

SUMMARY
Site-directed RNA editing approaches offer great potential to correct genetic mutations in somatic cells while avoiding permanent off-target genomic edits. Nuclease-dead RNA-targeting CRISPR-Cas systems recruit functional effectors to RNA molecules in a programmable fashion. Here, we demonstrate a Streptococcus pyogenes Cas9-ADAR2 fusion system that uses a 3′ modified guide RNA (gRNA) to enable adenosine-to-inosine (A-to-I) editing of specific bases on reporter and endogenously expressed mRNAs. Due to the sufficient nature of the 3′ gRNA extension sequence, we observe that Cas9 gRNA spacer sequences are dispensable for directed RNA editing, revealing that Cas9 can act as an RNA-aptamer-binding protein. We demonstrate that Cas9-based A-to-I editing is comparable in on-target efficiency and off-target specificity with Cas13 RNA editing versions. This study provides a systematic benchmarking of RNA-targeting CRISPR-Cas designs for reversible nucleotide-level conversion at the transcriptome level.
Graphical Abstract
In Brief
CRISPR-mediated transcriptome editing is an alternative strategy for genetic engineering. Marina et al. introduce a Cas9-directed method for RNA editing, which can be performed with a spacer-lacking guide RNA (gRNA). This system is comparable to other RNA-targeting Cas platforms both in on- and off-target capability.
Graphical Abstract
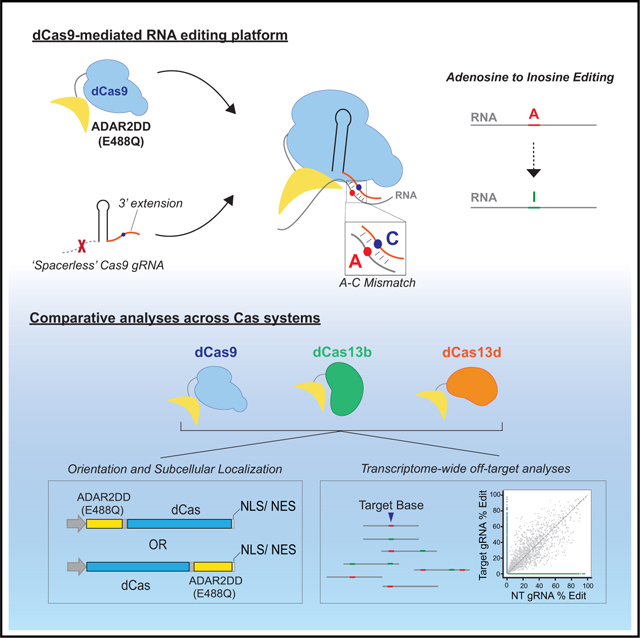
INTRODUCTION
Adenosine-to-inosine (A-to-I) RNA editing is an essential process that occurs naturally across approximately 2.5 million sites of the human transcriptome where the RNA sequence is chemically altered relative to that of the genome (Tan et al., 2017). In A-to-I editing, the adenosine deaminases acting on RNA (ADAR) family of enzymes catalyze the deamination of adenosine to form the base analog inosine (I), which is recognized as guanosine (G) by the splicing and translational machinery (Nishikura, 2010). ADAR proteins are a highly conserved family of proteins, containing a single deaminase domain (DD) as well as one or more double-stranded RNA (dsRNA)-binding domains (Phelps et al., 2015). The flexible nature of ADAR proteins, as well as the modular properties of their DDs, has made targeted RNA editing possible through either recruitment of endogenously expressed ADAR proteins to specific sites (Merkle et al., 2019; Qu et al., 2019) or fusion of these DDs to programmable protein modules (Montiel-Gonzalez et al., 2019). Both remain desirable strategies for achieving site-directed RNA editing in vivo.
The bacterial adaptive immune CRISPR-Cas9 system from Streptococcus pyogenes (SpCas9) has proven to be a powerful tool for manipulating eukaryotic genomes (Adli, 2018; Hsu et al., 2014). Most CRISPR-Cas9 genome editing applications use single guide RNAs (gRNAs) in complex with Cas9 nucleases to induce double-stranded DNA breaks at genomic target loci specified by a gRNA spacer sequence (Jinek et al., 2012; Mali et al., 2013). Such breaks can either be used to destroy gene function through the disruption of coding sequences or to facilitate specific gene edits through donor-template-driven homology-directed repair (Ran et al., 2013). These methods of genomic manipulation are irreversible, are potentially damaging to cells (Kosicki et al., 2018), and remain largely impractical for post-mitotic cell types that lack reliable DNA-break-repair mechanisms (Cox et al., 2015; Ran et al., 2013). Therefore, it has been of great interest to develop universally programmable methods to reversibly alter genetic information by targeting RNA instead.
Programmable targeting of cellular RNA by using RNA-targeting nuclease-dead SpCas9 (RCas9) was demonstrated in mammalian cells (Batra et al., 2017; Nelles et al., 2016) and in vivo to reverse disease-relevant phenotypes in a mouse model of myotonic dystrophy (Batra et al., 2020). This technology uses the capacity of Cas9 to bind specific RNAs when complexed with gRNAs that target nucleotide sequences that are absent of protospacer adjacent motifs (PAMs). Subsequent studies by other groups have demonstrated RNA-targeting capabilities for other Cas9 systems, including Campylobacter jejuni (CjCas9) (Dugar et al., 2018), Neisseria meningitidis (NmCas9) (Rousseau et al., 2018), and Staphylococcus aureus Cas 9 (SaCas9) (Strutt et al., 2018). All of these RNA-targeting Cas9 systems maintain RNA-binding capacity when DNA nuclease activity is inactivated, opening up the exciting possibility that RCas9 fusions to enzymatic RNA editing modules would allow programmable RNA editing at the single base level, analogous to Cas9-deaminase fusions acting on target DNA (Gaudelli et al., 2017; Kim et al., 2017b). Recent studies have demonstrated the natural and exclusive RNA-targeting capabilities of the Cas13 family of proteins, including Cas13a, Cas13b, and Cas13d, in vitro and in cells (Abudayyeh et al., 2017; Cox et al., 2017; Konermann et al., 2018). Similar to the RCas9 system, nuclease-dead versions of these Cas13 systems have been repurposed for effector module recruitment, including fluorescent proteins for dynamic RNA imaging (Yang et al., 2019), splicing factors for the regulation of alternative splicing and expression of specific protein isoforms (Konermann et al., 2018), and ADAR RNA DDs to dCas13b to direct A-to-I, as well as C-to-U, RNA editing (Cox et al., 2017; Abudayyeh et al., 2019).
CRISPR-Cas systems offer promise in the area of transcriptional editing, as the elements of these systems are genetically encodable and amenable to therapeutic viral delivery strategies (Ran et al., 2015; Konermann et al., 2018; Kim et al., 2017a). To date, dCas13b represents the only demonstrated example of an RNA-targeting CRISPR-Cas system capable of site-directed RNA base conversion, although the relative efficiency and specificity of this RNA editing approach compared to those of other CRISPR-Cas systems have not been systematically characterized (Vogel et al., 2018; Vogel and Stafforst, 2019). Moreover, it is still unclear if these or other potential Cas-directed RNA editing systems perform in a fully Cas-dependent manner (Qu et al., 2019). Here, we expand the Cas-mediated RNA editing toolbox by introducing an RCas9-ADAR2 platform capable of editing both reporter and endogenously expressed cellular transcripts in live cells. In characterizing this system, we also discovered a unique Cas9-dependent RNA-aptamer binding mechanism that is independent of a gRNA spacer sequence previously thought to be necessary for both DNA and RNA binding. To assess relative editing efficiencies and specificities of these Cas-based platforms, we assembled and tested several combinations of nuclease-dead Cas9 and Cas13-ADARDD fusions for a side-by-side comparison of RNA editing potential. Finally, we performed RNA sequencing (RNA-seq) for a subset of these platforms with respect to a shared mRNA target to assess transcriptome-wide off-target biases for each of these systems. Our results demonstrate the comparable efficacy of an RCas9-mediated RNA editing platform and define the parameters and limitations of currently available CRISPR-Cas-based RNA editing tools.
RESULTS
RNA-Targeting Cas9 Fused to ADAR2DD Supports A-to-I Editing in Live Cells
To evaluate if Cas9 can mediate RNA editing, we fused the DD of human ADAR2 (ADAR2DD) to the N terminus of catalytically inactive S. pyogenes Cas9 (dCas9), separated by an XTEN peptide linker (Figure 1A). To achieve site-specific A-to-I editing, we modified the gRNA to contain an additional 3′ terminal sequence designed to mimic the dsRNA substrate for ADAR2DD when base paired to the target RNA, presenting a mismatched bulged cytidine base opposite of the targeted adenosine (Figures 1A and 1B). A-C mispairings have been shown to facilitate ADAR editing in previously described Cas13 or aptamer-based systems (Wong et al., 2001; Cox et al., 2017; Vogel et al., 2018). These components together comprise an RCas9-ADAR2DD system that can be delivered to mammalian cells. To determine levels of Cas9-independent background editing, we also created a matched human ADAR2DD-only control (Figure 1A).
Figure 1. Editing Cellular Transcripts by using RCas9-ADAR2DD.

(A) Schematic of RCas9-ADAR2 deaminase domain (DD) fusion constructs and modified guide RNA (gRNA) constructs fused to an W58X EGFP reporter.
(B) Schematic design of RCas9-directed RNA editing strategy by using RCas9-ADAR2DD fusion proteins. A 3′ extension sequence forms a double-stranded RNA (dsRNA) substrate with the target RNA and forms a bulged residue to allow for directed adenosine (“A”) base editing.
(C) Sanger electropherograms and representative fluorescent images for EGFP+ cells measuring successful RNA editing of the W58X reporter in an RCas9-ADAR2DD, hyperactive mutant (RCas9-ADAR2DD(E488Q)) or ADAR2DD(E488Q) control expressing Flp-In T-REx 293 lines along with either a targeting or a reverse complement non-targeting (NT) 3′ extension (3′ex) sequence attached to the gRNA following doxycycline induction. As a transfection control, cells were also imaged for mCherry+ signal. Scale bar, 500 μm.
(D) Quantification of EGFP+ cells for Flp-In T-REx 293 cells following transfection with targeting or NT 3′ex gRNAs using fluorescence-activated cell sorting (FACS). EGFP+ percentages were calculated as a percentage of mCherry+ cells.
(E) Cells were transfected with a targeting 3′ ex gRNA in the presence of either an EGFP-targeting or scrambled, NT PAMmer sequence in both RCas9-ADAR2DD and RCas9-ADAR2DD(E488Q) expressing Flp-In T-REx 293 lines and quantified using FACS. Data are mean values ± SD with n = 1–3; unpaired two-tailed Student’s t test; *p < 0.05; *p < 0.01; ***p < 0.001; n.s., not significant. See also Figure S1.
To assess the efficacy of this RNA editing system, we generated stable Flp-In T-REx 293 cell lines containing single genomic copies of either an RCas9-ADAR2DD fusion or ADAR2DD-only control under an inducible Tet-On promoter. We then evaluated editing efficiency of these lines utilizing a previously established (Hanswillemenke et al., 2015) gain-of-function EGFP reporter containing a premature amber termination codon (UAG, W58X) which we cloned downstream of an mCherry cassette separated by a self-cleaving P2A peptide sequence (Figure 1A). Upon successful editing (UAG → UGG), EGFP signal is restored and detectable in cells (Figure S1A) (Hanswillemenke et al., 2015). Co-expression of RCas9-ADAR2DD with a targeting gRNA containing a complementary 3′ extension sequence led to successful EGFP editing at both mRNA and protein levels (Figure 1C), whereas a gRNA with a reverse complementary, non-targeting (NT) extension sequence or ADAR2DD-alone produced no detectable editing (Figures S1B and S1C). At the protein level, we observed comparable expression between RCas9-ADAR2DD and its hyperactive E488Q variant (Figure S1D). ADAR2DD-only controls exhibited protein expression (likely due to their shorter lengths) higher than Cas9-fusion proteins, and yet the editing efficiencies by them as measured by percentage of EGFP+ cells were non-existent (Figure 1D), further supporting that our on-target editing was RCas9-driven. Together, these results demonstrate that this directed editing system is dependent on both the specificity of the guide 3′ extension sequence and the Cas9 module. Surprisingly, an alternately designed gRNA with a 5′ extension sequence did not elicit this context-specific response, as the ADAR2DD control condition also resulted in editing with the gRNA (Figure S1E). Attempts to design a gRNA with a spacer sequence that could also serve as an editing substrate failed to achieve successful EGFP restoration as well (Figures S1F and S1G). We also evaluated a hyperactive E488Q ADAR2DD variant, which increased the editing level in the W58X EGFP reporter restoration at both RNA and protein levels, as expected (Montiel-González et al., 2016; Cox et al., 2017) (Figure 1C). The effects of this hyperactive mutant led to a ~2.5-fold increase in EGFP signal as measured by fluorescence-activated cell sorting (FACS) analysis (Figure 1D). Expression of only the ADAR2DD(E488Q) domain alone, in the presence of either targeting or non-targeting gRNAs, was not successful in restoring EGFP signal (Figures 1C and 1D), further highlighting the specificity of this interaction. Previous work showed that RNA-targeting using SpCas9 was augmented using a synthetic antisense oligonucleotide called a PAMmer, which carries a short DNA motif (the PAM of the form 5′-NGG-3′) (Nelles et al., 2016; Strutt et al., 2018). Similar to Batra et al. (2017), we observed that the PAMmer is dispensable for efficient on-target editing of the reporter RNA (Figure 1E).
RCas9 gRNA Spacer Sequence Is Dispensable for RNA Targeting
For initial design, we arbitrarily chose a spacer sequence targeting a region approximately 50 nt away from the targeted adenosine residue. To evaluate the optimal distance between where the spacer sequence recruits Cas9 on the target RNA and where the extension sequence directs ADAR2DD for editing, we tiled the mRNA transcript by using spacer sequences targeting sites up to 80 nt away from the extension sequence (Figure 2A). Surprisingly, the nucleotide distance between these gRNA sections did not show a detectable influence on target reporter editing efficiency, and although certain sites showed some target biases (e.g., 40 nt and 60 nt), modulation of the distance within a window of 5 to 80 bases between spacer and extension did not affect EGFP restoration (Figure 2B). Furthermore, removal of the spacer sequence from the gRNA (Figure S2A) conferred an editing efficiency comparable to that of spacer-containing guides (Figure 2B). This minimal “no spacer” (NS) gRNA was unaffected by the presence of a PAMmer sequence (Figure S2B) and failed to elicit background editing in the presence of the deaminating domain alone (Figure S2C). This result suggests that in the absence of a gRNA spacer sequence, Cas9’s interaction with the scaffold of the gRNA molecule is equivalent to an aptamer-binding effector fusion. Seeing that both target specificity and editing efficiency are driven by the 3′ RNA extension motif while still behaving in a Cas9-dependent manner, this led us to opine an “aptamer-binding model” in which RNA-RNA hybridization mediates RNA-protein interactions through dCas9-gRNA scaffold assembly (Figure 2C).
Figure 2. Spacer Sequence of the Modified gRNA Is Dispensable for RCas9-Mediated RNA Editing.

(A) Schematic of EGFP target mRNA and tiling gRNA designs. The 3′ex sequence (orange) remains constant, whereas the spacer sequence (blue) is variable in tiling EGFP mRNA reporter (black). The red box notes the original spacer sequence used in previous experiments.
(B) FACS quantification of EGFP+ cells for tiling gRNAs transfected into RCas9-ADAR2DD(E488Q) expressing Flp-In T-REx 293 cells. The number of nucleotides refers to spatial distance between spacer sequence target and edit site on EGFP mRNA. No spacer (NS) gRNA contains a targeting 3′ex sequence with no spacer complementary spacer sequence.
(C) Schematic for proposed “aptamer-binding complex” model that would feasibly allow for spacer-independent RNA targeting and editing.
(D and E) Heatmaps depicting targeted amplicon next-generation sequencing (NGS) of ACTB and CYFIP2 target and adjacent adenosine residues. Bar plots (right) summarizing A-to-I editing efficiency of target adenosine for both ACTB (A43) and CYFIP2 (A40). Data are mean values ± SD with n = 2–3; unpaired two-tailed Student’s t test; *p < 0.05; *p < 0.01; ***p < 0.001. See also Figure S2.
We next assessed the capacity for RCas9-ADAR2DD to modify endogenously expressed transcripts in cells. For our initial analysis, we chose to focus on a target site within the 3′ UTR of ACTB as well as a coding variant within the CYFIP2 gene, a known ADAR2 target transcript normally expressed but not edited in HEK293 cells. As we observed with reporter mRNA, we were able to achieve successful on-target editing within cellular targets ACTB and CYFIP2 by sing both spacer-containing and spacer-lacking gRNAs in 293XT cells through transient transfection (Figures 2D and 2E), which we quantified using next-generation sequencing (NGS). Editing for both of these targets were detected when both Cas9 and targeting gRNA were present, but gRNA alone or a non-targeting gRNA (NS-NT gRNA) failed to elicit detectible A-to-I editing. Moreover, ADAR2DD(E488Q) variants improve the on-target editing signal by 2- to 3-fold over the wild-type without dramatically increasing editing for adjacent adenosine residues. In this instance, NS gRNA performed as well if not slightly better than spacer-containing gRNAs for ACTB and CYFIP2 target sites. Although these editing rates were reported from transiently transfected cell lines, we were also able to verify successful editing in our stable Flp-In T-REx 293 cell lines (Figures S2D and S2E). This targeted editing is particularly noteworthy in the case of CYFIP2, as this process is normally mediated through exon-intron complementarity to allow the natural dsRNA substrate formation (Licht et al., 2016). We expanded this targeting mechanism to other target genes, including GAPDH and GUSB, which are not naturally edited by endogenous ADAR proteins (Figures S2F and S2G). We conclude that RCas9-ADAR2DD is capable of directing specific A-to-I edits on both reporter and cellular transcripts using a 3′ modified gRNA, highlighting a unique spacer-independent mechanism that can be used for RNA-targeting applications.
Comparison of RNA Targeting CRISPR Platforms Reveals Parameters Influencing On-Target Specificity
Studies have discovered other RNA-targeting CRISPR-Cas systems, of which some have been adapted as orthogonal A-to-I RNA editing platforms (Cox et al., 2017). However, these CRISPR-based platforms have not yet been compared to determine criteria that would make for an ideal RNA base editor. To investigate the site-directed RNA editing potentials for these systems, we cloned a representative panel of Cas-ADAR2DD(E488Q) fusions, in both ADAR2DD N-terminal (N-TERM ADAR2DD) and C-terminal (C-TERM ADAR2DD) orientations in the presence of either a nuclear localization signal (NLS) or nuclear export signal (NES), and co-transfected them into HEK293XT cells with respective W58X EGFP-targeting gRNA reporter constructs (Figures 3A and 3B). This diverse panel included SpCas9, SaCas9 (Strutt et al., 2018), Leptotrichia wadei Cas13a (LwaCas13a) (Abudayyeh et al., 2017), Prevotella sp. P5–125 Cas13b (PspCas13b) (Cox et al., 2017), and Ruminococcus flavefaciens XPD3002 Cas13d (RfxCas13d) (Konermann et al., 2018). For gRNA designs, Cas9 systems were outfitted with NS gRNAs with targeting or non-targeting 3′ extensions, and guides for Cas13 fusions were designed with the “bulge-containing” C-mismatch within the spacer sequence that determines the RNA targeting site. We decided to use these orientations for Cas13 modules (spacer-with-mismatch) based upon gRNA designs that were deemed to be optimal by the recently described RNA Editing for Programmable A to I Replacement (REPAIR) and RNA Editing for Specific C-to-U Exchange (RESCUE) methods (Cox et al., 2017; Abudayyeh et al., 2019; Figure 3B).
Figure 3. Comparative Analysis of Editing Efficiencies across Cas-ADAR2DD(E488Q) Platforms.

(A) Schematic depicting protein fusion orientations for editing comparisons. N-terminal ADAR2DD fusions were noted as being in the “N-TERM ADAR2DD” orientation, and C-terminal ADAR2 fusions were noted as being in the “C-TERM ADAR2DD” orientation. Domains were separated by an “XTEN” peptide linker, and subcellular localization was determined by a 2× SV40 nuclear localization signal (NLS) or HIV1 cytoplasmic localization signal (nuclear export signal [NES]).
(B–D) Visual representations of gRNAs being tested (B). The respective RNA-hybridization sequences (orange) are marked with the mismatched base (green). High-throughput EGFP+ FACS analysis was performed for both orientations on NLS (C) and NES (D), containing Cas-ADAR2DD fusions through co-transfection of HEK293XTs after 48 h. Samples were measured in both orientations by using specific EGFP-targeting or scrambled NT gRNAs for each Cas species. Under all conditions, n = 3. Data represented are mean values ± SD.
(E and F) Targeted amplicon NGS of ACTB (E) and CYFIP2 (F) target regions transiently transfected with SpCas9 (RCas9), Cas13b, and Cas13d along with targeting and NT gRNAs. RNA was collected, and libraries were prepared 48 h after transfection. Heatmaps (left) depict relative A-to-I editing ratios for both target and adjacent adenosine residues, and bar plots (right) represent A-to-I editing efficiency of target adenosine for both ACTB (A43) and CYFIP2 (A40) in replicate, with n = 2–3. Data represented are mean values ± SD. See also Figure S3.
Except for LwaCas13a, we found that editing efficiency as determined by EGFP correction was comparable across all nuclear-localized systems tested in the N-TERM ADAR2DD orientation (N-terminal ADAR2DD; Figure 3C). Importantly we found that C-TERM ADAR2DD fusion orientations (C-terminal ADAR2DD), although resulting in the highest editing rate, also yielded high background reporter editing with non-targeting gRNAs for all Cas9 and Cas13 fusions, sometimes even exceeding targeting gRNA editing. We conclude that certain orientations may be subject to non-specific background editing, at least when domains are connected through an XTEN linker. Of the N-TERM ADAR2DD fusions that reported >30% editing, the background-subtracted editing rate relative to non-targeting gRNA controls was comparable between SpCas9 and SaCas9, whereas PspCas13b seemed to have the highest signal for N-terminal ADAR2DD fusions among Cas13 proteins (Figure S3A). The addition of an NES to all fusions yielded similar results, in some cases raising editing rates by a few percentage points (SaCas9, which had the highest overall signal), whereas others experienced no change or a minor decrease in editing efficiency (SpCas9 and RfxCas13d; Figure 3D; Figure S3B). Seeing that editing rates were not appreciably different between nuclear and cytoplasmic localization, choosing between the two options will likely depend on the target and context, bearing in mind recent reports that off-target editing might be more prominent with cytoplasmic localization (Vallecillo-Viejo et al., 2018). Additionally, single-stranded RNA molecules of a certain length may be sufficient to trigger endogenous ADAR activity to result in editing though RNA-RNA hybridization kinetics alone (Qu et al., 2019). Although Cas13- and Cas9-driven fusions had comparable editing efficiency with respect to the EGFP reporter, expression of ADAR2DD(E488Q) alone was able to confer a noticeable editing signal for both Cas13b and Cas13d gRNAs (Figure S3C). We believe that this observation might be related to the length of the RNA-RNA hybridization region, as gRNAs with target hybridization lengths of 30 nucleotides (nt) or longer had increased background editing potential (Figures S3D and S3E) or local off-target edits (Figures S3F and S3H). Regardless of the mechanism, this result is consistent with findings of Vogel et al. (2018) who showed that 50-nt REPAIR RNA editing is in part Cas independent and conferred by expression of the guide/ADAR2DD combination alone (Vogel et al., 2018).
We next tested the performance of these Cas-based systems on endogenously expressed transcripts in live cells. To assess editing efficiency at a per-read level, we performed targeted-amplicon-specific NGS on target sites of endogenously expressed transcripts ACTB and CYFIP2 following co-transfection of N-terminal ADAR2-Cas fusions and respective gRNAs into 293XT cells. For this panel, we focused on characterizing SpCas9 (simply Cas9 going forward) alongside the two most successful Cas13 modules in our hands: Cas13b and Cas13d. Similar to what we had seen through EGFP restoration, Cas9 in both spacer-dependent and -independent contexts performed targeted A-to-I editing at a similar efficiency to both Cas13b and Cas13d (Figures 3E and 3F). Additionally, sequence edits called were seemingly confined to the targeted adenosine residues, even with the introduction of the hyperactive E488Q mutation to the Cas9-ADAR2DD fusion, although in the case of CYFIP2, there appeared to be instances of adjacent base editing in all targeting gRNA contexts (Figure 3F). This off-target editing may highlight a recurrent tradeoff between base specificity for editing efficiency that should be considered when using RNA ed- iting technologies.
Transcriptome-wide RNA-Seq Uncovers Consequences of RNA-Targeting CRISPR-Editing Platforms
Next, RNA-seq was used to systematically assess off-target consequences from the expression of Cas-ADAR2(E488Q)DD systems in HEK293XT cells. Cas9, Cas13b, and Cas13d fusions in the N-TERM ADAR2DD-NLS orientation were introduced with either a CYFIP2-targeting or scrambled NT gRNA control. After 48 h, RNA-seq libraries were prepared and sequenced to 42 million reads on average. Aligned reads were downsampled prior to variant calling to allow for comparisons unaffected by variable sequencing depth. We processed aligned reads using the SAILOR algorithm (Deffit et al., 2017) specifically designed to identify high-confidence A-to-I editing events from transcrip- tome-wide sequencing data (Figure 4A). SAILOR assigns a con- fidence score for A-to-G mismatches that differ from the genome using a beta distribution factoring in both site coverage and editing percent following removal of annotated hg19 SNPs that may be falsely identified as hits (Washburn et al., 2014). For our analyses, only events with a confidence score exceeding 90% detected in multiple replicates were considered consensus off-target events. This pipeline identified several thousand A-to-I editing events with a transient expression of Cas-ADAR2(E488Q)DD editing systems, ranging from as few as 3,336 events with Cas13b with a CYFIP2-targeting gRNA to as many as 5,545 events with Cas9 co-expressed with a NS CYFIP2-targeting gRNA (Figure 4B; Table S3). Although the number and percentage of editing events seemed to vary between Cas conditions, all unsurprisingly were elevated over an untransfected control condition (900 A-to-I edits). We also confirmed that the mRNA expression of each of the respective fusion proteins was similar by the transcripts per million (TPM) metric (Figure S4A). Although the distribution of editing percent was similar between all conditions, Cas-ADAR2(E488Q)DD fu- sions introduced more “subtly” edited events, whereas basally detected A-to-I edit events in untransfected cells were mostly above 10% (Figure 4B). Many of these events were “novel” in nature, meaning that only a minority were found to overlap pre- viously identified ADAR targets or naturally occurring editing events found in untransfected HEK293XT cells (Figures S4B and S4C). This result illustrates that off-target events detected occur largely de novo and are not simply an exacerbation of cellular RNA editing events.
Figure 4. Global RNA-Seq Reveals Transcriptome-wide Off-Target Consequences across Cas-ADAR2DD(E488Q) Platforms.

(A) Outline of RNA-seq processing pipeline to identify global A-to-I edits. Strip plots illustrating the number of global A-to-I identified under each experimental condition, as well as percent edited per site (y axis). Also represented are the total consensus editing events identified from untransfected 293T cells. Editing rate calculation for experimental RNA-seq samples consists of n = 2 individual replicates together, and untransfected control data were generated with n = 3 replicates.
(B) Pairwise scatterplots illustrating relative editing efficiencies between Cas proteins. The target edit site (CYFIP2 CDS) is colored orange and indicated with an arrow. Green and blue events symbolize those detected exclusively in sample 1 (x axis) or sample 2 (y axis).
(C) Three-way Venn diagram illustrating the number of reproducible edits between Cas conditions. The majority of off-target events appeared to be unique in the cases of Cas9 and Cas13d, which contained the highest number of global editing events.
(D) Sequence logos of edited adenosine residues identified from RNA-seq data in the presence of CYFIP2-targeting gRNA and Cas13b, Cas13d, or Cas9. See also Figure S4.
With respect to on-target editing, successful CYFIP2 editing was detected only in the presence of a targeting gRNA for each of the Cas constructs tested, again highlighting the gRNA- dependent nature of Cas-directed RNA editing (Figure S4D). In directly comparing on-target efficiencies of Cas-constructs, we noticed slightly higher on-target editing rates for Cas9 and Cas13d than those for Cas13b (Figure 4C); however, this effect is counterbalanced by the observation that these fusions tended to produce a higher number of global editing events as well. Although a subset of editing sites overlapped between Cas proteins, most sites seemed unique to experimental condition, indicating that there were few “hot spots” and that the majority of off-target edits introduced were randomly deposited across the transcriptome as a secondary consequence of Ca- s-ADAR2(E488Q)DD overexpression (Figure 4D). However, sequence preference was similar transcriptome wide among all conditions, with edits predominantly being sequestered into (U/A/C)AG trinucleotide sequences, which were previously described as being favorable edit motifs for ADAR family proteins (Figure 4E; Figure S4E; Kim et al., 2004). We also observed an inverse relationship between read coverage and editing percent per event, for which the extent of off-target events appeared to decrease with increasing coverage (Figure S4F). Although this observation is not a direct commentary on the severity of downstream transcriptional consequences, it does insinuate that highly edited events tend to be expressed at a relatively low level in relation to the remainder of the transcriptome.
In characterizing the nature of these off-target edits, we noticed that that majority of high-confidence edits detected were in the 3′ UTR (44%–49%) or protein-coding sequences (CDS) (22%–38%) of annotated genes (Figure 5A; Figure S5A). These edits are potentially consequential, with up to 70% of variant effects in the CDS corresponding to a predicted missense mutation in at least one protein-coding isoform (Figure S5B). This general phenomenon was observed across all editing platforms regardless of gRNA, indicating that off-target effects are largely a product of non-specific effects of ADAR2(E488Q)DD overexpression. It is worth noting that the majority (52.3%–69.7%) of global A-to-I edits tend to be low (less than 20% editing) for all the RNA editors tested (Figure 5B). Although off-target rates will inevitably decline with the discovery of improved base editors (Cox et al., 2017; Abudayyeh et al., 2019), it is important to note that all present Cas platforms tested behaved similarly transcriptome-wide and, thus, are seemingly prone to the same benefits and limitations with respect to tar- geted RNA editing.
Figure 5. Nature of Global RNA Edits Introduced through Cas-ADAR2DD(E488Q) Expression.

(A) Regional distribution of total edits per Cas condition with CYFIP2-targeting gRNA. The vast majority of edits identified were compiled in gene bodies, particularly in 3′ UTR and CDS for all Cas proteins. The number of events per annotated region are located in parentheses.
(B) Histograms illustrating the distribution of identified sites by editing rate. The vertical lines depicted are meant to signify the fraction of sites edited less than 20% under each condition. See also Figure S5.
DISCUSSION
Programmable transcriptome manipulation offers the ability to transiently alter genetic information without conferring permanent changes to the genome. Site-directed RNA editing tools are currently under rapid development to allow for a high-precision single-nucleotide editing capability. Due to the well-characterized biological function and modular structure of ADAR family proteins, most of these technologies direct ADAR DDs to a target adenosine for A-to-I conversion by using a gRNA bearing a mismatched cytidine base. There have been two major strategies in designing such base-editing systems: (1) recruitment of endogenous ADAR proteins by using synthetic oligonucleotides (Qu et al., 2019; Wettengel et al., 2017; Woolf et al., 1995) or( 2) exogenous expression of DDs fused to effector proteins or oligonucleotides (Azad et al., 2017; Cox et al., 2017; Montiel-González et al., 2016; Stafforst and Schneider, 2012; Vogel et al., 2018; Katrekar et al., 2019). Engineered ADAR fusions have an advantage in that they can be encoded genetically and are not dependent on the requirement for host cells to express ADAR proteins. Previously established ADAR fusion systems use the aptamer- binding lambda N (λN) and MS2 coat proteins, which bind BoxB and MS2 RNA hairpins adjacent to ADAR directing sequences within gRNAs (Azad et al., 2017; Montiel-González et al., 2016; Katrekar et al., 2019), to perform target-specific RNA editing in mammalian cells. Even more recently, RNA-tar- geting Cas13b proteins have been showcased as ADAR effector fusion systems capable of directing both A-to-I (REPAIR) and C- to-U (RESCUE) edits on target transcripts (Abudayyeh et al., 2019; Cox et al., 2017). Cas13b represents just one of many RNA-targeting Cas proteins with potential utility as programmable RNA editors, including the other characterized Cas13 family members Cas13a and Cas13d (Abudayyeh et al., 2016; Konermann et al., 2018), as well as dual DNA-RNA targeting Cas9 (RCas9) proteins, including SaCas9 and SpCas9 (Batra et al., 2017; Nelles et al., 2016; Strutt et al., 2018).
In this work, we demonstrate an additional function for the RCas9 system by fusing catalytically inactivated SpCas9 to ADAR2DD to perform programmable A-to-I RNA editing on both reporter and cellular mRNA targets. RCas9-ADAR2DD edting is Cas9 and gRNA dependent, as overexpression of ADAR2DD in the presence of a targeting gRNA alone is insufficient to trigger RNA editing (Figures 1C and 1D). Although previous reports have stressed the significance of a spacer sequence and complementary PAMmer for RCas9 RNA localization and binding (Nelles et al., 2016; O’Connell et al., 2014; Liu et al., 2019), we find that RCas9-ADAR2DD is not reliant on a synthetic PAMmer oligonucleotide (Figure 1E) or even spacer sequences (Figures 2A and 2B), which simplifies delivery and expands targeting capacity. We find that RCas9-ADAR2DD is only reliant upon 21-nt extension sequences located on the 3′ terminus of the gRNA. We hypothesize that RCas9-ADAR2DD editing is enabled through hybridization of this extension with the target RNA and recruitment of the ADAR2DD fusion to the forced dsRNA substrate by RCas9 (Figure 2C). This targeting approach is mechanistically reminiscent to the recently described CRISPR-Cas-inspired RNA targeting system (CIRTS), which re- lies only on Watson-Crick-Franklin base pairing through a gRNA to recruit functional ribonucleoprotein (RNP) complexes to specific sites on RNA (Rauch et al., 2019). Placing the A-C mismatch within the spacer region of the Cas9 gRNA could not induce successful targeting and RNA editing (Figures S1F and S1G), as has been observed with class 2 type VI RNA-guided RNA-targeting CRISPR-Cas systems (Abudayyeh et al., 2019; Cox et al., 2017), and hints at differences among various Cas systems in terms of the RNA-targeting mechanism.
The aptamer-binding model of catalytically inactive Cas9 to scaffold-containing gRNA for specific RNA-targeting could potentially be a useful tool for other applications requiring targeted protein tethering to specific RNA molecules bearing the gRNA scaffold sequence, analogous to the well-characterized MS2 coat protein and λN-BoxB systems (Keryer-Bibens et al., 2008). Seeing that the affinity for Cas9 binding to its gRNA scaffold is potentially tighter than the binding affinity of these other coat protein systems, with Cas9 being shown to bind gRNA hairpins in the picomolar range (Wright et al., 2015), it will be of great interest to determine the search mechanism used by Cas9- effector fusions targeted to RNA with 3′ gRNA extensions and whether this minimal design represents a universal Cas9-RNA- targeting rule. Furthermore, this alternative “spacerless” gRNA strategy expands the scope of RNA sites that can be targeted using RCas9, as this gRNA is no longer restricted to non-PAM adjacent target sequences, a constraint in designing target sites for RCas9 to prevent binding to genomic DNA. Notably, SaCas9 was also capable of RNA editing with a NS gRNA configuration (Figures 3C and 3D), reinforcing the proposed aptamer-binding model across bacterial species and illustrating the adaptability of this strategy to other Cas9 proteins.
We also evaluated the ability of RCas9-ADAR2DD fusions to edit endogenously expressed mRNAs in living cells. Like previously published RNA technologies, we show programmable and specific A-to-I editing within the 3′ UTRs of housekeeping genes not normally edited by endogenous ADAR (Figure 2D) and within the coding sequence of the ADAR target mRNA CYFIP2 (Fig- ure 2E), which is normally edited through co-transcriptional exon-intron pairing. Because this editing platform can perform specific RNA editing on a variety of transcripts across multiple genic regions, it can readily be used for biologically relevant appli- cations to study the effects of individual RNA editing events and to reverse G-to-A mutations associated with disease cause or risk.
Seeing that the RCas9 system functions by repurposing a naturally DNA-targeting protein to recognize cognate RNA se- quences, it is conceivable that that either natural RNA-targeting class 2 type VI Cas13 proteins would direct RNA editing with a higher efficiency or specificity than Cas9 family members or that Cas9 and Cas13 family members behave differently from one another in directed editing applications. To test this, we per- formed comparative analyses of available CRISPR-based RNA editing tools in various structural orientations and cellular localizations. Here, we found comparable on-target EGFP-reporter and cellular mRNA editing rates between Cas9 and several Cas13 ADAR2DD fusions (Figures 3C–3F). We observed that targeted RNA editing is context specific, as an alteration of orienta tion, and to a much lesser degree subcellular localization, could alter both on-target and non-specific RNA editing (Figures 3C and 3D). We also see some degree of background editing for Cas13b and Cas13d gRNAs, perhaps through recruitment of endogenous or overexpressed ADAR proteins (Figure S3C). This background editing may be caused by a number of factors, including hybridization length, as well as direct-repeat (DR) length or complexity, as gRNAs that have smaller DRs (Cas13b: 36 nt, Cas13d: 30 nt) tend to have higher backgrounds than those with longer scaffolds (SaCas9: 76 nt, SpCas9: 85 nt), although this mechanism is unclear. Background editing may also be due to hybridization of the duplex-forming portion of these bulge-containing guides, as there appears to be some length-dependent increases in both on-target and background editing rates in the case of Cas13b gRNAs (Figures S3D–S3I). Together, we were able to observe comparable on-target and off-target activity of our Cas9 RNA editors despite their secondary purpose as RNA-binding proteins.
In addition to evaluating the local, on-target editing rate with Cas-ADAR2DD systems by using targeted-amplicon NGS, we characterized the global off-target capacity of each of these systems by using poly(A)+ RNA-seq. Similar to our observation of comparable on-target efficiency between these Cas systems, we identified a widespread number of off-target edits for each of the systems we tested (Cas13b, Cas13d, and Cas9). Although we were able to identify our targeted editing event (CYFIP2) by sequencing (Figure S4C), all conditions generated several thou- sand potential off-target A-to-I editing events, with the highest numbers being detected in Cas13d (5,407) and Cas9 (5,545) (Figure 4B). The precise number of edits was variable among Cas protein and gRNA and was largely independent of gRNA design, as non-targeting gRNAs produced a comparable number of edits compared to those of their targeting counterparts (Figure 4B). These edits were largely random and artificial in nature, as most sites did not overlap with known editing loci or previously characterized Alu sites, which comprise >95% of endogenous A-to-I editing modifications (Levanon et al., 2004). Instead, most detected editing sites were deposited in the CDS and 3′ UTR of genes (Figure 5A; Figure S4F), which could spell downstream consequences for the protein-coding capacity and stability of target transcripts (Figure S4G). We find that most of the global editing events skewed low based upon percent editing (Figure 5B) and that events with the highest sequencing coverage tended to have the lowest editing rate (Figure S4E). Overall, both the number of editing events and degree of off- target editing must be considered limitations in these cases of post-transcriptional genome manipulation, although this approach may still be a viable alternative to DNA editing so long as RNA editing tools are deployed in a transient manner.
Due to their encodable and highly programmable nature, as well as demonstrated RNA-targeting capacity, CRISPR-Cas technologies remain tremendously promising for targeted RNA base editing. In this work, we expand and benchmark the list of orthogonal and available RNA editing CRISPR technologies. This study furthers our understanding of Cas-based technologies and helps to set the stage for further optimization. So far, rational, directed, and evolution-based approaches have been used to expand the sequence-targeting toolbox of both DNA and RNA base editors (Abudayyeh et al., 2019; Thuronyi et al., 2019). We believe that a combination of both of these engineering approaches, as well as a firm understanding of the sequence-based targeting “rules” of RNA-guided RNA-targeted CRISPR-Cas systems, will be necessary to further develop these powerful platforms in order to accomplish highly efficient and specific therapeutics for disease-relevant mutations.
STAR+METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gene W. Yeo (geneyeo@ucsd.edu).
Materials availability
Important plasmids described in this study will be deposited in the Addgene plasmid repository and available under a standard MTA.
Data and code availability
The accession number for the RNA sequencing data generated for this study is Gene Expression Omnibus: GSE152684. Uncropped western blots and original FACS data have been deposited to Mendeley Data: [https://doi.org/10.17632/99m44kn43w.1]. Software for SAILOR editing analysis is available on Github (https://github.com/YeoLab/sailor).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plasmid construction
For dCas9-ADAR2DD mammalian expression constructs, dCas9–2XNLS was amplified from pCDNA3.1- dCas9–2xNLS-EGFP (Addgene plasmid #74710) and fused to the human ADAR2 deaminase using Gibson assembly into a pcDNA(−)3.1 (Invitrogen) backbone, which had been digested using FastDigest EcoRI (Thermo Scientific). Domains were designed to be separated by an XTEN linker peptide, the sequence of which was included in amplification primers. The ADAR2DD-XTEN control plasmid was generated by eliminating the dCas9–2XNLS domain through inverse PCR using the primers ADAR2_CD_Inverse_F and ADAR2_CD_Inverse_R (primer sequences located in Table S1), followed by ligation with T4 DNA Ligase (New England Biolabs). Inverse PCR and ligation was also used to perform site-directed mutagenesis to generate the E488Q variants using the primers E488Q_Mut_F and E488Q_Mut_R. These fusions were then amplified and cloned into the pcDNA5/FRT/TO (Invitrogen) backbone using HindIII and NotI restriction sites for stable line generation. For comparison studies, sequences for an HA affinity tag in frame with either a 2XNLS or HIV1 NES localization sequence was cloned into the pcDNA(−)3.1 multiple cloning site using EcoRI and Hin- dIII restriction sites. These constructs were then digested with EcoRI and used as backbones into which both AXC and CXA fusions were integrated via Gibson assembly.
For each 3′ extension Cas9 guide RNA constructs, the gRNA scaffold sequence was amplified from pBluescriptSKII+ U6- gRNA(F+E) empty (Addgene plasmid #74707) with forward primer containing the desired spacer sequence with and reverse primer containing the 3′ extension sequence on the 5′ tail of the oligonucleotide (spacer and extension sequences located in Table S2), as well as complementary sequences for Gibson assembly (5′- AAAGGACGAAACACC-3′ for forward primer, 5′- CCCGGGCTGCAG GAAAAAAA-3′ for reverse primer). These products were then Gibson assembled into the pBluescriptSKII+ U6-gRNA(F+E) backbone which had been inversely amplified using gRNA_backbone_F and gRNA_backbone_R primers. Other Cas-construct gRNAs were assembled in this fashion using primers to amplify SaCas9 gRNA (IDT gBlock of 3F gRNA sequence described in Chen et al., 2013, LwaCas13a gRNA (a gift from Feng Zhang; Addgene plasmid # 91906), PspCas13b gRNA (a gift from Feng Zhang; Addgene plasmid #103854), and RfxCas13d gRNA (a gift from Patrick Hsu; Addgene plasmid # 10905).
For the dual fluorescence reporter, the W58X EGFP coding sequence was PCR amplified from the pCDNA3.1-EGFP(W58X) construct (a gift from Thorsten Stafforst’s lab, University of Tu€bingen), and fused to P2A-mCherry sequence using Gibson assembly downstream of an Ef1a constitutive promoter. This sequence was then inserted into the pBluescriptSKII+ U6-gRNA(F+E) backbone using the SmaI restriction site and Gibson assembly to allow for simultaneous gRNA and reporter delivery to cells.
Human cell culture conditions and maintenance
The Flp-In T-REx 293 cells (Invitrogen) and lenti-X HEK293T cells (HEK293XT, Takara Bio) are derived from transformed female hu- man embryonic kidney tissue. Cells were maintained in DMEM (4.5 g/L D-glucose) supplemented with 10% FBS (GIBCO) at 37°C with 5% CO2. Cells were periodically passaged once at 70%–90% confluency by dissociating with TrypLE Express Enzyme (GIBCO) at a ratio of 1:10. Additionally, Flp-In-293 cells were maintained under 5 ug/ml Blasticidin S (GIBCO) to ensure presence of an inte- grated FRT site. These lines were purchased directly from manufacturers.
Generation of Flp-In 293 cell lines
Generation of the Tet-inducible dCas9-ADAR2DD lines was performed according to the manufacturer’s instructions. Open reading frames were cloned into the pcDNA5/FRT/TO backbone as previously described. Once reaching 70%–90% confluency on a 10cm2 dish, Flp-In T-REx cells were transfected with 9ug of plasmid and pcDNA5 plasmid and 1ug of pOG44 Flp-Recombinase Expression Vector (Invitrogen) using polyethylenimine (PEI, Sigma Aldrich) in 250 ul Opti-MEM (GIBCO). Cells were passaged with TrypLE to 25% confluency 48 h after transfection and incubated for an additional 2–3 h at 37°C with 5% CO2. Cells were then selected with 200 ug/ml Hygromycin B (GIBCO) for 3–4 days until individual clones could be picked, expanded, and genotyped.
Western Blot
Cells were lysed in 1% NP-40 lysis buffer (20 mM Tris HCl pH 8, 137 mM NaCl, 1% Nonidet P-40, 2 mM EDTA) and quantified with the Pierce BCA Protein Assay Kit (Thermo Fisher). After quantification, 10 mg total protein lysate was loaded on a 4%–12% NuPAGE Bis- Tris gel (Thermo Fisher) and transferred onto a PVDF membrane. Membranes were blocked and probed with primary and secondary antibodies in 5% milk and imaged with Pierce ECL Western Blotting Substrate (Thermo Fisher). For immunoblotting, the following antibodies were used: anti-HA.11 Epitope Tag Antibody (Biolegend, # MMS-101R), anti-GAPDH (Abcam, # ab9485), Goat anti- Mouse IgG (H+L) Secondary Antibody, HRP (# 32430).
Transient transfection of human cell lines for EGFP restoration
For comparison experiments, cells were seeded in a 24-well plate 24 h prior to transfection. At approximately 70%–80% confluency, cells were transiently transfected with 500 ng gRNA/EGFP reporter plasmid for stable lines, or 300 ng gRNA/EGFP reporter and 200 ng Cas9-ADAR2 fusions for HEK293XT cells using Lipofectamine 3000 Reagent (Invitrogen) per well, according to manufac- turer’s instructions. For endogenously expressed targets, 500 ng gRNA plasmids were transfected per well. For experiments with PAMmers, 0.5 pmol was transfected with Lipofectamine RNAiMAX (Invitrogen) immediately after DNA transfection. For experiments involving Flp-In T-REx lines, fusion protein expression was induced by adding doxycycline (Sigma Aldrich) at a final concentration of 1 ug/ml to cell media. Experiments unless otherwise noted were performed in triplicate. After 48 h, cells were dissociated with TrypLE and harvested for both RNA and FACS analysis. Data were plotted and statistics were calculated using Prism 6 software.
RNA editing of W58X EGFP reporter using FACS
Following 48 h post transfection, cells were rinsed once with 1X DPBS (Corning) and dissociated with 400 ul TrypLE for 5–10 min at 37°C. Cells were then resuspended in FACS Buffer (10% FBS in 1X DPBS), strained over a 35 um nylon mesh, collected in round- bottom Falcon tubes and subjected to fluorescence-activated cell sorting (FACS) analysis using a BD LSRFortessa flow cytometer. After excluding cellular debris and doublets, transfected cells were first gated on mCherry-positive signal. The overall fraction of EGFP-positive cells in this mCherry+ population as calculated as a readout for successful RNA editing. FACS data were analyzed and plotted using the FlowJo software package.
For comparison experiments, HEK293XT cells were seeded on 48-well plates 24 h prior to transfection. When cells were 70%–80% confluent, they were co-transfected with 300 ng respective gRNA/EGFP reporter and an equimolar amount of Cas-ADAR2DD expression plasmid (for reference, SpCas9-ADAR2 2XNLS fusion: 0.007 pmol = ~50ng) in triplicate. After 48 h, cells were dissociated with TrypLE, resuspended in FACS buffer as previously describes, and transferred to flat-bottom 96-well plates. Samples were then processed on High Throughput Sampler (HTS) mode on a BD LSRFortessa X-20 instrument. Gating and subsequent analyses was performed as described above, where approximately 25,000 live cell events were counted for each sample.
Fluorescence visualization of live cells
After 48-hours following transfection and dCas9-ADAR2DD protein expression, live cells were subject to fluorescence imaging at 10–20X using a Zeiss Axio Vert.A1 fluorescence microscope equipped with an X-Cite 120Q illumination system. Images were captured at 20 ms for brightfield and mCherry, and 180 ms for EGFP fluorescence at 1X gain. Images were processed with compat- ible ZEN software, then exported and adjusted for brightness/contrast by matching Maximum/Minimum values across samples for fluorescent images in analysis software FIJI (Schindelin et al., 2012).
Extraction of RNA and RT-PCR
RNA isolations were carried out by resuspending cells 500 ul TRIzol reagent (Invitrogen) and extracting using the Direct-zol RNA Mini- prep kit (Zymo Research) according to manufacturer’s instructions. Samples were eluted in 25–50 ul nuclease-free H2O and concentrations were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific). For each sample, 500 ng to 1 ug of total RNA was reverse transcribed to cDNA with Superscript III Reverse Transcriptase (Invitrogen) using oligo(dT) and random hexamers ac- cording to manufacturer’s instructions. From this cDNA, 1 ul was taken and amplified using flanking primers for 35 cycles. Amplified products were extracted using QIAquick PCR Purification Kit (QIAGEN) and subjected to Sanger sequencing. When quantifying Sanger traces, editing efficiencies were calculated using FIJI software using the following metric: (height of ‘G’ peak) / ((height of ‘G’ peak) + (height of ‘A’ peak)) at each target site.
Targeted RNA editing analysis for endogenous transcripts
For targeted amplicon next-generation sequencing (NGS), HEK293XT cells were first seeded onto 24-well plate 24 h prior to transfection. When cells reached 70%–90% confluency, cells were co-transfected with 100 ng Cas-ADAR2DD plasmid and 400 ng of respective gRNA plasmid using Lipofectamine 3000. After 48 h, RNA was extracted using TRIzol reagent and 10 ng total RNA was reverse transcribed using Protoscript II Reverse Transcriptase (New England Biolabs) with target-specific reverse primers ac- cording to manufacturer’s instructions. 4 ul cDNA was amplified for 15 cycles using target-specific primers containing partial Illumina adapters using NEBNext Ultra II Q5 polymerase (New England Biolabs). PCR products were purified using AMPure XP beads (Beck- man Coulter) and 2ul of the first reaction was subjected to a second round of PCR for 15 cycles to affix Illumina-compatible indices (based upon TruSeq RNA adapters #1–14) using NEBNext. Reactions were purified again using AMPure XP beads, size-checked and quantified on the Agilent 2200 Tapestation system using D1000 ScreenTape reagents, pooled equally and diluted to 2nM final con- centration, spiked-into high complexity RNA-seq libraries at 1% of input material and read out on a 75-nt single-end run with an Illu- mina HiSeq4000 instrument.
Successfully sequenced libraries were first subject to quality control using FastQC, then aligned using Burroughs-Wheeler Aligner (BWA) (Version: 0.7.15-r1140) (Li and Durbin, 2009) using the command ‘bwa mem -t 8 (index.db) (sample.fq)’. Aligned BAMs were sorted using Samtools (Li et al., 2009). Editing percentage per base position was calculated at each annotated adenosine (A) residue by calculating:
Percent editing for each adenosine residue in each 75-nt single-end read for both CYFIP2 and ACTB mRNA is represented.
Transcriptome-wide RNA sequencing
RNA editing analysis was performed in transiently transfected HEK293XT cells. Cells were first seeded onto a 24-well plate format, 24 h prior to transfection. Upon reaching 70%–90% confluency, cells were co-transfected with 100 ng Cas-ADAR2DD plasmid along with 400 ng of either a scrambled, non-targeting or CYFIP2-targeting gRNA plasmid using Lipofectamine 3000. After 48 h incubation, RNA was extracted using TRIzol reagent and Direct-zol RNA purification reagents. Following elution, 100 ng of total RNA was quantified and used as input for poly(A)+ library preparation using TruSeq Stranded mRNA preps (Illumina) according to manufacturer’s instructions. All libraries were sequenced on an Illumina HiSeq 4000 platform (100bp, paired-end setting) with the exception of the untransfected control HEK293XT samples, which were sequenced on an Illumina NovaSeq 6000 platform (100bp, single-end setting) during a separate experimental run.
RNA-seq analysis
RNA-seq reads were adaptor-trimmed using Cutadapt (version 1.14) (Martin, 2011) and aligned to the hg19 (GRCh37) genome using STAR (version 2.5.2b) (Dobin et al., 2013)using default options, and subsequently sorted with samtools (version 1.5). Transcript-per- million (TPM) normalized counts were calculated following raw counts quantitation using featureCounts (version 1.5.0) (Liao et al., 2014). For quantification of Cas-ADAR2 fusions, raw reads were mapped to the hg19 reference genome with Cas-ADAR2 sequences added. For editing analysis, to accommodate sensitivity toward differing sequencing depths in variant calling, aligned reads were then randomly downsampled to 16–35 M mapped read pairs (32–70 M total mapped reads) using samtools view -bs to achieve desired sequencing depths for paired-end libraries. Aligned reads were then subjected to variant calling using the SAILOR (version 1.0.4) software program (Deffit et al., 2017) using default parameters. Candidate A-G variants (or T-C for negative strand) identified were furthermore filtered for read coverage (minimum 5 reads per site) and as well as naturally occurring variants using hg19 Com- mon SNPs (147). These candidate editing sites were assigned a confidence score using a previously described Bayesian model (Li et al., 2008) factoring in both read coverage and percent edited reads. Individual sites with less a 90% confidence score were not considered for downstream analysis. Consensus sites were for each experimental condition were determined as such by being called in both replicates. For each of these sites, per-site editing rate was recalculated by combining read coverage for both replicates and computing:
for positive strand and:
for negative strand. Gene region annotations were gathered from human hg19 gencode release 19 (GRCh37.p13). Intersecting sites were determined using bedtools/pybedtools software libraries (Quinlan and Hall, 2010; Dale et al., 2011), and sequence logos were generated using Weblogo 3 (Crooks et al., 2004). Protein coding consequences were predicted using Snpeff (Cingolani et al., 2012).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| One Shot E. Coli TOP10 | Thermo Scientific | Cat# C404003 |
|
| ||
| Antibodies | ||
|
| ||
| anti-HA.11 Epitope Tag Antibody | Biolegend | Cat# MMS-101R; RRID: AB_291262 |
| Anti-GAPDH | Abcam | Cat# ab9485; RRID: AB_307275 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Thermo Fisher | Cat# 32430; RRID: AB_1185566 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| FastDigest EcoRI | Thermo Scientific | Cat# FD0274 |
| FastDigest HindIII | Thermo Scientific | Cat# FD0504 |
| FastDigest SmaI | Thermo Scientific | Cat# FD0663 |
| FastDigest NotI | Thermo Scientific | Cat# FD0594 |
| T4 Polynucleotide Kinase | New England Biolabs | Cat# M0201S |
| T4 DNA Ligase | New England Biolabs | Cat# M0202L |
| DMEM | GIBCO | Cat# 11965092 |
| Fetal Bovine Serum | GIBCO | Cat# 16000044 |
| TrypLE Express | GIBCO | Cat# 12604013 |
| Opti-MEM Reduced Serum Medium | GIBCO | Cat# 31985070 |
| Hygromycin B | GIBCO | Cat# 10687010 |
| Blasticidin S HCl | GIBCO | Cat# A1113903 |
| Polyethylenimine (PEI) | Sigma Aldrich | Cat# 408727 |
| Doxycycline | Sigma Aldrich | Cat# D9891–5G |
| Lipofectamine 3000 | Invitrogen | Cat# L3000008 |
| Lipofectamine RNAiMAX | Invitrogen | Cat# 13778075 |
| Dulbecco’s Phosphate Buffered Saline | Corning | Cat# 20–031-CV |
| TRIzol Reagent | Invitrogen | Cat# 15596018 |
| Superscript III Reverse Transcriptase | Invitrogen | Cat# 18080044 |
| Oligo(dT)20 Primer | Invitrogen | Cat# 18418020 |
| Random Hexamers | Invitrogen | Cat# N8080127 |
| NEBNext Ultra II Q5 Master Mix | New England Biolabs | Cat# M0544S |
| Protoscript II Reverse Transcriptase | New England Biolabs | Cat# M0368L |
| Agencourt AMPure XP beads | Beckman Coulter | Cat# A63881 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat# 28706 |
| pcDNA5/FRT/TO Vector Kit | Invitrogen | Cat# V652020 |
| Direct-zol RNA Miniprep | Zymo | Cat# R2050 |
| TruSeq Stranded mRNA Library Prep | Illumina | Cat# 20020594 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 23227 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher | Cat# 32106 |
|
| ||
| Deposited Data | ||
|
| ||
| RNA sequencing data | This paper | GEO: GSE152684 |
| Uncropped western blots and primary FACS data | This paper | http://dx.doi.org/10.17632/99m44kn43w.1 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Lenti-X HEK293T Cell Line (Female) | Clonetech | Cat# 632180 |
| ADAR2DD-XTEN-dSpCas9 Flp-In T-REx 293 | This paper | Derived from Invitrogen # R78007 |
| ADAR2DD-XTEN Flp-In T-REx 293 | This paper | Derived from Invitrogen # R78007 |
| ADAR2DD(E488Q)-XTEN-dSpCas9 Flp-In T-REx 293 | This paper | Derived from Invitrogen # R78007 |
| ADAR2DD(E488Q)-XTEN Flp-In T-REx 293 | This paper | Derived from Invitrogen # R78007 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Oligonucleotides for PCR, cloning, and PAMmers; See Table S1 | This paper | N/A |
| Guide RNAs for mammalian RNA targeting; See Table S2 | This paper | N/A |
| Oligonucleotides for targeted NGS libraries; See Table S1 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmid: pOG44 Flp-Recombinase | Invitrogen | Cat# V600520 |
| Plasmid: pcDNA3.1 (−) | Invitrogen | Cat# V79520 |
| Plasmid: pCDNA3.1-W58X EGFP | Hanswillemenke et al., 2015 | N/A |
| Plasmid: pCDNA3.1-WT EGFP | Hanswillemenke et al., 2015 | N/A |
| Plasmid: pBluescript SKII U6-sgRNA-Ef1a-mCherry | This paper; See Table S2 for spacer and extension sequences | N/A |
| Plasmid: pBluescript SKII U6-sgRNA-Ef1a-mCherry-P2A-W58X-EGFP | This paper; See Table S2 for spacer and extension sequences | N/A |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dSpCas9-2XNLS | This paper | Adapted from Addgene plasmid #74710 |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-2XNLS | This paper | Adapted from Addgene plasmid #74710 |
| Plasmid: pcDNA3.1-dSpCas9-XTEN-ADAR2DD-2XNLS | This paper | Adapted from Addgene plasmid #74710 |
| Plasmid: pcDNA3.1-ADAR2DD(E488Q)-XTEN-dSpCas9-2XNLS | This paper | Adapted from Addgene plasmid #74710 |
| Plasmid: pcDNA3.1-ADAR2DD(E488Q)-XTEN-2XNLS | This paper | Adapted from Addgene plasmid #74710 |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dSaCas9-2XNLS | This paper | Adapted from Addgene plasmid #68495 from George Church |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dSaCas9-NES | This paper | Adapted from Addgene plasmid #68495 from George Church |
| Plasmid: pcDNA3.1-dSaCas9-XTEN-ADAR2DD-2XNLS | This paper | Adapted from Addgene plasmid #68495 from George Church |
| Plasmid: pcDNA3.1-dSaCas9-XTEN-ADAR2DD-NES | This paper | Adapted from Addgene plasmid #68495 from George Church |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dLwaCas13a-2XNLS | This paper | Adapted from Addgene Plasmid #91902 from Feng Zhang |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dLwaCas13a-NES | This paper | Adapted from Addgene Plasmid #91902 from Feng Zhang |
| Plasmid: pcDNA3.1-dLwaCas13a-XTEN-ADAR2DD-2XNLS | This paper | Adapted from Addgene Plasmid #91902 from Feng Zhang |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Plasmid: pcDNA3.1-dLwaCas13a-XTEN-ADAR2DD-NES | This paper | Adapted from Addgene Plasmid #91902 from Feng Zhang |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dPspCas13b-2XNLS | This paper | Adapted from Addgene Plasmid #91902 from Feng Zhang |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dPspCas13b-NES | This paper | Adapted from Addgene Plasmid #91902 from Feng Zhang |
| Plasmid: pcDNA3.1-dPspCas13b-XTEN-ADAR2DD-2XNLS | This paper | Adapted from Addgene Plasmid #91902 from Feng Zhang |
| Plasmid: pcDNA3.1-dPspCas13b-XTEN-ADAR2DD-NES | This paper | Adapted from Addgene Plasmid #91902 from Feng Zhang |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dRfxCas13d-2XNLS | This paper | Adapted from Addgene Plasmid #109049 from Patrick Hsu |
| Plasmid: pcDNA3.1-ADAR2DD-XTEN-dRfxCas13d-NEW | This paper | Adapted from Addgene Plasmid #109049 from Patrick Hsu |
| Plasmid: pcDNA3.1-dRfxCas13d-XTEN-ADAR2DD-2XNLS | This paper | Adapted from Addgene Plasmid #109049 from Patrick Hsu |
| Plasmid: pcDNA3.1-dRfxCas13d-XTEN-ADAR2DD-2XNES | This paper | Adapted from Addgene Plasmid #109049 from Patrick Hsu |
|
| ||
| Software and Algorithms | ||
|
| ||
| FastQC | Bioinformatics Group at the Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| BWA | Li and Durbin 2009 | http://bio-bwa.sourceforge.net/ |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| GraphPad Prism 8 | GraphPad Software, Inc. | http://graphpad.com/scientific-software/prism |
| ZEN | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software.html |
| FIJI | Schindelin et al., 2012 | https://imagej.net/Fiji |
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
| Python | Python Software Foundation | https://www.python.org/ |
| Cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| SAILOR | Deffit et al., 2017 | https://github.com/yeolab/sailor |
| Pybedtools | Dale et al., 2011; Quinlan and Hall, 2010 |
https://daler.github.io/pybedtools/
https://bedtools.readthedocs.io/en/latest/ |
| Snpeff | Cingolani et al., 2012 | http://snpeff.sourceforge.net/SnpEff.html |
| Subread | Liao et al., 2014 | http://subread.sourceforge.net/ |
| Weblogo 3 | Crooks et al., 2004 | http://weblogo.threeplusone.com/ |
|
| ||
| Other | ||
|
| ||
| Zeiss Axio Vert.A1 | Zeiss | N/A |
| LSRFortessa | BD Biosciences | N/A |
| LSRFortessa X-20 | BD Biosciences | N/A |
Highlights.
RNA-targeting Cas9-ADAR fusions direct specific A-to-I editing with modified gRNA
Cas9-ADAR targets exogenous and endogenous transcripts in a spacer-independent manner
Orientation of Cas9- and Cas13-ADAR fusions influences on-target efficacy
Cas-based ADAR strategies have distinct transcriptome-wide off-target edits
ACKNOWLEDGMENTS
We acknowledge members of the Yeo lab, particularly Dr. David Nelles and Dr. Frederick Tan for comments and assistance in initial construct design. We thank Dr. Thorsten Stafforst of the University of Tȕbingen, Germany, for the gift of the W58X EGFP reporter and Dr. Patrick Hsu of the University of California, Berkeley, for the gift of the RfxCas13d-expressing plasmid used in cloning. This work was partially supported by grants from the NIH (R01HG004659, R01EY029166, and R01NS103172) to G.W.Y. R.J.M. was supported in part by an institutional award to the UCSD Genetics Training Program from the National Institute of General Medical Sciences (T32GM008666) and by a NIH/NINDS Ruth L. Kirschstein National Research Service Award (F31NS111859). K.W.B. is a University of California President’s Postdoctoral Fellow and is supported by a NIH/NINDS Career Transition Award (K22NS112678). This publication includes data generated at the UC San Diego IGM Genomics Center using an Illumina NovaSeq 6000 instrument that was purchased with funding from a NIH SIG grant (S10OD026929).
DECLARATION OF INTERESTS
G.W.Y. is co-founder, member of the board of directors, equity holder, and paid consultant for Locanabio and Eclipse BioInnovations. G.W.Y. is a Distin-guished Visiting Professor at the National University of Singapore. The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies. R.J.M., K.W.B., and G.W.Y. are co-inventors on patent applications filed on technologies described in this manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108350.
REFERENCES
- Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L, et al. (2016). C2c2 is a single-component programmable RNA-guided RNA- targeting CRISPR effector. Science 353, aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Be- lanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, et al. (2017). RNA targeting with CRISPR-Cas13. Nature 550, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO, Gootenberg JS, Franklin B, Koob J, Kellner MJ, Ladha A, Joung J, Kirchgatterer P, Cox DBT, and Zhang F (2019). A cytosine deaminase for programmable single-base RNA editing. Science 365, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M (2018). The CRISPR tool kit for genome editing and beyond. Nat. Commun 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad MTA, Bhakta S, and Tsukahara T (2017). Site-directed RNA editing by adenosine deaminase acting on RNA for correction of the genetic code in gene therapy. Gene Ther. 24, 779–786. [DOI] [PubMed] [Google Scholar]
- Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, Chaim IA, Thomas JD, Zhang N, Nguyen V, et al. (2017). Elimination of Toxic Micro- satellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 170, 899–912.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra R, Nelles DA, Roth DM, Krach F, Nutter CA, Tadokoro T, Thomas JD, Sznajder LJ, Blue SM, Gutierrez HL, et al. (2020). The sus-tained expression of Cas9 targeting toxic RNAs reverses disease phenotypes in mouse models of myotonic dystrophy type 1. Nat. Biomed. Eng 10.1038/s41551-020-00607-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. (2013). Dynamic Im- aging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DB, Platt RJ, and Zhang F (2015). Therapeutic genome editing: pros- pects and challenges. Nat. Med 21, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, and Zhang F (2017). RNA editing with CRISPR-Cas13. Science 358, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale RK, Pedersen BS, and Quinlan AR (2011). Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformat- ics 27, 3423–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deffit SN, Yee BA, Manning AC, Rajendren S, Vadlamani P, Wheeler EC, Domissy A, Washburn MC, Yeo GW, and Hundley HA (2017). The C. elegans neural editome reveals an ADAR target mRNA required for proper chemotaxis. eLife 6, e28625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugar G, Leenay RT, Eisenbart SK, Bischler T, Aul BU, Beisel CL, and Sharma CM (2018). CRISPR RNA-Dependent Binding and Cleavage of Endogenous RNAs by the Campylobacter jejuni Cas9. Mol. Cell 69, 893–905.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, and Liu DR (2017). Programmable base editing of A T to G C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanswillemenke A, Kuzdere T, Vogel P, Jé kely G, and Stafforst T (2015). Site-Directed RNA Editing in Vivo Can Be Triggered by the Light-Driven As-sembly of an Artificial Riboprotein. J. Am. Chem. Soc 137, 15875–15881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, and Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrekar D, Chen G, Meluzzi D, Ganesh A, Worlikar A, Shih YR, Var-ghese S, and Mali P (2019). In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 16, 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keryer-Bibens C, Barreau C, and Osborne HB (2008). Tethering of proteins to RNAs by bacteriophage proteins. Biol. Cell 100, 125–138. [DOI] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, and Gabriel A (2004). Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 14, 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ,Jung MH, Kim S, et al. (2017a). In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun 8, 14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, and Liu DR (2017b). Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol 35, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, and Hsu PD (2018). Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Ef- fectors. Cell 173, 665–676.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, and Bradley A (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rear-rangements. Nat. Biotechnol 36, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. (2004). Systematic identification of abundant A-to-I editing sites in the human transcrip- tome. Nat. Biotechnol 22, 1001–1005. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Bur- rows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Sub- group (2009). The Sequence Alignment/Map format and SAMtools. Bioinfor- matics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ruan J, and Durbin R (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Research, 1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinfor- matics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Licht K, Kapoor U, Mayrhofer E, and Jantsch MF (2016). Adenosine to Inosine editing frequency controlled by splicing efficiency. Nucleic Acids Res. 44, 6398–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XM, Zhou J, Mao Y, Ji Q, and Qian SB (2019). Programmable RNA N6-methyladenosine editing by CRISPR-Cas9 conjugates. Nat. Chem. Biol 15, 865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high- throughput sequencing reads. EMBnet J. 17, 10–12. [Google Scholar]
- Merkle T, Merz S, Reautschnig P, Blaha A, Li Q, Vogel P, Wettengel J, Li JB, and Stafforst T (2019). Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol 37, 133–138. [DOI] [PubMed] [Google Scholar]
- Montiel-Gonzá lez MF, Vallecillo-Viejo IC, and Rosenthal JJ (2016). An efficient system for selectively altering genetic information within mRNAs. Nu- cleic Acids Res. 44, e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Gonzalez MF, Diaz Quiroz JF, and Rosenthal JJC (2019). Cur- rent strategies for Site-Directed RNA Editing using ADARs. Methods 156, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelles DA, Fang MY, O’Connell MR, Xu JL, Markmiller SJ, Doudna JA, and Yeo GW (2016). Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell 165, 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K (2010). Functions and regulation of RNA editing by ADAR deam- inases. Annu. Rev. Biochem 79, 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, and Doudna JA (2014). Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps KJ, Tran K, Eifler T, Erickson AI, Fisher AJ, and Beal PA (2015). Recognition of duplex RNA by the deaminase domain of the RNA edit- ing enzyme ADAR2. Nucleic Acids Res. 43, 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Yi Z, Zhu S, Wang C, Cao Z, Zhou Z, Yuan P, Yu Y, Tian F, Liu Z, et al. (2019). Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol 37, 1059–1069. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zet- sche B, Shalem O, Wu X, Makarova KS, et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch S, He E, Srienc M, Zhou H, Zhang Z, and Dickinson BC (2019). Programmable RNA-Guided RNA Effector Proteins Built from Human Parts. Cell 178, 122–134.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau BA, Hou Z, Gramelspacher MJ, and Zhang Y (2018). Pro- grammable RNA Cleavage and Recognition by a Natural CRISPR-Cas9 Sys- tem from Neisseria meningitidis. Mol. Cell 69, 906–914.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafforst T, and Schneider MF (2012). An RNA-deaminase conjugate selectively repairs point mutations. Angew. Chem. Int. Ed. Engl 51, 11166–11169. [DOI] [PubMed] [Google Scholar]
- Strutt SC, Torrez RM, Kaya E, Negrete OA, and Doudna JA (2018). RNA-dependent RNA targeting by CRISPR-Cas9. eLife 7, e32724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. (2017). Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuronyi BW, Koblan LW, Levy JM, Yeh WH, Zheng C, Newby GA, Wilson C, Bhaumik M, Shubina-Oleinik O, Holt JR, and Liu DR (2019). Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol 37, 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallecillo-Viejo IC, Liscovitch-Brauer N, Montiel-Gonzalez MF, Eisen- berg E, and Rosenthal JJC (2018). Abundant off-target edits from site- directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 15, 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel P, and Stafforst T (2019). Critical review on engineering deaminases for site-directed RNA editing. Curr. Opin. Biotechnol 55, 74–80. [DOI] [PubMed] [Google Scholar]
- Vogel P, Moschref M, Li Q, Merkle T, Selvasaravanan KD, Li JB, and Stafforst T (2018). Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods 15, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MC, Kakaradov B, Sundararaman B, Wheeler E, Hoon S, Yeo GW, and Hundley HA (2014). The dsRBP and inactive editor ADR-1 utilizes dsRNA binding to regulate A-to-I RNA editing across the C. elegans tran- scriptome. Cell Rep. 6, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettengel J, Reautschnig P, Geisler S, Kahle PJ, and Stafforst T (2017). Harnessing human ADAR2 for RNA repair - Recoding a PINK1 mutation res- cues mitophagy. Nucleic Acids Res. 45, 2797–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SK, Sato S, and Lazinski DW (2001). Substrate recognition by ADAR1 and ADAR2. RNA 7, 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf TM, Chase JM, and Stinchcomb DT (1995). Toward the therapeu- tic editing of mutated RNA sequences. Proc. Natl. Acad. Sci. USA 92, 8298–8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AV, Sternberg SH, Taylor DW, Staahl BT, Bardales JA, Korn- feld JE, and Doudna JA (2015). Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. USA 112, 2984–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LZ, Wang Y, Li SQ, Yao RW, Luan PF, Wu H, Carmichael GG, and Chen LL (2019). Dynamic Imaging of RNA in Living Cells by CRISPR- Cas13 Systems. Mol. Cell 76, 981–997.e7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA sequencing data generated for this study is Gene Expression Omnibus: GSE152684. Uncropped western blots and original FACS data have been deposited to Mendeley Data: [https://doi.org/10.17632/99m44kn43w.1]. Software for SAILOR editing analysis is available on Github (https://github.com/YeoLab/sailor).