

Abstract
Programmable C•G-to-G•C base editors (CGBEs) have broad scientific and therapeutic potential, but their editing outcomes have been difficult to predict and their editing efficiency and product purity are often low. We describe a suite of engineered CGBEs paired with machine learning models to enable efficient, high-purity C•G-to-G•C base editing. We performed a CRISPRi screen targeting DNA repair genes to identify factors that affect C•G-to-G•C editing outcomes and used these insights to develop CGBEs with diverse editing profiles. We characterized ten promising CGBEs on a library of 10,638 genomically integrated target sites in mammalian cells and trained machine learning models that accurately predict the purity and yield of editing outcomes (R=0.90) using these data. These CGBEs enable correction to the wild-type coding sequence of 546 disease-related transversion single-nucleotide variants with >90% precision (mean 96%) and up to 70% efficiency (mean 14%). Computational prediction of optimal CGBE-sgRNA pairs enables high-purity transversion base editing at >4-fold more target sites than can be achieved using any single CGBE variant.
Single-nucleotide variants (SNVs) represent approximately half of currently known human pathogenic gene variants1. Base editors, fusions of programmable DNA-binding proteins with base-modifying enzymes, enable conversion of individual target nucleotides in the genome2–10. The two major classes of base editors are cytosine base editors (CBEs), which convert C•G to T•A, and adenine base editors (ABEs), which convert A•T to G•C2,3,8. CBEs and ABEs can install transition mutations with high efficiency and product purity (the fraction of all edited alleles that contain only the desired edit), but in general, cannot efficiently install transversion mutations including C•G to G•C2,5,11,12.
We previously demonstrated that CBE editing byproducts, including C•G-to-G•C or C•G-to-A•T transversion outcomes, are inhibited by knockout of cellular uracil DNA N-glycosylase (UNG) or by fusion of uracil glycosylase inhibitor (UGI)2,7,8,11,12, suggesting that transversion byproducts result from an abasic intermediate that is generated by UNG-catalyzed excision of deaminated target cytosines (Fig. 1a). Consistent with this model, firstgeneration C•G-to-G•C base editors (CGBEs) were CBE derivatives that lack UGI domains11. These CGBEs, including editors with fusions to UNG and other DNA-repair proteins13–16, can provide efficient C•G-to-G•C editing but only at a minority of tested target sites with few criteria to identify sites amenable to CGBE editing13–15.
Figure 1. Development of prototype C•G-to-G•C base editors.
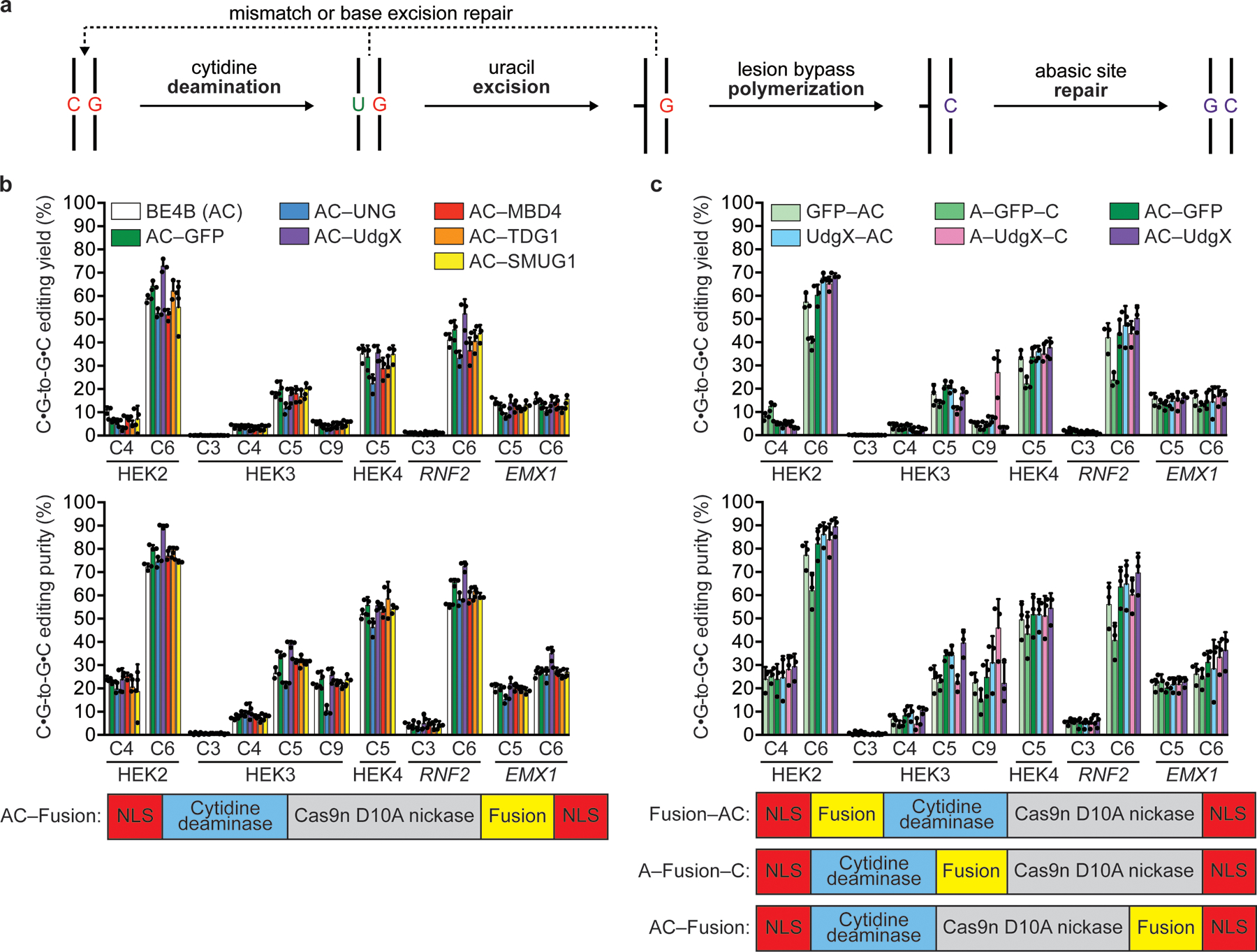
(a) Potential pathway for C•G-to-G•C conversion. (b) C•G-to-G•C editing outcomes in HEK293T cells for C-terminal fusions of DNA glycosylases to BE4B (AC, APOBEC1 cytidine deaminase–Cas9 nickase). (c) Different fusion protein architectures lead to different C•G-to-G•C editing properties in HEK293T cells at the HEK3 locus for the Apo-UdgX-Cas9n (AXC) architecture. Values and error bars reflect the mean and standard deviation of three biological replicates, shown as individual data points. HEK2=HEK site 2; HEK3=HEK site 3; HEK4=HEK site 4. C4, C6, and similar annotations indicate the in-window target nucleotides where the SpCas9 PAM is at positions 21–23.
Previously, we used libraries containing thousands of genomically integrated target sites and corresponding guide RNAs in mammalian cells to comprehensively characterize CBE and ABE base editing profiles. We used these data to train machine learning models (collectively named BE-Hive) that learned the sequence determinants driving CBE and ABE base editing outcomes12,17. We envisioned that broad characterization of the sequence determinants of CGBE editing outcomes could enable accurate prediction of editing efficiencies and product purities, and thus facilitate the broader use of CGBEs.
Here, we performed a focused CRISPR interference (CRISPRi) screen to identify DNA repair genes that impact cytosine base editing efficiency and purity. Guided by these data, we constructed various fusions proteins containing deaminases and Cas proteins fused to DNA repair components to engineer novel CGBEs with promising C•G-to-G•C editing activities. We characterized ten such CGBEs with diverse editing profiles using a “comprehensive context library” of 10,638 genomically integrated, highly variable target sites in mouse embryonic stem cells (mESCs)12. We used the resulting data to train machine learning models that successfully predict CGBE editing efficiency, purity, and bystander editing patterns with high accuracy (CGBE-Hive), enabling reliable identification of CGBE variants and target sites that together support high-purity C•G-to-G•C editing. Moreover, we show that editing activity is predicted with substantially higher accuracy by deep learning models compared to simpler models, indicating that CGBE-Hive has learned complex sequence features that play important roles in determining C-to-G editing activity. Notably, 247 cytosines predicted by CGBE-Hive to be edited by a CGBE with >80% C•G-to-G•C editing purity were indeed edited in mammalian cell experiments with an average of 83% purity.
The panel of CGBEs in this study offer diverse editing profiles that collectively expand the sequence landscape amenable to high-quality C•G-to-G•C editing by up to 4.1-fold over the number predicted to be amenable to editing by any single CGBE. Finally, we demonstrate CGBE-mediated correction of 546 disease-associated single-nucleotide variants (SNVs) with >90% precision among the resulting edited amino acid sequences. These findings advance our understanding of transversion base editing outcomes and provide new CGBEs that improve the scope and utility of base editing.
Results
Exploring the activity of DNA glycosylases in C•G-to-G•C transversion outcomes
Previous work suggested that excision of uracil from genomic DNA to generate an abasic lesion followed by error-prone polymerase activity on the strand opposite the abasic site results in C•G-to-G•C and C•G-to-A•T transversion outcomes (Fig. 1a)2,11,16. Motivated by this model, we sought to develop C•G-to-G•C base editors that enhanced uracil excision at CBE-edited nucleotides. We started with a CBE architecture lacking UGI (BE4B) (bpNLS–APOBEC1–Cas9 D10A–bpNLS; abbreviated AC), similar to other reported CGBEs13–15.
We fused a variety of known uracil excising and binding enzymes to the C-terminus of the BE4B (AC) scaffold and assessed the frequency of C•G-to-G•C edits across five genomic loci in HEK293T cells (Fig. 1b). Several glycosylases (i.e., SMUG1, MBD4, and TDG2) did not alter editing outcomes, and fusion to UNG led to a reduction of C•G-to-G•C editing yield and purity at three out of five targeted sites, consistent with a recent report13. Nevertheless, we found that fusion of a UNG orthologue from Mycobacterium smegmatis (UdgX) moderately improved C•G-to-G•C product purity by 1.2-fold on average18–20, with the largest improvement at the RNF2 locus (56±0.8% with BE4B to 72±2.1% with AC–UdgX; p=0.0002, Student’s two-sided t-test) and significant changes observed at HEK site 2 C6, HEK site 3 C5, and EMX1 C6 (p<0.01, Student’s two-sided t-test). However, we observed only modest changes to editing yield (1.1-fold relative to BE4B at the most efficiently edited C across the five tested genomic loci). These observations suggested that fusion partners may enhance C•G-to-G•C transversion base editing outcomes.
Next, we asked whether the orientation of the glycosylase fusion impacts editing outcomes. We constructed BE4B (AC) fusion variants with either UdgX (abbreviated X) or GFP in three orientations: at either the N- or C-terminus (e.g., XAC or ACX) or between the deaminase and Cas9 (e.g., AXC). We observed that C•G-to-G•C editing was similar or slightly improved for UdgX fusions compared to N- and C-terminal GFP fusions (Fig. 1c). However, the editing efficiency and purity of AXC was modestly higher than that of the best GFP fusion at a majority of sites (four out of five sites for efficiency; three out of five sites for purity). We chose to advance the AXC architecture since it offered similar or better performance than the XAC and ACX variants at these test loci.
CRISPRi screen for determinants of base editing outcomes
Next, we investigated whether other DNA repair or translesion synthesis factors impact C•G-to-G•C editing outcomes of AXC. We observed no significant changes in editing purity of AXC in individual UNG, APE1/APEX1, MLH1, REV1 knockout cell lines, and direct AXC fusions to mammalian polymerase domains did not consistently improve editing outcomes (Supplementary Figs. 1-2; Supplementary Discussion 1). We thus performed a much broader search for modulators of cytosine transversion editing by performing two high-throughput genetic screens.
Using a recently developed screening platform21 capable of reading out DNA repair outcomes by DNA sequencing (Fig. 2a-b, Supplementary Fig. 3a), we investigated how knockdown of each of 476 genes, a set enriched for regulators of DNA repair, impacts the activity of BE1 (deaminase–dCas9) and BE4B (AC) editors. Briefly, we transduced an sgRNA library (1,513 gene-targeting sgRNAs and 60 non-targeting controls, Supplementary Table 1) into HeLa cells stably expressing the CRISPRi effector dSpCas9–KRAB22. After allowing 5 days for gene knockdown, we transfected the cells with plasmids encoding SaCas9-based CBEs (either SaCas9-BE1 or SaCas9-BE4B) and an SaCas9 sgRNA that targets a sequence adjacent to the genomically integrated SpCas9 sgRNA sequences. Notably, we used SaCas9-based CBEs to avoid guide RNA exchange between the base editors and CRISPRi machinery. A key aspect of this approach was that the proximity of the target site and CRISPRi sgRNA enabled these features to be read out together by paired-end DNA sequencing, thus linking editing outcomes to CRISPRi perturbation identities (Fig. 2a). To prepare samples for sequencing, we isolated genomic DNA from treated cells, affixed unique molecular identifiers (UMIs) to DNA fragments containing both the sgRNA expression cassettes and edited target sites, and sequenced the linked sgRNA, target sites, and UMI sequences. Comparing frequencies of editing outcomes from each CRISPRi sgRNA with those from non-targeting sgRNAs (examples in Fig. 2b, Supplementary Fig. 3a) then identified genes that promote or suppress various editing outcomes (Supplementary Table 2).
Figure 2. CRISPRi knockdown screen across 476 genes enriched for those with roles in DNA repair identifies candidate regulators of C•G-to-G•C editing.
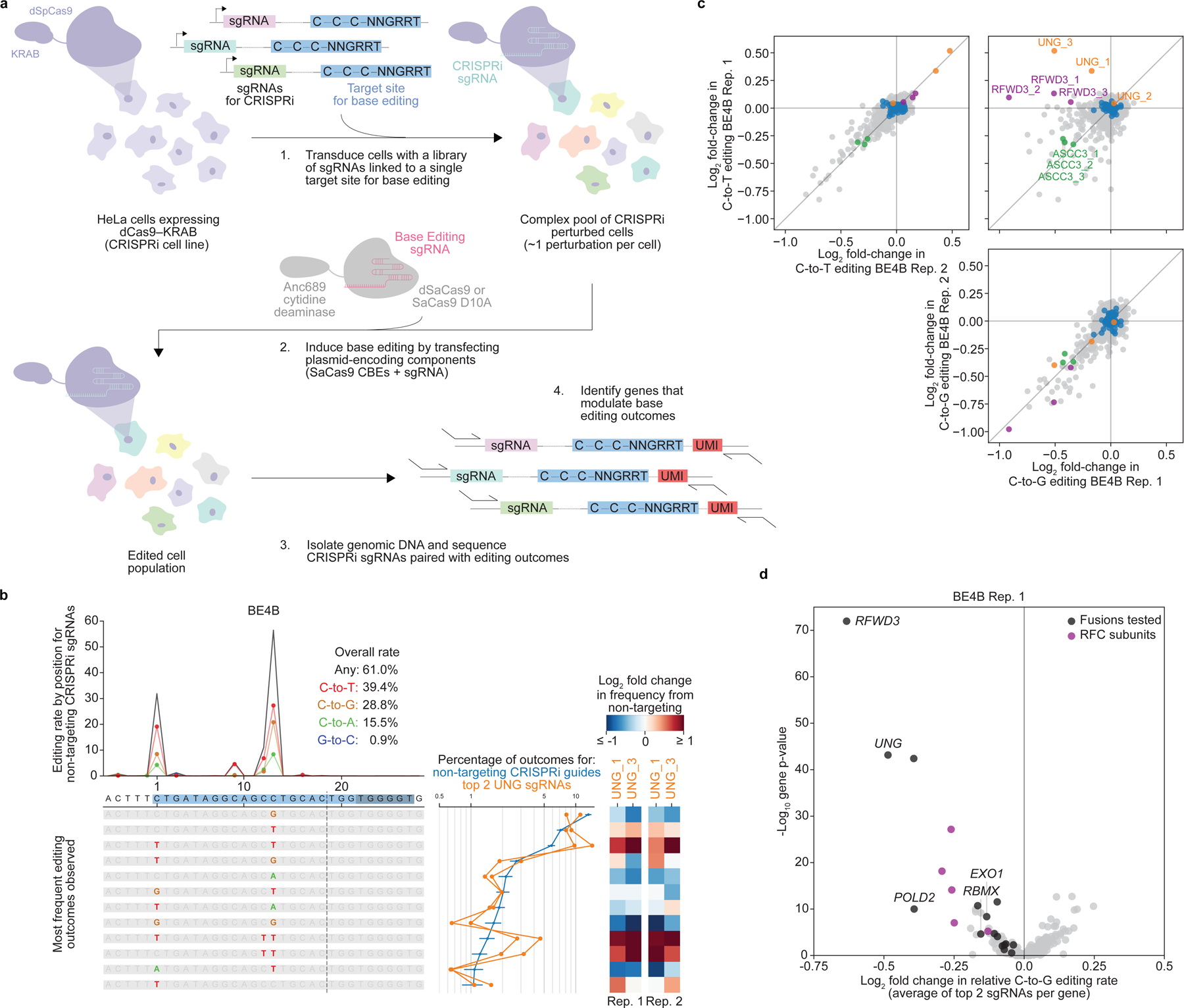
(a) Schematic of screen design. (b). Summary of base editing outcomes in BE4B (also AC) screen. Bottom left – all editing outcomes containing only point mutations present at >=1% frequency for non-targeting CRISPRi guide RNAs. Line plots above the individual outcomes show the total editing frequency (black line) and the frequencies of each single base edit (C-to-T=red, C-to-G=brown, C-to-A=green, and G-to-C=blue lines) at each position. Line plots to the right show frequencies of outcomes for specific CRISPRi guide RNAs (blue - average of all non-targeting guide +/− standard deviation across individual non-targeting guide RNAs; orange - top 2 most active UNG guide RNAs). Heatmaps show log2 fold changes in outcome frequencies for top 2 UNG guide RNAs relative to non-targeting guide RNAs. (c) Log2 fold changes in frequency of outcomes containing C-to-T or C-to-G edits for each CRISPRi guide compared to non-targeting guide RNAs. Upper left - comparison of changes in C-to-T editing between two biological replicates. Lower right - comparison of changes in C-to-G editing between replicates. Upper right - comparison of changes in C-to-G editing to changes in C-to-T editing in replicate 1. All guide RNAs with at least 500 recovered UMIs in each replicate are plotted. Blue dots: individual non-targeting guide RNAs, orange dots: UNG guide RNAs, green dots: ASCC3 guide RNAs, red dots: RFWD3 guide RNAs, grey dots: all other guide RNAs. (d) Effects of gene knockdown on relative C-to-G editing frequencies in BE4B screen. Each dot represents a gene, with the x-value representing the average of the two strongest Log2 fold changes in normalized C-to-G editing for guide RNAs targeting the gene from the average of all non-targeting guide RNAs, and the y-value representing a gene-level p-value summarizing the combined statistical significance of all guide RNAs targeting each gene (two-sided, uncorrected for multiple comparisons). Rep.=replicate.
Consistent baseline activity of BE1 and BE4B in the screens enabled quantitation of editing differences driven by CRISPRi sgRNAs (Fig. 2, Supplementary Fig. 3, Supplementary Fig. 4). To evaluate differences in point mutations, we calculated the effects of all CRISPRi sgRNAs on the frequencies of two major categories: outcomes containing any C•G-to-T•A point mutation and outcomes containing any C•G-to-G•C point mutation (Fig. 2c). For both classes, the effects of individual CRISPRi sgRNAs were consistent between replicates (Fig. 2c, upper left and lower right panels). Comparison between classes though revealed that some CRISPRi sgRNAs showed different effects on C•G-to-T•A versus C•G-to-G•C outcomes (Fig. 2c, upper right panel), indicating that specific genes influence partitioning between these outcomes. In the BE4B screen, the clearest differential effects resulted from sgRNAs targeting UNG (Fig. 2b, c). Consistent with the effects of UGI fusions and UNG loss2,11, UNG knockdown increased frequencies of C•G-to-T•A editing while decreasing frequencies of C•G-to-G•C editing. Notably, the effects of UNG repression on BE1 editing were not as significant or straightforward (Supplementary Fig. 3a,c), perhaps reflecting differences in how nicked versus unnicked target substrates are processed (Fig. 2b and Supplementary Fig. 3a).
One advantage to screening with sequencing-based readouts was that we could detect changes to a diverse range of editing products. For example, we also observed that CRISPRi-mediated depletion of double-strand breaks (DSB) repair genes affect the frequency of rare indels caused by base editing, though these pathway-phenotype relationships were not always straightforward (Supplementary Fig. 4a, Supplementary Table 2). Indeed, while knockdown of HDR factors BRCA1, BRCA2, and PALB2 increased AC-generated deletions, depletion of the HDR gene BLM decreased them. Interestingly, depletion of BRCA2 was also among the strongest reducers of C•G-to-T•A editing outcomes (Supplementary Fig. 4b). We also identified genes that affect the base editing window (Supplementary Figs. 4c, 5; Supplementary Discussion 2).
To identify genes that specifically promoted C•G-to-G•C editing, we calculated the relative fraction of outcomes containing any C•G-to-G•C edit among outcomes containing any point mutation for each CRISPRi sgRNA (Fig. 2d and Supplementary Fig. 4d). The gene whose knockdown most significantly reduced the C•G-to-G•C editing fraction compared to non-targeting sgRNAs was RFWD3, an E3 ligase with multiple roles in DNA repair recently identified as required for successful translesion synthesis across a variety of genomic lesions23. Other hits included UNG; multiple subunits of the replicative polymerase POLD and replicative clamp loader RFC; EXO1; translesion polymerases REV1 and REV3L; and RAD18, an E3 ubiquitin ligase involved in translesion synthesis (Supplementary Table 2). The different phenotypes for REV1 knockdown versus our individual knockout cell line may arise from compensatory mechanisms that could alter DNA repair outcomes in cells lacking REV1. We also identified genes whose knockdown reduced frequencies of both C•G-to-T•A and C•G-to-G•C base editing for both BE1 and BE4B (Supplementary Fig. 4e), including ASCC3, which may act by affecting accessibility of the target locus, a known determinant of base editing efficiency2,3,8. Together, these screen results suggest important roles for DNA replication processes, especially translesion synthesis, in modulating C•G-to-G•C base editing outcomes.
CBE fusion proteins can alter C•G-to-G•C transversion outcomes
To further advance the development of CGBEs, we generated new CGBE candidates by fusing AXC, the prototype CGBE described above, to proteins nominated by our CRISPRi screens. These included those encoded by genes that reduced C•G-to-G•C editing following knockdown, including DDX1, EXO1, POLD1, POLD2, POLD3, RAD18, RBMX, REV1, RFWD3, and TIMELESS, and several additional genes involved in DNA polymerization, some of which also affected editing outcomes in the CRISPRi screen (PCNA, POLH, POLK, UBE2I, and UBE2T, Supplementary Table 2).
We fused each of these proteins to the N- or C-terminus of AXC to assess their effect on C•G-to-G•C editing efficiency or purity and assessed their editing performance at five genomic loci in HEK293T cells. Three proteins increased C•G-to-G•C editing purity when fused to the N-terminus of AXC (Supplementary Fig. 6a): DNA polymerase D2 (POLD2), exonuclease 1 (EXO1), and RNA binding motif protein X-linked (RBMX). Editing improvements for fused constructs varied by site. The most pronounced effects were observed at the RNF2 locus, where editing purity significantly improved from 54±1.4% with AXC to 73±0.4% with RBMX–AXC, 74±1.4% for EXO1–AXC, and 77±0.8% for POLD2–AXC (p < 0.001, Student’s two-sided t-test). Marginal improvements in purity were also observed at HEK site 2, HEK site 3, and HEK site 4 loci. At RNF2 we also observed a significant increase in editing yield from 43±2.4% with AXC to 50±5.2% with RBMX–AXC, 53±3.6% with EXO1–AXC, and 55± 5.5% for POLD2–AXC (p < 0.05, Student’s two-sided t-test). C-terminal fusions typically did not perform as well as N-terminal fusions (Supplementary Data 1).
Encouraged by these improvements, we developed additional candidate CGBEs containing RBMX, EXO1, POLD2, and UdgX as fusions to AXC. We compared single and dual pairwise fusion architectures for these components, testing N- and C-terminal dual fusions as well as tandem N terminal fusions (N-, N-) using 32-residue linkers identified in a linker-testing experiment for these constructs (Supplementary Fig. 7). From a total of 28 single- and dual-fusion proteins tested, the four dual fusion architectures POLD2–deaminase–UdgX–nCas9–RBMX, POLD2–deaminase–UdgX–nCas9–UdgX, UdgX–deaminase–UdgX–nCas9–UdgX, and UdgX–deaminase–UdgX–nCas9–RBMX further increased C•G-to-G•C editor yield and purity at some sites (on average, by +10% and +13%, respectively) compared to single fusion architectures across nine cytosines in five genomic loci (Supplementary Fig. 6b).
Collectively, these results indicate that CGBEs, including fusions to proteins identified in the CRISPRi screen, can affect C•G-to-G•C editing outcomes in a site-dependent manner. Some base editing applications may prioritize protein size over other base editing characteristics. We therefore explored the use of trans-splicing split-inteins as a means to reduce the size of large CGBEs into two smaller protein components24, and observed no changes in editing outcomes of split-CGBEs compared to their full-length counterparts (Supplementary Fig. 8). When necessary, these split CGBE variants may support favorable cytosine transversion outcomes without requiring the expression of full-length proteins.
Base editor deaminase and Cas9 domains bias repair outcomes
We next sought to understand how different deaminase domains affect C•G-to-G•C editing in the AXC architecture. Since the base editing window may influence cytosine transversion outcomes2,11,12, we examined a panel of catalytically impaired deaminases that support different CBE editing windows25 and observed an increase in C•G-to-G•C editing purity at three of five tested loci (Fig. 3a). The APOBEC1 R126E R132E (EE)25 deaminase showed the greatest improvement, averaging 1.2-fold higher product purity at HEK site 2, HEK site 3, and RNF2. Editing yield with these deaminase alternatives varied by locus. We observed similar or reduced editing yield compared to AXC at four out of five loci that is likely due to the lower catalytic activity of these deaminases, though reduced yield did not correlate with altered C•G-to-G•C purity. Editing yield by EE-AXC at the RNF2 locus significantly improved (AXC=52±3.2% vs. EE-AXC=66±3.5%, p=0.007, Student’s two-sided t-test).
Figure 3. Effect of varying the cytidine deaminase and Cas9 components of CGBEs on C•G-to-G•C editing outcomes in HEK293T cells.
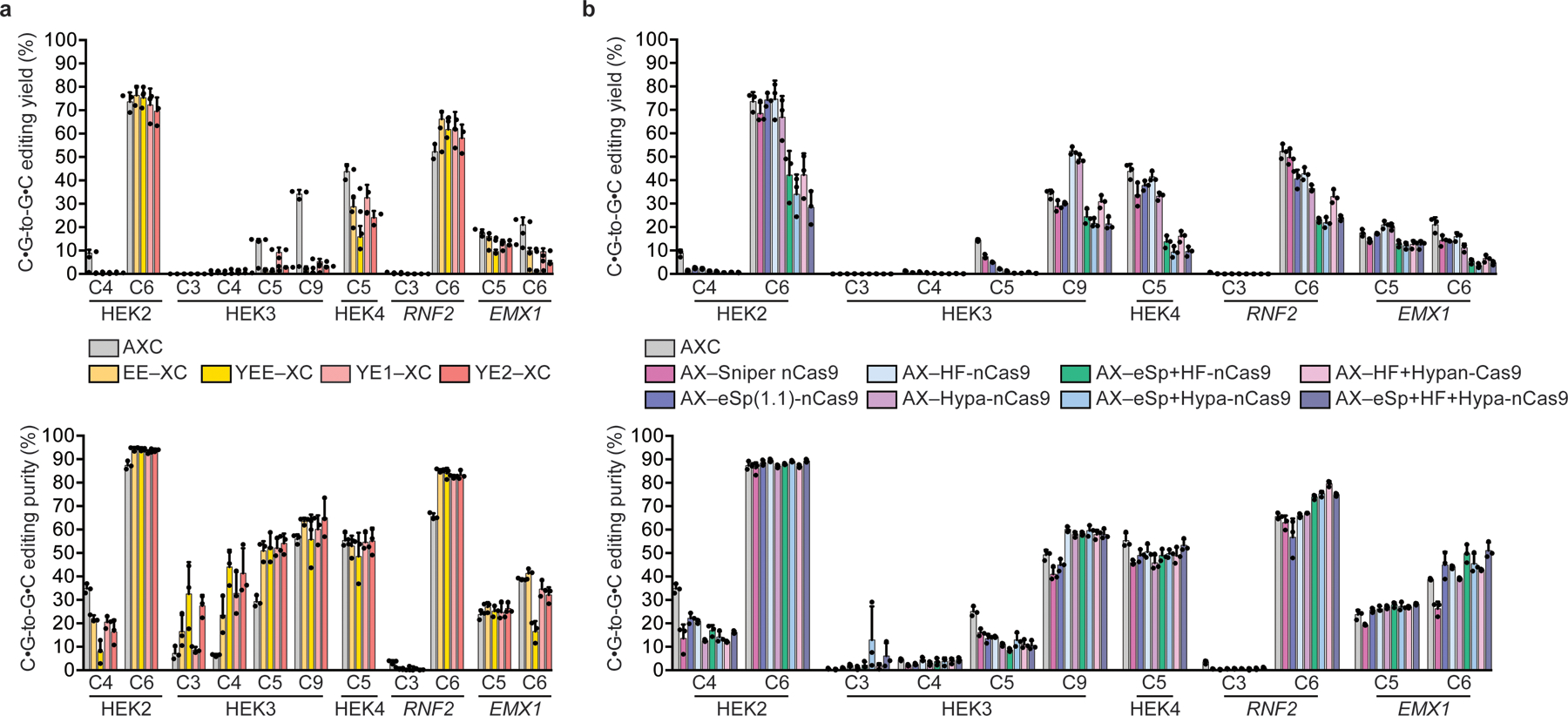
(a) C•G-to-G•C editing outcomes for catalytically impaired, narrow-window cytidine deaminases show higher editing purity at HEK2 and RNF2. (b) C•G-to-G•C editing outcomes for high-fidelity Cas9 variants show altered editing windows and improved CGBE performance at some positions. “Cas9” represents the Cas9 D10A nickase variant of each Cas effector. Values and error bars reflect the mean and standard deviation of three biological replicates, shown as individual data points. HEK2=HEK site 2; HEK3=HEK site 3; HEK4=HEK site 4. C4, C6, and similar annotations indicate the in-window target nucleotides where the SpCas9 PAM is at positions 21–23.
We also hypothesized that changes to the Cas9 binding domain of CGBEs could alter editing windows and C•G-to-G•C editing outcomes by altering the competition between Cas9 and repair machinery for access to the target locus. We assessed AXC editors that use Cas9 variants with different binding kinetics, including new variants with combinations of previously reported Cas9 mutations (Fig. 3b)26–29. AX–HF-nCas9 substantially improved C•G-to-G•C editing at the C9 position of the HEK site 3 locus, increasing yield (AXC=34±1.9% vs. AX–HF-nCas9=52±1.7%,) and purity (AXC=49±2.2% vs. AX–HF-nCas9=60±1.2%) (p < 0.005 for both, Student’s two-sided t-test) (Fig. 3b). AX-Hypa-nCas9 showed similar effects but AX-HF-nCas9 typically performed modestly better. These results suggest Cas protein binding parameters can affect C•G-to-G•C editing yield and purity of CGBEs at some target loci.
The balance of editing yield and purity among candidate CGBEs and the variability in these two measures across different loci suggests that different target sites will be best edited by different CGBEs. Therefore, a suite of CGBEs with different kinetics and substrate preferences would likely enable efficient and high-purity C•G-to-G•C editing across a broader range of diverse target sequences than could be achieved by any single CGBE variant alone.
Combining deaminase, Cas9 domain, and DNA repair fusion proteins into new CGBEs
We integrated the above findings from varying protein fusions, deaminases, and Cas domains into improved CGBEs. We evaluated the four most promising dual-fusion AXC editors (POLD2–AXC–RBMX, POLD2–AXC–UdgX, UdgX–AXC–RBMX, and UdgX–AXC–UdgX), four single-fusion AXC editors (POLD2–AXC, RBMX–AXC, EXO1–AXC, and UdgX–AXC), AXCs with deaminase variants of those same editors, and direct deaminase–nCas9 CGBEs without additional fusion proteins. The five cytidine deaminases tested in these 10 CGBE architectures included rAPOBEC1, EE, Anc689 (ancestrally-reconstructed APOBEC1 node 68930), eA3A, and eA3A-T31A12. In addition, we tested both SpCas9 nickase and HF-Cas9 nickase variants. In total, we evaluated 95 candidate CGBEs at eight genomic loci in HEK293T cells.
No single CGBE outperformed all other candidates at all sites (Fig. 4a). To identify a set of the most promising CGBEs, we selected 32 editors that demonstrated improved C•G-to-G•C editing outcomes at some sites for testing at eight additional genomic loci (Fig. 4b). We used these data to identify ten CGBEs with high purity, yield, and maximally distinct activities at different endogenous loci using quadratic programming and hierarchical clustering (Supplementary Methods): Anc689–nCas9, UdgX–Anc689–UdgX–nCas9–RBMX, eA3A–nCas9, RBMX–eA3A–UdgX–HF-nCas9, RBMX–eA3A–UdgX–nCas9, EE–nCas9, UdgX–EE–UdgX–nCas9–UdgX, APOBEC1–nCas9, UdgX–APOBEC1–UdgX–HF-nCas9, and POLD2–APOBEC1–UdgX–nCas9–UdgX.
Figure 4. Novel engineered CGBEs with various DNA repair proteins, deaminases, Cas proteins, and architectures offer diverse editing performance on different target sites.
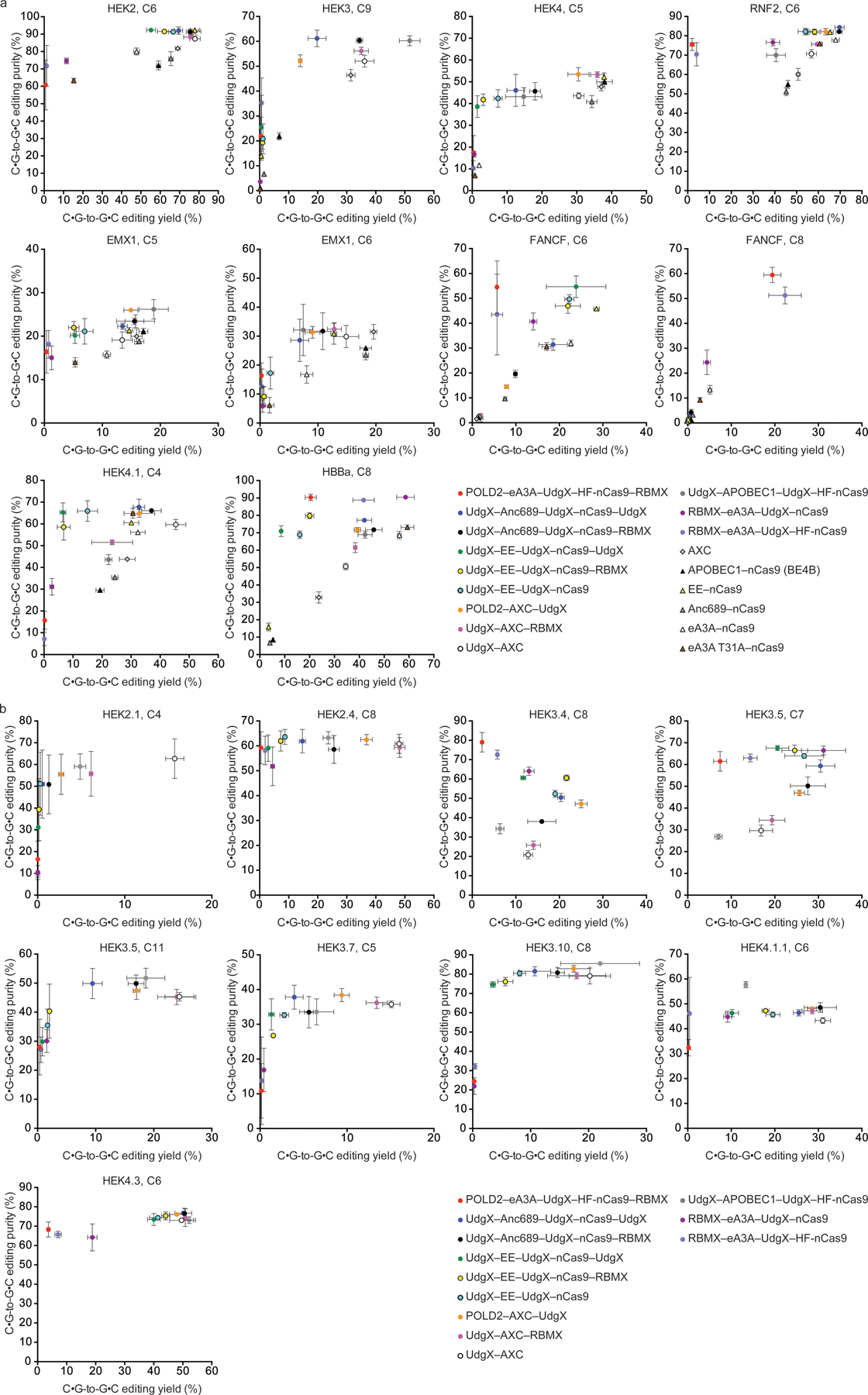
(a) C•G-to-G•C editing performance of CGBEs at eight genomic loci in HEK293T cells. (b) Further characterization of C•G-to-G•C editing outcomes for 12 variants from (a) at various genomic loci in HEK293T cells. Values and error bars reflect the mean and standard deviation of three biological replicates. HEK2=HEK293T cells site 2; HEK3=HEK293T cells site 3; HEK4=HEK293T cells site 4. C nucleotide annotations indicate the target nucleotide positions in the protospacer, where the SpCas9 PAM is at positions 21–23. Editing efficiencies, product purities, and indel frequencies for constructs that were tested but not shown in this figure can be found in Supplementary Data 1.
To test how this set of CGBEs performed in human cell lines other than HEK293T cells, we assayed the ability of each of these CGBEs to edit five target genomic sites in K562, U2OS, and HeLa (Supplementary Fig. 9). We observed that while CGBE outcomes vary modestly by cell type, the top-performing CGBE variants for each tested site were generally the same in all three additional cell lines. These results indicate that deaminase, Cas protein, and DNA repair protein variants can improve C•G-to-G•C editing in across different cell types.
Target library characterization of CGBEs
We observed that different target loci were best edited by different CGBEs, indicating that diverse CGBE sequence preferences may be strong determinants of C•G-to-G•C editing efficiency and purity. Previously, we used high-throughput analysis of base editing outcomes at thousands of genomically integrated target sequences to better understand CBE and ABE sequence-activity relationships, and we used these data to train machine learning models that facilitate the selection of target sequences amenable to C•G-to-G•C conversion by CBEs12. We envisioned that comprehensive characterization of our top ten promising and diverse CGBEs could similarly aid in the selection of targets amenable to efficient and high-purity C•G-to-G•C editing by specific CGBEs.
We characterized each of the ten CGBEs using a high-throughput genome-integrated library assay of 10,638 matched sgRNA and target pairs in mESCs, previously referred to as the “comprehensive context library”12. The target sequences in this library cover all possible sequence contexts surrounding the edited C•G with minimal sequence bias (Fig. 5a, Supplementary Methods). To detect editing outcomes with high sensitivity, we maintained an average coverage of ≥300x per library member throughout the course of the experiment and an average sequencing depth of ≥4,000x per target. We collected two biological replicates per CGBE characterization experiment. We previously validated that the library assay data has strong consistency between biological replicates and is concordant with data from base editing endogenous genomic loci12,31.
Figure 5. Target library characterization and machine learning modeling of 10 CGBE variants.
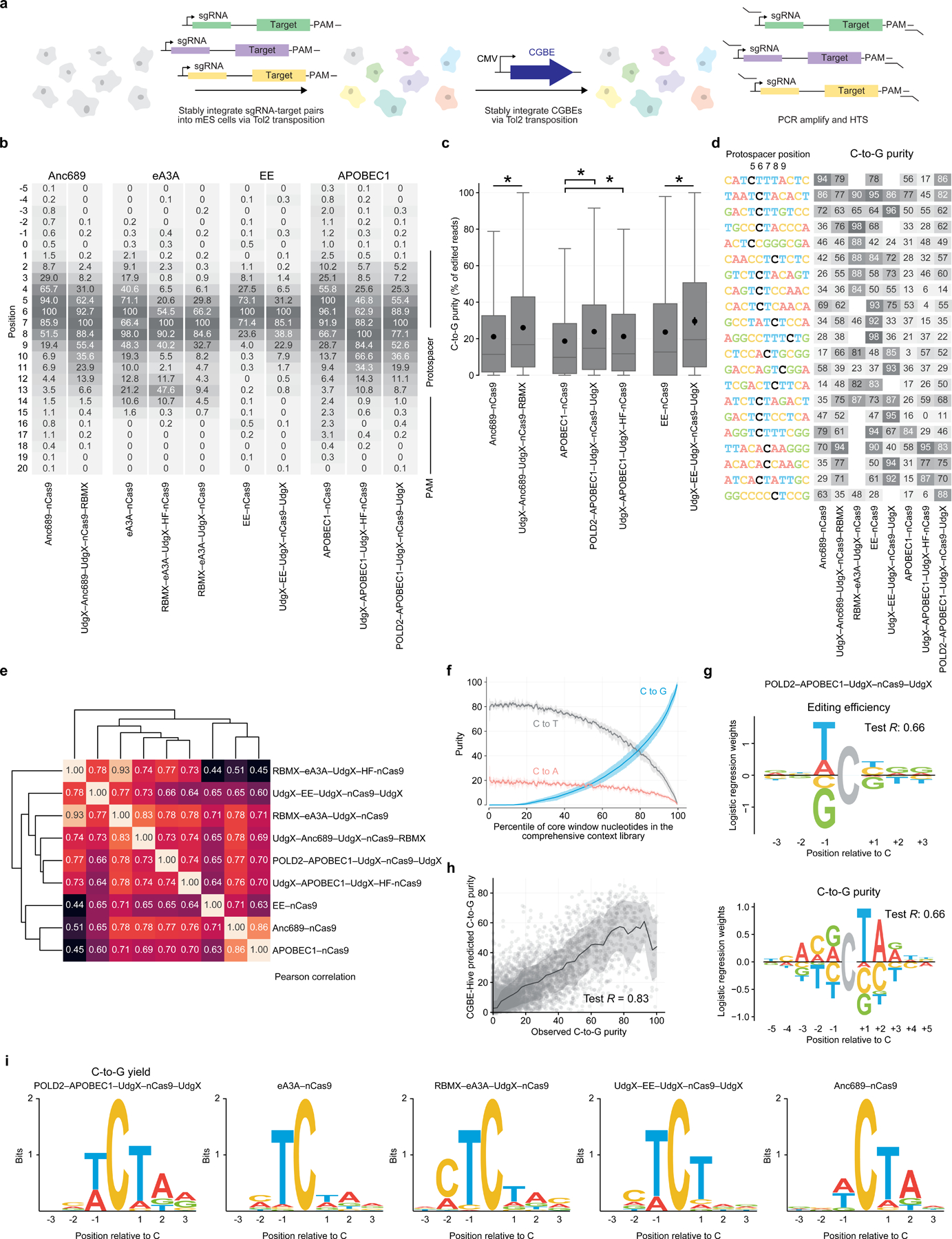
(a) Overview of genome-integrated target library assay. Libraries of 12,000 or 4,000 pairs of sgRNAs and corresponding target sites are integrated into the genomes of mammalian cells using Tol2 transposase and treated with base editors. Edited cells are enriched by antibiotic selection, and library cassettes are amplified for high-throughput sequencing. (b) Base editing windows. Values are C•G-to-G•C editing efficiencies normalized to a maximum of 100. The protospacer is at positions 1–20, with the SpCas9 PAM at positions 21–23. All data are in mES cells except for eA3A-nCas9, which is in HEK293T cells. (c) C•G-to-G•C editing purity in the comprehensive context library in mES cells. Box plots indicate median and interquartile range, whiskers indicate extrema, and black dots indicate mean. Two-sided Welch’s T-test * P≤5.1×10-9. (d) Heatmap of observed C•G-to-G•C purities by CGBE in target contexts from the comprehensive context library in mES cells. Black nucleotides indicate the cytosine for which purity is calculated. Target sites were sorted by outcome variance and manually selected. (e) Clustering of CGBEs based on measured C•G-to-G•C purity in core window cytosines across the comprehensive context library in mESCs. Values are Pearson correlation. (f) Purity of editing outcomes across core window nucleotides in the comprehensive context library, ranked by C•G-to-G•C purity, averaged across CGBEs in mESCs. Trend lines and shading show the rolling mean and standard deviation across 1% intervals. (g) Representative sequence motifs for editing efficiency and C•G-to-G•C purity from logistic regression models. The sign of each learned weight indicates a contribution above (positive sign) or below (negative sign) the mean activity. Logo opacity is proportional to the motif’s Pearson’s R on held-out sequence contexts. (h) Observed C•G-to-G•C purity across CGBEs in mESCs compared to CGBE-Hive predictions. Trend lines and shading show the rolling mean and standard deviation. (i) Sequence motifs for C•G-to-G•C editing yield.
We used the resulting library data to quantify editing windows and product purities for each CGBE (Fig. 5b, Supplementary Methods). CGBE editing activity was generally centered around protospacer position 6 with editing window widths ranging from 3 nt (EE–nCas9; positions 5–7) to 8 nt (UdgX–APOBEC1–UdgX–HF-nCas9 nickase; positions 4–11). The editing windows of CGBEs with additional components beyond Cas and deaminase domains were shifted by up to 3 nt compared to direct deaminase–Cas fusions, indicating that CGBE protein fusions can affect editing window size and position.
Engineered CGBE architectures showed significant improvements in C•G-to-G•C product purity compared to simple deaminase–nCas9 fusions. Across the 10,638 target sites in the comprehensive context library, the fusion CGBEs POLD2–APOBEC1–UdgX–nCas9–UdgX, UdgX–EE–UdgX–nCas9–UdgX, and UdgX–Anc689–UdgX–nCas9–RBMX showed 25% higher mean C•G-to-G•C purity than their corresponding deaminase–nCas9 counterparts within each editor’s editing window (P < 5.1×10−9; Welch’s t-test) (Fig. 5c). We observed large variation in CGBE editing efficiency, with mean efficiency ranging from 1.8% by UdgX–EE–UdgX–nCas9–UdgX to 23.0% by Anc689–nCas9 across the comprehensive context library within the same experimental batch. Notably, the protein fusion CGBEs exhibiting increased C•G-to-G•C purity also reduced editing yield by 1.4- to 1.6-fold on average.
C•G-to-G•C editing purity exceeded 90% for at least one of the tested CGBEs at 895 cytosines across the comprehensive context library. Some cytosines edited with purities as high as 90–100% by some CGBEs were edited with purity as low as 0–10% by other CGBEs, indicating that these CGBEs indeed offer complementary editing characteristics, and confirming that a panel of diverse CGBEs maximizes the utility of C•G-to-G•C base editing compared to using any single CGBE (Fig. 5d). We clustered CGBEs by C•G-to-G•C editing purity across the comprehensive context library and observed that engineered CGBEs did not cluster by deaminase (Fig. 5e), indicating that protein fusion engineering of CGBE architectures resulted in distinct sequence preferences governing C•G-to-G•C editing.
Sequence determinants and machine learning modeling of CGBE activity
C•G-to-G•C product purity of CGBEs varies substantially by sequence context (Fig. 5f). We observed 24.7±26.3% average C•G-to-G•C purity across all tested CGBEs for cytosines positioned near the center of the editing window, with substantial variation across target sequences: the top 5% had >79.6% C•G-to-G•C purity while the bottom 5% had <1.0%. To decipher the sequence determinants that underly CGBE activity, we computed simple motifs for editing efficiency and transversion purity using a logistic regression model that considers each nucleotide independently (Fig. 5g, Supplementary Methods)12. These motifs revealed that TC is strongly favored while GC is disfavored for editing efficiency across the tested CGBEs. We further trained gradient-boosted regression trees to predict CGBE editing efficiency sequence context, which achieved good accuracy with R=0.57–0.77 at held-out target sites. Consistent with our previous characterization of BE4 variants12, we observed sequence motifs that associated RCTA with higher C•G-to-G•C purity (R=A or G) across all characterized CGBEs. Cytosines in an ACTA motif were edited with an average C•G-to-G•C purity of 68.7% (N=1,760) across CGBEs, substantially higher than the 24.7% average across all sequence contexts, indicating a major role for sequence context in determining C•G-to-G•C editing outcomes. These simple target sequence motifs predicted 27.0%−53.3% of the variation in C•G-to-G•C purity.
Next, we trained BE-Hive models for these ten CGBEs (termed CGBE-Hive) and evaluated the models’ ability to predict C•G-to-G•C editing purity at held-out sequence contexts not seen during training. These models explained 58.3%−76.3% of the variance in C•G-to-G•C purity in the held-out dataset, a substantial improvement over logistic regression described above (27.0%−53.3%) (Fig. 5h). This performance improvement highlights that while C•G-to-G•C purity can be predicted using a simple motif such as RCTA that considers each nucleotide independently, higher-order interactions between nucleotides learned by deep neural networks substantially improve C•G-to-G•C editing purity predictions. Collectively, these observations establish that CGBE editing efficiency and purity can be accurately predicted by machine learning models.
To further investigate sequence determinants of CGBE editing outcomes, we calculated target sequence motifs for cytosines with the highest C•G-to-G•C efficiency for each CGBE (Supplementary Methods). While most CGBEs shared sequence preferences favoring TC for overall editing efficiency and RCTA for purity, different CGBEs had distinct motifs that correlated with C•G-to-G•C yield. POLD2–APOBEC1–UdgX–nCas9–UdgX favored RCTA for C•G-to-G•C yield, while eA3A–nCas9 simply favored TC (Fig. 5i). Interestingly, RBMX–eA3A–UdgX–nCas9 favored CTC, while UdgX–EE–UdgX–nCas9–UdgX favored TCT, and Anc689–nCas9 favored CTA (Fig. 5i). These observations reveal that different CGBEs show distinct sequence preferences that influence the yield of C•G-to-G•C outcomes.
We provide machine learning models trained on up to 10,638 sgRNA-target pairs for these ten CGBEs in our online interactive web app (www.crisprbehive.design)12. Users can query sgRNAs and target sequences for data-driven predictions on editing outcomes of all CGBEs characterized in this study.
Model-guided correction of pathogenic transversion SNVs
To extend the applicability of these CGBEs, we assessed their compatibility with PAM-variant Cas9 proteins. We evaluated editing at eight loci by CGBEs using Cas9-NG, an engineered SpCas9 variant with broadened PAM compatibility32, and observed similar editing purities to SpCas9 CGBEs at NGG PAM substrates (Supplementary Fig. 10, 11). The best performing NG-CGBEs at each locus retained >50% yield relative to SpCas9 CGBEs at targets with NGG PAMs (Supplementary Fig. 10).
Given the broadened targeting scope of NG-CGBEs we sought to characterize their performance on the “transversion-enriched SNV library”12 in mESCs, which contains 3,400 sgRNA-target pairs selected by BE-Hive from 18,523 disease-related G•C-to-C•G and A•T-to-C•G SNVs from the ClinVar and HGMD databases that are targetable by Cas9-NG1,33, predicted to be correctable by cytosine transversion base editing with high purity and yield. We generated the following NG-CGBEs based on their performance on the comprehensive context library: Anc689–nCas9-NG, APOBEC1–nCas9-NG, eA3A–nCas9-NG, UdgX– Anc689–UdgX–nCas9-NG –RBMX, and UdgX–APOBEC1–UdgX–HF-nCas9-NG. As Cas9-NG generally demonstrates reduced editing activity compared to wild-type SpCas932, similar to HF-Cas9, we included UdgX–APOBEC1–UdgX–nCas9-NG without the HF modifications as an alternative binding-impaired Cas9-fusion variant.
All six CGBEs tested on the transversion-enriched SNV library enabled high-purity C•G-to-G•C editing at disease-associated SNVs. At 247 cytosines predicted by CGBE-Hive to have >80% C•G-to-G•C editing purity, CGBEs demonstrated an average of 83% C•G-to-G•C editing purity (Fig. 6a). Each CGBE corrected > 200 SNVs to their wild-type coding sequence with >90% precision among edited amino acid sequences (amino acid correction precision; Fig. 6b), with a total of 546 unique SNVs across CGBEs. For example, in the genomeintegrated library, eA3A–nCas9-NG corrected the G•C-to-C•G SNV in COL3A1 associated with Ehlers-Danlos syndrome34 with 71.4% yield and 92.8% purity, and corrected an SNV in BRCA2 associated with familial breast and ovarian cancer35 with 66.5% yield and 82.5% purity. The fusion CGBE UdgX–APOBEC1–UdgX–nCas9-NG corrected an SNV in NSD1 associated with Sotos syndrome36 with 40.0% yield and 73.4% purity and corrected an SNV in NIPBL associated with Cornelia de Lange syndrome37 with 38.8% yield and 76.9% purity. Collectively, these results reveal efficient and high-purity correction of hundreds of disease-related SNVs by CGBEs.
Figure 6. Target library characterization and machine learning modeling of CGBE variants.
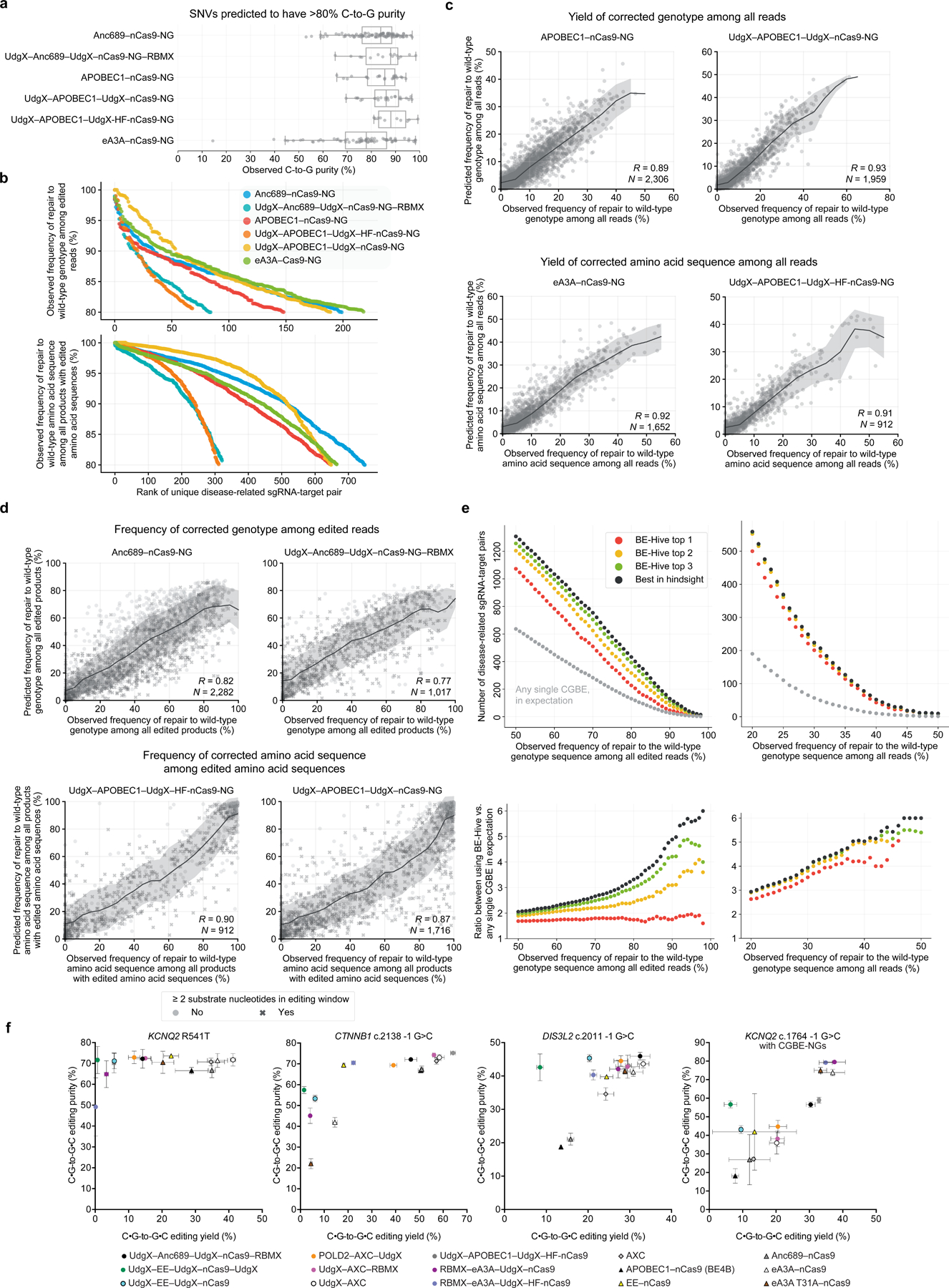
(a) Observed C-to-G purity by CGBE at SNVs predicted to have >80% C-to-G purity. Box plot indicates median and interquartile range, and whiskers indicate extrema. (b) Observed number of disease-related sgRNA-target pairs corrected at varying genotype precision and amino acid precision thresholds by various strategies for selecting CGBEs. See Supplementary Table 3. (c) Comparison of predicted versus observed correction yield of disease-related transversion SNVs in mES cells. Trend lines and shading show the rolling mean and standard deviation. (d) Comparison of predicted versus observed correction precision of disease-related transversion SNVs in mES cells. Trend lines and shading show the rolling mean and standard deviation. (e) Observed number of sgRNA-target pairs containing disease-related transversion SNVs corrected at various thresholds for genotype and amino acid precision. (f) Installation of disease-associated SNPs using CGBEs.
Notably, the UdgX–APOBEC1–UdgX–nCas9 CGBE maintained a similar high purity of C•G-to-G•C editing between HF-nCas9 and nCas9-NG variants. UdgX–APOBEC1–UdgX– nCas9-NG, however, offered substantially better yield of genotype and coding sequence corrected G•C-to-C•G SNVs (Fig. 6a,b). These results suggest that fusion of CGBEs to Cas9-NG variants may obviate the need to use HF-variant Cas9-proteins to alter their binding kinetics to promote C•G-to-G•C editing outcomes.
The best-edited targets in the transversion-enriched SNV library varied greatly by CGBE. Some SNVs edited with >90% purity by one CGBEs had purity below 5% for other CGBEs (Supplementary Fig. 12). CGBE-Hive models accurately accounted for this diversity in editing purity in the transversion-enriched SNV library, and accurately predicted the yield of exact genotype correction products and of alleles with corrected amino acid sequences (R=0.89–0.93 and R=0.91–0.94, respectively, Fig. 6c), as well as the DNA and amino acid correction precision (R=0.77–0.85 and R=0.82–0.90, respectively, Fig. 6d), including targets with multiple cytosines in the editing window. Since accurately predicting correction yield and precision requires accurate predictions for CGBE efficiency, C•G-to-G•C purity, and bystander editing patterns, these results establish that CGBE-Hive has learned important aspects of CGBE editing activity and can guide the use of CGBEs for high-purity correction of disease-related transversion SNVs.
Using CGBE-Hive to pick the best among the characterized CGBEs to correct each SNV should achieve greater C•G-to-G•C correction than applying any single CGBE to a set of targets. Indeed, we observed that using CGBE-Hive to choose the three CGBE variants predicted to best achieve the desired edit (top-3 performance) increased the number of targets corrected with ≥90% precision or to ≥40% efficiency by 4.1- and 5.0-fold, respectively, compared to the number of targets that are expected to be corrected with these precision and efficiency thresholds by picking any single CGBE (Fig. 6e). These improvements of 4.1- and 5.0-fold by using the top three CGBE-Hive choices were nearly identical to the performance from picking the best CGBE out of all six options in hindsight. CGBE-Hive also displayed strong top-1 performance: Using CGBE-Hive to choose just a single CGBE increased the number of targets corrected with ≥90% precision or to ≥40% efficiency to 1.7- and 4.0-fold, respectively, compared to picking a single CGBE in expectation.
For correction precision, CGBE-Hive recovered the best performing CGBE variant in its top choice in 43.3% of targets and in its top three choices in 84.2% of target sequences. For correction yield, CGBE-Hive recovered the best-performing CGBE variant in its top choice in 67.5% of targets and in its top three choices in 97.2% of targets. These results collectively demonstrate that this panel of CGBEs have diverse editing activities that CGBE-Hive has learned to predict, to optimize selection of the most promising CGBE variant to use for a desired edit. These improvements were also observed at endogenous loci in HEK293T cells (Fig. 6f, Supplementary Discussion 3). Thus, CGBE-Hive enables researchers to reap the benefits of the diversity of CGBEs developed in this study without the need to test all CGBE variants.
Comparisons with recently reported CGBEs, prime editing, and off-target profiling
Next, we determined whether the CGBE variants described in this work extend the scope of C•G-to-G•C base editing beyond those accessible with recently described CGBEs or PE. We were encouraged to find that the CGBEs developed in this study extend the scope of C•G-to-G•C genome editing by enabling higher yields and product purities at a wider array of target sequences compared to the use of previously described CGBEs alone except at loci already edited with high yield and purity by deaminase–nCas9 constructs (Supplementary Fig. 13; Supplementary Discussion 4). Furthermore, we observed that these novel CGBEs complement prime editing (PE) technology38. We found PE typically offers higher product purities while editing with CGBEs offers higher editing yields at some loci (Supplementary Fig. 14; Supplementary Discussion 5), consistent with recent reports13–15,38. Notably, prime editing currently requires extensive optimization of pegRNA features to achieve high-efficiency edits, while CGBE-Hive prediction obviates CGBE editor selection. CGBEs complement prime editing for efficient C•G-to-G•C editing, although additional optimization of both technologies may further improve their properties.
We also sought to characterize potential off-target editing outcomes of CGBEs. Since the genome-wide off-targets of base editors that use cytosine deaminase enzymes are known to be predominantly sgRNA dependent, we characterized Cas9-dependent off-target editing profiles of CGBEs by examining the activity of CGBEs at previously confirmed off-target loci of corresponding Cas9:sgRNA complexes8. The architectural changes and protein fusions used to develop the CGBEs in this study resulted in lower Cas9-dependent off-target editing compared to corresponding CGBEs lacking protein fusions (Supplementary Fig. 11, 15), despite their generally higher on-target editing, perhaps because the more complex fusions or architectural changes introduce additional conformational requirements in editor:DNA complexes that are not met by some off-target loci (see Supplementary Discussion 6). While DNA repair protein CGBE components may result in additional Cas-independent off-target effects, these are likely to differ by cell type and delivery method, and therefore are best assessed for each application.
Discussion
Understanding and controlling the outcomes of genome editing experiments are important challenges for achieving targeted, precise genome manipulation. We investigated molecular determinants of transversion base editing, including the effects of the deaminase and Cas effector domains, as well as many DNA repair proteins, and used these insights to engineer novel CGBEs. We characterized the editing outcomes and performance of these reagents using a high-throughput genome-integrated library assay in mammalian cells and identified sequence features that affect base editing outcomes of ten diverse CGBEs. We showed that C-to-G editing activity is predicted with substantially higher accuracy by deep learning models compared to simpler models, indicating that complex sequence features drive C•G-to-G•C editing activity.
We provide trained CGBE-Hive machine learning models which accurately predict CGBE efficiency, C•G-to-G•C editing purity, and bystander editing patterns (R=0.90) to enable predictable and consistently pure CGBE editing. We demonstrate a machine learning workflow using CGBE-Hive to identify optimal CGBE and sgRNA editing strategies to install a desired edit and show that this workflow expands high-efficiency and high-purity C•G-to-G•C editing to more loci than using any single CGBE by 5.0-fold and 4.1-fold with the top three CGBE-nominated choices. We demonstrate CGBE-mediated correction of the amino acid sequences of 546 disease-associated single nucleotide variants (SNVs) with >90% precision. Furthermore, we demonstrated efficient and pure installation of four disease-relevant SNPs and tested the performance of these tools in other mammalian cell lines. Collectively, the base editor and computational tools presented in this work substantially improve the targeting scope, effectiveness, and utility of CGBE-mediated transversion base editing.
Methods
General methods
DNA oligonucleotides were obtained from Integrated DNA Technologies (except where otherwise specified). All mammalian editor plasmids used in this work were cloned by Gibson assembly according to manufacturer’s protocols. Except for the CRISPRi library, plasmids expressing sgRNAs were constructed by ligation of annealed oligonucleotides into BsmBI-digested acceptor vector as previously described24,30. Plasmids expressing pegRNAs were constructed by Golden Gate assembly using a custom acceptor plasmid as previously described38. Protospacer sequences of sgRNAs used for non-library experiments in this work are listed in Supplementary Table 4. pegRNA protospacer and extension sequences are listed in Supplementary Table 4, tab #3. Vectors for low-throughput mammalian cell experiments were purified using Plasmid Plus Midiprep kits (Qiagen) or PureYield plasmid miniprep kits (Promega), which include endotoxin removal steps. Cloning of the CBE SaCas9 sgRNA for screening was conducted by KLD assembly according to the manufacturer’s protocol using BPK2660 (Addgene #70709) as a template with the following primers (protospacer is bolded): GGTGTTTCGTCCTTTCCACAAGATA, gCTGATAGGCAGCCTGCACTGGGTTTTAGTACTCTGTAATGAAAATTACAGAATCTAC.
General mammalian cell culture conditions
HEK293T (ATCC CRL-3216), U2OS (ATTC HTB-96), K562 (CCL-243), and HeLa (CCL-2) cells were cultured and passaged in Dulbecco’s Modified Eagle’s Medium (DMEM) plus GlutaMAX (ThermoFisher Scientific), DMEM (Gibco), McCoy’s 5A Medium (Gibco), RPMI Medium 1640 plus GlutaMAX (Gibco), or Eagle’s Minimal Essential Medium (EMEM, ATCC), respectively, each supplemented with ~10% (v/v) fetal bovine serum (Gibco, qualified) and 1x Penicillin Streptomycin (Corning). All cell types were incubated, maintained, and cultured at 37 °C with 5% CO2. Cell lines were authenticated by their respective suppliers or short tandem repeat profiling and tested negative for mycoplasma. Culturing conditions for library analyses are detailed below. Lentivirus was produced in HEK293T cells by co-transfection with packaging plasmids encoding gag and pol, rev, and tat from HIV-1 and VSVG envelope protein. For these transfections, we used either TransIT®-LT1 Transfection Reagent (Mirus) or Polyethylenimine (PEI; Polysciences, Inc.).
HEK293T tissue culture transfection (non-viral) protocol and genomic DNA preparation
HEK293T were cells grown, seeded, and transfected as previously described2,3,11,24,30,38. Briefly, cells were trypsinized and seeded on 48-well poly-D-lysine coated plates (Corning) to an approximated of 3 × 105 cells per well. 16–24 h post-seeding, cells were transfected at approximately 60% confluency with 1 µL of Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocols and 750 ng of base editor plasmid and 250 ng of sgRNA plasmid. For Prime editing experiments, non-nicking conditions were carried out with 750 ng of PE2 and 250 ng pegRNA while nicking experiments included an additional 83 ng of nicking sgRNA. 72 h post-transfection, media was removed, cells were washed with 1x PBS solution (Thermo Fisher Scientific), and genomic DNA was extracted by the addition of 150 µL of freshly prepared lysis buffer (10 mM Tris-HCl, pH 7.5; 0.05% SDS; 25 µg/mL Proteinase K (ThermoFisher Scientific)) directly into each well of the tissue culture plate. The genomic DNA•lysis buffer mixture was incubated at 37 °C for 1 h, followed by an 80 °C enzyme inactivation step for 30 min. Primers used for mammalian cell genomic DNA amplification are listed in Supplementary Table 4. Protospacer sequences used for each locus are listed in Supplementary Table 4.
High-throughput DNA sequencing of genomic DNA samples
Genomic sites of interest were amplified from genomic DNA prepared and sequenced on an Illumina MiSeq as previously described2,3,11,24,30,38 with minor modifications. Briefly, amplification primers containing Illumina forward and reverse adapters (Supplementary Table 4) were used for PCR 1, amplifying the genomic region of interest. PCR 1 reactions were performed with 0.5 µM of each forward and reverse primer, 1 µL of genomic DNA extract, 3% DMSO, 0.25 µL Phusion HS-II polymerase, 5 µL Phusion HF buffer, 0.5 µL 10mM dNTPs, and water to a final volume of 25 µL. PCR1 reactions were carried out as follows: 98 °C for 2 min, then 32 cycles of [98 °C for 10 s, 61 °C for 20 s, and 72 °C for 30 s], followed by a final 72 °C extension for 2 min. Unique Illumina barcoding primer pairs were added to each sample in a secondary PCR reaction (PCR 2). Specifically, 25 µL of a given PCR 2 reaction contained 0.5 µM of each unique forward and reverse Illumina barcoding primer pair, 1 µL of unpurified PCR 1 reaction mixture, 0.25 µL Phusion HS-II polymerase, 5 µL Phusion HF buffer, 0.5 µL 10mM dNTPs, and water to a final volume of 25 µL. The barcoding PCR 2 reactions were carried out as follows: 98 °C for 2 min, then 12 cycles of [98 °C for 10 s, 61 °C for 20 s, and 72 °C for 30 s], followed by a final 72 °C extension for 2 min. PCR products were evaluated by electrophoresis on 2% agarose gel. PCR 2 products (pooled by common amplicons) were purified by electrophoresis with a 2% agarose gel using a QIAquick Gel Extraction Kit (Qiagen), eluting with 40 µL of water. DNA concentration and library preparation was performed as previously described38 by fluorometric quantification (Qubit, ThermoFisher Scientific) and diluted to 4 nM final library concentration before sequencing on an Illumina MiSeq instrument according to the manufacturer’s protocols.
Sequencing reads were demultiplexed using MiSeq Reporter (Illumina). Alignment of amplicon sequences to a reference sequence was performed using CRISPResso239 which was run to calculate indels with a window size of 10. C•G-to-G•C editing purity was calculated as C•G-to-G•C editing yield ÷ [C•G-to-T•A yield + C•G-to-A•T yield + indels].
Nucleofection of HAP1, U2OS, K562, and HeLa cells
Nucleofection was performed on K562, HeLa, and U2OS cells as previously described38. 750ng of base editor-expression plasmid and 250ng sgRNA-expression plasmid were nucleofected in a final volume of 20uL in a 16-well nucleocuvette strip (Lonza). K562 cells were nucleofected using the SF Cell Line 4D-Nucleofector X Kit (Lonza) with 5 × 105 cells per sample (program FF-120), according to the manufacturer’s protocol. U2OS cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit (Lonza) with 3–4 × 105 cells per sample (program DN-100), according to the manufacturer’s protocol. HeLa cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit (Lonza) with 2 × 105 cells per sample (program CN-114), according to the manufacturer’s protocol. Nucleofiection of HAP1 cells was performed using the same amounts of DNA and final volume in a 16-well nucelocuvette strip; however, HAP1 cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit (Lonza) with 4 × 105 cells per sample (program DZ-113), according to the manufacturer’s protocol. Cells were harvested 72 hours after nucleofection for genomic DNA extraction.
Selection of ten CGBEs for target library characterization
We sought to select the most representative and diverse subset of CGBEs from endogenous base editing data for 72 CGBEs at eight or 16 endogenous target loci. Briefly, we used a convex relaxation of a quadratic program to find a subset of CGBEs with maximally diverse transversion editing purities and yields. Clustering analysis was used to suggest the number of unique CGBE families. Analytic results were curated manually. The six fusion CGBEs assayed were: PolD2–APOBEC1–UDGX–Cas9–UDGX, RBMX–eA3A–UDGX–Cas9, RBMX–eA3A–UDGX–HF-nCas9, UDGX–Anc689–UDGX–Cas9–RBMX, UDGX–APOBEC1–UDGX–HF-nCas9, and UDGX–EE–UDGX–Cas9–UDGX. The four simple CGBE editors were deaminase–nCas9 with eA3A, Anc689, APOBEC1, and EE deaminases. We also assayed eA3A-T31A–nCas9 and eA3A-BEN3—ΔN13-UGI. eA3A–nCas9, eA3A-T31A–nCas9 and eA3A-BEN3—ΔN13-UGI were characterized in the comprehensive context library only in HEK293T, while all other CGBEs were characterized in the comprehensive context library only in mESCs. eA3A–nCas9-NG and eA3A-T31A–nCas9-NG were further characterized in the transversion-enriched SNV library in mESCs.
To identify CGBEs with distinct activities, we used quadratic programming to identify a subset of CGBEs with maximum pairwise distances between vectors of C•G-to-G•C editing purity and yield across eight or 16 endogenous loci. We also performed hierarchical clustering, and observed that across these endogenous loci, CGBE editing activity primarily clustered by deaminase, though there were also substantial intra-cluster differences in editing activities due to variety in protein fusion architectures that were occasionally larger than inter-cluster differences, which indicates that CGBE editing activity is affected by both deaminase and protein fusion architectures. As our quadratic programming and clustering methods only consider numerical distances and do not propose subsets optimized for high purity or yield, we manually curated the quadratic programming results by replacing CGBEs with similar neighbors from hierarchical clustering when the neighbors had meaningfully higher purity or yield. Since deaminases, protein fusions, and high-fidelity Cas9 variants are known to alter base editing activity12–15,26, we also manually curated our final subset to ensure a diversity of these elements.
CRISPRi library construction
For our CRISPRi screen we used a platform called Repair-seq, which was developed by Hussmann et al. 2021 using a CRISPRi guide library described elsewhere21. This library contains 1513 gene-targeting sgRNAs selected from hCRISPRi-v2.140 and 60 non-targeting controls selected from hCRISPRi-v240 (Supplementary Table 1). Gene-targeted sgRNAs were against 476 genes enriched for ones involved in DNA metabolic processes (e.g., replication, repair, recombination). A minority of the spacer sequences for the gene-targeting sgRNAs in this library were repeated in hCRISPRi-v2.1 and are therefore annotated in Supplementary Table 1 as targeting multiple gene promoters, with multiple guide identifiers. Our 476 gene count considers only the first set of annotations. Oligonucleotides containing sgRNA targeting sequences were synthesized by Twist Bioscience (Supplementary Table 1).
CRISPRi library cloning
The guide library was cloned in pAX198 as described elsewhere21. This vector was derived from pU6-sgRNA EF1Alpha-puro-T2A-BFP41 (Addgene, 60955) through multi-step molecular cloning, as described elsewhere21. pAX198 contains a CRISPRi guide expression cassette driven by a modified mouse U6 promoter and ending with a termination signal consisting of 6 Ts. pAX198 also contains a ‘target region’ for genome editing derived from sequence at the human HBB gene, specifically the second and third exons of HBB (no intron) and part of the 3’UTR (ENST00000647020.1). This region is where we directed Anc689nCas9 and Anc689-dCas9 (see CRISPRi screen cell culture section of Methods). Prior to library cloning, a BstXI site was removed from the target region by site-directed mutagenesis. Library cloning was performed with standard protocols (details available at https://weissmanlab.ucsf.edu/CRISPR/Pooled_CRISPR_Library_Cloning.pdf). Briefly, library oligonucelotides were amplified by PCR (primers 5’-TATGAACCACTAAGGCGTCCAC, 5’-TCACCAGCAGACTTTACGCAGC), purified using MinElute Reaction Cleanup Kit (Qiagen), digested with BlpI and BstXI, isolated by gel purification, and ligated into a similarly digested expression vector (insert to backbone ratio of 1:1 for 16 hours at 16ºC). Ligation reactions were electroporated into MegaX DH10B T1R Electrocomp™ cells (ThermoFisher). Cells were grown on agar plates and then scraped into liquid for plasmid purification. The final sgRNA library (AX227) was verified by sequencing.
CRISPRi screen cell culture
The Repair-seq screens reported here were performed in previously described HeLa cells42, which stably express a dCas9-BFP-KRAB fusion (from pHR-SFFV-dCas9-BFP-KRAB; Addgene #46911), in two rounds. The first round of screening evaluated Anc689– nCas9. The second round evaluated Anc689-dCas9. Both rounds of screening were conducted as follows: Cells were transduced with guide library (AX227, see CRISPRi library cloning section of Methods) by lentiviral infection. The infections were carried out in DMEM supplemented with ~10% (v/v) fetal bovine serum, 1x Penicillin Streptomycin, and 8 µg/mL polybrene at an observed infection efficiency of ~5% for both Anc689–nCas9 and Anc689–dCas9, as determined by flow cytometry. Approximately 2 days post transduction, cells were selected in 3 µg/mL puromycin and then, 3 days later, transfected with plasmids for base editing. We performed each screen in replicates, each split one day prior to transfection onto 30 15 cm plates, each containing ~1.2e6 cells. The transfection procedure was as follows: (1) 25 ng plasmid DNA (75% editor plasmid; 25% sgRNA plasmid) was mixed with 3.5 mL of Opti-MEM (Gibco) and 4.6 mL Helafect Transfection Reagent (per 15 cm plate of cells). (2) This mixture was then incubated at room temperature for 20 minutes and (3) added to DMEM (Gibco) supplemented with ~10% (v/v) fetal bovine serum (20 mL per plate). (4) The prepared media was then used to replace non-transfection media on each plate of cells. Approximately 3 days later, cells were collected for sample preparation. For all arms of screening, ~100e6 cells or more were collected at a viability of >85%.
CRISPRi screen sample preparation
Sequencing libraries were prepared from cells collected at the end of the CRISPRi screens as follows: Genomic DNA was extracted from cell pellets (~200e6 cells for each replicate of Anc689–nCas9, and 125e6 and 098e6 cells for each of two replicates of Anc689-dCas9) using the NucleoSpin® Blood XL kit (Macherey-Nagel, up to 100e6 cells per column). We fragmented the genomic DNA by digestion with NotI-HF (NEB) and then enriched for edit-containing fragments (1447 bp) by size selecting each sample on a large 0.8% agarose gel (Owl™ A1 Large Gel System, Thermo Fisher Scientific). Gel electrophoresis was conducted at large-scale (i.e., with wells large enough to hold 1.5 mL volume per well) to maximize recovery of fragments containing both edited sequences and sgRNA expression cassettes (‘target’ fragments). Gel preparation details are available at https://weissmanlab.ucsf.edu/CRISPR/IlluminaSequencingSamplePrep_old.pdf. DNA was then isolated from excised regions of the gel using NucleoSpin® Gel and PCR Clean up kit (Macherey-Nagel) with columns placed on a vacuum manifold. Of note, large sample volumes were passed through individual columns using syringe barrels to increase capacity.
Next, size-selected target fragments were prepared for sequencing using custom adaptors compatible with next-generation sequencing technologies from Illumina. These adapters, which contained 12 nt unique molecular identifiers (UMIs), were made by annealing individual DNA oligonucleotides (obtained from Integrated DNA Technologies). The oligonucleotide components were oBA676 (5’-G*G*C*C*AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT, HPLC purified) and oBA677 (5’-CAAGCAGAAGACGGCATACGAGATNNNNNNNNNNNNGTGACTGGAGTTCAGACGTGTG CTCTTCCGATCT, HPLC purified), where * represents a phosphorothioated DNA base. Prior to ligation, DNA samples were digested with HindIII-HF (NEB). This step removed a 4 nt NotI overhang from one end of the target fragments, leaving only one side available for adaptor ligation. DNA was then purified using SPRIselect Reagent (Beckman Coulter) in a 0.8X reaction, quantified using Bioanalyzer High Sensitivity DNA Analysis (Agilent), and 1 µg of the product was ligated to adaptors using enzyme and buffer from the KAPA HyperPrep Kit (Roche) as follows: 30 µL ligation buffer, 10 µL ligase, adapter at 200:1 adaptor:insert ratio, and PCR-grade water to 110 µL total volume. These reactions were incubated at 4ºC overnight on a thermocycler with lid temperature set to 30ºC.
Following ligation, DNA was purified using SPRIselect Reagent (Beckman Coulter) in two reactions (0.65X followed by 0.8X) and target fragments were enriched by PCR as follows: 30 ng of template, amplification primers at 0.6 µM final concentration (each), 3% dimethyl sulfoxide, and 1X KAPA HiFi HotStart ReadyMix (50 µL total volume) run at 1 cycle of 3 minutes at 95ºC; 16 cycles of 15 seconds at 98ºC, followed by 15 seconds at 70ºC; 1 cycle of 1 minute at 72ºC; 4ºC hold. We performed enough PCR reactions to use nearly the entirety of each sample obtained from the ligation and subsequent clean-up reactions. Amplification primers used were oBA679 (5’-CAAGCAGAAGACGGCATACGAGAT) and 5’-AATGATACGGCGACCACCGAGATCTACAC-[index]-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTATCCCTTGGAGAACCACCTTGTTGG. Amplified DNA was purified using SPRIselect Reagent (Beckman Coulter) in a 0.8X reaction, and index samples were mixed for sequencing. Throughout sample preparation procedures, samples were checked for quality and yield using either a NanoDrop Spectrophotometers (Thermo Fisher Scientific), Agilent 2100 Bioanalyzer system, or by running on a Novex™ TBE Gel. Sample preparation procedures are also described elsewhere21.
CRISPRi screen analysis
Sequencing of CRISPRi screens, alignment and classification of screen sequencing data, statistical tests of gene significance in Fig. 2d, Supplementary Fig. 4, and Supplementary Table 2, and identification of the top two most active guide RNAs for relevant genes in Figure 2b and Supplementary Fig.5a and 5b were performed as described in Hussmann et al. 202121. Intervals in Supplementary Figure 3c are 95% Clopper-Pearson intervals of outcome fractions, converted to corresponding log2 fold changes. That is, given k observed UMIs for a given CRISPRi guide in a numerator outcome set out of n total UMIs in a denominator outcome superset, the bottom interval (vbottom) is the smallest value of the true population proportion of numerator to denominator outcomes such that there is <= 2.5% chance of observing >= k from Binomial(vbottom, n), and the top interval (vtop) is the largest value of the true population proportion of numerator to denominator outcomes such that there is <= 2.5% chance of observing <= k from Binomial(vtop, n).
Target library cloning
The target libraries used in this manuscript were previously generated in Arbab, Shen et al. 202012. All editors described in this paper were cloned between the N–terminal and C–terminal NLS sequences flanking the eA3A-BE4max (Addgene 152997).
Target library cell culture
mESC lines used have been described previously and were cultured as described previously43. For stable Tol2 transposon-mediated library integration, cells were transfected using Lipofectamine 3000 (Thermo Fisher) following standard protocols with equimolar amounts of Tol2 transposase plasmid (a gift from R. Sherwood) and transposon-containing plasmid. For library applications, 15-cm plates with 2×107 initial cells were used. To generate library cell lines with stable Tol2–mediated genomic integration, cells were selected with 150 µg/mL hygromycin starting the day after transfection and continued for >2 weeks. For editing experiments, CGBEs were transfected with Tol2 transposase plasmid using Lipofectamine 3000 and selected with 10 µg/mL blasticidin starting the day after transfection for 4 days before harvesting. We maintained an average coverage of ≥300x per library cassette throughout.
Target library high-throughput sequencing
Library preparation was performed as described in Arbab, Shen et al. 202012. Genomic DNA was collected from cells 5 days after transfection, after 4 days of antibiotic selection. For library samples, 20 µg gDNA was used for each sample and we maintained an average sequencing depth of ≥4,000x per target. All PCRs were performed using NEBNext Ultra II Q5 Master Mix. Samples were pooled using Tape Station (Agilent) and quantified using a KAPA Library Quantification Kit (KAPA Biosystems). The pooled samples were sequenced using Illumina NextSeq.
Target library analysis: data processing
Sequencing reads were assigned to designed library target sites by locality sensitive hashing12,31. Target contexts that were intentionally designed to be highly similar to each other were designed barcodes to assist accurate assignment. Sequence alignment was performed using Smith–Waterman with the parameters: match +1, mismatch −1, indel start −5, indel extend 0. Nucleotides with PHRED score below 30 were assumed to be the reference nucleotide.
For base editing analysis, aligned reads with no indels were retained for analysis and events were defined as the combination of all possible substitutions at all substrate nucleotides in the target site in a read, where a single sequencing read corresponds to an observation of a single event. Substrate nucleotides were defined as C and G for CBEs and A and C for ABEs.
For indel analysis, reads containing indels with at least one indel position occurring between protospacer positions −6 to 26 were retained, where position 1 is the 5’–most nucleotide of the protospacer, and 0 is used to refer to the position between −1 and 1. Reads containing indels without at least six nucleotides with at least 90% match frequency on both sides of each indel were discarded. Events were defined as indels identified by position, length, and inserted nucleotides occurring in a read. Combination indels were either not observed at all or only at exceedingly low frequencies in endogenous data and were therefore excluded from consideration when analyzing library data.
Target library analysis: base editing profiles
Base editing profiles were calculated using the same approach as Arbab, Shen et al. 202012, using a multi-step procedure to maximize sensitivity. Briefly, single-nucleotide mutation frequencies were tabulated at each target position from sequence alignments in treatment and control data. Treatment data was adjusted for 1) background mutations using untreated control data, 2) sequencing errors, 3) batch effects using other treatment data including published data from Arbab, Shen et al. 202012, which primarily helped adjust for rare substitution artifacts from library construction. We then identified mutations that occurred consistently for any editor across replicates to build base editing profiles with sufficient sensitivity to detect rare mutations. We defined cytosine base editing activity as C to A, G, or T at positions −9 to 20 and G to A or C at positions −9 to 5. For all analysis in this work that required tabulating reads with base editing activity, we discarded reads that did not have base editing activity according to these broad profiles. Window sizes were calculated at 50% or greater efficiency relative to the position–wise maximum.
Target library analysis: calculating efficiency and purity
We required a minimum of 100 reads for calculating editing efficiency, and a minimum of 100 edited reads to calculate purity of editing outcomes. Library members not satisfying these criteria were filtered. The resulting efficiency and purity values were reported as data in the manuscript, and used to train machine learning models. Calculated editing efficiencies and purities were not adjusted for batch effects: instead, our efficiency model is designed to account for batch variation in baseline editing efficiencies by taking it in as optional input. Bystander editing patterns were not found to vary substantially by batch (Arbab, Shen et al., 2020).
Target library analysis: clustering
CGBEs transversion purities at (target site, nucleotide) tuples in the comprehensive context library were tabulated, and pairwise distances between CGBEs were calculated as the variance explained (R2) between each pair of CGBEs. Clustering was performed using the L1 distance metric between vectors with the UPGMA clustering algorithm (average linkage).
Target library analysis: identifying targets with diverse editing outcomes
We calculated a “diversity score” for a target site and substrate nucleotide given observed editing activity values (yield or purity) by a panel of base editors. For a vector of observed values denoted x, our diversity score was defined as max(x) + 2*std(x). We included max(x) in the score function to encourage library members with very high and very low values to be considered diverse.
To explore the possibility that observed diversity of transversion purity could be explained by analyzing low-abundance outlier library members, we investigated the relationship between the diversity of transversion purity and library member abundance in the transversion-enriched SNV library. We computed a diversity score for each library member, where large values indicate that different CGBEs had different transversion editing purity at that target. We also calculated the relative abundance of each library member in the sequencing data. If library members with extremely high diversity scores were associated with low relative abundance (e.g., if they were explainable by low coverage bottlenecking outliers), their relative abundances should be shifted relative to the background distribution. We tested this hypothesis by comparing the distribution of relative abundance for the top 10 to top 50 library members ranked by diversity score to the full distribution of relative abundances. By Welch’s T-Test, we found no statistical evidence that high-diversity library members had shifted relative abundance (P>0.40, N=4,000). Furthermore, we observed a mildly positive Pearson correlation (R=0.14, P=4×10−14) between relative abundance and the diversity score, indicating that across the whole library, library members with higher relative abundance tend to have slightly higher diversity of base editing outcomes. Taken together with other analysis in our paper, we conclude that differences in editing purity by different CGBEs at the same target are better explained by their distinct sequence preferences.
Target library analysis: sequence motif models
For prediction tasks where the target variable is continuous and has range in (0, 1), we first applied a logistic transformation to the data, then used linear regression. For continuous data representing fractions, we discarded values equal to 0 or 1. For classification tasks, the target variables were either 0 or 1 indicating absence or presence of activity, and we used logistic regression. Target variables included the efficiency of C•G-to-T•A editing by CBEs and the purity of cytosine transversions by CBEs. Each of these statistics involves calculating a denominator corresponding to the total number of reads at a target sequence, or the total number of edited reads at a target sequence not including indels. Target sequences with fewer than 100 reads in the denominator were discarded to ensure the accuracy of estimated statistics in the training and testing data. Features were obtained by one–hot–encoding nucleotides per position relative to a substrate nucleotide or to the protospacer. When featurizing data relative to a single substrate nucleotide, each substrate nucleotide within a specified range of positions was used. Ranges used included position 6 only (for the comprehensive context library that contained all NNN–NNN–mers surrounding position 6) and positions 4–8, which was used only when exploratory data analysis indicated that the activity of interest did not vary substantially by position. All nucleotides within a 10–bp radius of the target position were one–hot–encoded. Position was not used as a feature. The data were randomly split into training and test sets at an 80:20 ratio. We note that sequence motifs described by these regression models consider each position independently and are intended primarily for visualization.
Motifs for yield were calculated from the top 150 cytosines ranked by C-to-G yield. Column sizes are scaled by their information content.
Target library analysis: base editing efficiency models
We observed that base editing efficiency varies by experimental batch. To combine replicates across batches, we first performed mean centering and logit transformation at up to 10,638 gRNA-target pairs in each experimental condition separately from the 12kChar library which includes all 4-mers surrounding A or C from protospacer positions 1 to 11. We discarded data at target sites with fewer than 100 total reads, then averaged values at matched target sites across experimental replicates. Values of negative or positive infinity (resulting from logit of 0 or 1) were discarded. The data were randomly split into training and test sets at a ratio of 90:10. Each target site had a single output value corresponding to the mean logit fraction of sequenced reads with any base editing activity. Data points comprising a single replicate were assigned weight=0.5. Data points comprising multiple replicates were assigned a weight of the median logit variance divided by the logit variance at that data point, or 1, whichever value was smaller. In this manner, exactly half of the data points comprising multiple replicates were assigned a weight of 1, and those with higher variance were assigned a lower weight. We obtained features from each target sequence using protospacer positions −9 to 21. Features included one-hot encoded single nucleotide identities at each position, one–hot encoded dinucleotides at neighboring positions, the melting temperature of the sequence and various subsequences, the total number of each nucleotide in the sequence, and the total number of G or C nucleotides in the sequence.
We used gradient-boosted regression trees from the python package scikit-learn and trained them with tuples of (x, y, weights) using the training data. We performed hyperparameter optimization as described in Arbab, Shen et al. 202012. We performed 5-fold cross–validation by splitting the training set into a training and validation set at a ratio of 8:1 and retained the combination of hyperparameters with the strongest average cross-validation performance as the final model. We trained models in this manner for each combination of cell-type and base editor. Models were evaluated on the test set which was not used during hyperparameter optimization.
Target library analysis: bystander editing models
Bystander models were designed and trained using the same approach as Arbab, Shen et al. 202012. Briefly, we designed and implemented a deep conditional autoregressive model that uses an input target sequence surrounding a protospacer and PAM to output a frequency distribution on combinations of base editing outcomes in the python package PyTorch44. The model predicts substitutions at cytosines and guanines for CBEs. The model transforms each substrate nucleotide and its local context using a shared encoder into a deep representation, then applies an autoregressive decoder that iteratively generates a distribution over base editing outcomes at each substrate nucleotide while conditioning on all previous generated outcomes. The encoder and decoder are coupled with a learned position–wise bias towards producing an unedited outcome. The model is trained on observed data by minimizing the KL divergence. Importantly, the conditional autoregressive design is sufficiently expressive to learn any possible joint distribution in the output space, thereby representing a powerful and general method for learning the editing tendencies of any base editor from data. We assembled a dataset where each sgRNA–target pair was matched with a table of observed base editing genotypes and their frequencies among reads with edited outcomes. We discarded data points with fewer than 100 edited reads. We discarded edited genotypes occurring at higher than 2.5% frequency with no edits at any substrate nucleotides (defined as C for CBEs and A for ABEs) in positions 1–10. Data from multiple experimental replicates were combined by summing read counts for each observed genotype.
Target library analysis: performance evaluation
We evaluated machine learning model performance using held-out data. For evaluating models at predicting yield, we used the efficiency model to predict a base editing efficiency score using efficiency summary statistics (mean, std) from the training set. We multiplied the predicted base editing efficiency with the predicted frequency of editing patterns from the bystander model.
Target library analysis: indel quantification
Indels were quantified using the same approach as Arbab, Shen et al. 202012. Indels have strong batch effects in our library assay which can be adjusted within each connected component in the graph defined with nodes representing base editors and edges connecting base editors measured in the same experimental batch. We were able to adjust batch effects for eA3A–nCas9 using two-way ANOVA as previously described since it was included in the same connected component as all BEs previously characterized in Arbab, Shen et al., 202012. We were not able to adjust batch effects for all other CGBEs as they were in a separate connected component.
CGBEs are expected to generate indels at higher frequency than canonical base editors as a consequence of generating abasic sites more efficiently. Consistent with this expectation, we previously observed lower base editing to indel (BE:indel) ratios at sites with higher transversion base editing activity. However, we were surprised to observe a positive correlation between BE:indel ratios and high C•G-to-G•C editing purity among target library editing outcomes. The geometric mean BE:indel ratio for eA3A–nCas9 was 15:1 across all target sequences, lower than canonical CBEs at 40:112; however, upon close inspection, we recognized that BE:indel ratios were split dependent upon whether the target sequence was edited with high or low purity. Indeed, the geometric mean BE:indel ratio was below this 15:1 ratio for sites with <40% C•G-to-G•C purity (decreases from 17:1 to 12:1 as editing purity increases from 0% to 40%) while the geometric average BE:indel ratio increased from 12:1 to 29:1 as C•G-to-G•C purity increased from 40% to 100%. This surprising positive correlation between BE:indel ratios and C•G-to-G•C purity was observed for 11 CGBEs across the comprehensive context and transversion-enriched libraries, with R=0.05 to 0.20 (P<2.4×10−6). No CGBE had a statistically significant negative correlation. This observation suggests that while abasic sites are a common precursor of both indel formation and C•G-to-G•C substitutions and that increased abasic site formation should lead to increases in both indels and C•G-to-G•C substitutions, target sites particularly amenable to highly pure C•G-to-G•C editing preferentially resolve abasic sites against indels. Taken together, these observations highlight the possibility of developing CGBEs with both highly pure C•G-to-G•C editing and high BE:indel ratios.
Target library analysis: evaluating CGBE-Hive optimization of CGBEs for SNVs
We used six CGBEs for this analysis: Anc689–nCas9-NG, APOBEC1–nCas9-NG, and eA3A–nCas9-NG, UdgX–Anc689–UdgX–nCas9-NG–RBMX, UdgX–APOBEC1–UdgX– nCas9-NG, and UdgX–APOBEC1–UdgX–HF-nCas9-NG. For each SNV, we used CGBE-Hive to identify which CGBE had the highest predicted genotype correction precision or amino acid correction precision among CGBEs that had data for that SNV, which was not always all six CGBEs, as some conditions had different SNVs filtered out due to low read counts or poor data quality. Only SNVs with data for at least three CGBEs were considered. The baseline used was the expectation of the statistic with respect to a uniform distribution over the six CGBEs for each SNV.
Obtaining biological materials
Plasmids encoding CGBEs and CRISPRi screening materials are available through Addgene.
Supplementary Material
Acknowledgements
This work was supported by U.S. NIH U01AI142756, UG3AI150551, RM1HG009490, R35GM118062, R35GM138167, P30CA072720, HHMI, and Princeton University. The authors acknowledge NSF Graduate Research Fellowships to L.W.K., M.W.S., and T.A.S.; a NWO Rubicon Fellowship to M.A.; a Jane Coffin Childs postdoctoral fellowship to A.V.A.; fellowship support from the NSF and Hertz Foundation to J.L.D.; a Helen Hay Whitney postdoctoral fellowship to G.A.N.; a Damon Runyon Postdoctoral Fellowship to D.Y.; a Singapore A*STAR NSS fellowship to B.M.; and NIH Ruth L. Kirschstein National Research Service Award F31NS115380 to J.M.R. J.A.H. was the Rebecca Ridley Kry Fellow of the Damon Runyon Cancer Research Foundation.
Footnotes
Data and code availability
The target library sequencing data generated during this study are available at the NCBI Sequence Read Archive database under PRJNA631290. Data from the Repair-seq screens are available under PRJNA721212. Processed target library data used for training machine learning models have been deposited under the following DOIs: 10.6084/m9.figshare.12275645 and 10.6084/m9.figshare.12275654.
Code availability
Code used for analyzing CRISPRi screens is available at https://github.com/jeffhussmann/repair-seq. Code used for target library data processing and analysis are available at https://github.com/maxwshen/lib-dataprocessing and https://github.com/maxwshen/lib-analysis. The machine learning models for CGBEs trained on target library data are available as a part of the BE-Hive interactive web application at https://crisprbehive.design and the BE-Hive Python package at https://github.com/maxwshen/be_predict_efficiency and https://github.com/maxwshen/be_predict_bystander.
References
- 1.Landrum MJ et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862–D868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaudelli NM et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gehrke JM et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nature Biotechnology 36, 977–982 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishida K et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729–aaf8729 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Richter MF et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nature Biotechnology 38, 883–891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rees HA & Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nature Reviews Genetics 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nature Biotechnology 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Gaudelli NM et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nature Biotechnology 38, 892–900 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Mok BY et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komor AC et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Science Advances 3, eaao4774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arbab M et al. Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning. Cell 182, 463–480.e430 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurt IC et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nature Biotechnology 39, 41–46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao D et al. Glycosylase base editors enable C-to-A and C-to-G base changes. Nature Biotechnology 39, 35–40 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Chen L et al. Programmable C:G to G:C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nature Communications 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu DR & Koblan LW Cytosine to Guanine Base Editor. World Intellectual Property Organization (2018).
- 17.Marquart KF et al. Predicting base editing outcomes with an attention-based deep learning algorithm trained on high-throughput target library screens. bioRxiv (2020). [DOI] [PMC free article] [PubMed]
- 18.Sang PB, Srinath T, Patil AG, Woo E-J & Varshney U A unique uracil-DNA binding protein of the uracil DNA glycosylase superfamily. Nucleic Acids Res 43, 8452–8463 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn W-C et al. Covalent binding of uracil DNA glycosylase UdgX to abasic DNA upon uracil excision. Nat Chem Biol 15, 607–614 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Tu J, Chen R, Yang Y, Cao W & Xie W Suicide inactivation of the uracil DNA glycosylase UdgX by covalent complex formation. Nat Chem Biol 15, 615–622 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Hussmann JA et al. Mapping the Genetic Landscape of DNA Double-strand Break Repair. BioRxiv (2021). [DOI] [PMC free article] [PubMed]
- 22.Gilbert LA et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallina I, Hendriks IA, Hoffmann S, Larsen NB, Johansen J, Colding-Christensen CS, Schubert L, Sellés-Baiget S, Fábián Z, Kühbacher U, Gao AO, Räschle M, Rasmussen S, Nielsen ML, Mailand N, Duxin JP The ubiquitin ligase RFWD3 is required for translesion DNA synthesis. Molecular Cell 81, 1–17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy JM et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat Biomed Eng 4, 97–110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YB et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nature Biotechnology 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinstiver BP et al. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slaymaker IM et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen JS et al. Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 550, 407–410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JK et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nature Communications 9, 3048 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koblan LW et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nature Biotechnology 36, 843–846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen MW et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishimasu H et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stenson PD et al. Human Gene Mutation Database: towards a comprehensive central mutation database. Journal of Medical Genetics 45, 124–126 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Frank M et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers–Danlos syndrome. European Journal of Human Genetics 23, 1657–1664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petrucelli N, Daly MB & Feldman GL Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genetics in Medicine 12, 245–259 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Douglas J et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. American journal of human genetics 72, 132–143 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luna-Peláez N et al. The Cornelia de Lange Syndrome-associated factor NIPBL interacts with BRD4 ET domain for transcription control of a common set of genes. Cell Death Dis 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nature Biotechnology 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horlbeck MA et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert Luke A. et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gilbert Luke A. et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 154, 442–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sherwood RI et al. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nature Biotechnology 32, 171–178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paszke A et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. Advances in Neural Information Processing Systems 32, 8024–8035 (2019). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.