

Abstract
Differential mobility spectrometry (DMS) is highly useful for shotgun lipidomic analysis because it overcomes difficulties in measuring isobaric species within a complex lipid sample and allows for acyl tail characterization of phospholipid species. Despite these advantages, the resulting workflow presents technical challenges, including the need to tune the DMS before every batch to update compensative voltages settings (COVs) within the method. The Sciex Lipidyzer platform uses a Sciex 5500 QTRAP with a DMS (SelexION), an LC system configured for direction infusion experiments, an extensive set of standards designed for quantitative lipidomics and a software package (Lipidyzer Workflow Manager) that facilitates the workflow and rapidly analyzes the data. Although the Lipidyzer platform remains very useful for DMS-based shotgun lipidomics, the software is no longer updated for current versions of Analyst and Windows. Furthermore, the software is fixed to a single workflow and cannot take advantage of new lipidomics standards or analyze additional lipid species. To address this multitude of issues, we developed Shotgun Lipidomics Assistant (SLA), a Python based application that facilitates DMS-based lipidomics workflows. SLA provides the user with flexibility in adding and subtracting lipid and standard MRMs. It can report quantitative lipidomics results from raw data in minutes, comparable to the Lipidyzer software. We show that SLA facilitates an expanded lipidomics analysis that measures over 1450 lipid species across 17 (sub)classes. Lastly, we demonstrate that the SLA performs isotope correction; a feature that was absent from the original software.
Keywords: Shotgun Lipidomics, DMS, Lipidyzer, Flow Injection, Lipids
Graphical Abstract
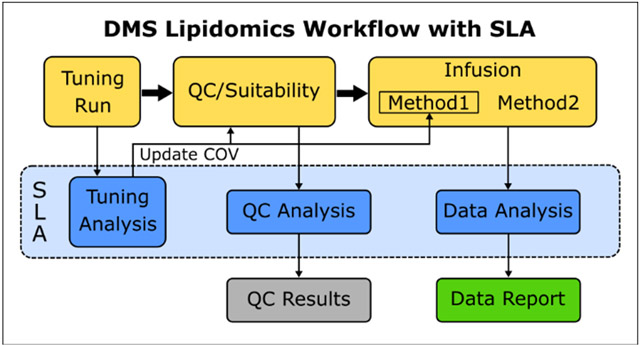
Summary: Schematic of the DMS Shotgun Lipidomics workflow and the data analysis processes within it that are automated by the SLA.
Introduction
Differential Mobility Spectrometry is a powerful tool in mass spectrometry to separate isobaric species based on a combination of mass, charge, and structure(1). Previous work established that DMS with chemical modifiers could be employed to reliably resolve isobaric phospholipid species that would otherwise require chromatographic separation(2). These studies established DMS settings and optimal chemical modifier (1-propanol) for analysis with the SelexION (DMS) device and Sciex QTRAP mass spectrometers (Sciex 5500 and 6500). Subsequently, Sciex created software that worked with their Analyst control software and licensed a set of lipid standards ideally suited for use with this SelexION (DMS)-based shotgun lipidomics workflow. This turnkey platform, marketed as the Lipidyzer(3), provided a rapid, analytically validated, targeted analysis capable of measuring up to 1100 lipid species commonly found in plasma or serum. The Lipidyzer software (Lipidyzer Workflow Manager or LWM) was established for plasma or serum samples and features rapid data analysis with no raw data processing required by the end user. In recent years, the Lipidyzer platform has been utilized in numerous published lipidomics projects analyzing plasma or serum(4, 5, 6), as well as other cells or tissue samples(7, 8, 9). The Lipidyzer platform is well-suited to experiments analyzing hundreds of samples and is compatible with large, multi-institution studies because it uses a standardized data acquisition method, lipid standards for quantification, and a robust computational analysis method.
There are, however, three principal problems with the Lipidyzer platform, all of which are centered around the software portion of the system. First, the Lipidyzer is restricted to a single assay that comprises a single set of acquisition methods, a single set and formulation of standards, and a single volume of plasma or serum. It cannot integrate new lipid MRMs or standards. As a consequence, the fixed Lipidyzer method does not include three phospholipid classes (Phosphatidylglycerol (PG), Phosphatidylinositol (PI) and Phosphatidylserine (PS)), despite reports that these lipids are resolvable with the DMS(2). Recently, new standards compatible with a DMS lipidomics workflow have been made commercially available, but unfortunately these cannot be used in the Lipidyzer’s fixed method. Furthermore, the Lipidyzer was developed for lipidomic analysis of plasma or serum, and as part of the analysis workflow, assumes it’s measuring a fixed volume. Thus, analysis of other types of samples requires additional backend calculations to re-normalize the reported concentrations.
Second, although the Lipidyzer data analysis method has been demonstrated to be quantitative with a high degree of accuracy, the exact method and calculations that the Lipidyzer employs has not been published. This is especially problematic because the LWM software was designed to only analyze data that it generated on the instrument it controls. As a consequence, Analyst data files from Lipidyzer experiments can be deposited in public databases, but that data cannot be re-analyzed by other researchers to reproduce the processed results. This inability to reanalyze the data is a serious problem for research transparency and is especially problematic for data sets that require further data processing beyond the initial analysis of the LWM.
Lastly, the LWM software is locked to Analyst 1.6.3 and has not been updated to function with the latest version of Analyst (version 1.7.1). Because Analyst 1.6.3 only runs on Windows 7, which is no longer supported, the LWM software cannot run on Windows 10. Sciex has discontinued the Lipidyzer platform, effectively sunsetting this platform despite the technical advantages and potential for this approach in performing shotgun lipidomics.
To address these limitations, we set out to develop an open-source alternative application to supervise and automate the DMS lipidomics workflow. The objective was to develop an application that provides the quantitative lipidomics and rapid data analysis capabilities of the current LWM, while including the versatility to oversee multiple methods that include additional lipid MRMs and standards. This application, termed Shotgun Lipidomics Assistant (SLA), is a Python software that works with the current version of Analyst and is being released as an open-source application. We show that the SLA can re-analyze data generated through the original LWM, providing this much needed capability. Further, we demonstrate that SLA can be used to perform markedly expanded DMS lipidomics experiments with the inclusion of additional lipid standards. SLA also provides an option to perform isotope contamination correction; an important feature to increase quantitative accuracy which is currently missing in the LWM. We expect that this open-source application will expand the capabilities of the current Lipidyzer community, and will become a useful application for the broader scientific community to facilitate DMS-based analysis of the lipidome.
Method
Hardware/System Setup
The Lipidyzer platform hardware has been described previously(10). The Turbo V source is outfitted with a 65μm ESI Electrode and the transfer line is plumbed with 50μm ID PEEKsil tubing from the Lipidyzer installation kit. For the SLA application method, a 100μl loop was installed.
SLA Overview
The Shotgun Lipidomics Assistant (SLA) oversees three processes in the DMS lipidomics workflow: 1) analyzing DMS tuning data to update methods utilizing the DMS, 2) analyzing QC/suitability test data to measure relative lipid standard response and 3) analyzing DMS lipidomics data files to yield processed quantitative results. This third process is subdivided into three parts 1) extracting data from Analyst data files, 2) merging extracted data with the sample map and performing normalization, and 3) creating plots and data sheets of the class total and triacylglycerol data. These five processes are carried out on separate tabs within the SLA GUI. We will describe each of these five processes and the computations performed in detail below.
While the LWM scripted the creation of Analyst sample batches and performed data analysis for these three processes, our workflow allows the user to setup the batches manually in Analyst and performs the analysis on the resulting data. This manual batch creation step requires a minimal amount of user time and gives the user flexibility in performing tuning, QC/suitability tests, and analyzing experimental data. The first step of analysis within each of the three primary processes is the conversion of the WIFF files to mzML format using Proteowizard’s MSconvertGUI application(11). The SLA reads the resulting mzML files utilizing the pyOpenMS package(12) and converts the data into readable data tables for further data processing.
SLA - Analyzing DMS tuning data
This step involves analyzing the data from a DMS tuning experiment; a process that must be performed prior to every DMS lipidomics experiment. The data is from a direct infusion experiment in which MRMs for defined lipid class standards are measured while the DMS ramps the compensation voltage (COV) at a fixed separation voltage. Within the LWM, a few such methods (which are separate for negative and positive mode tuning) are run consecutively until the DMS stabilizes. Within the SLA workflow, we ran each method three times and the third negative and positive mzML file was used for processing. The user also specifies a tuning dictionary Excel file that contains a list of the MRMs in the tuning program with their lipid class flag and a list of the MRMs within methods using the DMS with these lipid class flag. This allows the methods to be updated when the COV value is determined in the tuning run. The SLA selects this COV by identifying the COV value with the highest response or by finding the highest five, seven, or nine consecutive responses and selecting the COV of the center-most value. The SLA then lists this recommended COV value and provides the user with an interactive plot of the tuning data, such that the user can use the recommended COV value or choose one manually. After COV values have been selected, the SLA generates an Excel file containing the MRM lists with the newly specified COVs. This can be copy & pasted into said methods in Analyst.
SLA - Analyzing QC/Suitability Data
This step involves analyzing the data from a QC or suitability test method. This is a method in which the instrument infuses a known mixture of standards and measures MRMs for the standards utilized. The resulting data file contains 80 acquisitions of a small MRM list consisting of MRMs measuring representative lipids across various classes which utilize the DMS for resolution. The user can import the mzML file into SLA. The 80 acquisitions are averaged and reported in an Excel file containing the average values with the species names as noted in the Tune Dictionary file from the previous step.
SLA – Analyzing DMS Lipidomics Data Files
A DMS lipidomics experiment within this workflow consists of two direct infusion acquisitions, one which utilizes the DMS device (Method 1) and one that does not (Method 2). These runs each contain hundreds of MRMs that are acquired 20 times over the course of the infusion. The resulting WIFF file from each run is converted to mzML and analyzed in this step, in a similar manner to the LWM to yield quantitative results. When imported to a data frame, the first step is to identify MRMs with a significant number of zeros, as these will likely not be high quality measurements. The SLA removes any MRMs that have more than 2 zeros out of 20 acquisitions. If any standards are found to have more than 2 zeros (or have an average raw intensity below 100), they are removed along with all unknowns that use that standard. Any remaining zeros within the data set are dropped (if any MRM data sets have 1 or 2 zeros) and the remaining values are averaged. If the user has specified that isotope correction is to be performed, that calculation is performed at this step in the process on the averaged raw values (See below).
A species name dictionary is then used to apply lipid names to each MRM. As the mzML file does not contain this information, this dictionary must perfectly match the MRM order of the Analyst method. The SLA then draws upon a Standard dictionary Excel sheet that contains the assignment of standards to analytes, as well as the amount and concentration of each standard used. From this information, a quantification coefficient is calculated for each standard and used to calculate nanomolar concentrations of each analyte lipid using the following general calculation:
The calculation requires some dimensional analysis since all lipid standard documentation is in mg/mL (or μg/mL) concentrations and we have opted to report out all lipid concentrations in nmoles/mL(g)(Figure S1). This calculation assumes that the sample is a fixed volume of plasma (.025 mL), thus recapitulates the calculation of the LWM. This output is exported as Excel files with a merged Excel that is similar to the LWM output. Along with the species concentration, five additional tabs are created within this Excel sheet with various data presentations. These include various aggregations of species concentration (class concentration and class fatty acid concentration) and percentage-based analysis (species and fatty acids within class and class as percentage of total lipids). While this reporting works well for plasma lipidomics experiments (using the assumed fixed volume), these results will be re-normalized in the following steps for unique normalization (described below).
SLA – Isotope Correction
As lipids are large, carbon-rich molecules, there are potentially significant problems caused by isobaric overlap from heavy isotope-containing species. Different types of isotopic contamination have been well documented within lipidomics experiments. While cross-class contamination is minimized by the DMS for lipids measured with the SelexION device, isobaric overlap within a given class remains a significant problem that was not addressed in the commercial Lipidzyer software (LWM) and should be corrected. As such, we created a function within SLA that corrects for isobaric overlap by isotopomers of other species within the same class containing one additional double bond resulting in isobaric interference with a species containing one less double bond. The SLA both subtracts this signal from the contaminated species and corrects for the naturally occurring isotopes in all species. The basic methodology for lipid isotope correction has been described previously(13). The correction is complicated by the fact that the MRM fragmentation employed in the SLA DMS lipidomics technique that identifies acyl tails reduces the amount of contamination and makes the remaining isotope contamination more difficult to calculate. Within this MRM analysis, each acyl tail combination can experience isobaric overlap with corresponding species with one more double bond and exactly two heavy isotopes on the corresponding acyl tail and/or head group (none on the remainder of the molecule). To account for this overlap, one must calculate the probability of this specific isotope distribution.
As an example, the phosphatidylcholine species PC(16:0/18:1) is measured in negative mode with the MRM of 818.6/281.2 m/z; corresponding with the acetate adduct of the species and measuring the 18:1 fatty acid in Q3. This species experiences isobaric overlap with PC(16:0/18:2) with two heavy isotopes on the 18:2 acyl tail and PC(16:1/18:1) with two heavy isotopes on the 16:1 acyl tail or head group (Figure S2). The correction is calculated using the intensities of PC(16:0/18:2) and PC(16:1/18:1), which themselves should have been previously corrected for isotope overlap. For PC(16:0/18:2), the isobaric overlap is calculated as the intensity times the probability of there being two heavy isotopes on the 18:2 acyl tail and none on other remainder of the lipid (Figure S3). Likewise, the isobaric overlap from PC(16:1/18:1) is calculated using the probability of there being two heavy isotopes between the 16:1 and headgroup, but no heavy isotopes on the 18:1 acyl tail. In the final calculation, the isobaric overlaps are subtracted from PC(16:0/18:1) and then divided by the probability of no labels on PC(16:0/18:1). The intensity of the respective source species is corrected upward. The naturally occurring isotope distribution of each lipid species or fragmented parts used in the calculation are computed with R library Rdisop(14-17). We derived the chemical formula for each lipid species based on formula utilizing its nomenclature, weight and structure, which are verified on the LipidMap database. We are only considering n+1 as a source of contamination (this corrects for >99.9% of isobaric overlap, which is mainly caused by carbon-13 containing species).
To apply this correction to all lipid species, we built an Excel file with correction information; for each species, it flags the species that overlap with it. In general, monoacyl species have a single overlapping species and diacyl species have two possible overlapping species. For each species, the Excel dictionary contains the probabilities described above such that the isotope correction can be calculated using these values and the intensities of the species involved. The list is sorted by Q1 m/z value of the contaminated lipid species in ascending order. The SLA will go through the list in order and adjust the (averaged) intensities from the lowest m/z species (which by definition does not have other species overlapping with it) to the largest.
SLA – Merging Data/Normalization
This step has no equivalent in the LWM and provides the added functionality of assigning sample and group information to each sample as well as (re)normalization with a unique normalization factor. This allows for the analysis of cell and tissue samples in which different amounts of material are used in each sample. This step merges the two method data sets from the last step with the sample map. The species, fatty acid, and class quantification tabs are revised to normalization tabs in which the nanomolar amounts of each is normalized to the user specified map file. Lastly, new “average” sheets are created from the “Species Normalization” and “Class Normalization” tabs in which the average and standard deviation is calculated for each of the user specified sample groups within the map file.
SLA – Class Total/TAG Summary Plots
This step is also unique to the SLA. As hundreds of triacylglycerol species are measured in the average sample, we found it useful to create triacylglycerol profile plots in which all TAGs species are binned and summed by total acyl tail carbon number and double bound number. For each profile, the species of a given carbon or double bound number are binned together in separate data frames and reported in multiple sheets within an Excel file. The quantitative values for each bin are summed and divided by 3 because our MRM method (theoretically) quantifies each species 3 times when fragmenting on the various acyl tails. This summed data is plotted by calculating the average and standard deviation for the user-defined groups. Lastly, the Class Average data from the last step is plotted to provide a quick overview of large class changes within an experiment.
Bone Marrow-Derived Macrophage Lipidomics Collection
Macrophages were cultured and lipid extractions were carried out as previously described(10). In this experiment, Day 7 bone marrow-derived macrophages were either treated with TLR3 agonist poly (I:C), TLR4 agonist lipopolysaccharides (LPS), or vehicle and 8 replicates were collected after 48 hours for each condition (4 for each analysis).
Isotope Correction Experiment
The isotope correction test was performed on Control Plasma (Sciex 4386703) and QC Spike Standards (Sciex 5040408). All samples received 25μl of control plasma and then were divided into three groups that received 0, 10, or 25μl of QC Spike Standard. Lipid extractions were carried out as described above.
Results
SLA is capable of reanalyzing data files generated with Lipidyzer Workflow Manager
Two replicate experiments of 12 macrophage samples each were prepared. For this experiment, one quarter the amount of Lipidyzer standard recommended by the LWM was added to each sample, along with the Ultimate Splash Standards for PG, PI, and PS, as well as a single PA standard (Table S1). After Bligh & Dyer extraction, the resulting samples were analyzed on a single instrument utilizing the current LWM for the first experiment and in Analyst utilizing SLA for the second experiment. The LWM workflow was performed first, utilizing the tuning utility within the LWM to assign DMS compensation voltages (COV) and then performed the analysis. We allowed the LWM to process the data and then re-processed the raw data files through the SLA to compare results. Note that the LWM generated data was not properly normalized to cell number, and adjusted for the fact that we utilized one quarter the LWM-specified amount of standard. We adjusted the SLA standard dictionary to match the LWM settings for this test (no isotope correction was performed) to directly compare data analysis for the two programs.
Both methodologies reported out a comparable numbers of lipid species (LWM 687 vs SLA 688, with SLA reporting one additional lipid species, PE(22:0/18:1)), indicating a similar methodology in our detection cut off. A point-to-point comparison showed that only 4 out of 687 reported species were off by more than 0.1% (all of which were CE)(Figure 1). Taken together, these results indicate that the SLA and LWM are utilizing a comparable computational method and standard assignment.
Figure 1.
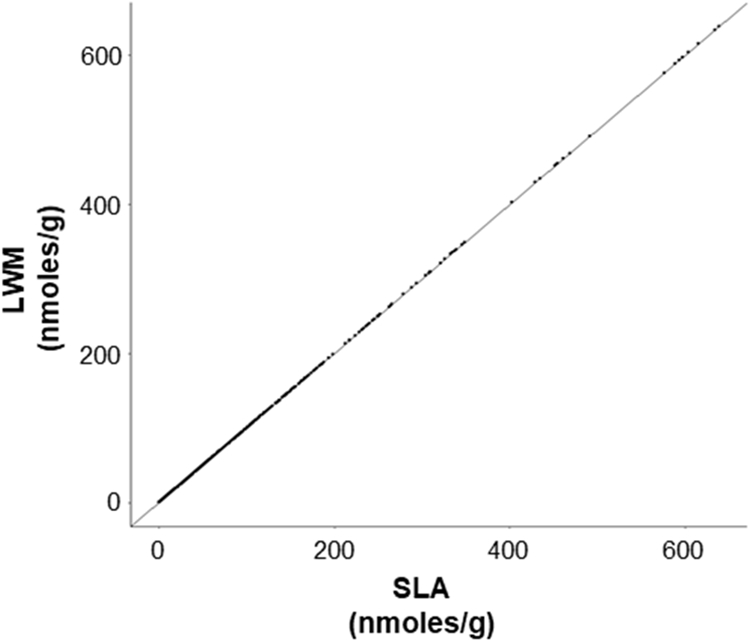
SLA recapitulates quantitative measurements made by the LWM. A scatter plot containing 6368 lipid measurements across 12 sample acquisitions supervised by the LWM and compared to re-analysis by the SLA. Pearson correlation coefficient was computed using all complete pairs of observations (r=1).
SLA Lipidomics Assay Yields Comparable Results to the LWM
Next, we set out to compare the data generated via the LWM to an expanded lipidomics assay supervised by the SLA. As part of the normal workflow for SLA, amounts of lipids detected are normalized to cell counts. Since, data generated by the LWM is not normalized we employed a post-hock normalization approach where the actual nanomoles in the sample was divided by the numbers of cell numbers in units of 107.
The SLA tuning was performed (to acquire COV values for PG, PI, and PS), but the COV values for all other classes common with the LWM run were retained from the previous LWM tune for consistency. These COV values were assigned to our expanded lipidomics method that incorporates the MRMs from the LWM method as well as comparably designed MRMs for PG, PI and PS classes, MRMs for bulk PA species, and a number of MRMs required for (optional) isotope correction. Following the SLA workflow, we analyzed the data and compared the data to the processed results generated by the LWM. As a first comparison, we examined the class totals values for the 13 lipid (sub)classes measured by the LWM, and the 17 subclasses measured by the SLA(Figure 2). The expanded lipidomics method was able to measure 901 lipid species in this experiment, compared to 687 lipid species within the original LWM method. Within the common 13 (sub)classes both methods detected a similar number of species (687 vs 693). Data generated by SLA and LWM for lipidomics of macrophages were comparable between the two experiments and consistent with previously published lipidomics data(9).
Figure 2.
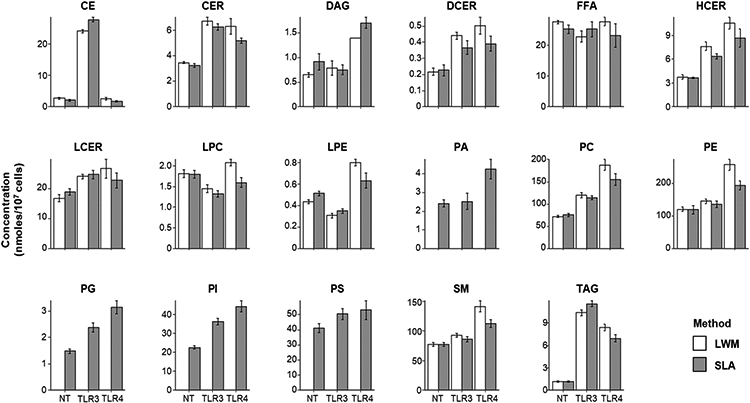
Independent experiments performed in LWM and SLA yield comparable quantitative results. Lipid subclass total concentration reported in nanomoles/107 cells. Totals calculated by summing the concentration of all species measured within subclass. Error bars represent standard deviation (n=4).
We then compared the averaged quantitative measurements within the three conditions common between the two experiments (1661 data points in total) and found strong correlation between the SLA and LWM method (r=.9947)(Figure S4). However, a careful comparison of measurements between treatments indicated experimental variation among the stimulated macrophages samples. This variation in the LPS stimulated samples can be clearly visualized in the comparison of the lipid subclass totals for the two experiments(Figure 2). Comparison of vehicle treated macrophages samples between the two experiments also showed a strong correlation (r=.9988)(Figure 3). The absolute quantification and order of abundance was highly comparable between the two experiments. The consistency in data generated by SLA indicates that the expanded lipidomics panel supervised by the SLA recapitulates the measurements made by the LWM and facilitates the measurement of hundreds of additional species with 4 additional lipid subclasses.
Figure 3.
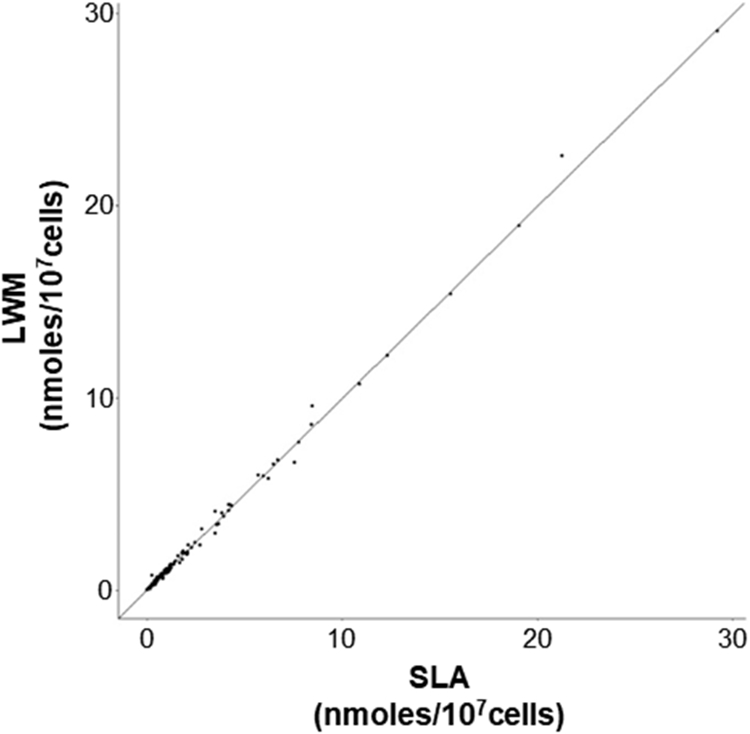
Independent experiments performed in LWM and SLA yield comparable quantitative results. Scatter plot containing 471 averaged lipid species quantitative values measured in the vehicle treated macrophages in both workflows, reported in nanomoles/107 cells. Pearson correlation coefficient was computed using all complete pairs (r=0.9988).
Development and testing of isotope correction function
Isobaric overlap remains a challenge in shotgun lipidomics experiments. The MRM data acquisition method utilized by the SLA and LWM creates added complications to the correction of that error. We created a function within the SLA that corrects for isobaric overlap. We acquire fragmentation data in the triple quad that requires the SLA correction method to take into account the probability of a specific distribution of heavy isotope atoms between the whole molecule and the fragment measured in Q3. This novelty prompted us to test whether the correction function was working as we would expect.
In most cases, the error introduced by isobaric overlap in the MRM experiments we have described is relatively small. As an example, we performed the SLA correction function on the previously describe macrophage experiment and calculated the percentage error for each species we measured. Of the 435 species detected in the first sample (NT_1), the vast majority (356 species) were corrected by 10% or less(Table S2). However, 31 species in this sample were corrected by more than 25%. It should be noted that the degree of isotope contamination varies from sample to sample. It is a function of both the formula/structure of the lipid species and their relative concentrations. The theoretical isobaric overlap between equimolar species ranges from 1-8%. The SLA correction key contains this theoretical percentage contamination and these percentages are largely a function of the number of carbons in a given lipid species (Table S3). More significant isotope contamination, like those seen in this experiment, occur when a high concentration species contaminates a low concentration species.
There are specific instances or types of experiment that can result in large systematic errors. One such case is where a biological sample was supplemented with a specific lipid or fatty acid which could overlap with lipids that are not actually changed by this perturbation. We experimentally created such a situation to test the SLA correction method by taking a control plasma sample and analyzing it alone and with a defined set of lipids spiked in that overlap with other detected species in the plasma sample. It should be noted that this experimental design is not intended to recapitulate the degree of correction required to actual biological samples, but to demonstrate the capability of the SLA to correct an extreme amount of specific isobaric overlap. To this end, we spiked in the Lipidyzer QC Spike Standards, which contain 52 lipid species, into a control plasma sample. Of those 52 lipids, 18 lipids had isobaric overlap with 19 lipid species that were not in the Spike Standard but were detected in the plasma sample(Table S4). One would expect increases in these 19 species in samples containing the Lipidyzer QC Spike that are corrected to baseline plasma concentration level following proper isotope correction. We analyzed control plasma with two different volumes of QC Spike (Figure 4). SLA analysis with and without the isotope correction was performed. Of these 19 species, our isotope correction option successfully corrected 16 measurements reasonably close to the baseline plasma concentration(Figure 4). Of the three remaining species, two were under-corrected (PC(16:0/16:0) and PE(18:0/22:4)) and one was over-corrected (PC(16:0/18:0)) (Figure 4). Thus, we conclude that the SLA isotope correction function works reasonably well for biological samples. Together, these studies show that utilization of SLA increases the capabilities of DMS-based lipidomics.
Figure 4.

SLA Isotope Correction function controls for isobaric overlap in complex biological samples. Quantitative measurement of 19 species experiencing isobaric overlap before and after isotope correction. Values are reported in control plasma in units of nanomoles/mL. The median and 25-75% interquartile range (IQR) are presented in box plots with whiskers representing 1.5x the IQR bounded by the highest and lowest samples (n=8).
Discussion
In this manuscript, we detail the development and application of the SLA, a software application designed to facilitate a DMS-based shotgun lipidomics workflow. Although other applications have been designed to facilitate LC-MS/MS and direct infusion MS/MS lipidomics(18), only the SLA and the original LWM are specifically designed for the unique challenges and advantages provided by DMS. We show that SLA adds unique functionalities not provided in the LWM including expanded lipid analysis, isotope correction and the ability to analyze data sets generated on other instruments. SLA also provides the user with the flexibility to modify acquisition methods and update standard assignments. We expect that SLA will enable a large number of labs using instruments with SelexION devices to perform Lipidyzer-style DMS lipidomics experiments utilizing the established methods. It should be noted that although SLA is designed for DMS shotgun lipidomics experiments, it can be used to analyze data from any direct infusion experiment with a comparable data acquisition method.
We have made several changes in this new workflow manager that result in improved capabilities for DMS-based lipidomics. We have improved the DMS tuning function with a display that accurately shows the quality of lipid class separation with the DMS, and the option to manually specify compensation voltages if desired. We have also made several changes to the data analysis portion of the application. We have added the ability to normalize quantification to unique normalization values for each sample (i.e., mg of tissue or cell number), making this technique easy-to-use with a variety of sample types. The ability to specify sample groups within an experiment that the SLA can use to calculate average and standard deviation values is also a convenient addition that minimizes manual data manipulation. Lastly the ability to perform isobaric overlap correction remedies a potentially serious weakness of the LWM. Together, these features and the open-source nature of the platform should give users a higher degree of confidence when performing DMS-based lipidomics experiments.
One of the greatest strengths of DMS lipidomics is its ability to provide acyl tail identification for diacyl species. However, this technique does not provide positional isomer information for diacyl species (i.e. Sn1/Sn2 position for each fatty acid). This point is confused by the fact that the LWM species reporting assumes positional isomer information according to LipidMaps nomenclature (i.e. PC(16:0/18:0) instead of PC 16:0_18:0, which doesn’t specify tail position). There are also a few other minor nomenclature disagreements between the Lipidyzer data formatting and LipidMaps nomenclature. The SLA is flexible in lipid nomenclature for reporting. Nomenclature is defined by the species dictionary and this document can be edited by the end user to match their desired format. For the purposes of this publication, we have kept the Lipidyzer nomenclature for ease of comparison with Lipidyzer data. However, the species dictionary for the expanded lipidomics panel is being released with LipidMaps nomenclature and we recommend that for all users moving forward.
The isotope correction function in the SLA also represents a significant step forward in DMS-based lipidomics. Isobaric overlap for this MRM technique necessitates a computationally complex correction strategy. Our test data shows that our correction strategy works well in most cases, however the correction isn’t perfect and there are limitations. The calculation assumes that all intensity measurements are linear, which may break down both on the high end and low end of lipid concentrations. Additionally, we have only designed our correction strategy to deal with isobaric overlap from varying double bond-containing species within the same subclass. While the DMS should prevent cross-class contamination, we cannot completely rule out the possibility of other isotope contamination effects among measurements that do not use the DMS.
Despite the advantages of SLA, there are several limitations and pitfalls to DMS lipidomics that should be kept in mind by users moving forward. The original Lipidyzer platform was described as having “accurate” quantification; that is to say that the experiment is able “to quantify lipid molecular species at the concentration level within an implicit quantitative bias.” (“Current State of Quantitation in Lipidomics Analysis”, Paul RS Baker, avantilipids.com). This requires a sufficient number of standards to represent the range of ionization/fragmentation efficiencies of each subclass being measured and that standards are properly matched to unknowns in order to minimize this bias in quantification. In the expanded lipidomics panel supervised by the SLA shown herein, we have adopted the Lipidyzer standards and standard assignments; thus, maintaining the quality of accurate quantification for those species. The new PG, PI, and PS standards utilized in the expanded run were designed in a similar manner to the Lipidyzer standards and we have assigned them using a strategy comparable to that used by the LWM in which standards are matched to unknowns based on total acyl tail carbon and double bond number, as well as the closest match for the acyl tail fragmented. While all due diligence has been given to establishing the quantification method for these lipid subclasses, we have not performed a direct comparison of our method with other established quantitative methods. We have observed that the ratios of phospholipid subclasses within BMDM samples were generally comparable to previous quantitative measurements(19, 20).
We are hopeful that others will create and validate additional acquisition/analysis methods to improve the application of SLA to DMS-based shotgun lipidomics. A logical next step would be to create a comparable method utilizing Avanti’s UltimateSplash ONE mix (containing 69 isotopically labeled standards). This is currently the only other standard mixture of comparable design and breath of coverage to the original Sciex Lipidyzer standards. We also expect that it will be possible to design methods for lipidomics experiments on bacteria, yeast and plant material that could be analyzed on the SLA, greatly expanding utility of this application. We are excited to see how the added features and capabilities of SLA will advance the field of DMS-based approaches and lipidomics in the future.
Supplementary Material
Acknowledgements/Grant Support
We are thankful to Baljit Ubhi and Steven Watkins for valuable conversation during software development. Special thanks to Shinobu Yoshida for connecting collaborators on this project.
This work has been partially been supported by NWO XOmics project 184.034.019 (MG), P30CA015704 (DR), P30AR074990 (LFB), P30DK035816 (DR), HL146358 (SJB)
Footnotes
Associated Content
SLA concentration calculation equation, schematic representation of isobaric overlap example, SLA isotope correction equation, correlation dot plot comparing SLA and LWM, table of lipid standards used in expanded lipidomics workflow, table of percentage isotope correction performed in an example macrophage sample, table of lipid species with isobaric overlap between QC spike and control plasma lipids (PDF).
Table containing the percentage of isobaric overlap between species at equimolar concentrations within the expanded lipidomics panel (Excel)
Data Availability Statement
Python application, Analyst methods, SLA dictionaries, user instructions, and sample data sets from this paper to be made available on GitHub [https://github.com/syjgino/SLA/tree/v1.2].
References
- (1).Kliman M, May JC, McLean JA: Lipid analysis and lipidomics by structurally selective ion mobility-mass spectrometry. Biochim Biophys Acta. 1811(11):935–45 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lintonen TP, Baker PR, Suoniemi M, Ubhi BK, Koistinen KM, Duchoslav E, Campbell JL, Ekroos K: Differential mobility spectrometry-driven shotgun lipidomics. Anal Chem. 86(19):9662–9 (2014) [DOI] [PubMed] [Google Scholar]
- (3).Ubhi BK: Direct Infusion-Tandem Mass Spectrometry (DI-MS/MS) Analysis of Complex Lipids in Human Plasma and Serum Using the Lipidyzer™ Platform. Methods Mol Biol. 1730:227–236 (2018) [DOI] [PubMed] [Google Scholar]
- (4).Contrepois K, Mahmoudi S, Ubhi BK, Papsdorf K, Hornburg D, Brunet A, Snyder M: Cross-Platform Comparison of Untargeted and Targeted Lipidomics Approaches on Aging Mouse Plasma. Sci Rep. 8(1):17747 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Middlekauff HR, William KJ, Su B, Haptonstall K, Araujo JA, Wu X, Kim J, Sallam T: Changes in lipid composition associated with electronic cigarette use. J Transl Med. 18(1):379 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Moghadam SD, Navarro SL, Shojaie A, Randolph TW, Bettcher LF, Le CB, Hullar MA, Kratz M, Neuhouser ML, Lampe PD, Raftery D, Lampe JW: Plasma lipidomic profiles after a low and high glycemic load dietary pattern in a randomized controlled crossover feeding study. Metabolomics. 16(12):121 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lepropre S, Kautbally S, Octave M, Ginion A, Onselaer MB, Steinberg GR, Kemp BE, Hego A, Wéra O, Brouns S, Swieringa F, Giera M, Darley-Usmar VM, Ambroise J, Guigas B, Heemskerk J, Bertrand L, Oury C, Beauloye C, Horman S: AMPK-ACC signaling modulates platelet phospholipids and potentiates thrombus formation. Blood. 132(11):1180–1192. (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhou QD, Chi X, Lee MS, Hsieh WY, Mkrtchyan JJ, Feng AC, He C, York AG, Bui VL, Kronenberger EB, Ferrari A, Xiao X, Daly AE, Tarling EJ, Damoiseaux R, Scumpia PO, Smale ST, Williams KJ, Tontonoz P, Bensinger SJ: Interferon-mediated reprogramming of membrane cholesterol to evade bacterial toxins. Nat Immunol. 21(7):746–755 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hsieh WY, Zhou QD, York AG, Williams KJ, Scumpia PO, Kronenberger EB, Hoi XP, Su B, Chi X, Bui VL, Khialeeva E, Kaplan A, Son YM, Divakaruni AS, Sun J, Smale ST, Flavell RA, Bensinger SJ: Toll-Like Receptors Induce Signal-Specific Reprogramming of the Macrophage Lipidome. Cell Metab. 32(1):128–143 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hsieh WY, Williams KJ, Su B, Bensinger SJ: Profiling of mouse macrophage lipidome using direct infusion shotgun mass spectrometry. STAR Protoc. 2(1):100235 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P: A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 30(10):918–20 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Röst HL, Sachsenberg T, Aiche S, Bielow C, Weisser H, Aicheler F, Andreotti S, Ehrlich HC, Gutenbrunner P, Kenar E, Liang X, Nahnsen S, Nilse L, Pfeuffer J, Rosenberger G, Rurik M, Schmitt U, Veit J, Walzer M, Wojnar D, Wolski WE, Schilling O, Choudhary JS, Malmström L, Aebersold R, Reinert K, Kohlbacher O: OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat Methods. 13(9):741–8 (2016) [DOI] [PubMed] [Google Scholar]
- (13).Ejsing CS, Duchoslav E, Sampaio J, Simons K, Bonner R, Thiele C, Ekroos K, Shevchenko A: Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal Chem. 78(17):6202–14 (2006) [DOI] [PubMed] [Google Scholar]
- (14).Böcker S, Letzel M, Lipták Z, Pervukhin A: SIRIUS: Decomposing isotope patterns for metabolite identification. Bioinformatics, 25(2), 218–224 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Böcker S, Lipták Z, Martin M, Pervukhin A, Sudek H: DECOMP—from interpreting Mass Spectrometry peaks to solving the Money Changing Problem. Bioinformatics, 24(4), 591–593 (2008) [DOI] [PubMed] [Google Scholar]
- (16).Böcker S, Letzel M, Lipták Z, Pervukhin A: Decomposing metabolomic isotope patterns. Proc. of Workshop on Algorithms in Bioinformatics. v4175 Lect. Notes Comput. Sci, 12–23 (2006) [Google Scholar]
- (17).Böcker S, Lipták Z: A fast and simple algorithm for the Money Changing Problem. Algorithmica, 48(4), 413–432 (2007) [Google Scholar]
- (18).Züllig T, Trötzmüller M, Köfeler HC: Lipidomics from sample preparation to data analysis: a primer. Anal Bioanal Chem. 412(10):2191–2209 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, Myers DS, Glass CK, Hardiman G, Reichart D, Merrill AH Jr, Sullards MC, Wang E, Murphy RC, Raetz CR, Garrett TA, Guan Z, Ryan AC, Russell DW, McDonald JG, Thompson BM, Shaw WA, Sud M, Zhao Y, Gupta S, Maurya MR, Fahy E, Subramaniam S: A mouse macrophage lipidome. J Biol Chem. 285(51):39976–85 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Andreyev AY, Fahy E, Guan Z, Kelly S, Li X, McDonald JG, Milne S, Myers D, Park H, Ryan A, Thompson BM, Wang E, Zhao Y, Brown HA, Merrill AH, Raetz CR, Russell DW, Subramaniam S, Dennis EA: Subcellular organelle lipidomics in TLR-4-activated macrophages. J Lipid Res. 51(9):2785–97 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Python application, Analyst methods, SLA dictionaries, user instructions, and sample data sets from this paper to be made available on GitHub [https://github.com/syjgino/SLA/tree/v1.2].