

SUMMARY
A unifying feature of the RAS superfamily is a conserved GTPase cycle by which these proteins transition between active and inactive states. We demonstrate that autophosphorylation of some GTPases is an intrinsic regulatory mechanism that reduces nucleotide hydrolysis and enhances nucleotide exchange, altering the on/off switch that forms the basis for their signaling functions. Using X-ray crystallography, nuclear magnetic resonance spectroscopy, binding assays, and molecular dynamics on autophosphorylated mutants of H-RAS and K-RAS, we show that phosphoryl transfer from GTP requires dynamic movement of the switch II region and that autophosphorylation promotes nucleotide exchange by opening the active site and extracting the stabilizing Mg2+. Finally, we demonstrate that autophosphorylated K-RAS exhibits altered effector interactions, including a reduced affinity for RAF proteins in mammalian cells. Thus, autophosphorylation leads to altered active site dynamics and effector interaction properties, creating a pool of GTPases that are functionally distinct from their non-phosphorylated counterparts.
In brief
Johnson et al. identify a group of GTPases that undergo autophosphorylation via a conserved active site substitution. Using RASA59T as a prototypical autophosphorylating GTPase, they show that autophosphorylation is a stable post-translational modification that inhibits GTP hydrolysis and enhances nucleotide exchange. Despite promoting cell transformation, autophosphorylation inhibits K-RAS effector interactions.
Graphical Abstract
INTRODUCTION
Proteins of the RAS GTPase superfamily coordinate cellular behaviors in response to extracellular signals. The current paradigm is that these signaling hubs are inactive when bound to guanine nucleotide diphosphate (GDP) and active when bound to guanine nucleotide triphosphate (GTP), with cycling between these states regulated by intrinsic mechanisms of GTP hydrolysis and nucleotide exchange or by mechanisms facilitated by GTPase activating proteins (GAPs) and guanine nucleotide exchange proteins (GEFs) (Cherfils and Zeghouf, 2013). When bound to GTP, the active sites of GTPases undergo conformational changes that allow interaction and activation of effector proteins. Furthermore, GTPases undergo a number of post-translational modifications (PTMs) that regulate their dynamics, subcellular localization, and activity. Dysregulation of the GTPase cycle by mutations in either GTPases or their regulators can lead to cancer, neurological diseases, or developmental syndromes (Qu et al., 2019; Simanshu et al., 2017). While this aspect of GTPases is well studied, there is less known about how pathogenic mutations and PTMs interact to modulate the function of GTPases.
Much of what we know about the core biochemical properties of GTPases comes from early studies of oncogenic RAS encoded by the Harvey and Kirsten RAS tumor viruses. Intriguingly, while the viral proteins exhibit a high degree of sequence identity with their cellular homologs, substitution of alanine at amino acid 59 for threonine (A59T) is the only shared difference, suggesting that the change in biochemical function resulting from this substitution provides a selective advantage for viral tumorigenesis. The Thr59 substitution is buried in the active site of RAS and undergoes autophosphorylation when RAS is bound to GTP (Shih et al., 1980). For this reason, H-RAS was initially thought to be a serine/threonine kinase. We now know that the primary biochemical activity of cellular RAS proteins is GTP hydrolysis, which is defective in the viral oncoproteins mutations at codon 12 (G12R in v-H-RAS and G12S in v-K-RAS).
The biological advantage of Thr59 in v-RAS, and the relevance of its associated phosphorylation, is still not understood. Most members of the small GTPase superfamily have alanine at residue 59, however, a small number of family members carry an autophosphorylation motif at this position (Table 1). In some of these, autophosphorylation has been observed, including H-RAS (Chung et al., 1993), RALA (Frech et al., 1990), RAB1B (Touchot et al., 1989), and elongation factor Tu (Cool et al., 1990). Thus, autophosphorylation appears to be possible when either a threonine or serine nucleophile is present in the active site at this position, suggesting that autophosphorylation is a conserved enzymatic function in small GTPases.
Table 1.
Small GTPases with autophosphorylation motif
| Subfamily | GTPase | Switch II (G3 motif) | PDB | Biochemical features | Biological and disease associations | Cancer associated mutationsa |
|---|---|---|---|---|---|---|
|
| ||||||
| RAS | K-RAS A59T | LDTTGQEEYSAM RDQYMRTG | - | High-exchange activity, low-GTPase activity, predominantly GTP-bound | Oncogene | Numerous |
| RAS | RASD1 | LDTSGNHPFPA xxRLSILTG | - | Not studied | Circadian clock synchronization with light-dark cycles, neurogenesis, steroid response, tumor suppression, adipogenesisb | S80P Ewing Sarcoma |
| RAS | RASD2 | LDTSGNHPFPA MRRLSILTG | - | Predominantly GTP-boundc | Huntington disease, Parkinson’s disease, neurological function, mitophagyd | S71Y Breast |
| RAS | DIRAS1 | TDTTGSHQFPAM QRLSISKG | 2GF0 | High-exchange activity, low-GTPase activity, predominantly GTP-bound, GAP-sensitivee | Tumor suppression, Autophagy, Epilepsyf | - |
| RAS | DIRAS2 | TDTTGSHQFPAM QRLSISKG | 2ERX | Low-GTPase activity, predominantly GTP-bound, GAP-sensitiveg | Attention Deficit Hyperactive Disorder (ADHD), Tumor suppression, Autophagyh | T63M Colon and Stomach |
| RAB | RAB40A | WDTSGQGRFCT IFRSYSRGA | - | Not studied | Protein ubiquitination, cell deadhesioni | S71del Lung |
| RAB | RAB40B | WDxSGQGRFCTI FRSYSRGA | - | Not studied | Regulation of invadopodia, Gastric cancerj | - |
| RAB | RAB40C* | WDTSGQGRFCT IFRSYSRxA | - | Hydrolysis and GAP-sensitivity conservedk | Protein ubiquitination, noncanonical WNT-signaling, endocytosis, lipid droplet homeostasisl | - |
| RHO | RND1 | WDTSGSPYYDN VRPLCYSDS | 2CLS | Lacks intrinsic GTPase activity, has increased nucleotide exchange in favor of GTPm | Cell motility, smooth muscle contractility, Neurogenesis, Tumor suppressionn | |
| RHO | RND2 | WDTSGSxYYDN VRPLAYPDS | - | Not studied | Neurogenesiso | S63del Cervix |
| RHO | RND3 | WDTSGSPYYDNV RPLSYPDS | 1M7B | Lacks intrinsic GTPase activity and GAP-resistantp | Cell motility, smooth muscle contractility, Neurogenesis, Tumor suppression and cell cycle inhibition, oncogeneq | - |
| ARF | ARL6 | FDMSGQGxYRXL WEHYYKxx | 2H57 | Predominantly GDP boundr | Ciliopathies, Bardet-Biedl syndrome, BBSome regulations | - |
Consensus sequence was generated by an 85% similarity cutoff, instead of a 95% cutoff for all others.
Bouchard-Cannon et al., 2018; Cha et al., 2013; Cheng et al., 2004; Kemppainen and Behrend, 1998; Tang et al., 2019.
Substitutions at Ala59 of K-, H-, and N-RAS—including threonine (A59T) and glutamate (A59E) substitutions—represent approximately 0.2% of oncogenic RAS mutations, implicating autophosphorylation in RAS oncogenicity, yet the molecular and cellular properties of these mutants are poorly characterized (Haigis, 2017). In this study, we sought to understand how autophosphorylation changes H- and K-RAS function. Furthermore, our studies suggest that other small GTPases with an autophosphorylation motif share this unique mechanism of activation.
RESULTS
Autophosphorylation alters the GTPase cycle of K-RAS
In a colon cancer cell line carrying endogenous K-RASA59T (SNU-175), we noted that K-RAS protein exhibited an electrophoretic shift (Figure 1A), as did purified protein expressed in E. coli (Figure 1B). We confirmed by mass spectroscopy of purified protein that Thr59 was phosphorylated, indicating that phosphorylation is an intrinsic property of K-RASA59T (Figures S1A and S1B), and subsequently showed in two ways that autophosphorylation resulted in the electrophoretic shift. First, K-RASA59E—a phosphomimetic substitution—migrated at a similar speed as the upper K-RASA59T band (Figure 1B). Second, the upper K-RASA59T band could be removed by lambda phosphatase treatment, albeit inefficiently and only after the protein was denatured (Figure 1C). This was an important observation because it suggested that phosphorylated Thr59 is protected by the tertiary structure of K-RAS. Using the electrophoretic shift as a readout, we measured the kinetics of autophosphorylation of purified K-RASA59T (Figures 1D and S1C), which produced results similar to historical studies on H-RASA59T, affirming that autophosphorylation occurs via an intramolecular reaction (John et al., 1988).
Figure 1. T59 phosphorylation alters K-RAS cycling.

(A) Western blot of K-RAS in LIM1215 (WT) and SNU-175 (A59T) cells.
(B) SDS-PAGE of purified K-RAS with different residues at position 59.
(C) K-RASA59T incubated in the presence of GDP (lane 2) or GTP (lane 3). Dephosphorylation of K-RASA59T incubated with GTP with or without lambda phosphatase (LMP) for different times at 30°C after pre-incubation of protein at 30°C (lanes 4–6) or 95°C (lanes 7–9). Band quantification is shown below.
(D) Summary of K-RASA59T autophosphorylation kinetics alone or with regulatory proteins. Autophosphorylation rate constants (k) were calculated as v•[E0]−1.
(E) Serum-starved and hEGF-stimulated SNU-175 cells showing phosphorylated ERK1/2 (pERK) or K-RAS. Replicates are labeled above the gel. Band quantification was normalized to the average “0” replicate and is shown on the right.
(F) Intrinsic and GAP-catalyzed hydrolysis (k) for K-RAS and mutants. Each bar or data point represents the average k (n = 3–4).
(G) Exchange of GDP-loaded K-RAS4B for mant-GTP or mant-GDP (n = 3–4).
* denotes p < 0.05 and ** denotes p < 0.005 by Student’s t test. Error bars represent ± SD.
The relationship between GTP hydrolysis and autophosphorylation is not defined. One possibility is that RAS autophosphorylation is a passive byproduct of the hydrolysis reaction and that Thr59 acts as a nucleophile that substitutes for the catalytic water present in the active site. If this were true, we would expect GAP, which binds to RAS proteins and enhances hydrolysis, to likewise enhance the rate of autophosphorylation. To the contrary, we found that autophosphorylation was not enhanced by GAP (Figures 1D and S1C; Table S1). Thus, autophosphorylation is not a byproduct of hydrolysis, but is sensitive to active site conformation and is mechanistically independent of GTP hydrolysis. Furthermore, enhancement of nucleotide exchange by addition of SOS1 did not significantly affect the rate of autophosphorylation (Figures 1D and S1C; Table S1). Because active site dynamics of K-RAS are modulated by growth signals received by cells, we tested whether the levels of autophosphorylated K-RASA59T (K-RASA59Tp) changed in response to epidermal growth factor (EGF) or insulin stimulation (Figures 1E and S1D; Table S1). We observed no changes in the relative amount of K-RASA59Tp in these experiments, suggesting that autophosphorylation is not dynamically regulated by upstream signals.
A59T and A59E are cancer-associated mutations of RAS. Because cancer-associated mutations hyperactivate RAS proteins by altering the GTPase cycle, we examined how mutation and/or Thr59 phosphorylation affect cycling. We found that residue 59 mutants of K-RAS exhibited impaired intrinsic and GAP-mediated GTP hydrolysis, like the strongly activated mutant K-RASG12D (Figure 1F and Table S1). The effects on nucleotide exchange were more complex (Figures 1G, S1E, and S1F; Table S1). First, mutation and phosphorylation strongly enhanced the rate of intrinsic exchange. Second, K-RASA59T remained sensitive to GEF-induced exchange, while K-RASA59Tp was less sensitive and K-RASA59E was entirely resistant. Third, while K-RASA59T/E showed no preference for GDP or GTP exchange, K-RASA59Tp preferentially exchanged GDP for GTP.
Our experiments demonstrate that K-RASA59Tp and K-RASA59E have a significant impact on the dynamics of nucleotide exchange and hydrolysis, which leads to functional activation by increasing the steady-state levels of GTP-bound K-RAS. We noted that many of the GTPases in Table 1 share the biochemical characteristics of residue 59 mutants, including high rates of nucleotide exchange coupled with low rates of GTP hydrolysis and dominance of the active GTP-bound state. Furthermore, mutation of Ser64 of RhoE/RND3 to alanine rescues GTP hydrolysis and limits intrinsic nucleotide exchange of this GTPase (Foster et al., 1996). Altogether, these observations indicate that autophosphorylation may be a normal regulatory function of these GTPases.
Switch II mobility promotes autophosphorylation
Our data suggest that the mechanism of autophosphorylation is not a byproduct of GTP hydrolysis. A previous crystal structure of H-RASG12V/A59T (PDB: 521P) shows an active site unfavorable for autophosphorylation because Thr59 is oriented away from GTP (Figure S2A) (Krengel et al., 1990). To help explain the mechanism of phosphorylation, we solved two crystal structures of H-RASA59T bound to a non-hydrolyzable GTP analog (GppNHp) (Figure S2B and Table 2). We chose H-RAS because it favors a closed active site, while K-RAS favors an open active site (Johnson et al., 2019; Parker et al., 2018). We reasoned that active site closure was necessary for γ-phosphate de-solvation to favor phosphoryl transfer to Thr59. Our crystal structures revealed that the A59T mutation disconnects switch II from both the switch I and the P loop and, in agreement with the observed reduction in GTP hydrolysis, displaces the nucleophilic water out of the active site (Figures 2A and S2C). Thr59 rearranges the active site through a clash between its methyl group and Tyr64 while its hydroxyl group forms optimal H-bonds with the backbone of Thr35 and Gln61 (black bonds (1) in Figure 2A). This results in switch II shifting away from the active site (black and gray bonds (2) in Figure 2A) and breaking the H-bond between Gly60 and Gly12 in the P loop (gray bonds (3) in Figure 2A) that normally stabilizes active site closure upon GTP binding. Next, the Thr35-Thr59 interaction breaks the β sheet H-bond between Thr58 and Ile36, causing destabilization of switch I. The β sheet Thr58-Ile36 interaction is characteristic of wild-type H-RAS, but is absent in other fast-exchange mutants of RAS (Johnson et al., 2019).
Table 2.
Data collection and structure refinement statistics
| PDB ID | H-RAS A59T GppNHp crystal 1 7JIF | H-RAS A59T GppNHp crystal 2 7JIG | H-RAS A59E GppNHp 7JIH | H-RAS A59E GDP 7JII | K-RAS A59E GDP 7KMR |
|---|---|---|---|---|---|
|
| |||||
| Data collection and processing | |||||
|
| |||||
| Resolution range | 33.49–1.757 (1.82–1.757) | 33.74–2.322 (2.405–2.322) | 29.17–1.989 (2.06–1.989) | 36.14–1.532 (1.587–1.532) | 55.57–1.51 (1.564–1.51) |
| Space group | P3221 | P3221 | P1211 | P1 | P321 |
|
| |||||
| Unit cell dimensions | |||||
|
| |||||
| a, b, c (Å) | 39.567 39.567 158.302 | 38.964 38.964 159.048 | 55.675 49.832 57.32 | 38.593 37.946 56.254 | 78.4 78.4 55.57 |
| α, β, γ (°) | 90 90 120 | 90 90 120 | 90 117.812 90 | 107.362 107.185 95.319 | 90 90 120 |
| Total reflections | 45757 | 41434 | 50242 | 60862 | 311129 |
| Unique reflections | 15450 | 6642 | 19261 | 43101 | 31274 |
| Multiplicity | 4.3 | 6.5 | 3.3 | 2.2 | 9.9 (10.4) |
| completeness (%) | 69.07 (7.99) | 94.04 (61.02) | 80.00 (9.58) | 65.82 (6.14) | 99.91 (99.93) |
| I/sigma(I) | 29.0 (1.9) | 9.5 (1.5) | 6.6 (0.5) | 38.3 (9.2) | 14.39 (1.22) |
| Wilson B-factor | 24.6 | 29.12 | 33.07 | 16.83 | 20.83 |
| Rmerge | 0.032 | 0.076 | 0.083 | 0.02 | 0.158 |
| Rmeas | 0.036 | 0.082 | 0.098 | 0.028 | 0.167 |
| Rpim | 0.016 | 0.031 | 0.051 | 0.018 | 0.054 |
| CC1/2 | (1) | (0.765) | (0.597) | (0.952) | (0.998) |
| CC* | (1) | (0.931) | (0.865) | (0.988) | (0.999) |
|
| |||||
| Crystal structure refinement | |||||
|
| |||||
| Used reflections | 10481 (119) | 6204 (393) | 15420 (182) | 28213 (263) | 31274 (3066) |
| Reflections Rfree | 1047 (11) | 611 (43) | 1575 (22) | 2005 (16) | 1542 (168) |
| Rwork | 0.1708 (0.4107) | 0.1807 (0.2688) | 0.1960 (0.3317) | 0.1577 (0.2410) | 0.1615 (0.2452) |
| Rfree | 0.2456 (0.6982) | 0.2625 (0.3255) | 0.2553 (0.3940) | 0.2021 (0.2879) | 0.1967 (0.2950) |
|
| |||||
| Atom information | |||||
|
| |||||
| macromolecules | 1339 | 1294 | 2425 | 2640 | 1357 |
| ligands | 39 | 33 | 78 | 62 | 28 |
| solvent | 155 | 61 | 102 | 336 | 136 |
| Protein residues | 168 | 168 | 322 | 336 | 172 |
| RMS (bonds, Å) | 0.007 | 0.007 | 0.008 | 0.007 | 0.013 |
| RMS (°) | 1.21 | 1.19 | 1.29 | 1.23 | 2.03 |
|
| |||||
| Ramachandran (%) | |||||
|
| |||||
| favored | 98.78 | 95.12 | 96.73 | 97.56 | 98.2 |
| allowed | 1.22 | 4.27 | 2.94 | 2.13 | 1.8 |
| outliers | 0 | 0.61 | 0.33 | 0.3 | 0.0 |
| Clashscore | 4.77 | 6.56 | 4.71 | 4.16 | 0.0 |
|
| |||||
| B-factors | |||||
|
| |||||
| Average | 23.26 | 30.85 | 35.24 | 21.06 | 28.99 |
| macromolecules | 22.74 | 30.89 | 35.32 | 20.39 | 28.16 |
| ligands | 20.45 | 26.19 | 33.74 | 17.14 | 20.97 |
| solvent | 28.40 | 32.58 | 34.57 | 27.06 | 38.87 |
Parentheses represent highest resolution averages.
Figure 2. Conserved mechanism of autophosphorylation in GTPases.

(A) Active site comparison of H-RASA59T crystal 1 (green) and WT H-RAS (PDB: 3K8Y, gray). Black and gray dashed lines are H-bonds made in H-RASA59T and WT structures. Thr59 is shown in yellow.
(B) Active site similarities between H-RASA59T and other small GTPases. Black sticks are from the H-RASG12V/A59T structure (PDB: 521P) with an alternate Thr59 orientation.
(C) Bond distances made during simulation of K-RASA59T bound to GTP. Inset on the right shows measured bonds.
(D) Frequency distribution of nucleophile to γ-phosphate distances from MD simulations.
(E) Proposed mechanism of autophosphorylation. The N-H group of switch II represents the backbone carbonyl of Gln61.
Our H-RASA59T structures suggest that movement of Thr59 toward the γ-phosphate of GTP, a necessary step for autophosphorylation, is possible (Figure S2D). Comparison of available crystal structures of GTP-bound small GTPases showed similar orientations and distances between their corresponding residue and the γ-phosphate, supporting their potential to undergo autophosphorylation (Figures 2B and S2E). Likewise, purified K-RASA59S, K-RASA59H, and K-RASA59Y proteins demonstrate a weak capacity to undergo autophosphorylation under the same reaction conditions as K-RASA59T (Figure S2F). Nevertheless, it was unclear how residue 59 reaches the γ-phosphate. To answer this question, we used molecular dynamics (MD) simulations to monitor active site motions in K-RASA59T and other GTPases. Throughout the K-RASA59T simulation, Thr59 closely associated with the γ-phosphate of GTP while maintaining its interaction with Gln61 (Figure 2C). In contrast, the Thr35-Thr59 interaction was commensurate with Thr59 pulling away from GTP and Q61 in switch II (Figure 2C). Consistently, simulations with the other GTPases showed strong association of residue 59 with GTP that was regulated by residue 35 in switch I (Figures 2D and S2G).
Gly12 co-mutations can enhance the steady-state phosphorylation of H-RASA59T in NIH 3T3 cells by enhancing the rate of autophosphorylation (Gibbs et al., 1984). We tested the potentially competitive nature of the Thr59-Thr35 interaction with autophosphorylation by examining the ratio of phosphorylated protein in the presence of different co-mutations. We found that K-RASG12V/A59T, and the fast-exchange mutations (i.e., K-RASG13D/A59T and K-RASA59T/A146T), increased autophosphorylation relative to K-RASA59T and K-RASG12R/A59T (Figure S2H). These results are consistent with a role for switch I in dictating autophosphorylation because these mutants favor opening of the active site (Johnson et al., 2019; Poulin et al., 2019). In contrast, Arg12 directly interacts with switch II in crystal structures of K-RASG12R, which suggests that in K-RASG12R/A59T, movement of Thr59 toward the γ-phosphate is unfavourable (Hobbs et al., 2020).
Our MD analysis validates that serine and/or threonine nucleophiles in the proper active site position are poised for phosphoryl transfer after switch II movement toward the γ-phosphate (Figure 2E). The simple mechanism presented in Figure 2E is consistent with the observed effect on K-RAS autophosphorylation by GAP and SOS1. GAP makes significant interactions with switch II, essentially locking it into a particular conformation for duration of the complex and preventing movement of Thr59 to GTP (Figure 1D). Likewise, while the crystal structure of H-RAS bound to SOS1 shows that the catalytic domain of SOS1 would move Thr59 toward GTP (Boriack-Sjodin et al., 1998), which is a major determinant of autophosphorylation rate (Chung et al., 1993), our data suggest that the hydroxyl nucleophile of Thr59 is activated and carefully oriented by intramolecular interactions with switch II.
A “Mg2+ extraction” mechanism for hyperexchange
We next addressed how alterations at position 59 promote high rates of nucleotide exchange (hyperexchange) and why K-RASA59Tp and K-RASA59E have exchange properties that are different from K-RASA59T. First, we explored the dynamics of GDP-bound K-RASA59T and K-RASA59E using 1H-15N heteronuclear single quantum coherence (HSQC) nuclear magnetic resonance (NMR) spectroscopy in reference to wild-type K-RAS (Figure 3A). Consistent with our nucleotide exchange data, we observed chemical shift perturbation in backbone amide protons around the active sites of both K-RASA59T and K-RASA59E. Overall, Glu59 displayed a more global effect on K-RAS backbone chemical shifts than Thr59, indicating a more significant effect on K-RAS tertiary structure. Notably, while the switch I region (residues 28–40) showed the largest chemical shift changes in K-RASA59T, the same region experienced further peak broadening in K-RASA59E, suggesting that a conformational coupling between Thr59 and the active site in switch I is enhanced by Glu59 substitution. These chemical shift changes are consistent with Thr59 motions and associated local changes around this residue in switch II, and are consistent with our crystal structures and MD simulations.
Figure 3. The molecular mechanism of hyperexchange.

(A) Chemical shift perturbations induced by mutation of Ala59 plotted by K-RAS residue. 1H-15N cross-peaks for K-RASA59E or K-RASA59T are referenced to WT K-RAS-GppNHp. K-RASA59T versus WT chemical shift perturbations are shown as negative values to compare changes to K-RASA59E. Resonances detected in WT, but not in mutants, due to perturbations or severe peak broadening (p/b), are shown as dashed bars with an arbitrary value of 1 ppm. Residues not assigned for WT are marked with X. Horizontal lines show threshold of mean chemical perturbation plus one SD for K-RASA59T (gray, dashed) and K-RASA59E (solid black).
(B) Comparison of K-RASA59E crystal structures bound to GppNHp (purple) and GDP (pink) to WT H-RAS bound to GDP or GppNHp (gray). Dashed circle indicates Glu59.
(C) Molecule B of H-RASA59E bound to GDP (purple). Glu59 rearranges the active site to favor GDP release, unlike the WT reference structure (PDB: 4OBE, gray). Gray dashes are shared H-bonds between structures. Colored dashes are described in text.
(D) Structure of GDP-bound K-RASA59E lacking Mg2+ compared to the WT reference structure. Dashed circle shows junction between switch I and helix 1.
(E) Glu59 and Asp57 stabilize a pro-exchange active site in K-RASA59E.
(F) Chemical shift perturbations (A) were mapped to the K-RAS surface. Color intensity represents magnitude of chemical shift changes relative to WT as defined by the scale. Resonances detected in WT, but not in the mutants, due to perturbation or broadening are shown in purple. Residues without assignments are colored in gray.
(G) Frequency distribution of bond distances during MD simulations of K-RASA59T and K-RAST59p bound to GDP.
(H) Cluster analysis of K-RAST59p MD simulation.
(I) Frequency distribution of bond distances during MD simulations of phosphorylated GTPases bound to GDP.
To better understand the structural changes induced by autophosphorylation and the chemical shift perturbations seen in K-RASA59E, we solved crystal structures of both H-RASA59E and K-RASA59E in different nucleotide-bound states (Figure S2B and Table 2). Our crystals represented different stages of nucleotide exchange induced by Glu59. As demonstrated by H-RA-SA59E, Glu59 repels switches I and II from the active site and weakens inter-switch β sheet interactions, regardless of the nucleotide (dashed circle in Figure 3B; Figure S3A). Moreover, Glu59 alters active site solvation, which in turn influences Mg2+ and GDP stability (gray dashes in Figure 3C). First, repulsion of switch I by Glu59 breaks a canonical Tyr32-Tyr40 H-bond observed in wild-type H-RAS (magenta dashes at (1) in Figure 3C), allowing Glu59 to interact with Mg2+ and drawing switch II toward Lys16 and the P loop (orange dashes at (2) in Figure 3C). This creates a network that stabilizes active site opening and draws Lys16 and Mg2+ away from GDP (magenta dashes at (3) in Figure 3C).
The impact of Mg2+ release is demonstrated by K-RASA59E, which appeared to be an intermediate of intrinsic exchange and which was similar to the crystal structure of K-RASA146T, a mutant that also exhibits fast GEF-independent nucleotide exchange (Poulin et al., 2019). The active site of K-RASA59E is completely open and lacks Mg2+, with switches I and β2 peeled away from the globular domain to form a novel β sheet (orange arrow in Figure 3D). Mg2+ dissociation allows β3 to extend toward switch II and Glu59 and Asp57 to make salt bridges with the P loop and K16, respectively (magenta dashes in Figure 3E). From the novel β sheet, β2 leads into the inter-switch loop 3 with rearrangement of salt bridges between Arg164, Asp47, and Glu49 at the junction of switch I and helix 1 (dashed circle in Figures 3D and S3B). Ultimately, these changes remove pi-stacking interactions between Phe28 in switch I and the guanine base of GDP, increasing solvent exposure of the nucleotide (Figures 3E and S3C). The details of our A59E crystal structures are consistent with the peak broadening of switch I and II resonances observed in our HSQC spectra of K-RASA59E, and this was particularly obvious after mapping the mutation-induced chemical shift perturbations onto the crystal structure of K-RASA59E (Figure 3F).
Our data suggest a mechanism of “Mg2+ extraction” that supports hyperexchange by autophosphorylated K-RASA59T and, possibly, other autophosphorylating GTPases. To explore this possibility, we used MD simulations to examine the dynamic relationship of position 59, Mg2+, and GDP. Residue 59 Cβ atom motions in K-RASA59T showed that both the mutation and the charge associated with phosphorylation shift residue 59 and switch II into the active site (Figures 3G, 3H and S3D). These results are consistent with simulations for other autophosphorylating GTPases (Figures 3I and S3E). Together, our data show that negative charge at position 59 destabilizes the Mg2+-GDP interaction.
“Mg2+ extraction” appeared to represent an intermediate step in the process of adopting the conformation seen in our crystal structure of K-RASA59E, as the overall changes in K-RAS structure and dynamics seen in our NMR experiments were more consistent with the K-RASA59E crystal structure than the H-RASA59E crystal structures (Figure 3F). This explains why K-RASA59Tp and K-RASA59E have different kinetics of nucleotide exchange compared to K-RASA59T. While K-RASA59T locally influences switch I and II dynamics around the nucleotide to enhance exchange (upper versus lower, Figure 3F), the overall conformation is still recognizable to SOS1. In contrast, the extended and open active site conformation of K-RASA59E, and presumably K-RASA59Tp, is resistant to recognition and complex formation by SOS1.
Because of the extensive broadening of resonances, our NMR experiments do not provide direct evidence for the β sheet structure seen in the K-RASA59E crystal; however, the extended and open active site conformation stabilized by Glu59 shares the same global conformation as crystal structures of K-RASA146T and wild-type K-RAS (Dharmaiah et al., 2019; Poulin et al., 2019). Taken together, these observations suggest that the extended and open conformation is likely present in wild-type K-RAS, but is less favored and in conformational equilibrium with other active site states. Furthermore, Glu59 directly stabilizes the extended and open conformation in K-RASA59E through an additional salt bridge (Figure 3E).
Finally, our structural analysis suggests two important points regarding the mechanism of RAS nucleotide exchange. First, the active site of RAS opens more than is necessary for intrinsic exchange of GDP for GTP, as K-RASA59T, K-RASA59Tp, and K-RASA59E all converge on similar rate constants for this reaction (Figure 1G). Second, the mechanism of SOS1-catalyzed nucleotide exchange is similar, but not identical, to the intrinsic mechanism of nucleotide exchange. Thus, our different biophysical and theoretical approaches argue that introduction of a Ser or Thr functional group at “residue 59” alters the enzymatic function of small GTPases to enable autophosphorylation, which permanently alters active site dynamics to favor intrinsic nucleotide exchange.
Autophosphorylation functionally activates K-RAS
The active site dynamics of K-RASA59Tp and K-RASA59E necessary for nucleotide exchange appear to be at odds with GTPase function, as small GTPases undergo closure of the active site to interact with their known effectors (Vetter, 2017). Nevertheless, the fact that Ala59 mutations occur in cancer suggest that they functionally activate K-RAS. To address this paradox, we first examined K-RASA59T function in SNU-175 cells expressing a doxycycline-inducible short hairpin RNA (shRNA) targeting the KRAS transcript. Knockdown of K-RAS in SNU-175 cells reduced proliferation and ERK phosphorylation, indicating that Thr59 does promote a proliferative function for K-RAS (Figures 4A, 4B, and S4A). At the same time, knockdown of K-RAS resulted in an increase in the fraction of K-RASA59T that was phosphorylated because phosphorylated KRASA59T accumulates over time as translation is inhibited (Figure 4C), as has been demonstrated for v-H-RAS (Ulsh and Shih, 1984). This result confirms the independent and passive nature of K-RASA59T autophosphorylation. To validate these observations in an independent system, we used CRISPR editing to insert the A59T mutation into the Kras locus of mouse embryonic stem (mES) cells (Figures S4B and S4C) and found that endogenous expression of K-RASA59T increases the phosphorylated state of MEK, ERK, and AKT (Figure 4D).
Figure 4. Roles of K-RASA59T and K-RASA59E in cellular transformation.

(A) Effect of KRAS shRNA on proliferation in SNU-175 cells (n = 2–3).
(B) Effect of KRAS shRNA on ERK1/2 phosphorylation in SNU-175 cells (n = 2–3). Data are normalized to the average ERK1/2 phosphorylation on day 0.
(C) Effect of KRAS shRNA on K-RAS expression and autophosphorylation in SNU-175 cells (n = 2–3). Data are normalized to the average K-RAS expression on day 0 (upper).
(D) Baseline phosphorylation of Mek, Erk, and Akt in WT and K-RasA59T mouse embryonic stem cells after 6 h of serum starvation. The red arrow denotes autophosphorylated K-RAS. Quantification is shown on the right and normalized to average phosphorylation for WT cells.
(E) Growth of NIH3t3 cells with ectopic K-RAS on plastic (x axis, n = 4–5) and in soft agar (y axis, n = 3) in 10% serum.
(F and G) Representative western blots from precipitation of GTP-bound K-RAS from NIH3t3 cells (F and G) by C-RAF-RBD-GST. Note that the upper band representing K-RAST59p is absent.
(H) Quantification of Akt (Ser473) and Rps6 (Ser235/236) phosphorylation (n = 4).
(I) Comparison of Mek phosphorylation (x axis, n = 4) and K-RAS-GTP (y axis, n = 4). Phosphorylated was normalized to total Mek. K-RAS-GTP was scaled to protein input and normalized to the average in cells expressing WT K-RAS.
(J) Quantification of C-Raf phosphorylation on Ser289/296/301 (n = 4).
(K) Quantification of Erk1/2 phosphorylation on Thr202/Tyr204 (n = 4).
In (A–C), errors bars represent ± SD and statistical analyses used Student’s t test. For (H–K), errors bars represent ± SD and statistical analyses were performed with Mann-Whitney test. *, **, and *** represent P values of < 0.05, < 0.005, and < 0.0005, respectively. In (E–K), statistical comparisons and representative western blots are in Figure S4.
While our knockdown and knockin experiments demonstrated the ability of A59T mutation to activate K-RAS, we next tested the relative activity and transforming potential of Ala59 mutants, with and without co-mutation of G12V, by ectopic expression in NIH 3T3 fibroblasts (Figure S4D). Ectopic expression of each of the K-RAS mutants caused fibroblasts to exhibit a spindle-shaped morphology with elongated processes and refractile cytoplasm (Figure S4E), consistent with transformation. We also found that K-RASA59E showed a greater ability to transform fibroblasts than K-RASA59T (Figures 4E, S4F, and S4G), suggesting that negative charge, and not just Ala59 mutation, conferred transformation. Likewise, we noted that transformation by K-RASG12V/A59T was enhanced compared to K-RASA59T. Nevertheless, both Ala59 mutations alone promoted cell proliferation to a lesser extent than K-RASG12V, which was surprising given the strong effect that A59T and A59E have on GTP hydrolysis and nucleotide exchange (Figures 1F and 1G).
To understand these differences in fibroblast transformation, we examined activation of the MAPK and AKT signaling pathways. We first measured the ability of K-RAS to interact with the RAS binding domain of C-RAF (C-RAF-RBD), which recognizes and binds to GTP-bound RAS. C-RAF binding by K-RASA59T was more similar to wild-type K-RAS, and K-RASA59Tp failed to interact with C-RAF-RBD at all (Figure 4F). Likewise, co-mutation of A59T with G12V selectively enhanced the pull-down of the non-phosphorylated protein only (Figure 4G). Consistent with our pull-down experiments, activation of AKT and phosphorylation of its downstream target Rps6 was enhanced by co-mutation with G12V (Figures 4H and S4H–S4J). In contrast, K-RASA59T showed an intermediate activation of AKT and Rps6, while K-RASA59E expression induced no change at all (Figures 4H and S4I). However, we found that while all the K-RAS mutants induced similar levels of MEK phosphorylation in dividing fibroblasts, this activity poorly correlated with binding of C-RAF-RBD to Ala59 mutants (Figures 4I and S4I–S4L). Thus, K-RASA59T and K-RASA59Tp appear disparate in their ability to interact with C-RAF, alter cell signaling, and initiate fibroblast transformation.
Because K-RASA59E binds C-RAF-RBD, but K-RASA59Tp does not, we examined the dynamics of MAPK signaling in more detail. Strong activation of the MAPK signaling pathway can be detected by measuring hyperphosphorylation of C-RAF (Dougherty et al., 2005). Unlike fibroblasts expressing K-RASA59T and K-RASG12V, K-RASA59E does not induce C-RAF hyperphosphorylation (Figures 4J and S4I), and ERK phosphorylation was not significantly upregulated by K-RASA59E in dividing fibroblasts (Figures 4K and S4I). Thus, while our experiments broadly demonstrate cell transformation by K-RASA59T and K-RASA59E, their oncogenic activity is potentially limited by active site dynamics that alter their abilities to interact with specific effectors, essentially creating hypomorphic activating mutations.
Autophosphorylation influences K-RAS effector interactions
Our data indicated that residue 59 phosphorylation might inhibit effector binding because of its influence on switches I and II, which participate in effector binding (yellow surfaces in Figure 5A). To test this, we preloaded mutant K-RAS protein from our NIH 3T3 lysates with nucleotide and again tested them for interaction with C-RAF-RBD or, additionally, with full-length RASSF5 protein. These effector interactions provide complementary information because C-RAF-RBD interacts exclusively with switch I, while RASSF5 interacts with both switches I and II. We found that Thr59 phosphorylation inhibits binding to C-RAF and RASSF5, while A59E selectively inhibits binding to RASSF5 (Figure 5B). Notably, under these experimental conditions K-RASA59T was able to bind C-RAF and RASSF5 (red versus black arrows in Figure 5B).
Figure 5. GTP induces phosphoryl movement to discriminate effector interactions.

(A) Binding interfaces of C-RAF-RBD (PDB: 4G0N) and RASSF5-RBD (PDB: 3DDC). Blue arrows indicate Ala59 of RAS.
(B) Pull-down of ectopic K-RAS, preloaded with GTP or GDP, by C-RAF-RBD-GST or RASSF5-GST. Arrows denote non-phosphorylated (black) and phosphorylated (or A59E) (red) K-RAS.
(C) Frequency distribution of bond distances during MD simulations of mutant K-RAS bound to GTP.
(D) MD simulation cluster analysis of K-RAST59p.
(E) BRET saturation curves showing interaction between RAF isoform regulatory domains and K-RAS mutants in HEK293FT cells.
(F) Western blot of RAF isoforms co-immunoprecipitating with Venus-tagged K-RAS from serum-starved HeLa cells.
(G) Affinity changes of K-RAS for the RAF-RBDs determined by BLI. Data normalized to WT K-RAS (KDWT/KDMut). Error bars represent ± SD (n = 2–4)
(H) Co-immunoprecipitation of B-RAF by C-RAF with different K-RAS mutants from serum-starved HeLa cells.
(I) Comparison of K-RAS conformations (green, purple) generated by cluster analysis of MD simulations to H-RAS (gray) bound to C-RAF (PDB: 4G0N, cyan). Nucleotide is shown in black.
Because our GppNHp-bound H-RASA59E crystal structures have disordered active sites, we used MD simulations to explain why K-RASA59Tp and K-RASA59E appeared to behave differently. We noted that simulations of GDP-bound K-RASA59Tp exhibited low frequency conformations with the phosphoryl group facing away from the active site (arrow in Figures 3G and 3H). Likewise, while GTP-bound wild-type K-RAS and K-RASA59T had similar overall dynamic profiles (Figures S5A and S5B), the phosphoryl group of K-RASA59Tp bound to GTP showed a clear transition out of the active site, repulsing Glu37 (arrows in Figures 5C and 5D). In contrast, GTP-bound K-RASA59E did not make this transition (Figures 5C and S5C), and instead switch interactions are enhanced, as Glu37 in switch I and Arg68 in switch II interactduring the simulation. Except for ARL6 and DIRAS1, phosphoryl transitions in the other GTPases and their overall effects on switches I and II were like phosphorylated K-RASA59T (Figure S5D).
Thus, the GTP-bound states of K-RASA59T and K-RASA59E were likely underestimated in our initial RAS activity assays because autophosphorylation impacts the affinity of RAS-RAF interaction (Figure 4F). To address this directly, we tested RAF binding activity of K-RASA59T and K-RASA59E using bioluminescence energy transfer (BRET) in cells with the regulatory domains of the RAF isoforms (Terrell et al., 2019). The regulatory domain of each RAF isoform consists of a variable N-terminal segment followed by the RBD and a cysteine-rich domain (CRD), which together bind RAS (Cookis and Mattos, 2021; Tran et al., 2021). Like our binding experiments with C-RAF-RBD, K-RASA59T and K-RASA59E exhibited reduced affinity for the regulatory domains of C-RAF, A-RAF, and B-RAF relative to K-RASG12V and K-RASG12D (Figures 5E, 5F, S6A and Table S2). The status of MEK phosphorylation in serum-starved cells expressing K-RASA59T or K-RASA59E trended with C-RAF co-immunoprecipitation (Figure 5F), reflecting the graded differences in fibroblast proliferation induced by K-RASA59T and K-RASA59E and altered kinetics of MAPK signaling seen in our dividing NIH 3T3 cells (Figures 4E, 4J, 4K, and S4L).
Recent crystal structures of RAS in complex with C-RAF-RBD-CRD (Cookis and Mattos, 2021; Tran et al., 2021), as well as NMR-data-driven models of K-RAS and the RBD-CRD tethered to nanodiscs (Fang et al., 2020), suggest Ala59 mutations would predominantly affect interactions with the RBD domains of the three RAF isoforms (Figures S6B and S6C). Using biolayer interferometry (BLI), we found that each RAF-RBD has a reduced affinity for K-RASA59T and K-RASA59E (Figures 5G and S6D).However, the affinities of Ala59 mutants for A-RAF and C-RAF measured by BRET (i.e., BRET50) were not consistent with our BLI and co-immunoprecipitation data. While differences in B-RAF colocalization with K-RASA59T and K-RASA59E also reflect differences in their GTP-bound state (Figure 4F), colocalization with C-RAF, and more so A-RAF, appears less dependent on K-RASA59T or K-RASA59E interacting with their respective RBDs. Because BRET can be induced by distances of <100Å (Boute et al., 2002), it is possible that the association of K-RAS with C-RAF or A-RAF may be facilitated by other factors.
While B-RAF exhibits weak association with K-RASA59T and K-RASA59E, it could still be activated through heterodimerization with C-RAF (Freeman et al., 2013). We tested this possibility and found that K-RASA59T and K-RASA59E showed a barely detectable increase in C-RAF/B-RAF heterodimerization compared to wildtype K-RAS (Figure 5H). Our data suggest that K-RASA59T and K-RASA59E likely do not activate MAPK signaling through B-RAF/C-RAF heterodimerization, but rather through weak activation of A-RAF and C-RAF. Further, these data are consistent with the altered kinetics of MAPK signaling seen in our dividing fibroblasts (Figures 4E, 4J, 4K, and S4L).
The reduction in RASSF5 binding in response to Ala59 mutation was also noteworthy. RASSF5 is a member of a putative tumor suppressor family associated with growth inhibition downstream of activated RAS (Volodko et al., 2014). In agreement with a recent report (Dhanaraman et al., 2020), we discovered that only RASSF1 and RASSF5 exhibit affinity for active K-RAS and that K-RASA59Tp and K-RASA59E appeared to specifically affect the interaction with RASSF5 (Figures S7A and S7B). The functional activation of K-RAS—i.e., its induction of proliferation—by Ala59 mutations might be due to its weak activation of pro-proliferation signaling and its lack of activation of anti-proliferation signaling.
For small GTPases, increased active site dynamics in the GTP-bound state, along with phosphorylation that changes active site compaction and organization, will necessarily alter effector affinity and kinetics of complex formation. This is demonstrated by comparison of our MD simulations to crystal structures of RAS bound to C-RAF-RBD (Figure 5I) and RASSF5 (Figure S7C). For instance, in solution, K-RASA59T shows weak affinity for the RAF isoforms and RASSF5 (Figures 5G and S7A). For RAF, this makes sense because our MD simulations of K-RASA59T show that Thr59 association with GTP is disruptive to Thr35 packing in the active site (Figures 2C and S2F), and it is well known that Thr35 mutants of RAS have reduced affinity for C-RAF due to enhanced switch I dynamics (Spoerner et al., 2001). After Thr59 phosphorylation, dynamics in switches I and II increase, preventing RAF (and presumably RASSF5) binding to K-RASA59Tp (Figures 5I and S7C). Changes in RAF affinity appear to be specific for K-RASA59T and K-RASA59E because H-RASA59G and K-RASA59G, which have similar changes in GTP hydrolysis and nucleotide exchange, maintain high affinity for RAF (Hall et al., 2001; Lu et al., 2018). It remains unclear why the Ala59 mutants studied here disrupt binding to B-RAF more so than A-RAF or C-RAF (Figure 5G).
A notable difference between K-RASA59E and K-RASA59Tp was that K-RASA59E was capable of binding C-RAF (Figure 5B). This is likely due to the Glu59 inducing less dynamical changes in switch I and changes in switch II being less important for RAF interaction (Figures 5I, S6B, and S6C). Differences in the movement of Glu37 in our MD simulations of K-RASA59E and K-RASA59Tp are also consequential because Glu37 mutants inhibit binding to C-RAF (Hamad et al., 2002).
Nevertheless, changes in switch dynamics also contribute to impaired binding of K-RASA59E and K-RASA59Tp to RASSF5. For the K-RASA59T/RASSF5 interaction, weakened affinity can be explained by the expanded binding interface of RASSF5, which is very sensitive to changes in active site compaction (Stieglitz et al., 2008). The addition of negative charge at the RASSF5 binding interface also plays an important role. For instance, the E37G mutation, which reduces the affinity of RAS proteins for RalGDS, improves binding of RAS to RASSF5 (Hamad et al., 2002; Khokhlatchev et al., 2002). Thus, repulsion of Glu37 by Glu59, or phosphorylated Thr59, further inhibits binding of RASSF5 to K-RASA59E and K-RASA59Tp (Figures 5B, S7A, and S7B). This rationale can be extended to interactions with PI3K and Sin1, part of the mTORC2 complex, because, like RASSF5, they form extended interactions with switch II (Figures S6E and S6F). Thus, differences in AKT phosphorylation in our fibroblast transformation experiments (Figure 4I) could be due to a failure of Ala59 mutants to recruit and associate with PI3K and mTORC2 (Castel et al., 2021; Castellano and Downward, 2011; Kovalski et al., 2019). Together, these data demonstrate that a consequence of GTPase autophosphorylation is to alter effector engagement.
DISCUSSION
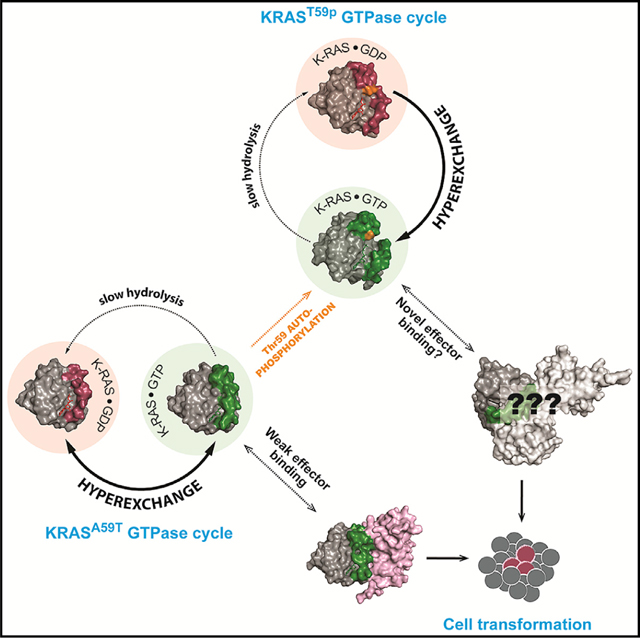
Here, we elucidate the mechanistic basis for the autophosphorylation of small GTPases and determine the effect of this modification on the GTPase cycle. The functional state of a GTPase is presumed to be a function of its nucleotide binding state (Figure 6A). In the canonical GTPase cycle, intrinsic and GEF-induced nucleotide exchange activate the protein by loading GTP into the active site, while intrinsic and GAP-induced GTP hydrolysis serve to inactivate. Using K-RASA59T as an architype for small GTPases that are capable of autophosphorylation, we show that phosphorylation at position 59 alters active site dynamics, nucleotide exchange, and effector interaction to dramatically reorganize the GTPase cycle (Figure 6B). Although the ability of H-RASA59T to autophosphorylate has been known for some time, our study disproves one of the initial presumptions of this biochemical activity—that it is simply a byproduct of the hydrolysis reaction.
Figure 6. Influence of autophosphorylation on nucleotide cycling and effector engagement.

(A) The normal GTPase cycle of K-RAS.
(B) Thr59 inhibits nucleotide hydrolysis and promotes intrinsic nucleotide exchange, activating K-RAS. Subsequently, the mutant shows weak binding to RAF and RASSF5 proteins.
(C) When Thr59 becomes phosphorylated (orange), K-RAS enters an alternative cycle where it becomes insensitive to GEF and loses the ability to bind to RAF and RASSF5 but opens up the possibility of interacting with novel effectors.
An intriguing outcome of our study is that autophosphorylation enhances intrinsic exchange but desensitizes K-RAS to SOS1-catalyzed nucleotide exchange (Figure 1G), and this may explain why K-RASA59T shows weak cellular activation that is overcome by co-mutation of G12V (Figures 4F–4H). Moreover, phosphorylation of Thr59 creates a preference for GTP exchange over GDP exchange (Figure 1G). After GTP binding, K-RASA59Tp and K-RASA59E do undergo necessary dynamics to take on an active GTP-bound state, but with altered effector binding (Figure 6C). Altered interaction with RAF proteins likely limits pro-proliferation signaling downstream of K-RASA59T and K-RASA59E.
It is also possible that autophosphorylation allows for novel effector interactions, perhaps overlapping with the other small GTPases that potentially autophosphorylate. Indeed, DIRAS subfamily members can compete with K-RAS to bind to effector proteins (Bergom et al., 2016). Likewise, K-RASA59T may become susceptible to new regulation, as DIRAS1/2 are regulated by RapGAPs (Gasper et al., 2010). Thus, the unexpectedly low activation of K-RASA59T, compared to K-RASA59E, could be due to stimulation of GTP hydrolysis by alternate GAPs, whose activity is suppressed by co-mutation of Gly12. In support of this, a recent report shows that RGS3, a GAP of G-protein-coupled receptors, can catalyze hydrolysis of GTP bound to K-RASG12C (Li et al., 2021).
Our observations explain why Ala59 mutations are so rare in cancer despite their inhibition of GTP hydrolysis and increase of nucleotide exchange. Functional activation of K-RASA59T and K-RASA59E is likely limited by the mechanism of hyperexchange and its altered effector interactions, ultimately creating weakly activated mutants. Indeed, the attenuating function of Thr59 might be necessary for v-K-RAS and v-H-RAS to avoid oncogene-induced senescence due to hyperactivated MAPK signaling or through binding to RASSF5 (Donninger et al., 2016). Consistently, we found that co-mutation of Ala59 and Gly12 enhanced Akt phosphorylation and activation of non-phosphorylated K-RAS in mouse fibroblasts, suggesting that autophosphorylation has a specific function. How K-RASA59T and K-RASA59E change the kinetics of activation and feedback regulation of the MAPK signaling pathway and their reliance on evasion of RASSF5 to promote cell transformation and tumorigenesis are questions that remain to be answered.
Because the field has yet to identify GEFs for many of the GTPases listed in Table 1, it is possible that autophosphorylation of threonine or serine at position 59 might replace these regulatory proteins (Foster et al., 1996). For GTPases capable of autophosphorylation, altered nucleotide binding cycle is likely an aspect of their normal regulation, creating a pool that is functionally distinct from the non-phosphorylated pool. We also noted that some of these small GTPases have cancer-associated mutations at their analogous residue 59 (Table 1), which would render them unable to autophosphorylate and perhaps alter their GTPase cycles back toward the more canonical form.
The most thoroughly characterized RAS modifications (e.g., phosphorylation, di-ubiquitination) regulate its subcellular localization, typically pushing it from the plasma membrane to organellar endomembranes (Ahearn et al., 2018). But others (e.g., monoubiquitination, acetylation, AMPylation, tyrosyl phosphorylation) are reported to affect the GTPase cycle by influencing nucleotide exchange (Barthelmes et al., 2020; Kano et al., 2019; Sasaki et al., 2011; Ting et al., 2015; Yang et al., 2012). Indeed, our work has parallels with a recent proposal that Src shifts K-RAS into a “dark state” through phosphorylation of Tyr32 and Tyr64. In vitro characterization demonstrated that Tyr phosphorylation enhances the GTP-bound population of K-RAS but simultaneously impairs binding to RAF kinases (Kano et al., 2019). However, our study differs from that of Kano et al. in that phosphorylation of residue 59 still promotes functional activation of K-RAS. Thus, our work illuminates a new paradigm for regulation of the GTPase cycle via autophosphorylation. This discovery opens the door to a deeper look at the PTM landscape on GTPases and more detailed studies of their functional ramifications.
Limitations of the study
Our studies used K-RASA59E to mimic K-RASA59Tp. While neither glutamate nor aspartate can fully recapitulate the charge and shape of phospho-threonine, we did find that K-RASA59Tp and K-RASA59E shared a couple of key traits. First, both prefer intrinsic over SOS1-mediated nucleotide exchange. Second, both affect switch II conformation and dynamics such that RASSF5, and likely PI3K and Sin1, are incompatible binding partners. Unlike K-RASA59E, however, K-RASA59Tp prefers GTP over GDP and cannot bind RAF. Thus, more work will need to be done to elucidate the subtle differences between Ala59 mutations.
While several GTPases share the ability to autophosphorylate when mutated to serine or threonine at the correct active site position, experimental validation of autophosphorylation by the GTPases in Table 1 was not included in this work. However, this study shows that most of these GTPases should be capable of this reaction, as their respective residue 59 nucleophiles move toward the γ-phosphate. Further studies of the GTPases in Table 1 need to be performed.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for data and resources should be directed to the lead contact, Dr. Kevin Haigis (mailto:Kevin_haigis@DFCI.harvard.edu).
Materials availability
Plasmids generated by this study are available upon request.
Data and code availability
Original scans of western blot data have been deposited at Mendeley Data and are publicly available as of the date of publication. Validation reports of X-ray crystallography structures have been deposited at the PDB and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| A-RAF | Santa Cruz | Cat#: sc-408; RRID: AB_630882 |
| AKT (pan) (40D4) | Cell Signaling Technologies | Cat#: 2920; RRID:AB_1147620 |
| Anti-GFP | MBL International | Cat#:D153–3; RRID:AB_591817 |
| Anti-GST (91G1) | Cell Signaling Technologies | Cat#: 2625; RRID:AB_490796 |
| Anti-HA-tag (6E2) | Cell Signaling Technologies | Cat#: 2367; RRID:AB_10691311 |
| Anti-Venus | Roche | Cat#: 118114460001 |
| B-RAF | Santa Cruz | Cat#: sc-5284; RRID:AB_2721130 |
| C-RAF | BD Pharmagen | Cat#: 610152; RRID:AB_397553 |
| c-RAF (D5X6R) Mouse mAb | Cell Signaling Technologies | Cat#: 12552S; RRID:AB_2728706 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Invitrogen | Cat#: A-21058; RRID:AB_2535724 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 800 | Invitrogen | Cat#: A-32735; RRID:AB_2633284 |
| MEK1 | BD Biosciences | Cat#: 610122; RRID:AB_397528 |
| MEK1/2 (L38C12) | Cell Signaling Technologies | Cat#: 4694; RRID:AB_10695868 |
| p44/42 MAPK (ERK1/2) (L34F12) | Cell Signaling Technologies | Cat#: 4696; RRID:AB_390780 |
| pC-RAF (s289/296/301) | Cell Signaling Technologies | Cat#: 9431; RRID:AB_561402 |
| Phospho-Akt (Ser473) (D9E) XP® | Cell Signaling Technologies | Cat#: 4060; RRID:AB_2315049 |
| Phospho-MEK1/2 (Ser217/221) | Cell Signaling Technologies | Cat#: 9121; RRID:AB_331648 |
| Phospho-MEK1/2 (Ser217/221) (41G9) | Cell Signaling Technologies | Cat#: 9154; RRID:AB_2138017 |
| Phospho-p44/42 MAPK (ERK1/2) (Thr202/ Tyr204) (197G2) | Cell Signaling Technologies | Cat#: 4377; RRID:AB_331775 |
| Phospho-S6 Ribosomal Protein (Ser235/236) | Cell Signaling Technologies | Cat#: 2211; RRID:AB_331679 |
| polyclonal KRAS antibody | Proteintech | Cat#: 12063–1-AP; RRID:AB_878040 |
| RAS10 | Millipore-Sigma | Cat#: 05–516; AB_2121151 |
| S6 Ribosomal Protein (54D2) | Cell Signaling Technologies | Cat#: 2317; RRID:AB_2238583 |
| Vinculin (E1E9V) XP® | Cell Signaling Technologies | Cat#: 13901; RRID:AB_2728768 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| BL21 (DE3 Codon+) | Agilent | Cat#: 230280 |
| BL21 (DE3) | Thermo Scientific | Cat#: C600003 |
| BL21-A1 | Thermo Fisher Scientific | Cat#: C607003 |
| DH5α-T1R | Invitrogen | Cat#: 12297–016 |
| pCL-Eco | Addgene | Cat#: 12371 |
| Tet-pLKO-puro shKRAS | Addgene | Cat#: 116871 |
| TOP10 E. coli strain | Fisher Scientific | Cat#: C404003 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 1.0 M Calcium acetate hydrate | Hampton Research | Cat#: HR2–567 |
| 1.0 M HEPES pH 7.5 | Hampton Research | Cat#: HR2–729 |
| 1.0 M Magnesium acetate tetrahydrate | Hampton Research | Cat#: HR2–561 |
| 100% glycerol | Hampton Research | Cat#: HR2–623 |
| 100% PEG400 | Hampton Research | Cat#: HR2–603 |
| 100X Glutamax supplement | Thermo Fisher Scientific | Cat#: 35050061 |
| 100X MEM Non-Essential Amino Acids Solution | Life Technologies | Cat#: 11140–050 |
| 10X CutSmart buffer | New England Biolabs | Cat#: R3195T |
| 1X Knockout DMEM | Life Technologies | Cat#: 10829–018 |
| 2-Mercaptoethanol | Millipore-Sigma | Cat#: M3148–250ML |
| 2’/3′-O-(N-Methyl-anthraniloyl)-guanosine-5′-diphosphate | Fisher Scientific | Cat#: M12414 |
| 2’/3′-O-(N-Methyl-anthraniloyl)-guanosine-5′-triphosphate | Fisher Scientific | Cat#: M12415 |
| 4.0 M Magnesium chloride hexahydrate | Hampton Research | Cat#: HR2–803 |
| 5.0 M Sodium chloride | Hampton Research | Cat#: HR2–637 |
| 50% PEG3350 mono disperse | Hampton Research | Cat#: HR2–527 |
| 5X Phusion HF Buffer | New England Biolabs | Cat#: M0530S |
| Accucore C18 resin | ThermoFisher Scientific | N/A |
| Antibiotic Antimycotic Solution (PenStrep) | Sigma-Aldrich | Cat#: A5955 |
| Chymotrypsin | Promega | Cat #V106A |
| Coelenterazine-h | N/A | N/A |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | Cat#: N0447S |
| DMSO | Sigma-Aldrich | Cat#: D2650–100mL |
| Dulbeccos Modification of Eagles Medium (DMEM) | TC-Corning | Cat#: 10–013-CV |
| EcoRV-HF® restriction enzyme | New England Biolabs | Cat#: R3195T |
| ESGRO ®Leukemia Inhibitory Factor (LIF), 1 million units/1mL | Millipore-Sigma | Cat#: ESG1106 |
| Fetal Bovine Serum (FBS) | GE Healthcare | Cat#: SH30070.03 |
| Gelatin from bovine skin | Sigma-Aldrich | Cat#: G9391–100G |
| GreenGlo™ Safe DNA Dye, 20,000X in Water | Denville Scientific | Cat#: ca3600 |
| Guanosine 5′-[β,γ-imido]triphosphate trisodium salt hydrate | Sigma-Aldrich | Cat#: G0635–100MG |
| HyClone Defined Fetal Bovine Serum (FBS), US Origin | Cytiva | Cat#: SH30070.03 |
| Intercept ®(TBS) Blocking Buffer | LI-COR Biosciences | Cat#: 927–6003 |
| Intercept ®T20 (TBS) Antibody Diluent | LI-COR Biosciences | Cat#: 927–65001 |
| Iodoacetamide | ThermoFisher Scientific | Cat #A39271 |
| Lambda Phosphatase | New England BioLabs | Cat#: P0753S |
| Modified Terrific Broth | Fisher Scientific | Cat#: BP9729–600 |
| Phusion ®High-Fidelity DNA Polymerase | New England Biolabs | Cat#: M0530S |
| Radioimmunoprecipitation assay (RIPA) buffer | Boston Bioproducts | Cat#: BP-115–500 |
| RPMI 1640 | Fisher Scientific | Cat#: MT10040CV |
| Tris(2-carboxyethyl)phosphine | ThermoFisher Scientific | Cat #77720 |
| 0.05% Trypsin EDTA | Invitrogen | Cat#: 25300–120 |
| UltraPure™ DNase/RNase-Free Distilled Water | Invitrogen | Cat#: 10977023 |
|
| ||
| Critical commercial assays | ||
|
| ||
| EnzChek™ Phosphate Assay Kit | Thermo Fisher Scientific | Cat#: E6646 |
| Mouse ES Cell Nucleofector®Kit | Lonza | Cat#: VPH-1001 |
| PureLink® HiPure Plasmid Filter Maxiprep kit | Thermo Fisher Scientific | Cat#: K2100–17 |
| Quick-DNA 96 kit | Zymo Research | Cat#: D3012 |
| QuikChange site-directed mutagenesis | Agilent | Cat#: 200519 |
| XtremeGENE9 transfection Kit | Millipore-Sigma | Cat#: 6365779001 |
|
| ||
| Deposited data | ||
|
| ||
| H-RAS A59E GDP | https://www.rcsb.org | PDB: 7JII |
| H-RAS A59T GppNHp crystal 1 | https://www.rcsb.org | PDB: 7JIF |
| H-RAS A59T GppNHp crystal 2 | https://www.rcsb.org | PDB: 7JIG |
| H-RAS A59E GppNHp | https://www.rcsb.org | PDB: 7JIH |
| K-RAS A59E GDP | https://www.rcsb.org | PDB: 7KMR |
| Raw data western blot data from figures were deposited on Mendeley at https://dx.doi.org/10.17632/9n7bryshg3.1 | https://data.mendeley.com | Mendeley data: https://doi.org/10.17632/9n7bryshg3.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| EmbryoMax® Primary Mouse Embryonic Fibroblasts | Millipore-Sigma | Cat#: PMEF-CFX |
| HEK293FT cells | Thermo Fisher Scientific | Cat#: R70007; RRID: CVCL_6911 |
| HEK293t cells | ATCC | Cat#: CRL-3216; RRID: CVCL_0063 |
| HeLa cells | ATCC | Cat#: CCL-2; RRID: CVCL_0030 |
| LIM1215 cells | Whitehead et al., 1985 | N/A; RRID: CVCL_2574 |
| K-Ras(+/LSL-wildtype) Mouse Embryonic Stem cells | Poulin et al., 2019 | N/A |
| K-Ras(A59T/LSL-A59T) Mouse Embryonic Stem cells | This study | N/A |
| NIH 3T3 cells | ATCC | Cat#: CRL-1658; RRID: CVCL_0594 |
|
| ||
| Oligonucleotides | ||
|
| ||
| 5′-CTTGGATATTCTC GACACAACAGGTCA AGAGGAGTACAG-3′ | Integrated DNA Technologies | pBABE KRAS4B A59T Forward |
| 5′-CTGTACTCCTCT TGACCTGTTGTGTCG AGAATATCCAAG-3′ | Integrated DNA Technologies | pBABE KRAS4B A59T Reverse |
| 5′-GGATATTCTCG ACACAGAAGGTCAA GAGGAGTACAG-3′ | Integrated DNA Technologies | pBABE KRAS4B A59E Forward |
| 5′-CTGTACTCCTCT TGACCTTCTGTGTCGA GAATATCC-3′ | Integrated DNA Technologies | pBABE KRAS4B A59E Reverse |
| 5′-GTAGTTGGAGC TGTTGGCGTA GGCAAG-3′ | Integrated DNA Technologies | pBABE KRAS4B G12V Forward |
| 5′-CTTGCCTACGC CAACAGCTCC AACTAC-3′ | Integrated DNA Technologies | pBABE KRAS4B G12V Reverse |
| 5′-GTGGTAGTTGGAGCTCGTGG CGTAGGCAAG-3′ | Integrated DNA Technologies | pBABE KRAS4B G12R Forward |
| 5′-CTTGCCTACGCCACGAGCTCCA ACTACCAC-3′ | Integrated DNA Technologies | pBABE KRAS4B G12R Reverse |
| 5′-GGTGGTTGGTGCCGATGGTG TGGGTAAAAG-3′ | Integrated DNA Technologies | pET21 KRAS4B G12D Forward |
| 5′-CTTTTACCCACACCATCGGCA CCAACCACC-3′ | Integrated DNA Technologies | pET21 KRAS4B G12D Reverse |
| 5′-CATTCTGGATACCACCGGCC AGGAAG-3′ | Integrated DNA Technologies | pET21 KRAS4B A59T Forward |
| 5′-CTTCCTGGCCGGTGGTAT CCAGAATG-3′ | Integrated DNA Technologies | pET21 KRAS4B A59T Reverse |
| 5′-GACATTCTGGATACCGAAGGC CAGGAAGAGTATAG-3′ | Integrated DNA Technologies | pET21 KRAS4B A59E Forward |
| 5′-CTATACTCTTCCTGGCCTTCGG TATCCAGAATGTC-3′ | Integrated DNA Technologies | pET21 KRAS4B A59E Reverse |
| 5′-CACCGTTCTCGACACAGCAGGT CAAG-3′ | Integrated DNA Technologies | SaCas9 gRNA 1 Forward |
| 5′-AAACCTTGACCTGCTGTGTC GAGAAC-3′ | Integrated DNA Technologies | SaCas9 gRNA 1 Reverse |
| 5′-TGTGACCATTAGCATTGT TTGC-3′ | Integrated DNA Technologies | mES Ex2 genotyping Forward primer |
| 5′-CTTAAACCCACCTATAAT GGTG-3′ | Integrated DNA Technologies | mES Ex2 genotyping Reverse primer |
| 5′-AGTAATTGATGGAGAAA CCTG-3′ | Integrated DNA Technologies | mES Ex2 Sequencing Forward primer |
| 5′-ATAATGGTGAATATCTT CAAA-3′ | Integrated DNA Technologies | mES Ex2 Sequencing Reverse primer |
| 5′-CTGTAATAAT CCAGACTGTG TTTCTCCCTT CTCAGGACTCC TACAGGAAAC AAGTAGTAAT TGATGGAGAA ACCTGTCTCT TGGATATCCT CGACACAACA GGTCAAGAAG AGTACAGTGC AATGAGGGAC CAGTACATGA GAACTGGGGA GGGCTTTCTT TGTGTATT TGCCATAAA TAATACTAA ATCATTTG AAGAT-3′ | Integrated DNA Technologies | Repair template for CRISPR experiments |
|
| ||
| Recombinant DNA | ||
|
| ||
| 100 bp DNA ladder | New England Biolabs | Cat#: N3231L |
| 6xHis-GAP (715–1047) | This study | N/A |
| 6xHis-K-RAS4B | This study | N/A |
| 6xHis-SOS11 (564–1049) | This study | N/A |
| Gateway pDEST15 vector | Thermo Scientific | Cat#: 11802014 |
| H-RAS (1–166) | Buhrman et al., 2010 | N/A |
| pBABE-hygro vector | Addgene | Cat#: 1765 |
| pCMV5-Venus-K-Ras4B | Terrell et al., 2019 | N/A |
| pET-21a(+) vector | EMD Biosciences | https://www.emdmillipore.com/US/en/product/pET-21+-DNA-Novagen,EMD_BIO-69770www.emdmillipore.com/US/en/product/pET-21+-DNA-Novagen,EMD_BIO-69770 |
| pET28 vector | EMD Biosciences | https://www.emdmillipore.com/US/en/product/pET-28a+-DNA-Novagen,EMD_BIO-69864www.emdmillipore.com/US/en/product/pET-28a+-DNA-Novagen,EMD_BIO-69864 |
| pGEX-4T2-A-RAF(17–94) | Smith and Ikura, 2014 | N/A |
| pGEX-4T2-B-RAF(150–233) | Smith and Ikura, 2014 | N/A |
| pGEX-4T2-C-RAF(53–137) | Fang et al., 2020 | N/A |
| pGEX-4T2-RASSF5(199–367) | Smith and Ikura, 2014 | N/A |
| pLHCX-WT-RafReg-Rluc8 (A-, B-, C-Raf) | Terrell et al., 2019 | N/A |
| pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA | Addgene | Cat#: 61591 |
| Raf1-RBD | Addgene | Cat#: 13338 |
| RAS pathway collection v2.0 | Addgene | Kit#: 1000000070 |
| RASSF1 | Addgene | Cat#: 70535 |
| RASSF10 | Addgene | Cat#: 70537 |
| RASSF2 | Addgene | Cat#: 70539 |
| RASSF3 | Addgene | Cat#: 70541 |
| RASSF4 | Addgene | Cat#: 70543 |
| RASSF5 | Addgene | Cat#: 70545 |
| RASSF6 | Addgene | Cat#: 70547 |
| RASSF7 | Addgene | Cat#: 70549 |
| RASSF8 | Addgene | Cat#: 70551 |
| RASSF9 | Addgene | Cat#: 70553 |
|
| ||
| Software and algorithms | ||
|
| ||
| CHARM-GUI (solution builder) | Brooks et al., 2009; Jo et al., 2008; Lee et al., 2016 | https://charmm-gui.org/?doc=input/solution |
| Chimera (ver. 1.15) | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Coot | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| GraphPad (ver. 9.1.0) | Prism | https://www.graphpad.com |
| GROMACS (ver. 2020.1) | Abraham et al., 2015 | http://www.gromacs.org |
| HKL3000 package | Otwinowski and Minor, 1997 | https://hkl-xray.com/hkl-3000 |
| Image Studio lite (ver. 5.2.5) | LI-COR | https://www.licor.com/bio/image-studio/ |
| NMRPipe | Delaglio et al., 1995 | https://groups.io/g/nmrpipe |
| CcpNmr | Vranken et al., 2005 | N/A |
| PHENIX (ver. 1.11.1–2575) | Adams et al., 2010 | https://phenix-online.org |
| PyMOL (ver. 2.4.0) | Schrodinger | https://pymol.org/2/ |
| Sequest | Eng et al., 1994 | http://fields.scripps.edu/yates/wp/?page_id=17 |
| SnapGene (ver. 5.2.4) | GSL Biotech | N/A |
| VMD (ver. 1.9.4a38) | Humphrey et al., 1996 | http://www.ks.uiuc.edu/Research/vmd/ |
| XDS | Kabsch, 2010 | https://xds.mr.mpg.de |
|
| ||
| Other | ||
|
| ||
| 96-Well Clear Bottom Black or White Polystyrene Microplates | Corning | Cat#: 3917 |
| 96-well black-walled plate (OptiPlate) | Perkin Elmer | Cat#: 6005270 |
| 384-well white-walled plate (CulturePlate) | Perkin Elmer | Cat#: 6007680 |
| Accela600 liquid chromatography pump | Thermo Fisher Scientific | N/A |
| AKTA Pure | Cytiva | http://www.cytivalifesciences.com/en/us/shop/chromatography/chromatographysystems/akta-pure-p-05844 |
| Cellometer ®AutoT4 cell counter | Nexcelom Bioscience | https://www.nexcelom.com/nexcelomproducts/automated-cell-counters/cellometer-auto-t4-automated-cellcounter/ |
| ChemiDoc XRS+ System | Bio-Rad | Cat#: 1708265 |
| Epson Perfection V600 Photo Scanner | Epson | Cat#: B11B198011 |
| Famos Autosampler | LC Packings | N/A |
| HiTrap QHP column | Cytiva | Cat#: 17115401 |
| HiTrap TALON column | Cytiva | Cat#:29048565 |
| MicroMax007HF | Rigaku | N/A |
| Mono-Q column | Cytiva | Cat#: 17516601 |
| NEO III HD 800MHz spectrometer | Bruker | N/A |
| Ni-NTA beads | QIAGEN | Cat#: 30210 |
| NT8 Crystallization robot | Formulatrix | https://formulatrix.com/proteincrystallization-systems/nt8crystallization-robot/ |
| Nucleofector® II Device | Lonza | N/A |
| Octet RED-384 biolayer interferometry biosensor instrument | Sartorius | https://www.sartorius.com/en/products/protein-analysis/octet-label-freedetection-systems |
| Odyssey ®CLx | LI-COR | https://www.licor.com/documents/8prh6ps2abjbemx68412 |
| PD-10 desalting column | Cytiva | Cat#: 17085101 |
| PHERAstar Plus plate reader | BMG Labtech | https://www.bmglabtech.com/pherastardetection-system/ |
| Phoenix liquid handler | ArtRobbins | https://www.artrobbins.com/crystalphoenix |
| Protein G Sepharose | Cytiva | Cat#: GE17–0618-01 |
| Q Exactive mass spectrometer | Thermo Fisher Scientific | N/A |
| R-Axis IV2+ | Rigaku | N/A |
| RockImager Protein Crystallization Imager | Formulatrix | https://formulatrix.com/proteincrystallization-systems/rock-imagercrystallization-imagers/ |
| StageTip | N/A | N/A |
| Superdex 75 10/300 column | Cytiva | Cat#: 17517401 |
| Synergy H1 Hybrid Multi-Mode Reader microplate | BioTek | https://www.biotek.com/products/detection-hybrid-technology-multi-modemicroplate-readers/synergy-h1-hybridmulti-mode-reader/ |
| Tecan Infinite M1000 plate reader | Thermo Fisher Scientific | N/A |
| Ultra-15 Centrifugal Filter Units (10kDa MWCO) | Fisher Scientific | Cat#: UFC901008 |
| Zeba™ Spin Desalting Columns | Thermo Fisher Scientific | Cat#: PI89892 |
| Protein model: Wildtype H-RAS GppNHp | Buhrman et al., 2010 | 3K8Y |
| Protein model: Wildtype H-RAS GDP | Scheidig et al., 1999 | 1CTQ |
| Protein model: Wildtype H-RAS bound to GAP and AlFx | Scheffzek et al., 1997 | 1WQ1 |
| Protein model: H-RAS G12V/A59T bound to GTP | Krengel et al., 1990 | 521P |
| Protein model: RND3 bound to GTP | Fiegen et al., 2002 | 1M7B |
| Protein model: RND1 bound to GTP | N/A | 2CLS |
| Protein model: DIRAS2 bound to GDP | N/A | 2ERX |
| Protein model: DIRAS1 bound to GDP | N/A | 2GF0 |
| Protein model: ARL6 bound to GTP | Structural Genomics Consortium | 2H57 |
| Protein model: Wildtype K-RAS bound to GDP | Hunter et al., 2014 | 4OBE |
| Protein model: Wildtype H-RAS bound to the RBD domain of RASSF5 | Stieglitz et al., 2008 | 3DDC |
| Protein model: Wildtype H-RAS bound to the RBD of C-RAF | Fetics et al., 2015 | 4G0N |
| Protein model: Wildtype H-RAS bound to the RBD-CRD of C-RAF | Cookis and Mattos, 2021 | 7JHP |
| Protein model: Wildtype K-RAS bound to the RBD-CRD of C-RAF | Tran et al., 2021 | 6XI7 |
| Protein model: H-RAS G12V bound to the RBD of PI3K | Pacold et al., 2000 | 1HE8 |
| Protein model: Wildtype K-RAS bound to the RBD-PH domain of SIN1 | Castel et al., 2021 | 7LC1 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All cell lines were cultured at 37°C, ~95% humidity, and 5% CO2. Short tandem repeat (STR) genotyping, performed by LabCorp, was done to ensure cell line authenticity.
SNU-175 and LIM1215 cells
Cells were cultured in RPMI containing 10% FBS and 1% PenStrep. Cells were maintained according to published protocols (Ku and Park, 2005; Oh et al., 1999; Whitehead et al., 1985).
HEK293t, HEK293FT, and NIH 3T3 cells
Cells with cultured in DMEM containing 10% FBS and 1% PenStrep. Cells were split every 2–3 days.
HeLa cells
Cells with cultured in EMEM containing 10% FBS and 1% PenStrep. Cells were split every 2–3 days.
Mouse embryonic stem (mES) cells
K-Ras+/LSL-wildtype and K-RasA59T/LSL-A59T mES cells were cultured on both commercial and homemade irradiated feeder MEFs (1.5–2.0×106 cells per gelatin coated 10cm dish). Cells were grown in the following media: 425 mL of GIBCO Knockout DMEM, 75 mL of heat inactivated HyClone Fetal Bovine Serum, 5 mL 100x Glutamax, 3.5 μL of 14.3 M Mercaptoethanol, 5 mL 100X MEM non-essential amino acid solution, 5 mL 100X PenStrep, and 1 μL/mL of ESGRO Leukemia inhibitory factor (LIF). mES colonies were grown until visible to the naked eye before splitting. Cell media was refreshed every day, and always 2–3 h before sub-culture, clone picking, or nucleofection.
METHOD DETAILS
Sequence alignment of small GTPases with autophosphorylation motif
Sequences for K-RAS, ARL6, DIRAS1, DIRAS2, RAB40A, RAB40B, RAB40C, RASD1, RASD2, RND1, RND2, and RND3 were extracted from the NCBI database (Coordinators, 2016). Alignments were done using the T-COFFEE algorithm provided by SnapGene® software (GSL Biotech).
Bottom-up mass spectrometry
Approximately 20 μg of purified 6xHis-tagged K-RAS4BA59T were reduced with 5 mM tris(2-carboxyethyl)phosphine for 30 min at room temperature, alkylated with 10 mM iodoacetamide for 30 min at room temperature in the dark, and quenched with 10 mM dithiothreitol for 15 min at room temperature. The sample was divided into two replicates, precipitated by trichloroacetic acid precipitation, and digested with 1:50 (w/w) chymotrypsin in 100 mM Tris-HCl and 10 mM CaCl2 for 18 h at 37°C with shaking. The samples were desalted using a StageTip, dried by vacuum centrifugation and solubilized in 5% acetonitrile and 5% formic acid. LC-MS/MS analysis was performed on a Q Exactive mass spectrometer coupled with a Famos Autosampler and an Accela600 liquid chromatography pump. Peptides were separated on a 100 μm inner diameter microcapillary column packed with :25 cm of Accucore C18 resin (2.6 μm, 150 Å). For each analysis we loaded ~1 μg onto the column.
Peptides were separated using a one-h method of 5%–25% acetonitrile in 0.125% formic acid with a flow rate of :300 nL/min. The scan sequence began with an Orbitrap MS1 spectrum with the following parameters: resolution 70,000, scan range 300–1500 Th, automatic gain control target 1 × 105, maximum injection time 250 ms, and centroid spectrum data type. We selected the top twenty precursors for MS2 analysis which consisted of high-energy collision dissociation with the following parameters: resolution 17,500, AGC 1 × 105, maximum injection time 60 ms, isolation window 2 Th, normalized collision energy 30, and centroid spectrum data type. The underfill ratio was set at 1%, which corresponds to a 1.1 × 104 intensity threshold. Unassigned and singly charged species were excluded from MS2 analysis and dynamic exclusion was set to automatic.
Spectra were searched using Sequest with a 3 ppm precursor mass tolerance, 0.03 fragment ion tolerance, and no limit on internal cleavage sites (Eng et al., 1994). Cysteine alkylation was set as a fixed modification, methionine oxidation and phosphorylation of serine, threonine, and tyrosine were included as variable modifications. Spectra were searched against a custom database containing the K-RAS4BA59T sequence, common contaminants, and the reversed peptide sequences. False discovery rate was estimated by linear discriminant analysis and applied at one percent at the peptide level (Elias and Gygi, 2007; Peng et al., 2003).
Molecular dynamics simulations
Starting coordinates for molecular dynamic simulation were generated from the crystal structures published here and from the PDB. For simulations of GDP-bound or GTP-bound K-RAS, we used the starting wild-type structures 6MBU (Dharmaiah et al., 2019) and 4DSO (Maurer et al., 2012). Starting PDB coordinates for RND1, RND3, DIRAS1, DIRAS2, and ARL6 were 2CLS, 1M7B (Fiegen et al., 2002), 2GF0, 2ERX, and 2H57, respectively. Starting structures went through an extra round of modeling and refinement to add in missing residues and remove alternate conformations. Structures were converted into the GDP- or GTP-bound states, or mutated, in silico. Preparation of starting files, including ‘residue 59’ phosphorylation was done using ‘solution builder’ available from CHARM-GUI (Brooks et al., 2009; Jo et al., 2008; Lee et al., 2016). Charged residues, including protein termini, were protonated or deprotonated in accordance with neutral pH. A cubic box with edges 10 Å from each protein was created and filled with TIP3P water molecules and neutralized with Cl− and Na+ ions to 150 mM. Minimizations, equilibrations, and simulations were done using GROMACS (ver. 2020.1) and a GPU server featuring 8x Tesla v100 workstation, on the O2 High Performance Compute Cluster, supported by the Research Computing Group, at Harvard Medical School. Solvated systems were energy minimized by steep integration for 5000 steps or at a maximum force of 1000 kJ/mol/nm or less. The Verlet cutoff scheme was used for nonbonded atoms and the LINCS algorithm was applied to covalent H-bonds. Short-range van der Waals interactions were switched off from 1.0–1.2 nm, and long-range interactions were computed using the Particle Mesh Ewald method. Simulation temperatures were maintained at 310K using Nose-Hoover extended ensemble. The isothermal-isobaric ensemble (NPT) was generated using the Parrinello-Rahman barostat method with periodic boundary conditions. Simulations were done for 800–900 ns using the GROMOS force-field.
Validation and analyses steps were done in GROMACS, including cluster analysis using the GROMOS algorithm (Daura et al., 1999). Distance measurements and visual analyses were done using PyMOL and VMD (Humphrey et al., 1996). Frequency distributions that depict the distances between atoms throughout the simulations were calculated using a 0.20Å bin cutoff in PRISM.
Protein purification for biochemical assays
Human K-RAS4B and the catalytic domains of GAP (715–1047) and SOS11 (564–1049) were synthesized with N-terminal hexa-histidine tags (6xHis) and inserted in the pET21a plasmid for E. coli expression by GENEWIZ. K-RAS mutants (i.e., G12D, A59T, A59E, A59S, A59H, and A59Y) were made by site-directed mutagenesis. All expressed K-RAS proteins were full-length with the C-terminal two amino acids removed to mimic post-translational processing (Hancock et al., 1991). Protein expression was done using chemically transformed BL21 (DE3) strain of E. coli and Terrific Broth media. A 30mL culture of broth was inoculated with BL21 E. coli cells with the appropriate tagged protein and allowed to grow overnight at 37°C with agitation. The following morning 25mL of culture was applied to a 1L culture of terrific broth, and protein expression was induced with 120mg/L of IPTG after E. coli reached an O.D. 0.8. After 6 h at 37°C the cell solution was centrifuged at 10,000RPM at 4°C and pellets were stored at −80°C. His-tagged protein was purified from E. coli paste as follows. Frozen pellets were re-suspended in sterile filtered buffer TA (20 mM Tris pH 7.5, 50 mM NaCl, 5 mM MgCl2, 5% glycerol, 10 μM GDP, 156 mL/L 2-mercaptoethanol) containing 4 mM PMSF at < 0.5mg/mL by agitation at room temperature for 15–30 min. E. coli cells were lysed by sonication on ice. Lysate was then clarified for 30 min at 4°C and 14,000 RPM. Chromatography of supernatant was done using an AKTA FPLC. Supernatant was first subjected to immobilized metal affinity chromatography (IMAC) using a 5 mL Hi-Trap TALON column. The running buffer for IMAC was buffer TA, and sterile buffer TB (20 mM Tris pH 7.5, 50 mM NaCl, 5 mM MgCl2, 200 mM Imidazole, 5% glycerol, 10 μM GDP, 156 μL/L of 2-mercaptoethanol) was used for gradient elution. Post-elution, positive fractions were screened by SDS-PAGE, pooled and concentrated to < 1mL using a 10,000 kDa Amicon Ultra spin concentrator. Concentrated protein was then buffer exchanged into QA buffer (20 mM Tris pH 8.0, 50 mM NaCl, 5 mM MgCl2, 5% glycerol, 10 μM GDP, 156 μL/L 2-mercaptoethanol) using a ZEBA spin column per manufacturer protocol. Buffer exchanged protein was applied to a 5 mL HiTrap QHP column using a 30% gradient of QB buffer (20 mM Tris pH 8.0, 1.0 M NaCl, 5 mM MgCl2, 5% glycerol, 10 μM GDP, 156 μL/L 2-mercaptoethanol) over 60 mL. Positive fractions were then pooled and subjected to buffer exchange twice using ZEBA spin columns. The first exchange was into protein into stabilization buffer (20 mM Tris pH 7.5, 10 mM NaCl, 1 mM MgCl2, 1% glycerol, 1 mM DTT) containing 5 mM EDTA and then protein was re-exchanged into stabilization buffer without EDTA. Protein concentration was then determined, and equimolar GDP was added to KRAS4B protein. Protein for nucleotide hydrolysis and exchange were diluted to approximately 100μM, including GAP334 and SOS1cat, flash frozen in liquid nitrogen in 50–150 μL aliquots for single use, and then stored at −80°C. To obtain phosphorylated K-RAS4B, after the IMAC step, K-RAS4BA59T was incubated overnight or longer in the presence of GTP, and then purification was continued. Post-QHP, phosphorylated protein was identified by SDS-PAGE and pooled separately from the non-phosphorylated form.
Crystallization of Ala59 mutants of H-RAS and K-RAS
Purification and crystallization of truncated (residues 1–166) and untagged H-RASA59T and H-RASA59E was done using a previously published protocol (Johnson et al., 2015). H-RASA59T bound to GppNHp was concentrated to 12.1 mg/mL, flash frozen, and stored at −80°C before crystallization. H-RASA59E bound to GppNHp and GDP were stored the same way but concentrated to 18.1 and 15.2 mg/mL, respectively. All crystals were grown using reagents from Hampton Research. Crystals were grown using the hanging drop vapor diffusion method in 24-well plates sealed with Vaseline at 18°C. Mother liquor constitution was unique for each crystal grown, and each crystal was grown against a mother liquor well volume of 402 μL. Crystals were harvested at 2–4 weeks and flash frozen using mother liquor with 30% glycerol. The first crystal of H-RASA59T bound to GppNHp was crystallized in 2 μL by2 μL drops using the following reservoir solution: 2.6 mM NaCl, 1.0 mM MgCl2, 15.7 mM HEPES pH 7.5, 2.5 mM DTT, 37.3 mM Ca(OAc)2 and 20.5% PEG 3350. The second crystal of H-RASA59T was crystallized in 1 μL by 1 μL drops using the following reservoir solution: 10 mM Mg(OAc)2, 44.8% PEG400, 15.7 mM HEPES pH 7.5, 1.0 mM MgCl2, 3.4 mM NaCl, and 2.5 mM DTT. H-RASA59T crystals contained one molecule of H-RASA59T in the asymmetric unit and exhibited P3221 symmetry. H-RASA59E bound to GDP was crystallized in 1 μL by 1 μL drops using the following reservoir solution: 2.6 mM NaCl, 1.0 mM MgCl2, 15.7 mM HEPES pH 7.5, 2.5 mM DTT, 43.5 mM Ca(OAc)2, and 20.5% PEG 3350. H-RASA59E bound to GppNHp was crystallized in 1 μL by 1 μL drops using the following reservoir solution: 2.6 mM NaCl, 1.0 mM MgCl2, 15.7 mM HEPES pH7.5, 2.5 mM DTT, 9.95 mM Ca(OAc)2, and 19.9% PEG 3350. Both H-RASA59E crystals contained two molecules in the asymmetric unit, but GppNHp crystals grew with P1211 symmetry while the GDP crystals grew with P1 symmetry. Data collection for all crystals was done on a home source MicroMax007HF with Cu2+ anode and tungsten filament, and a R-Axis IV2+ detector from Rigaku. Indexing, integration and scaling data processing steps were done using the HKL3000 package (Otwinowski and Minor, 1997). For molecular replacement, a previously refined model of wild-type H-RAS (PDB code 1CTQ) was used for both H-RASA59T and H-RASA59E crystals bound to GppNHp (Klink and Scheidig, 2010). H-RASA59E bound to GppNHp was used as starting models for the H-RASA59E GDP structures. A single round of simulated annealing was performed before refinement. Molecular replacement and structure refinement was done use the PHENIX program (version 1.11.1–2575) (Adams et al., 2010).
Protein for crystallization of K-RASA59E bound to GDP was acquired by a different purification scheme than above. His-tagged K-RAS4BA59E was overexpressed in E. coli BL21 (DE3) and purified in the presence of 20 μM GDP. Briefly, cells were grown at 37°C in TB medium in the presence of 100 μg/mL of ampicillin to an OD of 0.8, cooled to 17°C, induced with 500 μM IPTG, incubated overnight at 17°C, collected by centrifugation, and stored at −80°C. Cell pellets were lysed in buffer A (25 mM HEPES, pH 7.5, 500 mM NaCl, 5 mM MgCl2, 5% glycerol, 20 mM GDP, 7 mM 2-mercaptoethanol, and 20 mM Imidazole), and the resulting lysate was centrifuged at 30,000 g for 40 min. Ni-NTA beads (QIAGEN) were mixed with cleared lysate for 30 min and washed with buffer A. Beads were transferred to an FPLC-compatible column, and the bound protein was washed further with buffer A for 10 column volumes and eluted with buffer B (25 mM HEPES, pH 7.5, 500 mM NaCl, 5 mM MgCl2, 5% glycerol, 20 μM GDP, 7 mM 2-mercaptoethanol, and 400 mM Imidazole). The eluted sample was concentrated, then 10-fold diluted in buffer C (20 mM Tris, pH 8.0, 200 mM NaCl, 5 mM MgCl2, 5% glycerol, 20 μM GDP, and 1 mM DTT), applied to Mono-Q column, and eluted by using 50–500 mM NaCl gradient. K-RAS containing fractions were concentrated and passed through a Superdex 75 16/600 column in a buffer containing 25 mM HEPES, pH 8.0, 200 mM NaCl, 5 mM MgCl2, 5% glycerol, 20 μM GDP, and 1 mM DTT. HRV 3C protease was added to KRAS mutants containing fractions and incubated overnight at 4°C, followed by passing through Ni-NTA column to remove His-tag and 3C protease. Fractions were pooled, concentrated to approximately 20 μg/mL (for LC-ESI-MS) or 40 mg/mL (for crystallization), and frozen at −80°C. For crystallization, a Formulatrix NT8, RockImager and ArtRobbins Phoenix liquid handler was used to dispense a 100 nL sample of 800 μM K-RASA59E with an equal volume of 1.5 M NaMalonate and 0.1 M HEPES pH 7.5 for crystallization by sitting-drop vapor diffusion. Crystals were grown at 27°C for three days before harvest. Diffraction data were collected at beamline 24ID-E of the NE-CAT at the Advanced Photon Source (Argonne National Laboratory). Datasets were integrated and scaled using XDS (Kabsch, 2010). Structures were solved by molecular replacement using the program Phaser and the search model PDB entry 5TAR (Dharmaiah et al., 2016; McCoy et al., 2007). Iterative manual model building and refinement using Phenix and Coot led to a model with excellent statistics (Adams et al., 2010; Emsley and Cowtan, 2004). Statistics for all crystal data can be found in Table 1.
NMR spectroscopy
Isotopically 15N labeled K-RAS (residues 1–185 of K-RAS4B with a C118S substitution bearing A59T or A59E substitutions or wild-type A59) was expressed with an N-terminal His-tag from the pET28 vector in the E. coli strain BL21 (DE3 Codon+). A codon-optimized sequence encoding K-RAS4B was synthesized (GenScript) and the A59 codon was mutated using QuikChange site-directed mutagenesis (Agilent). Transformed bacteria was cultured at 37°C in M9 minimal media in the presence of kanamycin and chloramphenicol and supplemented with 1 g/L 15N ammonium chloride until the O.D.600 nm reached 0.6. Protein expression was then induced with 0.2 mM IPTG (Isopropyl β-D-1-thiogalactopyranoside) at 155C overnight.
After centrifugation of the culture, the cell pellets were re-suspended with lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% NP-40, 10% Glycerol, 10 mM Imidazole, 5 mM MgCl2, 1 mM PMSF, 10mM β-mercaptoethanol and lysozyme at pH 8.0) and lysed by sonication. Following centrifugation, His-tagged K-RAS proteins were purified by Ni2+-NTA column affinity chromatography from the soluble fraction, the buffer was exchanged to reduce the imidazole concentration, then K-RAS was further purified by size exclusion chromatography (Superdex™ S75 26–60 column) in a running buffer comprising 20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, and 2 mM tris(2-carboxyethyl)phosphine (TCEP), pH 7.4. RAS proteins naturally co-purify bound to GDP (Feuerstein et al., 1987), but the purity of K-RASA59T (i.e., lack of K-RASA59Tp) was confirmed by the NMR spectra (data not shown).
1H-15N HSQC spectra were collected with 8 scans at 25°C on a Bruker NEO III HD 800MHz spectrometer equipped with a 5 mm TXO CryoProbe. K-RAS samples were concentrated (wild-type: 500 μM; K-RASA59T: 360 μM; K-RASA59E: 500 μM) in size exclusion buffer plus 5% (vol/vol) D2O. NMR data were processed and analyzed using NMRPipe (Delaglio et al., 1995) and CcpNmr (Vranken et al., 2005).
Chemical shift perturbations (CSPs, chemical shift changes of the NH cross-peaks of the A59T/E mutants relative to wild-type), were calculated using the formula ΔδNH.N(ppm) = Ö ([(ΔH)]2+[(ΔN/5)]2) and plotted against residue number using GraphPad 9.1.0. The NH resonances of the mutants were assigned based on previous assignment of wild-type K-RAS (BMRB entry 27720) together with a 3D 15N-edited NOESY HSQC spectrum (mixing time 120ms). Chemical shift changes were mapped onto the structure of wild-type K-RAS bound to GDP (PDB 6MBU) as well as our new structure of K-RASA59E using Chimera 1.15.
Autophosphorylation and dephosphorylation experiments
Phosphorylated β-casein was used as a positive control for Lambda Phosphatase (LMP) activity for dephosphorylation experiments. A solution of 90 μL of tagged K-RASA59T (or β-casein) at 10 mg/mL and 10 μL of GTP or GDP at 50 mg/mL were incubated overnight at 37°C. After overnight incubation, 10μL of the GTP and K-RASA59T mixture (or β-casein) was incubated at 30°C or 95°C for 10 min, and subsequent dephosphorylation was tested by adding 1.0 μL of this solution to 49 μL of 1x phosphatase buffer supplemented with MnCl2 and either 400 units of LMP or water. Dephosphorylation was allowed to proceed for 2, 4, and 6 h at 30°C.
The kinetics of autophosphorylation were also done using purified K-RASA59T with SOS1 or GAP334. Autophosphorylation reactions were done using a 20X reaction buffer (1.0 M Tris-HCL, 20 mM MgCl2, pH 7.5). Purified proteins were diluted to initial starting concentrations of 250 μM K-RASA59T and 100 μM GAP and SOS1. For the K-RASA59T reaction without regulators, reactions were run at the following conditions: 2.5 μL of 20X reaction buffer, 5 μL GTP (25 mg/mL) dissolved in water, 1–16 μL of 250μM KRAS4BA59T and finally balanced to 50 μL with Ultrapure water. For autophosphorylation in the presence of GAP and SOS1, the above reaction mixture was used, except 4 μL of 250 μM K-RASA59T was fixed for each reaction and 0.5–20 μL of purified regulator was added to each reaction and balanced with water to 50 μL. Each autophosphorylation experiment was done over 8 h at 37°C. Each h, a 2 μL sample was taken from each reaction and then added to 2 μL of 6X SDS-PAGE sample buffer and 8 μL of water. Samples were then boiled for 5 min at 70°C. Changes in phosphorylation were detected by 12.5% SDS-PAGE. While the dephosphorylation experiments were done using Coomassie blue staining, the kinetics of autophosphorylation were measured using western blot. Quantification was performed by measuring changes in the upper band versus the sum of the upper and lower bands.
To detect and compare autophosphorylation in K-RASA59S, K-RASA59H, and K-RASA59Y compared to wildtype K-RAS and K-RASA59T, proteins were purified using the Ni-NTA bead method (above). Purified wildtype K-RAS and K-RASA59T, K-RASA59S, K-RASA59H, and K-RASA59Y, at a concentration of 100 μM, were incubated at 37°C for six h with and without 5mM GTP in a reaction buffer containing 50 mM Tris-pH7.5 and 1 mM MgCl2 and analyzed by LC-ESI-MS as previously described (Zhang et al., 2012).
RAS hydrolysis and nucleotide exchange assays
Assays were done with a BioTek microplate reader using black-walled non-TC treated 96-well plates. The GTP hydrolysis assay described here is a modified form of the EnzChek Phosphate Assay Kit assay. For GTP hydrolysis, both preloading of GTP and measurement of inorganic phosphate were done in a 96-well plate format. First, a GTP preloading mixture consisting of 72 μM K-RAS, 10 mM GTP, 4 mM EDTA, and 2 mM DTT in a final volume of 25 μL was stored on ice until use. Second, a GTP hydrolysis mixture consisting of 60 mM Tris, 60 mM NaCl, 6 mM MgCl2, 1.6 mM DTT, 240 μM of 2-amino-6-mercapto-7-methylpurine riboside (MESG), 1.2 U/mL of purine nucleoside phosphorylase (PNP), and varying concentrations of GAP was brought to a final volume of 125 μL. The GTP exchange and reaction mixtures were incubated at 37°C for 35 min in the microplate reader in separate wells in the black-walled 96-well plate. After 35 min, the GTP preloading mixture was added to the hydrolysis mixture and nucleotide hydrolysis was measured over 90 min at 37°C. A change in inorganic phosphate due to GTP hydrolysis was measured by the difference in absorbance at 360nm from a reference reaction without K-RAS protein.
Nucleotide exchange assays were performed for 2’/3′-O-(N-Methyl-anthraniloyl)-guanosine-5′-triphosphate (mant-GTP) or mant-GDP. Mant-GTP and GDP stocks were purchased as 5 mM stocks from Invitrogen. To measure nucleotide exchange at 37°C, we generated a protein mixture containing 1.6 μM K-RAS, 52.5 mM Tris pH 7.5, 52.5 mM NaCl, 5.25 mM MgCl2, 4.2 mM DTT and varying concentrations of SOS1 at a final volume of 140 μL, and a second mixture containing 225 μM of mant-GTP or GDP, 48.6 mM Tris, 48.6 mM NaCl, 4.9 mM MgCl2, 1.9 mM DTT at pH7.5 at a final volume of 10 μL. The two mixtures were placed in the black-walled 96-well plate, wrapped in foil, and incubated in the dark for 30–40 min. Once combined, measurement of nucleotide exchange was taken every 65 s for 2 h at 37°C. Measurement of mant-nucleotide association with K-RAS was done by excitation at 360nm, detection of fluorescent emission at 440nm, and subtraction of a reference reaction lacking K-RAS protein. Replicate measurements of hydrolysis and nucleotide exchange were used to determine an apparent first-order rate constant (kobs) using the program GraphPad Prism (Notredame et al., 2000).
Measurement of K-RASA59T in cells and transformation experiments
Measurement of autophosphorylation in response to human Epidermal Growth Factor (hEGF) or insulin signaling was done using SNU-175 cells seeded at a starting density of 7.5×105 cells/well in a 6-well plate, allowed to adhere overnight, and then serum starved for 24 h. After serum starvation, induction was done by either addition of vehicle (BSA suspended in HEPES), 1.5 μg/mL of hEGF, or 5 μg/mL of insulin. To measure changes in K-RAS autophosphorylation, cells were lysed at different times in the following manner. First, cells were gently washed once with 1 mL of phosphate buffered saline solution (PBS). Next, 150 μL of radioimmunoprecipitation assay (RIPA) buffer was applied to the well and lysis was allowed to proceed for 15 min at 4°C with gentle rocking. Cell lysate was collected and clarified by high-speed centrifugation at 4°C for 15 min before flash freezing in liquid nitrogen and storage at −80°C. For K-RAS knockdown experiments, shRNA in the pLKO-tet plasmid vector targeting KRAS, or GFP as a negative control, was obtained from Addgene and lentivirus was produced as in Ref. (Shao et al., 2014) using HEK293t cells. SNU-175 cells were infected for 24 h and allowed to expand for 1 week before selection with puromycin. For K-RAS knockdown experiments, puromycin selected cells were seeded at a starting density of 5×104 cells/well in a 24-well plate. Cells were allowed to adhere overnight before induction of shRNA against KRAS using RPMI 1640 media supplemented with 2 μg/mL of doxycycline. To monitor changes in K-RAS autophosphorylation in response to KRAS knockdown, cells were lysed every 24 h using the same method as above. To measure the effect of K-RAS knockdown on SNU-175 cell proliferation, parental SNU-175 cells, or cells infected with shRNA suppressing K-RAS or GFP, were grown for 1 week at 2 μg/mL of doxycycline. After one week, 50,000 cells/well were seeded in 24-well plates and allowed to grow for 13 days. A doxycycline positive and negative set of proliferation experiments were performed in parallel.
Ectopic expression of mutant HA-tagged K-RAS4B was done in NIH 3T3 cells using the pBABE-hygro vector and the retroviral pCL packaging system in HEK293t cells (Morgenstern and Land, 1990; Naviaux et al., 1996). First, HEK293t cells were lipofectamine transfected at 70% confluence. On the third day, virus was collected and used to infect NIH 3T3 cells at 20% confluence. Infection proceeded overnight in the TC incubator, and after 48 h, infected cells underwent selection with 300 μg/mL of hygromycin for one week.
Cell lysate for effector binding assays were collected in the following manner. Infected NIH 3T3 cells were grown to high density in 175 mm dishes, and then washed twice with ice-cold 10 mL of PBS and lysed with 400–600μL of MLB solution (25 mM HEPES pH 7.5, 150 mM NaCl, 1% IGEPAL-CA630, 0.25% sodium deoxycholate, 10% glycerol, and 10 mM MgCl2). Lysate and cell debris were immediately scrapped into Eppendorf tubes, rotated for 30 min and then clarified for 30 min using high-speed centrifugation at 4°C. Cell proliferation was measured over three days at the indicated times at 10% FBS in at least quadruplicate by seeding T25 culture flasks with 5000 cells. Cells were collected and counted using a Nexcelom Bioscience Cellometer® AutoT4 cell counter.
Anchorage independent growth assays were done in triplicate, with quadruplicate repeats, in 12-well plates using only the top and bottom rows. For each well, 2.35×104 cells were suspended in 1 mL of soft-agar media (0.83X DMEM, 8.3% FBS, 83 units/mL penicillin, 83 μg/mL of streptomycin, and 0.3% bacto-agar) and carefully placed on top of 1mL of set dense soft-agar (0.67X DMEM, 6.7% FBS, 67 units/mL penicillin, 67μg/mL of streptomycin, and 0.6% bacto-agar). The following day, and every third day after that for three weeks, cells were fed with 250 μL of cell media (1X DMEM, 10%FBS, 100 units/mL penicillin, 100 μg/mL of streptomycin). Colony growth was measured by first carefully removing media from each well, applying 1 mg/mL of MTT dissolved in PBS, and allowing MTT reduction to occur overnight in the TC incubator. The next day, excess solution was removed from each well and plates were scanned using an Epson V600 Photo Scanner. Colonies were counted and analyzed using ImageJ and GraphPad Prism (Notredame et al., 2000; Schneider et al., 2012).
CRISPR mutagenesis of mES cells and related experiments
mES cells with the genotype K-Ras+/LSL-wildtype were modified using the SaCas9 CRISPR system to be K-RasA59T/LSL-A59T. The SaCas9 plasmid was modified to contain gRNA sequence targeting exon 2 and codon 59 according to the protocol shown in (Ran et al., 2015). Before nucleofection, mES cells and feeders were washed twice with PBS, and then dissociated by incubation for 10–15 min at 37°C with 0.05% Trypsin-EDTA. Cell solution was collected, excess media was added to inactivate trypsin, and then cells were spun down for 5 min at 4°C at 80xg. Cells were resuspended in media and then applied to a gelatin coated 10 cm dish at 37°C. After 30 min, the non-adherent mES cells were collected and spun down for 5 min at 4°C at 80xg. The cell pellet was resuspended in PBS and cel number was determined using a Nexcelom Bioscience Cellometer® AutoT4 cell counter. Using the same centrifuge conditions, a cell pellet of 3.0×106 K-Ras+/LSL-wildtype mES cells was collected and then redissolved in 90 μL of supplemented nucleofector solution prepared per the Lonza Mouse ES cell Nucleofector® kit protocol. Next, the cell solution was gently mixed with 10 μL of 1X TAE containing 40 μg of single-stranded DNA repair template, purchased from Integrated DNA Technologies, and 5 μg of modified SaCas9 plasmid that was purified from E.coli using the PureLink® HiPure Plasmid Filter Maxiprep kit.
Nucleofection was performed according to the Lonza Mouse ES cell Nucleofector® kit protocol. Briefly, the mES cell and DNA solution was carefully added to the supplied cuvette using a plastic pipette. Care was taken to limit air bubbles. The cuvette was then placed in a Nucleofector® II device and the program A-013 was run. Immediately after nucleofection, 500 μL of pre-warmed media was added to the cuvette and gently mixed using the plastic pipette. The entire solution was then added to single well of a gelatin coated 6-well dish with feeders. Cells were fed fresh media every day for 2–3 days. After 2–3 days, cells were collected, diluted 1:1000, and plated on three gelatin coated 10 cm dishes with feeders. The unused cells were spun down at 45C for 5 min at 80xg, resuspended in chilled mES freezing media (80% media, plus an extra 10% HyColone FBS and 10%DMSO), and then stored for later use at −80°C.
Colonies were grown until visible to the naked eye and then collected in 96-well plates containing 100 μL trypsin/well. After 5–10 min at 37°C, another 100 μL of media was added to each well, cells were resuspended, and then split between two gelatin coated 96-well plates containing feeders. mES clones were then transferred back to 37°C. After 2–4 days, mES clones in one plate were frozen and stored at −80°C by first washing with PBS, dissociating the cells with 50 μL 0.05% trypsin-EDTA for 10 min, and then mixing dissociated cells with 50 μL of 2X mES freezing media (60% media, plus 20% HyClone FBS and 20% DMSO). The other 96-well plate underwent genomic DNA extraction using the Quick-DNA kit from Zymo Research.
Besides the A59T mutation, a silent EcoRV restriction site was included in the DNA repair template for genotyping positive mES clones. To genotype clones, 5 μL of extracted genomic DNA was mixed with 0.25 μL of 100 μM of forward and reverse mES Ex2 genotyping primers each, 1 μL of dNTP,10 μL of 5X Phusion HF Buffer, and 0.5 μL of Phusion® High-Fidelity DNA Polymerase. PCR reactions were diluted to a final concentration of 50 μL using Ultrapure water and touchdown PCR, using a melting temperature Tm of 58°C, was performed (Korbie and Mattick, 2008). Once the PCR was complete, 12 μL of the PCR product was removed and mixed with 0.3 mL of EcoRV-HF, 2.5 μL of 10x CutSmart buffer, and 10.2 μL of Ultrapure water. These samples were digested for 1 h at 37°C, enzyme inactivated for 20 min at 65°C, and then run on 1.8%–2.0% TAE agarose gels containing GreenGlo. DNA bands were visualized using a ChemiDoc XRS+ System. Undigested PCR product migrates at ~850 bp, while a positive clone produces a band around ~700 bp. Positive clones were further validated by sequencing using the same PCR recipe, but with the forward and reverse mES Ex2 sequencing primers and a touchdown protocol using a Tm of 56°C. The PCR product was sequenced by Genewiz using the Forward sequencing primer. Finally, K-RasA59T/LSL-A59T mES cells were validated at the protein level by detection of an apparently higher migrating band visible by western blot, similar to what we observe in our SNU-175 cells, NIH 3T3 cells, and purified protein.
Detection of MAPK and Akt signaling by western blot was done in mES cells that were expanded in the presence of feeders, but then removed from feeders as above, before being plated in triplicate on gelatin coated dishes. For these experiments mES cells were serum starved for 6 h before lysis.
K-RAS effector binding and precipitation assays
The RAS binding domain of C-RAF kinase (RAF-RBD) (Brtva et al., 1995) and full-length human RASSF5 from the RAS clone collection were obtained from Addgene. The RAS clone collection was a gift from Dominic Esposito (Addgene kit 1000000070). GST tagged RAF1-RBD (GST-RAF-RBD) was expressed and purified from BL21 cells as in ref. (Taylor et al., 2001). GST-tagged RASSF5 (GST-RASSF5) sequence was transferred into the pDEST15 expression vector, and then expressed and purified from BL21-A1 cells. Our protocol for measuring effector-K-RAS interactions was done as in (Taylor et al., 2001), with the following changes. After protein induction, by either isopropyl β-d-1-thiogalactopyranoside (for GST-RAF-RBD) or arabinose (for GST-RASSF5), cells were washed once in HBS buffer (25 mM HEPES pH 7.5, 150 mM NaCl), resuspended in RLB solution (20 mM HEPES in pH 7.5, 120 mM NaCl, 10% glycerol) and then lysed by sonication. Lysate was clarified by high-speed centrifugation at 4°C, flash frozen and stored at −80°C until use. Before measuring mutant HA-K-RAS4B precipitation by RAF-RBD or RASSF5, HA-K-RAS4B in NIH 3T3 lysates were preloaded with nucleotide by diluting NIH 3T3 lysate in MLB to 2 μg/μL in the presence of 1.0 mg/mL of nucleotide, 15 mM EDTA, and incubated at 32°C for 30 min. After nucleotide preloading, 200 μL of nucleotide exchanged lysate was added to an equal volume of MLB buffer containing 10 μL of GST-RAF1-RBD or GST-RASSF5 loaded glutathione beads. Binding of K-RAS mutants to the different effectors was allowed to proceed for 2 h at 4°C with gentle rotation. After the precipitation reaction was complete, beads were washed three times (> 20,000xg at 4°C) with equal volumes of MLB. The activated state of HA-K-RAS4B in NIH 3T3 cells (i.e., cellular GTP-bound state) was done as above except cell lysates were prepared fresh, not subjected to chemical loading of nucleotide, and precipitation was done in a volume of 300 μL. K-RAS-effector precipitation was detected by western blot and detection of HA-tag. Affinity precipitation of other RASSF proteins was done as above for RASSF5 using plasmids provided by Addgene RAS clone collection (RASSF1: #70535, RASSF2: #70539, RASSF3: #70541, RASSF4: #70543, RASSF6: #70547, RASSF7: #70549, RASSF8: #70551, RASSF9: #70553, RASSF10: #70537).
BRET assay
293FT cells were seeded into 12-well dishes at a concentration of 1×105 cells/well. 16 h after plating, pCMV5-Venus-K-Ras4B and pLHCX-CMV-Raf-Rluc8 constructs were transfected into cells using a calcium phosphate protocol (Terrell et al., 2019). Duplicate 12-point saturation curves were generated in which the concentration of the energy donor construct (Rluc8) was held constant (62.5 ng) as the concentration of the energy acceptor plasmid (Venus) increased (0–1.0 μg). 48 h after transfection, cells from each well were collected, washed, and resuspended in PBS (500 μL). 30 μL of the cell suspension was plated in duplicate into wells of a 384-well white-walled plate and coelenterazine-h was added to a final concentration of 3.375 μM. The BRET and Rluc8 emissions were measured simultaneously using a PHERAstar Plus plate reader, with BRET monitored at 535 nm (bandwidth 30 nm) and Rluc8 measured at 475 nm (bandwidth 30 nm). To monitor the increasing levels of acceptor expression, 90 μL of the cell suspension was also plated in duplicate into wells of a 96-well black-walled plate (PerkinElmer OptiPlate), and Venus fluorescence was determined using an excitation wavelength of 485 nm (5 nm bandwidth) and fluorescence read at 530 nm (5 nm bandwidth) using a Tecan Infinite M1000 plate reader. The BRET ratio was determined by calculating the 535/475 ratio for a given sample (sample BRET) and was normalized to the BRET signal from cells expressing the donor construct alone to enable comparisons between experiments. The normalized mBRET signal was calculated as follows: (1000 X (sample BRET ratio/background BRET ratio)) – 1000. The acceptor/donor ratio (Venus emission/Rluc8 emission) for each data point was background corrected and equalized against a control where equal quantities of the Venus (acceptor) and Rluc8 (donor) constructs were transfected to allow for comparisons between experiments. Data was analyzed using GraphPad Prism. Non-linear regression was used to plot the best fit hyperbolic curve, and values for BRETmax and BRET50 were obtained from the calculated best fit curves.
Co-immunoprecipitation experiments
HeLa cells were plated at a concentration of 8 × 105 per 10 cm dish 18–24 h prior to transfection. pCMV5-Venus-K-RAS constructs were then transfected into cells using the XtremeGENE9 transfection reagent per the manufacturer’s instructions, using a 2:1 ratio of XtremeGENE9 to DNA. 48 h after transfection, serum-starved cells were washed twice with ice cold PBS and lysed in1% NP-40 lysis buffer (20mM Tris [pH 8.0], 137 mM NaCl, 10% glycerol, 1% NP-40 alternative, 0.15 U/mL aprotinin, 1 mM PMSF, 0.5 mM sodium vanadate, 20 μM leupeptin; 500mL/10 cm dish) for 15 min at 4°C on a rocking platform. Lysates were clarified by centrifugation at 14,000 RPM for 10 min at 4°C, after which the protein content was determined by Bradford assays. Lysates containing equivalent amounts of protein were incubated with either anti-GFP/Venus or anti-C-RAF and protein G Sepharose beads for 2 h at 4°C on a rocking platform. Complexes were washed extensively with 1% NP-40 buffer and then examined by western blot analysis along with aliquots of equalized total cell lysate.
Biolayer interferometry
The binding affinities for the RBDs of A-RAF (residues 17–94), B-RAF (residues 150–233), C-RAF (residues 54–137) and the RA domain of RASSF5 (residues 199 –367) of K-RAS-GMPPNP (wild-type, G12V and A59T/E) were measured using an Octet RED-384 biolayer interferometry biosensor instrument (BLI) running Octet Data Acquisition 9.0.0.37 and analyzed with Forté Bio Data analysis software. To prepare GppNHp-loaded K-RAS, protein was incubated in the presence of excess nucleotide analog, EDTA and calf intestinal phosphatase, which were then removed by passage of exchanged protein through size exclusion chromatography (Superdex™ S75 10/300 GL). The RBD/RA of B-RAF, C-RAF, A-RAF and RASSF5 were subcloned into pGEX-4T2 to produce N-terminal GST fusion proteins, which were expressed and purified as described previously (Smith and Ikura, 2014). These GST-tagged proteins were immobilized by incubating anti-GST-conjugated biosensors with GST-RBD proteins (10 μg/mL) for 10 min. The coated sensors were dipped into wells containing a range of concentrations of wild-type K-RAS, K-RASG12V, K-RASA59T, or K-RASA59E (54.7 nM to 20 μM K-RAS) in 20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 2 mM TCEP, 0.01% BSA and 0.005% Tween-20) for 30 or 60 s to monitor association, then transferred to buffer alone to monitor dissociation for 30 or 60 s. These binding assays were performed at 25°C in 96-well plates with agitation (1000 RPM). Analogous experiments were performed using GST-RASSF5-RA (immobilized at 5 μg/mL) with K-RAS concentrations ranging from 62 nM to 50 μM K-RAS. Sensors with immobilized GST-RBD/RAs were dipped into buffer alone as a control. Binding data were fitted to a 1:1 binding stoichiometry model using both kinetic and steady state analyses. GraphPad 9.1.0 was used to perform t tests and prepare graphs.
Western blotting and antibodies
All samples were run on either homemade 12.5% polyacrylamide gels or criterion pre-cast TGX gels from Bio-Rad. Wet transfer was performed overnight at 20V and at 4°C. Membranes were blocked for 1 h at room temp, with gentle agitation, using Intercept TBS buffer. Primary antibodies were diluted in Intercept T20 antibody diluent buffer, per manufacturer protocol, and precipitation was allowed to proceed overnight at 4°C with gentle agitation. The following day, blots were washed 3–4 times at room temperature in 5–15 min intervals using homemade 1X TBST, before the addition of secondary antibodies dissolved in Intercept T20 antibody diluent buffer. Secondary precipitation was allowed to proceed for 1 h at room temperature with gentle agitation. Blots were again washed 3–4 times at room temperature in 5–15 min intervals using homemade 1X TBST. Western blot bands were visualized and quantified using a Li-COR Biosciences imaging system.
The antibodies used in experiments involving the SNU-175, mES and NIH 3T3 cell lines are as follows: anti-GST (Cell Signaling Technologies, 2625), anti-HA-tag (Cell Signaling Technologies, 2367), pC-Raf (Cell Signaling Technologies, 9431), phospho-MEK1/2 (Cell Signaling Technologies, 9121), polyclonal KRAS antibody (Proteintech, 12063–1-AP), RAS10 (Millipore-Sigma, 05–516), vinculin (Cell Signaling Technologies, 13901), p44/42 MAPK (Erk1/2) (Cell Signaling Technologies, 4696), phospho-p44/42 MAPK (Erk1/2) (Cell Signaling Technologies, 4377), MEK1/2 (Cell Signaling Technologies, 4694), phospho-MEK1/2 (Cell Signaling Technologies, 9154), Akt (Cell Signaling Technologies, 2920), phospho-Akt (Cell Signaling Technologies, 4060), S6 ribosomal protein (Cell Signaling Technologies, 2317), phospho-S6 ribosomal protein (Cell Signaling Technologies, 2211), and C-Raf (Cell Signaling Technologies, 12552S).
The antibodies used in the co-immunoprecipitation experiments are as follows: A-Raf (Santa Cruz, sc-408), B-Raf (Santa Cruz, sc-5284), C-Raf (BD Pharmagen, 610152), GFP (MBL International, D153–3), Venus (Roche, 118114460001), MEK1 (BD Biosciences, 610122), and phospho-MEK1/2 (Cell Signaling Technologies, 9121).
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to predetermine sample size. The experiments were not randomized, nor were investigators blinded to sample allocation and experimental assessment. For biochemical or enzymology experiments, t tests were used to determine significance. For biological experiments, the Mann-Whitney U-test was used to determine significance, unless otherwise indicated.
Supplementary Material
Highlights.
Small GTPases undergo autophosphorylation through active site Ser/Thr substitution
K-RAS autophosphorylation inhibits GTP hydrolysis and promotes nucleotide exchange
K-RAS autophosphorylation promotes cellular transformation
Autophosphorylation inhibits K-RAS binding to effectors
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (R01CA178017 and R01CA232372 to K.M.H.) and an award from the Cancer Research UK Grand Challenge and the Mark Foundation to the SPECIFICANCER team. C.W.J. was supported by postdoctoral fellowship 130428-PF-17-066-01-TBG from ACS. S.D.P. acknowledges funding from the Linde Family Foundation, the Doris Duke Charitable Foundation, Deerfield 3DC, and Taiho Pharmaceuticals. Work from the Princess Margaret Cancer Centre was supported by the Canadian Cancer Society Research Institute (Grant # 706696 to M.I.) and the Princess Margaret Cancer Foundation. The NMR spectrometer and Octet biosensor were funded by CFI. Work done at the NCI was supported by federal funds from the NCI (ZIA BC 010329). Synchrotron data collection was done at the Northeastern Collaborative Access Team beamlines, which were funded by the NIGMS from the NIH (P30 GM124165). The Eiger 16M detector on the 24-ID-E beam line was funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a DOE Office of Science User Facility at Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The MicroMax007HF used to collect the X-ray data at Northeastern University was purchased in part with funds from the NSF MRI-1228897 grant to C.M. We thank Isidoro Tavares and Scott Ficarro from the Blais Proteomics Center for conducting mass spectrometry analyses. Molecular dynamics simulations were conducted on the HMS O2 High Performance Compute Cluster (https://it.hms.harvard.edu/our-services/research-computing).
Footnotes
DECLARATION OF INTERESTS
K.M.H. is married to a member of Molecular Cell’s Advisory Board.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2022.02.011.
REFERENCES
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25. [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahearn I, Zhou M, and Philips MR (2018). Posttranslational Modifications of RAS Proteins. Cold Spring Harb. Perspect. Med 8, a031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelmes K, Ramcke E, Kang HS, Sattler M, and Itzen A. (2020). Conformational control of small GTPases by AMPylation. Proc. Natl. Acad. Sci. USA 117, 5772–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergom C, Hauser AD, Rymaszewski A, Gonyo P, Prokop JW, Jennings BC, Lawton AJ, Frei A, Lorimer EL, Aguilera-Barrantes I, et al. (2016). The tumor-suppressive small GTPase DiRas1 binds the noncanonical guanine nucleotide exchange factor SmgGDS and antagonizes SmgGDS interactions with oncogenic small GTPases. J. Biol. Chem 291, 10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, and Kuriyan J. (1998). The structural basis of the activation of Ras by Sos. Nature 394, 337–343. [DOI] [PubMed] [Google Scholar]
- Bouchard-Cannon P, Lowden C, Trinh D, and Cheng HM (2018). Dexras1 is a homeostatic regulator of exercise-dependent proliferation and cell survival in the hippocampal neurogenic niche. Sci. Rep 8, 5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boute N, Jockers R, and Issad T. (2002). The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol. Sci 23, 351–354. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL 3rd, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. (2009). CHARMM: the biomolecular simulation program. J. Comput. Chem 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brtva TR, Drugan JK, Ghosh S, Terrell RS, Campbell-Burk S, Bell RM, and Der CJ (1995). Two distinct Raf domains mediate interaction with Ras. J. Biol. Chem 270, 9809–9812. [DOI] [PubMed] [Google Scholar]
- Buhrman G, Holzapfel G, Fetics S, and Mattos C. (2010). Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. USA 107, 4931–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel P, Dharmaiah S, Sale MJ, Messing S, Rizzuto G, Cuevas-Navarro A, Cheng A, Trnka MJ, Urisman A, Esposito D, et al. (2021). RAS interaction with Sin1 is dispensable for mTORC2 assembly and activity. Proc. Natl. Acad. Sci. USA 118, e2103261118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano E, and Downward J. (2011). RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2, 261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JY, Kim HJ, Yu JH, Xu J, Kim D, Paul BD, Choi H, Kim S, Lee YJ, Ho GP, et al. (2013). Dexras1 mediates glucocorticoid-associated adipogenesis and diet-induced obesity. Proc. Natl. Acad. Sci. USA 110, 20575–20580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HY, Obrietan K, Cain SW, Lee BY, Agostino PV, Joza NA, Harrington ME, Ralph MR, and Penninger JM (2004). Dexras1 potentiates photic and suppresses nonphotic responses of the circadian clock. Neuron 43, 715–728. [DOI] [PubMed] [Google Scholar]
- Cherfils J, and Zeghouf M. (2013). Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev 93, 269–309. [DOI] [PubMed] [Google Scholar]
- Chung HH, Benson DR, and Schultz PG (1993). Probing the structure and mechanism of Ras protein with an expanded genetic code. Science 259, 806–809. [DOI] [PubMed] [Google Scholar]
- Cookis T, and Mattos C. (2021). Crystal Structure Reveals the Full Ras-Raf Interface and Advances Mechanistic Understanding of Raf Activation. Biomolecules 11, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool RH, Jensen M, Jonák J, Clark BF, and Parmeggiani A. (1990). Substitution of proline 82 by threonine induces autophosphorylating activity in GTP-binding domain of elongation factor Tu. J. Biol. Chem 265, 6744–6749. [PubMed] [Google Scholar]
- Coordinators NR; NCBI Resource Coordinators (2016). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 44 (D1), D7–D19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart AE, Box GM, Court W, Gale ME, Brown JP, Pinder SE, Eccles SA, and Wells CM (2015). PAK4 promotes kinase-independent stabilization of RhoU to modulate cell adhesion. J. Cell Biol 211, 863–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, and Mark AE (1999). Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. Engl 38, 236–240. [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A. (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Dhanaraman T, Singh S, Killoran RC, Singh A, Xu X, Shifman JM, and Smith MJ (2020). RASSF effectors couple diverse RAS subfamily GTPases to the Hippo pathway. Sci. Signal 13, eabb4778. [DOI] [PubMed] [Google Scholar]
- Dharmaiah S, Bindu L, Tran TH, Gillette WK, Frank PH, Ghirlando R, Nissley DV, Esposito D, McCormick F, Stephen AG, and Simanshu DK (2016). Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc. Natl. Acad. Sci. USA 113, E6766–E6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmaiah S, Tran TH, Messing S, Agamasu C, Gillette WK, Yan W, Waybright T, Alexander P, Esposito D, Nissley DV, et al. (2019). Structures of N-terminally processed KRAS provide insight into the role of N-acetylation. Sci. Rep 9, 10512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donninger H, Schmidt ML, Mezzanotte J, Barnoud T, and Clark GJ (2016). Ras signaling through RASSF proteins. Semin. Cell Dev. Biol 58, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Müller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, and Morrison DK (2005). Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 17, 215–224. [DOI] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fan Y, Esmail MA, Ansley SJ, Blacque OE, Boroevich K, Ross AJ, Moore SJ, Badano JL, May-Simera H, Compton DS, et al. (2004). Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat. Genet 36, 989–993. [DOI] [PubMed] [Google Scholar]
- Fang Z, Lee KY, Huo KG, Gasmi-Seabrook G, Zheng L, Moghal N, Tsao MS, Ikura M, and Marshall CB (2020). Multivalent assembly of KRAS with the RAS-binding and cysteine-rich domains of CRAF on the membrane. Proc. Natl. Acad. Sci. USA 117, 12101–12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, and Mattos C. (2015). Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 23, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein J, Goody RS, and Wittinghofer A. (1987). Preparation and characterization of nucleotide-free and metal ion-free p21 “apoprotein”. J. Biol. Chem 262, 8455–8458. [PubMed] [Google Scholar]
- Fiegen D, Blumenstein L, Stege P, Vetter IR, and Ahmadian MR (2002). Crystal structure of Rnd3/RhoE: functional implications. FEBS Lett. 525, 100–104. [DOI] [PubMed] [Google Scholar]
- Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J, and Settleman J. (1996). Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol. Cell. Biol 16, 2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frech M, Schlichting I, Wittinghofer A, and Chardin P. (1990). Guanine nucleotide binding properties of the mammalian RalA protein produced in Escherichia coli. J. Biol. Chem 265, 6353–6359. [PubMed] [Google Scholar]
- Freeman AK, Ritt DA, and Morrison DK (2013). Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 49, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasper R, Sot B, and Wittinghofer A. (2010). GTPase activity of Di-Ras proteins is stimulated by Rap1GAP proteins. Small GTPases 1, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglieri V, Napolitano F, Pelosi B, Schepisi C, Migliarini S, Di Maio A, Pendolino V, Mancini M, Sciamanna G, Vitucci D, et al. (2015). Rhes influences striatal cAMP/PKA-dependent signaling and synaptic plasticity in a gender-sensitive fashion. Sci. Rep 5, 10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JB, Ellis RW, and Scolnick EM (1984). Autophosphorylation of v-Ha-ras p21 is modulated by amino acid residue 12. Proc. Natl. Acad. Sci. USA 81, 2674–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis KM (2017). KRAS Alleles: The Devil Is in the Detail. Trends Cancer 3, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BE, Yang SS, Boriack-Sjodin PA, Kuriyan J, and Bar-Sagi D. (2001). Structure-based mutagenesis reveals distinct functions for Ras switch 1 and switch 2 in Sos-catalyzed guanine nucleotide exchange. J. Biol. Chem 276, 27629–27637. [DOI] [PubMed] [Google Scholar]
- Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, Der CJ, and Counter CM (2002). Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 16, 2045–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock JF, Cadwallader K, and Marshall CJ (1991). Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). EMBO J. 10, 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng JI, Nguyen L, Castro DS, Zimmer C, Wildner H, Armant O, Skowronska-Krawczyk D, Bedogni F, Matter JM, Hevner R, and Guillemot F. (2008). Neurogenin 2 controls cortical neuron migration through regulation of Rnd2. Nature 455, 114–118. [DOI] [PubMed] [Google Scholar]
- Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, Anderson AO, Waters AM, Diehl JN, Papke B, et al. (2020). Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 10, 104–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K. (1996). VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38, 27–28. [DOI] [PubMed] [Google Scholar]
- Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, and Westover KD (2014). In situ selectivity profiling and crystal structure of SML-8–73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA 111, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Linklater E, Bayless BA, Lyons T, and Prekeris R. (2016). The role and regulation of Rab40b-Tks5 complex during invadopodia formation and cancer cell invasion. J. Cell Sci 129, 4341–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, and Im W. (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- John J, Frech M, and Wittinghofer A. (1988). Biochemical properties of Ha-ras encoded p21 mutants and mechanism of the autophosphorylation reaction. J. Biol. Chem 263, 11792–11799. [PubMed] [Google Scholar]
- Johnson CW, Buhrman G, Ting PY, Colicelli J, and Mattos C. (2015). Expression, purification, crystallization and X-ray data collection for RAS and its mutants. Data Brief 6, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CW, Lin YJ, Reid D, Parker J, Pavlopoulos S, Dischinger P, Graveel C, Aguirre AJ, Steensma M, Haigis KM, and Mattos C. (2019). Isoform-Specific Destabilization of the Active Site Reveals a Molecular Mechanism of Intrinsic Activation of KRas G13D. Cell Rep. 28, 1538–1550.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano Y, Gebregiworgis T, Marshall CB, Radulovich N, Poon BPK, St-Germain J, Cook JD, Valencia-Sama I, Grant BMM, Herrera SG, et al. (2019). Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun 10, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen RJ, and Behrend EN (1998). Dexamethasone rapidly induces a novel ras superfamily member-related gene in AtT-20 cells. J. Biol. Chem 273, 3129–3131. [DOI] [PubMed] [Google Scholar]
- Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, Seed B, and Avruch J. (2002). Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol 12, 253–265. [DOI] [PubMed] [Google Scholar]
- Klink BU, and Scheidig AJ (2010). New insight into the dynamic properties and the active site architecture of H-Ras p21 revealed by X-ray crystallography at very high resolution. BMC Struct. Biol 10, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Hori Y, Ueda N, Kajiho H, Muraoka S, Shima F, Kataoka T, Kontani K, and Katada T. (2009). Biochemical characterization of missense mutations in the Arf/Arl-family small GTPase Arl6 causing Bardet-Biedl syndrome. Biochem. Biophys. Res. Commun 381, 439–442. [DOI] [PubMed] [Google Scholar]
- Kontani K, Tada M, Ogawa T, Okai T, Saito K, Araki Y, and Katada T. (2002). Di-Ras, a distinct subgroup of ras family GTPases with unique biochemical properties. J. Biol. Chem 277, 41070–41078. [DOI] [PubMed] [Google Scholar]
- Korbie DJ, and Mattick JS (2008). Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc 3, 1452–1456. [DOI] [PubMed] [Google Scholar]
- Kovalski JR, Bhaduri A, Zehnder AM, Neela PH, Che Y, Wozniak GG, and Khavari PA (2019). The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol Cell 73, 830–844.e812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krengel U, Schlichting I, Scherer A, Schumann R, Frech M, John J, Kabsch W, Pai EF, and Wittinghofer A. (1990). Three-dimensional structures of H-ras p21 mutants: molecular basis for their inability to function as signal switch molecules. Cell 62, 539–548. [DOI] [PubMed] [Google Scholar]
- Ku JL, and Park JG (2005). Biology of SNU cell lines. Cancer Res. Treat 37, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RH, Iioka H, Ohashi M, Iemura S, Natsume T, and Kinoshita N. (2007). XRab40 and XCullin5 form a ubiquitin ligase complex essential for the noncanonical Wnt pathway. EMBO J. 26, 3592–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, et al. (2016). CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jia Q, Wang Y, Li F, Jia Z, and Wan Y. (2015). Rab40b upregulation correlates with the prognosis of gastric cancer by promoting migration, invasion, and metastasis. Med. Oncol 32, 126. [DOI] [PubMed] [Google Scholar]
- Li C, Vides A, Kim D, Xue JY, Zhao Y, and Lito P. (2021). The G protein signaling regulator RGS3 enhances the GTPase activity of KRAS. Science 374, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Bera AK, Gondi S, and Westover KD (2018). KRAS Switch Mutants D33E and A59G Crystallize in the State 1 Conformation. Biochemistry 57, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Li C, Tan R, Xu X, Wu WKK, Satoh A, Wang T, and Yu S. (2017). A RasGAP, DAB2IP, regulates lipid droplet homeostasis by serving as GAP toward RAB40C. Oncotarget 8, 85415–85427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, et al. (2012). Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 109, 5299–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern JP, and Land H. (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouly L, Gilhodes J, Lemarié A, Cohen-Jonathan Moyal E, Toulas C, Favre G, Sordet O, and Monferran S. (2019). The RND1 Small GTPase: Main Functions and Emerging Role in Oncogenesis. Int. J. Mol. Sci 20, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV (2018). The molecular machines that traffic signaling receptors into and out of cilia. Curr. Opin. Cell Biol 51, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano F, D’Angelo L, de Girolamo P, Avallone L, de Lange P, and Usiello A. (2018). The Thyroid Hormone-target Gene Rhes a Novel Crossroad for Neurological and Psychiatric Disorders: New Insights from Animal Models. Neuroscience 384, 419–428. [DOI] [PubMed] [Google Scholar]
- Naviaux RK, Costanzi E, Haas M, and Verma IM (1996). The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol 70, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, and Chardin P. (1998). A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J. Cell Biol 141, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, and Heringa J. (2000). T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol 302, 205–217. [DOI] [PubMed] [Google Scholar]
- Ogita Y, Egami S, Ebihara A, Ueda N, Katada T, and Kontani K. (2015). Di-Ras2 Protein Forms a Complex with SmgGDS Protein in Brain Cytosol in Order to Be in a Low Affinity State for Guanine Nucleotides. J. Biol. Chem 290, 20245–20256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JH, Ku JL, Yoon KA, Kwon HJ, Kim WH, Park HS, Yeo KS, Song SY, Chung JK, and Park JG (1999). Establishment and characterization of 12 human colorectal-carcinoma cell lines. Int. J. Cancer 81, 902–910. [DOI] [PubMed] [Google Scholar]
- Okada T, Sinha S, Esposito I, Schiavon G, López-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M, et al. (2015). The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat. Cell Biol 17, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W. (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, and Williams RL (2000). Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 103, 931–943. [DOI] [PubMed] [Google Scholar]
- Parker JA, Volmar AY, Pavlopoulos S, and Mattos C. (2018). K-Ras Populates Conformational States Differently from Its Isoform H-Ras and Oncogenic Mutant K-RasG12 D. Structure 26, 810–820.e814. [DOI] [PubMed] [Google Scholar]
- Paysan L, Piquet L, Saltel F, and Moreau V. (2016). Rnd3 in Cancer: A Review of the Evidence for Tumor Promoter or Suppressor. Mol. Cancer Res 14, 1033–1044. [DOI] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, and Gygi SP (2003). A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol 21, 921–926. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, et al. (2019). Tissue-Specific Oncogenic Activity of KRASA146T. Cancer Discov. 9, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Pan C, He SM, Lang B, Gao GD, Wang XL, and Wang Y. (2019). The Ras Superfamily of Small GTPases in Non-neoplastic Cerebral Diseases. Front. Mol. Neurosci 12, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif A, Nguyen TT, Weissflog L, Jacob CP, Romanos M, Renner TJ, Buttenschon HN, Kittel-Schneider S, Gessner A, Weber H, et al. (2011). DIRAS2 is associated with adult ADHD, related traits, and co-morbid disorders. Neuropsychopharmacology 36, 2318–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou P, Villalonga P, and Ridley AJ (2010). Rnd proteins: multifunctional regulators of the cytoskeleton and cell cycle progression. BioEssays 32, 986–992. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gabin AG, Almazan G, and Larocca JN (2004). Vesicle transport in oligodendrocytes: probable role of Rab40c protein. J. Neurosci. Res 76, 758–770. [DOI] [PubMed] [Google Scholar]
- Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, Haviv S, Asara JM, Pandolfi PP, and Cantley LC (2011). Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal 4, ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmu€ller L, Lautwein A, Schmitz F, and Wittinghofer A. (1997). The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338. [DOI] [PubMed] [Google Scholar]
- Scheidig AJ, Burmester C, and Goody RS (1999). The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure 7, 1311–1324. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahani N, Swarnkar S, Giovinazzo V, Morgenweck J, Bohn LM, Scharager-Tapia C, Pascal B, Martinez-Acedo P, Khare K, and Subramaniam S. (2016). RasGRP1 promotes amphetamine-induced motor behavior through a Rhes interaction network (“Rhesactome”) in the striatum. Sci. Signal 9, ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et al. (2014). KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Ramírez Jarquín UN, Rivera O, Kazantzis M, Eshraghi M, Shahani N, Sharma V, Tapia R, and Subramaniam S. (2019). Rhes, a striatal-enriched protein, promotes mitophagy via Nix. Proc. Natl. Acad. Sci. USA 116, 23760–23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih TY, Papageorge AG, Stokes PE, Weeks MO, and Scolnick EM (1980). Guanine nucleotide-binding and autophosphorylating activities associated with the p21src protein of Harvey murine sarcoma virus. Nature 287, 686–691. [DOI] [PubMed] [Google Scholar]
- Simanshu DK, Nissley DV, and McCormick F. (2017). RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, and Ikura M. (2014). Integrated RAS signaling defined by parallel NMR detection of effectors and regulators. Nat. Chem. Biol 10, 223–230. [DOI] [PubMed] [Google Scholar]
- Spoerner M, Herrmann C, Vetter IR, Kalbitzer HR, and Wittinghofer A. (2001). Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc. Natl. Acad. Sci. USA 98, 4944–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieglitz B, Bee C, Schwarz D, Yildiz O, Moshnikova A, Khokhlatchev A, and Herrmann C. (2008). Novel type of Ras effector interaction established between tumour suppressor NORE1A and Ras switch II. EMBO J. 27, 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MN, Yang H, Huang GY, Fu C, Pontikos M, Wang Y, Mao W, Pang L, Yang M, Liu J, et al. (2018). RAS-related GTPases DIRAS1 and DIRAS2 induce autophagic cancer cell death and are required for autophagy in murine ovarian cancer cells. Autophagy 14, 637–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarnkar S, Chen Y, Pryor WM, Shahani N, Page DT, and Subramaniam S. (2015). Ectopic expression of the striatal-enriched GTPase Rhes elicits cerebellar degeneration and an ataxia phenotype in Huntington’s disease. Neurobiol. Dis 82, 66–77. [DOI] [PubMed] [Google Scholar]
- Tang YJ, Huang J, Tsushima H, Ban GI, Zhang H, Oristian KM, Puviindran V, Williams N, Ding X, Ou J, et al. (2019). Tracing Tumor Evolution in Sarcoma Reveals Clonal Origin of Advanced Metastasis. Cell Rep 28, 2837–2850.e2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47 (D1), D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SJ, Resnick RJ, and Shalloway D. (2001). Nonradioactive determination of Ras-GTP levels using activated ras interaction assay. Methods Enzymol. 333, 333–342. [DOI] [PubMed] [Google Scholar]
- Terrell EM, Durrant DE, Ritt DA, Sealover NE, Sheffels E, Spencer-Smith R, Esposito D, Zhou Y, Hancock JF, Kortum RL, et al. (2019). Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol Cell 76, 872–884.e875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting PY, Johnson CW, Fang C, Cao X, Graeber TG, Mattos C, and Colicelli J. (2015). Tyrosine phosphorylation of RAS by ABL allosterically enhances effector binding. FASEB J. 29, 3750–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchot N, Zahraoui A, Vielh E, and Tavitian A. (1989). Biochemical properties of the YPT-related rab1B protein. Comparison with rab1A. FEBS Lett. 256, 79–84. [DOI] [PubMed] [Google Scholar]
- Tran TH, Chan AH, Young LC, Bindu L, Neale C, Messing S, Dharmaiah S, Taylor T, Denson JP, Esposito D, et al. (2021). KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun 12, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulsh LS, and Shih TY (1984). Metabolic turnover of human c-rasH p21 protein of EJ bladder carcinoma and its normal cellular and viral homologs. Mol. Cell. Biol 4, 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargiu P, De Abajo R, Garcia-Ranea JA, Valencia A, Santisteban P, Crespo P, and Bernal J. (2004). The small GTP-binding protein, Rhes, regulates signal transduction from G protein-coupled receptors. Oncogene 23, 559–568. [DOI] [PubMed] [Google Scholar]
- Vetter IR (2017). Interface analysis of small GTP binding protein complexes suggests preferred membrane orientations. Biol. Chem 398, 637–651. [DOI] [PubMed] [Google Scholar]
- Volodko N, Gordon M, Salla M, Ghazaleh HA, and Baksh S. (2014). RASSF tumor suppressor gene family: biological functions and regulation. FEBS Lett. 588, 2671–2684. [DOI] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, and Laue ED (2005). The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696. [DOI] [PubMed] [Google Scholar]
- Whitehead RH, Macrae FA, St John DJ, and Ma J. (1985). A colon cancer cell line (LIM1215) derived from a patient with inherited nonpolyposis colorectal cancer. J. Natl. Cancer Inst 74, 759–765. [PubMed] [Google Scholar]
- Wielaender F, Sarviaho R, James F, Hytönen MK, Cortez MA, Kluger G, Koskinen LL, Arumilli M, Kornberg M, Bathen-Noethen A, et al. (2017). Generalized myoclonic epilepsy with photosensitivity in juvenile dogs caused by a defective DIRAS family GTPase 1. Proc. Natl. Acad. Sci. USA 114, 2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang MH, Nickerson S, Kim ET, Liot C, Laurent G, Spang R, Philips MR, Shan Y, Shaw DE, Bar-Sagi D, et al. (2012). Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. USA 109, 10843–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, et al. (2012). Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol 19, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original scans of western blot data have been deposited at Mendeley Data and are publicly available as of the date of publication. Validation reports of X-ray crystallography structures have been deposited at the PDB and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| A-RAF | Santa Cruz | Cat#: sc-408; RRID: AB_630882 |
| AKT (pan) (40D4) | Cell Signaling Technologies | Cat#: 2920; RRID:AB_1147620 |
| Anti-GFP | MBL International | Cat#:D153–3; RRID:AB_591817 |
| Anti-GST (91G1) | Cell Signaling Technologies | Cat#: 2625; RRID:AB_490796 |
| Anti-HA-tag (6E2) | Cell Signaling Technologies | Cat#: 2367; RRID:AB_10691311 |
| Anti-Venus | Roche | Cat#: 118114460001 |
| B-RAF | Santa Cruz | Cat#: sc-5284; RRID:AB_2721130 |
| C-RAF | BD Pharmagen | Cat#: 610152; RRID:AB_397553 |
| c-RAF (D5X6R) Mouse mAb | Cell Signaling Technologies | Cat#: 12552S; RRID:AB_2728706 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Invitrogen | Cat#: A-21058; RRID:AB_2535724 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 800 | Invitrogen | Cat#: A-32735; RRID:AB_2633284 |
| MEK1 | BD Biosciences | Cat#: 610122; RRID:AB_397528 |
| MEK1/2 (L38C12) | Cell Signaling Technologies | Cat#: 4694; RRID:AB_10695868 |
| p44/42 MAPK (ERK1/2) (L34F12) | Cell Signaling Technologies | Cat#: 4696; RRID:AB_390780 |
| pC-RAF (s289/296/301) | Cell Signaling Technologies | Cat#: 9431; RRID:AB_561402 |
| Phospho-Akt (Ser473) (D9E) XP® | Cell Signaling Technologies | Cat#: 4060; RRID:AB_2315049 |
| Phospho-MEK1/2 (Ser217/221) | Cell Signaling Technologies | Cat#: 9121; RRID:AB_331648 |
| Phospho-MEK1/2 (Ser217/221) (41G9) | Cell Signaling Technologies | Cat#: 9154; RRID:AB_2138017 |
| Phospho-p44/42 MAPK (ERK1/2) (Thr202/ Tyr204) (197G2) | Cell Signaling Technologies | Cat#: 4377; RRID:AB_331775 |
| Phospho-S6 Ribosomal Protein (Ser235/236) | Cell Signaling Technologies | Cat#: 2211; RRID:AB_331679 |
| polyclonal KRAS antibody | Proteintech | Cat#: 12063–1-AP; RRID:AB_878040 |
| RAS10 | Millipore-Sigma | Cat#: 05–516; AB_2121151 |
| S6 Ribosomal Protein (54D2) | Cell Signaling Technologies | Cat#: 2317; RRID:AB_2238583 |
| Vinculin (E1E9V) XP® | Cell Signaling Technologies | Cat#: 13901; RRID:AB_2728768 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| BL21 (DE3 Codon+) | Agilent | Cat#: 230280 |
| BL21 (DE3) | Thermo Scientific | Cat#: C600003 |
| BL21-A1 | Thermo Fisher Scientific | Cat#: C607003 |
| DH5α-T1R | Invitrogen | Cat#: 12297–016 |
| pCL-Eco | Addgene | Cat#: 12371 |
| Tet-pLKO-puro shKRAS | Addgene | Cat#: 116871 |
| TOP10 E. coli strain | Fisher Scientific | Cat#: C404003 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 1.0 M Calcium acetate hydrate | Hampton Research | Cat#: HR2–567 |
| 1.0 M HEPES pH 7.5 | Hampton Research | Cat#: HR2–729 |
| 1.0 M Magnesium acetate tetrahydrate | Hampton Research | Cat#: HR2–561 |
| 100% glycerol | Hampton Research | Cat#: HR2–623 |
| 100% PEG400 | Hampton Research | Cat#: HR2–603 |
| 100X Glutamax supplement | Thermo Fisher Scientific | Cat#: 35050061 |
| 100X MEM Non-Essential Amino Acids Solution | Life Technologies | Cat#: 11140–050 |
| 10X CutSmart buffer | New England Biolabs | Cat#: R3195T |
| 1X Knockout DMEM | Life Technologies | Cat#: 10829–018 |
| 2-Mercaptoethanol | Millipore-Sigma | Cat#: M3148–250ML |
| 2’/3′-O-(N-Methyl-anthraniloyl)-guanosine-5′-diphosphate | Fisher Scientific | Cat#: M12414 |
| 2’/3′-O-(N-Methyl-anthraniloyl)-guanosine-5′-triphosphate | Fisher Scientific | Cat#: M12415 |
| 4.0 M Magnesium chloride hexahydrate | Hampton Research | Cat#: HR2–803 |
| 5.0 M Sodium chloride | Hampton Research | Cat#: HR2–637 |
| 50% PEG3350 mono disperse | Hampton Research | Cat#: HR2–527 |
| 5X Phusion HF Buffer | New England Biolabs | Cat#: M0530S |
| Accucore C18 resin | ThermoFisher Scientific | N/A |
| Antibiotic Antimycotic Solution (PenStrep) | Sigma-Aldrich | Cat#: A5955 |
| Chymotrypsin | Promega | Cat #V106A |
| Coelenterazine-h | N/A | N/A |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | Cat#: N0447S |
| DMSO | Sigma-Aldrich | Cat#: D2650–100mL |
| Dulbeccos Modification of Eagles Medium (DMEM) | TC-Corning | Cat#: 10–013-CV |
| EcoRV-HF® restriction enzyme | New England Biolabs | Cat#: R3195T |
| ESGRO ®Leukemia Inhibitory Factor (LIF), 1 million units/1mL | Millipore-Sigma | Cat#: ESG1106 |
| Fetal Bovine Serum (FBS) | GE Healthcare | Cat#: SH30070.03 |
| Gelatin from bovine skin | Sigma-Aldrich | Cat#: G9391–100G |
| GreenGlo™ Safe DNA Dye, 20,000X in Water | Denville Scientific | Cat#: ca3600 |
| Guanosine 5′-[β,γ-imido]triphosphate trisodium salt hydrate | Sigma-Aldrich | Cat#: G0635–100MG |
| HyClone Defined Fetal Bovine Serum (FBS), US Origin | Cytiva | Cat#: SH30070.03 |
| Intercept ®(TBS) Blocking Buffer | LI-COR Biosciences | Cat#: 927–6003 |
| Intercept ®T20 (TBS) Antibody Diluent | LI-COR Biosciences | Cat#: 927–65001 |
| Iodoacetamide | ThermoFisher Scientific | Cat #A39271 |
| Lambda Phosphatase | New England BioLabs | Cat#: P0753S |
| Modified Terrific Broth | Fisher Scientific | Cat#: BP9729–600 |
| Phusion ®High-Fidelity DNA Polymerase | New England Biolabs | Cat#: M0530S |
| Radioimmunoprecipitation assay (RIPA) buffer | Boston Bioproducts | Cat#: BP-115–500 |
| RPMI 1640 | Fisher Scientific | Cat#: MT10040CV |
| Tris(2-carboxyethyl)phosphine | ThermoFisher Scientific | Cat #77720 |
| 0.05% Trypsin EDTA | Invitrogen | Cat#: 25300–120 |
| UltraPure™ DNase/RNase-Free Distilled Water | Invitrogen | Cat#: 10977023 |
|
| ||
| Critical commercial assays | ||
|
| ||
| EnzChek™ Phosphate Assay Kit | Thermo Fisher Scientific | Cat#: E6646 |
| Mouse ES Cell Nucleofector®Kit | Lonza | Cat#: VPH-1001 |
| PureLink® HiPure Plasmid Filter Maxiprep kit | Thermo Fisher Scientific | Cat#: K2100–17 |
| Quick-DNA 96 kit | Zymo Research | Cat#: D3012 |
| QuikChange site-directed mutagenesis | Agilent | Cat#: 200519 |
| XtremeGENE9 transfection Kit | Millipore-Sigma | Cat#: 6365779001 |
|
| ||
| Deposited data | ||
|
| ||
| H-RAS A59E GDP | https://www.rcsb.org | PDB: 7JII |
| H-RAS A59T GppNHp crystal 1 | https://www.rcsb.org | PDB: 7JIF |
| H-RAS A59T GppNHp crystal 2 | https://www.rcsb.org | PDB: 7JIG |
| H-RAS A59E GppNHp | https://www.rcsb.org | PDB: 7JIH |
| K-RAS A59E GDP | https://www.rcsb.org | PDB: 7KMR |
| Raw data western blot data from figures were deposited on Mendeley at https://dx.doi.org/10.17632/9n7bryshg3.1 | https://data.mendeley.com | Mendeley data: https://doi.org/10.17632/9n7bryshg3.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| EmbryoMax® Primary Mouse Embryonic Fibroblasts | Millipore-Sigma | Cat#: PMEF-CFX |
| HEK293FT cells | Thermo Fisher Scientific | Cat#: R70007; RRID: CVCL_6911 |
| HEK293t cells | ATCC | Cat#: CRL-3216; RRID: CVCL_0063 |
| HeLa cells | ATCC | Cat#: CCL-2; RRID: CVCL_0030 |
| LIM1215 cells | Whitehead et al., 1985 | N/A; RRID: CVCL_2574 |
| K-Ras(+/LSL-wildtype) Mouse Embryonic Stem cells | Poulin et al., 2019 | N/A |
| K-Ras(A59T/LSL-A59T) Mouse Embryonic Stem cells | This study | N/A |
| NIH 3T3 cells | ATCC | Cat#: CRL-1658; RRID: CVCL_0594 |
|
| ||
| Oligonucleotides | ||
|
| ||
| 5′-CTTGGATATTCTC GACACAACAGGTCA AGAGGAGTACAG-3′ | Integrated DNA Technologies | pBABE KRAS4B A59T Forward |
| 5′-CTGTACTCCTCT TGACCTGTTGTGTCG AGAATATCCAAG-3′ | Integrated DNA Technologies | pBABE KRAS4B A59T Reverse |
| 5′-GGATATTCTCG ACACAGAAGGTCAA GAGGAGTACAG-3′ | Integrated DNA Technologies | pBABE KRAS4B A59E Forward |
| 5′-CTGTACTCCTCT TGACCTTCTGTGTCGA GAATATCC-3′ | Integrated DNA Technologies | pBABE KRAS4B A59E Reverse |
| 5′-GTAGTTGGAGC TGTTGGCGTA GGCAAG-3′ | Integrated DNA Technologies | pBABE KRAS4B G12V Forward |
| 5′-CTTGCCTACGC CAACAGCTCC AACTAC-3′ | Integrated DNA Technologies | pBABE KRAS4B G12V Reverse |
| 5′-GTGGTAGTTGGAGCTCGTGG CGTAGGCAAG-3′ | Integrated DNA Technologies | pBABE KRAS4B G12R Forward |
| 5′-CTTGCCTACGCCACGAGCTCCA ACTACCAC-3′ | Integrated DNA Technologies | pBABE KRAS4B G12R Reverse |
| 5′-GGTGGTTGGTGCCGATGGTG TGGGTAAAAG-3′ | Integrated DNA Technologies | pET21 KRAS4B G12D Forward |
| 5′-CTTTTACCCACACCATCGGCA CCAACCACC-3′ | Integrated DNA Technologies | pET21 KRAS4B G12D Reverse |
| 5′-CATTCTGGATACCACCGGCC AGGAAG-3′ | Integrated DNA Technologies | pET21 KRAS4B A59T Forward |
| 5′-CTTCCTGGCCGGTGGTAT CCAGAATG-3′ | Integrated DNA Technologies | pET21 KRAS4B A59T Reverse |
| 5′-GACATTCTGGATACCGAAGGC CAGGAAGAGTATAG-3′ | Integrated DNA Technologies | pET21 KRAS4B A59E Forward |
| 5′-CTATACTCTTCCTGGCCTTCGG TATCCAGAATGTC-3′ | Integrated DNA Technologies | pET21 KRAS4B A59E Reverse |
| 5′-CACCGTTCTCGACACAGCAGGT CAAG-3′ | Integrated DNA Technologies | SaCas9 gRNA 1 Forward |
| 5′-AAACCTTGACCTGCTGTGTC GAGAAC-3′ | Integrated DNA Technologies | SaCas9 gRNA 1 Reverse |
| 5′-TGTGACCATTAGCATTGT TTGC-3′ | Integrated DNA Technologies | mES Ex2 genotyping Forward primer |
| 5′-CTTAAACCCACCTATAAT GGTG-3′ | Integrated DNA Technologies | mES Ex2 genotyping Reverse primer |
| 5′-AGTAATTGATGGAGAAA CCTG-3′ | Integrated DNA Technologies | mES Ex2 Sequencing Forward primer |
| 5′-ATAATGGTGAATATCTT CAAA-3′ | Integrated DNA Technologies | mES Ex2 Sequencing Reverse primer |
| 5′-CTGTAATAAT CCAGACTGTG TTTCTCCCTT CTCAGGACTCC TACAGGAAAC AAGTAGTAAT TGATGGAGAA ACCTGTCTCT TGGATATCCT CGACACAACA GGTCAAGAAG AGTACAGTGC AATGAGGGAC CAGTACATGA GAACTGGGGA GGGCTTTCTT TGTGTATT TGCCATAAA TAATACTAA ATCATTTG AAGAT-3′ | Integrated DNA Technologies | Repair template for CRISPR experiments |
|
| ||
| Recombinant DNA | ||
|
| ||
| 100 bp DNA ladder | New England Biolabs | Cat#: N3231L |
| 6xHis-GAP (715–1047) | This study | N/A |
| 6xHis-K-RAS4B | This study | N/A |
| 6xHis-SOS11 (564–1049) | This study | N/A |
| Gateway pDEST15 vector | Thermo Scientific | Cat#: 11802014 |
| H-RAS (1–166) | Buhrman et al., 2010 | N/A |
| pBABE-hygro vector | Addgene | Cat#: 1765 |
| pCMV5-Venus-K-Ras4B | Terrell et al., 2019 | N/A |
| pET-21a(+) vector | EMD Biosciences | https://www.emdmillipore.com/US/en/product/pET-21+-DNA-Novagen,EMD_BIO-69770www.emdmillipore.com/US/en/product/pET-21+-DNA-Novagen,EMD_BIO-69770 |
| pET28 vector | EMD Biosciences | https://www.emdmillipore.com/US/en/product/pET-28a+-DNA-Novagen,EMD_BIO-69864www.emdmillipore.com/US/en/product/pET-28a+-DNA-Novagen,EMD_BIO-69864 |
| pGEX-4T2-A-RAF(17–94) | Smith and Ikura, 2014 | N/A |
| pGEX-4T2-B-RAF(150–233) | Smith and Ikura, 2014 | N/A |
| pGEX-4T2-C-RAF(53–137) | Fang et al., 2020 | N/A |
| pGEX-4T2-RASSF5(199–367) | Smith and Ikura, 2014 | N/A |
| pLHCX-WT-RafReg-Rluc8 (A-, B-, C-Raf) | Terrell et al., 2019 | N/A |
| pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA | Addgene | Cat#: 61591 |
| Raf1-RBD | Addgene | Cat#: 13338 |
| RAS pathway collection v2.0 | Addgene | Kit#: 1000000070 |
| RASSF1 | Addgene | Cat#: 70535 |
| RASSF10 | Addgene | Cat#: 70537 |
| RASSF2 | Addgene | Cat#: 70539 |
| RASSF3 | Addgene | Cat#: 70541 |
| RASSF4 | Addgene | Cat#: 70543 |
| RASSF5 | Addgene | Cat#: 70545 |
| RASSF6 | Addgene | Cat#: 70547 |
| RASSF7 | Addgene | Cat#: 70549 |
| RASSF8 | Addgene | Cat#: 70551 |
| RASSF9 | Addgene | Cat#: 70553 |
|
| ||
| Software and algorithms | ||
|
| ||
| CHARM-GUI (solution builder) | Brooks et al., 2009; Jo et al., 2008; Lee et al., 2016 | https://charmm-gui.org/?doc=input/solution |
| Chimera (ver. 1.15) | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Coot | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| GraphPad (ver. 9.1.0) | Prism | https://www.graphpad.com |
| GROMACS (ver. 2020.1) | Abraham et al., 2015 | http://www.gromacs.org |
| HKL3000 package | Otwinowski and Minor, 1997 | https://hkl-xray.com/hkl-3000 |
| Image Studio lite (ver. 5.2.5) | LI-COR | https://www.licor.com/bio/image-studio/ |
| NMRPipe | Delaglio et al., 1995 | https://groups.io/g/nmrpipe |
| CcpNmr | Vranken et al., 2005 | N/A |
| PHENIX (ver. 1.11.1–2575) | Adams et al., 2010 | https://phenix-online.org |
| PyMOL (ver. 2.4.0) | Schrodinger | https://pymol.org/2/ |
| Sequest | Eng et al., 1994 | http://fields.scripps.edu/yates/wp/?page_id=17 |
| SnapGene (ver. 5.2.4) | GSL Biotech | N/A |
| VMD (ver. 1.9.4a38) | Humphrey et al., 1996 | http://www.ks.uiuc.edu/Research/vmd/ |
| XDS | Kabsch, 2010 | https://xds.mr.mpg.de |
|
| ||
| Other | ||
|
| ||
| 96-Well Clear Bottom Black or White Polystyrene Microplates | Corning | Cat#: 3917 |
| 96-well black-walled plate (OptiPlate) | Perkin Elmer | Cat#: 6005270 |
| 384-well white-walled plate (CulturePlate) | Perkin Elmer | Cat#: 6007680 |
| Accela600 liquid chromatography pump | Thermo Fisher Scientific | N/A |
| AKTA Pure | Cytiva | http://www.cytivalifesciences.com/en/us/shop/chromatography/chromatographysystems/akta-pure-p-05844 |
| Cellometer ®AutoT4 cell counter | Nexcelom Bioscience | https://www.nexcelom.com/nexcelomproducts/automated-cell-counters/cellometer-auto-t4-automated-cellcounter/ |
| ChemiDoc XRS+ System | Bio-Rad | Cat#: 1708265 |
| Epson Perfection V600 Photo Scanner | Epson | Cat#: B11B198011 |
| Famos Autosampler | LC Packings | N/A |
| HiTrap QHP column | Cytiva | Cat#: 17115401 |
| HiTrap TALON column | Cytiva | Cat#:29048565 |
| MicroMax007HF | Rigaku | N/A |
| Mono-Q column | Cytiva | Cat#: 17516601 |
| NEO III HD 800MHz spectrometer | Bruker | N/A |
| Ni-NTA beads | QIAGEN | Cat#: 30210 |
| NT8 Crystallization robot | Formulatrix | https://formulatrix.com/proteincrystallization-systems/nt8crystallization-robot/ |
| Nucleofector® II Device | Lonza | N/A |
| Octet RED-384 biolayer interferometry biosensor instrument | Sartorius | https://www.sartorius.com/en/products/protein-analysis/octet-label-freedetection-systems |
| Odyssey ®CLx | LI-COR | https://www.licor.com/documents/8prh6ps2abjbemx68412 |
| PD-10 desalting column | Cytiva | Cat#: 17085101 |
| PHERAstar Plus plate reader | BMG Labtech | https://www.bmglabtech.com/pherastardetection-system/ |
| Phoenix liquid handler | ArtRobbins | https://www.artrobbins.com/crystalphoenix |
| Protein G Sepharose | Cytiva | Cat#: GE17–0618-01 |
| Q Exactive mass spectrometer | Thermo Fisher Scientific | N/A |
| R-Axis IV2+ | Rigaku | N/A |
| RockImager Protein Crystallization Imager | Formulatrix | https://formulatrix.com/proteincrystallization-systems/rock-imagercrystallization-imagers/ |
| StageTip | N/A | N/A |
| Superdex 75 10/300 column | Cytiva | Cat#: 17517401 |
| Synergy H1 Hybrid Multi-Mode Reader microplate | BioTek | https://www.biotek.com/products/detection-hybrid-technology-multi-modemicroplate-readers/synergy-h1-hybridmulti-mode-reader/ |
| Tecan Infinite M1000 plate reader | Thermo Fisher Scientific | N/A |
| Ultra-15 Centrifugal Filter Units (10kDa MWCO) | Fisher Scientific | Cat#: UFC901008 |
| Zeba™ Spin Desalting Columns | Thermo Fisher Scientific | Cat#: PI89892 |
| Protein model: Wildtype H-RAS GppNHp | Buhrman et al., 2010 | 3K8Y |
| Protein model: Wildtype H-RAS GDP | Scheidig et al., 1999 | 1CTQ |
| Protein model: Wildtype H-RAS bound to GAP and AlFx | Scheffzek et al., 1997 | 1WQ1 |
| Protein model: H-RAS G12V/A59T bound to GTP | Krengel et al., 1990 | 521P |
| Protein model: RND3 bound to GTP | Fiegen et al., 2002 | 1M7B |
| Protein model: RND1 bound to GTP | N/A | 2CLS |
| Protein model: DIRAS2 bound to GDP | N/A | 2ERX |
| Protein model: DIRAS1 bound to GDP | N/A | 2GF0 |
| Protein model: ARL6 bound to GTP | Structural Genomics Consortium | 2H57 |
| Protein model: Wildtype K-RAS bound to GDP | Hunter et al., 2014 | 4OBE |
| Protein model: Wildtype H-RAS bound to the RBD domain of RASSF5 | Stieglitz et al., 2008 | 3DDC |
| Protein model: Wildtype H-RAS bound to the RBD of C-RAF | Fetics et al., 2015 | 4G0N |
| Protein model: Wildtype H-RAS bound to the RBD-CRD of C-RAF | Cookis and Mattos, 2021 | 7JHP |
| Protein model: Wildtype K-RAS bound to the RBD-CRD of C-RAF | Tran et al., 2021 | 6XI7 |
| Protein model: H-RAS G12V bound to the RBD of PI3K | Pacold et al., 2000 | 1HE8 |
| Protein model: Wildtype K-RAS bound to the RBD-PH domain of SIN1 | Castel et al., 2021 | 7LC1 |