skewing microglial activation towards the proinflammatory M1 phenotype, promoting release of pro‐inflammatory cytokines (IL‐1β, IL‐6, TNFα), and suppressing the release of anti‐inflammatory cytokines (IL‐3, IL‐4, IL‐10, IL‐13). In turn this promotes neurotoxic tau aggregation (with microtubule instability) and mitochondrial damage leading to increased membranotoxcic reactive oxygen species (peroxides, superoxide, hydroxyl radical, singlet oxygen). Finally, this culminates in cellular death by both necrosis and apoptosis with associated symptom expression in the form of disordered cognition and short term memory information processing.
FIGURE 2.
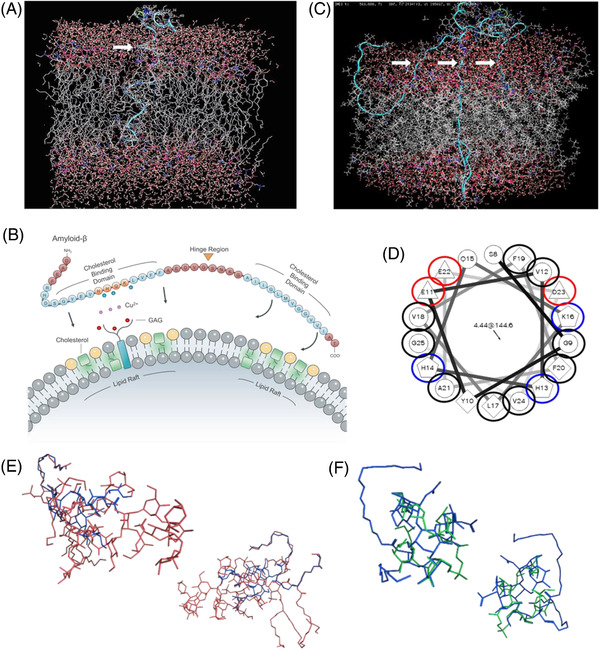
In silico calculations of amyloid beta (Aβ) and membrane dynamics. (A) Simulation of a single Aβ peptide inserting into a model neuronal membrane after 450 ps. Insertion occurred only in the presence of cholesterol, irrespective of variations in computational parameters. Aβ favorably inserted adjacent to cholesterol with the C‐terminus (residues 28–42) inserting first. B) Proposed model of Aβ membrane insertion in which HHQK motif facilitates anchoring (aided by glycosaminoglycans [GAGs] and metal ions), with insertion occurring at the C‐terminus via twisting about the hinge region. (C) Simulation of Aβ oligomer (3–5 peptides) inserting into membrane after 450 ps, upon pre‐equilibration outside the membrane for 30 ns. Oligomeric insertion formed a loosely organized membrane‐disrupting aggregate. (D) Schiffer‐Edmundson helical wheel conformation of Aβ. Negatively charged residues (red) align on a single face of the helix, compatible with the proposed HHQK motif anchored membrane insertion model (blue: positive; black: hydrophobic; unmarked: polar, uncharged). (E) Molecular modeling demonstrates notable overlap between GM1 and lipopolysaccharide, and (F) lipoteichoic acid, upon energy minimization calculations, suggesting molecular mimicry among markers of infection and necrosis toward Aβ production
These simulations also showed that Aβ’s hydrophobic C‐terminus (Aβ28‐42) was energetically unstable lying on the aqueous‐bathed extracellular membrane surface (supported by Schiffer‐Edmundson helical wheel representation; Figure 2D). Although insertion into the membrane's lipophilic interior would resolve this thermodynamically unstable situation, repeated simulations failed to demonstrate facile insertion. However, when cholesterol was incorporated into the membranes, specifically as microdomains (rafts) mimicking eukaryotic membranes, insertion did occur—a process facilitated by Aβ’s flexible G25S26 “hinge.” Insertion occurred adjacent to cholesterol rafts, indicating the cholesterol–phospholipid interface yields improved Aβ penetration susceptibility; such insertions occurred approximately once every fifteenth Aβ/membrane collision, proceeding to completion once initiated. This was also true for Aβ oligomers, which collectively inserted, ultimately creating membrane‐breaching pores. Control studies with scrambled Aβ (AIAEGDSHVLKEGAYMEIFDVQGHVFGGKIFRVVDLGSHNVA) revealed no membrane insertion.
These in silico simulations suggest that one of Aβ’s fundamental cytotoxic mechanisms is via membrane insertion/disruption identical to an antimicrobial peptide; this result is compatible with our previous calculations indicating that Aβ has AMP properties analogous to melittin. 5 To further predict AMP‐likeness, we computed Boman indices (BI[Aβ1‐40] = 4.10 kJ/mol, BI[Aβ1‐42] = 3.22 kJ/mol), which indicate Aβ’s AMP‐analogous capacity to bind to bacterial membranes. 6 However, beyond simple bacteriocidal activity, AMPs are also known fundamental regulators of innate immunity via multiple pathways, including GAG‐binding. 7 Supporting a similar immunomodulatory/antimicrobial duality for Aβ, our calculations demonstrate that Aβ interacts with GAGs via the HHQK domain, a region recognized by Williamson et al. 8 as providing an in vitro binding site for GAGs and gangliosides and by Giulian et al. 9 as providing the structural basis for Aβ‐mediated immunopathology, which comprises activation of microglia and the release of neurotoxic pro‐inflammatory cytokines. In computationally assessing Aβ’s AMP‐like immunoregulatory role, we further identified the immunomodulatory GAG(heparan)‐binding BBXB/BXBB sequence (where B is a basic amino acid) not only in Aβ(HHQK16) and tau(KKAK144) but also in multiple cytokines and immunopeptides that participate in the Aβ‐initiated neurotoxic cascade (interleukin [IL]‐1R1[HKEK80], IL‐1β[KLRK76], IL‐4[HHEK85], IL‐6[KKAK159], IL‐10[RRCH128], IL‐12[HKLK219], IL‐13[HLKK138], C1qA[KKGH225], IFN‐γ[KKKR112], ICAM‐1[RDHH180], MIP‐1α[KRSR47], MIP‐1β[KRSK69], S100β[HKLK29], SDF‐1[KHLK46] and RANTES[RKNR70]), implying the HHQK/BBXB motif may be a promiscuous receptor in the proteo‐immunopathogenesis of AD. 10 Therefore, in silico simulations predict that Aβ has AMP‐like activity and is a molecular trigger of innate immunity.
3.2. Aβ is an innate immune system effector immunopeptide
Subsequent in vitro studies confirmed Aβ is an AMP‐type immunopeptide, exhibiting activity against viruses (herpes simplex virus 1) and bacteria (E. coli, S. aureus, S. marcescens, K. pneumonia; previously described by Soscia et al. 11 ; Figures 3A‐3F). Conversely, we demonstrated using a 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) cell viability assay, that conventional AMPs (LL‐37, Cecropin A) are toxic to SK‐N‐AS human neuroblastoma and rat primary neuronal cells (Figures 3G‐3H). In addition, Aβ’s AMP activity is influenced by metal ions (Cu2+, Zn2+; similar to conventional AMPs 12 ); against E. coli, for example, Aβ proved more bacteriotoxic in the presence of Zn2+ (IC50 = 42μM) and Cu2+ (IC50 = 43μM; IC50 = 51μM for H2O control). (Clinically, AD is accompanied by localized Zn2+/Cu2+ dyshomeostasis, and a higher metallic burden in the AD brain augments Aβ neurotoxicity. 13 )
FIGURE 3.
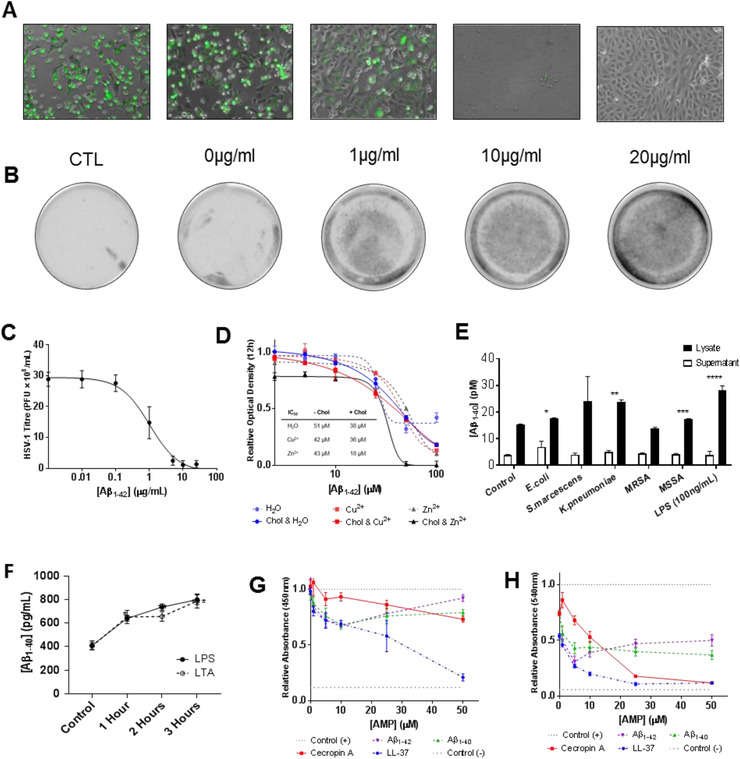
Antibacterial and antiviral functionality of amyloid beta (Aβ). (A) Fluorescence microscopy of Vero cells infected with Vesicular Stomatitis Virus tagged with green fluorescent protein (VSV‐GFP) pretreated overnight with varying Aβ1‐42 concentrations. Control (CTL) samples were diluted and infected immediately, without overnight treatment; cells imaged 24 hours post‐infection and reveal a dose‐dependent antiviral response. (B) Typhoon scans of culture plates, where plaques are visible as dark‐gray spots, show the same. (C) Dose‐response curve of Aβ against herpes simplex virus 1 (HSV‐1), derived from fluorescence experiments, demonstrating clear antiviral functionality. Calculated EC50 = 1 μg/ml or 0.2 μM. (D) Toxicity/antimicrobial peptides (AMP) activity of Aβ1‐42 against E.coli, in the presence of cholesterol and metal ions (Cu2+ and Zn2+), as determined by relative 12‐hour optical density (OD12H); and calculated IC50 of Aβ1‐42 with/out cholesterol and metal ions from fitted nonlinear regressions (sigmoidal) to data. Cholesterol and Zn2+ enhance Aβ1‐42 toxicity against bacteria. (E) Aβ production in lysate and supernatant of SK‐N‐AS incubated with liopopolysaccharide (LPS), lipoteichoic acid, and bacterial fragments. Aβ production quantified by enzyme‐linked immunosorbent assay; significant increases observed in cell lysates only, exposed to E.coli (*P = 0.03), K. pneumoniae (**P < 0.01), Methicillin‐susceptible S. aureus (MSSA, *** P = 0.02), LPS (100 ng/ml, **** P < 0.01). MRSA denotes Methicillin‐resistant S. aureus. (F) Time‐dependent production of Aβ in lysates of SK‐N‐AS showing upregulation within 1 hour of initial exposure, suggesting innate (as opposed to adaptive) immune response; P < 0.01 (compared to control) in all cases. (G) Relative neurotoxicity of Aβ (1‐40 and 1‐42) and AMPs (Ceropin and LL‐37) against SK‐N‐AS cells, assayed by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT). (H) Relative neurotoxicity of Aβ and AMPs against primary cultured rat neurons, assayed by MTT
We also observed an increase in AMP efficacy after addition of cholesterol. Aβ’s IC50 against E. coli was reduced to 38 μM (from 51 μM) with supplementary cholesterol, and was further reduced to 18 μM with both Zn2+ and cholesterol. Although elevated cholesterol is widely reported in AD cohorts, it is uncharacteristic for an AMP to gain cytotoxicity in the presence of cholesterol; indeed, it has been suggested that the presence of cholesterol is a key factor by which physiologic AMPs distinguish between microbial and non‐neuronal eukaryotic membranes. 14 However, our in silico simulations suggest that in electrically active neuronal membranes, cholesterol paradoxically enhances Aβ’s AMP‐like destructive membrane insertion.
Beyond demonstrating that Aβ has antimicrobial activity, it is also necessary to show that neural tissue produces Aβ in response to bacterial exposures known to elicit AMP release. When we exposed SK‐N‐AS neuroblastoma cells to either Gram‐negative (E. coli) or Gram‐positive (S. aureus) bacteria, significant concentrations of Aβ were produced, indicating that bacteria trigger Aβ production. To identify the molecular basis of this observation, lipopolysaccharide (LPS) from lysed Gram‐negative bacterial membranes and lipoteichoic acid (LTA) from Gram‐positive bacteria exhibited the same ability to elicit increased Aβ production; when SK‐N‐AS neuroblastoma cells were incubated with 1 μg/ml of LPS or LTA, Aβ concentrations reached 800 pg/ml in the cell lysate within 3 h (Figure 3E), consistent with previous observations by Lee et al. 15
However, infections are not the only trigger for AMP release. Trauma, 16 ischemia, 17 environmental pollution, 18 and depression 19 are other well‐described sterile stimuli for AMP release via pathogen‐associated/damage‐associated molecular pattern (PAMP/DAMP) overlaps. When we exposed neuroblastoma cells to breakdown products from mechanically injured neurons, significant Aβ concentrations were produced. This observation is compatible with the role of repetitive head trauma, ranging from sports to domestic abuse, as an AD risk factor. 20
3.3. AD is a chronic autoimmune disorder
These data, though interesting, fail to explain why AD is a chronic disorder. The evolutionary older innate immune system is a short‐term acute line of defense; it is the adaptive immune system which permits chronic immunological memory. To investigate a mechanism for chronicity within innate immunity, we sought to identify stimuli leading to sustained pathology. Because Aβ induces neuronal death by both apoptotic and necrotic mechanisms, we explored if one of these processes could yield propagative Aβ release. Accordingly, we produced neuronal death by apoptosis and necrosis independently (verified by caspase‐3/7 activation), and administered the breakdown products to healthy SK‐N‐AS neurons (Figure 4). These results demonstrated that only necrotic (not apoptotic) breakdown products triggered a significant release of Aβ.
FIGURE 4.
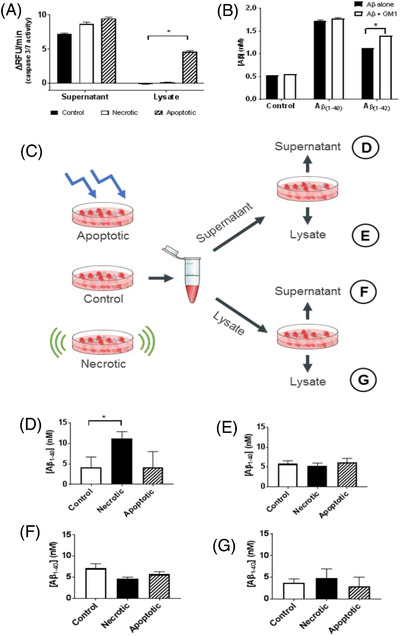
Amyloid beta (Aβ) production in response to innate immune activation. (A) To specify the signaling leading to upregulation of Aβ production, cells were treated with mechanical agitation (scraping) to induce necrosis and ultraviolet light (10 minutes with 254 nm light) to induce apoptosis. Evaluation of caspase‐3/7 activity, by relative fluorescence per minute (ΔRFU/min), verified which treatments led to a specified mode of cell death, with only the apoptosis treatment significantly elevating caspase activity (P < 0.01, in lysate). (B) Aβ production in response to incubation with fluorescently labeled Aβ (10 μM), and GM1 (molecular marker of neural necrosis, 5 μM). Significant elevation in Aβ production followed exposure to Aβ1‐40. Aβ1‐42 induces less Aβ production, though addition of GM1 significantly elevates Aβ levels (P > 0.01)—indicating synergy between neural necrosis and self‐perpetuated release of Aβ. (C) Scheme to quantify origin of elevated Aβ signaling, exposing control/necrotic/apoptotic cells’ supernatant and lysates to fresh SK‐N‐AS cells, with subsequent quantification of Aβ production in supernatant and lysates. (D) Aβ production in supernatant of cells exposed to supernatant of noxious cells; necrotic supernatant significantly (P = 0.03) elevated Aβ production. E, Aβ production in lysates of cells exposed to supernatant of noxious cells; no significant difference. (F) Aβ production in supernatants of cells exposed to noxious lysates; no significant difference. (G) Aβ production in lysates of cells exposed to noxious lysates; no significant difference
To identify the molecular effector of this chronicity, we showed that the damaged membranes of necrotic Aβ‐killed neurons release soluble GM‐1 and GM1‐Aβ complexes (previously reported by Michikawa et al. 21 and Blennow et al. 22 ), which can induce adjacent neurons to produce/release oligomeric Aβ (previously reported by Zha et al. 23 ). Thus, LPS (acutely from bacteria) or GM1 (chronically from necrotic neurons) analogously induce Aβ release from nearby neurons—a molecular mimicry described for these molecules in other neuro‐immunological disorders (Guillain‐Barré syndrome), and involving a sialosyl‐galactose (Galβ1‐3GalNAcβ1‐4[NeuAcα2‐3]Galβ)–dependent carbohydrate similarity (Figure 2E‐2F). 24
Therefore, in response to PAMP/DAMP‐stimulating events (e.g., infection, trauma, ischemia, pollution), Aβ is produced as an early immunopeptidic responder with combined immunomodulatory/antimicrobial duality that results in a misdirected attack upon “self” neurons, arising from the unique electrophysiological similarities between neurons and bacteria in terms of transmembrane potential gradients (–80 mV) and anionic charges on outer leaflet membrane macromolecules (gangliosides in neurons; lipopolysaccharides in bacteria). The subsequent breakdown products of necrotic neurons elicit further release of Aβ leading to a chronic, self‐perpetuating cycle—this is autoimmunity. Considering AD an autoimmune disorder of innate immunity raises the possibility of identifying endogenous factors with which to ameliorate disease progression (analogous to the use of glucocorticoid‐based therapeutics in the treatment of classical adaptive autoimmune disease).
3.4. Identifying endogenous anti‐AD molecules
Immunological processes such as AMP signaling are subject to homeostatic regulation; likewise, because many proteins are susceptible to amyloid‐like aggregation (“amylome” 25 ), there is evolutionary pressure to physiologically control protein folding. Accordingly, it is reasonable to hypothesize the existence of molecules endogenous to the human brain as regulators of innate immunity that could modulate AD's immunopathic–proteopathic neurotoxicity.
To identify a compound endogenous to the human brain that could be exploited as a starting point in the development of an orally active, brain‐penetrant therapeutic, we performed an unbiased screen of 1137 molecules (molecular weight < 650 g/mol; Table S1 in supporting information) occurring normally in the human brain, first using biotinylated Aβ1‐42 anti‐oligomerization and kinetic thioflavin T (ThT) Aβ1‐40 anti‐aggregation assays, and second using cytokine‐based anti‐inflammatory assays.
The initial screening identified multiple “hits” against Aβ aggregation within the tryptophan metabolic pathway, including both indole‐based and anthranilate‐based metabolites (less active hits were also obtained from the arginine metabolic pathway). Although tryptophan was inactive, metabolites from its principal catabolic pathways (Figure 5A) including tryptamine, 5‐hydroxytryptamine (5‐HT), 5‐methoxytryptamine and 3‐hydroxyanthranilate inhibited Aβ1‐42 oligomerization and Aβ1‐40 aggregation (IC50’s: 20‐30 μM; Figure 5B). In secondary anti‐immunopathy screens, these same metabolites modified the release of pro‐inflammatory cytokines. 5‐HT, for example, modulated IL‐1β production and release from microglia at 50 and 100μM (compatible with observations by Krause et al. 26 and Dürk et al. 27 ). These dual screens suggest tryptophan metabolites as a single molecular platform for the design of agents targeting both of AD's immunopathic–proteopathic pathologies.
FIGURE 5.
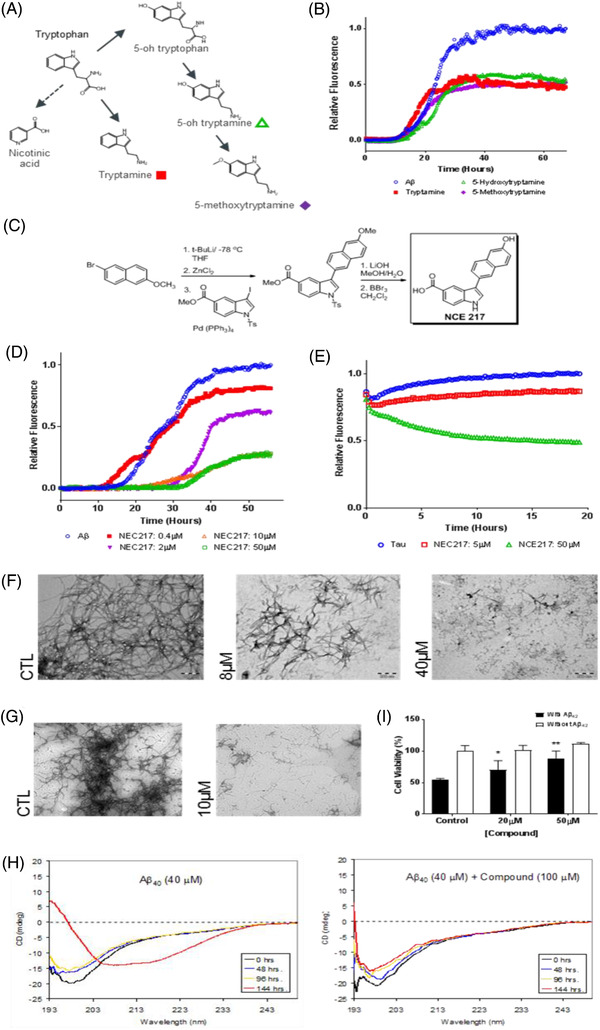
Identification and optimization of endogenous anti‐immunopathic/anti‐proteopathic molecules. (A) In silico screen of endogenous brain molecules (see Table S1) identified numerous metabolites of tryptophan as potential multifunctional agents against Alzheimer's disease (AD). Molecules representing intermediates in tryptophan's major metabolic pathways (shown: tryptamine, 5‐OH tryptamine, 3‐OH anthranilic acid) were selected for in vitro screening. (B) Thioflavin T (ThT) measured aggregation of amyloid beta (Aβ; in which ThT fluorescence increases when bound to β‐sheets of fibrils, rather than α‐helices of monomers) revealed multiple viable inhibitors of Aβ aggregation. (C) Chemical synthesis of tryptophan‐based analogue: NCE217. (D) ThT measured aggregation of Aβ in the presence of varying concentrations of NCE217, demonstrating dose‐dependent inhibition of aggregation from 0.4 μM to 10 μM. (E) ThS measured aggregation of tau observed considerable inhibition of fibrillization with 50μM of NCE217—evidenced by decreased fluorescence of ThS in the treatment group over time. (F) Transmission electron microscopy (TEM) of Aβ1‐42 in the absence and presence of NCE217 (8μM and 40μM), demonstrating substantial inhibition of Aβ fibril formation, and preponderance of smaller species upon exposure to compound. (G) TEM at 6000X of tau (control and treated with 10μM of NCE217) showing disruption of large tangles, and preponderance of smaller species after 72‐hour exposure. (H) Circular dichroism (CD) spectra of Aβ40 in the absence (left) and presence (right) of NCE217 (100μM), demonstrating preservation of α‐helical conformation past 144 hours, in contrast to uninhibited Aβ. (I) SH‐SY5Y neuroblastoma cell viability (assayed by 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide [MTT]) upon exposure to Aβ (10 μM). NCE217 significantly increases cell viability (*P = 0.05 at 20μM; ** P = 0.01 at 50 μM) after Aβ exposure, suggesting it may alleviate Aβ‐induced toxicity
Though tryptophan‐kynurenine metabolism proved anti‐proteopathic and anti‐immunopathic, other downstream metabolites (e.g., 3‐hydroxykynurenine, quinolinic acid) are neurotoxic—promoting lipid peroxidation and amplified neuroflammation. 28 Accordingly, simple supplementation with tryptophan metabolites may not be an optimal therapeutic strategy. To enable de novo molecular design of new chemical entities not susceptible to transformation into neurotoxic metabolites, we next studied the molecular basis of the combined anti‐proteopathic/immunopathic effects of tryptophan metabolites.
Commencing with tryptamine as a prototypic anti‐aggregation indole‐based metabolite, we used molecular mechanics calculations to discern its atomistic interactions with Aβ1‐42. We modelled tryptamine docking to Aβ at 126 different starting positions, using three random starting points at each Aβ1‐42 residue. Tryptamine bound with the greatest affinity at H13H14 (within Aβ’s HHQK domain), forming aromatic–cationic interactions (via tryptamine's indole ring); additional binding at F19F20 via aromatic–aromatic stacking interactions was also demonstrated. These interactions were verified using higher‐level density functional theory quantum mechanics calculations, which revealed tryptamine bound to H13H14 with a 25‐28 kcal/mol binding energy at a separation of 2.1 Å.
We next evaluated the molecular basis of tryptophan metabolite‐mediated anti‐immunopathic effects. Although the relationship between tryptophan metabolism and innate immunity is complex, IDO‐1–mediated catabolism of tryptophan is the earliest step in the metabolic cascade and is a recognized immunoregulatory target, working in concert with IL‐1β, tumor necrosis factor alpha, and nuclear factor kappa B. 29 It is also well established that IDO‐1 inhibition downregulates immuntoxicity and ameliorates cognitive impairment in the APP/PS1 AD mouse model; 30 moreover, tryptophan analogues, including 1‐methyltryptophan, are known IDO‐1 inhibitors. 31 Additionally, IDO‐1 inhibition may contribute to reducing neurotoxicity by diverting tryptophan to the hydroxylase‐initiated serotoninergic pathway, thereby increasing the biosynthesis of anti‐oligomeric tryptophan metabolites, such as 5‐HT. Accordingly, we selected IDO‐1 inhibition for further molecular modelling studies. Using force field calculations, in conjunction with known crystal structures, 32 we identified a tripartite binding pocket within the IDO‐1 active site: part A – IDO‐1163‐168 [FFLVSL], part B – IDO‐1226‐231 [FFSVLR], part C – heme group; (FFLVSL and FFSVLR exhibit reverse‐direction homology with Aβ’s HHQKLVFF sequence). An optimal IDO‐1 inhibitor must engage all three binding pocket regions for sustained inhibition. Tryptamine (shown to bind to Aβ13‐20) also demonstrated an in silico binding affinity for the tripartite IDO‐1 binding pocket.
Exploiting these computational results, we next sought to optimize this tryptophan‐based platform via the design of a de novo therapeutic.
3.5. De novo design of anti‐AD therapeutics
Although tryptophan‐methylpropionate dipeptide has Aβ anti‐aggregative activities, 33 and a tryptophan–quinine bicyclic compound elicited phenotypic recovery in a Drosophila AD model, 34 peptidic compounds tends to be poor central nervous system drugs. Therefore, to transform tryptophan metabolites into a drug‐like molecular platform, we implemented a sequential bioisosteric substitution approach based on systematic manipulation of electron density topologies (i.e., a carboxylate “fragment” is replaced by a bio‐actively equivalent tetrazole “fragment,” which in turn is replaced by other heteroaromatic ring systems, and so on, such that multiple fragments within the initial molecule are replaced piece‐by‐piece with chemically different but biologically equivalent fragments to yield a drug‐like new chemical entity 35 ). By this procedure, tryptophan and related metabolites were transformed into a synthetically tractable bi‐aromatic series based on bi‐indole and indole‐naphthol scaffolds. From this, we synthesized a focused 330 compound library (Figures S2, S3; Table S2; Appendix 4 in supporting information). Among these, NCE217 proved most instructive (Figures 5C‐5H). It demonstrated an ability to: inhibit the oligomerization of biotinylated Aβ1‐42 (IC50 = 9 μM); block Aβ1‐40 fibrillization, assayed by ThT (IC50 = 7.5 μM); block tau aggregation, assayed by thioflavin S (IC50 = 24 μM); dose‐dependently prevent the aggregation of Aβ1‐40 and tau‐441 in electron microscopy studies (at 8, 20, and 40 μM); prevent the conformational change in Aβ1‐40 from α‐helical to β‐sheet in CD studies; bind in silico to both the HHQK (Aβ13‐16) and LVFF (Aβ17‐20) motifs of Aβ, and to the corresponding BBXB motifs within tau, IL‐4, IL‐12, IL‐13, IFN‐γ, ICAM‐1, RANTES, and C1qA (Figure S4 in supporting information); bind to Aβ13‐16 in 2D‐NMR and DOSY studies; and prevent Aβ1‐40 toxicity against SH‐SY5Y neuroblastoma cells, assayed by MTT (Figure S5) in supporting information. Moreover, Caco‐2 and MDR1‐MDCK in vitro assays suggest that NCE217 is orally bioavailable and blood‐brain barrier penetrant, further evidenced by in vivo microdialysis studies that confirmed the presence of NCE217 in mouse brain.
We also observed NCE217 protects doubly transgenic APP/PS1‐derived neurons from Aβ1‐40 induced toxicity in hippocampal slice studies (P = 0.04; Figure 6). NCE217 improved memory in APP/PS1 mice, with performance in AD cohorts recovering to levels comparable to wild‐types, both in radial arm (20 mg/kg/day, P = 0.04) and Morris water maze studies (20 mg/kg/day, P < 0.01). Post mortem, Aβ plaque burden was reduced by 30% in the cortex (P = 0.024), but to a lesser extent in the hippocampus, with subsequent dot blot studies revealing a 41% reduction in oligomeric Aβ throughout the brain (P = 0.04); these findings combined with the prevention of tau aggregation raise the possibility that decreased tau might also play a role in improved learning. NCE217 demonstrated no behavioural or histopathological toxicity at 300 mg/kg in mice. Mechanistically, NCE217 displayed no in vitro effect against Aβ‐induced microglial activation, inflammatory cytokine release or IDO‐1 enzyme activity; that is, NCE217 is anti‐proteopathic but not anti‐immunopathic.
FIGURE 6.
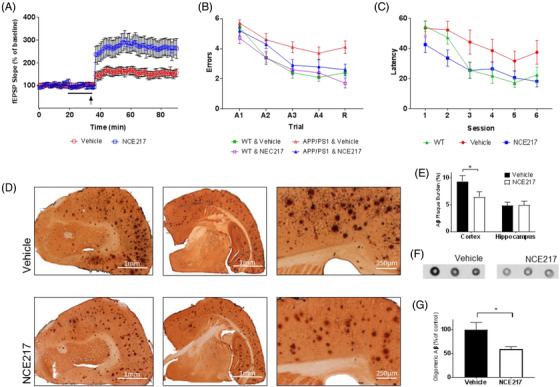
In vivo efficacy of tryptophan‐based anti‐Alzheimer's disease (AD) therapeutic compound. (A) Long‐term potentiation is strengthened in APP/PS1 transgenic murine hippocampal slices (400 μm) upon exposure to 50 μM of NCE217 (indicated by arrow)—as evidenced by heightened field excitatory postsynaptic potential (fEPSP) slope upon θ‐burst stimulation (P < 0.01). (B) In radial arm water maze testing, APP/PS1 transgenic mice treated with NCE217 (20mg/kg/day) made statistically fewer errors than mice treated with a vehicle (P = 0.04). Performance during retention (R) proved comparable to wild‐type (WT) mice, suggesting possible preservation of memory in APP/PS1 murine models. (C) In Morris water maze testing, APP/PS1 murine models treated with NCE217 (20mg/kg/day) exhibited a lower latency (time to locate a hidden platform) than vehicle‐treated models. Performance was again comparable to WT mice, suggesting possible preservation of memory (P < 0.01). (D) Amyloid beta (Aβ) plaque burden in brains of APP/PS1 mice treated with vehicle (top) and NCE217 (bottom). Plaque burden is visibly diminished with administration of NCE217, across rostral coronal sections (left), coronal sections near bregma (center), and in the cerebral cortex (right). E, Computational plaque quantification (by ImageJ software) observed significant reduction (*P = 0.024) of amyloid plaques in the cortex. F, Immunohistochemistry of Aβ in the brain, assayed by dot blots using the A11 anti‐oligomeric Aβ antibody, revealed a lower preponderance of neurotoxic oligomers in the brain upon NCE217 treatment. G, Quantification (by ImageJ) revealed a statistically significant decrease in the prevalence of oligomers (P = 0.04)
To incorporate efficacy against immunopathic components of AD, we performed additional IDO‐1 molecular modelling simulations. 36 Based upon these calculations, the NCE217‐like naphthol‐indole platform was transformed into a family of styryl‐indoles; additional in silico simulations led to specific 3‐styrylindole compounds. To synthesize a library of these compounds, the Doebner modification of the Knoevenagel condensation 37 was used to produce an additional 61 compounds as inhibitors of both IDO‐1 and Aβ aggregation (Table S3 in supporting information). IDO‐1 inhibition was measured in human‐recombinant IDO‐1 assays using IDO‐1‐transfected HEK293 cells and interferon‐γ stimulated human microglia cells. Inhibition of Aβ oligomerization and aggregation were assayed as previously.
Multiple styryl‐indole analogues were inhibitors of both IDO‐1 metabolic activity and Aβ aggregation. The most potent inhibitors possessed: an indole substituted with a small, hydrophobic group at the 5‐position; a rigid linker, such as an alkene, connected from the 3‐position; and a monocyclic aryl ring with substituents placed to interact with elements of the heme group. Within this compound series, (E)‐4‐(2‐[5‐methoxy‐1H‐indol‐3‐yl]vinyl)phenol inhibited Aβ oligomerization (IC50 = 6.5 μM), inhibited IDO‐1 enzyme activity (IC50 = 2.8 μM), and inhibited INF‐γ‐induced IDO‐1 activity (IC50 = 10.8μM). Thus, tryptophan metabolite analogues achieved combined anti‐oligomeric/anti‐IDO activities, confirming this platform's therapeutic utility for targeting multiple aspects of AD's immunopathic–proteopathic spectrum.
3.6. Alternative platforms for therapeutics discovery
As an alternative to synthetic tryptophan metabolite analogues, we next performed an in silico screen of 2950 natural products (BioCyc/MetaCyc database; 38 Table S4 in supporting information), to identify natural products capable of energetically favorable interactions with the HHQK/BBXB motif. The highest ranking Aβ13‐16‐binding hits were obtained from plant‐based bi‐aromatic or aromatic‐anionic phenylalanine metabolites. 39 These structurally diverse phenylpropanoid metabolites categorize into four classes: phenolates, coumarates, stilbenes, and flavonoids, with salicylate, ferulate, resveratrol, and tannate being class‐specific prototypes, respectively. These compounds favorably bound in silico to Aβ13‐16 and to the BBXB domains of IL‐4, IL‐12, IL‐13, IFN‐γ, ICAM‐1, and C1qA. In ThT and CD anti‐aggregation assays, salicylate, ferulate, sinapate, 4‐coumarate, caffeiate, tannate, and resveratrol exhibited Aβ anti‐aggregant activities. To demonstrate their utility as a molecular platform, 30 analogues of ferulate were synthesized and demonstrated varying Aβ1‐40 anti‐aggregant activities (Table S5 in supporting information). 40 Ferulate has also been shown to inhibit IDO‐1 enzymatic activity. 41
Additionally, these plant‐based hits support the concept of AD as an innate autoimmune disorder. The phenylpropanoid pathway is an endogenous regulator of plant innate immunity (mirroring the role of tryptophan metabolism in mammals). Salicylates for example are protectively co‐released with AMPs to ensure plant cell viability during innate immune system activation and to decrease AMP‐induced autotoxicity. 42 Resveratrol and salicylate also have the capacity to modulate human innate immunity. 43
Extending this natural product screening to a drug repurposing screening campaign, we next similarly evaluated 1768 known drugs (Table S6 in supporting information) identifying indomethacin (2‐[1‐(4‐chlorobenzoyl)‐5‐methoxy‐2‐methylindole‐3‐yl]acetic acid), fluvastatin ([E]‐7‐[3‐(4‐fluorophenyl)‐1‐propan‐2‐ylindole‐2‐yl]‐3,5‐dihydroxyhept‐6‐enoate), mefanamic acid (N‐[2,3‐xylyl]‐anthranilate), and furosemide (4‐chloro‐N‐[2‐furylmethyl]‐5‐sulfamoyl‐anthranilate); all contain either indole or anthranilate tryptophan‐like moieties; all have previously documented anti‐oligomeric or anti‐inflammatory activities, or both. 44 We synthesized 50 furosemide analogues demonstrating the combined anti‐proteopathic and anti‐immunopathic bioactivities of this known platform. 45
4. DISCUSSION
Although others have postulated AD to be an autoimmune disease, 46 , 47 previous work has typically focused on a conventional approach to autoimmunity invoking autoantibodies and adaptive immune responses. Our data are compatible with AD as a brain‐centric autoimmune disorder of innate immunity. In response to PAMP/DAMP‐stimulating events (e.g., microbes, trauma, ischemia, pollution, depression), Aβ is produced as an early molecular component of the innate immunity cascade. However, Aβ’s immunomodulatory/antimicrobial duality results in a misdirected attack upon “self” neurons, arising mechanistically from the similarities between neurons and bacteria in terms of transmembrane potential gradients (≈ –80 mV) and anionic charges on outer leaflet membrane macromolecules (gangliosides in neurons; lipopolysaccharides and cardiolipin in bacteria), enhancing the probability of interaction with a cationic AMP. The subsequent breakdown products of necrotic neurons elicit further release of Aβ leading to a chronic, self‐perpetuating cycle with concomitant microglial activation, pro‐inflammatory cytokine release, and tau aggregation.
Considering AD an autoimmune disorder raises the possibility of identifying endogenous factors to ameliorate disease progression. Tryptophan metabolites modulate key cytokines associated with innate immunity and inhibit proteopathic oligomerization, thereby functioning as putative endogenous anti‐AD molecules. A pathogenic‐therapeutic role for tryptophan metabolism in AD is compatible with various clinical data: serum tryptophan levels show a statistically significant reduction with age, people with AD have reduced plasma levels of tryptophan, and acute tryptophan depletion in people with AD causes increased cognitive dysfunction. 48 , 49 From these observations, multiple therapeutic approaches emerge. The important role of the gut microbiome in producing neuro‐active tryptophan metabolites represents one novel approach for therapeutic intervention. 50 More conventionally, as evidenced by this study, novel small‐molecule analogues of tryptophan metabolites may represent an avenue to globally available inexpensive drugs. Although administering analogues of endogenous compounds (e.g., isoproterenol as a congener of norepinephrine) are time‐honored therapeutic strategies, this direction has not been extensively evaluated for AD.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
D.F.W., F.S.M‐S., M.D.C., and V.C.M‐S. designed the experimental approach. V.C.M.‐S., A.R.M., V.F., C.J.B., H.D.S.C., V.C.‐S., D.F.W. performed in silico modelling, computational studies, and MM/QM calculations. M.D.C., S.J., F.W., E.L., L.F., A.J.M., A.Y., M.H., M.R., B.K., E.D.‐C., I.K., and D.F.W. completed all medicinal chemistry and synthetic organic chemistry, including synthesis, purification, and characterization of all small molecules. F.S.M.‐W., M.D.C., Y.W., L.P., Q.C., T.N.G., B.S., P.S., R.B., R.S.N., M.T., L.S., E.D‐C., and C.D.R. completed aspects of experimental in vitro work, including antibacterial, antiviral, ThT, CD, cell‐based, and Aβ oligomerization/aggregation studies. G.A.S. performed peptide NMR studies. M.R.V.S.M., P.L., S.C., V.G., and E.D.P. carried out peptide mass spectrometry studies. A.S., H.Z., R.B., and O.A. carried out in vivo studies. F.S.M.‐S., M.D.C., G.A.R., and S.D. completed histopathology studies. F.S.M.‐S., C.S., I.L., and D.F.W. edited the paper and assembled the manuscript.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
This research was supported by the BrightFocus Foundation, Canadian Institutes of Health Research, Alzheimer's Society of Canada, Ontario Brain Institute, Canada Foundation for Innovation, Krembil Foundation, Toronto General and Western Hospital Foundation, Sobey Family and Sobey Foundation, Weston Brain Institute, Michael Albert Garron Foundation, Dalhousie Medical Research Foundation, and the Atlantic Canada Opportunities Agency. D.F.W. acknowledges salary support from a Canada Research Chair, Tier 1.
Meier‐Stephenson FS, Meier‐Stephenson VC, Carter MD, et al. Alzheimer's disease as an autoimmune disorder of innate immunity endogenously modulated by tryptophan metabolites. Alzheimer's Dement. 2022;8:e12283. 10.1002/trc2.12283
REFERENCES
- 1. Kurkinen M. Alzheimer's trials: a cul‐de‐sac with no end in sight. Adv Clin Exp Med. 2021;30(7):653‐654. 10.17219/acem/139501 [DOI] [PubMed] [Google Scholar]
- 2. Frisoni GB, Altomare D, Thal DR, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci. 2022;23(1):53‐66. 10.1038/s41583-021-00533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luo JE, Li YM. Turning the tide on Alzheimer's disease: modulation of γ‐secretase. Cell Biosci. 2022;12(1):2. 10.1186/s13578-021-00738-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weaver DF. Amyloid beta is an early responder cytokine and immunopeptide of the innate immune system. Alzheimers Dement (N Y). 2020;6(1):e12100. 10.1002/trc2.12100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim ST, Weaver DF. Theoretical studies on Alzheimer's disease: structures of β‐amyloid aggregates. J Mol Struct (Theochem). 2000;527:127‐138. [Google Scholar]
- 6. Boman HG. Antibacterial peptides: basic facts and emerging concepts. J Intern Med. 2003;254:197‐215. [DOI] [PubMed] [Google Scholar]
- 7. Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005;16:625‐636. [DOI] [PubMed] [Google Scholar]
- 8. Williamson MP, Suzuki Y, Bourne NT, et al. Binding of amyloid beta‐peptide to ganglioside micelles is dependent on histidine‐13. Biochem J. 2006;397:483‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giulian D, Haverkamp LJ, Yu J, et al. The HHQK domain of beta‐amyloid provides a structural basis for the immunopathology of Alzheimer's disease. J Biol Chem. 1998;273:29719‐29726. [DOI] [PubMed] [Google Scholar]
- 10. Stephenson VC, Heyding RA, Weaver DF. The promiscuous drug concept with applications to Alzheimer's disease. FEBS Lett. 2005;579:1338‐1342. [DOI] [PubMed] [Google Scholar]
- 11. Soscia SJ, Kirby JE, Washicosky KJ et al. The Alzheimer's disease‐associated amyloid β‐protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kulon K, Valensin D, Kamysz W, et al. The His‐His sequence of the antimicrobial peptide demegen P‐113 makes it very attractive ligand for Cu2+. J Inorg Biochem. 2008;102:960‐972. [DOI] [PubMed] [Google Scholar]
- 13. Sensi SL, Granzotto A, Siotto M, et al. Copper and zinc dysregulation in Alzheimer's disease. Trends Pharmacol Sci. 2018;39:1049‐1063. [DOI] [PubMed] [Google Scholar]
- 14. McHenry AJ, Sciacca MF, Brender JR, et al. Does cholesterol suppress the antimicrobial peptide induced disruption of lipid raft containing membranes? Biochim Biophys Acta. 2012;1818:3019‐3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee JW, Lee YK, Yuk DY, et al. Neuro‐inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta‐amyloid generation. J Neuroinflammation. 2008;5:37‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klüter T, Fitschen‐Oestern S, Lippross S, et al. The antimicrobial peptide lysozyme is induced after multiple trauma. Mediators Inflamm. 2014;2014:303106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu K‐X, Chen S‐Q, Zhang H, et al. Ischemia/reperfusion upregulates beta‐defensin‐2 expression and causes acute lung injury in the rat. Injury. 2009;40:950‐955. [DOI] [PubMed] [Google Scholar]
- 18. Rogan MP, Geraghty P, Greene CM, et al. Antimicrobial proteins and polypeptides in pulmonary innate defence. Respir Res. 2006;7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kozłowska E, Wysokiński A, Brzezińska‐Błaszczyk E. Serum levels of peptide cathelicidin LL‐37 in elderly patients with depression. Psychiatry Res. 2017;255:156‐160. [DOI] [PubMed] [Google Scholar]
- 20. Leung FH, Thompson K, Weaver DF. Evaluating spousal abuse as a potential risk factor for Alzheimer's disease: rationale, needs and challenges. Neuroepidemiology. 2006;27;13‐16. [DOI] [PubMed] [Google Scholar]
- 21. Michikawa M, Gong JS, Fan QW, et al. A novel action of Alzheimer's amyloid beta‐protein (Abeta): oligomeric abeta promotes lipid release. J Neurosci. 2001;21:7226‐7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blennow K, Davidsson P, Wallin A, et al. Differences in cerebrospinal fluid gangliosides between “probable Alzheimer's disease” and normal aging. Aging Clin Exp Res. 1992;4:301‐306. [PubMed] [Google Scholar]
- 23. Zha Q, Ruan Y, Hartmann T, et al. GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Mol Psychiatry. 2004;9:946‐952. [DOI] [PubMed] [Google Scholar]
- 24. Yuki N, Susuki K, Koga M, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain‐Barre syndrome. Proc Natl Acad Sci USA. 2004;101:11404‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid‐like fibrils. Proc Natl Acad Sci USA. 2010;107:3487‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krause D, Suh HS, Tarassishin L, et al. The tryptophan metabolite 3‐hydroxyanthranilic acid plays anti‐inflammatory and neuroprotective roles during inflammation: role of hemeoxygenase‐1. Am J Pathol. 2011;179:1360‐1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dürk T, Panther E, Müller T, et al. 5‐Hydroxytryptamine modulates cytokine and chemokine production in LPS‐primed human monocytes via stimulation of different 5‐HTR subtypes. Int Immunol. 2005;17:599‐606. [DOI] [PubMed] [Google Scholar]
- 28. Sharma R, Razdan K, Bansal Y, Kuhad A. Rollercoaster ride of kynurenines: steering the wheel towards neuroprotection in Alzheimer's disease. Expert Opin Ther Targets. 2018;22:849‐867. [DOI] [PubMed] [Google Scholar]
- 29. Huang YS, Ogbechi J, Clanchy FI, et al. IDO and kynurenine metabolites in peripheral and CNS disorders. Front Immunol. 2020;11:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu D, Tao BB, Yang YY, et al. The IDO inhibitor coptisine ameliorates cognitive impairment in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2015;43:291‐302. [DOI] [PubMed] [Google Scholar]
- 31. Cady SG, Sono M. 1‐Methyl‐DL‐tryptophan, beta‐(3‐benzofuranyl)‐DL‐alanine (the oxygen analog of tryptophan), and beta‐3‐benzo[b]thienyl‐DL‐alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3‐dioxygenase. Arch Biochem Biophys. 1991;291:326‐33. [DOI] [PubMed] [Google Scholar]
- 32. Sugimoto H, Oda S, Otsuki T, et al. Crystal structure of human indoleamine 2,3‐dioxygenase: catalytic mechanism of O2 incorporation by a heme‐containing dioxygenase. Proc Natl Acad Sci USA. 2006;103:2611‐2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ano Y, Yoshino Y, Uchida K, Nakayama H. Preventive effects of tryptophan‐methionine dipeptide on neural inflammation and Alzheimer's pathology. Int J Mol Sci. 2019;20:3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scherzer‐Attali R, Pellarin R, Convertino M, et al. Complete phenotypic recovery of an Alzheimer's disease model by a quinone‐tryptophan hybrid aggregation inhibitor. PLoS One. 2010;5:e11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matta CF, Arabi AA, Weaver DF. The bioisosteric similarity of the tetrazole and carboxylate anions: clues from the topologies of the electrostatic potential and of the electron density. Eur J Med Chem. 2010;45:1868‐1872. [DOI] [PubMed] [Google Scholar]
- 36. Zheng Y, Stafford PM, Stover KR, et al. A series of 2‐((1‐Phenyl‐1H‐imidazol‐5‐yl)methyl)‐1H‐indoles as indoleamine 2,3‐dioxygenase 1 (IDO1) inhibitors. ChemMedChem. 2021;16(14):2195‐2205. [DOI] [PubMed] [Google Scholar]
- 37. Doebner O. Ueber die der Sorbinsäure homologen, ungesättigten Säuren mit zwei Doppelbindungen. Berichte der deutschen chemischen Gesellschaft. 1902;35:1136–36. [Google Scholar]
- 38. Caspi R, Altman T, Dreher K, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40(Database issue):D742‐D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kolaj I, Imindu Liyanage S, Weaver DF. Phenylpropanoids and Alzheimer's disease: a potential therapeutic platform. Neurochem Int. 2018;120:99‐111. [DOI] [PubMed] [Google Scholar]
- 40. Kolaj I, Wang Y, Ye K, et al. Ferulic acid amide derivatives with varying inhibition of amyloid‐β oligomerization and fibrillization. Bioorg Med Chem. 2021;43:116247. 10.1016/j.bmc.2021.116247 [DOI] [PubMed] [Google Scholar]
- 41. Koshiguchi M, Komazaki H, Hirai S, Egashira Y. Ferulic acid suppresses expression of tryptophan metabolic key enzyme indoleamine 2, 3‐dioxygenase via NFκB and p38 MAPK in lipopolysaccharide‐stimulated microglial cells. Biosci Biotechnol Biochem. 2017;81(5):966‐971. [DOI] [PubMed] [Google Scholar]
- 42.La Camera S, Gouzerh G, Dhondt S, et al. Metabolic reprogramming in plant innate immunity: the contributions of phenylpropanoid and oxylipin pathways. Immunol Rev. 2004;198:267‐84. [DOI] [PubMed] [Google Scholar]
- 43. Das S, Das K. Anti‐inflammatory responses of resveratrol. Inflamm Allergy Drug Targets. 2007;6:168‐173. [DOI] [PubMed] [Google Scholar]
- 44. Joo Y, Kim HS, Woo RS, et al. Mefenamic acid shows neuroprotective effects and improves cognitive impairment in in vitro and in vivo Alzheimer's disease models. Mol Pharmacol. 2006;69:76‐84. [DOI] [PubMed] [Google Scholar]
- 45. Wang Z, Wang Y, Pasangulapati JP, et al. Design, synthesis, and biological evaluation of furosemide analogs as therapeutics for the proteopathy and immunopathy of Alzheimer's disease. Eur J Med Chem. 2021;222:113565. 10.1016/j.ejmech.2021.113565 [DOI] [PubMed] [Google Scholar]
- 46. Arshavsky YI. Alzheimer's disease: from amyloid to autoimmune hypothesis. Neuroscientist. 2020;26(5‐6):455‐470. [DOI] [PubMed] [Google Scholar]
- 47. D'Andrea MR. Add Alzheimer's disease to the list of autoimmune diseases. Med Hypotheses. 2005;64(3):458‐463. [DOI] [PubMed] [Google Scholar]
- 48. Bonaccorso S, Lin A, Song C, et al. Serotonin‐immune interactions in elderly volunteers and in patients with Alzheimer's disease (DAT): lower plasma tryptophan availability to the brain in the elderly and increased serum interleukin‐6 in DAT. Aging Clin Exp Res. 1998;10:316‐323. [DOI] [PubMed] [Google Scholar]
- 49. Murphy F, Smith K, Cowen P, et al. The effects of tryptophan depletion on cognitive and affective processing in healthy volunteers. Psychopharmacology. 2002;163:42‐53. [DOI] [PubMed] [Google Scholar]
- 50. Doifode T, Giridharan VV, Generoso JS, et al. The impact of the microbiota‐gut‐brain axis on Alzheimer's disease pathophysiology. Pharmacol Res. 2021;164:105314. 10.1016/j.phrs.2020.105314 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information