

Abstract
Leber congenital amaurosis (LCA) is a severe, genetically heterogeneous recessive eye disease in which ~ 35% of gene mutations are in-frame nonsense mutations coding for loss-of-function premature termination codons (PTCs) in mRNA. Nonsense suppression therapy allows read-through of PTCs leading to production of full-length protein. A limitation of nonsense suppression is that nonsense-mediated decay (NMD) degrades PTC-containing RNA transcripts. The purpose of this study was to determine whether inhibition of NMD could improve nonsense suppression efficacy in vivo. Using a high-throughput approach in the recessive cep290 zebrafish model of LCA (cep290;Q1223X), we first tested the NMD inhibitor Amlexanox in combination with the nonsense suppression drug Ataluren. We observed reduced retinal cell death and improved visual function. With these positive data, we next investigated whether this strategy was also applicable across species in two mammalian models: Rd12 (rpe65;R44X) and Rd3 (rd3;R107X) mouse models of LCA. In the Rd12 model, cell death was reduced, RPE65 protein was produced, and in vivo visual function testing was improved. We establish for the first time that the mechanism of action of Amlexanox in Rd12 retina was through reduced UPF1 phosphorylation. In the Rd3 model, however, no beneficial effect was observed with Ataluren alone or in combination with Amlexanox. This variation in response establishes that some forms of nonsense mutation LCA can be targeted by RNA therapies, but that this needs to be verified for each genotype. The implementation of precision medicine by identifying better responders to specific drugs is essential for development of validated retinal therapies.
Keywords: precision medicine, Ataluren, Amlexanox, nonsense suppression, RPE65, CEP290, RD3
Introduction
The extensive molecular genetic heterogeneity observed in ocular diseases such as Leber congenital amaurosis (LCA) is a barrier to development of new therapeutics. More than 400 mutations in at least 21 different genes cause LCA.1,2 Some animal models of LCA have been successfully corrected with gene-based therapies,3–7 and clinical trials have been initiated for one LCA gene (RPE65).8–10 Only recently has one RPE65 therapeutic (Luxturna®) reached clinical practice and then for only a subset of patients.11,12 It took more than 10 years from identification of RPE65 gene mutations to development of a therapeutic, and there are still 20 other genes causing LCA for which there are no therapeutics.
A nonsense mutation leading to a premature termination codon (PTC) is a common type of gene defect that occurs in about 35% of LCA patients,1 thus targeting this type of mutation could benefit a large number of patients. Most mRNA templates that contain a PTC are recognized by a surveillance mechanism called nonsense-mediated decay (NMD), which prevents synthesis of truncated proteins with potential dominant-negative or toxic gain-of-function activities.13 However, altering the efficiency of the termination process during mRNA translation can result in replacement of a PTC with a near-cognate aminoacyl tRNA (nonsense suppression), so in effect would produce either the correct protein or a protein with one amino acid sequence change.14 If the PTC is not in a critical position for protein activity, then a functional protein could be produced. Using a variety of small molecule drugs, suppression of nonsense mutations has been demonstrated in mammalian and patient-derived cell lines, and in a variety of pre-clinical animal models.15 Although several studies have questioned the mechanism of action of one of these drugs (Ataluren),16,17 this has now been refuted in a number of follow-up studies.18,19 We have therefore used this approach to treat zebrafish20 and rodent21 models of inherited retinal degenerations, and have reversed the congenital eye malformation defects in Pax6 mutant mice using a topical eye drop preparation in the early postnatal period.22
Based on evidence from animal studies23 and clinical trials using current nonsense suppression drugs,24,25 their efficacy is limited in part because NMD is still active and reduces the levels of nonsense-containing transcripts.26 Hence the drugs may not be able to make enough protein to provide a therapeutic benefit. Several studies have shown that inhibition of NMD leads to an increase in nonsense-containing mRNAs and increased synthesis of full-length protein, in cells and animal models carrying nonsense mutations.27–29 Therefore, in this study we compared nonsense suppression alone or in combination with NMD inhibition in three genetic models of LCA caused by mutations in cep290,30rpe65,31 and rd3.32
Materials and methods
Animals
All studies were carried out with the approval of the Animal Care Committee at the University of British Columbia, Canada and in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Mice were housed under cyclic light (14 hours on: 10 hours off) and had access to food and water ad libitum. Mice were maintained on the C57BL/6 background strain as recessive Rd12 homozygotes (JAX Lab, stock #005397; rpe65 R44X mutation [UGA]) or Rd3 homozygotes (JAX Lab, stock #008627; rd3 R107X mutation [UGA]). Wildtype AB33 and heterozygous cep290 (Zebrafish Mutation Project Repository, stock #sa1383; cep290 Q1223X mutation [UAG])30 zebrafish strains were maintained as inbred stocks and staged according to morphological criteria as previously described.34 Embryos were raised at 28.5°C on a 14 hour light/10 hour dark cycle in 100 mm2 Petri dishes containing E2 or E3 medium.35
Drug administration
In cep290 zebrafish, after dechorionation of embryos at 10 hours post-fertilization (hpf), a range of doses of Amlexanox or Ataluren (0.3–60 μM) were added directly to the aquarium water and then embryos were raised to 6 days post-fertilization (dpf). Each day the aquaria water containing the drug was refreshed. For each drug dose, 20 embryos were used in three independent experiments. The number of embryos surviving at 6 days was counted and quantitative data were expressed as mean ± SEM. The cep290 mutant embryos were treated with either 1 μM Amlexanox or combined 1 μM Amlexanox/3 μM Ataluren from 10 hpf with assessment of the phenotypic effect at 6 dpf by histology. For postnatal drug treatment, mice received daily subcutaneous injections of Ataluren (30 μg/g; Selleckchem), or a combination of 30 μg/g Ataluren plus Amlexanox (25 μg/g; Bio-Techne Canada) from postnatal (P) day 4 to P120. For prenatal treatment, time-mated mice received daily subcutaneous injections of combined Ataluren plus Amlexanox from E12 until birth. This was followed by the postnatal treatment regimen as described above to P120. Efficacy of the treatment was assessed by optokinetic tracking (OKT) followed by histology, enzyme-linked immunosorbent assay (ELISA), western blotting, and qRT-PCR.
Optokinetic tracking
We assessed behavioral responses in mice by measuring the spatial frequency threshold during optokinetic tracking as previously described22 using rodent-specific OptoMotry software (Cerebral Mechanics Inc.). Briefly, a virtual cylinder was created that comprised a vertical sine wave grating projected onto four computer monitors that surrounded a platform on which the mice were placed. The cylinder was rotated at 12 deg/sec and head tracking was monitored via a video camera. The highest spatial frequency capable of driving the head tracking response was adopted as the threshold (photopic intensity, 142 cd/m2).
Histology and immunocytochemistry
Eyes were enucleated from euthanized mice and then fixed in Karnovsky’s fixative for 2 hours prior to embedding in paraffin wax. Zebrafish embryos were fixed by immersion in 4% paraformaldehyde overnight at 4°C. Fixed embryos were washed in phosphate-buffered saline (PBS) and then dehydrated through a graded ethanol series (50%, 70%, 90% and 100%), before being transferred to xylene and embedded in paraffin wax. Sections of mouse eyes and zebrafish heads were stained with hematoxylin and eosin and photographed using an Aperio ScanScope digital scanning system (Leica Biosystems). To count the number of rows of photoreceptor nuclei, we standardized the method by measuring a region two optic disc diameters in length from each side of the optic nerve head, and counting the rows in that region only. For immunohistochemistry, eyes were fixed in 4% paraformaldehyde overnight and then infiltrated with 30% sucrose at 4°C, before embedding in Polyfreeze medium (Polysciences Inc.). Immunolabelling was carried out with 6–10 μm thick frozen sections using a primary monoclonal antibody to RPE65 (1:1000, Abcam, ab513826) and detected with Alexa Fluor 488 goat anti-mouse IgG (Thermo Fisher Scientific) secondary antibody. Sections were incubated overnight at 4°C with primary antibody diluted in blocking buffer (2% normal goat serum, 0.1% Triton-X-100 in PBS). After extensive washes in PBS-Tween 20, localization of antibody labelling was detected after a 60 minute incubation at room temperature (RT) with the secondary antibody diluted in PBS containing 2% normal goat serum. Nuclei were counterstained with DAPI and then images acquired using confocal scanning laser microscopy.
Protein analysis methodologies
Eyes were opened at the limbus and retina/RPE tissue was removed and placed into 100 μl 1X phosphate buffered saline (PBS). Tissue samples were then homogenized, followed by two freeze-thaw cycles. Homogenates were centrifuged for 5 minutes at 5000 × g. The amount of RPE65 in the supernatant was compared to a standard curve using a mouse RPE65 ELISA kit (MyBioSource) according to the manufacturer’s instructions. For immunoprecipitation, protein from homogenate supernatants was immunoprecipitated with antibodies to REN1/hUPF1, phosphor-Upf1 and β-actin using SureBeads Protein G Magnetic Beads (Bio-Rad) according to the manufacturer’s instructions. A 10% w/v SDS-PAGE gel was used to separate proteins, which were then transferred to Immobilon-FL membrane (Millipore). Blocking of membranes was carried out in 5% non-fat dry milk powder in PBS/0.1% Tween-20 (PBST) for 1 hour at RT. Following three washes in PBST, the membranes were incubated with REN1/hUPF1 monoclonal (1:1000, Abcam, ab109363), phosphor-Upf1 (Ser1127, EMD Millipore, 07–1016), β-actin monoclonal (1:1000, Proteintech, 60008–1-Ig) at 4°C overnight. The membranes were washed three times 20 minutes each with PBST in the dark. Anti-rabbit IgG DyLight 800 (1:1000, Rockland, 611–145-002) and anti-mouse IgG DyLight 680 (1:2000, Rockland, 610–144-002) were used as the secondary antibodies. Visualization of protein was performed with an Odyssey LI-COR imaging system. The molecular weight markers used were from the BLUelf pre-stained protein ladder (GeneDireX).
TUNEL apoptosis assay
The TUNEL staining assay for mouse eyes was carried out on 12 μm thick frozen sections according to the manufacturer’s instructions (TUNEL Assay Kit, Abcam, ab66110). The slides were incubated with 3% Triton-100 in PBS for 15 minutes, and then incubated with wash buffer for two times 5 minutes. A DNA labeling solution containing TdT enzyme, Br-dUTP in TdT reaction buffer was added to each section and the slides were incubated at 37°C for 1 hour. Slides were washed two times 5 minutes in PBS, and then added with Anti-BrdU-Red antibody and incubated in the dark for 30 minutes. Nuclei were counterstained with DAPI. Slides were detected by fluorescence microscopy within 3 hours of staining. Cell counting was completed in Photoshop. An area of 10 mm × 20 mm was selected to count the number of total cells and the dead cells. TUNEL staining in wholemount zebrafish eyes was carried out as previously described36 using ApopTag Fluorescence In Situ Apoptosis Detection Kit (Millipore-Sigma, S7110) according to the manufacturer’s instructions.
Quantitative real-time RT-PCR
Total RNA was isolated from retinal/RPE tissues using TRI Reagent (Sigma). The RNA was purified with RNA Clean & Concentrator −25 (ZYMO research). From each sample, 200 ng of total RNA was reverse-transcribed to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) as per the manufacturer’s protocol. Gene expression was quantified using the TaqMan primer/labeled probe system and the 7500 Fast Real-Time PCR system (Applied Biosystems). All reactions were performed using the TaqMan Fast Advanced Master Mix (2X), FAM-labeled TaqMan pre-validated gene expression assays for rpe65, VIC-labelled TaqMan endogenous control Gapdh, and 10 ng of cDNA. All reactions were run in triplicates of three, with N = 3. Thermocycling parameters were as follows: 2 minutes at 50°C, 20 seconds at 95°C, 40 cycles of 1 second at 95°C, plus 20 seconds at 60°C. Real-time PCR data were analyzed by the comparative CT method.37
Visual background adaptation assay
At 5 dpf, treated or untreated zebrafish embryos were transferred to a Petri dish and placed inside a box for dark-adaptation lasting 30 minutes. The dish was then placed on a stereomicroscope stand and the embryos were exposed to a bright light. Images were taken as soon as the light was switched on, and again 15 minutes later to allow for full contraction of the skin melanophores as previously described.38
Statistical analysis
Analyses were performed with GraphPad Prism 5.0. For parameter comparisons between groups, an unpaired Student’s t test was used. P values of less than 0.05 were considered significant. Results are reported as mean ± SEM.
Multiple group comparison was performed by one-way ANOVA followed by Tukey post hoc tests. Differences were considered significant at P < 0.05. Results are reported as mean ± SEM.
Results
Therapeutic dose for Amlexanox and Ataluren in zebrafish
To determine the highest dose of drugs that could be tolerated, survival experiments were carried out. The survival rates of wildtype embryos dosed continuously from 10 hpf with increasing concentrations of Amlexanox or Ataluren from 0.3 to 60 μM, were measured at 6 dpf (Fig. 1A), and behavioral and gross morphological defects were noted in surviving larvae. Embryos treated with up to 1 μM Amlexanox or 3 μM Ataluren displayed normal morphology, feeding and motor behaviors with 100% survival rates at 6 dpf, and retinal sections had a normal histological appearance at 6 dpf similar to untreated embryos (Fig. 1B). Doses of ≥3 μM for Amlexanox or >10 μM Ataluren yielded toxic side effects including pericardial edema, bent tails, spinal curvature, shorter body length, and abnormal swimming behavior (Fig. 1C). Doses of ≥30 μM Amlexanox or ≥60 μM Ataluren were lethal in 100% of embryos. The LD50 for Amlexanox was 16 μM and for Ataluren was 20 μM estimated from the survival curves.
Figure 1.

Toxicity testing for Amlexanox in wildtype (wt) zebrafish embryos. (A) Survival curves for increasing concentrations of Amlexanox (solid line) and Ataluren (dotted line) after 6 days continuous exposure. (B) At 6 dpf, Amlexanox (1 μM) and Ataluren (3 μM) had no adverse effects on ocular histology or body shape. Size bar = 50 μm. (C) At 48 hpf, 5 μM Amlexanox resulted in a curved tail, whereas at 10 μM a short, bent tail, pericardial edema (arrow) and abnormal brain and eye development was present. Exposure to 30 μM Ataluren resulted in curved tail and pericardial edema (arrow).
Phenotype of cep290 zebrafish embryos
The cep290 protein plays an important role in centrosome function and for ciliogenesis in tissues such as the brain, retina, ear, and kidney.39 Morphological analysis of cep290 embryos revealed abnormal brain development at 24 hpf (Fig. 2A). The telencephalon was projected forwards and the top of the head appeared flattened when viewed laterally. In addition, the dorsal view showed that the forebrain and midbrain ventricles were smaller compared to wildtype embryos and the neuroepithelium was not smooth. From 48 hpf onwards hydrocephalus in the cep290 embryos was observed particularly in the hindbrain compared to wildtype (Fig. 2B and C). At 72 hpf neural cells in the midbrain were tightly packed in wildtype embryos, whereas in cep290 embryos there were loosely packed cells, and hydrocephalus appeared as a dorsal gap in cross-section (Fig. 2C). Also, the retina of the cep290 eyes did not show the typical inner (INL) and outer nuclear layers (ONL) like wildtype retina, instead it appeared immature as a single neuroblastic layer (Fig. 2C). At 6 dpf the midbrain was underdeveloped with hydrocephalus, and additionally pericardial edema. The eyes were also smaller and misshapen, and the swim bladder did not form in the cep290 mutants (Fig. 2B and C). The eye and brain phenotype observed in these cep290 embryos could be interpreted as suggesting that this model is a form of Joubert syndrome that is also caused by CEP290 mutations in humans.39 Such a designation would require further assessment. Recently, a different cep290fh297/fh297 allele was characterized exhibiting a slow progressive cone degeneration and scoliosis,40 demonstrating that a range of phenotypes are caused by different mutations in zebrafish cep290 as also seen in human CEP290 disease.
Figure 2.

cep290 morphology (Mt) compared to wildtype (Wt) embryos. (A) Lateral and dorsal views of wholemount morphology in embryos at 24 hpf. Telencephalon (arrows) is projected forward in cep290 embryos. F, forebrain ventricle; M, midbrain ventricle. (B) Wildtype brain development at 48 hpf, 72 hpf, and 6 dpf. Left panels, wholemount morphology; right panels, transverse H and E sections through the midbrain. sb, swim bladder; mb, midbrain; onl, outer nuclear layer of the retina; inl, inner nuclear layer. Size bar = 100 μm. (C) cep290 brain development at 48 hpf, 72 hpf, and 6 dpf. Left panels, wholemount morphology; right panels, transverse H and E sections through the midbrain. h, hydrocephalus; nbl, neuroblastic layer; *periocular edema. Size bar = 100 μm.
Effect of RNA therapy on ocular phenotype of cep290 mutants
The effect of nonsense suppression in the cep290 mutants was tested using Ataluren alone or in combination with Amlexanox to inhibit NMD. As the size of the eyes appeared smaller in cep290 mutants, we used TUNEL staining to assess whether cell death contributed to this phenotype. At 6 dpf a few TUNEL-positive cells in the retina were observed in wholemount sections of wildtype eyes representing normal histogenic cell death, whereas the whole retina was TUNEL-positive in the cep290 eyes indicating significant cell death (Fig. 3A). Treatment of cep290 embryos with 3 μM Ataluren from 10 hpf to 6 dpf resulted in greatly reduced TUNEL staining; TUNEL-positive cells were only seen in the outer nuclear layer of the retina. Treatment of cep290 embryos with 3 μM Ataluren plus 1 μM Amlexanox resulted in a just few TUNEL-positive cells in the retina, comparable to the wildtype control. We counted the number of TUNEL-positive cells in the retina in each treatment group (Ataluren: 68 ± 9 cells/section versus Ataluren plus Amlexanox: 8 ± 2 cells per section) indicating that the drug combination was significantly more effective in inhibiting cell death (P < 0.001, N = 4). To test whether the eyes could perceive light, we used the visual background adaptation (VBA) assay.38 This is a visually mediated endocrine camouflage response that in the light results in contraction of melanophores in the skin, whereas in the dark they are dispersed. When cep290 embryos were exposed to bright light, this did not result in contraction of melanophores compared to wildtype embryos (Fig. 3B). However, treatment with either Ataluren or combined Ataluren/Amlexanox resulted in melanophore contraction, demonstrating that light perception was normalized in the cep290 mutant. With these positive data, we next tested this combination nonsense suppression approach in two LCA mouse models carrying nonsense mutations.
Figure 3.

Ocular phenotype in cep290 mutants. (A) Wholemount TUNEL staining (green) comparing Wt eyes, untreated cep290 eyes (Mt), and eyes from Mt embryos treated with either 3 μM Ataluren (AT) or 3 μM AT plus 1 μM Amlexanox (AM). Sections were counterstained with DAPI. Size bar = 20 μm. (B) Visual background adaptation assay. In Wt embryos the melanophores (arrows) contract after 15 minutes of bright light, but not in cep290 embryos (Mt). Treatment with AT or AT/AM resulted in contracted melanophores in Mt embryos.
Effect of RNA therapy on retinal cell death in Rd3 and Rd12 mice
The RD3 protein is expressed in both rod and cone photoreceptors where it interacts with and stabilizes the expression of guanylate cyclase.41 The retina develops normally until P14 when the eyes open, but then undergoes rapid photoreceptor degeneration so that by 4 months of age (P120) very few photoreceptors remain.32 Therefore, we treated Rd3 mice from P4-P120 with subcutaneous injections of either Ataluren or Ataluren plus Amlexanox. At P120 there was a single row of photoreceptors in the ONL of untreated Rd3 mice (Fig. 4A). Treatment with Ataluren alone had no effect on the histological phenotype, neither did combining Ataluren with Amlexanox.
Figure 4.

Ataluren and Amlexanox treatment in Rd3 and Rd12 mice assessed at P120. (A) H&E sections of wildtype (Wt) retina compared to Rd3 retina when treated with either Ataluren (AT) or AT plus Amlexanox (AM). ONL, outer nuclear layer; INL, inner nuclear layer. Size bar = 50 μm. (B) H&E sections of Wt retina compared to Rd12 retina when treated with combined AT plus AM postnatally (P4-P120) and prenatally and continued postnatally (E12-P120). RPE, retinal pigment epithelium; OS, outer segments; IS, inner segments. Size bar = 50 μm. (C) Cryosections with TUNEL-staining (red) in Wt and Rd12 retina treated with combined AT plus AM either postnally from P4-P120 or from E12-P120. Sections were counterstained with DAPI. Size bar in all panels = 25 μm.
RPE65 is a retinoid isomerase localized to the retinal pigment epithelium (RPE) that is involved in the conversion of all-trans-retinal to 11-cis-retinal, before the chromophore is shuttled back to the photoreceptors.42 In the absence of RPE65 there is a slow degeneration of photoreceptors beginning at about 3 months of age (P90) and continuing past 2 years of age.31 In Rd12 retina at P120 during the most active phase of photoreceptor degeneration, the ONL consisted of seven rows of photoreceptor nuclei compared to 12 rows in wildtype retina (Fig. 4B). In addition the inner and outer segment (IS and OS) boundary of the photoreceptors had begun to degenerate. In Rd12 mutant mice treated with either Ataluren or Amlexanox alone no benefit was observed (data not shown). However, when both drugs were given in combination from P4-P120, the IS/OS distinction was clearly preserved (Fig. 4B). When the combined treatment was given to pregnant female mice at E12 and continued in offspring from P4-P120, no additional benefit was observed by giving the drugs earlier. In the treated mice there were eight to nine rows of nuclei compared to seven rows in untreated animals, which was consistent between mice. To reduce variability, rows of nuclei were counted in all histology sections from a standardized region measuring two optic disc diameters in length from each side of the optic nerve head. At P120 during active retinal degeneration TUNEL-positive cells were present in the INL, ONL, and retinal pigment epithelium (RPE) (Fig. 4C). In mice treated postnatally from P4-P120 or from E12-P120 with the combination of Ataluren/Amlexanox, the presence of TUNEL-positive cells was greatly reduced (Fig. 4C). A few TUNEL-positive cells were observed in the RPE cells in treated retina. The number of TUNEL-positive cells was quantified relative to the total number of nuclei in the ONL and INL (Fig. 5A). In untreated Rd12 retina approximately 30% of nuclei in the ONL and 41% of nuclei in the INL were TUNEL-positive. In comparison, after postnatal treatment with combined Ataluren/Amlexanox, TUNEL-positive cell counts in the ONL and INL were significantly reduced to ~ 6% and 5% of nuclei, respectively (N = 5, P < 0.01). No TUNEL-positive cells were detected in the ONL or INL when mice were treated prenatally/postnatally with the Ataluren/Amlexanox combination, which was comparable to wildtype retina.
Figure 5.

Efficacy of combined drug treatment in the Rd12 mouse eyes (Mt) compared to wildtype eyes (Wt). (A) Quantitation of the number of TUNEL-positive nuclei in the ONL or INL relative to the total number of nuclei in each layer. Drugs were given either postnatally (Post At+AM), or prenatally and then postnatally (Pre AT+AM). *P < 0.01; **P < 0.001; N = 5. (B) Relative expression of Rpe65 assessed by qRT-PCR. Data plotted as mean ± SEM. Significance comparing treatment groups with wildtype levels determined by one-way ANOVA with Tukey post hoc tests (*P = 0.94, N = 6). (C) Quantitation of Rpe65 protein in retinal/RPE extracts. Data plotted as mean ± SEM. Significance comparing treatment groups with wildtype levels determined by one-way ANOVA with Tukey post hoc tests (*P = 0.78, N = 6). (D) Immunohistochemical localization of Rpe65 protein (green) in RPE cells in different treatment groups. Sections were counterstained with DAPI. Size bar in all panels = 25 μm.
Effect of RNA therapy on rpe65 gene and protein expression
To determine whether the levels of rpe65 mRNA were increased with drug treatment as a result of nonsense suppression, total retinal/RPE mRNA was isolated and real-time RT-PCR analysis carried out. Very little mRNA was detectable in the rpe65 untreated group as would be expected; however, both postnatal and prenatal/postnatal treatment resulted in increased rpe65 mRNA levels (Fig. 5B). One-way ANOVA with Tukey post hoc comparisons between treatment groups showed no statistically significant difference between treatment groups and wildtype controls (*P = 0.94, N = 6). ELISA was used to quantify expression of RPE65 protein levels (Fig. 5C). In untreated retina very little RPE65 protein was detectable, whereas both treatment groups showed greatly increased RPE65 protein expression. One-way ANOVA with Tukey post hoc comparisons between treatment groups showed no statistically significant difference between treatment groups and wildtype controls (P = 0.78, N = 6). This increase in RPE65 protein expression was also analyzed by immunohistochemistry (Fig. 5D). RPE65 protein was localized to the RPE in wildtype mouse eyes and was undetectable in Rd12 eyes. However, in the presence of combined drugs, RPE65 protein was observed in the RPE.
Effect of Amlexanox on inhibition of NMD
NMD decay is associated with binding of UPF (up-frameshift factors) to the ribosome when it reaches a nonsense mutation in the mRNA sequence, which stimulates ribosome release from the mutant transcript.43 This process is regulated by phosphorylation of UPF1.44 UPF1 is ubiquitously expressed and we did not observe any significant differences in protein expression between wildtype, mutant, or treated groups (Fig. 6A). However, phosphorylated UPF1 (p-UPF1) was only observed in mutant Rd12 retinal/RPE extracts confirming activation of NMD in the disease process (Fig. 6B). In both postnatal and prenatal/postnatal treatment groups p-UPF1 was decreased compared to untreated controls, suggesting activation of NMD was down-regulated. Quantification by densitometry relative to β-actin revealed that postnatal treatment reduced p-UPF1 to 27% of the mutant level, whereas prenatal/postnatal treatment reduced p-UPF1 to 51% of mutant levels (Fig. 6D).
Figure 6.
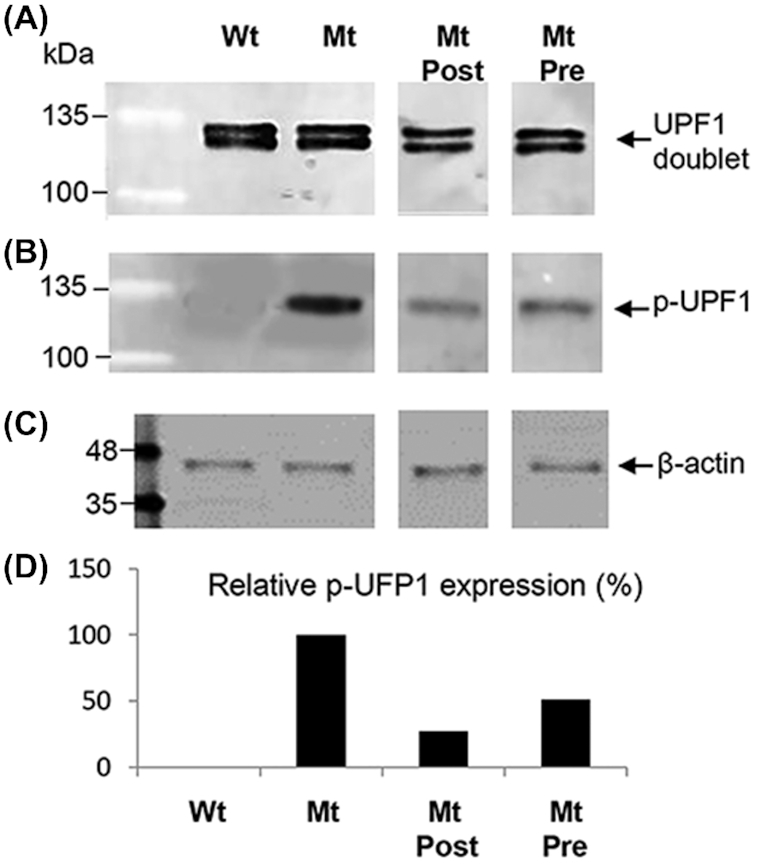
UPF1 phosphorylation in mouse Rd12 retina/RPE extracts analyzed by western blotting. (A) UPF1 protein expression in wildtype (Wt), untreated Rd12 mutants (Mt), Mt treated postnatally (Mt Post), and Mt treated prenatally plus postnatal treatment (Mt Pre). UPF1 is seen as a doublet on western blotting. (B) Phosphorylated UPF1 (p-UPF1) in Wt, Mt, Mt Post, and Mt Pre protein extracts. (C) β-actin loading control. Protein marker is BLUelf ladder. (D) Quantification of p-UPF1 expression between treatment groups relative to β-actin. Untreated Mt set as 100% p-UPF1expression level.
Behavioral testing in treated mice
To determine whether improved histological and biochemical changes related to drug treatment led to a functional benefit, we initially tested electrical activity in the retina by electroretinography (ERG) in both Rd12 and Rd3 models. In both mouse models, however, there were no detectable scotopic or photopic ERG responses above background noise at P14, the time of eye opening, or at subsequent times (data not shown). This has also been reported in other studies on the Rd12 mouse where scotopic and photopic ERG responses were barely above background noise.45,46 This is consistent with identifying these mice as animal models of LCA because ERG responses are absent from birth in human patients.10 Therefore, optokinetic tracking (OKT) analysis was carried out instead as a quantifiable measure of retinal function. This tests the ability of an animal’s eye to track a moving stimulus, which approximates visual acuity.47 At P120 Rd12 mutant mice have a reduced spatial frequency threshold of 0.34 cycles/degree (c/d) compared to wildtype mice (0.5 c/d), representing a 32% decrease in tracking ability (Fig. 7). It has been suggested that in Rd12 mutant mice enough 11-cis-retinal for this residual visual function could be obtained from photo-conversion of all-trans-retinal in the retina in rod photoreceptors instead of the RPE.48 In addition Müller cells contribute small amounts of 11-cis-retinal to the retina using an alternative isomerase.49 Both postnatal and prenatal/postnatal treatment groups had significantly improved spatial frequency thresholds of 0.59 and 0.58 c/d (P < 0.001), respectively, that were similar to wildtype mice, despite having three fewer rows of nuclei in the ONL. This is consistent with several studies indicating that behavioral tests of vision in some animal models of retinal degeneration do not show a linear correlation with loss of photoreceptors, although eventually the OKT response is lost when all the photoreceptors have degenerated.50,51
Figure 7.
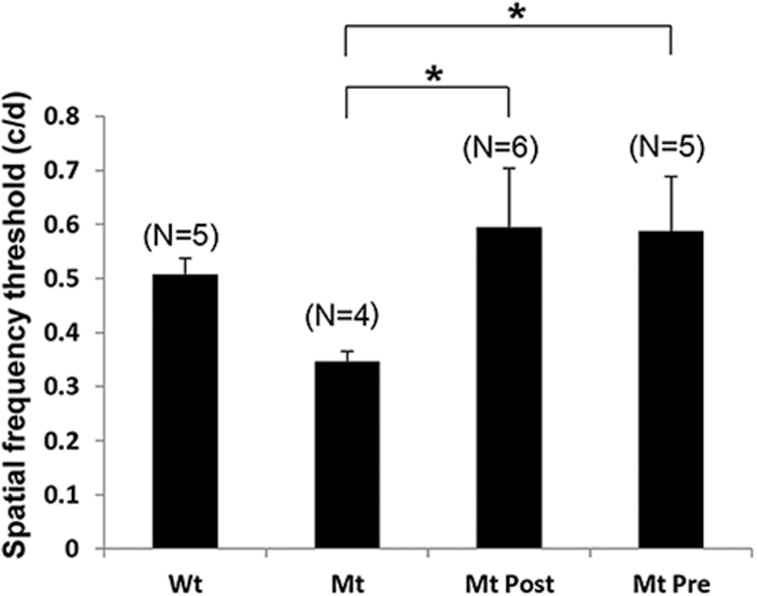
Behavioral optokinetic tracking (OKT) assessments in Rd12 mice (Mt) compared to wildtype mice (Wt). Spatial frequency threshold measured in cycles/degree (c/d) for Wt mice, untreated Mt mice, Mt treated postnatally (Mt Post), and Mt treated prenatally plus postnatal treatment (Mt Pre). Data plotted as mean ± SEM, number mice per group in parentheses. *P < 0.001.
Discussion
Nonsense suppression is a validated approach to reduce the effect of nonsense mutation disease genes resulting in full-length protein production, and rescue of a disease phenotype.52 This approach has been tested in many different animal model systems and patient-derived cell lines using a number of different nonsense suppression drugs.15,20–23,43 One nonsense suppression drug (Translarna®, PTC Therapeutics Inc.) has been granted conditional approval in Europe for the treatment of Duchenne muscular dystrophy,53,54 and it is currently being tested in clinical trials for aniridia (ClinicalTrials.gov: NCT02647359), Dravet syndrome (NCT02758626), and Duchenne muscular dystrophy in the USA (NCT03648827). As in-frame nonsense mutations account for 35% of mutations in LCA genes, we tested whether nonsense suppression alone or in combination with NMD inhibition could be a relevant therapeutic approach. We found positive benefit in the cep290 model with a reduction in retinal cell death and improved responses to light. In the Rd12 model, combined therapy slowed retinal degeneration, inhibited retinal cell death, resulted in new RPE65 protein and improved visual tracking responses. However, in the Rd3 model we found no benefit with individual or combined drug treatment. This could be caused, in part, by the rapid retinal degeneration that would be difficult to overcome with low levels of read-through, similar to that seen in the Rd1 mouse where nonsense suppression was unable to rescue the rapid retinal degeneration caused by a Pde6b nonsense mutation (S. Pittler, unpublished data). Thus, each genotype needs to be assessed independently to determine the validity of nonsense suppression therapy in the disease context, and whether adjunct NMD inhibitors would be advantageous.
The efficacy of response to nonsense therapy varies depending on multiple factors that could explain the different responses we observed in our models. First, the efficiency of translational read-through depends on the nonsense codon and context of the codon mutation relative to surrounding sequences: the ranking of read-through efficiency is UGA > UAG > UAA.55 In addition, if the nonsense codon is UGA, then the base immediately after it affects efficiency ranked as C > A > G > U.56,57 There is also evidence that the six nucleotides [CA(A/G)N(U/C/G)A] following the stop signal also confer efficient read-through.58 Finally, the 5′ context also modulates read-through: the presence of two adenine residues immediately prior to the nonsense codon showed translational read-through of 6%–16%.59 The mutations and surrounding sequence context in the animal models we tested are compared in Fig. 8. Despite the rd3 sequence having more of the highest ranking consensus residues (eight) than the rpe65 (six) and cep290 sequences (three), suggesting that read-through would be most efficient in the rd3 mRNA, this did not correlate with drug effect in vivo. Therefore, other factors could be at play such as penetrance of the drug to the tissue, the amino acid replacing the PTC, and availability of baseline mutant transcript (i.e. the efficiency of NMD).
Figure 8.
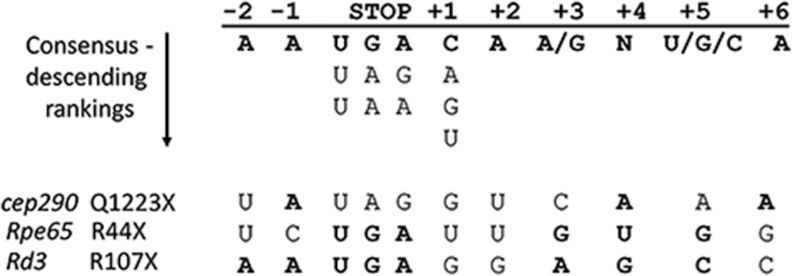
Consensus sequence for nonsense suppression. The nucleotide positions surrounding the stop codon are indicated at the top and underneath are the consensus nucleotides conferring the best read-through capability. The RNA sequences for cep290, Rpe65, and Rd3 are at the bottom. Nucleotides matching the consensus sequence are in bold.
Regarding tissue accessibility in the cep290 zebrafish model, Ataluren and Amlexanox had direct access to the developing eye through the ocular surface. This may explain why the drug effects in vivo were efficacious, even though the model had the middle ranking nonsense codon (UAG). We have previously used nonsense suppression drugs in three other zebrafish ocular disease models, and in each case the disease phenotype was rescued.20 As small molecule drugs are able to cross the zebrafish cornea and reach the retina, we predicted that a positive result would be achieved for Ataluren in the cep290 model. However, this is the first time that Amlexanox has been used in vivo to inhibit NMD and promote read-through in an inherited disease model. We found that there were fewer TUNEL-positive cells in the Ataluren/Amlexanox-treated retina compared to Ataluren treatment alone, suggesting that inhibition of NMD allowed more transcript availability for nonsense suppression. In comparison, both mouse models received the drugs systemically. Although an eye drop preparation of Ataluren has been previously used successfully in a mouse model of aniridia with a UGA codon,22 systemic injection into this mouse was also highly effective. As we have not validated an eye drop version of Amlexanox, we used both drugs systemically and predicted that a positive benefit would be observed. However, a positive benefit was only demonstrated when a combination of the two drugs was used in the Rd12 model. A topical version of the drug combination could be tested in future studies in both models to potentially improve efficacy.
The mode of inheritance, read-through efficiency and the amino acid replacing the stop codon could also be key factors in drug efficacy. In other animal models of disease such as the Mdx mouse model of Duchenne muscular dystrophy,60 the Cftr mouse model of cystic fibrosis,61 the Cln1 mouse model of infantile neuronal ceroid lipofuscinosis,62 and a mouse model of Usher syndrome,63 nonsense suppression therapy resulted in 5%–30% read-through efficiency with new protein production, which correlated with functional benefit. In a previous study, Ataluren was especially beneficial in the haploinsufficient Pax6+/− aniridia model, perhaps because additional functional protein on top of the 50% already present is sufficient to rescue the phenotype.22 However, the lack of any benefit in the recessive Rd3 model in this study may be because a minimum of 50% RD3 protein is required to see functional benefit, whereas a lower amount of protein is needed to replace RPE65 function. A further contributing factor to success of nonsense suppression is which amino acid is inserted into the protein instead of the PTC, as this would act as a missense mutation and may not fully recapitulate wildtype protein stability or function. A number of in vitro studies have demonstrated that Arg, Cys, or Trp residues are preferentially inserted at UGA codons, whereas Tyr and Gln were inserted at UAG and UAA codons.64–66 In the rd3 mouse sequence the UGA replaces an Arg residue. Thus, under nonsense suppression the stop codon could be replaced by another Arg residue producing wildtype protein. This would be predicted to occur only 4% of the time.19 The most frequently inserted residue at a UGA codon is Trp (86% of the time) compared to Cys (9% of the time). Thus, if the Arg107 residue was replaced by either Cys or Trp the consequence would likely be detrimental, because this conserved part of the protein binds to retinal guanylate cyclase and inhibits enzyme activity.67 The mutated residue in the Rd12 mouse is also an Arg so the same amino insertions could apply. However, position Arg44 is outside of the catalytic domain of the RPE65 retinoid isomerase68 and therefore insertion of Cys or Trp may have a less detrimental effect to protein function.
A final factor to consider is the inherent NMD activity and baseline transcript levels. The magnitude of NMD varies between individuals even if they carry the same nonsense mutation, as demonstrated by the different clinical phenotypes. For example, the same nonsense mutation causes typical Duchenne muscular dystrophy in one patient and mild Becker muscular dystrophy in another.69 Similarly, cystic fibrosis patients with the same nonsense mutation have highly variable mRNA levels, and the best responders to nonsense suppression drugs were those with the highest transcript levels.26 There are a number of known factors that influence NMD efficiency, including cellular stress, microRNAs, negative feedback regulation, and competition with the Staufen-mediated mRNA decay pathway.70,71 Thus, in any one individual the ‘net’ amount of NMD could account for the variable clinical phenotypes and individual drug responses in patients carrying the same nonsense mutations.
It has been suggested that there could be a clinically relevant ‘threshold’ of NMD that will determine how well a patient will respond to nonsense suppression.67 NMD inhibition drugs to enhance nonsense suppression have been tested in human cells in vitro27,72 and in a mouse model of Hurler syndrome in vivo.28 It has also been found that NMD-enhancement therapy is tissue-specific72 and that NMD might have detrimental effects, by degrading mRNA that encodes a partially functioning protein, so could make the condition worse if it was degraded.73 Thus, the efficacy of the NMD drug would need to be titrated against the native NMD efficiency rate to determine the appropriate dose. Prior to clinical trials, patients would need to be tested for levels of the specific mutant mRNA transcript, whether a truncated protein is being produced, and levels of UPF1 to quantitate the relative amount of transcript that is escaping degradation, as a prognostic indicator of therapeutic effect. Using this type of precision medicine approach in individual patients, it is more likely that promising research studies will be effectively translated into clinical practice.
Acknowledgements
This work was supported by Fighting Blindness Canada (grant No. 20R23299). We thank Olena Sivak for contributions to zebrafish experiments and Dr. Robert Molday for provision of the Rd3 mouse model.
Contributor Information
Xia Wang, Department of Ophthalmology and Visual Sciences, University of British Columbia, Vancouver BC V5Z 3N9, Canada.
Xianghong Shan, Department of Ophthalmology and Visual Sciences, University of British Columbia, Vancouver BC V5Z 3N9, Canada.
Kevin Gregory-Evans, Department of Ophthalmology and Visual Sciences, University of British Columbia, Vancouver BC V5Z 3N9, Canada.
Cheryl Y Gregory-Evans, Department of Ophthalmology and Visual Sciences, University of British Columbia, Vancouver BC V5Z 3N9, Canada.
Conflict of interest statement
None declared.
References
- 1. Wang X, Wang H, Sun V, et al.. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50:674–88. doi: 10.1136/jmedgenet-2013-101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guo Y, Prokudin I, Yu C, et al.. Advantage of whole exome sequencing over allele-specific and targeted segment sequencing in detection of novel TULP1 mutation in Leber congenital amaurosis. Ophthalmic Genet. 2015;36:333–8. doi: 10.3109/13816810.2014.886269. [DOI] [PubMed] [Google Scholar]
- 3. Acland GM, Aguirre GD, Ray J, et al.. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–5. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- 4. Bemelmans AP, Kostic C, Crippa MSV, et al.. Lentiviral gene transfer of RPE65 rescues survival and function of cones in a mouse model of Leber congenital amaurosis. PLoS Med. 2006;3:e347. doi: 10.1371/journal.pmed.0030347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pawlyk BS, Smith AJ, Buch PK, et al.. Gene replacement therapy rescues photoreceptor degeneration in a murine model of Leber congenital amaurosis lacking RPGRIP. Invest Ophthalmol Vis Sci. 2005;46:3039–45. doi: 10.1167/iovs.05-0371. [DOI] [PubMed] [Google Scholar]
- 6. Williams ML, Coleman JE, Haire SE, et al.. Lentiviral expression of retinal guanylate cyclase-1 (RetGC1) restores vision in an avian model of childhood blindness. PLoS Med. 2006;3: e201. doi: 10.1371/journal.pmed.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Molday LL, Djajadi H, Yan P, et al.. RD3 gene delivery restores guanylate cyclase localization and rescues photoreceptors in the Rd3 mouse model of Leber congenital amaurosis 12. Hum Mol Genet. 2013;22:3894–905. doi: 10.1093/hmg/ddt244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bainbridge JW, Smith AJ, Barker SS, et al.. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–9. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 9. Jacobson SG, Cideciyan AV, Ratnakaram R, et al.. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: Safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Le Meur G, Lebranchu P, Billaud F, et al.. Safety and long-term efficacy of AAV4 gene therapy in patients with RPE65 Leber congenital amaurosis. Mol Ther. 2018;26:256–68. doi: 10.1016/j.ymthe.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Russell S, Bennett J, Wellman JA, et al.. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–60. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miraldi UV, Coussa RG, Antaki F, et al.. Gene therapy for RPE65-related retinal disease. Ophthalmic Genet. 2018;39:671–7. doi: 10.1080/13816810.2018.1533027. [DOI] [PubMed] [Google Scholar]
- 13. Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–56. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- 14. Baxter-Roshek JL, Petrov AN, Dinman JD. Optimization of ribosome structure and function by rRNA base modification. PLoS One. 2007;2:e174. doi: 10.1371/journal.pone.0000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peltz SW, Morsy M, Welch EM, et al.. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med. 2013;64:407–25. doi: 10.1146/annurev-med-120611-144851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Auld DS, Lovell S, Thorne N, et al.. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc Natl Acad Sci U S A. 2010;107:4878–83. doi: 10.1073/pnas.0909141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Auld DS, Thorne N, Maguire WF, et al.. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci U S A. 2009;106:3585–90. doi: 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Green L, Goff SP. Translational readthrough-promoting drugs enhance pseudoknot-mediated suppression of the stop codon at the Moloney murine leukemia virus gag-pol junction. J Gen Virol. 2015;96:3411–21. doi: 10.1099/jgv.0.000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roy B, Friesen WJ, Tomizawa Y, et al.. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc Natl Acad Sci U S A. 2016;113:12508–13. doi: 10.1073/pnas.1605336113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moosajee M, Gregory-Evans K, Ellis CD, et al.. Translational bypass of nonsense mutations in zebrafish rep1, pax2.1 and lamb1 highlights a viable therapeutic option for untreatable genetic eye disease. Hum Mol Genet. 2008;17:3987–4000. doi: 10.1093/hmg/ddn302. [DOI] [PubMed] [Google Scholar]
- 21. Guerin K, Gregory-Evans CY, Hodges MD, et al.. Systemic aminoglycoside treatment in rodent models of retinitis pigmentosa. Exp Eye Res. 2008;87:197–207. doi: 10.1016/j.exer.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 22. Gregory-Evans CY, Wang X, Wasan KM, et al.. Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J Clin Invest. 2014;124:111–6. doi: 10.1172/JCI70462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bidou L, Hatin I, Perez N, et al.. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther. 2004;11:619–27. doi: 10.1038/sj.gt.3302211. [DOI] [PubMed] [Google Scholar]
- 24. Wagner KR, Hamed S, Hadley DW, et al.. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol. 2001;49:706–11. doi: 10.1002/ana.1023. [PubMed] [Google Scholar]
- 25. Kerem E, Konstan MW, De Boeck K, et al.. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539–47. doi: 10.1016/S2213-2600(14)70100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Linde L, Boelz S, Nissim-Rafinia M, et al.. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117:683–92. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gonzalez-Hilarion S, Beghyn T, Jia J, et al.. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012;7:58. doi: 10.1186/1750-1172-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keeling KM, Wang D, Dai Y, et al.. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS One. 2013;8:e60478. doi: 10.1371/journal.pone.0060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Banning A, Schiff M, Tikkanen R. Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder aspartylglucosaminuria. Biochim Biophys Acta Mol Basis Dis. 2018;1864:668–75. doi: 10.1016/j.bbadis.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 30. Zebrafish Mutation Project. Wellcome Sanger Institute UK; https://www.sanger.ac.uk/resources/zebrafish/zmp/.
- 31. Pang JJ, Chang B, Hawes NL, et al.. Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA). Mol Vis. 2005;11: 152–62. PMID: 15765048. [PubMed] [Google Scholar]
- 32. Friedman JS, Chang B, Kannabiran C, et al.. Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am J Hum Genet. 2006;79:1059–70. doi: 10.1086/510021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnson SL, Midson CN, Ballinger EW, et al.. Identification of RAPD primers that reveal extensive polymorphisms between laboratory strains of zebrafish. Genomics. 1994;19:152–6. doi: 10.1006/geno.1994.1026. [DOI] [PubMed] [Google Scholar]
- 34. Kimmel CB, Ballard WW, Kimmel SR, et al.. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 35. Nusslein-Volhard C, Zebrafish DR. A Practical Approach. UK: Oxford University Press, 2005, [Google Scholar]
- 36. Gregory-Evans CY, Moosajee M, Shan X, et al.. Gene-specific differential response to anti-apoptotic therapies in zebrafish models of ocular coloboma. Mol Vis. 2011;17:1473–84. PMID: 21677791. [PMC free article] [PubMed] [Google Scholar]
- 37. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 38. Fleisch VC, Neuhauss SC. Visual behavior in zebrafish. Zebrafish. 2006;3:191–201. doi: 10.1089/zeb.2006.3.191. [DOI] [PubMed] [Google Scholar]
- 39. Craige B, Tsao C-C, Diener DR, et al.. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–40. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lessieur EM, Song P, Nivar GC, et al.. Ciliary genes arl13b, ahi1 and cc2d2a differentially modify expression of visual acuity phenotypes but do not enhance retinal degeneration due to mutation of cep290 in zebrafish. PLoS One. 2019;14:e0213960. doi: 10.1371/journal.pone.0213960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Azadi S, Molday LL, Molday RS. RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc Natl Acad Sci U S A. 2010;107:21158–63. doi: 10.1073/pnas.1010460107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Redmond TM, Yu S, Lee E, et al.. Rpe65 is necessary for production of 11-cis-vitamin a in the retinal visual cycle. Nat Genet. 1998;20:344–51. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 43. Wang X, Gregory-Evans CY. Nonsense suppression therapies in ocular diseases. Cell Mol Life Sci. 2015;72:1931–8. doi: 10.1007/s00018-015-1843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ohnishi T, Yamashita A, Kashima I, et al.. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol Cell. 2003;12:1187–200. doi: 10.1016/s1097-2765(03)00443-x. [DOI] [PubMed] [Google Scholar]
- 45. Seeliger MW, Grimm C, Stahlberg F, et al.. New views on RPE65 deficiency: the rod system is the source of vision in a mouse model of Leber congenital amaurosis. Nat Genet. 2001;29:70–4. doi: 10.1038/ng712. [DOI] [PubMed] [Google Scholar]
- 46. Cachafeiro M, Bemelmans AP, Canola K, et al.. Remaining rod activity mediates visual behavior in adult Rpe65−/− mice. Invest Ophthalmol Vis Sci. 2010;51:6835–42. doi: 10.1167/iovs.09-3870. [DOI] [PubMed] [Google Scholar]
- 47. Wang X, Gregory-Evans K, Wasan KM, et al.. Efficacy of postnatal in vivo nonsense mutation suppression therapy in a Pax6 mouse model of congenital aniridia. Mol Ther Nucl Acids. 2017;7:417–28. doi: 10.1016/j.omtn.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Hooser JP, Liang Y, Maeda T, et al.. Recovery of visual functions in a mouse model of Leber congenital amaurosis. J Biol Chem. 2002;277:19173–82. doi: 10.1074/jbc.M112384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kaylor JJ, Yuan Q, Cook J, et al.. Identification of DES1 as a vitamin a isomerase in Muller glial cells of the retina. Nat Chem Biol. 2013;9:30–6. doi: 10.1038/nchembio.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McGill TJ, Douglas RM, Lund RD, et al.. Quantification of spatial vision in the Royal College of surgeons rat. Invest Ophthalmol Vis Sci. 2004;45:932–6. doi: 10.1167/iovs.03-0964. [DOI] [PubMed] [Google Scholar]
- 51. McGill TJ, Prusky GT, Douglas RM, et al.. Discordant anatomical, electrophysiological, and visual behavioral profiles of retinal degeneration in rat models of degenerative disease. Invest Ophthalmol Vis Sci. 2012;53:6232–44. doi: 10.1167/iovs.12-9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Asiful Islam M, Alam F, Kamal MA, et al.. Therapeutic suppression of nonsense mutation: an emerging target in multiple diseases and thrombotic disorders. Curr Pharm Des. 2017;23:1598–609. doi: 10.2174/1381612823666161122142950. [DOI] [PubMed] [Google Scholar]
- 53. Ryan NJ. Ataluren: First global approval. Drugs. 2014;74: 1709–14. doi: 10.1007/s40265-014-0287-4. [DOI] [PubMed] [Google Scholar]
- 54. Hoffman EP. Pharmacotherapy of Duchenne muscular dystrophy. Handb Exp Pharmacol. 2019. doi: 10.1007/164_2019_256 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 55. Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–55. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Beznoskova P, Gunisova S, Valasek LS. Rules of UGA-N decoding by near cognate tRNAs and analysis of readthrough on short uORFs in yeast. RNA. 2016;22:456–66. doi: 10.1261/rna.054452.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dabrowski M, Bukowy-Bieryllo Z, Zietkiewicz E. Translational readthrough potential of natural termination codons in eukaryotes-the impact of RNA sequence. RNA Biol. 2015; 12:950–8. doi: 10.1080/15476286.2015.1068497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Namy O, Hatin I, Rousset JP. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001;2:787–93. doi: 10.1093/embo-reports/kve176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tork S, Hatin I, Rousset J-P, et al.. The major 5′ determinant in stop codon read-through involves two adjacent adenines. Nucleic Acids Res. 2004;32:415–21. doi: 10.1093/nar/gkh201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Welch EM, Barton ER, Zhuo J, et al.. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 61. Du M, Jones JR, Lanier J, et al.. Aminoglycoside suppression of a premature stop mutation in a Cftr−/− mouse carrying a human CFTR-G542X transgene. J Mol Med. 2002;80:595–604. doi: 10.1007/s00109-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 62. Miller JN, Kovacs AD, Pearce DA. The novel Cln1(R151X) mouse model of infantile neuronal ceroid lipofuscinosis (INCL) for testing nonsense suppression therapy. Hum Mol Genet. 2015;24:185–96. doi: 10.1093/hmg/ddu428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goldmann T, Overlack N, Möller F, et al.. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol Med. 2012;4:1186–99. doi: 10.1002/emmm.201201438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roy B, Leszyk JD, Mangus DA, et al.. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc Natl Acad Sci U S A. 2015;112:3038–43. doi: 10.1073/pnas.1424127112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xue X, Mutyam V, Thakerar A, et al.. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum Mol Genet. 2017;26:3116–29. doi: 10.1093/hmg/ddx196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Feng YX, Copeland TD, Oroszlan S, et al.. Identification of amino acids inserted during suppression of UAA and UGA termination codons at the gag-pol junction of Moloney murine leukemia virus. Proc Natl Acad Sci U S A. 1990;87:8860–3. doi: 10.1073/pnas.87.22.8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Peshenko IV, Olshevskaya EV, Azadi S, et al.. Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry. 2011;50:9511–9. doi: 10.1021/bi201342b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jin M, Li S, Moghrabi WN, et al.. Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell. 2005;122:449–59. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nguyen LS, Wilkinson MF, Gecz J. Nonsense-mediated mRNA decay: Inter-individual variability and human disease. Neurosci Biobehav Rev. 2014;46:175–86. doi: 10.1016/j.neubiorev.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Maquat LE, Gong C. Gene expression networks: competing mRNA decay pathways in mammalian cells. Biochem Soc Trans. 2009;37:1287–92. doi: 10.1042/BST0371287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Durand S, Cougot N, Mahuteau-Betzer F, et al.. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J Cell Biol. 2007;178:1145–60. doi: 10.1083/jcb.200611086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Huang L, Lou CH, Chan W, et al.. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell. 2011;43:950–61. doi: 10.1016/j.molcel.2011.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Holbrook JA, Neu-Yilik G, Hentze MW, et al.. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004; 36: 801–8. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]