

Abstract
Inflammasomes are multiprotein complexes that assemble upon detection of danger signals to activate the inflammatory enzyme caspase-1, trigger secretion of the highly proinflammatory cytokine IL-1β, and induce an inflammatory cell death called pyroptosis. Distinctiveness of the NLRP3 inflammasome resides in the diversity of molecules that induce its activation, indicating a certain intricacy. Furthermore, besides the canonical activation of NLRP3 in response to various stimuli, caspase-11-dependent detection of intracellular LPS activates NLRP3 through a non-canonical pathway. Several aspects of the NLRP3 inflammasome are not characterized or remain unclear. In this review, we summarize the different modes of NLRP3 activation. We describe recent insights into post-translational and cellular regulation that confer further complexity to NLRP3 inflammasomes.
Keywords: Inflammasome, NLRP3, ASC, Caspase-1, Caspase-11, IL-1β, ubiquitination, phosphorylation
Summary sentence
Precise NLRP3 inflammasome regulation relies on post-translational modifications, altered subcellular localization, and dynamic interactions with other proteins
Graphical Abstract
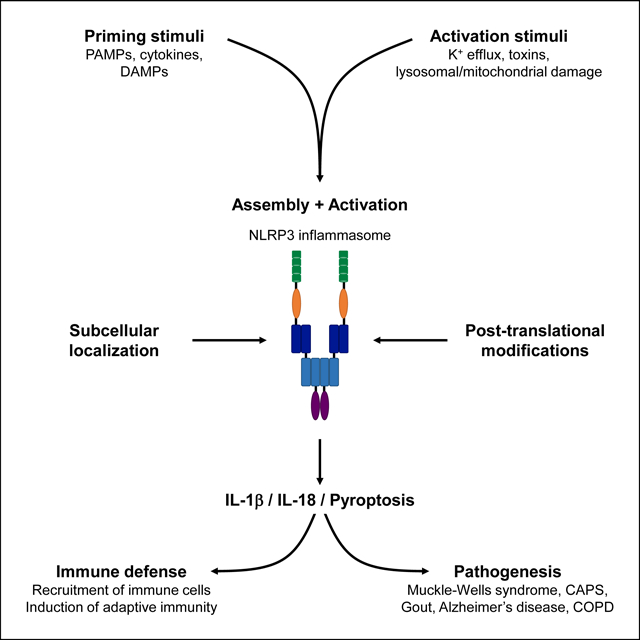
Introduction
Microbes are sensed upon detection of Pathogen Associated Molecular Patterns (PAMPs) by Pattern Recognition Receptors (PRRs) expressed by the innate immune cells [1, 2]. PRRs include a variety of molecules either expressed at the cell surface, such as Toll-like receptors (TLRs) or C-type Lectin receptors (CLRs), or localized in the cytoplasm, such as NOD-like receptors (NLRs) or RIG-I-like receptors (RLRs). Each PRR recognizes particular microbial components and subsequently mobilizes specific signaling cascades that modulate phagocytosis and degradation of the microbe, regulation of gene expression to promote an innate immune response including production of inflammatory cytokines, and eventually the establishment of an adaptive immune response to fight the microbial infection at the scale of the organism [3, 4].
Some members of the cytoplasmic NLRs are associated with macromolecular complexes called inflammasomes, which mediate immune responses to microbial infection [5]. Inflammasomes are composed of three different types of proteins: a sensor (most often a member of the NLR family), an adaptor (ASC, apoptosis-associated speck-like protein containing a caspase-recruitment domain (CARD)), and an effector protein, the cysteine protease pro-caspase-1. To date, five PRRs (NLRP1, NLRP3, NLRC4, Pyrin, and AIM2) have been shown to form inflammasomes [6, 7]. Most NLRs contain three domains: a N-terminal pyrin domain (PYD), a centrally located NACHT or nucleotide-binding domain (NBD) that mediates self-oligomerization, and carboxy-terminal Leucine Rich Repeats (LRRs) that are involved in the recognition of PAMPs [8]. Activation of inflammasomes triggers proteolytic cleavage of pro-caspase-1 into its catalytically active form. Caspase-1 then processes the cytokine precursors pro-IL-1β and pro-IL-18 into mature and biologically active IL-1β and IL-18 [9]. IL-1β is a potent proinflammatory cytokine, which notably elicits the recruitment of innate immune cells to the site of infection and regulates adaptive immune cells. IL-18 promotes the activation of cytotoxic natural killer (NK) cells and T cells [10]. Caspase-1 also induces a proinflammatory form of cell death called pyroptosis [11].
The NLRP3 inflammasome has been extensively studied in the last two decades, primarily because of its potential involvement in the pathogenesis of several diseases. The NLRP3 inflammasome mediates host immune defense against infectious microbes of either viral or bacterial origin, and is therefore beneficial to the organism [12]. Yet, dysregulation or hyperactivation of the NLRP3 inflammasome results in hyperinflammation and causation of several inherited or acquired inflammatory diseases in sterile conditions [13, 14]. For instance, NLRP3 has initially been associated with the Muckle-Wells syndrome, an autoinflammatory disease where mutations in the NALP3/Cryopyrin gene lead to hyperactivation of inflammasome [15]. Activity of the NLRP3 inflammasome is also involved in the cryopyrin-associated periodic fever syndrome (CAPS), where mutations in the NLRP3 NACHT domain lead to hyperactivation of the NLRP3 inflammasome [16]. NLRP3 is associated with the development of gout and pseudogout where monosodium urate and calcium pyrophosphate dihydrate crystals, deposited within the joint and periarticular tissues, were shown to activate the NLRP3 inflammasome [17]. NLRP3 has also been associated with the pathogenesis of Alzheimer’s disease where fibrillar amyloid-β deposits activate the NLRP3 inflammasome and drive Tau pathology [18]. Reversely, activation of inflammasome in microglia, and particularly formation and release of ASC specks, was shown to promote the formation of amyloid-β deposits [19], highlighting a complex amplification relationship between the NLRP3 inflammasome and amyloid-β plaques formation [20]. Furthermore, NLRP3 inflammasome activation underlies numerous lung inflammatory diseases such as chronic obstructive pulmonary disease (COPD), silicosis and asbestosis [21]. Thus, the NLRP3 inflammasome appears as a central pathway during infection or inflammatory disease. It is of particular importance to characterize the various mechanisms that regulate the successive stages of NLRP3 inflammasome activation, and potentially identify molecules to target specific aspects of its function. Recent advances on this aspect have enabled the development of NLRP3-targeting molecules which are being tested for treatment of inflammatory diseases, especially in the context of abnormal activation of NLRP3 in sterile, chronic inflammatory disease [12]. There is great interest in pharmacological targeting of “unwanted” NLRP3 activity in the context of chronic inflammatory diseases because of their impact on the well-being of afflicted individuals and the associated healthcare costs to society.
In this review, we discuss the different steps for activation of the NLRP3 inflammasome, either canonical or non-canonical. We highlight a complex regulation at different levels of NLRP3 inflammasome activation. From the very initial stimulation of NLRP3 by a wide variety of stimuli to the many post-translational modifications that modulate inflammasome assembly and activation, it appears that several aspects of NLRP3 inflammasome biology remain to be clarified and may reflect the context of its activation.
Activation of the canonical NLRP3 inflammasome
Assembly of the NLRP3 inflammasome relies on homotypic interactions between the N-terminal PYD domain of NLRP3 and the PYD domain of the adaptor ASC, as well as between the respective CARD domains of ASC and pro-caspase-1. The resulting macromolecular complexes, which contain NLRP3, ASC and caspase-1, are visualized as ASC specks by microscopy, and they enable catalytic inflammasome activity [6]. Activation of the NLRP3 inflammasome requires two steps: 1) a priming step, which promotes the expression of inflammasome components, and 2) an activation step, which is triggered by several types of molecules that specifically activate NLRP3.
Priming of the NLRP3 inflammasome
The hallmark response of inflammasome activation includes the activation/proteolytic cleavage of caspase-1, secretion of the biologically active cytokines IL-1β and IL-18, as well as induction of pyroptosis. The requirement for priming prior to effective NLRP3 inflammasome activation was revealed by the observation that phagocytes such as macrophages, do not directly activate the inflammasome in response to treatment with NLRP3 inducers [22]. However, treatment of phagocytes with PAMPs (such as LPS) prior to NLRP3 inducers, leads to a robust inflammasome activation [22]. Priming is triggered by different extracellular signals such as PAMPs (TLRs ligands or NOD2 ligands for instance) or cytokines (TNF-α, IL-1β, IFN-I), and the activation of their respective receptors that all converge on NF-κB activation [22, 23] (Figure 1). Furthermore, detection of DAMPs – or alarmins, such as double stranded DNA, mitochondrial DNA, ATP, reactive oxygen species (ROS), heme or uric acid released by neighboring necrotic cells or damaged tissues – by various receptors also primes immune cells in non-infectious contexts, notably via activation of NF-κB pathway [24]. It results in transcriptional and translational induction of various innate immune effectors, among which are the inflammasome sensors, including NLRP3, and the pro-form of IL-1β, pro-IL-1β (Figure 1). Priming by DAMPs underlies mechanisms at the heart of sterile, chronic inflammatory syndromes involving NLRP3. The levels of the adaptor ASC and the effector pro-caspase-1, common to all types of inflammasomes, are unchanged and sufficient for efficient inflammasome activation.
Figure 1. Activation of the canonical NLRP3 inflammasome.

Activation of the NLRP3 inflammasome first requires the priming of NLRP3. Activation of membrane TLR4 by LPS from Gram-negative bacteria, for instance, or detection of DAMPs result in downstream activation of NF-kB, which promotes the transcription of NLRP3. Specific activation of NLRP3 occurs mostly downstream of K+ efflux, which can be generated by bacterial pore-forming toxins or ATP-induced activation of P2X7 purinergic receptor P2X7R, although how NLRP3 is activated by reduction of intracellular K+ concentration remains unknown. Lysosomal or mitochondrial damage can also lead to activation of NLRP3 via uncharacterized pathways. After sensing of these specific stimuli, the sensor NLRP3 assembles together with the adaptor ASC and the effector pro-caspase-1, via homotypic interactions between the N-terminal PYD domain of NLRP3 and the PYD domain of ASC, as well as between the respective CARD domains of ASC and pro-caspase-1. The kinase NEK7 is required for assembly of the NLRP3 inflammasome as well. Assembly of the NLRP3 inflammasome leads to activation of caspase-1, which then cleaves the pro-forms of IL-1β and IL-18, resulting in the secretion of biologically active cytokines, as well as Gasdermin D, resulting in pyroptosis via the formation of pores at the plasma membrane.
Activation of the NLRP3 inflammasome: a wide range of inducers
Once expressed in phagocytes, NLRP3 can be activated by a wide variety of inducers, including microbes and microbial molecules (from either bacterial, viral or fungal origins, for instance bacterial RNA or bacterial muramyl dipeptide), DAMPs (for instance ATP or uric acid), as well as pore-forming toxins, ionophores (such as Nigericin) or crystals (for example Hemozoin) [15, 22, 25–30].
The diversity of these molecules likely excludes the ability of NLRP3 to interact with and directly detect each of them, and rather suggests that NLRP3 would sense and respond to one – or a limited number of – common cellular component(s) or event(s). Several mechanisms have been proposed to activate NLRP3:
Potassium K+ efflux: The role of K+ efflux from cytosol to the extracellular medium is the most largely accepted mechanism for NLRP3 activation, and also seems to be associated with other types of NLRP3 triggers such as lysosomal leakage or Ca2+ signaling. Several NLRP3 inducers such as ATP and Nigericin lower the cytosolic concentration of K+ as a trigger for NLRP3 activation [31, 32]. Consistently, high cytoplasmic concentrations of K+ specifically inhibit the NLRP3 inflammasome but not the other inflammasomes [33, 34]. The membrane purinergic receptor P2X7R that senses extracellular ATP was notably shown to induce a K+ efflux responsible for NLRP3 activation, which is proposed as the mechanism for ATP-induced activation of NLRP3 [33]. However, how K+ efflux is sensed by NLRP3, and allows assembly and activation of the NLRP3 inflammasome, has not been determined (Figure 1).
Ca2+ signaling, potentially by inducing NLRP3 complex formation or by causing mitochondrial dysfunction and successive NLRP3 activation [35, 36].
Translocation of NLRP3 to mitochondria via remodeling of microtubules network, enabling interaction of NLRP3 with mitochondrial ASC and inflammasome assembly [37–39].
Mitochondrial damage and dysfunction, which releases both mitochondrial DNA (mtDNA) and reactive oxygen species (mtROS) that were shown to activate NLRP3 [39–42].
Leakage of lysosomes, notably in response to large particulate activators, which results in the release of cathepsins that activate NLRP3 [43].
However, these processes do not occur in response to all NLRP3 inducers, and contradictory findings show that their involvement is still a matter of debate [33, 36, 44, 45].
Upon stimulation, notably in response to K+ efflux, the sensor NLRP3 indeed undergoes a conformational change that enable its oligomerization via the self-oligomerization NACHT domain. The NLRP3 oligomers then recruit the adaptor ASC, which also polymerize, and the effector caspase-1, forming the characteristic inflammasome macromolecular complex [46, 47]. Assembly of the NLRP3 inflammasome was also shown to require the kinase NEK7 (NIMA-related kinase 7). NEK7 binds to NLRP3 upon activation, and is essential for NLRP3 and subsequent ASC oligomerization and activation of caspase-1 catalytic activity [48–50]. Of note, NLRP3 exhibits an ATP-binding and ATP-hydrolysis motif within its NACHT domain, called Walker B, which is required for NLRP3 inflammasome activity [51]. Recent studies have further characterized that MCC950, a common inhibitor of NLRP3, binds to the Walker B motif, prevents ATP binding and hydrolysis, resulting in the inability of NLRP3 to gain an active conformation [52, 53]. Besides its assembly, the regulation of NLRP3 inflammasome activity remains an intriguing question as well.
Beyond the various inducers of NLRP3, it has been recently shown that the NLRP3 inflammasome is connected to cell death pathways. Notably, the protein FADD, the key adaptor for the death receptors of the TNF-Receptor family, was shown to activate the NLRP3 inflammasome in a process involving the apoptotic protein caspase-8, both at the priming and activation steps [54]. Alternatively to apoptosis, Caspase-8 can serve as a scaffold to recruit the protein kinase RIPK3 and drive MLKL-dependent necroptosis [55], a pathway that was also shown to activate NLRP3 inflammasome downstream of TLR [56]. Importantly, in human monocytes, FADD and caspase-8 were also identified as activators of a so-called alternative K+ efflux-insensitive NLRP3 inflammasome, downstream of TLR4-TRIF-RIPK1 signaling axis [57]. In contexts where both necroptosis and apoptosis are impaired in intestinal epithelial cells, recent studies have shown that Caspase-8 participates in mediating ASC oligomerization, in activation of the inflammasome, and in the promotion of pyroptosis. Thus, Caspase-8 is considered as a determinant molecule that controls all three cell death pathways: apoptosis, necroptosis and pyroptosis [58, 59].
Activation of the non-canonical NLRP3 inflammasome
Activation of the NLRP3 inflammasome can also be triggered by distinct inflammatory caspases, murine caspase-11 and its human orthologs caspase-4 and caspase-5. Caspase-11 was first identified as an interactor of caspase-1, which promotes caspase-1 activation [60]. Caspase-11 has been shown to induce pyroptosis, similarly but independently of caspase-1, via cleavage of the pore-forming protein Gasdermin D [61–68]. Caspase-11, caspase-4 and caspase-5 are not able to cleave pro-IL-1β and pro-IL-18 and to release the biologically active cytokines [69, 70]. Yet, caspase-11 was shown to be required for activation of the NLRP3 inflammasome and release of IL-1β in response to several Gram-negative bacteria, including Escherichia coli and Citrobacter rodentium, highlighting a novel inflammasome pathway: the non-canonical NLRP3 inflammasome [70].
Unlike caspase-1, but similarly to the sensor NLRP3, the levels of caspase-11 in phagocytes are low, and activation of caspase-11 requires a priming step. It depends on IFN-I signaling via its membrane receptor IFNAR, which promotes expression of the pro-form of caspase-11, pro-caspase-11. In this model, primary sensing of LPS by TLR4 and subsequent TRIF-dependent expression and secretion of IFN-I are required for caspase-11 priming [71–73] (Figure 2). Although commonly accepted, the role of IFN signaling in priming pro-caspase-11 expression has been a matter of debate since earlier work demonstrated that levels of pro-caspase-11 after infection of macrophages with Salmonella typhimurium were unaffected in the absence of IFNAR. Yet, IFNAR signaling was still required for caspase-11 activation [69]. Importantly, priming does not appear to be required in human monocytes, where caspase-4 is constitutively expressed and can lead to activation of the non-canonical NLRP3 inflammasome without further induction [74]. Besides priming of pro-caspase-11, IFN signaling was also shown to promote the expression of an important family of GTPases, guanylate binding proteins (GBPs), which are involved in the destabilization of phagosomal membranes. Therefore, GBPs enable the release of phagosomal contents, including LPS, a process required for activation of caspase-11 and the non-canonical inflammasome by Gram-negative bacteria [72, 75].
Figure 2. Activation of the non-canonical NLRP3 inflammasome.

Activation of the non-canonical NLRP3 inflammasome involves interferon-inducible caspase-11. Induction of IFN-β expression by Gram-negative bacteria relies on activation of endosomal/phagosomal TLR4 and the signaling pathway downstream of the adaptor TRIF. Secreted IFN-β then activates IFNAR, notably in an autocrine manner, which promotes expression of pro-caspase-11 and additional IFN-inducible proteins such as GBPs. Caspase-11 is activated by direct binding of intracellular LPS, released from the phagosomes, to the CARD domain of caspase-11. GBPs are recruited to the phagosome and participate in phagosomal lysis and release of LPS in the cytoplasm. Active caspase-11 directly cleaves Gasdermin D to induce pyroptosis, and also leads to activation of the NLRP3 inflammasome via uncharacterized mechanisms.
Activation of caspase-11 occurs in response to Gram-negative but not Gram-positive bacteria, suggesting that LPS, not expressed by Gram-positive bacteria, is important for non-canonical NLRP3 inflammasome activation. Several studies have indeed characterized the crucial role of caspase-11 in sensing intracellular LPS that is delivered by transfection or released during infection by virulent cytosol-invasive Gram-negative bacteria [76, 77]. Pro-caspase-11 acts as a PRR for intracellular LPS, of which the most conserved Lipid A moiety binds to the CARD domain of pro-caspase-11, initiates its oligomerization and promotes proximity-induced activation of caspase-11 catalytic activity [68](Figure 2). Importantly, LPS also binds to human caspase-4 and caspase-5, but not to other inflammatory or apoptotic caspases [68]. Thus, the current paradigm stipulates that LPS is the unique and sufficient trigger for caspase-11 activation, induction of pyroptosis via Gasdermin D, and subsequent activation of the non-canonical NLRP3 inflammasome by Gram-negative bacteria (Figure 2).
Besides LPS, several findings have identified additional bacterial components involved in the activation of NLRP3 inflammasome. Notably, only live but not dead Gram-negative bacteria, both of which express LPS, can activate the NLRP3 inflammasome and upon detection of the vita-PAMP bacterial RNA present in live and lost from dead bacteria [30]. The helicase DHX33 was later shown to be involved in the recognition of bacterial RNA and activation of NLRP3 inflammasome [78]. Whether these molecules are involved in the non-canonical NLRP3 inflammasome pathway remains unknown. Furthermore, outer membrane vesicles (OMVs) produced by live Gram-negative bacteria were shown to contain LPS and to be endocytosed by phagocytes, thus delivering cytosolic LPS and resulting in activation of caspase-11 [79]. Lastly, distinct mitochondrial electron transport chain adaptations are initiated by bacterial RNA or live bacteria [80] and mediated by innate immune receptor-mediated endosomal ROS production during bacterial infection [81]. The result is increased mitochondrial electron transport capacity [80] and potentially a byproduct of electron transport chain activity, mitochondrial ROS that has been reported to be critical for NLRP3 inflammasome activation in some contexts [81].
The details of NLRP3 inflammasome activation by caspase-11 remain poorly defined. Caspase-11 can form a heterodimer with caspase-1 and promote its activation [60, 82], although the requirements for NLRP3 and ASC in this process have not been assessed. Alternatively, caspase-11-dependent cleavage of Gasdermin D is thought to be a prerequisite for activation of the NLRP3 inflammasome via the K+ efflux engendered by the plasma membrane pores it generates [12, 64]. We could also hypothesize that a functional interaction between caspase-11 and the NLRP3 inflammasome, and perhaps involving additional partners, could promote non-canonical activation of the NLRP3 inflammasome and processing of IL-1β and IL-18.
Regulation of the NLRP3 inflammasome
Post-translational ubiquitination and phosphorylation
Post-translational modifications of NLRP3, including phosphorylation and ubiquitination, have emerged as essential regulators of NLRP3 inflammasome activation.
Ubiquitination: inhibition of the NLRP3 inflammasome?
Ubiquitination corresponds to the conjugation of ubiquitin moieties to substrate proteins via isopeptidic linkage on internal Lysine residues of the substrate, a process catalyzed by a series of enzymes including E3 ubiquitin ligases [83]. Proteins can be monoubiquitinated (a single ubiquitin on a single Lysine), multi-ubiquitinated (single ubiquitins on various Lysine residues scattered over the substrate) or polyubiquitinated (ubiquitin chains on one or several Lysine residues). Moreover, deubiquitinating enzymes (DUBs) counteract ubiquitination of proteins. These various modifications constitute signals that can impart a large panel of fates on a given protein including trafficking, modification of interacting protein networks, activation/inhibition of enzymatic activity, change of subcellular localization, as well as proteasomal, lysosomal or autophagic degradation.
The first evidence for involvement of ubiquitination in NLRP3 inflammasome biology came from inhibition of deubiquitination in macrophages with the isopeptidase inhibitor G5, where the NLPR3 inflammasome was inhibited without affecting the AIM2 and NLRC4 inflammasomes [84]. The DUB BRCC3 was further shown to deubiquitinate the LRR region of NLRP3 prior to NLRP3 assembly and activation [84]. These findings not only suggest that NLRP3 is ubiquitinated via K63-linked polyubiquitin chains – which is the specificity of BRCC3 – but also indicate that NLRP3 ubiquitination can prevent its activation.
Several E3 ubiquitin ligases have been identified in NLRP3 inflammasome inhibition. The E3 ligase TRIM31 was reported to interact with NLRP3, promote NLRP3 polyubiquitination, and result in proteasomal degradation of NLRP3. Of note, TRIM31 expression was induced by LPS priming concomitantly to NLRP3 expression, suggesting that ubiquitination by TRIM31 could act as a fine-tuning inhibitory mechanism to prevent hyperactivation of the NLRP3 inflammasome, hyperinflammation and pyroptotic cell death [85]. Additional E3 ligases, such as MARCH7 [86], F-Box L2 (FBXL2) [87] or Ariadne homolog 2 (ARIH2) [88], inhibit NLRP3 via a similar process of polyubiquitination and proteasomal degradation of NLRP3. In contrast, ubiquitination of NLRP3 by the E3 ligase Pellino2, which catalyzes K63-linked polyubiquitination, promotes NLRP3 inflammasome activation [88], although the exact mechanisms by which this ubiquitination activates the NLRP3 inflammasome remain unclear. Besides activation of NLRP3, ubiquitination was also shown to be involved in the regulation of NLRP3 priming. For instance, the DUB A20 was shown to inhibit NLRP3 by preventing its expression, presumably via its well-characterized inhibitory effect on the NF-κB pathway [89, 90] (Figure 3A).
Figure 3. Regulation of the NLRP3 inflammasome by post-translational modifications and sub-cellular localization.

A. Ubiquitination and deubiquitination, as well as phosphorylation and dephosphorylation, regulate NLRP3 inflammasome activation. Post-translational events targeting the sensor NLRP3 and the adaptor ASC are summarized, including the name of the enzymes involved (when identified), the type of modification and the consequence for NLRP3 inflammasome activity. PolyUb = polyubiquitination, DUB = deubiquitination, P- = phosphorylation, De-P- = dephosphorylation.
B. The sub-cellular localization of NLRP3 may determine NLRP3 inflammasome activation. Different possible localizations of NLRP3 are indicated, including cytosolic or Endoplasmic Reticulum (ER) localizations in the absence of activation. When NLRP3 is activated, several relocalization have been described, including translocation to mitochondria where NLRP3 functionally interacts with MAVS, the Golgi apparatus where NLRP3 physically interacts with SREBP2 and SCAP, or the Trans-Golgi network (TGN) where NLRP3 binds to phosphatidylinositol-4-phosphate (PtdIns4P). How these localizations enable the activation of the NLRP3 inflammasome remain unclear.
NLRP3 is not the only component of the inflammasome targeted by ubiquitination. The adaptor ASC can also be polyubiquitinated via K63-linked ubiquitin chains. This K63 polyubiquitination can be induced by NLRP3 or AIM2 inducers, and was initially shown to target ASC for autophagic degradation resulting in inflammasome inhibition [91]. It also highlighted the reverse correlation between inflammasome activity and autophagy induction [91]. In contrast, K63 polyubiquitination of ASC can be catalyzed by the E3 ubiquitin ligase TRAF3 in response to VSV infection – known to induce NLRP3 inflammasome – and promotes inflammasome activation and production of IL-1β [92]. ASC is also subjected to linear polyubiquitination by the linear ubiquitin chain assembly complex LUBAC, which enhances NLRP3 inflammasome activity [93] (Figure 3A).
Furthermore, pro-IL-1β was also shown to be ubiquitinated on Lysine K133, and deubiquitination of pro-IL-1β by A20 was shown to repress activity of the NLRP3 inflammasome [94]. Thus, ubiquitination of pro-IL-1β supports NLRP3 inflammasome assembly and activity, while its deubiquitination by A20 prevents excessive activation of the inflammasome. Notably, mature IL-1β is not ubiquitinated, suggesting that an additional deubiquitination step is also involved during the processing of pro-IL-1β by the inflammasome.
Phosphorylation in regulation of the NLRP3 inflammasome
Comparable to ubiquitination and deubiquitination, phosphorylation and dephosphorylation of proteins can have multiple consequences on the fate of proteins, and the two types of post-translational modifications are often linked [95]. NLRP3 is a substrate for the kinase PKA, which binds and phosphorylates NLRP3 within the NACHT domain, more precisely on Serine 291 (or Serine 295 in human NLRP3). This phosphorylation was shown to promote ubiquitination of NLRP3, via both K48 and K63-linked ubiquitin chains, subsequent degradation of NLRP3 by the proteasome, and therefore inhibition of the NLRP3 inflammasome [96–98]. Interestingly, the same residue of NLRP3 can also be phosphorylated by the Golgi-associated kinase PKD. In this case, phosphorylation promotes NLRP3 assembly and activation [99], showing the complexity of NLRP3 regulation by phosphorylation, which may depend on interacting partners or subcellular localization of NLRP3. Similarly, phosphorylation of NLRP3 on another Serine residue, Serine 194, also promotes activation of the NLRP3 inflammasome. This phosphorylation is catalysed by the kinase JUN N-terminal kinase (JNK) and occurs during the priming step prior to inflammasome assembly and activation [100].
Another Serine residue of NLRP3, Serine 5 within the PYD domain, is modified by phosphorylation and dephosphorylation. While the phosphatase 2A (PP2A) dephosphorylates Serine 5 of NLRP3, a phosphomimetic residue abrogates NLPR3 inflammasome activation by impairing NLRP3 – ASC interaction [101], demonstrating a critical balance between NLRP3 phosphorylation and dephosphorylation.
Besides Serine phosphorylation, NLRP3 can undergo Tyrosine phosphorylation and dephosphorylation as evidenced by the interaction of NLRP3 with the phosphatase protein tyrosine phosphatase non-receptor 22 (PTPN22), which dephosphorylates Tyrosine 861 of NLRP3 and promotes activation of the NLRP3 inflammasome [102](Figure 3A).
Phosphorylation also targets the adaptor ASC in response to several stimuli including NLRP3 inducers. The kinases spleen tyrosine kinase (SYK) and JUN N-terminal kinase (JNK) phosphorylate several residues within the CARD domain of ASC, events which are required for the processing of caspase-1 [103, 104](Figure 3A).
Studies have revealed many ubiquitination and phosphorylation related processes that target NLRP3 and ASC, and therefore modulate activation of the NLRP3 inflammasome. Such a diversity may result from the different contexts in which experimentation was performed, and may depend on the type of immune cells studied, the stimulus used, or the stage of NLRP3 activation considered. As such, the abundance of post-translational modifications highlights the importance of the NLRP3 inflammasome pathway, which must be fine-tuned to ensure proper and efficient function and avoid deleterious dysfunction.
Regulation by Sub-cellular localization
Activation of inflammasomes relies on the assembly of three different proteins via homodomain interactions, and presumes that all three molecules colocalize at least momentarily during the process of activation. Recent studies have highlighted the regulation of subcellular localization of these molecules as an important parameter that facilitates inflammasome assembly and activation.
In the absence of activation, NLRP3 was shown to be cytoplasmic but also associated with the Endoplasmic Reticulum (ER) [39]. After stimulation, NLRP3 was proposed to relocalize to mitochondria, where it may functionally interact with the mitochondria-associated PRR MAVS. In that case, both MAVS and subcellular relocalization were required for NLRP3 inflammasome activation [37–39]. Changes in subcellular localization may depend on microtubule-mediated transport of mitochondria to the ER, a process which specifically regulates the NLRP3 but not AIM2 or NLRC4 inflammasomes [37]. Yet, whether NLRP3 is recruited specifically to mitochondria via protein-protein interaction has not been characterized.
Besides translocation to the mitochondria, NLRP3 has been shown to relocate to the Golgi apparatus where it interacts with two proteins involved in cholesterol biosynthesis: SREBP2 (Sterol regulatory element-binding protein 2) and SCAP (SREB cleavage-activating protein) [105]. This localized tripartite interaction is required for both activation of NLRP3 inflammasome and cholesterol synthesis via maturation of SREBP2.
Stimulation of NLRP3 has also been shown to disrupt the trans-Golgi network (TGN) and result in the relocalization of NLRP3 to the dispersed TGN [106]. Recruitment of NLRP3 to the dispersed TGN depends on ionic bonding with phosphatidylinositol-4-phosphate (PtdIns4P), and is required for assembly and activation of the NLRP3 inflammasome. Thus, subcellular localization of NLRP3 has emerged as a crucial parameter to regulate activation of the NLRP3 inflammasome. Several different subcellular sites may be involved depending on the stimulus that engages NLRP3 (Figure 3B). Subcellular localization as a mode of regulation may also be involved in non-canonical NLRP3 activation by caspase-11.
Localization of the adaptor ASC may also be of importance. ASC is localized to the cytosol but also to the nucleus at steady state, and can be associated with the ER [107]. Upon inflammasome activation, ASC is observed in characteristic cytosolic ASC specks, which correspond to the oligomerized and assembled inflammasomes [108]. It is presently not known whether ASC co-localizes with inflammasome sensors, including NLRP3, in various subcellular sites and how this relates to the assembly and localization of ASC specks.
Conclusion
Although the outcomes of NLRP3 inflammasome activation are well characterized, the processes involved in its activation and regulation provoke debate and are under intense study. Many regulatory mechanisms of the NLRP3 inflammasome have recently been identified including post-translational modifications and subcellular relocalization of inflammasome components. Such a variety and layering of regulators demonstrate the requirement for precise control of the NLRP3 inflammasome. The NLRP3 inflammasome must be efficiently activated to cope with the microbial threat and prevent propagation of the infection, but activation must be controlled to prevent hyperinflammation and deleterious consequences for the host organism.
Acknowledgements
This work was supported by institutional seed funds to J.M.B. J.M. was partly supported by an award from the Philippe Foundation. J.M.B. and her laboratory are supported by NIH grants DK072201, AI095245 and AI123284, the Burroughs Wellcome Fund, and a Leukemia and Lymphoma Society Scholar Award.
Abbreviations list
- ASC
Apoptosis-associated speck-like protein containing a caspase-recruitment domain
- CARD
Caspase-recruitment domain
- CLR
C-type lectin receptor
- DAMP
Damage-associated molecular pattern
- ER
Endoplasmic reticulum
- IFN-I
type I Interferon
- IFNAR
Interferon-α/β receptor
- IL-1β
Interleukin-1β
- IL-18
Interleukin-18
- JNK
c-Jun N-terminal kinase
- LPS
Lipopolysaccharides
- LRR
Leucine rich repeats
- LUBAC
Linear ubiquitin chain assembly complex
- MAVS
Mitochondrial antiviral-signaling protein
- NEK7
NIMA-related kinase 7
- NLR
NOD-like receptor
- NOD
Nucleotide-binding oligomerization domain
- OMV
Outer membrane vesicles
- PAMP
Pathogen-associated molecular pattern
- PKA
Protein kinase A
- PKD
Protein kinase D
- PP2A
Protein phosphatase 2A
- PTPN22
Protein tyrosine phosphatase, non-receptor type 22
- PRR
Pattern recognition receptor
- PYD
Pyrin domain
- RIG-I
Retinoic acid-inducible gene-I
- RLR
RIG-I-like receptor
- SCAP
SREB cleavage-activating protein
- SREBP2
Sterol regulatory element-binding protein 2
- SYK
Spleen tyrosine kinase
- TGN
Trans Golgi network
- TNF-α
Tumor necrosis factor-α
- TRIF
TIR-domain-containing adapter-inducing interferon-β
Footnotes
Conflict of interest disclosure
The authors declare no conflict of interest.
References
- 1.Janeway CA Jr., Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol, 1989. 54 Pt 1: p. 1–13. [DOI] [PubMed] [Google Scholar]
- 2.Janeway CA Jr., The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today, 1992. 13(1): p. 11–6. [DOI] [PubMed] [Google Scholar]
- 3.Moretti J and Blander JM, Insights into phagocytosis-coupled activation of pattern recognition receptors and inflammasomes. Curr Opin Immunol, 2014. 26: p. 100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson MR, et al. , Pattern recognition receptors and the innate immune response to viral infection. Viruses, 2011. 3(6): p. 920–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franchi L, Munoz-Planillo R, and Nunez G, Sensing and reacting to microbes through the inflammasomes. Nat Immunol, 2012. 13(4): p. 325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamkanfi M and Dixit VM, Mechanisms and functions of inflammasomes. Cell, 2014. 157(5): p. 1013–22. [DOI] [PubMed] [Google Scholar]
- 7.Man SM and Kanneganti TD, Regulation of inflammasome activation. Immunol Rev, 2015. 265(1): p. 6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franchi L, et al. , Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev, 2009. 227(1): p. 106–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinon F, Burns K, and Tschopp J, The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell, 2002. 10(2): p. 417–26. [DOI] [PubMed] [Google Scholar]
- 10.Dinarello CA, Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol, 2009. 27: p. 519–50. [DOI] [PubMed] [Google Scholar]
- 11.Miao EA, Rajan JV, and Aderem A, Caspase-1-induced pyroptotic cell death. Immunol Rev, 2011. 243(1): p. 206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mangan MSJ, et al. , Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov, 2018. 17(8): p. 588–606. [DOI] [PubMed] [Google Scholar]
- 13.Lamkanfi M and Dixit VM, Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol, 2012. 28: p. 137–61. [DOI] [PubMed] [Google Scholar]
- 14.Strowig T, et al. , Inflammasomes in health and disease. Nature, 2012. 481(7381): p. 278–86. [DOI] [PubMed] [Google Scholar]
- 15.Martinon F, et al. , Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol, 2004. 14(21): p. 1929–34. [DOI] [PubMed] [Google Scholar]
- 16.Keddie S, et al. , Cryopyrin-Associated Periodic Fever Syndrome and the Nervous System. Curr Treat Options Neurol, 2018. 20(10): p. 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinon F, et al. , Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature, 2006. 440(7081): p. 237–41. [DOI] [PubMed] [Google Scholar]
- 18.Ising C, et al. , NLRP3 inflammasome activation drives tau pathology. Nature, 2019. 575(7784): p. 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venegas C, et al. , Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature, 2017. 552(7685): p. 355–361. [DOI] [PubMed] [Google Scholar]
- 20.Heneka MT, McManus RM, and Latz E, Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci, 2018. 19(10): p. 610–621. [DOI] [PubMed] [Google Scholar]
- 21.Sayan M and Mossman BT, The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol, 2016. 13(1): p. 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauernfeind FG, et al. , Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol, 2009. 183(2): p. 787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franchi L, Eigenbrod T, and Nunez G, Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol, 2009. 183(2): p. 792–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bortolotti P, Faure E, and Kipnis E, Inflammasomes in Tissue Damages and Immune Disorders After Trauma. Front Immunol, 2018. 9: p. 1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dowling JK and O’Neill LA, Biochemical regulation of the inflammasome. Crit Rev Biochem Mol Biol, 2012. 47(5): p. 424–43. [DOI] [PubMed] [Google Scholar]
- 26.Franchi L, et al. , Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol, 2014. 193(8): p. 4214–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mariathasan S, et al. , Cryopyrin activates the inflammasome in response to toxins and ATP. Nature, 2006. 440(7081): p. 228–32. [DOI] [PubMed] [Google Scholar]
- 28.Sha W, et al. , Human NLRP3 inflammasome senses multiple types of bacterial RNAs. Proc Natl Acad Sci U S A, 2014. 111(45): p. 16059–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanneganti TD, et al. , Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature, 2006. 440(7081): p. 233–6. [DOI] [PubMed] [Google Scholar]
- 30.Sander LE, et al. , Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature, 2011. 474(7351): p. 385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munoz-Planillo R, et al. , K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity, 2013. 38(6): p. 1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piccini A, et al. , ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A, 2008. 105(23): p. 8067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franchi L, et al. , Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem, 2007. 282(26): p. 18810–8. [DOI] [PubMed] [Google Scholar]
- 34.Petrilli V, et al. , Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ, 2007. 14(9): p. 1583–9. [DOI] [PubMed] [Google Scholar]
- 35.Lee GS, et al. , The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature, 2012. 492(7427): p. 123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murakami T, et al. , Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A, 2012. 109(28): p. 11282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Misawa T, et al. , Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol, 2013. 14(5): p. 454–60. [DOI] [PubMed] [Google Scholar]
- 38.Subramanian N, et al. , The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell, 2013. 153(2): p. 348–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou R, et al. , A role for mitochondria in NLRP3 inflammasome activation. Nature, 2011. 469(7329): p. 221–5. [DOI] [PubMed] [Google Scholar]
- 40.Iyer SS, et al. , Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity, 2013. 39(2): p. 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakahira K, et al. , Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol, 2011. 12(3): p. 222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimada K, et al. , Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity, 2012. 36(3): p. 401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hornung V, et al. , Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol, 2008. 9(8): p. 847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He Y, Hara H, and Nunez G, Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci, 2016. 41(12): p. 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swanson KV, Deng M, and Ting JP, The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol, 2019. 19(8): p. 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Compan V, et al. , Cell volume regulation modulates NLRP3 inflammasome activation. Immunity, 2012. 37(3): p. 487–500. [DOI] [PubMed] [Google Scholar]
- 47.Lu A, et al. , Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell, 2014. 156(6): p. 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He Y, et al. , NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature, 2016. 530(7590): p. 354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmid-Burgk JL, et al. , A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem, 2016. 291(1): p. 103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi H, et al. , NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol, 2016. 17(3): p. 250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duncan JA, et al. , Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A, 2007. 104(19): p. 8041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coll RC, et al. , MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol, 2019. 15(6): p. 556–559. [DOI] [PubMed] [Google Scholar]
- 53.Tapia-Abellan A, et al. , MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol, 2019. 15(6): p. 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gurung P, et al. , FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol, 2014. 192(4): p. 1835–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mocarski ES, Upton JW, and Kaiser WJ, Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol, 2011. 12(2): p. 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang S, et al. , Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun, 2015. 6: p. 7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaidt MM, et al. , Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity, 2016. 44(4): p. 833–46. [DOI] [PubMed] [Google Scholar]
- 58.Fritsch M, et al. , Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature, 2019. 575(7784): p. 683–687. [DOI] [PubMed] [Google Scholar]
- 59.Newton K, et al. , Activity of caspase-8 determines plasticity between cell death pathways. Nature, 2019. 575(7784): p. 679–682. [DOI] [PubMed] [Google Scholar]
- 60.Wang S, et al. , Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell, 1998. 92(4): p. 501–9. [DOI] [PubMed] [Google Scholar]
- 61.Aachoui Y, et al. , Caspase-11 protects against bacteria that escape the vacuole. Science, 2013. 339(6122): p. 975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aglietti RA, et al. , GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A, 2016. 113(28): p. 7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding J, et al. , Pore-forming activity and structural autoinhibition of the gasdermin family. Nature, 2016. 535(7610): p. 111–6. [DOI] [PubMed] [Google Scholar]
- 64.Kayagaki N, et al. , Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature, 2015. 526(7575): p. 666–71. [DOI] [PubMed] [Google Scholar]
- 65.Liu X, et al. , Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature, 2016. 535(7610): p. 153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sborgi L, et al. , GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J, 2016. 35(16): p. 1766–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi J, et al. , Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature, 2015. 526(7575): p. 660–5. [DOI] [PubMed] [Google Scholar]
- 68.Shi J, et al. , Inflammatory caspases are innate immune receptors for intracellular LPS. Nature, 2014. 514(7521): p. 187–92. [DOI] [PubMed] [Google Scholar]
- 69.Broz P, et al. , Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature, 2012. 490(7419): p. 288–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kayagaki N, et al. , Non-canonical inflammasome activation targets caspase-11. Nature, 2011. 479(7371): p. 117–21. [DOI] [PubMed] [Google Scholar]
- 71.Gurung P, et al. , Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem, 2012. 287(41): p. 34474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meunier E, et al. , Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature, 2014. 509(7500): p. 366–70. [DOI] [PubMed] [Google Scholar]
- 73.Rathinam VA, et al. , TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell, 2012. 150(3): p. 606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang J, Zhao Y, and Shao F, Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol, 2015. 32: p. 78–83. [DOI] [PubMed] [Google Scholar]
- 75.Pilla DM, et al. , Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A, 2014. 111(16): p. 6046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hagar JA, et al. , Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science, 2013. 341(6151): p. 1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kayagaki N, et al. , Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science, 2013. 341(6151): p. 1246–9. [DOI] [PubMed] [Google Scholar]
- 78.Mitoma H, et al. , The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity, 2013. 39(1): p. 123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vanaja SK, et al. , Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell, 2016. 165(5): p. 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Garaude J, et al. , Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol, 2016. 17(9): p. 1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sander LE and Garaude J, The mitochondrial respiratory chain: A metabolic rheostat of innate immune cell-mediated antibacterial responses. Mitochondrion, 2018. 41: p. 28–36. [DOI] [PubMed] [Google Scholar]
- 82.Sanders MG, et al. , Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death Dis, 2015. 6: p. e1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Komander D and Rape M, The ubiquitin code. Annu Rev Biochem, 2012. 81: p. 203–29. [DOI] [PubMed] [Google Scholar]
- 84.Py BF, et al. , Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell, 2013. 49(2): p. 331–8. [DOI] [PubMed] [Google Scholar]
- 85.Song H, et al. , The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun, 2016. 7: p. 13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yan Y, et al. , Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell, 2015. 160(1–2): p. 62–73. [DOI] [PubMed] [Google Scholar]
- 87.Han S, et al. , Lipopolysaccharide Primes the NALP3 Inflammasome by Inhibiting Its Ubiquitination and Degradation Mediated by the SCFFBXL2 E3 Ligase. J Biol Chem, 2015. 290(29): p. 18124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawashima A, et al. , ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J Immunol, 2017. 199(10): p. 3614–3622. [DOI] [PubMed] [Google Scholar]
- 89.Catrysse L, et al. , A20 in inflammation and autoimmunity. Trends Immunol, 2014. 35(1): p. 22–31. [DOI] [PubMed] [Google Scholar]
- 90.Vande Walle L, et al. , Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature, 2014. 512(7512): p. 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shi CS, et al. , Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol, 2012. 13(3): p. 255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guan K, et al. , MAVS Promotes Inflammasome Activation by Targeting ASC for K63-Linked Ubiquitination via the E3 Ligase TRAF3. J Immunol, 2015. 194(10): p. 4880–90. [DOI] [PubMed] [Google Scholar]
- 93.Rodgers MA, et al. , The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med, 2014. 211(7): p. 1333–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Duong BH, et al. , A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity, 2015. 42(1): p. 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hunter T, The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell, 2007. 28(5): p. 730–8. [DOI] [PubMed] [Google Scholar]
- 96.Guo C, et al. , Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity, 2016. 45(4): p. 802–816. [DOI] [PubMed] [Google Scholar]
- 97.Hughes MM and O’Neill LAJ, Metabolic regulation of NLRP3. Immunol Rev, 2018. 281(1): p. 88–98. [DOI] [PubMed] [Google Scholar]
- 98.Mortimer L, et al. , NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol, 2016. 17(10): p. 1176–86. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Z, et al. , Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med, 2017. 214(9): p. 2671–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Song N, et al. , NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell, 2017. 68(1): p. 185–197 e6. [DOI] [PubMed] [Google Scholar]
- 101.Stutz A, et al. , NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med, 2017. 214(6): p. 1725–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Spalinger MR, et al. , NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest, 2016. 126(5): p. 1783–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hara H, et al. , Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol, 2013. 14(12): p. 1247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lin YC, et al. , Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J Leukoc Biol, 2015. 97(5): p. 825–835. [DOI] [PubMed] [Google Scholar]
- 105.Guo C, et al. , Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity, 2018. 49(5): p. 842–856 e7. [DOI] [PubMed] [Google Scholar]
- 106.Chen J and Chen ZJ, PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature, 2018. 564(7734): p. 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bryan NB, et al. , Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol, 2009. 182(5): p. 3173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stutz A, et al. , ASC speck formation as a readout for inflammasome activation. Methods Mol Biol, 2013. 1040: p. 91–101. [DOI] [PubMed] [Google Scholar]