

Abstract
Mossy cells (MCs) of the dentate gyrus are key components of an excitatory associative circuit established by reciprocal connections with dentate granule cells (GCs). MCs are implicated in place field encoding, pattern separation, and novelty detection, as well as in brain disorders such as temporal lobe epilepsy and depression. Despite their functional relevance, little is known about the determinants that control MC activity. Here, we examined whether MCs express functional kainate receptors (KARs), a subtype of glutamate receptors involved in neuronal development, synaptic transmission, and epilepsy. Using mouse hippocampal slices, we found that bath application of submicromolar and micromolar concentrations of the KAR agonist kainic acid induced inward currents and robust MC firing. These effects were abolished in GluK2 KO mice, indicating the presence of functional GluK2-containing KARs in MCs. In contrast to CA3 pyramidal cells, which are structurally and functionally similar to MCs and express synaptic KARs at mossy fiber (MF) inputs (i.e., GC axons), we found no evidence for KAR-mediated transmission at MF–MC synapses, indicating that most KARs at MCs are extrasynaptic. Immunofluorescence and immunoelectron microscopy analyses confirmed the extrasynaptic localization of GluK2-containing KARs in MCs. Finally, blocking glutamate transporters, a manipulation that increases extracellular levels of endogenous glutamate, was sufficient to induce KAR-mediated inward currents in MCs, suggesting that MC-KARs can be activated by increases in ambient glutamate. Our findings provide the first direct evidence of functional extrasynaptic KARs at a critical excitatory neuron of the hippocampus.
SIGNIFICANCE STATEMENT Hilar mossy cells (MCs) are an understudied population of hippocampal neurons that form an excitatory loop with dentate granule cells. MCs have been implicated in pattern separation, spatial navigation, and epilepsy. Despite their importance in hippocampal function and disease, little is known about how MC activity is recruited. Here, we show for the first time that MCs express extrasynaptic kainate receptors (KARs), a subtype of glutamate receptors critically involved in neuronal function and epilepsy. While we found no evidence for synaptic KARs in MCs, KAR activation induced strong action potential firing of MCs, raising the possibility that extracellular KARs regulate MC excitability in vivo and may also promote dentate gyrus hyperexcitability and epileptogenesis.
Keywords: CA3, dentate gyrus, epilepsy, GluK2, kainic acid, mossy fiber
Introduction
Hilar mossy cells (MCs) in the hilus of the dentate gyrus (DG) are major excitatory neurons that widely project onto dentate granule cells (GCs) to control their activity (Buckmaster and Schwartzkroin, 1994; Hashimotodani et al., 2017; Scharfman, 2018; Botterill et al., 2019). Within the DG, MCs form an associative network with GCs, in which MCs receive extensive convergent excitatory inputs from GCs (Patton and McNaughton, 1995; Acsády et al., 1998; Buckmaster and Jongen-Rêlo, 1999; Ribak and Shapiro, 2007) and then send excitatory feedback projections to up to ∼30,000 GCs along the dorsoventral axis of the ipsilateral and contralateral hippocampus (Ribak et al., 1985; Frotscher et al., 1991; Buckmaster et al., 1996; Wenzel et al., 1997). Thus, the activity of a single MC can significantly impact the activity of numerous GCs, and ultimately, DG–CA3 information transfer. MCs contribute to hippocampal-dependent computations and behaviors such as pattern separation, spatial navigation, and other cognitive functions such as novelty detection (Duffy et al., 2013; Danielson et al., 2017; GoodSmith et al., 2017; Senzai and Buzsáki, 2017; Fredes et al., 2021). In addition, aberrant MC function has been linked to brain disorders such as temporal lobe epilepsy (TLE), depression, anxiety, and schizophrenia (Scharfman, 2016). Despite the important role that MCs play in brain function and disease, the mechanisms through which MC activity is recruited are still largely unexplored.
MCs share many structural and functional properties with CA3 pyramidal cells. Both CA3 pyramidal cells and MCs receive in their proximal dendrites a major excitatory input from GCs via the mossy fiber (MF) axons, which impinge on complex spines called thorny excrescence (TEs) via giant presynaptic boutons (Amaral and Dent, 1981; Acsády et al., 1998). Functionally, the MF–MC synapse expresses robust forms of short-term and long-term plasticity (Lysetskiy et al., 2005), similar to those reported at the MF–CA3 pyramidal cell synapse (Henze et al., 2002; Nicoll and Schmitz, 2005). In addition, glutamate release at both synapses is inhibited by activation of presynaptic group 2/3 metabotropic glutamate receptors (mGluR2/3; Kamiya et al., 1996; Lysetskiy et al., 2005). While excitatory transmission is mainly mediated by AMPA and NMDA ionotropic glutamate receptors (AMPARs and NMDARs), a slow component of MF–CA3 synaptic transmission is mediated by kainate receptors (KARs; Castillo et al., 1997). A similar, albeit modest KAR-mediated component has recently been reported at the MF–MC synapse (Hedrick et al., 2017). KARs are ionotropic glutamate receptors expressed in several brain areas, which have been implicated in neuronal development, neuronal excitability, and synaptic transmission and plasticity (Lerma and Marques, 2013). In vivo injection of the KAR agonist kainic acid (KA), a widely used animal model of TLE (Rusina et al., 2021), strongly activates not only CA3 pyramidal neurons (Westbrook and Lothman, 1983; Crépel and Mulle, 2015), but also MCs (Nasrallah et al., 2021), followed by extensive MCs loss (Buckmaster and Jongen-Rêlo, 1999; Sloviter et al., 2003), suggesting that MCs are particularly sensitive to the activation of KARs. Intriguingly, while KAR-mediated responses are much weaker at MF–MC synapses (Hedrick et al., 2017) than at MF–CA3 synapses (Castillo et al., 1997; Mulle et al., 1998), transcriptome profiling revealed that MCs and CA3 pyramidal cells display comparable levels of transcripts for the functional KAR subunit GluK2 (Cembrowski et al., 2016), raising the possibility that KARs may have additional roles at MCs.
In this study, we combined in vitro electrophysiology in acute rat and mouse hippocampal slices, of wild-type (WT) and Grik2 knock-out (KO) mice (here referred as GluK2 KO), with anatomic approaches such as immunofluorescence and immunoelectron microscopy, to determine the role and subcellular localization of KARs in MCs. We found that submicromolar and micromolar concentrations of KA induced robust inward currents and strong MC firing, and both effects were absent in GluK2 KO mice. Surprisingly, unlike in CA3 pyramidal neurons (Castillo et al., 1997), MF activation did not elicit any measurable KAR-mediated synaptic response in MCs. Consistent with these observations, immunofluorescence and immunoelectron microscopy revealed GluK2-containing KARs in the soma and dendrites of MCs, but were nearly absent from MC TEs. Last, blocking glutamate uptake by excitatory amino acid transporters (EAATs) elicited KAR-mediated inward currents in MCs. Altogether, our findings support the notion that MCs express functional extrasynaptic KARs whose activation by pharmacological agents (e.g., KA) and ambient glutamate may play an important role in engaging the GC–MC–GC recurrent circuit.
Materials and Methods
Animals.
Experiments were performed on postnatal Sprague Dawley rats [postnatal day 18 (P18) to P28] of both sexes, and on C57BL/6 mice of both sexes for electrophysiological recordings. Animals were group housed in a standard 12 h light/dark cycle. WT, GluK2 KO, and GAD67+/GFP mice (Tamamaki et al., 2003) were obtained from author Y.Y. Handling and use of animals adhered to a protocol approved by the Animal Care and Use Committee at the Albert Einstein College of Medicine, at Yale University, and at the Faculty of Medicine at Hokkaido University; and in accordance with guidelines provided by the National Institutes of Health.
Hippocampal slice preparation.
Animals were deeply anesthetized with isoflurane and then decapitated. The brain was then rapidly removed from the skull, and hippocampi were dissected. Hippocampi were included in agar supports and acute transverse hippocampal slices (400 µm thick for Sprague Dawley rats, 300 µm thick for C57BL/6 mice) were cut using a vibratome (model VT1200s, Leica Microsystems) in a sucrose-based cutting solution containing the following (in mm): 215 sucrose, 2.5 KCl, 26 NaHCO3, 1.6 NaH2PO4, 1 CaCl2, 4 MgCl2, 4 MgSO4, and 20 d-glucose. After 15 min in recovery postsectioning, the solution was replaced by extracellular artificial CSF (ACSF) recording solution containing the following (in mm): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, and 10 d-glucose. Slices were incubated for a minimum of 30 min in the ACSF before recording. Solutions were equilibrated with 95% O2 and 5% CO2, pH 7.4.
Electrophysiology.
All experiments were performed in a submersion-type recording chamber perfused at ∼2 ml/min, at 28 ± 1°C, except those seen in Figure 6 where the temperature was raised to 34 ± 1°C. Whole-cell patch-clamp recordings using an amplifier (model Multiclamp 700A, Molecular Devices) were performed from MCs and GCs in voltage-clamp configuration (Vhold, −60 mV) or current-clamp configuration (Vrest, approximately −65 mV) using borosilicate pipette electrodes (∼3–4 MΩ). Recordings were performed using a K+-based internal solution containing the following (in mm): 135 KMeSO4, 5 KCl, 1 CaCl2, 5 NaOH, 10 HEPES, 5 MgATP, 0.4 Na3GTP, 5 EGTA, 10 d-glucose, at pH 7.2 (280–290 mOsm). In some recordings, a Cs+-based internal solution was used containing the following (in mm): 131 Cs-gluconate, 8 NaCl, 1 CaCl2, 10 EGTA, 10 d-glucose, and 10 HEPES, at pH 7.2 (285–290 mOsm). Series resistance (∼7–25 MΩ) was monitored throughout all experiments with a −5 mV, 80 ms voltage step, and cells that exhibited a significant change (>20%) were excluded from analysis.
Figure 6.

MC-KARs can be activated by endogenous glutamate. A, Representative experiments showing the effect of TBOA (100 μm; left), DHK (100 μm; middle) mice, and DHK in GluK2 KO mice on MC holding current. Recording conditions were as in Figure 1. Activation of KARs was confirmed by application of the AMPAR/KAR antagonist NBQX (25 μm) at the end of the recording. NBQX was not applied in GluK2 KO as no inward current was detected. B, Quantification of the effect of TBOA (left) and DHK (right) on MC holding current. Dots represent the average current of 2 min of recording taken 2 min before DHK/TBOA application (baseline), 2 min before NBQX application (+DHK/TBOA), and 2 min at the end of NBQX application (+NBQX). Connected dots represent the same cell. C, Top, Representative traces showing the NBQX lack of effect on MC holding current in normal (2.5 mm; left) and high (8.5 mm; right) extracellular K+. Bottom, Representative traces showing the effect of 100 μm DHK in the presence of high extracellular K+ (left) and the same manipulation in the presence of 25 μm NBQX (right). D, Summary bar graph of the experiments shown in A–C. Each circle corresponds to one cell. Data are presented as the mean ± SEM. *p < 0.05; **p < 0.01; n.s., nonsignificant.
MCs were identified using previously established criteria (Larimer and Strowbridge, 2008). Specifically, firing properties and membrane time constant were measured by the injection of a step of depolarizing current while in current-clamp configuration. Cells were confirmed as MCs by exhibiting elevated spontaneous synaptic activity, little to no afterhyperpolarization, and non-burst firing patterns on depolarizing pulses (duration, 5 s; 60–120 pA). Additional confirmation was performed post hoc through morphologic analysis of biocytin-filled cells where MCs were identified by the presence of distinctive complex TEs in their proximal dendrites. To isolate KAR-mediated currents and EPSCs, the following cocktail of antagonists was used: LY303070 (15 μm) or GYKI 53655 (GYKI, –30 μm), d-APV (25 μm), picrotoxin (50 μm), and CGP35348 (3 μm) to block AMPA, NMDA, GABAA, and GABAB receptors, respectively. The cocktail was applied right after the target cell was identified as an MC. For the isolation of KAR-EPSC, AMPAR-EPSCs were first monitored in the presence of the above-specified cocktail, without LY303070, which was bath applied after a stable baseline was acquired.
To evoke MF synaptic responses in MCs and CA3 pyramidal cells, a bipolar stimulating theta-glass pipette was filled with ACSF and placed in the subgranular zone of the DG. Only EPSCs that showed a <2 ms 20–80% rise time and robust paired-pulse facilitation (EPSC2/EPSC1, >2) were considered MF-derived and included in the analysis. To increase the probability of detecting a KAR-mediated EPSC, two stimuli were delivered (interstimulus interval, 5 ms; duration, 100 µs; amplitude, ∼100 µA) using a stimulus isolator unit (Isoflex, AMPI). Typically, stimulation was adjusted to obtain comparable magnitude synaptic responses across experiments. To activate multiple MF inputs onto MCs, two patch-type micropipettes (monopolar stimulation) with a broken tip (diameter, ∼50 µm) were placed in the subgranular zone of the DG ∼200 µm apart from each other. To stimulate CA3 backprojections to MC, the stimulating micropipette was placed in the pyramidal cell body layer of the CA3c area, and CA3 pyramidal cells axons were activated in the presence of 1 μm DCG-IV to prevent the activation of MF inputs via antidromic stimulation of GCs.
Data analysis for electrophysiology experiments.
Electrophysiological data were acquired at 5 kHz filtered at 2.4 kHz and analyzed using custom-made software for IgorPro (WaveMetrics). The change in the holding current (ΔI holding) was calculated by subtracting the baseline holding current value (average, 50 s before KA application) from the average holding current post-drug application (average, 50 s before washout). To calculate firing rate in the current-clamp configuration, spikes were detected using a custom-written MATLAB script, which detected all voltage increases above a threshold value established by the experimenter (i.e., 0 mV). When 3 μm KA was used to depolarize MCs, the peak of the action potentials gradually decreased (likely because of inactivation of voltage-gated sodium channels) and became undistinguishable from spontaneous activity. This led to an underestimation of the effect of 3 μm KA on the firing of MCs. Firing rate was quantified as the number of spikes per second. The investigators were blind to genotype during both data acquisition and analysis.
Reagents.
Reagents were bath applied following dilution into ACSF from stock solutions stored at −20°C prepared in water or DMSO, depending on the manufacturer recommendation. The final DMSO concentration was <0.01% total volume. All chemicals and drugs used for the electrophysiology experiments were purchased from Sigma-Aldrich except NBQX, CGP-55 845, DCG-IV, and GYKI 53655, which were obtained from Tocris-Cookson, and tetrodotoxin (TTX), which was obtained from HelloBio. LY 303070 was obtained from ABX Advanced Biochemical Compounds.
Quantification and statistical analysis.
Statistical analysis was performed using OriginPro software (OriginLab). The normality of distributions was assessed using the Shapiro–Wilk test. In normal distributions, Student's unpaired and paired t tests were used to assess between-group and within-group differences, respectively. The nonparametric paired-sample Wilcoxon signed-rank test and Mann–Whitney U test were used in non-normal distributions. Statistical significance was set to p < 0.05 (***p < 0.001, **p < 0.01, and *p < 0.05). All values are reported as the mean ± SEM.
Fixation and sections.
We used glyoxal fixative containing 9% glyoxal and 8% acetic acid (v/v), pH 4.0 adjusted with 5N NaOH, which is modified from the original glyoxal fixative (Richter et al., 2018). Under deep pentobarbital anesthesia (100 mg/kg body weight, i.p.), mice were fixed by transcardial perfusion with ∼60 ml of glyoxal solution for 10 min at room temperature. Brains were postfixed in the same fixative for 3 h and cryoprotected with 30% sucrose in 0.1 m PB, pH 7.2, for 2 d. For immunofluorescence and immunoelectron microscopy, 50-μm-thick coronal sections through the ventral hippocampus (3.0–3.7 mm posterior to bregma) were prepared on a cryostat (model CM1900, Leica Microsystems) and subjected to free-floating incubation.
Antibodies.
We used the following antibodies: mouse anti-calretinin (catalog #MAB1568, Millipore; RRID:AB_94259); goat anti-EGFP (Takasaki et al., 2010; RRID:AB_2571574); rabbit anti-GluK2/3 (Straub et al., 2011); guinea pig anti-Neto1(Straub et al., 2011); and guinea pig anti-PSD-95 (Fukaya and Watanabe, 2000; RRID:AB_2571612).
Immunofluorescence.
All immunohistochemical procedures for immunofluorescence were performed at room temperature, and PBS containing 0.1% Triton X-100 was used as a dilution and washing buffer. Sections were incubated with 10% normal donkey serum for 20 min, a mixture of primary antibodies overnight (1 μg/ml each), and a mixture of Alexa Fluor 405-, 488-, 647-labeled, or Cy3-labeled species-specific secondary antibodies for 2 h at a dilution of 1:200 (Thermo Fisher Scientific/Jackson ImmunoResearch). To avoid cross talk between multiple fluorophores, images were taken with a confocal laser-scanning microscope equipped with 405, 473, 559, and 647 nm diode laser lines, and UPLSAPO 10× [numerical aperture (NA), 0.4], and PLAPON 60×OSC2 (NA, 1.4; oil immersion) objective lenses (model FV1200, Olympus). Image and pinhole size were 800 × 800 pixels and 1 airy unit, respectively. To compare genotypic and regional difference, images were taken at the same condition.
Pre-embedding immunoelectron microscopy.
All incubations were performed at room temperature, and PBS containing 0.1% Tween 20 (PBST) was used as a dilution and washing buffer. Sections were incubated in 10% normal goat serum (for 20 min; Nichirei) and with primary antibody against GluK2/3 (1 µg/ml) overnight and then with secondary antibodies linked to 1.4 nm gold particles (1:100; Nanogold, Nanoprobes) for 4 h. After extensive washing with PBST and HEPES buffer (200 mm sucrose, 50 mm HEPES, pH 8.0), immunogold was intensified with a silver enhancement kit (R-GENT SE-EM, Aurion) for 45–60 min. Sections were further treated with 1% osmium tetroxide for 15 min, stained with 2% uranyl acetate for 20 min, dehydrated with graded ethanol series, and embedded in Epon 812 (TAAB). After polymerization at 60°C for 48 h, ultrathin sections were prepared with an ultramicrotome (model Ultracut, Leica), mounted on copper-mesh grids, and stained with 2% uranyl acetate for 5 min and Reynold's lead citrate solution for 1 min. Photographs were taken with an electron microscope (model JEM1400, JEOL). Electron micrographs were randomly taken within ∼5 µm from the surface to avoid false-negative areas. To quantify metal particle labeling, 3 × 3 montage images (∼6 × 6 µm) were randomly taken at a magnification of 15,000×.
For quantitative analysis, plasma membrane-attached immunogold particles, being defined as those <35 nm apart from the cell membrane, were counted and analyzed using MetaMorph software (Molecular Devices). The mean number of membrane-attached gold particles per 1 μm of the plasma membrane was counted for each neuronal compartment (dendritic spine, dendritic shaft, and soma). Measurements were made from three WT and two GluK2 KO mice and pooled together, because there was no significant difference in the labeling density in the same genotype. In each neuronal compartment, labeling density was calculated for individual profile. Statistical analyses were performed using GraphPad Prism 9.0 (GraphPad Software). All data are given as the mean ± SEM. Data were analyzed using Kruskal–Wallis test followed by Dunn's post-test (*p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
Results
Kainate receptors mediate inward currents and action potential firing in hilar mossy cells
To test whether MCs expressed functional KARs, we first performed whole-cell patch-clamp recordings from MCs in acute rat hippocampal slices and bath applied the KAR agonist KA. MCs were identified based on the high frequency of spontaneous EPSCs, non-burst firing pattern on depolarization, and action potentials with almost no afterhyperpolarization (see Materials and Methods; Larimer and Strowbridge, 2008; Fig. 1A). To confirm the identity of the recorded cell, we loaded putative MCs with biocytin and stained with Alexa Fluor 594-conjugated streptavidin, and confirmed the presence of TEs, a hallmark of MCs (Fig. 1A; Scharfman and Schwartzkroin, 1988). We examined whether KA bath application induced KAR-mediated inward currents in MCs, as previously shown in KAR-expressing CA3 pyramidal cells (Castillo et al., 1997; Mulle et al., 1998) To isolate these currents, recordings were performed in the presence of LY303070 (15 μm), d-APV (25 μm), picrotoxin (50 μm) and CGP35348 (3 μm) to block AMPARs, NMDARs, and GABAA and GABAB receptors, respectively; and MCs were voltage clamped at −60 mV. Under these recording conditions, KA bath application (0.3 and 3 μm) induced large, concentration-dependent inward currents in MCs [Fig. 1B,C; ΔI holding MC: 0.3 μm KA: 165.3 ± 28.6 pA; n = 5a/5c; 3 μm KA: 736.2 ± 82.3 pA; n = 4a/4c (one of the cells died after 0.3 μm application; please note that in all figure citations, “a” and “c” represent the number of animals and cells, respectively]. In contrast, the same concentrations of KA induced modest currents in GCs (ΔI holding GC: 0.3 μm KA: 30.7 ± 1.4 pA; n = 3a/3c; 3 μm KA: 72.22 ± 6.3 pA; n = 3a/3c). By activating KARs in CA3 pyramidal cells, KA application could recruit CA3 pyramidal cells (Robinson and Deadwyler, 1981; Westbrook and Lothman, 1983; Castillo et al., 1997), which make synaptic contacts with MCs (Scharfman, 1994) and could indirectly activate KARs in MCs. To test this possibility, action potential generation was prevented by perfusing the voltage-gated sodium channel blocker tetrodotoxin (TTX) (0.5 μm) in the bath. In the presence of TTX, KA-induced currents were not significantly different from control conditions (Fig. 1D,E; ΔI holding MC + TTX: 0.3 μm KA: 117.3 ± pA; n = 5a/6c; 3 μm KA: 765.2 ± 79.1 pA; n = 5a/6c; 0.3 μm control vs TTX: n.s., p = 0.12045, two-sample t test; 3 μm control vs TTX: n.s., p = 0.81 287, two-sample t test), indicating that these currents do not result from indirect activation from CA3 pyramidal neurons. In addition, the competitive AMPAR/KAR antagonist NBQX (25 μm) abolished KA-mediated inward currents, strongly suggesting that these currents were mediated by KAR activation in MCs (Fig. 1D,E; ΔI holding MC: 0.3 μm KA control: 182.8 ± 25.7 pA; n = 3a/4c; 3 μm KA + 25 μm NBQX: 13.0 ± 3.3; n = 3a/4c; 0.3 μm KA vs 3 μm KA + NBQX: ***p = 0.000012, two-sample t test). Last, KA bath application induced currents in MCs of mouse hippocampal slices, which were abolished in GluK2 KO mice (Fig. 1F,G; ΔI holding MC WT: 0.1 μm KA, 58.3 ± 19.5 pA; 0.3 μm KA, 75.4 ± 13.9; 1 μm KA, 233.9 ± 15.7 pA; 3 μm KA, 494.3 ± 85.0 pA; n = 4a/7c; ΔI holding GluK2 KO: 0.1 μm KA, 19.8 ± 9.3 pA; 0.3 μm KA, 13.4 ± 4.4 pA; 1 μm KA, 24.7 ± 10.6 pA; 3 μm KA, 14.4 ± 5.0 pA; n = 4a/6c; WT vs GluK2 KO: F(1,3) = 146.98 864, **p = 0.00,121; two-way repeated-measures ANOVA), indicating that these currents are mediated by GluK2-containing KARs.
Figure 1.

Activation of KARs mediates inward currents in hilar mossy cells. A, Identification of hilar MCs in acute hippocampal slices. Left, Schematic of MC recordings in the hilus of the DG. Middle, Patched neurons were depolarized to analyze their firing properties. Right, Post hoc staining of an MC using Alexa Fluor 594-conjugated streptavidin. The yellow boxed area is magnified on the right-hand side. Patched cells were confirmed to be MCs if evoked spikes displayed no evident afterhyperpolarization and by the presence of TEs (white arrowheads). B, Representative experiment showing that bath application of 0.3 and 3 μm KA in the same cell induced concentration-dependent inward currents in MCs (top), but only a negligible inward current in dentate gyrus GCs (bottom). C, Summary plot of the amplitude of the inward currents (ΔI holding) induced by KA application. Here and in all figures, “a” and “c” represent number of animals and cells, respectively. D, Representative experiments showing that KA-induced inward current was not affected by the coapplication of TTX (0.5 μm; top), but it was abolished by the AMPAR/KAR antagonist NBQX (25 μm; bottom). A low concentration of KA (0.3 μm) was previously tested to verify the presence of a normal, fully reversible KA-induced current in the same cell. E, Summary plot. F, KA-induced currents in MCs were robust in WT mice (top) but were abolished in GluK2 KO mice (bottom). G, Concentration–response curve in WT and GluK2 KO mice. In this and all figures, summary data are presented as the mean ± SEM.
We hypothesized that KAR-mediated currents can produce enough depolarization to drive MC action potential firing. To test this possibility, we recorded MCs in current-clamp mode before and after KA application. Given that hippocampal interneurons impinging on MCs could express functional KARs (Frerking et al., 1998), these experiments were performed with intact excitatory and inhibitory components of synaptic transmission to assess the net effect of KAR activation on MC firing. Under these recording conditions, bath application of 0.3 and 3.0 μm KA induced strong MC firing (Fig. 2A; WT average firing rate: 0.3 μm KA: 0.333 ± 0.025 spikes/s; n = 2a/3c; 3 μm KA: 5.54 ± 0.28; n = 2a/3c), and this effect was abolished in GluK2 KO mice (Fig. 2B). These results indicate that the activation of GluK2-containing KARs with low concentrations of the agonist KA can powerfully drive MCs.
Figure 2.

GluK2-containing KARs mediate KA-induced robust action potential firing of MCs. A, Top, Effect of KA bath application (0.3 and 3 μm) on the membrane potential of MCs in current-clamp configuration. The application of 3 μm KA induced firing with faster onset and higher frequency than 0.3 μm KA. Firing in 3 μm KA eventually disappeared, likely because of excessive depolarization and action potential refractoriness. Bottom, Average firing rate histogram (expressed in spikes per second) for the experiments in the top panel. Black and gray traces represent the mean and SEM of the MC firing rate. B, Representative traces showing no effect of KA application on MCs firing in GluK2 KO mice.
KARs-mediated EPSCs are undetectable at MF–MC synapse
We next sought to determine the subcellular localization of KARs on MCs. Because of the strong structural and functional similarities between CA3 pyramidal cells and MCs, we tested whether MF activation elicits KAR-EPSCs in MCs, as previously shown at MF inputs onto CA3 pyramidal cells (Castillo et al., 1997). To this end, we evoked AMPAR-EPSCs by stimulating MF axons with two stimuli to boost glutamate release from MFs (interstimulus interval, 5 ms) in the presence of a cocktail of NMDARs, and GABAA and GABAB receptor antagonists (see Materials and Methods). We then attempted to isolate the KAR-mediated component of the MF-EPSC by applying the selective, noncompetitive AMPAR antagonist GYKI 53655 (30 μm). GYKI application abolished the AMPAR-EPSCs, but, surprisingly, it failed to uncover a KAR-mediated EPSC (Fig. 3A; EPSC amplitude post-GYKI application: 2.4 ± 0.5% of baseline; n = 4a/5 s). In addition, increasing the number of stimuli (from two to five), a manipulation expected to increase the likelihood of detecting KAR-EPSCs at the MF–CA3 synapse (Castillo et al., 1997), also did not generate any detectable current (data not shown; five pulses: 2.1 ± 0.4% of baseline; n = 4a/5c). To verify that the EPSCs were MF mediated, in a separate set of interleaved experiments, we applied the mGluR2/3 agonist DCG-IV (1 μm), which selectively blocks glutamate release at MF–MC synapses (Fig. 3B; Lysetskiy et al., 2005; Hedrick et al., 2017). DCG-IV reduced synaptic responses by ∼70%, indicating our stimulation mainly recruited MF inputs onto MCs (EPSC amplitude post-DCG-IV: 29.4 ± 7.1% of baseline; n = 3a/4c). As a positive control, and as previously reported (Castillo et al., 1997), MF stimulation elicited GYKI-resistant, NBQX-sensitive KAR-EPSCs in CA3 pyramidal neurons (Fig. 3C; EPSC amplitude post-GYKI application: 15.9 ± 6.6% of baseline; n = 2a/4c; KAR-EPSC amplitude post-NBQX application: 0.5 ± 0.3% of baseline; n = 2a/4c). These results indicate that in contrast to MF–CA3 synapses, KARs do not mediate synaptic transmission at MF–MC synapses. A previous report suggested the presence of a putative KAR-mediated component of the MF–MC EPSC (Hedrick et al., 2017). Since we did not observe a GYKI-resistant component with a single extracellular stimulator, we tried to recruit multiple MF inputs using two different stimulation sites (see Materials and Methods), which likely represents a more physiological approach, given the convergence of MF inputs onto MCs (Patton and McNaughton, 1995; Acsády et al., 1998; Buckmaster and Jongen-Rêlo, 1999; Ribak and Shapiro, 2007). While this multisite stimulation uncovered a GYKI-resistant component of the MF–MC EPSC, this component was equally observed in the GluK2 KO mice (Fig. 3D; GYKI-resistant vs AMPAR-mediated component: WT: 0.027 ± 0.004; n = 4a/5c; GluK2 KO: 0.018 ± 0.005; n = 5a/6c; n.s. p = 0.22, two-sample t test), suggesting that it was not mediated by GluK2-containing KARs. Last, we tested whether activation of the CA3 backprojection, the other main known input to MCs (Scharfman, 2007), could elicit a GYKI-resistant, KAR-mediated EPSC, but GYKI abolished CA3–MC synaptic transmission (Fig. 3E; AMPAR, −394.1 ± 107.2 pA; GYKI, −4.7 ± 1.6 pA; n = 3a/4c), indicating that KARs do not contribute to transmission at this synapse. Given the robust activation of MCs by low concentrations of KA (Figs. 1, 2), our findings thus far suggest KARs in MCs are extrasynaptic.
Figure 3.
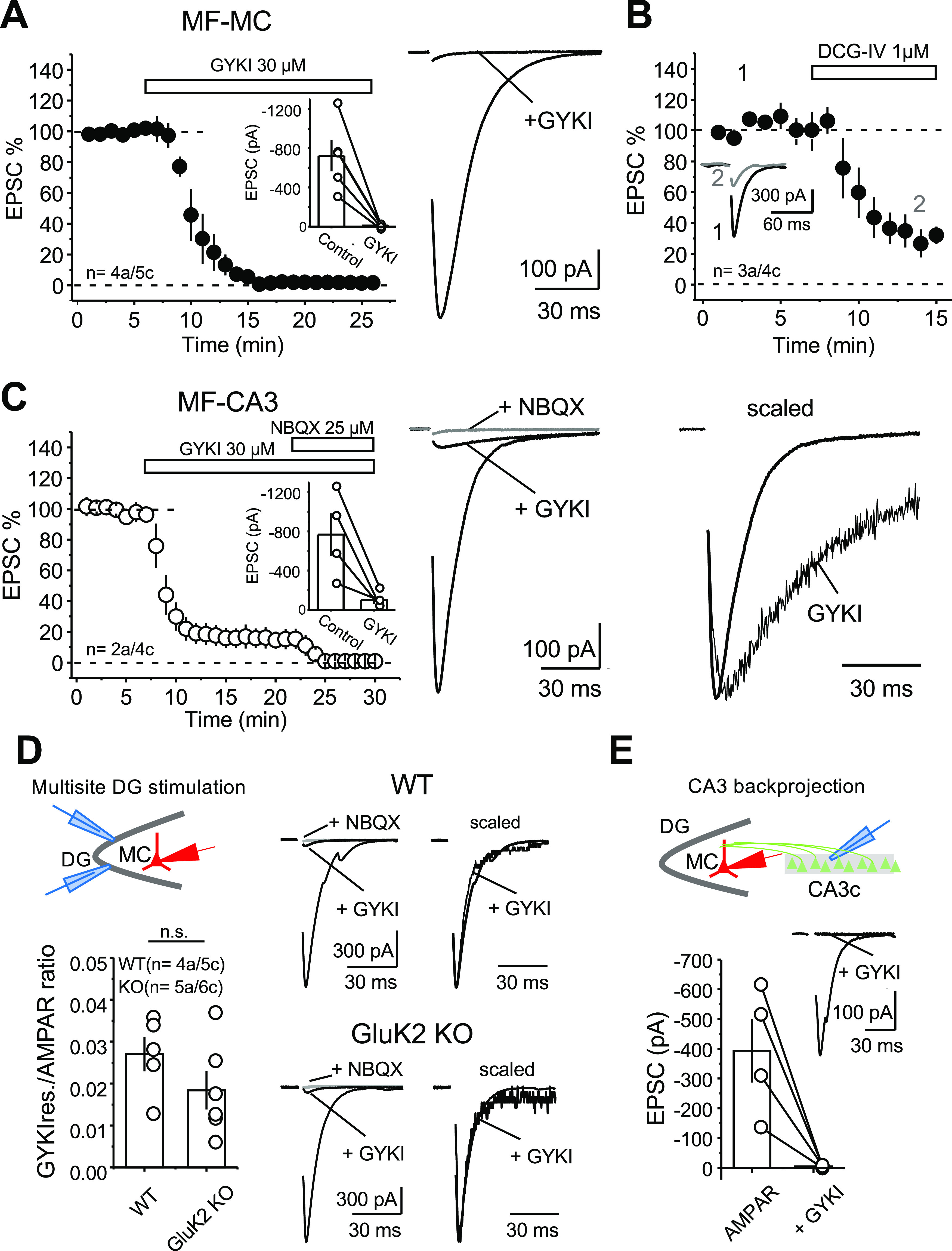
KAR-mediated transmission at MF–CA3 but not MF–MC synapses. A, Left, Bath application of the selective AMPAR antagonist GYKI 53655 (30 μm) abolished synaptic transmission at MF–MC synapses. Right, Representative average traces (30 consecutive responses). Inset, Raw values of the MF–MC AMPAR-EPSC: control, −723.1 ± 160.0 pA; GYKI, −9.9 ± 8.8 pA. B, Bath application of the mGluR2/3 agonist DCG-IV (1 μm) significantly reduced MF–MC EPSCs (∼70%). C, Left, GYKI 53655 application (30 μm) blocked AMPAR-mediated transmission at the MF–CA3 synapse, thus revealing a KAR-mediated component that was abolished by 25 μm NBQX. Inset, Raw values of the MF–MC AMPAR-EPSC: control, −767.2 ± 217.2 pA; GYKI, −99.5 ± 42.5 pA. Middle, Representative average traces (30 consecutive responses) before and after GYKI application, and after subsequent application of NBQX. Right, Normalized AMPAR and KAR components highlighting the slow kinetics of the KAR-EPSC. In all traces included in this figure, stimulus artifacts were deleted for clarity. Data are presented as the mean ± SEM. D, Left Top, Diagram illustrating the multisite stimulation of MF inputs onto MCs. Left bottom, Summary plot of the MF–MC GYKI-resistant (GYKI-res.) and AMPAR-mediated components represented as GYKI-res. versus AMPAR ratio. Right, Representative average traces of the AMPAR EPSC before and after GYKI application, and after subsequent application of NBQX in WT mice (top) and GluK2 KO mice (bottom). Scaled traces of AMPAR and GYKI-res. currents are shown on the right side. E, Top, Diagram illustrating the stimulation arrangement of the CA3 backprojection to MCs. Bottom, Summary plot of the CA3-MC AMPAR-EPSC before and after GYKI application. Inset, Representative average traces of the AMPAR-EPSC before and after GYKI application.
Distinct subcellular localization of KARs in CA3 pyramidal cells and mossy cells
To determine the anatomic localization of KARs in MCs, we applied immunostaining to tissue sections fixed with a glyoxal-based fixative (see Materials and Methods), which is effective for detection of both nonsynaptic and synaptic molecules. First, we confirmed the specificity of the antibody against GluK2/3 by blank labeling in GluK2 KO mouse hippocampi (Fig. 4A,B). In WT mice, the antibody yielded a contrasting pattern of labeling across hippocampal subregions: intense and coarse punctate labeling in the CA3 stratum lucidum, and moderate and diffuse labeling in the hilus (Fig. 4A). Further quadplex immunofluorescence using GAD67+/GFP mice (Tamamaki et al., 2003) allowed us to distinguish excitatory MCs from inhibitory calretinin-positive interneurons, and to examine whether MCs express GluK2/3 (Fig. 4C,D) together with or without the excitatory postsynaptic marker PSD-95 (Fig. 4C), or the KAR auxiliary subunit Neto1 (Fig. 4D). In CA3 stratum lucidum, GluK2/3-positive puncta were intense and aggregated into large clusters and were almost perfectly overlapped with PSD-95 (Fig. 4E,F), suggesting exclusive localization in MF–CA3 pyramidal cell synapse. Compared with CA3, GluK2/3 labeling was smaller and less frequent in the hilus of the DG (Fig. 4G,H). MC soma and dendrites, which can be unequivocally identified as calretinin-positive and GFP-negative structures (Fig. 4G), were associated with GluK2/3 puncta that did not colocalize with PSD-95 (Fig. 4H). Similarly, while Neto1 staining was overlapped with GluK2/3-positive puncta in stratum lucidum (Fig. 4I,J), it was not found around MC soma and dendrites (Fig. 4K,L). Together, these findings suggest that GluK2/3-containing KARs in MCs are expressed at extrasynaptic sites.
Figure 4.
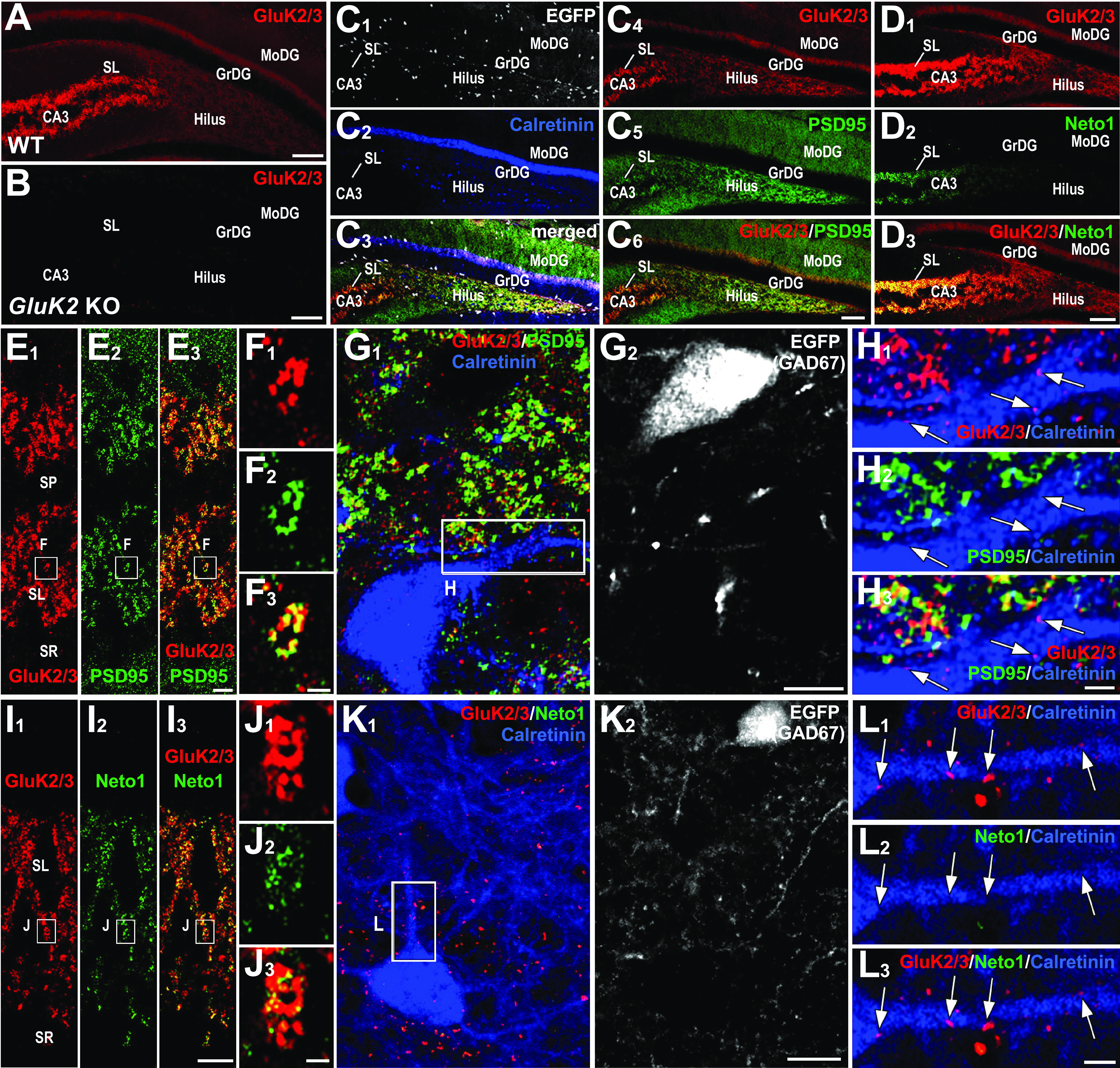
Contrasting localization of GluK2/3 and its molecular partners in CA3 stratum lucidum and DG hilus. A, B, In WT mice, GluK2/3 labeling is intense in CA3 stratum lucidum, while it is moderate and diffuse in the dentate gyrus (A); note the lack of GluK2/3 staining in GluK2 KO mice, indicating the specificity of the GluK2/3 antibody and exclusive expression of GluK2 in these hippocampal regions (B). C1–C6, Quadplex immunofluorescence for GFP (C1, C3, white), calretinin (C2, C3, blue), GluK2/3 (C3, C4, C6, red), and PSD-95 (C5, C6, green) in GAD67+/GFP mice. D1, D2, Double immunofluorescence for GluK2/3 (D1, red) and Neto1 (D2, green). Note that intense signal for Neto1 is almost limited to CA3 stratum lucidum. E1–H3, Distinct GluK2/3 and PSD-95 localization between CA3 pyramidal cells and hilar MCs. E1–F3, GluK2/3 and PSD-95 labeling are intense in the CA3 stratum lucidum (E1–E3), and a high-magnification image confirms their extensive overlap (F1–F3). G1–H3, An MC, which is identified as a calretinin-positive (G1, blue) and GFP-negative (G2, white) cell in GAD67+/GFP mice, shows weak labeling for GluK2/3 on its dendrites (red, arrows in H1–H3). Note that such GluK2/3 puncta are neither overlapped nor associated with PSD-95 signal (H2, green). I1–L3, Distinct GluK2/3 and Neto1 localization between CA3 pyramidal cells and hilar MCs. I1–I3, Intense GluK2/3 and Neto1 labeling in the CA3 stratum lucidum (l). J1–J3, A high-magnification image shows that Neto1 labeling is observed only on GluK2/3-positive puncta. K1–L3, An MC, which is calretinin-positive(K1, blue) GFP-negative (L2, white) cell inGAD67+/GFP mice, shows weak labeling for GluK2/3 on its dendrites (K1, L2, L3, red arrows) but lacks Neto1 labeling (K1, L2, L3, green). GrDG, GC layer of the DG; MoDG, molecular layer of the DG; SL, stratum lucidum; SP, stratum pyramidale; SR, stratum radiatum. Scale bars: A, B, C1–C6, D1–D3, 100 µm; E1–G2, I1–K2, 10 µm; H1–H3, L1–L3, 2 µm.
For a more accurate assessment of the subcellular localization of KARs in MCs, we performed pre-embedding immunoelectron microscopy for GluK2/3. In WT mice, metal particles for GluK2/3 were observed on the postsynaptic membrane of TEs (Fig. 5A,G, blue) of CA3 pyramidal cells facing large MF boutons (12.10 ± 0.95 particles/mm). In GluK2 KO mice, immunolabeling was essentially absent on the postsynaptic membrane of CA3 spines (Fig. 5B,G, blue), confirming the specificity of the immunolabeling (0.07 ± 0.07 particles/mm; Dunn's multiple-comparison test, p < 0.0001, compared with WT). In the DG hilus, MCs can be identified as having spiny dendrites contacted with large MF terminals (Acsády et al., 1998). In contrast to MF–CA3 synapses, MF–MC synapses were not labeled for GluK2/3 (Fig. 5C,G; 0.06 ± 0.04 particles/μm in WT vs 0.01 ± 0.01 particles/μm in GluK2 KO; p > 0.99). Instead, occasional weak labeling was observed on the nonsynaptic membrane of dendritic shaft and spines of MCs (Fig. 5E,F, green). The density of nonsynaptic particles was much lower than that at MF–CA3 synapses (p = 0.0137), but was significantly higher than their counterparts in GluK2 KO (Fig. 5D,G; 4.1 ± 0.8 particles/μm in WT vs 0.02 ± 0.01 particles/μm in GluK2 KO; p = 0.008). These results not only demonstrate the presence of KARs in MCs but also show that, in contrast to CA3 pyramidal neurons, GluK2-containing KARs are exclusively expressed at nonsynaptic sites in proximity of MF–MC synapses.
Figure 5.

GluK2/3 in hilar MCs are enriched at nonsynaptic sites. A, B, A large MF terminal characteristically forms multiple asymmetric synapses (arrowheads) with TEs (blue) of CA3 pyramidal cells. A, In WT mice, metal particles for GluK2/3 (arrows) are prevalent on PSD. B, Postsynaptic labeling is absent in GluK2 KO mouse. C–F, MCs (green) contact with large MF terminals in the hilus of the DG. C, In WT mice, MF synapses on MC spines are rarely labeled for GluK2/3; occasionally, nonsynaptic membrane on the dendritic shaft has low but significant labeling. D, Neither synaptic nor extrasynaptic labeling is observed in GluK2 KO mice. E, F, MCs occasionally have thin dendrites originating from soma (E) and contact with large MF terminal via multiple spines (F). Note that metal particles for GluK2/3 (arrows) leave postsynaptic membrane unlabeled. G, Average and individual data points for the density of metal particles for GluK2/3 on CA3 pyramidal cells (blue symbols, left axis) and hilar MCs (green symbols, right axis). Note the low but significant nonsynaptic labeling on MCs. The edges of PSD are indicated by pairs of arrowheads. The number of measured profiles (#) and membrane length (mm) are indicated in parentheses. Dunn's post-test: *p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Scale bars: A–D, F, 200 nm; E, 1 µm.
Activation of KARs in mossy cells by increase in ambient glutamate
Given the extracellular location of KARs in MCs, we hypothesized that these receptors are activated by ambient glutamate. To test this possibility, we blocked EAATs, a manipulation that can raise extracellular glutamate and activate extrasynaptic NMDARs (Le Meur et al., 2007). We first examined the effect of the nonselective EAAT blocker DL-TBOA (100 μm) on MC holding current in the presence of antagonists of AMPARs, NMDAR, and GABAA and GABAB receptors (see Materials and Methods). Bath application of TBOA mediated a significant NBQX-sensitive inward current in MCs, suggesting that KARs could be activated by endogenous glutamate (Fig. 6A,B,D; ΔI holding MC + TBOA: 53.7 ± 17.9; n = 5a/7c; p = 0.024 one-sample t test). We next used the more selective blocker dihydrokainic acid (DHK), which selectively blocks EAAT2 (GLT-1), a glutamate transporter that accounts for ∼90% of glutamate uptake (Rose et al., 2017) and is enriched in the telencephalon including the hippocampus (Chaudhry et al., 1995). Bath application of 100 μm DHK also induced NBQX-sensitive inward currents in MCs (Fig. 6A,B,D), strongly suggesting that DHK-induced currents are mediated by the activation of KARs (ΔI holding MC + DHK: 28.1 ± 7.4 pA; n = 3a/6c; p = 0.01292, one-sample t test). To determine whether DHK-induced currents occurred because of the activation of KARs, we repeated the experiment in GluK2 KO mice and found that in these mice DHK failed to induce inward currents in MCs (Fig. 6A,B,D; ΔI holding MC + DHK GluK2 KO: −8.5 ± 7.5; n = 3a/4c; p = 0.338, one-sample t test; DHK vs DHK GluK2 KO mice: p = 0.0105, two-sample t test). These findings suggest that increases in ambient glutamate can activate extrasynaptic MC-KARs.
Extrasynaptic glutamate receptors can be tonically activated by low concentrations of ambient glutamate (Le Meur et al., 2007; Rose et al., 2017). To determine whether MC-KARs could also be tonically activated by ambient glutamate, we tested the effect of NBQX on MC holding current. Bath application of NBQX (25 μm) had no effect on MC holding current, suggesting that MC-KARs, at least under our recording conditions, are not activated in a tonic manner by ambient glutamate (Fig. 6C,D; ΔI holding MC + NBQX: −18.4 ± 25.1; n = 4a/7c; p = 0.49, one-sample t test). We next tested whether we could engage extrasynaptic MC-KARs by artificially increasing ambient glutamate. To this end, we increased the extracellular K+ concentration from 2.5 to 8.5 mm, a manipulation that is expected to enhance ambient glutamate by facilitating glutamate release and neuronal activity. However, increasing extracellular K+ concentration did not significantly change the effect of NBQX on the MC holding current (Fig. 6C,D; ΔI holding MC + NBQX/High K+: −0.6 ± 30.6; n = 3a/6c; p = 0.98, one-sample t test; NBQX control vs NBQX/high K+: p = 0.65705, two-sample t test), suggesting that EAATs were effectively buffering increases in extracellular glutamate. To test this possibility, we bath applied the EAAT2 blocker DHK (100 μm) in the presence of 8.5 mm K+, and found that DHK elicited a larger current than when applied in control conditions (i.e., normal extracellular K+ concentration; ΔI holding MC + DHK/high K+: −63.5 ± 11.9 pA; n = 4a/8c; p = 0.00108, one-sample t test; DHK vs DHK/high K+: p = 0.0386, two-sample t test). Moreover, the large DHK-mediated current in high K+ was abolished in the presence of NBQX (Fig. 6C,D; ΔI holding MC + DHK/High K+/NBQX: −12.3 ± 4.8 pA; n = 3a/3c; p = 0.12, one-sample t test). Altogether, these results suggest that EAATs tightly control extracellular glutamate concentrations and thus prevent MC-KAR activation by ambient glutamate.
Discussion
In this study, we provide functional and anatomic evidence that MCs express extrasynaptic KARs whose activation in the rodent hippocampus can drive MCs. Specifically, we show that low concentrations of KA induced inward currents and action potential firing of MCs. In contrast, KA-induced currents were nearly absent in GCs, indicating that KARs have a unique pattern of expression among excitatory cells in the DG. Unexpectedly, MF activation failed to evoke a KAR-EPSCs in MCs, indicating that MF–MC synapses, unlike MF–CA3 synapses, do not normally express synaptic KARs. Our immunofluorescence and immunoelectron microscopy data confirmed that KARs in MCs are sparsely distributed at extrasynaptic sites and are mainly excluded from postsynaptic compartments. Finally, blockade of the astrocytic glutamate transporter EAAT2 revealed that MC-KARs can be activated by increasing ambient glutamate.
The presence of extrasynaptic KARs has previously been suggested in hippocampal CA1 pyramidal neurons (Bureau et al., 1999), striatal medium spiny neurons (Chergui et al., 2000), and cortical layer V pyramidal neurons (Eder et al., 2003). These functional studies inferred the presence of extrasynaptic KARs given the robust effects on bath-applied KAR agonist (e.g., membrane depolarization, inward current, action potential firing) with little evidence for KAR-mediated EPSCs. Of note, none of these studies provided ultrastructural evidence in support of extrasynaptic KARs. To the best of our knowledge, our immunoelectron microscopy data together with our electrophysiological characterization are the first direct evidence of a selective extrasynaptic localization of functional KARs in the mammalian brain.
Given the similarities between MF–CA3 and MF–MC synapses, the absence of KARs at MF–MC synapses is intriguing. Based on the presence of a GYKI-resistant component following MF stimulation, a previous study reported the presence of postsynaptic KARs in MCs (Hedrick et al., 2017). However, these currents were not validated in GluK2 KO mice and showed relatively fast kinetics, which is unusual for KAR-EPSCs. Our study does not discard the possibility that KARs could be expressed at MF–MC synapses early during development (Lauri and Taira, 2011; Lerma and Marques, 2013). The molecular mechanisms that target KARs to the synapse remain unclear, but several KAR interacting proteins, such as Neto1 and Neto2 (Straub et al., 2011; Tomita and Castillo, 2012; Wyeth et al., 2014), N-cadherins (Coussen et al., 2002; Fièvre et al., 2016), and presynaptic C1ql family proteins (Matsuda et al., 2016; Straub et al., 2016), may contribute. Consistent with data derived from a population-level transcriptomics study in the hippocampus (Cembrowski et al., 2016), we found that a Neto1 signal was absent from MCs (Fig. 4), suggesting that lack of Neto1 could contribute to the low expression of KARs at the synapse (Wyeth et al., 2014). However, lack of GluK2-containing KARs also leads to reduced levels of Neto1 (Straub et al., 2011). The C-terminal domain of KARs themselves is important for the synaptic stabilization of KARs in the cerebellum (Straub et al., 2016). Additionally, phosphorylation of specific residues in the C-terminal and other intracellular regions of KARs has been implicated in the modulation of KARs function and trafficking (Wang et al., 1993; Kornreich et al., 2007; Carta et al., 2013; Zhu et al., 2014). Further studies are required to clarify the molecular mechanisms that determine the exclusion of KARs from MF–MC synapses.
Like other extrasynaptic receptors, KARs in MCs could be engaged by a rise in ambient glutamate, which can occur as a result of glutamate spillover during sustained synaptic activity (Le Meur et al., 2007; Rose et al., 2017). Glutamate could also arise from MC dendrites, as previously reported in neocortical and cerebellar neurons (Zilberter, 2000; Shin et al., 2008), and activate extrasynaptic KARs. We found that blockade of EAAT2 induces KAR-mediated inward currents likely because of the increase in extracellular glutamate. In the CA1 area of the hippocampus, activation of extrasynaptic receptors by synaptically released glutamate is limited by efficient astrocytic EAAT2 activity (Diamond and Jahr, 2000), consistent with a neuroprotective role of this transporter (Kong et al., 2012; Pajarillo et al., 2019). However, EAAT2 may saturate in other brain areas (Armbruster et al., 2016; Pinky et al., 2018). EAAT2 saturation during neuronal hyperactivity could enable the activation of extrasynaptic KARs at MCs. In addition, the expression of EAAT2/GLT1 is reduced in animal models of epilepsy (Hubbard et al., 2016), a condition that could increase extracellular glutamate and activate extrasynaptic KARs. Alternatively, extrasynaptic KARs could be activated by glutamate released from astrocytes (Araque et al., 2014; Pal, 2015), as previously reported in CA1 GABAergic interneurons (Liu et al., 2004). There is evidence that glutamate released from astrocytes can also activate extrasynaptic NMDARs in CA1 pyramidal neurons (Fellin et al., 2004), and depolarize both hilar GABAergic interneurons and MCs (Pabst et al., 2016). These depolarizations were blocked by nonselective antagonism of all ionotropic glutamate receptors, raising the possibility that extrasynaptic KARs could be implicated. Future work is required to determine whether astrocytic processes (Gavrilov et al., 2018) could release glutamate in proximity to extrasynaptic KARs, thereby avoiding glutamate uptake by EAAT2.
Although the precise role for extrasynaptic KARs in MCs is unclear, they might detect changes in the levels of ambient glutamate and mediate tonic depolarization. In vivo, MCs display a high level of activity compared with neighboring GCs (Danielson et al., 2017; GoodSmith et al., 2017; Senzai and Buzsáki, 2017). In standard home-cage rats, MCs stain positive for the activity-dependent immediately early gene c-fos (Duffy et al., 2013), suggesting that even at the basal level, MCs are remarkably active. It is possible that in behaving animals, where spontaneous activity is most likely higher than in vitro, glutamate might escape reuptake by EAATs and activate extrasynaptic KARs. During periods of particularly high activity and potential EAAT2 saturation, KARs might act as nonlinear integrators of synaptic inputs and enhance MC output. KARs can also work in a metabotropic fashion and could potentially affect the excitability of MCs by suppressing the slow afterhyperpolarization (Melyan et al., 2002; Ruiz et al., 2005). Ultimately, MC-KARs could contribute to the promiscuous activity of MCs in multiple locations and environments (Danielson et al., 2017; GoodSmith et al., 2017; Senzai and Buzsáki, 2017).
Both MCs and KARs have been linked to several neurologic and psychiatric disorders (Ratzliff et al., 2002; Lerma and Marques, 2013; Scharfman, 2016). Of particular relevance is the strong link between KARs and MCs with TLE. KARs have been strongly implicated in epilepsy (Crépel and Mulle, 2015; Falcón-Moya et al., 2018), and KA-induced TLE is one of the most widely used models of TLE (Lévesque and Avoli, 2013; Rusina et al., 2021). The importance of KARs in KA-induced TLE is highlighted by the fact that the loss of GluK2-containing KARs strongly reduces the susceptibility to KA-induced seizures (Mulle et al., 1998). Similarly, MCs have been proposed to have a proepileptogenic role in the early phases of TLE (Ratzliff et al., 2002; Botterill et al., 2019) and to undergo prominent cell death in both animal models of epilepsy (Blümcke et al., 2000) and in human patients (Margerison and Corsellis, 1966; Seress et al., 2009). Recently, MCs have been shown to be robustly activated in vivo by intraperitoneal KA injections (Nasrallah et al., 2021). However, the precise mechanism through which KARs and MCs are involved in TLE is still unclear. The expression of KARs in MCs strongly suggests that MCs could be a direct target of KA in KA-induced TLE. KA-induced MCs firing could contribute to hyperexcitability of the associative GC–MC–GC network and to the generation of seizures. Moreover, sustained KAR-mediated MC firing could be a critical trigger for long-lasting forms of plasticity in the DG associative network (Hashimotodani et al., 2017), which could contribute to the prolongation of epileptic activity. A major limitation for the study of MC function in behavior is the lack of molecular tools that specifically target MCs. Thus far, the manipulation of MC activity in vivo relied on viral delivery of constructs under the control of promoters that are not highly specific for MCs (Jinde et al., 2012; Puighermanal et al., 2015). Establishing the precise role of MC-KARs on hippocampal function and TLE will require novel strategies such as intersectional genetics approaches (Dymecki et al., 2010; Graybuck et al., 2021) that will allow more selective targeting of MCs while sparing neighboring KAR-expressing cells such as CA3 pyramidal neurons and hilar interneurons.
Footnotes
This research was supported by National Institutes of Health Grants R01-NS-113600, R01-MH-116673, and R01-MH-125772 to P.E.C; Grant-in-Aid for Specially Promoted Research from Japan Society for the Promotion of Science (JSPS; 20H05628) and for Scientific Research B from JSPS (21H02589) to M.W.; and JSPS KAKENHI Grants 17H0631310 and 20H03410 to M.Y. We thank the members of the Castillo laboratory (in particular Dr. Kaoutsar Nasrallah) for constructive feedback and for reading and commenting on the manuscript.
The authors declare no competing financial interests.
References
- Acsády L, Kamondi A, Sík A, Freund T, Buzsáki G (1998) GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci 18:3386–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, Dent JA (1981) Development of the mossy fibers of the dentate gyrus: I. A light and electron microscopic study of the mossy fibers and their expansions. J Comp Neurol 195:51–86. 10.1002/cne.901950106 [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A (2014) Gliotransmitters travel in time and space. Neuron 81:728–739. 10.1016/j.neuron.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster M, Hanson E, Dulla CG (2016) Glutamate clearance is locally modulated by presynaptic neuronal activity in the cerebral cortex. J Neurosci 36:10404–10415. 10.1523/JNEUROSCI.2066-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümcke I, Suter B, Behle K, Kuhn R, Schramm J, Elger CE, Wiestler OD (2000) Loss of hilar mossy cells in Ammon's horn sclerosis. Epilepsia 41:S174–S180. 10.1111/j.1528-1157.2000.tb01577.x [DOI] [PubMed] [Google Scholar]
- Botterill JJ, Lu YL, LaFrancois JJ, Bernstein HL, Alcantara-Gonzalez D, Jain S, Leary P, Scharfman HE (2019) An excitatory and epileptogenic effect of dentate gyrus mossy cells in a mouse model of epilepsy. Cell Rep 29:2875–2889.e6. 10.1016/j.celrep.2019.10.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Jongen-Rêlo AL (1999) Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J Neurosci 19:9519–9529. 10.1523/JNEUROSCI.19-21-09519.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Schwartzkroin PA (1994) Hippocampal mossy cell function: a speculative view. Hippocampus 4:393–402. 10.1002/hipo.450040402 [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Wenzel HJ, Kunkel DD, Schwartzkroin PA (1996) Axon arbors and synaptic connections of hippocampal mossy cells in the rat in vivo. J Comp Neurol 366:271–292. [DOI] [PubMed] [Google Scholar]
- Bureau I, Bischoff S, Heinemann SF, Mulle C (1999) Kainate receptor-mediated responses in the CA1 field of wild-type and GluR6-deficient mice. J Neurosci 19:653–663. 10.1523/JNEUROSCI.19-02-00653.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Opazo P, Veran J, Athané A, Choquet D, Coussen F, Mulle C (2013) CaMKII-dependent phosphorylation of GluK5 mediates plasticity of kainate receptors. EMBO J 32:496–510. 10.1038/emboj.2012.334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA (1997) Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388:182–186. 10.1038/40645 [DOI] [PubMed] [Google Scholar]
- Cembrowski MS, Wang L, Sugino K, Shields BC, Spruston N (2016) Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. Elife 5:e14997. 10.7554/eLife.14997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J (1995) Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15:711–720. 10.1016/0896-6273(95)90158-2 [DOI] [PubMed] [Google Scholar]
- Chergui K, Bouron A, Normand E, Mulle C (2000) Functional GluR6 kainate receptors in the striatum: indirect downregulation of synaptic transmission. J Neurosci 20:2175–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussen F, Normand E, Marchal C, Costet P, Choquet D, Lambert M, Mège RM, Mulle C (2002) Recruitment of the kainate receptor subunit glutamate receptor 6 by cadherin/catenin complexes. J Neurosci 22:6426–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crépel V, Mulle C (2015) Physiopathology of kainate receptors in epilepsy. Curr Opin Pharmacol 20:83–88. 10.1016/j.coph.2014.11.012 [DOI] [PubMed] [Google Scholar]
- Danielson NB, Turi GF, Ladow M, Chavlis S, Petrantonakis PC, Poirazi P, Losonczy A (2017) In vivo imaging of dentate gyrus mossy cells in behaving mice. Neuron 93:552–559.e4. 10.1016/j.neuron.2016.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (2000) Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol 83:2835–2843. 10.1152/jn.2000.83.5.2835 [DOI] [PubMed] [Google Scholar]
- Duffy AM, Schaner MJ, Chin J, Scharfman HE (2013) Expression of c-fos in hilar mossy cells of the dentate gyrus in vivo. Hippocampus 23:649–655. 10.1002/hipo.22138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymecki SM, Ray RS, Kim JC (2010) Mapping cell fate and function using recombinase-based intersectional strategies. Methods Enzymol 477:183–213. 10.1016/S0076-6879(10)77011-7 [DOI] [PubMed] [Google Scholar]
- Eder M, Becker K, Rammes G, Schierloh A, Azad SC, Zieglgänsberger W, Dodt HU (2003) Distribution and properties of functional postsynaptic kainate receptors on neocortical layer V pyramidal neurons. J Neurosci 23:6660–6670. 10.1523/JNEUROSCI.23-16-06660.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcón-Moya R, Sihra TS, Rodríguez-Moreno A (2018) Kainate receptors: role in epilepsy. Front Mol Neurosci 11:217. 10.3389/fnmol.2018.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G (2004) Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43:729–743. 10.1016/j.neuron.2004.08.011 [DOI] [PubMed] [Google Scholar]
- Fièvre S, Carta M, Chamma I, Labrousse V, Thoumine O, Mulle C (2016) Molecular determinants for the strictly compartmentalized expression of kainate receptors in CA3 pyramidal cells. Nat Commun 7:12738. 10.1038/ncomms12738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredes F, Silva MA, Koppensteiner P, Kobayashi K, Joesch M, Shigemoto R (2021) Ventro-dorsal hippocampal pathway gates novelty-induced contextual memory formation. Curr Biol 31:25–38.e5. 10.1016/j.cub.2020.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA (1998) Synaptic activation of kainate receptors on hippocampal interneurons. Nat Neurosci 1:479–486. 10.1038/2194 [DOI] [PubMed] [Google Scholar]
- Frotscher M, Seress L, Schwerdtfeger WK, Buhl E (1991) The mossy cells of the fascia dentata: a comparative study of their fine structure and synaptic connections in rodents and primates. J Comp Neurol 312:145–163. 10.1002/cne.903120111 [DOI] [PubMed] [Google Scholar]
- Fukaya M, Watanabe M (2000) Improved immunohistochemical detection of postsynaptically located PSD-95/SAP90 protein family by protease section pretreatment: a study in the adult mouse brain. J Comp Neurol 426:572–586. [DOI] [PubMed] [Google Scholar]
- Gavrilov N, Golyagina I, Brazhe A, Scimemi A, Turlapov V, Semyanov A (2018) Astrocytic coverage of dendritic spines, dendritic shafts, and axonal boutons in hippocampal neuropil. Front Cell Neurosci 12:248. 10.3389/fncel.2018.00248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GoodSmith D, Chen X, Wang C, Kim SH, Song H, Burgalossi A, Christian KM, Knierim JJ (2017) Spatial representations of granule cells and mossy cells of the dentate gyrus. Neuron 93:677–690.e5. 10.1016/j.neuron.2016.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybuck LT, Daigle TL, Sedeño-Cortés AE, Walker M, Kalmbach B, Lenz GH, Morin E, Nguyen TN, Garren E, Bendrick JL, Kim TK, Zhou T, Mortrud M, Yao S, Siverts LA, Larsen R, Gore BB, Szelenyi ER, Trader C, Balaram P, et al. (2021) Enhancer viruses for combinatorial cell-subclass-specific labeling. Neuron 109:1449–1464.e13. 10.1016/j.neuron.2021.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Nasrallah K, Jensen KR, Chávez AE, Carrera D, Castillo PE (2017) LTP at hilar mossy cell-dentate granule cell synapses modulates dentate gyrus output by increasing excitation/inhibition balance. Neuron 95:928–943.e3. 10.1016/j.neuron.2017.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick TP, Nobis WP, Foote KM, Ishii T, Chetkovich DM, Swanson GT (2017) Excitatory synaptic input to hilar mossy cells under basal and hyperexcitable conditions. eNeuro 4:ENEURO.0364-17.2017. 10.1523/ENEURO.0364-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsáki G (2002) Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci 5:790–795. 10.1038/nn887 [DOI] [PubMed] [Google Scholar]
- Hubbard JA, Szu JI, Yonan JM, Binder DK (2016) Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp Neurol 283:85–96. 10.1016/j.expneurol.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinde S, Zsiros V, Jiang Z, Nakao K, Pickel J, Kohno K, Belforte JE, Nakazawa K (2012) Hilar mossy cell degeneration causes transient dentate granule cell hyperexcitability and impaired pattern separation. Neuron 76:1189–1200. 10.1016/j.neuron.2012.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C (1996) Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol 493:447–455. 10.1113/jphysiol.1996.sp021395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q, Takahashi K, Schulte D, Stouffer N, Lin Y, Lin CL (2012) Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis 47:145–154. 10.1016/j.nbd.2012.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornreich BG, Niu L, Roberson MS, Oswald RE (2007) Identification of C-terminal domain residues involved in protein kinase A-mediated potentiation of kainate receptor subtype 6. Neuroscience 146:1158–1168. 10.1016/j.neuroscience.2007.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer P, Strowbridge BW (2008) Nonrandom local circuits in the dentate gyrus. J Neurosci 28:12212–12223. 10.1523/JNEUROSCI.3612-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Taira T (2011) Role of kainate receptors in network activity during development. Adv Exp Med Biol 717:81–91. 10.1007/978-1-4419-9557-5_8 [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E (2007) Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol 580:373–383. 10.1113/jphysiol.2006.123570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J, Marques JM (2013) Kainate receptors in health and disease. Neuron 80:292–311. 10.1016/j.neuron.2013.09.045 [DOI] [PubMed] [Google Scholar]
- Lévesque M, Avoli M (2013) The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev 37:2887–2899. 10.1016/j.neubiorev.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QS, Xu Q, Arcuino G, Kang J, Nedergaard M (2004) Astrocyte-mediated activation of neuronal kainate receptors. Proc Natl Acad Sci U S A 101:3172–3177. 10.1073/pnas.0306731101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysetskiy M, Földy C, Soltesz I (2005) Long- and short-term plasticity at mossy fiber synapses on mossy cells in the rat dentate gyrus. Hippocampus 15:691–696. 10.1002/hipo.20096 [DOI] [PubMed] [Google Scholar]
- Margerison JH, Corsellis JA (1966) Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain 89:499–530. 10.1093/brain/89.3.499 [DOI] [PubMed] [Google Scholar]
- Matsuda K, Budisantoso T, Mitakidis N, Sugaya Y, Miura E, Kakegawa W, Yamasaki M, Konno K, Uchigashima M, Abe M, Watanabe I, Kano M, Watanabe M, Sakimura K, Aricescu AR, Yuzaki M (2016) Transsynaptic modulation of kainate receptor functions by C1q-like proteins. Neuron 90:752–767. 10.1016/j.neuron.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B (2002) Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron 34:107–114. 10.1016/s0896-6273(02)00624-4 [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Pérez-Otaño I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF (1998) Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature 392:601–605. 10.1038/33408 [DOI] [PubMed] [Google Scholar]
- Nasrallah K, Frechou MA, Yoon YJ, Persaud S, Gonçalves T, Castillo PE (2021) Activity-dependent LTP in the dentate gyrus promotes epileptic seizures. bioRxiv. 10.1101/2021.06.30.450628. [DOI] [Google Scholar]
- Nicoll RA, Schmitz D (2005) Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci 6:863–876. 10.1038/nrn1786 [DOI] [PubMed] [Google Scholar]
- Pabst M, Braganza O, Dannenberg H, Hu W, Pothmann L, Rosen J, Mody I, van Loo K, Deisseroth K, Becker AJ, Schoch S, Beck H (2016) Astrocyte intermediaries of septal cholinergic modulation in the hippocampus. Neuron 90:853–865. 10.1016/j.neuron.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Pajarillo E, Rizor A, Lee J, Aschner M, Lee E (2019) The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: potential targets for neurotherapeutics. Neuropharmacology 161:107559. 10.1016/j.neuropharm.2019.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal B (2015) Astrocytic actions on extrasynaptic neuronal currents. Front Cell Neurosci 9:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton PE, McNaughton B (1995) Connection matrix of the hippocampal formation: I. The dentate gyrus. Hippocampus 5:245–286. 10.1002/hipo.450050402 [DOI] [PubMed] [Google Scholar]
- Pinky NF, Wilkie CM, Barnes JR, Parsons MP (2018) Region- and activity-dependent regulation of extracellular glutamate. J Neurosci 38:5351–5366. 10.1523/JNEUROSCI.3213-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puighermanal E, Biever A, Espallergues J, Gangarossa G, De Bundel D, Valjent E (2015) drd2-cre:ribotag mouse line unravels the possible diversity of dopamine d2 receptor-expressing cells of the dorsal mouse hippocampus. Hippocampus 25:858–875. 10.1002/hipo.22408 [DOI] [PubMed] [Google Scholar]
- Ratzliff A, Santhakumar V, Howard A, Soltesz I (2002) Mossy cells in epilepsy: rigor mortis or vigor mortis? Trends Neurosci 25:140–144. 10.1016/s0166-2236(00)02122-6 [DOI] [PubMed] [Google Scholar]
- Ribak CE, Shapiro LA (2007) Ultrastructure and synaptic connectivity of cell types in the adult rat dentate gyrus. Prog Brain Res 163:155–166. 10.1016/S0079-6123(07)63009-X [DOI] [PubMed] [Google Scholar]
- Ribak CE, Seress L, Amaral DG (1985) The development, ultrastructure and synaptic connections of the mossy cells of the dentate gyrus. J Neurocytol 14:835–857. 10.1007/BF01170832 [DOI] [PubMed] [Google Scholar]
- Richter KN, Revelo NH, Seitz KJ, Helm MS, Sarkar D, Saleeb RS, D'Este E, Eberle J, Wagner E, Vogl C, Lazaro DF, Richter F, Coy-Vergara J, Coceano G, Boyden ES, Duncan RR, Hell SW, Lauterbach MA, Lehnart SE, Moser T, et al. (2018) Glyoxal as an alternative fixative to formaldehyde in immunostaining and super-resolution microscopy. EMBO J 37:139–159. 10.15252/embj.201695709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JH, Deadwyler SA (1981) Kainic acid produces depolarization of CA3 pyramidal cells in the vitro hippocampal slice. Brain Res 221:117–127. 10.1016/0006-8993(81)91067-2 [DOI] [PubMed] [Google Scholar]
- Rose CR, Felix L, Zeug A, Dietrich D, Reiner A, Henneberger C (2017) Astroglial glutamate signaling and uptake in the hippocampus. Front Mol Neurosci 10:451. 10.3389/fnmol.2017.00451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F, Mulle C (2005) Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci 25:11710–11718. 10.1523/JNEUROSCI.4041-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusina E, Bernard C, Williamson A (2021) The kainic acid models of temporal lobe epilepsy. eNeuro 8:ENEURO.0337-20.2021. 10.1523/ENEURO.0337-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1994) Evidence from simultaneous intracellular recordings in rat hippocampal slices that area CA3 pyramidal cells innervate dentate hilar mossy cells. J Neurophysiol 72:2167–2180. 10.1152/jn.1994.72.5.2167 [DOI] [PubMed] [Google Scholar]
- Scharfman HE (2007) The CA3 “backprojection” to the dentate gyrus. Prog Brain Res 163:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (2016) The enigmatic mossy cell of the dentate gyrus. Nat Rev Neurosci 17:562–575. 10.1038/nrn.2016.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (2018) Advances in understanding hilar mossy cells of the dentate gyrus. Cell Tissue Res 373:643–652. 10.1007/s00441-017-2750-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Schwartzkroin PA (1988) Electrophysiology of morphologically identified mossy cells of the dentate hilus recorded in guinea pig hippocampal slices. J Neurosci 8:3812–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senzai Y, Buzsáki G (2017) Physiological properties and behavioral correlates of hippocampal granule cells and mossy cells. Neuron 93:691–704.e5. 10.1016/j.neuron.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seress L, Abrahám H, Horváth Z, Dóczi T, Janszky J, Klemm J, Byrne R, Bakay RA (2009) Survival of mossy cells of the hippocampal dentate gyrus in humans with mesial temporal lobe epilepsy. J Neurosurg 111:1237–1247. 10.3171/2008.11.JNS08779 [DOI] [PubMed] [Google Scholar]
- Shin JH, Kim YS, Linden DJ (2008) Dendritic glutamate release produces autocrine activation of mGluR1 in cerebellar Purkinje cells. Proc Natl Acad Sci U S A 105:746–750. 10.1073/pnas.0709407105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA, Frotscher M (2003) “Dormant basket cell” hypothesis revisited: relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J Comp Neurol 459:44–76. 10.1002/cne.10630 [DOI] [PubMed] [Google Scholar]
- Straub C, Hunt DL, Yamasaki M, Kim KS, Watanabe M, Castillo PE, Tomita S (2011) Distinct functions of kainate receptors in the brain are determined by the auxiliary subunit Neto1. Nat Neurosci 14:866–873. 10.1038/nn.2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub C, Noam Y, Nomura T, Yamasaki M, Yan D, Fernandes HB, Zhang P, Howe JR, Watanabe M, Contractor A, Tomita S (2016) Distinct subunit domains govern synaptic stability and specificity of the kainate receptor. Cell Rep 16:531–544. 10.1016/j.celrep.2016.05.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki C, Yamasaki M, Uchigashima M, Konno K, Yanagawa Y, Watanabe M (2010) Cytochemical and cytological properties of perineuronal oligodendrocytes in the mouse cortex. Eur J Neurosci 32:1326–1336. 10.1111/j.1460-9568.2010.07377.x [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T (2003) Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol 467:60–79. 10.1002/cne.10905 [DOI] [PubMed] [Google Scholar]
- Tomita S, Castillo PE (2012) Neto1 and Neto2: auxiliary subunits that determine key properties of native kainate receptors. J Physiol 590:2217–2223. 10.1113/jphysiol.2011.221101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Taverna FA, Huang XP, MacDonald JF, Hampson DR (1993) Phosphorylation and modulation of a kainate receptor (GluR6) by cAMP-dependent protein kinase. Science 259:1173–1175. 10.1126/science.8382377 [DOI] [PubMed] [Google Scholar]
- Wenzel HJ, Buckmaster PS, Anderson NL, Wenzel ME, Schwartzkroin PA (1997) Ultrastructural localization of neurotransmitter immunoreactivity in mossy cell axons and their synaptic targets in the rat dentate gyrus. Hippocampus 7:559–570. [DOI] [PubMed] [Google Scholar]
- Westbrook GL, Lothman EW (1983) Cellular and synaptic basis of kainic acid-induced hippocampal epileptiform activity. Brain Res 273:97–109. 10.1016/0006-8993(83)91098-3 [DOI] [PubMed] [Google Scholar]
- Wyeth MS, Pelkey KA, Petralia RS, Salter MW, McInnes RR, McBain CJ (2014) Neto auxiliary protein interactions regulate kainate and NMDA receptor subunit localization at mossy fiber–CA3 pyramidal cell synapses. J Neurosci 34:622–628. 10.1523/JNEUROSCI.3098-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu QJ, Kong FS, Xu H, Wang Y, Du CP, Sun CC, Liu Y, Li T, Hou XY (2014) Tyrosine phosphorylation of GluK2 up-regulates kainate receptor-mediated responses and downstream signaling after brain ischemia. Proc Natl Acad Sci U S A 111:13990–13995. 10.1073/pnas.1403493111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter Y (2000) Dendritic release of glutamate suppresses synaptic inhibition of pyramidal neurons in rat neocortex. J Physiol 528:489–496. 10.1111/j.1469-7793.2000.00489.x [DOI] [PMC free article] [PubMed] [Google Scholar]