

Abstract
Introduction
Benign lymph nodes have been considered the hubs of immune surveillance in cancer patients. The microenvironment of these lymphoid tissues can be immune suppressed, hence allowing for tumor progression. Understanding the spectrum of benign findings in bystander lymph nodes in immune checkpoint blockade therapy could prove to be key to understanding the mechanism and assessing treatment response.
Methods
Benign lymph nodes and spleen were evaluated from patients treated with immunotherapy who subsequently received postmortem examination. We used quantitative immunofluorescence (QIF) to assess tumor infiltrating lymphocytes (TIL) and macrophage marker expression and characterized activation status using a novel multiplexed QIF assay including CD3, GranzymeB, and Ki67. We performed immunohistochemistry to correlate results of QIF.
Results
Benign lymph nodes from non-responders to immunotherapy showed significantly higher expression of cytotoxic markers and proliferation index (Ki67) in T cells compared to responders. Higher expression of PD-L1 in macrophages was also observed. There was no significant difference in CD3+ expression, but higher levels of CD8+ T cells as well as CD20+ B cells were seen in lymph nodes of non-responders. No significant differences were seen between responder and non-responder splenic tissue. Findings were supported by traditional immunostaining methods.
Conclusions
While most studies in biomarkers for immunotherapy focus on tumor microenvironment, we show that benign lymph node microenvironment may predict response to immunotherapy. In responding patients, bystander lymph nodes appear to have been mobilized, resulting in reduced cytotoxic T cells. Conversely, patients whose disease progressed on immunotherapy demonstrate higher levels of macrophages that express increased PD-L1, and activated T cells not recruited to the tumor site.
Keywords: response to immunotherapy, lymph node microenvironment, quantitative immunofluorescence
Introduction
The discovery of immune checkpoints and the introduction of immunotherapy in clinical practice have completely changed the treatment landscape of many types of malignancies. A major mechanism of immune system inhibition is activation of programmed death 1 (PD-1) by its activator ligand programmed death ligand-1 (PD-L1) on the surface of tumor infiltrating lymphocytes (TILs) in the tumor microenvironment (TME).1,2 Expression of PD-L1 on tumor and immune cells is widely used as a biomarker for selection of patients who would respond to immune checkpoint inhibitors (ICIs) and more specifically to PD-1/PD-L1 pathway blockade. Although PD-L1 expression has been successfully used to predict response,3,4 there is a group of patients in the low expression group who would still benefit from immunotherapy,5 making the role of PD-L1 expression as a biomarker controversial. The lack of consistency in predicting response to ICIs has led to ongoing efforts for discovery of new markers and assays to improve patient selection. T cell gene expression signatures have been recently developed and can select subgroups of patients that would respond to immune checkpoint blockade.6–9 Similarly, tumor mutation burden (TMB) has been shown to be a new and promising predictive biomarker that is independent and potentially complementary to PD-L1 tumor expression.10–12
Although the need for assays and biomarkers that can accurately select patients who will respond to ICIs is urgent, as potential toxicity and the high cost of treatment have become important criteria for this treatment option selection,13–17 most efforts are focused on expression of immune-related markers on the tumor and the TME. However, immune response to tumor neoantigens is a complex mechanism that cannot solely be reflected by the TME cell populations. It starts at the regional lymph nodes, where antigen presentation takes place and is regulated by multiple cell types and mechanisms. As a result, studying and understanding the underlying mechanisms that lead to recruitment of cytotoxic T cells from lymphoid organs to the sites of disease, including the TME, is likely to be important.
To explore the hypothesis that benign lymph nodes may display detectable and quantifiable differences, we used a cohort of eight patients with a history of ICIs and collected postmortem tissue from regional benign lymph node and spleen sites. The goal of this study is to evaluate the morphology and phenotype of these lymphoid tissues and assess any differences in the immune cell populations and their activation status across responders and non-responders to immunotherapy. We test the underlying hypothesis that, although not contiguous with the tumor, the lymph node represents an extension of the TME and, as such, shows characteristics that may be associated with response or resistance to immune therapy.
Methods
Patient cohort
The Yale Pathology Autopsy database was searched for postmortem specimens from 2011 to 2017 for cancer cases with a history of PD-1/PD-L1 pathway blockade treatment. A total of eight cases was selected after evaluation for sufficient benign nodal and splenic tissue and good tissue quality. H&E stained sections of benign lymph nodes and spleen were reviewed for specimen integrity by two pathologists. The lymph nodes examined were all regional/draining lymph nodes from the patient’s last known site of disease. Clinicopathological information from cohort patients were collected from clinical records and pathology reports. Response was defined by Response Evaluation Criteria In Solid Tumors (RECIST) v1.1. Detailed characteristics of the patient cohort are presented in Table 1.
Table 1 .
Patient cohort information.
| Case | Age | Sex | Diagnosis | Immunotherapy | Cycles | Response | Time from treatment to death (days) | Cause of death | Other concurrent treatment |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 69 | F | Renal cell carcinoma | Anti PD-1 | 2 | No | 157 | Status epilepticus | Axitinib, bevacizumab, interferon-⍺ |
| 2 | 66 | M | Malignant melanoma | Anti PD-1 | 6 | No | 116 | Pulmonary embolism | Radiation, gp-100-CD3 (immunocore), ulixertinib |
| 3 | 73 | M | Non small cell carcinoma lung | Anti PD-L1 | 4 | Yes | 16 | Pericarditis with heart failure | None |
| 4 | 65 | F | Non small cell carcinoma lung | Anti PD-1, anti CTLA4 | 4 | Yes | 33 | Cardiac tamponade | None |
| 5 | 60 | M | Non small cell carcinoma lung | Anti PD-1, anti CTLA4 | 18 | Yes | 139 | Cardiomyopathy with heart failure | None |
| 6 | 70 | M | Oral squamous cell carcinoma | Anti PD-1 | 70 | Yes | 18 | Squamous cell carcinoma with hemorrhagic complications | None |
| 7 | 64 | F | Non small cell carcinoma lung | Anti PD-1 | 3 | Yes | 23 | Myocarditis and heart failure | None |
| 8 | 47 | F | Malignant melanoma | Anti PD-1 | 8 | Yes | 22 | Cardiomyopathy with heart failure | None |
Immunofluorescence staining: multiplexed TILs and TILs activation
The multiplexing TIL protocol has been presented before.18 Briefly, fresh TMA sections were deparaffinized and subjected to antigen retrieval using EDTA buffer (Sigma-Aldrich, St Louis, MO, USA) pH 8.0 for 20 minutes at 97 °C in a pressure-boiling container (PT module, Lab Vision, Fremont, CA, USA). Slides were then incubated in 30% hydrogen peroxide in methanol for 30 minutes at room temperature and subsequently with a blocking solution containing 0.3% bovine serum albumin in 0.05% Tween solution for 30 minutes. Staining for pan-cytokeratin, CD4, CD8, and CD20 was performed using a sequential multiplexed immunofluorescence protocol with isotype-specific primary antibodies to detect epithelial tumor cells (cytokeratin: clone Z0622, Agilent, Santa Clara, CA, USA), helper T cells (CD4 IgG, 1:100, clone SP35, Spring Bioscience, Pleasanton, CA, USA), cytotoxic T cells (CD8 IgG1, 1:250, clone C8/144B, Agilent), and B lymphocytes (CD20 IgG2a, 1:150, clone L26, Agilent). Nuclei were highlighted using 4′,6-diamidino-2-phenylindole (DAPI). Secondary antibodies and fluorescent reagents used were goat anti-rabbit Alexa546 (Molecular Probes, Eugene, OR, USA), anti-rabbit Envision (K4009, Agilent) with biotynilated tyramide/streptavidine-Alexa750 conjugate (Perkin-Elmer, Waltham, MA, USA), anti-mouse IgG1 antibody (1:100, eBioscience, San Diego, CA, USA) with fluorescein-tyramide (Perkin-Elmer), anti-mouse IgG2a antibody (1:200, Abcam, Cambridge, MA) with Cy5-tyramide (Perkin-Elmer). Residual horseradish peroxidase activity between incubations with secondary antibodies was eliminated by exposing the slides twice for seven minutes to a solution containing benzoic hydrazide (0.136 g) and hydrogen peroxide (50 μl).
Staining for T cell activation panel19 included pan-cytokeratin, CD3, Ki67, and Granzyme B, and was performed using a similar sequential multiplexed immunofluorescence protocol with isotype-specific primary antibodies to detect epithelial tumor cells (cytokeratin, clone Z0622, 1:100, Agilent), T lymphocytes (CD3 IgG, 1:100, clone SP7, Novus Biologicals, Littleton, CO, USA), Ki67 (IgG1, 1:100, clone MIB-1, Agilent), and Granzyme B (IgG2a, 1:2000, clone 4E6, Abcam). For the macrophage panel, primary antibodies were used to detect epithelial tumor cells (cytokeratin, clone Z0622, 1:100, Agilent), macrophages (IgG3, 1:200, clone PGM1, Agilent), and PD-L1 (1:1000, clone SP142, Spring Bioscience, Pleasanton, CA, USA). Fresh control slides from morphologically normal human tonsil were included in each staining batch as positive controls and to ensure reproducibility.
Immunohistochemistry staining
Tissue sections were subjected to the same deparaffinization, antigen retrieval, and blocking protocol mentioned above. The lymph node and spleen sections were stained with primary antibodies for CD3 (clone 2GV6, Ventana), CD4 (Clone SP35, Ventana), CD8 (clone 144B, Agilent), CD20 (Clone L26, Agilent), and TIA-1 (clone TIA1, Biocare).
Quantitative immunofluorescence
Quantitative measurement of the immune markers and TILs activation markers was performed using an AQUA (Automated Quantitative Analysis) method (NavigateBP, Carlsbad, CA, USA), quantifying fluorescent signal within subcellular compartments, as previously described.20 For CD3, CD4, CD8, CD20, and CD68, the area in which the signal was measured was defined by positive DAPI staining. TILs activation markers, Ki67 and Granzyme B, were measured in a dilated CD3 positive compartment, defining predominantly T lymphocytes. PD-L1 was measured in the CD68 positive compartment.
QIF scores were calculated by dividing the target pixel intensity by the area of the compartment, and were then normalized to the exposure time and bit depth at which the images were captured, allowing scores collected at different exposure times to be comparable. All fields of view (FOV) were visually evaluated and those with staining artifacts or presence of <2% compartment area were systematically excluded.
Statistical analysis
Immunohistochemistry scoring of the markers was performed by two pathologists on lymph node and spleen sections. For each slide evaluated, pathologists interpreted staining at 40× high power field and counted positive cells per background cell were appropriate (such as Ki67 quantification and CD4/CD8 ratio). For cytotoxic marker evaluation on immunohistochemistry, which is more difficult to ascertain on a per cell basis, approximate level of positivity was recorded (−, −/+, +, ++). Comparison of QIF scores for different markers across responders and non-responders was performed by Mann–Whitney analysis. Each patient case was represented by the 10 highest scored FOV for both lymph nodes and spleen. All P values were based on two-sided tests and any P value < 0.05 was considered statistically significant. Statistical analysis was performed using JMP Pro software (version 11.2.0, 2014, SAS Institute Inc, Cary, NC, USA) and GraphPad Prism v6.0 for Windows (GraphPad Software, Inc, San Diego, CA, USA).
Ethics approval
The collection of patients’ tissue/data and subsequent analysis was approved by the University Human Subjects Institutional Review Board.
Results
Histopathological analysis of chromogenic assays
Postmortem specimens of eight patients with a history of treatment with PD-1/PD-L1 pathway inhibitors were collected and examined to assess whether there is a difference in morphology and common immune marker expression in benign regional lymph nodes associated with response to immunotherapy. Similarly, matched spleen specimens were assessed to determine whether the effect of immune checkpoint inhibition is systemic and reflected on all lymphatic system organs. The cohort included patients with solid tumors with an average age of 64 years and was balanced in terms of sex. Most patients received anti PD-1/PD-L1 pathway inhibitors, with two non-small cell lung cancer (NSCLC) patients receiving combination treatment with anti-CTLA4 and anti-PD-1 inhibitors. Out of the eight patients, only two did not respond to immunotherapy, as defined by RECIST V1.1 criteria.
H&E stained sections of benign lymph nodes and spleen were reviewed for morphological differences by two pathologists independently, both blinded to clinical response. On microscopic examination, the lymph nodes of responders showed marked architectural abnormalities, including lack of defined follicle formation, overall reduction in lymphocytes, and frequent replacement by fibrosis. However, the lymph nodes from non-responders were cellular but showed greater infiltration by macrophages.
Lymph node and matched spleen sections were subsequently stained and scored by two pathologists for CD4, CD8, and T cell intracytoplasmic antigen (TIA-1) expression to assess the immune cell populations and their activation status (Table 2). Representative images are shown in Fig. 1A–H. The agreement between Pathologist 1 (P1) and Pathologist 2 (P2) scoring was high. Benign lymph node scoring for CD4:CD8 ratio did not show any consistent differences across responders and non-responders to immunotherapy. Interestingly, lymph node TIA-1 expression was found to be high in non-responders, whereas in responders there were substantially fewer cells positive for TIA-1. In spleen sections, there was no difference in the CD4:CD8 ratio or TIA-1 expression.
Table 2 .
Pathologist scoring of lymph node and spleen tissue for CD4:CD8 ratio and TIA-1 expression.
| Case | Lymph node CD4:CD8 | Spleen CD4:CD8 | Spleen CD4:CD8 | Spleen TIA-1 | ||||
|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P1 | P2 | P1 | P2 | P1 | P2 | |
| 1 | 1:1 | 2:1 | (++) | (++) | 1:2 | 1:3 | (+) | (++) |
| 2 | 3:1 | 2:1 | (++) | (++) | 1:8 | 1:10 | (++) | (++) |
| 3 | 3:1 | 3:1 | (−) | (−) | 4:1 | 4:1 | (+) | (+) |
| 4 | 3:1 | 3:1 | (−) | (−) | 1:3 | 1:3 | (+) | (+) |
| 5 | 6:1 | 8:1 | (−) | (+) | 1:8 | 1:10 | (+) | (+) |
| 6 | 6:1 | 4:1 | (−) | (−/+) | 5:1 | 5:1 | (+) | (++) |
| 7 | 3:1 | 2:1 | (−/+) | (−/+) | 1:1 | 1:1 | (+) | (+) |
| 8 | 1:1 | 1:1 | (−) | (−) | 3:1 | 2:1 | (+) | (+) |
Figure 1 .
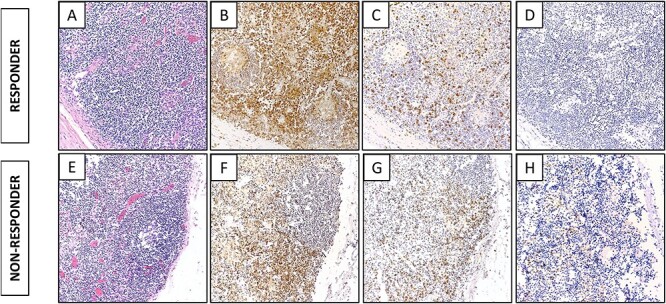
Representative immunohistochemistry images of marker expression in regional lymph nodes of a responder and a non-responder to immunotherapy. In a responder: (A) H&E; (B) CD4; (C) CD8; (D) TIA expression. In a non-responder: (E) H&E; (F) CD4; (G) CD8; (H) TIA expression.
Quantitative assessment of immune marker expression
Benign lymph node and spleen sections from the eight cohort patients were stained with three multiplex panels to quantitatively assess the expression of CD4, CD8, CD20, and CD3 and the T cell activation status. Additionally, we measured CD68 and expression of PD-L1 in macrophages. Representative QIF images of the multiplex panels are shown in Fig. 2. Benign lymph node analysis revealed that there was statistically significantly higher expression of CD4 (P = 0.0001), CD8 (P < 0.0001), and CD20 (P < 0.0001) in immunotherapy non-responders compared to responders (Fig. 3). While there was no difference in the expression of CD3, T cells in the non-responders had a high expression of Granzyme B (P < 0.0001) and Ki67 (P < 0.0001). Granzyme B and Ki67 have been previously used in this multiplex panel as markers of T cell activation,20 reflecting cytotoxicity and proliferation, respectively. Consistent with the morphological features previously presented, CD68 expression was higher in the non-responders (P < 0.0001). Additionally, PD-L1 expression measured in the macrophage compartment was similarly found to be higher in non-responders compared to responders (P < 0.0001). Analysis of the spleen sections revealed an inverse correlation for CD4 and CD8 expression, with the responders showing higher expression (P < 0.0001 and P = 0.0135 respectively) (Fig. 4). Conversely, similar to the lymph node expression, non-responders had a higher CD20 (P = 0.0035) and T cell Granzyme B (P = 0.0258) expression. No difference was seen for CD3, Ki67, CD68, and PD-L1 expression across patient groups.
Figure 2 .
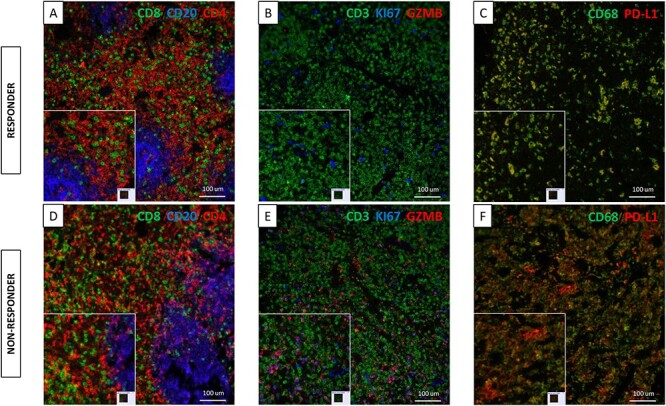
Representative multiplexed QIF images of marker expression in regional lymph nodes of a responder and a non-responder to immunotherapy. (A) TILs marker expression in a responder; CD8 (green), CD20 (blue), CD4 (red). (B) TILs activation marker expression in a responder; Granzyme B (red), Ki67 (blue), CD3 (green), cytokeratin (green). (C) Macrophage marker expression in a responder; PD-L1 (red), CD68 (green). (D) TILs marker expression in a non-responder; CD8 (green), CD20 (blue), CD4 (red). (E) TILs activation marker expression in a non-responder; Granzyme B (red), Ki67 (blue), CD3 (green), cytokeratin (green). (F) Macrophage marker expression in a non-responder; PD-L1 (red), CD68 (green).
Figure 3 .

Immune marker expression in benign lymph nodes by response to immunotherapy. (A) CD4; (B) CD8; (C) CD20; (D) CD3; (E) Granzyme B in CD3; (F) Ki67 in CD3; (G) CD68; (H) PD-L1 in CD68. Mann–Whitney two-tailed U test comparing the expression of immune markers in 10 FOV hotspots of lymph node tissue from patients treated with immune checkpoint inhibitors.
Figure 4 .
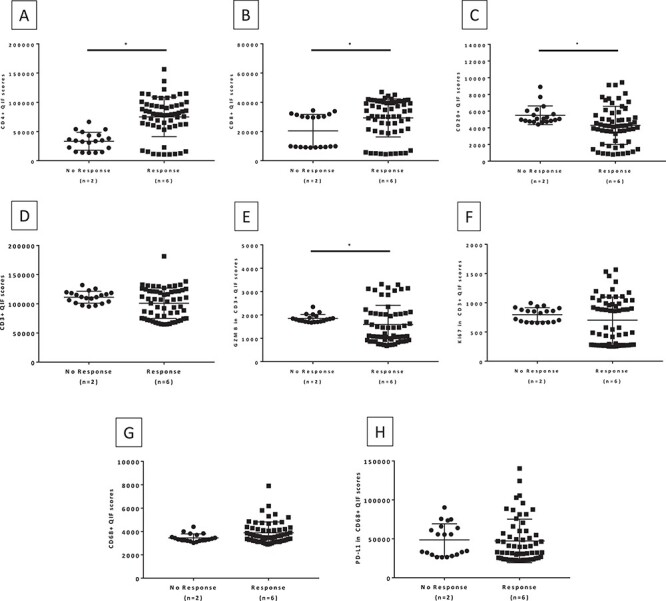
Immune marker expression in spleen by response to immunotherapy. (A) CD4; (B) CD8; (C) CD20; (D) CD3; (E) Granzyme B in CD3; (F) Ki67 in CD3; (G) CD68; (H) PD-L1 in CD68. Mann–Whitney two-tailed U test comparing the expression of immune markers in 10 FOV hotspots of spleen tissue from patients treated with immune checkpoint inhibitors.
Discussion
Benign lymph nodes have been considered the hubs of immune surveillance in cancer patients. The structure of lymph nodes is complex as there are distinct compartments with specific functions. Lymph node cortex contains B cell-enriched follicles, while T lymphocytes are located in the interfollicular areas of the cortex and paracortical zones in the medulla. High-endothelium venules of the paracortex form a site at which naive T cells interact with antigen presenting cells (APCs) to generate T cell-dependent immune responses.
The immune system protects the host against tumor development, growth, and metastasis21,22 and lymph nodes are involved in anti-tumor immune responses as the site of tumor antigen presentation by various APCs like dendritic cells.23–25 Their interaction with T cells sensitizes the latter to tumor antigens and initiates the cellular immune response.26 The activated T cells are then recruited to the TME to destroy the tumor. The importance of regional lymph nodes as the immune sensitization sites to tumor antigens rather than the primary tumor was underlined in the guinea pig line-10 hepatoma model, showing that removal of the draining lymph node (LN) to the tumor immunization skin site, resulted in an abrogation in the development of systemic immunity.27
Over the past decade, the TME has been extensively studied as a site of immune suppression. The presence of intratumoral CD8+ and CD4+ T cells has been independently associated with a good clinical outcome,28 while the expression of immune checkpoints by tumor cells is a well characterized immune evasion mechanism.29 An additional mechanism limiting the anti-tumor immune response is deregulation of immune-cell recruitment. In particular, recruitment of suppressive myeloid cells, such as myeloid-derived suppressor cells, tumor-associated macrophages, immature dendritic cells (DCs), and immunosuppressive neutrophils, has been described as a mechanism of immunosuppression.30,31
In this study, we used postmortem benign lymph node and spleen tissue from patients with a history of ICIs to assess the predictive value of immune cell populations residing in those lymphoid structures. Our main finding was the retained proliferative cytotoxic CD8+ T cells in uninvolved lymph nodes with a compensatory increase in macrophages in those who failed treatment. Additionally, non-recruited T cells in lymph nodes from patients that did not respond to ICIs had a higher expression of Granzyme B, a marker reflecting the levels of the cytotoxic granules, and Ki67, a marker of proliferation.20 The QIF findings were supported by TIA-1 chromogenic assays scored by two pathologists. TIA-1 is a marker that is mainly expressed by activated cytotoxic lymphocytes and natural killer cells.32–34 The elevation in levels of Granzyme B in the non-responders appears to be constant regardless of time interval from treatment, suggesting a prolonged defect in appropriate recruitment of these cytotoxic elements to the site of action. It may be that a compensatory overproliferation exists to explain the increase in Ki67. Also of interest, although difficult to demonstrate quantitatively, is the more often depleted morphology of bystander lymph nodes in the responder category. All of these findings together argue for an inability to recruit necessary cytotoxic elements from benign lymph nodes in the patients who did not respond to immunotherapy.
The mechanism involved in the defective recruitment of activated T cells in TME is still unknown. It has been reported that the microenvironment of these lymphoid tissues can be immune suppressed, hence allowing for tumor progression. Recent studies have shown that the presence of regulatory T cells (Tregs) has a suppressive effect on anti-tumor immunity, and their depletion enhances effector T cell responses in tumor draining lymph nodes.35,36 Tregs in regional draining lymph nodes have been shown contribute to the immunosuppressive environment of colorectal cancer patients,37 but their association to clinical outcome is still controversial.38–42
Although there have been a few studies reporting contradicting results regarding the prognostic value of immune cells in regional LN,43,44 here we use an alternative approach to explore T cell populations and their activation status and associate these findings to response to ICIs. This approach gives a new insight on the importance of uninvolved regional lymph nodes in predicting outcome to immunotherapy. Tissue from LN is usually widely available after surgical excision in most malignancies. Morphological differences that can be identified by pathologists without the need of specialized assays can aid in therapeutic decisions, and identification of the activation status of immune cells residing in the regional LN can be a complementary assay in the effort to predict response to ICIs. Although we also attempted to study the same cellular components in the spleen, the data in postmortem spleen appears to be confounded by greater autolysis than in most other tissues. While a couple of the lymphoid markers mimic the finding in lymph nodes, many of the markers were similar across patient cohorts. These markers, such as Ki67 and CD68, are known to be highly affected by tissue viability.
There are a number of limitations in this study. This is a descriptive hypothesis-generating study with a small number of patients and no validation set to confirm our findings. Based on our current progress, efforts are under way to include multiple centers in the country with a standardized protocol that will allow us to have definite answers on the mechanism of immune cell recruitment in the TME and the role of benign lymph nodes in predicting response to ICIs. Secondly, although standard immunohistochemistry (IHC) assays are common, the use of multiplex QIF is not yet implemented in clinical practice. However, similar to this study we have shown that immune markers can be easily measured using QIF on whole sections45 and can be easily adopted as a predictive assay. Finally, no matching tumor tissue was evaluated to assess the TME. Most of the patients in this cohort had remarkable response to immunotherapy, with only residual fibrosis and no viable tumor remaining. Assessing the correlation of lymph node and TME immune cell populations is a direction that our efforts will follow.
In conclusion, in this study we found that benign regional lymph nodes had morphological differences with unique residing immune cell populations across responders and non-responders to immunotherapy. Strong expression of activation markers was seen in the lymph nodes of non-responding patients, implicating that their recruitment in the TME was deficient. Additionally, higher macrophage infiltration and high PD-L1 expression could result in poor recruitment of immune cells in the TME. Understanding the spectrum of benign findings in these bystander lymph nodes in immune checkpoint blockade therapy could prove to be a valuable component in understanding the mechanism and assessing the treatment response.
Acknowledgments
The authors acknowledge the expert assistance of Lori Charette and her staff in the Yale Pathology Tissue Services. This work was funded by Yale Department of Pathology.
Conflict of interest
None declared.
References
- 1. Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sharpe AH, Wherry EJ, Ahmed R, et al. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 2007;8:239–45. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 3. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shien K, Papadimitrakopoulou VA, Wistuba II. Predictive biomarkers of response to PD-1/PD-L1 immune checkpoint inhibitors in non-small cell lung cancer. Lung Cancer 2016;99:79–87. doi: 10.1016/j.lungcan.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Socinski MA, Jotte RM, Cappuzzo F, et al. Overall survival (OS) analysis of IMpower150, a randomized Ph 3 study of atezolizumab (atezo) + chemotherapy (chemo) ± bevacizumab (bev) vs chemo + bev in 1L nonsquamous (NSQ) NSCLC. J Clin Oncol 2018;36:9002–2. doi: 10.1200/JCO.2018.36.15_suppl.9002. [DOI] [Google Scholar]
- 7. Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 2018;378:2288–301. doi: 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 8. Kowanetz M, Zou W, McCleland M, et al. MA 05.09 pre-existing immunity measured by Teff gene expression in tumor tissue is associated with Atezolizumad efficacy in NSCLC. J Thorac Oncol 2017;12:S1817–8. doi: 10.1016/j.jtho.2017.09.485. [DOI] [Google Scholar]
- 9. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016;387:1837–46. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 10. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909–20.doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018;378:2093–104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with Ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus Ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Daud AI, Wolchok JD, Robert C, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody Pembrolizumab in melanoma. J Clin Oncol 2016;34:4102–9. doi: 10.1200/JCO.2016.67.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with Lambrolizumab (anti–PD-1) in melanoma. N Engl J Med 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J, Chmielowski B, Pellissier J, et al. Cost-effectiveness of pembrozilumab versus ipilimumab in ipilimumab-naive patients with advanced melanoma in the United States. J Manag Care Spec Pharm 2017;23:184–94. doi: 10.18553/jmcp.2017.23.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown JR, Wimberly H, Lannin DR, et al. Multiplexed quantitative analysis of CD3, CD8, and CD20 predicts response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res 2014;20:5995–6005. doi: 10.1158/1078-0432.CCR-14-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gettinger SN, Choi J, Mani N, et al. A dormant TIL phenotype defines non-small cell lung carcinomas sensitive to immune checkpoint blockers. Nat Commun 2018;9:3196. doi: 10.1038/s41467-018-05032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med 2002;8:1323–7. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- 21. Nanni P, Nicoletti G, Palladini A, et al. Antimetastatic activity of a preventive cancer vaccine. Cancer Res 2007;67:11037–44. doi: 10.1158/0008-5472.CAN-07-2499. [DOI] [PubMed] [Google Scholar]
- 22. Moriya K, Wakabayashi A, Shimizu M, et al. Induction of tumor-specific acquired immunity against already established tumors by selective stimulation of innate DEC-205(+) dendritic cells. Cancer Immunol Immunother 2010;59:1083–95. doi: 10.1007/s00262-010-0835-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol 2007;7:543–55. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 24. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 25. Wilson NS, Villadangos JA. Regulation of antigen presentation and cross-presentation in the dendritic cell network: facts, hypothesis, and immunological implications. Adv Immunol 2005;86:241–305. doi: 10.1016/S0065-2776(04)86007-3. [DOI] [PubMed] [Google Scholar]
- 26. Cahalan MD, Parker I. Imaging the choreography of lymphocyte trafficking and the immune response. Curr Opin Immunol 2006;18:476–82. doi: 10.1016/j.coi.2006.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanna MG Jr, Bucana CD, Pollack VA. Immunological stimulation in situ: the acute and chronic inflammatory responses in the induction of tumor immunity. Contemp Top Immunobiol 1980;10:267–96. doi: 10.1007/978-1-4684-3677-8_13. [DOI] [PubMed] [Google Scholar]
- 28. Fridman WH, Pages F, Sautes-Fridman C, et al. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 29. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 2008;8:467–77. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 30. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12:253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 32. Anderson P, Nagler-Anderson C, O'Brien C, et al. A monoclonal antibody reactive with a 15-kDa cytoplasmic granule-associated protein defines a subpopulation of CD8+ T lymphocytes. J Immunol 1990;144:574–82. PMID: 2104899. [PubMed] [Google Scholar]
- 33. Anderson P. TIA-1: structural and functional studies on a new class of cytolytic effector molecule. Curr Top Microbiol Immunol 1995;198:131–43. doi: 10.1007/978-3-642-79414-8_8. [DOI] [PubMed] [Google Scholar]
- 34. Tian Q, Streuli M, Saito H, et al. A polyadenylate binding protein localized to the granules of cytolytic lymphocytes induces DNA fragmentation in target cells. Cell 1991;67:629–39. doi: 10.1016/0092-8674(91)90536-8. [DOI] [PubMed] [Google Scholar]
- 35. Tanaka H, Tanaka J, Kjaergaard J, et al. Depletion of CD4+ CD25+ regulatory cells augments the generation of specific immune T cells in tumor-draining lymph nodes. J Immunother 2002;25:207–17. doi: 10.1097/00002371-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 36. Munn DH, Mellor AL. The tumor-draining lymph node as an immune-privileged site. Immunol Rev 2006;213:146–58. doi: 10.1111/j.1600-065X.2006.00444.x. [DOI] [PubMed] [Google Scholar]
- 37. Deng L, Zhang H, Luan Y, et al. Accumulation of foxp3+ T regulatory cells in draining lymph nodes correlates with disease progression and immune suppression in colorectal cancer patients. Clin Cancer Res 2010;16:4105–12. doi: 10.1158/1078-0432.CCR-10-1073. [DOI] [PubMed] [Google Scholar]
- 38. Yaqub S, Henjum K, Mahic M, et al. Regulatory T cells in colorectal cancer patients suppress anti-tumor immune activity in a COX-2 dependent manner. Cancer Immunol Immunother 2008;57:813–21. doi: 10.1007/s00262-007-0417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clarke SL, Betts GJ, Plant A, et al. CD4+CD25+FOXP3+ regulatory T cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS One 2006;1:e129. doi: 10.1371/journal.pone.0000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ling KL, Pratap SE, Bates GJ, et al. Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun 2007;7:7. PMID: 17388261. [PMC free article] [PubMed] [Google Scholar]
- 41. Frey DM, Droeser RA, Viehl CT, et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer 2010;126:2635–43. doi: 10.1002/ijc.24989. [DOI] [PubMed] [Google Scholar]
- 42. Salama P, Phillips M, Grieu F, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol 2009;27:186–92. doi: 10.1200/JCO.2008.18.7229. [DOI] [PubMed] [Google Scholar]
- 43. Kemp RA, Black MA, McCall J, et al. T cell subpopulations in lymph nodes may not be predictive of patient outcome in colorectal cancer. J Exp Clin Cancer Res 2011;30:78. doi: 10.1186/1756-9966-30-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kohrt HE, Nouri N, Nowels K, et al. Profile of immune cells in axillary lymph nodes predicts disease-free survival in breast cancer. PLoS Med 2005;2:e284. doi: 10.1371/journal.pmed.0020284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McLaughlin J, Han G, Schalper KA, et al. Quantitative assessment of the heterogeneity of PD-L1 expression in non-small-cell lung cancer. JAMA Oncol 2016;246–54. doi: 10.1001/jamaoncol.2015.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]