

Abstract
Dysregulation of autophagic pathways leads to accumulation of abnormal proteins and damaged organelles in many neurodegenerative disorders, including Parkinson's disease (PD) and Lewy body dementia (LBD). Autophagy-related dysfunction may also trigger secretion and spread of misfolded proteins, such as α-synuclein (α-syn), the major misfolded protein found in PD/LBD. However, the mechanism underlying these phenomena remains largely unknown. Here, we used cell-based models, including human induced pluripotent stem cell-derived neurons, CRISPR/Cas9 technology, and male transgenic PD/LBD mice, plus vetting in human postmortem brains (both male and female). We provide mechanistic insight into this pathologic pathway. We find that aberrant S-nitrosylation of the autophagic adaptor protein p62 causes inhibition of autophagic flux and intracellular buildup of misfolded proteins, with consequent secretion resulting in cell-to-cell spread. Thus, our data show that pathologic protein S-nitrosylation of p62 represents a critical factor not only for autophagic inhibition and demise of individual neurons, but also for α-syn release and spread of disease throughout the nervous system.
SIGNIFICANCE STATEMENT In Parkinson's disease and Lewy body dementia, dysfunctional autophagy contributes to accumulation and spread of aggregated α-synuclein. Here, we provide evidence that protein S-nitrosylation of p62 inhibits autophagic flux, contributing to α-synuclein aggregation and spread.
Keywords: α-synuclein, autophagy, Lewy body dementia, p62, Parkinson's disease
Introduction
p62, also known as SQSTM1, was the first protein identified as an autophagy adaptor molecule, a group that now includes four other proteins with similar function: NBR1, TAX1BP1, NDP52, and OPTN (Moscat et al., 2016). Among the various domains of p62, the ubiquitin-associated domain and the LC3-interacting region (LIR) are closely associated with the autophagy-lysosomal degradation pathway (Pankiv et al., 2007; Kraft et al., 2010), which is critical for cellular removal of dysfunctional proteins and organelles. In neurons and possibly other cell types, impairment of basal macroautophagy (hereafter designated simply as autophagy) is thought to contribute to the pathogenesis of neurodegenerative disorders, such as Parkinson's disease (PD), Lewy body dementia (LBD), Alzheimer's disease (AD), Huntington's disease, and amyotrophic lateral sclerosis by allowing accumulation of potentially toxic products (Menzies et al., 2017).
A neuropathological hallmark of PD/LBD in human brain is the presence of intracytoplasmic proteinaceous inclusions termed Lewy bodies (LBs) (Spillantini et al., 1997; Shults, 2006), which are predominantly composed of misfolded α-synuclein (α-syn) in addition to other proteins, such as p62 and ubiquitin (Zatloukal et al., 2002; Kuusisto et al., 2003). Rare missense mutations or triplication of the SNCA gene, which encodes α-syn, cause early-onset PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Singleton et al., 2003; Zarranz et al., 2004), and mouse models of the disease that overexpress α-syn develop LB-like pathology and can spread through the brain to resemble LBD (Rockenstein et al., 2002). Intriguingly, these mouse models show enhanced α-syn pathology under the condition of p62 deficiency (Tanji et al., 2015), suggesting a neuroprotective function of p62 by controlling α-syn homeostasis.
Our group and others have shown that genetic mutation and environmental toxins can contribute to neurodegenerative diseases, including PD/LBD, in part by generating excessive levels of reactive oxygen and reactive nitrogen species (RNS), such as nitric oxide (NO) (Chung et al., 2004; Yao et al., 2004; Ryan et al., 2013). Such overproduction of RNS can result in aberrant protein S-nitrosylation, often mimicking the effect of rare genetic mutations and enhancing neuronal cell death (T. Nakamura et al., 2013), partly by inhibiting the autophagy-lysosomal pathway (Sarkar et al., 2011; Montagna et al., 2016). Along these lines, we noted that p62 contained a partial motif that can facilitate S-nitrosylation, indicating its susceptibility to this post-translational modification under disease conditions when RNS/NO are elevated. Indeed, in the present study, we report evidence for S-nitrosylation of p62 (forming SNO-p62) in various PD/LBD model systems, including postmortem human brain. The resulting levels of SNO-p62 inhibit autophagic flux, thus contributing to aberrant α-syn accumulation and synucleinopathy. Moreover, considerable in vitro and in vivo evidence has been mounted that pathologically misfolded/oligomerized α-syn is released from neurons and then spreads to other brain regions (Vargas et al., 2019). This α-syn “transmission hypothesis” predicts that pathologically released α-syn can be transmitted from cell to cell, and the resultant pathology can lead to additional neuronal dysfunction and degeneration. Our findings show that SNO-p62 can trigger this spread of disease by increasing neuronal release of pathologic α-syn that is not otherwise degraded by autophagy.
Materials and Methods
Cell culture and transfection
SH-SY5Y (ATCC; CRL-2266) neural cells, HEK293TN (System Biosciences; LV900A-1) cells, and p62 KO mouse embryonic fibroblast (MEF) cells (Linares et al., 2013) were maintained in DMEM with GlutaMAX (DMEM Media-GlutaMAX-I, Thermo Fisher Scientific; 10566016) supplemented with 10% FBS (Sigma, F7524), 100 IU/ml, and 100 µg/ml penicillin-streptomycin (Thermo Fisher Scientific, 10378016) in a 5% CO2 incubator at 37°C. Transfections were performed in 6-well plates using 1 mg/ml polyethyleneimine solution (Sigma, P3143) with DMEM; after 3 h, mixtures were replaced with fresh culture medium. After 2 d, cells were analyzed by immunoblotting. For Venus-α-syn plasmids, transfections were conducted with Lipofectamine 2000 (Thermo Fisher Scientific, 11668019) using the protocol recommended by the manufacturer.
Human induced pluripotent stem cell (hiPSC) dopaminergic neuron induction
hiPSCs were cultured, maintained, and differentiated into hiPSC-DA neurons as we previously described (Ryan et al., 2013). In brief, hiPSCs were maintained in a feeder-free system in MEF-conditioned hiPSC medium (DMEM/F12 with GlutaMAX [Thermo Fisher Scientific, 10565018] plus KO serum replacement [KSR] medium [20%, Thermo Fisher Scientific, 10828028], nonessential amino acid solution [1%, Thermo Fisher Scientific, 11140050], antibiotic-antimycotic additives [1%, Thermo Fisher Scientific,15240062], 2-mercaptoethanol [110 μm, Thermo Fisher Scientific, 21985023], and FGF2 [8 ng/ml, 233-FB]). For dopaminergic neural induction, hiPSCs were disaggregated using TrypLE Express for 3 min, washed with hiPSC media, and replated at a density of 3.5 × 104 to 4.0 × 104 cells/cm2 on Matrigel (BD Biosciences, 345230)-coated plates in MEF-conditioned hiPSC medium supplemented with 10 ng/ml of FGF2 and thiazovivin (Stem Cell Technologies, 72252). After 24 h, thiazovivin was withdrawn, and hiPSCs were allowed to expand for 3 d or until they were nearly confluent. Initial differentiation conditions consisted of KSR medium, including LDN193189 (100 nm, Stemgent, 04-0074-02), SB431542 (10 μm, Tocris Bioscience, 1614), sonic hedgehog C25II (100 ng/ml, R&D Systems, 464-SH), purmorphamine (2 μm, Stemgent, 04-0009), and CHIR99021 (CHIR; 3 μm, Stemgent, 04-0004-02). At day 5 of differentiation, KSR medium was gradually shifted to N2 medium, with increasing amounts of N2 medium (25%, 50%, 75%) added to KSR medium every 2 d. At day 11, progenitor cells were isolated by dissociating with accutase (Innovative Cell Technologies, AT104) and replated at a density of 3 × 105 cells/cm2 in DMEM/F12 + B27 (Thermo Fisher Scientific, 12587010) and N2 supplemented (Thermo Fisher Scientific, 17502048) with LDN193189, CHIR, FGF (20 ng/ml) on dishes precoated with polyornithine (15 μg/ml, P4638)/laminin (1 μg/ml, R&D Systems, 3400-010-02)/fibronectin (2 μg/ml, 3420-001-01). Thiazovivin (2 μm) was added during the first isolation. These progenitor cells were expanded once weekly. To induce terminal differentiation, the culture medium was then switched to Neurobasal/B27/GlutaMAX supplemented with CHIR and DAPT (10 μm; Tocris Bioscience, 2634), BDNF (20 ng/ml; R&D Systems, 248-BDB), GDNF (20 ng/ml; R&D Systems, 212-GD), TGFβ3 (1 ng/ml; R&D Systems, 243-B3), ascorbic acid (0.2 mm, Sigma, A4544), and dibutyryl cAMP (0.5 mm; Sigma, D0260) between days 11 and 13, and then to Neurobasal/B27/GlutaMAX supplemented with BDNF, GDNF, TGFβ3, ascorbic acid, and dibutyryl cAMP between days 13-35. After 21 d of differentiation, DA neurons and conditioned media were harvested for biotin-switch assays and α-syn secretion assays, respectively.
α-syn transgenic mice and human LBD brain tissue
Transgenic mice overexpressing human WT α-syn under the Thy1 promoter were used (Rockenstein et al., 2002; Chesselet et al., 2012); male mice were used here because PD/LBD predominates in males over females. To detect SNO-p62, brains were collected from 4-month-old littermate control (C57BI/6) or α-syn transgenic mice (n = 4) and homogenized in HENTS buffer (100 mm HEPES, pH 7.4, 1 mm EDTA, 0.1 mm neocuproine, 0.1% SDS, 1% Triton X-100, and 10 mm methyl methanethiosulfonate). Brain lysates were subjected to biotin-switch assay. All animal experiments were approved by The Scripps Research Institute or Sanford Burnham Prebys Medical Discovery Institute Animal Care and Use Committee, where the brains were collected. Deidentified human patient brain tissues were obtained postmortem from subjects whose age, postmortem interval, and sex (both female and male) are described in Extended Data Figure 6-1. Human brain samples were analyzed with institutional permission under the state of California and National Institutes of Health guidelines. Informed consent was obtained according to procedures approved by Institutional Review Boards at the University of California, San Diego and The Scripps Research Institute.
CRISPR/Cas9 knock-in of p62(C331A) mutant cell lines
Human p62 sgRNA guide A, B plasmids, and single-stranded donor oligodeoxynucleotide (ssODN) template were cotransfected into SH-SY5Y cells using polyethyleneimine. After 1 d, cells were dissociated by TrypLE Express and resuspended with basic sorting buffer (1× PBS [Ca2+ and Mg2+ free], 2.5 mm EDTA, 25 mm HEPES, pH 7.0, 1% heat-inactivated FBS, and 1% penicillin-streptomycin), sterilized with a 0.2 µm prefilter. GFP-positive cells were sorted by flow cytometry (MoFlo Astrios EQ, Beckman Coulter). Green clones were screened by Mlu I-HF restriction enzyme digestion (New England Biolabs, R3198S) after genomic DNA purification using QuickExtract DNA Extraction Solution (Lucigen, QE09050) and amplification by PCR. The PCR reactions were performed with Platinum PCR SuperMix (Thermo Fisher Scientific, 11306016) using the following protocol: 95°C for 5 min, followed by 35 cycles of denaturing at 95°C for 30 s, annealing at 59°C for 30 s, extension at 68°C for 60 s, and then final extension at 68°C for an additional 10 min. mRNAs were extracted using a Quick-RNA MiniPrep Plus kit (Zymo Research, R1055). After cDNA synthesis using a QuantiTect Reverse Transcription Kit (QIAGEN, 205311), p62 cDNA fragments containing the Mlu I digestion site were amplified and digested with Mlu I-HF restriction enzyme. Human p62 sequencing primers are described in Extended Data Figure 3-3. We generated three heterozygous mutant p62(C331A) cell lines among 98 clones that we grew.
Plasmid constructs and sgRNAs
WT HA-tagged pcDNA3 p62 plasmid was produced in the laboratory of J.M., as previously described (Linares et al., 2013). HA-tagged C-terminal fragment of p62 (amino acids 230-440) was cloned from the HA-p62 plasmid. The C331A-mutated C-terminal fragment of p62 mutant was generated using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies, 210514) according to the manufacturer's protocol. GFP-LC3 plasmid was gifted from Yang-Hee Kim (Sejong University). The nV5-tagged pcDNA α-syn construct was cloned from a pcDNA3-α-syn plasmid, which was a gift of Zhuohua Zhang (Sanford Burnham Prebys Medical Discovery Institute). The mNeonGreen (mNG)-tagged α-syn construct was cloned from a nV5-tagged pcDNA α-syn plasmid into a pmNeonGreen-NT plasmid, a gift of Nathan Shaner (UCSD). VN-α-syn (Addgene, plasmid #89470; http://n2t.net/addgene:89470; RRID:Addgene_89470) and α-syn-VC (Addgene, plasmid #89471; http://n2t.net/addgene:89471; RRID:Addgene_89471) were a gift from Bradley Hyman & Tiago Outeiro (Outeiro et al., 2008). Human p62 sgRNAs were designed from http://crispr.mit.edu/ and cloned into pSpCas9n(BB)-2A-GFP (PX461), a gift from Feng Zhang (Addgene plasmid #48140; http://n2t.net/addgene:48140; RRID:Addgene_48140) (Ran et al., 2013). The ssODN template was designed by adding Mlu I restriction enzyme sequence to select gene-edited clones. Human p62 sgRNA target sequences and ssODN template sequence are described in Extended Data Figure 3-3.
Biotin-switch assay to detect p62 S-nitrosylation
The biotin-switch method for detecting S-nitrosylated proteins was used as described previously (Uehara et al., 2006; Oh et al., 2017). In brief, cells or brain samples were lysed with HENTS buffer. SDS solution (25% w/v) was added to lysed samples to a final concentration of 1% and incubated for 20 min at 50°C with frequent vortexing to facilitate blockade of free thiol groups. After removing excess methyl methanethiosulfonate by acetone precipitation, S-nitrosothiols were reduced to thiols with 20 mm ascorbate. Newly formed thiols were then linked with the sulfhydryl-specific biotinylating reagent N-[6-biotinamido]-hexyl]-3′-(2′pyridyldithio) propionamide (Biotin-HPDP). Unreacted biotin-HPDP was removed by acetone precipitation, and the pellet was resuspended in HENS buffer (100 mm HEPES, pH 7.4, 1 mm EDTA, 0.1 mm neocuproine, 1% SDS), neutralized, and centrifuged to clear any undissolved debris. Five percent of the supernatant was used as the input for the loading control. Biotinylated proteins were pulled down with Streptavidin-Agarose beads and analyzed by immunoblotting for SNO-p62. In some experiments, exogenous NO donors were used to supply NO for S-nitrosothiol formation (Lei et al., 1992).
Immunoblotting
Cells were harvested and lysed in RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS) for 10 min at 4°C. Equivalent protein quantities were subjected to Bolt Bis-Tris Plus (Thermo Fisher Scientific, NW00102BOX) gel electrophoresis and transferred to PVDF membranes (Millipore, IPFL00010). Membranes were blocked with Odyssey blocking buffer (Li-Cor, 927-40 000) for 30 min at room temperature and then probed with primary antibodies against p62 (1:3000, BD Transduction Laboratories, 610833; Proteintech, 18 420-1-AP; or Progen, GP62-C), LC3B (1:3000, Sigma, L7543), actin (1:5000, Millipore, MAB1501), or flotillin-1 (1:2000, BD Transduction Laboratories, 610820). After incubation with secondary antibodies (IR-dye 680LT-conjugated goat anti-mouse [1:20,000; Li-Cor, 926-68 020], IR-dye 800CW-conjugated goat anti-rabbit [1:15,000; Li-Cor, 926-32 211] or IR-dye 800CW-conjugated donkey anti-guinea pig [1:15,000; Li-Cor, 926-32 411]), membranes were scanned with an Odyssey infrared imaging system (Li-Cor). ImageJ program (http://imagej.nih.gov/ij/download.html) and Image Studio (Li-Cor) software were used for densitometric analysis of immunoblots, and quantified results were normalized to loading controls.
Immunoprecipitation and in vitro binding assay
SH-SY5Y cells were cotransfected with GFP-LC3 and WT HA-p62 or non-nitrosylatable HA-p62(C331A). After 1 d, cells were harvested and lysed in 1% Triton X-100 in PBS, and equimolar protein immunoprecipitated for 2 h at 4°C with anti-HA antibody (F-7; Santa Cruz Biotechnology, sc-7392)-conjugated magnetic beads (Dynabeads Protein G; Thermo Fisher Scientific, 10004D). Immunoprecipitants were eluted and subjected to immunoblotting with anti-HA antibody (F-7; 1:3000, Santa Cruz Biotechnology, sc-805) and anti-GFP antibody (1:3000, Cell Signaling, 2956). For the in vitro binding assay, SH-SY5Y cells were transfected with WT HA-p62 or HA-p62(C331A). One day later, transfected cells were lysed in lysis buffer and immunoprecipitated overnight at 4°C with anti-HA antibody (F-7; Santa Cruz Biotechnology, sc-7392)-conjugated magnetic beads (Dynabeads Protein G). Concomitantly, p62 KO MEF cells were treated with 100 nm BafA1 (Sigma, B1793) for 16 h. The next day, immunoprecipitants were washed 3 times with lysis buffer containing 500 mm NaCl. In parallel, BafA1-treated p62 KO MEF cells were harvested and lysed in lysis buffer. Next, equimolar amounts of these cell lysates were mixed with immunoprecipitated HA-p62 or HA-p62(C331A) and incubated for 2 h at 4°C on a rocking table. p62-bound proteins were then eluted and subjected to immunoblotting with anti-HA antibody (F-7; 1:3000, Santa Cruz Biotechnology, sc-805) and anti-LC3 antibody (1:3000, Sigma, L7543).
Immunocytochemistry
SH-SY5Y cells were fixed with 4% PFA for 20 min, washed 3 times with PBS, and blocked (3% BSA, 0.3% Triton X-100 in PBS) for 30 min, all at room temperature. Cells were incubated with anti-LC3B (1:200, Cell Signaling, 2775) and anti-LAMP1 (1:100, Santa Cruz Biotechnology, sc-20011) antibodies overnight at 4°C, followed by incubation with AlexaFluor-488 or -555-conjugated secondary antibodies. Cell images were acquired with a Nikon A1 Confocal Microscope using a 60×/1.40 oil objective (0.5 μm Z stack). Volume projection images were generated by NIS-Elements AR software (Nikon). Colocalization of LC3 and LAMP1 was monitored using the Fiji program colocalization test module (https://imagej.nih.gov/ij/download.html), and the data were analyzed with Pearson's correlation coefficient, as previously described (Oh et al., 2017).
Homology modeling of the LC3–p62 complex
The published structure of the LC3-p62 complex (PDB ID: 2ZJD) (Ichimura et al., 2008) consists of the LC3 protein and a short fragment of p62 (a decamer: G333GDDDWTHLS342) that does not contain Cys331. To build a homology model of the p62 structure including Cys331, we identified sensor protein PhoQ (6A8U_A) (Yoshitani et al., 2019) as a structural template (sequence identity 19% with the p62 region between amino acids 312 and 358) using the FFAS sequence alignment server (Jaroszewski et al., 2011) together with the Modeller program (Webb and Sali, 2017). The initial version of the p62 model was computationally docked to the binding region of LC3 using the existing p62 decamer in the 2ZJD structure. In this modeled complex, the D335DDWTHLS342 region of the p62 model superimposed with the corresponding region of the x-ray crystal structure (2ZJD) with the root-mean-square deviation of 0.53 Å. We then made adjustments to the ϕ/psi angles of Gly333 and Gly334 in the p62 model structure for a better alignment with the p62 decamer from 2ZJD, yielding 0.22 Å root-mean-square deviation between Gly333 to Ser342. The alignment and dihedral angle manipulation were performed with the PyMOL program. For p62(C331A) homology modeling, the model-built structures were minimized and subjected to molecular dynamics simulations (5 ns long) to optimize interatomic distances as well as intermolecular interactions. Molecular simulations were performed with Amber16 molecular dynamics package and ff14SB force field (Maier et al., 2015) in a periodic box of TIP3P water molecules (Jorgensen et al., 1983).
Transmission electron microscopy (TEM)
Cells grown to 60%-70% confluency in Permanox culture plates were fixed in oxygenated, 2.5% glutaraldehyde plus 4% PFA in 0.1 m sodium cacodylate buffer, pH 7.34, for 5 min at 37°C and then fixed for 1 h at 4°C (Johnson, 1987). After washing in 0.1 m sodium cacodylate buffer, the samples were postfixed in buffered 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 h at 4°C, rinsed in the same buffer, and then stained en bloc with 0.5% uranyl acetate overnight at 4°C. Samples were washed in double-distilled (dd)H2O and dehydrated through a graded series of ethanol followed by acetone and infiltrated with LX-112 (Ladd) epoxy resin and polymerized at 60°C. Ultrathin sections (silver) were imaged at 80 kV with a Thermo Fisher Scientific Talos L120C transmission electron microscope, and images were acquired with a CETA 16 M CMOS camera.
Autophagic flux assay
WT or mutant p62(C331A) knock-in cells were plated on 96-well plates (CellCarrier-96 Ultra Microplates, PerkinElmer, 60055550). After 2 d, 0.5 μm DALGreen (Dojindo Molecular Technologies, D675-10) and 0.1 μm DAPRed (Dojindo Molecular Technologies, D677-10) were added to the culture medium and incubated in a 5% CO2 incubator at 37°C. After 6 h, live cells were stained with Hoechst 33258 for 15 min, washed twice with BrightCell NEUMO Photostable Media (Millipore, SCM146), and imaged with an ImageXpress Micro Confocal High Content Imaging System (Molecular Devices). Five randomly selected fields from each of three wells were analyzed per condition, and MetaXpress software (Molecular Devices) was used to quantify DALGreen and DAPRed puncta.
Measurement of vesicular α-syn by vesicle flow cytometry
mNG-α-syn plasmids were transfected into WT or mutant p62(C331A) knock-in cells. After 4 d, CM was collected and centrifuged at 300 × g for 5 min at room temperature; the supernatant was collected and centrifuged again at 2000 × g for 20 min at 4°C. Supernatants were then filtered using an Amicon Ultra-15 Centrifugal Filter (Ultracel-100K, Millipore, UFC910024), and centrifuged a third time at 3220 × g for 20 min at 4°C. The concentrated CM was subsequently analyzed by vesicle flow cytometry using a commercial kit (vFC, Cellarcus Biosciences, CBS4) and flow cytometer (Beckman Coulter CytoFlexS) to quantify mNG-α-syn-positive EVs. The specificity of single vesicle analysis was demonstrated via essential controls and calibration, including serial dilution to exclude coincidence, detergent treatment to determine vesicular nature of the detected events, and fluorescence calibration to support vesicle size estimates and mNG intensities.
Measurement of α-syn by ELISA
CM was collected from V5-α-syn transfected WT cells versus knock-in mutant p62(C331A) cells, and from isogenic corrected hiPSC-DA neurons versus mutant α-syn(A53T) hiPSC-DA neurons cultured in the absence or presence of l-nitro-arginine (l-NAME). CM was centrifuged at 300 × g for 5 min at room temperature; the supernatant was collected and centrifuged again at 2000 × g for 10 min at 4°C. Supernatants were then filtered using 0.2 µm syringe filter and fractionated for EVs by ultracentrifugation at 100,000 × g for 90 min at 4°C. The remaining supernatants were collected as the “EV-free fraction,” and the pellets were collected as the “EV fraction,” which was then washed with cold PBS. The EV and EV-free fractions were mixed with 1% Triton X-100 in PBS buffer at a 1:1 ratio and subjected to ELISA using a LEGEND MAX Human α-Synuclein ELISA Kit (BioLegend, SIG-38974) according to the manufacturer's instructions. Luminescence was quantified using a Luminoskan Ascent plate leader (Thermo Fisher Scientific).
Cell-to-cell spread of α-syn by live in vitro monitoring
WT or CRISPR/Cas9-mediated mutant p62(C331A) knock-in cells were plated on transwell inserts (Thermo Fisher Scientific, Nunc Polycarbonate Cell Culture Inserts, 140620) or coverslips after transfection with hemi-Venus-α-syn (V1S) or hemi-Venus-α-syn (SV2) Venus-α-syn. One day later, the transwell was placed on the coverslip to allow transfer and uptake of α-syn to be monitored. After 3 d, the recipient cells on the coverslip were fixed with 4% PFA for 20 min. Fixed cells were then stained with Hoechst 33258 for 15 min, mounted, and imaged with an ImageXpress Micro Confocal High Content Imaging System. Five randomly selected fields were analyzed per condition using MetaXpress software to quantify Venus-positive puncta and fluorescence intensity.
Quantification and statistical analysis
Experiments were performed in a blinded fashion. All data represent at least three independent experiments, presented as mean ± SEM. Unless otherwise stated, statistical significance between two samples was analyzed by a Student's t test, and for multiple samples by ANOVA with a post hoc Tukey's or Sidak's multiple comparison test (GraphPad Prism).
Results
S-Nitrosylation of p62 in models of PD/LBD
Initially, we examined the ability of the physiological NO donor and nitrosylating compound, S-nitrosocysteine (SNOC), to S-nitrosylate p62 (thus forming SNO-p62). SNOC induced S-nitrosylation of both endogenous and HA-tagged p62 in the SH-SY5Y dopaminergic neuroblastoma cell line, as detected by biotin-switch assay (Uehara et al., 2006; Oh et al., 2017) (Fig. 1A,B). We then asked whether p62 can be S-nitrosylated by endogenous NO in a pathophysiologically relevant manner. To this end, we exposed SH-SY5Y cells to rotenone, a mitochondrial complex I inhibitor known to increase RNS/NO and to be associated with PD (Yao et al., 2004; Ozawa et al., 2013; Ryan et al., 2013; Kumar et al., 2017). A 3 h exposure to rotenone induced significant SNO-p62 levels (Fig. 1C,D). Additionally, we detected endogenous SNO-p62 in vivo in the α-syn-overexpressing mouse model of PD/LBD (Fig. 1E,F) (Rockenstein et al., 2002; Oh et al., 2017).
Figure 1.

S-Nitrosylation of p62 in PD/LBD models. A, B, S-Nitrosylation of exogenous and endogenous p62 by the NO donor SNOC. HA-p62 transfected SH-SY5Y cells (A) or untransfected SH-SY5Y cells (B) were exposed to 100 μm freshly prepared SNOC or old SNOC (from which NO had been dissipated). After 20 min, cell lysates were subjected to the biotin-switch assay. The “ascorbate minus” sample served as a negative control. SNO-p62 and total (input)-p62 detected by immunoblot with anti-p62 antibody. C, S-Nitrosylation of p62 by endogenously generated NO. SH-SY5Y cells exposed to 1 μm rotenone (Rot) in the presence of 1 mm l-arginine and subjected to biotin-switch assay. D, Ratio of SNO-p62/input p62 (lanes 1 and 3). Data are mean ± SEM; n = 3. **p < 0.01, Student's t test. E, S-Nitrosylation of p62 in transgenic PD/LBD mouse model. Brain lysates from 4-month-old control (WT) or human Thy1 promoter-driven α-syn-overexpressing mice subjected to biotin-switch assay. F, Ratio of SNO-p62/input p62 from WT or α-syn-overexpressing mice. Data are mean ± SEM; n = 4 mice in each group. *p < 0.05, Student's t test. G, S-Nitrosylation of p62 in hiPSC-DA neurons. A53T mutant α-syn or isogenic control (corr) hiPSC-DA neurons in the presence or absence of 1 μm rotenone were subjected to the biotin-switch assay. The corr and A53T blots are derived from the same gel. H, Ratio of SNO-p62/input p62 (lanes 1, 3, and 5). For each histogram, data are mean ± SEM; n = 3. **p < 0.01, ***p < 0.001, ANOVA with Tukey's correction.
Next, to test whether S-nitrosylation of p62 occurs in a human disease context, we used hiPSCs bearing a mutation in the gene encoding α-syn that is known to cause PD (A53T SNCA) compared with isogenic, gene-corrected control cells (Soldner et al., 2011). For this purpose, using methods we previously published (Kriks et al., 2011; Ryan et al., 2013), we differentiated the hiPSCs into A9-type dopaminergic neurons (designated hiPSC-DA neurons), the initial cell type in the substantia nigra pars compacta in the brainstem to be affected by PD. We found that A53T hiPSC-DA neurons expressed significantly higher levels of endogenous SNO-p62 than WT isogenic control hiPSC-DA neurons. Notably, exposing isogenic control hiPSC-DA neurons to rotenone also increased SNO-p62 to levels comparable to those observed in untreated A53T neurons (Fig. 1G,H).
S-Nitrosylation of C-terminal p62 affects its binding to LC3
p62 has been shown to have a number of binding partners, and chief among these is LC3, an ATG8 protein family member known to be critical for autophagy substrate selection and autophagosome biogenesis, although LC3 may subserve other cell functions as well (Galluzzi and Green, 2019). LC3 binds to p62 via its LIR motif (Pankiv et al., 2007; Ichimura et al., 2008; Moscat and Diaz-Meco, 2009), and this interaction reportedly increases in PD (Lim et al., 2011). Therefore, we asked whether S-nitrosylation of p62 affects its interaction with LC3. To test this, we coexpressed HA-tagged p62 and GFP-tagged LC3 in SH-SY5Y cells, exposed the cells to SNOC, collected cell lysates, and performed coimmunoprecipitation. We found that SNOC increased the interaction between HA-p62 and GFP-LC3 (Fig. 2A,B). Potentially accounting for this effect, the LIR motif contains a single cysteine residue (Cys331) that resides in a partial motif for protein S-nitrosylation (Stamler et al., 1997). To test this premise, we constructed a C-terminal fragment of p62 (amino acids 230-440) that includes the LIR motif and a total of three cysteine residues. We found that SNOC could S-nitrosylate the C-terminal p62 fragment (Extended Data Fig. 2-1), and mutation of Cys331 to alanine abrogated the reaction by ∼75% (Fig. 2C,D). This finding indicates that Cys331 is the predominant site of S-nitrosylation in the C-terminal region of p62.
Figure 2.
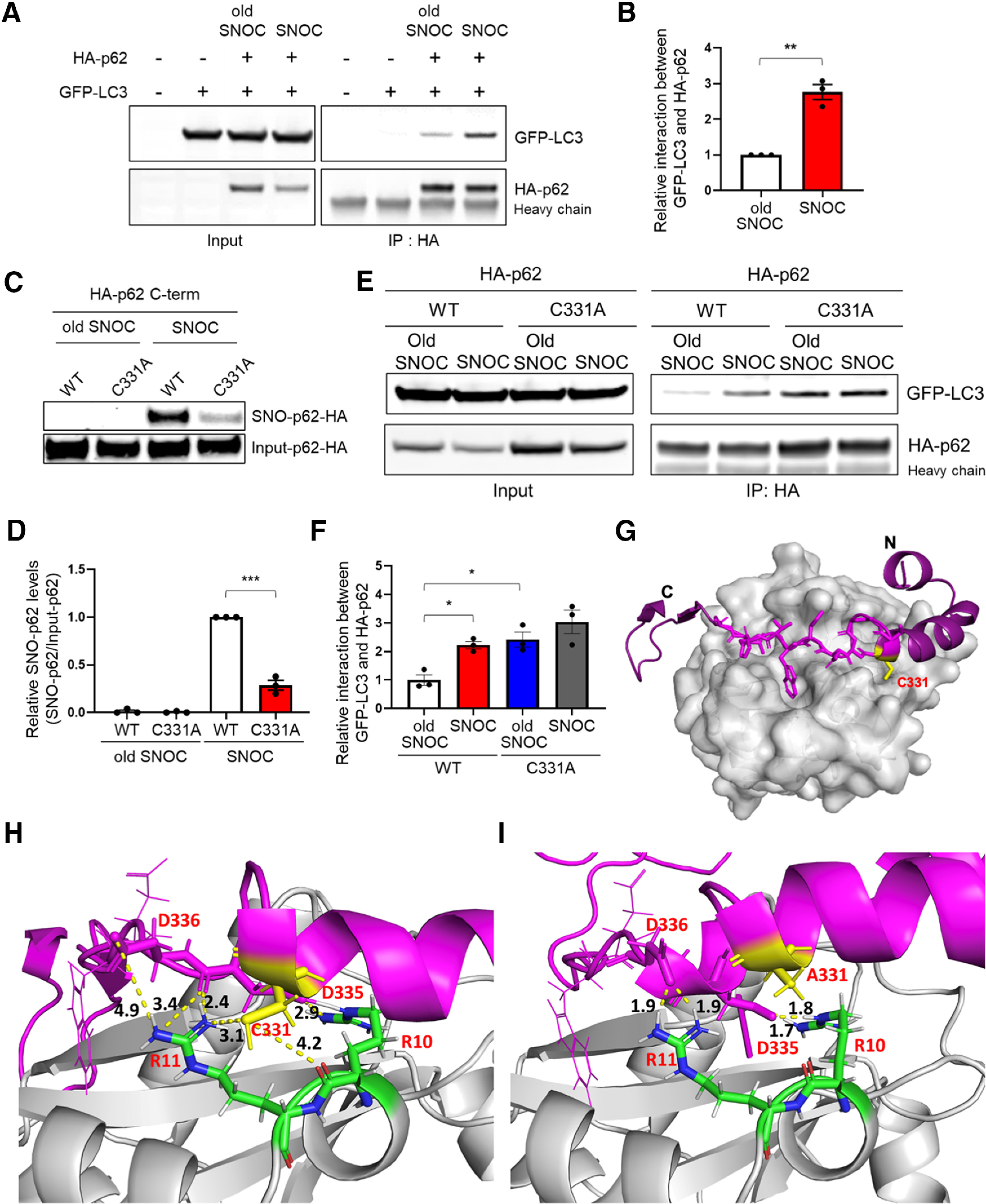
Non-nitrosylatable p62(C331A) mutant mimics SNO-p62 to increase LC3 binding. A, SH-SY5Y cells were transfected with HA-p62 and GFP-LC3. After 1 d, cells were exposed to 200 μm old or fresh SNOC, and 30 min later, lysed and immunoprecipitated with anti-HA antibody. Total cell lysates (Input) and immunoprecipitates (IP) were probed with anti-GFP and anti-HA antibodies on immunoblots. “Heavy chain” shows presence of antibody. B, Coimmunoprecipitated GFP-LC3 levels normalized to immunoprecipitated HA-p62. Data are mean ± SEM; n = 3. **p < 0.01, Student's t test. C, SH-SY5Y cells were transfected with C-terminal HA-tagged WT p62 or mutant p62(C331A). Cells were exposed to 100 μm old or fresh SNOC and biotin-switch assay performed 30 min later. D, Ratio of SNO-p62/input p62 for C-terminal WT HA-p62 or mutant p62(C331A) transfected SH-SY5Y cells. Data are mean ± SEM; n = 3. ***p < 0.001 by Student's t test. E, SH-SY5Y cells were transfected with HA-tagged WT p62 or mutant p62(C331A). The next day, cells were exposed to 200 μm old or fresh SNOC; 30 min later, lysates were prepared and immunoprecipitated with anti-HA antibody. Total cell lysates (Input) and immunoprecipitates (IP) were immunoblotted with anti-GFP and anti-HA antibodies. F, Coimmunoprecipitated GFP-LC3 levels normalized to immunoprecipitated HA-p62. Data are mean ± SEM; n = 3. *p < 0.05, ANOVA with Tukey's correction. G-I, Homology model of the p62-LC3 complex. G, Overall structure of the p62-LC3 complex. Homology modeling of the p62 fragment (magenta) was docked to the crystal structure of LC3 (gray [2ZJD]). A network of Asp-Arg interactions (shown as dotted lines with distance between the atoms shown in Å) is present around p62(Cys331); these interactions facilitate binding of p62 to LC3 (H) or p62(Ala331) (I). See Extended Data Figures 2-1 and 2-2.
S-Nitrosylation of C-terminal p62. A, Schematic domain structures of p62. Arrows indicate cysteine residues in p62. B, SH-SY5Y cells were transfected with HA-tagged full-length p62 or C-terminal p62 (amino acids 230-440). After 1 d, we exposed the cells to 200 μm old or fresh SNOC and performed a biotin-switch assay 30 min later. SNO-HA-p62 and total (input)-HA-p62 detected by immunoblot with anti-p62 antibody. Download Figure 2-1, TIF file (7.6MB, tif) .
Non-nitrosylatable mutant p62(C331A) shows increased binding to endogenous LC3 in vitro. A, Anti-HA antibody immunoprecipitates from lysates were prepared from HEK293 cells 1 d after transfection with HA-tagged WT p62 or mutant p62(C331A). Immunoprecipitated p62 was mixed with lysates from p62 KO MEF cells that had been pre-treated with 100 nm BafA1. An in vitro pull-down assay was then performed, and subsequently immunoblotted with anti-p62 and anti-LC3 antibodies (right). Total cell lysates from BafA1-exposed p62 KO MEF cells are immunoblotted with anti-p62 and anti-LC3 antibodies (left). B, Endogenous LC3 levels, pulled-down from lysates, were normalized to immunoprecipitated HA-p62. Data are mean ± SEM; n = 3, **p < 0.01 by Student's t test. Download Figure 2-2, TIF file (3.5MB, tif) .
Non-nitrosylatable p62(C331A) mutant mimics SNO-p62 interaction with LC3
Next, to test the effect of the p62(C331A) non-nitrosylatable mutant on LC3 interaction, we cotransfected SH-SY5Y cells with GFP-LC3 plus WT or C331A mutant HA-p62, exposed the cells to SNOC (or control “old” SNOC from which NO had been dissipated), and performed immunoprecipitation. We found increased binding of non-nitrosylatable HA-p62(C331A) to GFP-LC3 in the presence of “old” SNOC, similar to levels observed when WT HA-p62 was exposed to SNOC. Moreover, addition of SNOC did not further increase the interaction between GFP-LC3 and HA-p62(C331A) in the coimmunoprecipitate (Fig. 2E,F).
In general, similar in some respects to phosphorylation, S-nitrosylation of a given protein at a critical cysteine residue can either increase or decrease that protein's activity. In an analogous fashion, introduction of a mutation to the target Cys to prevent S-nitrosylation can either mimic or suppress the effect of S-nitrosylation, in this case mimicking the effect of S-nitrosylation just like mutations of phosphorylation site serine to glutamate can mimic the effect of phosphorylation (T. Nakamura et al., 2013; Oh et al., 2017; Otto et al., 2017). In the case of non-nitrosylatable p62(C331A), we found that, by increased binding to LC3, this mutant phenocopies (rather than prevents) the SNO-p62 effect (Palmer et al., 2008; Feng et al., 2013; Oh et al., 2017).
To gain insight into the potential molecular mechanism of the effect of SNO-p62 or p62(C331A) mutation on LC3 binding, we generated an atomic structure of the LC3/p62 complex using homology modeling (Fig. 2G). The model structure shows electrostatic interactions between amino acid residues p62-Asp335 and LC3-Arg10, as well as between p62-Asp336 and LC3-Arg11, consistent with prior findings (Pankiv et al., 2007; Ichimura et al., 2008) that these two Asp residues of p62 are required for efficient binding to LC3. Notably, the side chain of p62-Cys331 is structurally in the vicinity of LC3-Arg11, positioned to perturb the noncovalent interaction of LC3-Arg11 and p62-Asp336 (Fig. 2H,I). Moreover, our model predicts that the interaction between p62-Cys331 and LC3-Arg11 becomes less favorable on post-translational modification of p62-Cys331 by a more hydrophobic SNO group (or replacement by Ala in non-nitrosylatable mutant p62(C331A)). This interference with the interaction between p62-331 and LC3-Arg11 would allow the side chain of LC3-Arg11 to form an enhanced electrostatic interaction with the COO- group of p62-Asp336 and potentially with the backbone of p62-Asp335 (Fig. 2H,I). Therefore, S-nitrosylation of p62-Cys331 (or substitution of Cys331 with an Ala in the non-nitrosylatable mutant) would be expected to increase the binding of p62 to LC3, as observed empirically.
One potential caveat to the conclusions based on our binding assays concerns the possibility that overexpressed GFP-LC3 could skew our assays that analyze binding to SNO-p62 or p62(C331A) mutant protein. Along these lines, while p62 increases under neurodegenerative conditions to levels similar to those used in these experiments (Bartlett et al., 2011), it could be argued that the use of exogenous, labeled LC3 might introduce artifacts. Therefore, we next examined the binding of p62(C331A) to endogenous LC3 in a pull-down assay. For this experiment, we transfected WT HA-p62 or mutant HA-p62(C331A) into HEK293 cells, immunoprecipitated these HA-tagged proteins, and intermixed them in a cell-free system with whole-cell lysates containing endogenous LC3 prepared from p62 KO MEF cells; we used p62 KO MEFs to ensure that the endogenous LC3 was not bound to endogenous p62. Then, the use of BafA1 in this in vitro binding assay allowed us to monitor p62 binding to both LC3-I and LC3-II. We pulled down the mixture with anti-HA antibody to test binding of WT p62 or mutant p62(C331A) to LC3. We observed increased binding of LC3 to p62(C331A) mutant, consistent with the notion that non-nitrosylatable mutant p62 indeed phenocopies the effect of SNO-p62 by manifesting increased binding to LC3 (Extended Data Fig. 2-2).
p62(C331A) knock-in mutant inhibits autophagic flux
Since we found that non-nitrosylatable p62(C331A) mutant mimics the effect of SNO-p62, we generated knock-in p62(C331A) SH-SY5Y cell lines using a CRISPR/Cas-9 homology-direct repair system. This approach allowed us to compare the effect of WT p62 and non-nitrosylatable mutant p62(C331A) in a cell-based model system in the absence of stimulation by NO, which might otherwise affect many other proteins by S-nitrosylation or other mechanisms. Endogenously expressed mutant p62(C331A) should also overcome potential artifacts introduced by transient overexpression of p62 (e.g., possibly forming inclusion bodies) (Bjorkoy et al., 2005). To generate p62(C331A) knock-in cell lines, we used two guide RNAs (gRNA) and an ssODN-containing C331A mutant nucleotide sequence which overlaps an Mlu I restriction enzyme sequence for the selection marker (Extended Data Fig. 3-1A). We confirmed that we had generated three heterozygous p62(C331A) knock-in clones by DNA sequencing, but no cells homozygous for mutant p62 were generated (Extended Data Fig. 3-1B). Accordingly, we found that both WT and mutant mRNAs were concurrently expressed in the cells (Extended Data Fig. 3-1C).
Next, we investigated how expression of endogenous non-nitrosylatable mutant p62 in these knock-in cell lines affected autophagy. Under normal physiological conditions, we found that both p62 and LC3-II levels were greater in the p62(C331A) knock-in cells than in WT (Fig. 3A–C). This result is consistent with the notion that p62(C331A) either increases autophagy or inhibits autophagic flux at a step downstream from LC3 lipidation/autophagosome formation. To distinguish between these possibilities, we exposed the cells to BafA1, which inhibits vacuolar H+ ATPase (V-ATPase) activity, resulting in blockade of fusion between autophagosomes and lysosomes, and hence inhibits autophagic flux (Yamamoto et al., 1998). We found that BafA1 did not affect autophagic flux in mutant p62(C331A) cells, while autophagic flux was impaired in WT cells (Fig. 3A–C). Moreover, BafA1 failed to increase colocalization between LC3 and the lysosomal marker LAMP1 in mutant p62(C331A) cells (Extended Data Fig. 3-2). These results are consistent with the notion that the non-nitrosylatable p62 mutant blocks autophagic flux.
Figure 3.
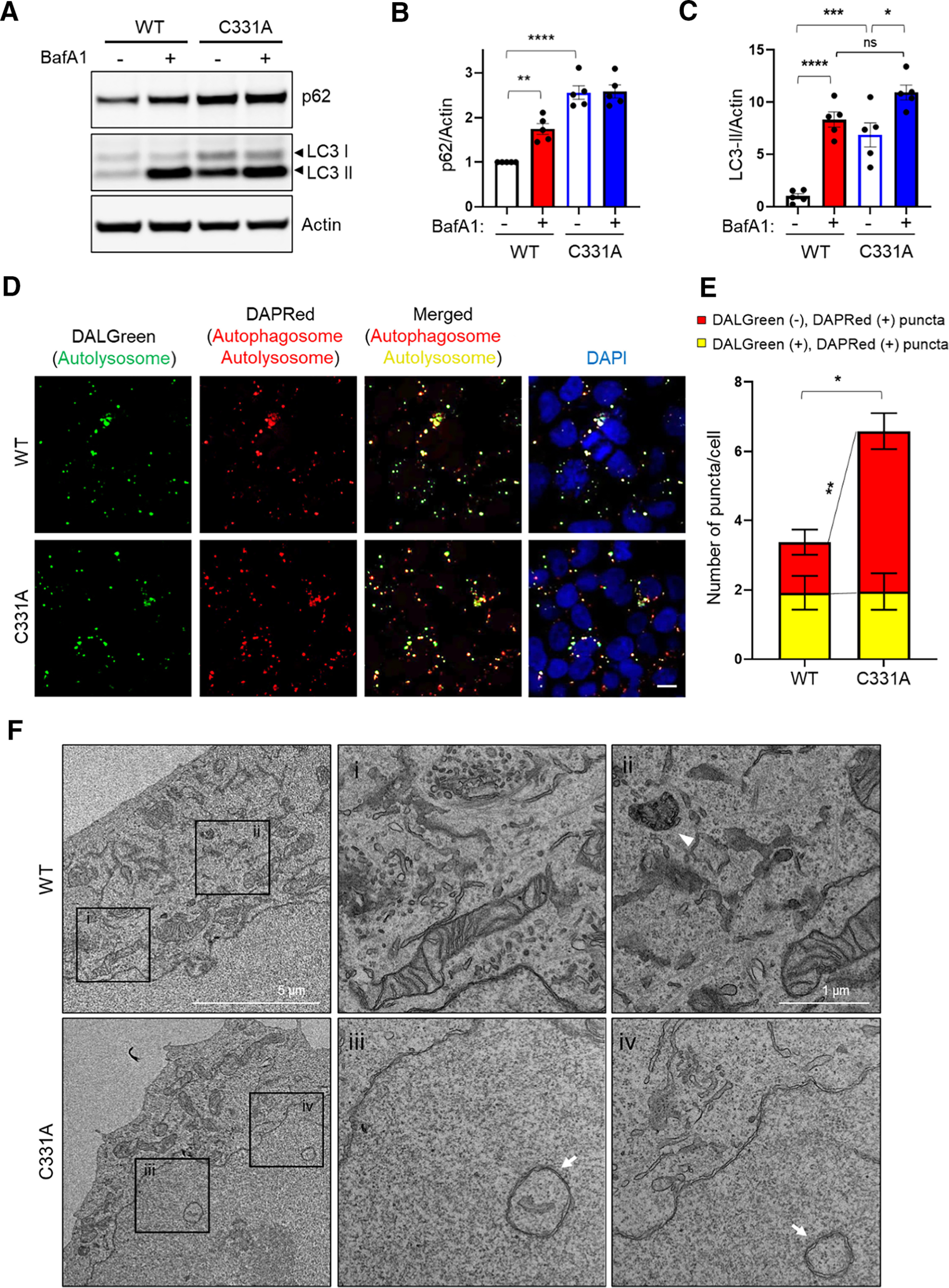
Knock-in p62(C331A) mutant inhibits autophagic flux. A, WT or mutant p62(C331A) knock-in cells exposed to 400 nm BafA1 for 4 h were lysed and immunoblotted with anti-p62, anti-LC3, and anti-actin antibodies. B, C, Immunoblot quantification of relative p62 expression levels normalized to actin (B), and LC3-II/LC3-I ratio (C). Data are mean ± SEM; n = 5. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ANOVA with Tukey's correction. D, Representative confocal images of WT or mutant p62(C331A) in knock-in cells taken 6 h after prestaining with 1 μm DALGreen and 0.1 μm DAPRed dye for 30 min. Scale bar, 20 µm. E, Quantification of DALGreen and DAPRed puncta. Data are mean ± SEM; n = 3. *p < 0.05 (total puncta, WT vs C331A) by Student's t test. **p < 0.01 (DAPRed puncta, WT vs C331A) by ANOVA with Tukey's correction. F, Representative TEM images showing ultrastructure of autophagic vacuoles in WT (boxed areas; i, ii) or mutant p62(C331A) knock-in cells (boxed areas; iii, iv). Arrowhead indicates autolysosome. Arrows indicate autophagosomes. See Extended Data Figures 3-1, 3-2, and 3-3.
CRISPR/Cas9-mediated knock-in of mutant p62(C331A). A, Design of the ssODN template for p62(C331A) mutation. The human p62 nucleotide sequence encoding the cysteine residue at position 331 was substituted for an alanine-coding sequence, which also introduced a new Mlu I restriction enzyme digestion site for screening (top panel). Screening by genomic DNA PCR followed by Mlu I restriction enzyme digestion to identify heterozygous p62(C331A) mutant (bottom panel). B, Sanger sequencing of mutant p62 genomic DNA confirmed Cys (TGT) and Ala (GCG), indicating a heterozygous knock-in mutation. C, Identification of WT p62 and mutant p62(C331A) mRNA by RT-PCR and Mlu I restriction enzyme digestion. Download Figure 3-1, TIF file (10.3MB, tif) .
BafA1 does not affect co-localization of LC3 and lysosomal marker LAMP1 in mutant p62(C331A) cells. A, WT or mutant p62(C331A) knock-in cells exposed to 400 nm BafA1 for 4 h were fixed and immunostained with anti-LC3 and anti-LAMP1 antibodies. Representative and merged images of LC3 and LAMP1 are shown. Scale bar, 20 µm. B, Plot of correlation coefficient for LC3 and LAMP1 co-localization. Data are mean ± SEM; n = 3, ns: not significant by ANOVA with Tukey's correction. Download Figure 3-2, TIF file (11.9MB, tif) .
List of sgRNA, ssODN, and primers. Download Figure 3-3, DOCX file (13KB, docx) .
To confirm this conclusion, we next monitored autophagic flux more directly in mutant p62(C331A) and WT cells under fluorescence microscopy, using DALGreen and DAPRed sensors. These two fluorescent probes are designed to monitor autophagic flux by differentially labeling autophagosomes and autolysosomes. DALGreen fluorescence is enhanced at acidic pH, which can be used to monitor the latter phases of autophagy, indicating formation of autolysosomes. In contrast, DAPRed fluorescence remains at approximately constant brightness throughout the autophagic process, labeling at both the autophagosome stage and at the subsequent autolysosome stage (Iwashita et al., 2018). After loading with both DALGreen and DAPRed, we monitored the staining patterns of the fluorescent probes in WT and mutant p62(C331A) cells. Red and green puncta remained relatively equal in the WT cells, indicating normal autophagic flux. However, autophagic flux was significantly blocked in the mutant p62(C331A) cells as evidenced by increased DAPRed-positive and DALGreen-negative puncta (producing more red puncta); this represents the accumulation of autophagosomes that could not merge with lysosomes to form autolysosomes (Fig. 3D,E). Additionally, TEM confirmed the accumulation of autophagosomes, represented by double-membraned structures, in mutant p62(C331A) cells compared with WT (Fig. 3F) (Mizushima et al., 2010). Together, these results are consistent with the notion that non-nitrosylatable mutant p62(C331A) inhibits autophagic flux.
Mutant p62(C331A)/SNO-p62 promotes α-syn secretion
Recent evidence has suggested that inhibition of autophagic flux might cause the buildup of abnormally folded α-syn in intracellular vesicles, thus promoting their secretion as extracellular vesicles (EVs), resulting in cell-to-cell transmission of aberrant α-syn (Fussi et al., 2018; Minakaki et al., 2018). It has been suggested that α-syn can be secreted by a variety of different mechanisms, including an EV-release pathway via exosomes (Alvarez-Erviti et al., 2011; Danzer et al., 2012), nonclassical exocytosis (Jang et al., 2010; Lee et al., 2013), and exophagy (Ejlerskov et al., 2013). However, the trigger for this release and whether it occurs under pathophysiologically relevant conditions remain unknown. Here, to answer these critical questions, we used knock-in cells with mutant p62 expressed at endogenous levels. This approach afforded us the opportunity to simulate the effects of SNO-p62, as found in PD/LBD brain, by using the p62(C331A) mutant without increasing NO levels, thereby avoiding nitrosylating extraneous proteins and thus introducing confounding factors. In this manner, we determined whether inhibition of autophagic flux in knock-in cells bearing mutant p62(C331A) results in increased α-syn secretion. We examined the levels of extracellular α-syn, p62, and LC3, which have previously been shown to be contained in exosomes (Hessvik et al., 2016). We found that conditioned medium (CM) from p62(C331A) mutant knock-in cells contains high levels of LC3 but fairly low levels of p62, detectable only after immunoprecipitation, in which case the two proteins remained bound to one another (Extended Data Fig. 4-1A).
Next, we investigated the ramifications of this result using mutant p62 knock-in cells transfected with a V5-tagged α-syn. We found that these cells manifested an increase in α-syn secretion (Extended Data Fig. 4-1B). To determine the mechanism(s) for α-syn secretion, we separated CM by ultracentrifugation into EVs and vesicle-free proteins. Interestingly, we detected both p62 and LC3-II, the membrane-bound/lipidated form of LC3, in the EV fraction, but only LC3-I, the cytosolic/free form of LC3, in the supernatant (Fig. 4A). Using an ELISA, we then measured the V5-α-syn levels in each fraction and found significantly increased α-syn in both EVs and supernatants from the mutant p62 knock-in cell CM (Fig. 4B,C). These findings suggest that both EV release and exocytosis play a role in α-syn secretion. However, >99.99% of the secreted V5-α-syn was found in the EV-free fraction from WT or mutant p62 knock-in cell CM. This result is consistent with the notion that only a very minor fraction of secreted α-syn is associated with EVs (Danzer et al., 2012; Shi et al., 2014; Gustafsson et al., 2018).
Figure 4.
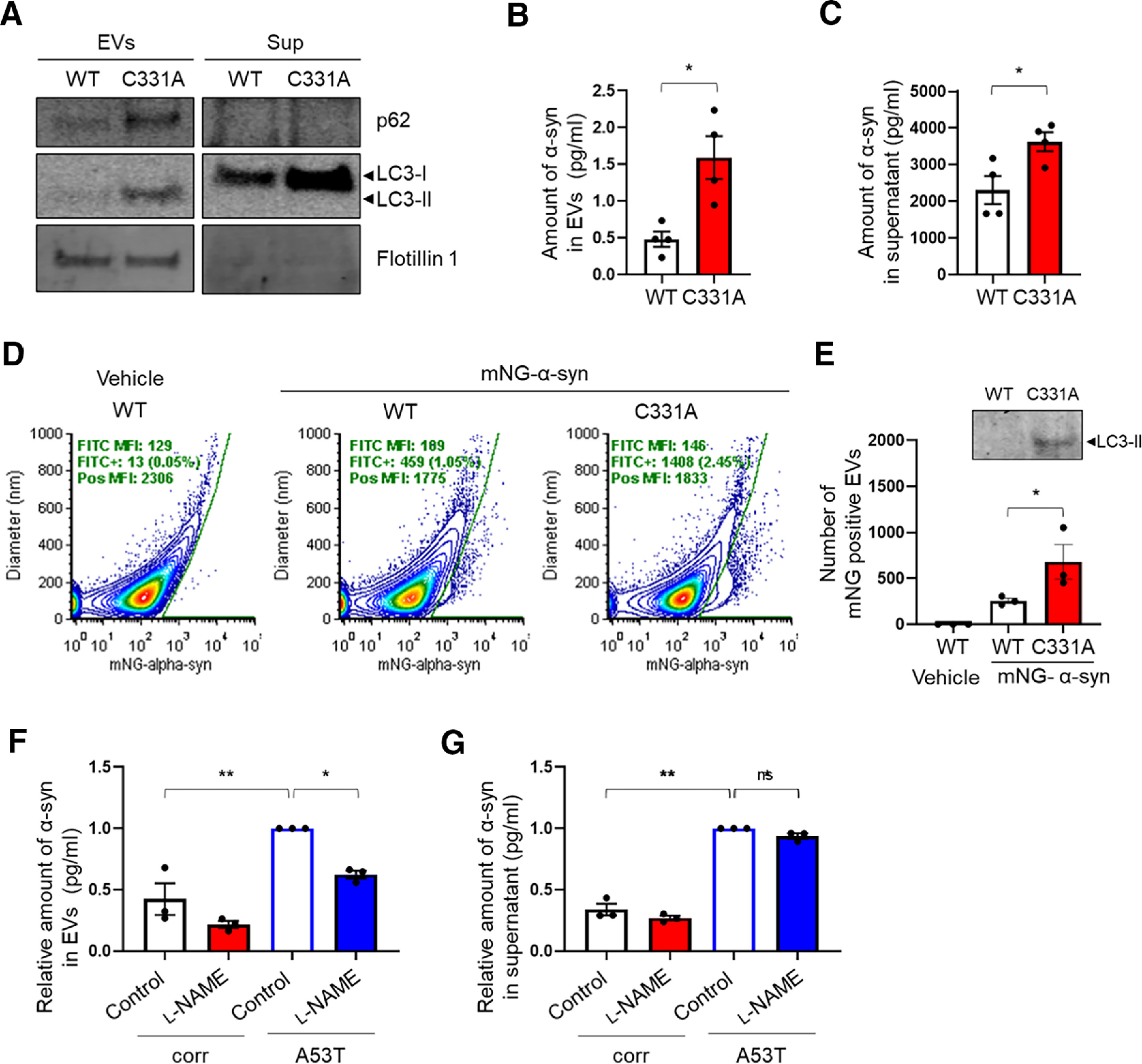
Mutant p62(C331A) increases α-syn and p62/LC3 secretion. A, Immunoblots for p62, LC3, and Flotillin1 in supernatants or EVs obtained after ultracentrifugation from CM collected from V5-α-syn-transfected WT p62 or mutant p62(C331A) knock-in cells. LC3-I was the predominant form in supernatants, while LC3-II was the major form in EVs. B, C, V5-α-syn levels in EVs (B) and supernatants (C) measured by ELISA. Data are mean ± SEM; n = 4. *p < 0.05, Student's t test. D, Vesicle flow cytometry of CM (concentrated with 100 kDa cutoff filter) from mNeonGreen-tagged human α-syn (mNG-α-syn)-transfected WT p62 or mutant p62(C331A) knock-in cells. mNG-α-syn+ events were determined using a gate set on a bivariate plot of diameter versus mNG fluorescence for untransfected WT cell-derived EVs. E, Quantification of mNG-α-syn-positive EVs by flow cytometry. Inset, Immunoblot of CM used for flow cytometry. Data are mean ± SEM; n = 3. *p < 0.05, Student's t test. F, G, α-syn levels by ELISA in EVs (F) and supernatants (G) ultracentrifuge fractions of conditioned media from isogenic control or A53T hiPSC-DA neuronal cultures in the presence or absence of 1 mm l-NAME. Data are mean ± SEM; n = 3. *p < 0.05, **p < 0.01, Sidak's correction. See Extended Data Figures 4-1, 4-2, and 4-3.
Knock-in mutant p62(C331A) increases α-syn and p62/LC3 secretion. A, Immunoblots of co-immunoprecipitated (IP) p62 and LC3 from CM collected from WT p62 or mutant p62(C331A) knock-in cells cultured for 5 d. B, Immunoblot for IP'd anti-V5-α-syn in CM collected from V5-α-syn-transfected WT p62 or mutant p62(C331A) knock-in cells. C, Vesicle flow cytometry of CM (concentrated with a 100 kDa cutoff filter) from mNeonGreen-tagged human α-syn (mNG-α-syn)-transfected WT p62 or mutant p62(C331A) knock-in cells. vFluorRed (vFRed) was used to stain vesicles; vFRed-H refers to fluorescence pulse height and vFRed-A to fluorescence pulse area; VSCC is violet side-scatter. vFRed-triggered events were gated by time (to eliminate background during the first 20 s after sample introduction), by vFRed-H versus vFRed-A (to eliminate background events with pulse widths characteristic of noise), and by VSSC versus vFRed-A (to select events with light scatter and fluorescence characteristic of EVs). Download Figure 4-1, TIF file (5.6MB, tif) .
A53T mutant α-syn hiPSC-DA neurons secrete α-syn. A, B, LC3 levels by immunoblot (A) and α-syn levels by ELISA (B) in CM of A53T hiPSC-DA neuron cultures or isogenic control (corr). Data are mean ± SEM; n = 3, **p < 0.01 by Student's t test. C, Immunoblots of p62, LC3 and Flotilin1 from EV and supernatant ultracentrifuge fractions of CM from isogenic control or A53T hiPSC-DA neuronal cultures. D, E, α-syn levels in EVs (D) and supernatants (E) measured by ELISA. Data are mean ± SEM; n = 3, **p < 0.01 by Student's t test. Download Figure 4-2, TIF file (7.6MB, tif) .
l-NAME decreases S-nitrosylation of p62 in A53T hiPSC-DA neurons. A, S-Nitrosylation of p62 in A53T mutant α-syn hiPSC-DA neurons in the presence or absence of 1 mm l-NAME. Cell lysates were subjected to the biotin-switch assay. B, Ratio of SNO-p62/input p62. Data are mean ± SEM; n = 4, **p < 0.01 by Student's t test. Download Figure 4-3, TIF file (2.4MB, tif) .
Nonetheless, although the EV-associated fraction of extracellular α-syn is small, this mode of transfer has been claimed to mediate the majority of cell-to-cell transfer (Danzer et al., 2012; Gustafsson et al., 2018). To study this process more mechanistically, rather than using ELISA of bulk CM, we turned to directly counting the number of α-syn-positive EVs by flow cytometry. For this purpose, we transfected WT and mutant p62(C331A) knock-in cells with mNeonGreen tagged α-syn (mNG-α-syn); we used mNeonGreen because it arguably manifests the highest quantum yield among available fluorophores, and thus increased our detection limits beyond previously used techniques. We then counted the number of mNG-α-syn-positive EVs in concentrated CM. By vesicle flow cytometry analysis (Nolan and Duggan, 2018), we could detect and quantify mNG-α-syn positive EVs (Fig. 4D; Extended Data Fig. 4-1C). Using this approach, we found a striking increase in mNG-α-syn-positive EVs in mutant p62 knock-in cells relative to WT (Fig. 4E), similar to our ELISA findings (Fig. 4B). We also detected an increase in endogenous LC3 in these EVs by immunoblotting with anti-LC3 antibody (Fig. 4E). Together, these findings support the notion that mutant p62(C331A) increases α-syn secretion via both EV-dependent and EV-independent mechanisms. However, in mutant p62(C331A) knock-in cell CM compared with WT, we found a 3.5-fold increase in EV-mediated α-syn secretion, but only a 1.6-fold increase in EV-independent α-syn secretion, suggesting that EV-mediated secretion increases the most under pathophysiologically relevant conditions, that is, using CRISPR/Cas9 knock-in of mutant p62(C331A) to simulate the formation of SNO-p62 that occurs in PD/LBD brain.
Since we had shown that A53T hiPSC-DA neurons manifested increased levels of SNO-p62 compared with their isogenic, gene-corrected controls (Fig. 1G,H), we next examined CM from these cells to quantify the secreted levels of LC3 and α-syn. We found increased levels of LC3-I by immunoblot (Extended Data Fig. 4-2A) and α-syn by ELISA (Extended Data Fig. 4-2B) in A53T hiPSC-DA neuron CM compared with isogenic control. Additionally, we found increased levels of p62 and LC3-II in the EV fraction of CM from A53T hiPSC-DA neuron compared with isogenic control (Extended Data Fig. 4-2C). Consistent with our mutant p62(C331A) knock-in cell data, we measured higher levels of extracellular α-syn in both the CM supernatant and EV fractions with a ∼3-fold increase for A53T hiPSC-DA neurons compared with isogenic (WT) controls (Extended Data Fig. 4-2D,E). Notably, incubation of A53T hiPSC DA neurons with the NO synthase inhibitor l-NAME not only decreased S-nitrosylation of p62 (Extended Data Fig. 4-3A,B), but also abrogated the increase in release of EV-mediated α-syn into the CM of A53T hiPSC-DA neurons (Fig. 4F,G). Together with the mutant p62(C331A) knock-in data, these results are consistent with the notion that protein S-nitrosylation of p62 is causally related to the increased release of α-syn associated with p62/LC3 from hiPSC-derived neurons bearing a patient-related mutation in α-syn.
Mutant p62(C331A)/SNO-p62 promotes cell-to-cell spread of α-syn
Emerging evidence suggests that secreted/extracellular α-syn is taken up by neighboring cells, acting to seed aggregation of endogenous α-syn in recipient cells (Luk et al., 2009; Nonaka et al., 2010). Here, we investigated whether SNO-p62-triggered α-syn release might contribute to this cell-to-cell spread. To monitor cell-to-cell transmission, we monitored uptake of extracellular α-syn using a dual-cell bimolecular fluorescence complementation (BiFC) platform (Fig. 5A) (Outeiro et al., 2008; Kim et al., 2018). This system is composed of two different α-syn expression vectors: the first expresses V1S, representing α-syn conjugated to the N-terminal fragment of Venus; the other expresses SV2, α-syn conjugated to the C-terminal fragment of Venus. When α-syn aggregates, the two Venus fragments dimerize, and the fluorescence therefore increases. We transfected WT or mutant p62(C331A) knock-in cells with V1S and plated them on transwell inserts as α-syn “donor cells.” We then plated WT cells transfected with the SV2 on a coverslip as the “recipient cells.” The coverslip was placed in the transwell and incubated for 3 d with the insert (Fig. 5B). We found that the recipient WT cells cocultured with mutant p62(C331A) knock-in donor cells in the transwell insert, compared with WT donor cells in the insert, showed significantly increased cell-to-cell transmission of α-syn, as evidenced by more Venus-α-syn-positive puncta as well as total fluorescence intensity (Fig. 5C–E). Collectively, these results indicate that the p62(C331A) mutant, which phenocopies SNO-p62 by inhibiting autophagic flux to increase α-syn secretion, promotes cell-to-cell transmission of α-syn.
Figure 5.

p62(C331A) mutant increases cell-to-cell transmission of α-syn. A, Schematic diagram of dual-cell BiFC system. In this assay, the hemi-Venus-α-syn (V1S) construct expresses α-syn conjugated to the N-terminal fragment of Venus, and α-syn-hemi-Venus (SV2) expresses α-syn conjugated to the C-terminal fragment of Venus. Upon α-syn dimerization/aggregation, the combination of V1S and SV2 emits green fluorescence. B, Scheme of analysis of α-syn cell-to-cell spread using the BiFC system. V1S-transfected WT or mutant p62(C331A) knock-in cells were plated on transwell inserts (donor cells), and SV2-transfected WT cells were plated on coverslips (recipient cells). One day after transfection, the donor and recipient cells were incubated for 3 d. C, Representative BiFC images (top panels) plus Hoechst staining (bottom panels). Scale bar, 20 µm. D, E, Quantification of green Venus-α-syn puncta (D) and relative intensity (E). Data are mean ± SEM; n = 3. *p < 0.05, **p < 0.01, Student's t test.
Pathophysiologically relevant levels of SNO-p62 in human LBD brains
LBD is a prominent form of cognitive dysfunction occurring in the context of PD and is thought to be associated with the spread of misfolded or mutant α-syn. Since we had found that mutant p62(C331A)/SNO-62 inhibits autophagic flux, consequently triggering α-syn release and spread, we reasoned that high levels of SNO-p62 should be present in human LBD brains. Indeed, we detected significantly increased SNO-p62 in human LBD patient postmortem cortical brain samples compared with controls (Fig. 6A; Extended Data Fig. 6-1).
Figure 6.

S-Nitrosylation of p62 in human LBD brains and schema for α-syn cell-to-cell spread. A, S-Nitrosylation of human LBD and control brains. Human brain tissues with relatively short postmortem times (described in Extended Data Fig. 6-1) were subjected to biotin-switch assay. B, Ratio of SNO-p62/input p62 from control or LBD brains. Data are mean ± SEM; n = 5 brains in each group. **p < 0.01, Student's t test. C, Combined data for ratio of SNO-p62/Input p62 from Figures 1D, F, H and 6B (asterisks are from the original figures and retain their original meaning; vertical dotted line at “1” indicates the control for each condition from data shown in each of the other figures listed above). D, Schema of mechanism for S-nitrosylated p62 increasing cell-to-cell transmission of α-syn. Cytoplasmic misfolded α-syn can be directly released via nonclassical exocytosis or exophagy, yielding free α-syn in the extracellular medium (Ejlerskov et al., 2013). Alternatively, misfolded α-syn can be relegated to multivesicular bodies in the cell and then released into the extracellular space via enriched exosomes or EVs (Jang et al., 2010; Alvarez-Erviti et al., 2011; Danzer et al., 2012; Lee et al., 2013). SNO-p62 enhances the release of misfolded α-syn via both of these pathways by enhancing protein buildup because of blockade of autophagy at the stage of autolysosome formation. Released α-syn is then internalized by neighboring cells, and can seed endogenous α-syn to form aggregates (Luk et al., 2009; Nonaka et al., 2010). EV-mediated uptake occurs through direct endocytosis of the vesicle (Lee et al., 2008), while EV-independent uptake of α-syn may be mediated by binding to a receptor on the cell surface of the cell with subsequent endocytosis and endosome formation of the complex (Mao et al., 2016). See Extended Data Figure 6-1.
List of human brains used for assessing SNO-p62 expression. Download Figure 6-1, DOCX file (14.4KB, docx) .
Next, we measured the relative levels of SNO-p62 in human LBD brain versus control (i.e., SNO-p62/total p62 ratio from biotin-switch and immunoblots), using a technique that we have previously published (Uehara et al., 2006; Cho et al., 2009) (Fig. 6B). Critically, we found that the relative level of SNO-p62 in human LBD brain was similar (∼2-fold increase) to that found in our cell-based models and α-syn transgenic mice in which we characterized the effects of SNO-p62 on autophagic flux inhibition, and consequent α-syn release and spread (Fig. 6C). This finding indicates that S-nitrosylated p62 is present in human LBD brain at pathophysiologically relevant levels to the disease process.
Discussion
Increasing evidence suggests that autophagic dysfunction contributes to the accumulation of abnormal proteins and damaged organelles, as well as to synaptic disruption in various neurodegenerative and neuropsychiatric disorders, including PD/LBD, AD, Huntington's disease, and amyotrophic lateral sclerosis (Banerjee et al., 2010; Tomoda et al., 2020). Recently, autophagy-related dysfunction has also been posited to lead to secretion of misfolded proteins, thus contributing to abnormal protein spreading to other regions of the brain (Menzies et al., 2017; Vargas et al., 2019). However, the mechanism triggering autophagy-mediated secretion and spread of misfolded proteins remains unknown. Here, we provide the first mechanistic insight into this pathway, presenting evidence that S-nitrosylation of the autophagic adaptor protein p62 mediates inhibition of autophagic flux, leading to the intracellular buildup and consequent secretion of misfolded α-syn both directly into the extracellular medium by exocytosis and via EVs (Fig. 6D). Moreover, we find that EV-related secretion, rather than exocytosis, appears to be the predominant form of α-syn released from neurons that underlies subsequent cell-to-cell transmission despite the fact that most of the released α-syn is not confined to EVs.
Concerning the trigger for this autophagic dysfunction, abnormal protein release, and cell-to-cell spread, our group and others have shown that buildup of misfolded/mutated proteins, including α-syn, or exposure to mitochondrial toxins, such as the complex I inhibitors, rotenone, N-methyl-4-phenylpyridinium, or 1, 1′-dimethyl-4,4′-bipyridinium (paraquat) plus manganese ethylene bisdithiocarbamate (maneb), increases the intracellular level of RNS/NO (Lim et al., 2011; Cui et al., 2012; Ryan et al., 2013). In the present study, we find that this increase in NO results in S-nitrosylation of p62 (forming SNO-p62), consequently increasing p62 binding to LC3 and impairing autophagic flux. These events occur in both transgenic mouse models of PD as well as in a human context in hiPSC-DA neurons and in LBD postmortem brains. Additionally, as α-syn continues to misfold, it can trigger further increases in NO/RNS (Ryan et al., 2013), resulting in dramatically enhanced formation of SNO-p62, forming a positive feedforward of pathogenic events. Notably, the NO synthase blocker l-NAME abrogated the increase in α-syn secretion from A53T mutant hiPSC-derived neurons, further supporting our hypothesis that formation of SNO-p62 because of nitrosative stress is largely responsible for α-syn release.
Our data also show that a critical cysteine residue susceptible to S-nitrosylation in p62 is located at Cys331. This cysteine is located near the LIR motif of the protein, which, as the name implies, binds to LC3 located on newly formed autophagosomes (Moscat et al., 2016). Interestingly, we show that CRISPR/Cas9-mediated knock-in of mutant p62(C331A) mimics the effects of SNO-p62, increasing its interaction with LC3 and inhibiting autophagic flux. Collectively, these results indicate that abnormally high levels of NO inhibit autophagic flux at least in part by S-nitrosylating p62 at Cys331.
The process of autophagic flux comprises autophagosome formation, fusion with lysosomes, and degradation of proteins and organelles (Zhang et al., 2013). During autophagosome formation, the Atg8/LC3 family of proteins is integrated into the autophagosome membrane and recruits various receptor proteins, including p62, NBR1, and OPTN (Birgisdottir et al., 2013). Recent studies have also found that LC3-II plays a critical role in autophagosome-lysosome fusion via recruitment of PLEKHM1 (Pleckstrin homology domain-containing family M member 1) to the outer membrane of the autophagosome (Nguyen et al., 2016); PLEKHM1 directly interacts with LC3-II via its LIR motif to facilitate autophagosome-lysosome fusion (McEwan et al., 2015; S. Nakamura and Yoshimori, 2017). Along these lines, our autophagic flux assay demonstrated in p62(C331A) knock-in cells that autophagosomes could not merge with lysosomes, thereby inhibiting autophagic flux (Fig. 3D,E). Mechanistically, we posit that the increased affinity of SNO-p62 [and also its mimetic, p62(C331A)] for LC3-II competitively interferes with PLEKHM1 recruitment by LC3-II to the outer membrane of the autophagosome to inhibit fusion and thus autolysosome formation.
α-syn is known to be the primary component of LBs. Recent evidence suggests that aberrant forms of α-syn are not only a hallmark of PD/LBD but can also contribute to neuronal dysfunction and death. Moreover, pathologic spread of α-syn may mediate disease progression in PD and LBD (Minakaki et al., 2018; Henderson et al., 2019). Importantly, autophagy can facilitate degradation of misfolded α-syn, thus preventing α-syn secretion into the extracellular space. Accordingly, inhibition of autophagy, through the silencing of Atg5 or by the addition of BafA1, has been reported to increase α-syn secretion via EVs (e.g., exosomes) as well as by exocytosis, contributing to α-syn-induced toxicity (Fussi et al., 2018; Minakaki et al., 2018). Our new data demonstrate that, in the context of PD/LBD pathology, formation of SNO-p62 inhibits autophagic flux to increase α-syn secretion and spreading. Importantly, we also provide evidence for SNO-p62 in human LBD brains at levels similar to those found in our transgenic mouse and cell-based models, indicating that sufficient nitrosylation of p62 occurs in the human brain to trigger inhibition of autophagic flux with consequent release and spread of misfolded α-syn.
In the present study, we provide compelling evidence that overall secretion of aberrant α-syn increases following inhibition of autophagic flux by SNO-p62, but only a minor amount is found within EVs. Prior results, however, suggested that propagation of α-syn pathology is predominantly mediated through EVs (Danzer et al., 2012; Shi et al., 2014; Gustafsson et al., 2018). As a possible explanation for this observation, it has been reported that EV-associated α-syn is preferentially internalized into cells (Danzer et al., 2012; Shi et al., 2014; Gustafsson et al., 2018). Critically, we also found by quantitative analysis that SNO-p62-mediated α-syn spread is greater in the EV-associated fraction compared with the EV-free fraction. Together, these data indicate that EVs are taken up more efficiently into neighboring cells, but this occurs only following the key triggering event of S-nitrosylation of p62.
In conclusion, we found aberrantly elevated levels of SNO-p62 in both cell-based and animal models of PD/LBD, including hiPSC-DA neurons. Most importantly, SNO-p62 is also greatly elevated in human LBD patient brains. We describe a unique cellular mechanism by which SNO-p62 inhibits autophagic flux, resulting in increased α-syn secretion and cell-to-cell transmission. Thus, SNO-p62 may represent a novel therapeutic target for α-synucleinopathy, including PD and LBD, as well as other neurodegenerative diseases known to manifest both α-syn and pathologically high levels of NO because of genetic or environmental mechanisms.
Footnotes
This work was supported in part by National Institutes of Health Grants R01 NS086890, DP1 DA041722, R01 DA048882, RF1 AG057409, R35 AG071734, and R01 AG056259 to S.A.L., and R01 AG061845, R61 NS122098, and RF1 NS123298 to T.N. Additional funding was provided by a Distinguished Investigator Award from the Brain & Behavior Research Foundation to S.A.L. and the Michael J. Fox Foundation to S.A.L. and T.N. We thank Scott R. McKercher (The Scripps Research Institute) for critical reading of the manuscript; Robert Rissman (UC San Diego) for providing human brain tissue; and Scott Henderson, Kimberly Vanderpool, and Theresa Fassel (Microscopy Core Facility at The Scripps Research Institute) for expert assistance with the electron microscopy experiments.
The authors declare no competing financial interests.
References
- Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM (2011) Lysosomal dysfunction increases exosome-mediated α-synuclein release and transmission. Neurobiol Dis 42:360–367. 10.1016/j.nbd.2011.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Beal MF, Thomas B (2010) Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci 33:541–549. 10.1016/j.tins.2010.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert DR, Simonsen A, Finley KD (2011) p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 7:572–583. 10.4161/auto.7.6.14943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif-crucial for selective autophagy. J Cell Sci 126:3237–3247. 10.1242/jcs.126128 [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614. 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-Nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324:102–105. 10.1126/science.1171091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR (2012) A progressive mouse model of Parkinson's disease: the Thy1-aSyn ('Line 61') mice. Neurotherapeutics 9:297–314. 10.1007/s13311-012-0104-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM (2004) S-Nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304:1328–1331. 10.1126/science.1093891 [DOI] [PubMed] [Google Scholar]
- Cui W, Zhang Z, Li W, Mak S, Hu S, Zhang H, Yuan S, Rong J, Choi TC, Lee SM, Han Y (2012) Unexpected neuronal protection of SU5416 against 1-methyl-4-phenylpyridinium ion-induced toxicity via inhibiting neuronal nitric oxide synthase. PLoS One 7:e46253. 10.1371/journal.pone.0046253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell-to-cell transmission of α-synuclein oligomers. Mol Neurodegener 7:42. 10.1186/1750-1326-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejlerskov P, Rasmussen I, Nielsen TT, Bergstrom AL, Tohyama Y, Jensen PH, Vilhardt F (2013) Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem 288:17313–17335. 10.1074/jbc.M112.401174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Wang C, Chen Q, Chen H, Ren B, Li X, Zuo J (2013) S-Nitrosylation of phosphotransfer proteins represses cytokinin signaling. Nat Commun 4:1529. 10.1038/ncomms2541 [DOI] [PubMed] [Google Scholar]
- Fussi N, Hollerhage M, Chakroun T, Nykanen NP, Rosler TW, Koeglsperger T, Wurst W, Behrends C, Hoglinger GU (2018) Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis 9:757. 10.1038/s41419-018-0816-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Green DR (2019) Autophagy-independent functions of the autophagy machinery. Cell 177:1682–1699. 10.1016/j.cell.2019.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson G, Loov C, Persson E, Lazaro DF, Takeda S, Bergstrom J, Erlandsson A, Sehlin D, Balaj L, Gyorgy B, Hallbeck M, Outeiro TF, Breakefield XO, Hyman BT, Ingelsson M (2018) Secretion and uptake of α-Synuclein via extracellular vesicles in cultured cells. Cell Mol Neurobiol 38:1539–1550. 10.1007/s10571-018-0622-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson MX, Trojanowski JQ, Lee VM (2019) α-Synuclein pathology in Parkinson's disease and related α-synucleinopathies. Neurosci Lett 709:134316. 10.1016/j.neulet.2019.134316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessvik NP, Overbye A, Brech A, Torgersen ML, Jakobsen IS, Sandvig K, Llorente A (2016) PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell Mol Life Sci 73:4717–4737. 10.1007/s00018-016-2309-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M (2008) Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 283:22847–22857. 10.1074/jbc.M802182200 [DOI] [PubMed] [Google Scholar]
- Iwashita H, Sakurai HT, Nagahora N, Ishiyama M, Shioji K, Sasamoto K, Okuma K, Shimizu S, Ueno Y (2018) Small fluorescent molecules for monitoring autophagic flux. FEBS Lett 592:559–567. 10.1002/1873-3468.12979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ (2010) Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 113:1263–1274. 10.1111/j.1471-4159.2010.06695.x [DOI] [PubMed] [Google Scholar]
- Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A (2011) FFAS server: novel features and applications. Nucleic Acids Res 39:W38–W44. 10.1093/nar/gkr441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TJ (1987) Glutaraldehyde fixation chemistry: oxygen-consuming reactions. Eur J Cell Biol 45:160–169. [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. 10.1063/1.445869 [DOI] [Google Scholar]
- Kim C, Spencer B, Rockenstein E, Yamakado H, Mante M, Adame A, Fields JA, Masliah D, Iba M, Lee HJ, Rissman RA, Lee SJ, Masliah E (2018) Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating α-synuclein transmission and neuroinflammation. Mol Neurodegener 13:43. 10.1186/s13024-018-0276-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Peter M, Hofmann K (2010) Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12:836–841. 10.1038/ncb0910-836 [DOI] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480:547–551. 10.1038/nature10648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat Genet 18:106–108. 10.1038/ng0298-106 [DOI] [PubMed] [Google Scholar]
- Kumar R, Jangir DK, Verma G, Shekhar S, Hanpude P, Kumar S, Kumari R, Singh N, Sarovar Bhavesh N, Ranjan Jana N, Kanti Maiti T (2017) S-Nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci Rep 7:44558. 10.1038/srep44558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuusisto E, Parkkinen L, Alafuzoff I (2003) Morphogenesis of Lewy bodies: dissimilar incorporation of α-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol 62:1241–1253. 10.1093/jnen/62.12.1241 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ (2008) Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int J Biochem Cell Biol 40:1835–1849. 10.1016/j.biocel.2008.01.017 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ (2013) Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 45:e22. 10.1038/emm.2013.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA (1992) Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 8:1087–1099. 10.1016/0896-6273(92)90130-6 [DOI] [PubMed] [Google Scholar]
- Lim J, Kim HW, Youdim MB, Rhyu IJ, Choe KM, Oh YJ (2011) Binding preference of p62 towards LC3-ll during dopaminergic neurotoxin-induced impairment of autophagic flux. Autophagy 7:51–60. 10.4161/auto.7.1.13909 [DOI] [PubMed] [Google Scholar]
- Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT (2013) K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 51:283–296. 10.1016/j.molcel.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM (2009) Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci USA 106:20051–20056. 10.1073/pnas.0908005106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11:3696–3713. 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Gonçalves RA, Liang Y, Zhang S, et al. (2016) Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374. 10.1126/science.aah3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I (2015) PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57:39–54. 10.1016/j.molcel.2014.11.006 [DOI] [PubMed] [Google Scholar]
- Menzies FM, et al. (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93:1015–1034. 10.1016/j.neuron.2017.01.022 [DOI] [PubMed] [Google Scholar]
- Minakaki G, et al. (2018) Autophagy inhibition promotes SNCA/α-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 14:98–119. 10.1080/15548627.2017.1395992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140:313–326. 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna C, Rizza S, Maiani E, Piredda L, Filomeni G, Cecconi F (2016) To eat, or NOt to eat: S-nitrosylation signaling in autophagy. FEBS J 283:3857–3869. 10.1111/febs.13736 [DOI] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT (2009) p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137:1001–1004. 10.1016/j.cell.2009.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Karin M, Diaz-Meco MT (2016) p62 in cancer: signaling adaptor beyond autophagy. Cell 167:606–609. 10.1016/j.cell.2016.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Yoshimori T (2017) New insights into autophagosome-lysosome fusion. J Cell Sci 130:1209–1216. 10.1242/jcs.196352 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA (2013) Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron 78:596–614. 10.1016/j.neuron.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215:857–874. 10.1083/jcb.201607039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan JP, Duggan E (2018) Analysis of individual extracellular vesicles by flow cytometry. Methods Mol Biol 1678:79–92. 10.1007/978-1-4939-7346-0_5 [DOI] [PubMed] [Google Scholar]
- Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M (2010) Seeded aggregation and toxicity of α-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem 285:34885–34898. 10.1074/jbc.M110.148460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh CK, Sultan A, Platzer J, Dolatabadi N, Soldner F, McClatchy DB, Diedrich JK, Yates JR, Ambasudhan R, Nakamura T, Jaenisch R, Lipton SA (2017) S-Nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiPSC-based Parkinson's disease models. Cell Rep 21:2171–2182. 10.1016/j.celrep.2017.10.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto NM, McDowell WG, Dickey DM, Potter LR (2017) A glutamate-substituted mutant mimics the phosphorylated and active form of guanylyl cyclase-A. Mol Pharmacol 92:67–74. 10.1124/mol.116.107995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ (2008) Formation of toxic oligomeric α-synuclein species in living cells. PLoS One 3:e1867. 10.1371/journal.pone.0001867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Komatsubara AT, Nishimura Y, Sawada T, Kawafune H, Tsumoto H, Tsuji Y, Zhao J, Kyotani Y, Tanaka T, Takahashi R, Yoshizumi M (2013) S-Nitrosylation regulates mitochondrial quality control via activation of parkin. Sci Rep 3:2202. 10.1038/srep02202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer ZJ, Duncan RR, Johnson JR, Lian LY, Mello LV, Booth D, Barclay JW, Graham ME, Burgoyne RD, Prior IA, Morgan A (2008) S-Nitrosylation of syntaxin 1 at Cys(145) is a regulatory switch controlling Munc18-1 binding. Biochem J 413:479–491. 10.1042/BJ20080069 [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145. 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E (2002) Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res 68:568–578. 10.1002/jnr.10231 [DOI] [PubMed] [Google Scholar]
- Ryan SD, et al. (2013) Isogenic human iPSC Parkinson's model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155:1351–1364. 10.1016/j.cell.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, Garcia-Arencibia M, Rose C, Luo S, Underwood BR, Kroemer G, O'Kane CJ, Rubinsztein DC (2011) Complex inhibitory effects of nitric oxide on autophagy. Mol Cell 43:19–32. 10.1016/j.molcel.2011.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Liu C, Cook TJ, Bullock KM, Zhao Y, Ginghina C, Li Y, Aro P, Dator R, He C, Hipp MJ, Zabetian CP, Peskind ER, Hu SC, Quinn JF, Galasko DR, Banks WA, Zhang J (2014) Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson's disease. Acta Neuropathol 128:639–650. 10.1007/s00401-014-1314-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shults CW (2006) Lewy bodies. Proc Natl Acad Sci USA 103:1661–1668. 10.1073/pnas.0509567103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, et al. (2003) α-Synuclein locus triplication causes Parkinson's disease. Science 302:841. 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, Zhang L, Guschin D, Fong LK, Vu BJ, Meng X, Urnov FD, Rebar EJ, Gregory PD, Zhang HS, Jaenisch R (2011) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146:318–331. 10.1016/j.cell.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) α-Synuclein in Lewy bodies. Nature 388:839–840. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ, Lipton SA, Sucher NJ (1997) (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18:691–696. 10.1016/S0896-6273(00)80310-4 [DOI] [PubMed] [Google Scholar]
- Tanji K, Odagiri S, Miki Y, Maruyama A, Nikaido Y, Mimura J, Mori F, Warabi E, Yanagawa T, Ueno S, Itoh K, Wakabayashi K (2015) p62 deficiency enhances α-synuclein pathology in mice. Brain Pathol 25:552–564. 10.1111/bpa.12214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda T, Yang K, Sawa A (2020) Neuronal autophagy in synaptic functions and psychiatric disorders. Biol Psychiatry 87:787–796. 10.1016/j.biopsych.2019.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA (2006) S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441:513–517. 10.1038/nature04782 [DOI] [PubMed] [Google Scholar]
- Vargas JY, Grudina C, Zurzolo C (2019) The prion-like spreading of α-synuclein: from in vitro to in vivo models of Parkinson's disease. Ageing Res Rev 50:89–101. 10.1016/j.arr.2019.01.012 [DOI] [PubMed] [Google Scholar]
- Webb B, Sali A (2017) Protein structure modeling with MODELLER. Methods Mol Biol 1654:39–54. 10.1007/978-1-4939-7231-9_4 [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23:33–42. 10.1247/csf.23.33 [DOI] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA (2004) Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA 101:10810–10814. 10.1073/pnas.0404161101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitani K, Ishii E, Taniguchi K, Sugimoto H, Shiro Y, Akiyama Y, Kato A, Utsumi R, Eguchi Y (2019) Identification of an internal cavity in the PhoQ sensor domain for PhoQ activity and SafA-mediated control. Biosci Biotechnol Biochem 83:684–694. 10.1080/09168451.2018.1562879 [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. 10.1002/ana.10795 [DOI] [PubMed] [Google Scholar]
- Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H (2002) p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 160:255–263. 10.1016/S0002-9440(10)64369-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XJ, Chen S, Huang KX, Le WD (2013) Why should autophagic flux be assessed? Acta Pharmacol Sin 34:595–599. 10.1038/aps.2012.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S-Nitrosylation of C-terminal p62. A, Schematic domain structures of p62. Arrows indicate cysteine residues in p62. B, SH-SY5Y cells were transfected with HA-tagged full-length p62 or C-terminal p62 (amino acids 230-440). After 1 d, we exposed the cells to 200 μm old or fresh SNOC and performed a biotin-switch assay 30 min later. SNO-HA-p62 and total (input)-HA-p62 detected by immunoblot with anti-p62 antibody. Download Figure 2-1, TIF file (7.6MB, tif) .
Non-nitrosylatable mutant p62(C331A) shows increased binding to endogenous LC3 in vitro. A, Anti-HA antibody immunoprecipitates from lysates were prepared from HEK293 cells 1 d after transfection with HA-tagged WT p62 or mutant p62(C331A). Immunoprecipitated p62 was mixed with lysates from p62 KO MEF cells that had been pre-treated with 100 nm BafA1. An in vitro pull-down assay was then performed, and subsequently immunoblotted with anti-p62 and anti-LC3 antibodies (right). Total cell lysates from BafA1-exposed p62 KO MEF cells are immunoblotted with anti-p62 and anti-LC3 antibodies (left). B, Endogenous LC3 levels, pulled-down from lysates, were normalized to immunoprecipitated HA-p62. Data are mean ± SEM; n = 3, **p < 0.01 by Student's t test. Download Figure 2-2, TIF file (3.5MB, tif) .
CRISPR/Cas9-mediated knock-in of mutant p62(C331A). A, Design of the ssODN template for p62(C331A) mutation. The human p62 nucleotide sequence encoding the cysteine residue at position 331 was substituted for an alanine-coding sequence, which also introduced a new Mlu I restriction enzyme digestion site for screening (top panel). Screening by genomic DNA PCR followed by Mlu I restriction enzyme digestion to identify heterozygous p62(C331A) mutant (bottom panel). B, Sanger sequencing of mutant p62 genomic DNA confirmed Cys (TGT) and Ala (GCG), indicating a heterozygous knock-in mutation. C, Identification of WT p62 and mutant p62(C331A) mRNA by RT-PCR and Mlu I restriction enzyme digestion. Download Figure 3-1, TIF file (10.3MB, tif) .
BafA1 does not affect co-localization of LC3 and lysosomal marker LAMP1 in mutant p62(C331A) cells. A, WT or mutant p62(C331A) knock-in cells exposed to 400 nm BafA1 for 4 h were fixed and immunostained with anti-LC3 and anti-LAMP1 antibodies. Representative and merged images of LC3 and LAMP1 are shown. Scale bar, 20 µm. B, Plot of correlation coefficient for LC3 and LAMP1 co-localization. Data are mean ± SEM; n = 3, ns: not significant by ANOVA with Tukey's correction. Download Figure 3-2, TIF file (11.9MB, tif) .
List of sgRNA, ssODN, and primers. Download Figure 3-3, DOCX file (13KB, docx) .
Knock-in mutant p62(C331A) increases α-syn and p62/LC3 secretion. A, Immunoblots of co-immunoprecipitated (IP) p62 and LC3 from CM collected from WT p62 or mutant p62(C331A) knock-in cells cultured for 5 d. B, Immunoblot for IP'd anti-V5-α-syn in CM collected from V5-α-syn-transfected WT p62 or mutant p62(C331A) knock-in cells. C, Vesicle flow cytometry of CM (concentrated with a 100 kDa cutoff filter) from mNeonGreen-tagged human α-syn (mNG-α-syn)-transfected WT p62 or mutant p62(C331A) knock-in cells. vFluorRed (vFRed) was used to stain vesicles; vFRed-H refers to fluorescence pulse height and vFRed-A to fluorescence pulse area; VSCC is violet side-scatter. vFRed-triggered events were gated by time (to eliminate background during the first 20 s after sample introduction), by vFRed-H versus vFRed-A (to eliminate background events with pulse widths characteristic of noise), and by VSSC versus vFRed-A (to select events with light scatter and fluorescence characteristic of EVs). Download Figure 4-1, TIF file (5.6MB, tif) .
A53T mutant α-syn hiPSC-DA neurons secrete α-syn. A, B, LC3 levels by immunoblot (A) and α-syn levels by ELISA (B) in CM of A53T hiPSC-DA neuron cultures or isogenic control (corr). Data are mean ± SEM; n = 3, **p < 0.01 by Student's t test. C, Immunoblots of p62, LC3 and Flotilin1 from EV and supernatant ultracentrifuge fractions of CM from isogenic control or A53T hiPSC-DA neuronal cultures. D, E, α-syn levels in EVs (D) and supernatants (E) measured by ELISA. Data are mean ± SEM; n = 3, **p < 0.01 by Student's t test. Download Figure 4-2, TIF file (7.6MB, tif) .
l-NAME decreases S-nitrosylation of p62 in A53T hiPSC-DA neurons. A, S-Nitrosylation of p62 in A53T mutant α-syn hiPSC-DA neurons in the presence or absence of 1 mm l-NAME. Cell lysates were subjected to the biotin-switch assay. B, Ratio of SNO-p62/input p62. Data are mean ± SEM; n = 4, **p < 0.01 by Student's t test. Download Figure 4-3, TIF file (2.4MB, tif) .
List of human brains used for assessing SNO-p62 expression. Download Figure 6-1, DOCX file (14.4KB, docx) .