

ABSTRACT
As of early 2022, the coronavirus disease 2019 (COVID-19) pandemic remains a substantial global health concern. Different treatments for COVID-19, such as anti-COVID-19 neutralizing monoclonal antibodies (mAbs), have been developed under tight timelines. Not only mAb product and clinical development but also chemistry, manufacturing, and controls (CMC) process development at pandemic speed are required to address this highly unmet patient need. CMC development consists of early- and late-stage process development to ensure sufficient mAb manufacturing yield and consistent product quality for patient safety and efficacy. Here, we report a case study of late-stage cell culture process development at pandemic speed for mAb1 and mAb2 production as a combination therapy for a highly unmet patient treatment. We completed late-stage cell culture process characterization (PC) within approximately 4 months from the cell culture process definition to the initiation of the manufacturing process performance qualification (PPQ) campaign for mAb1 and mAb2, in comparison to a standard one-year PC timeline. Different strategies were presented in detail at different PC steps, i.e., pre-PC risk assessment, scale-down model development and qualification, formal PC experiments, and in-process control strategy development for a successful PPQ campaign that did not sacrifice quality. The strategies we present may be applied to accelerate late-stage process development for other biologics to reduce timelines.
KEYWORDS: Process characterization (PC), process performance qualification (PPQ), in-process control (IPC), Chinese hamster ovary (CHO) cell culture platform, scale-down model (SDM), design of experiment (DOE), failure mode and effects analysis (FMEA), quality by design (QbD), COVID-19 monoclonal antibodies (mAbs)
Introduction
Since the onset of the pandemic in late 2019, more than 434 million confirmed cases of coronavirus disease 2019 (COVID-19) and about 6 million deaths have been reported by the World Health Organization as of February 27, 2022.1 Effective treatments against COVID-19 would represent a substantial advance in scientific, technological, and health-care system innovation around monoclonal antibody (mAb) treatments for infectious diseases.2 To accommodate this urgency, US Food and Drug Administration (FDA) has issued a guidance for chemistry, manufacturing, and controls (CMC) to provide recommendations to sponsors on the fast development of mAb products targeting COVID-19.3 Different mAbs have been intensively researched for an effective treatment for COVID-19 patients.4–6 Several mAb products, including anti-SARS-CoV-2 antibodies casirivimab + imdevimab (Ronaprev),7 bamlanivimab + etesevimab,8 sotrovimab (Xevudy®),9 bebtelovimab,10 regdanvimab (Regkirona), tixagevimab + cilgavimab (Evusheld), and amubarvimab + romlusevimab,11 were developed at pandemic speed and they are either approved for emergency use or marketing applications are under consideration by numerous regulatory agencies. Many more mAbs for COVID-19 are under development at different clinical stages.11,12
In parallel to product and clinical development, CMC process development of mAbs for COVID-19 at pandemic speed is also required to meet urgent patient need. Process development for mAb manufacturing using Chinese hamster ovary (CHO) cells is relatively mature and has a long history of producing products with demonstrated safety to patients, high process yield, and suitable product quality requirements for human use. The entire product and process development from DNA for cell-line generation to biologics license application (BLA) submission usually takes 10 to 15 years.13,14 MAb process development is divided into upstream and downstream portions. The focus of this report is the upstream portion, which starts from vial thaw of a cell bank in a small shake flask and proceeds to seed culture expansion and final large-scale bioreactor production. Cell culture upstream process development includes two different stages: 1) the early-stage process development for investigational new drug (IND) application to the start of first-in-human (FIH) Phase 1 clinical trials, and 2) the late-stage development toward BLA submission for commercial use.
Early-stage process development consists of cell-line generation, lead clone selection, initial cell culture process development with the lead clone research cell bank (RCB), master cell bank (MCB) creation from the lead clone RCB and MCB release, and the early-stage process definition or lock for the good manufacturing practice (GMP) FIH campaign. Unmet patient need and a highly competitive biopharmaceutical industry have encouraged a reduction of the early-stage process development timeline, which is on the critical path of mAb product development, to about 12–16 months from DNA to IND.15 Different strategies have been implemented, including robust cell culture platform,16 targeted integration for cell-line generation,17 and use of a non-clonal pool for toxicology (Tox) studies.18–20 To address COVID-19 pandemic needs, multiple reports have demonstrated that the early-stage process development can be shortened further from 12–16 to approximately 4–6 months for the development of neutralizing mAbs.15,21,22
Late-stage upstream process development consists of commercial process development, process characterization (PC), and in-process control strategy development. In comparison to the early-stage upstream process, the major goals of commercial process development, usually using MCB, working cell bank (WCB), or their cell age equivalent development cell banks (DCBs), are to improve upstream titer for cost reduction, process robustness, and scale-up manufacturing for a much larger quantity of commercial drug substance generation, while maintaining comparable product quality profile.23,24 Then, the commercial process is locked for pivotal GMP campaign to generate drug substance for Phase 2 and 3 clinical trials, long-term stability study (LTSS), PC, and defining an in-process control strategy (IPC) for process performance qualification (PPQ), which are regulatory requirements for BLA submission.24 Quality by design (QbD) principles have been used throughout all stages of process development in the industry. Pharmaceutical QbD is a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.25–28
Late-stage process development is usually not on the critical path for standard mAb product development, since late-stage clinical trials take much more time than process development. For a standard mAb product, it usually takes one year from the initiation of formal PC studies after the commercial process lock to the initiation of the PPQ campaign. Upstream PC studies to develop effective IPC strategies supporting a PPQ campaign include Failure Mode and Effects Analysis (FMEA), scale-down model (SDM) development and qualification, multiple PC studies using a qualified SDM, and PC and IPC report writing.29–31
Here, we present a case study on late-stage upstream process development within 4 months from upstream process lock to the start of PPQ campaign for a combination therapy using two mAbs (referred to as mAb1 and mAb2). This report describes different strategies to shorten the CMC development timeline for mAb1 and mAb2.
Results
Overall CMC timeline for mAb1 and mAb2 process development and characterization
As shown in Figure 1, the CMC timeline for a standard mAb is 12–16 months for the early-stage process development from DNA to IND. To address highly unmet patient needs, we shortened the early-stage development to approximately 6 months for mAb1 and mAb2 in parallel as a combination therapy. The main strategies are shown as follows (Figure 1). Targeted integration was used for cell-line generation of both mAb1 and mAb2. Both Tox study and FIH Phase 1 study materials were generated using non-clonal pools. The strategy was reported in detail in our previous studies on how to shorten the CMC timeline while achieving a good yield with comparable product quality.19,20 The 6-month timeline from DNA to IND filing in this study agrees with another recent report using similar strategies.21
Figure 1.
Standard mAb CMC timeline versus the accelerated development timeline of mAb1 and mAb2 together for early-stage CMC development towards IND filing and late-stage CMC development towards BLA submission.Non-clonal pools were used to generate Tox study and FIH Phase 1 study materials simultaneously. The early-stage timeline from DNA to IND was shortened from 12-16 to 6 months for the accelerated timeline. Late-stage process development including scale-up run was accelerated by using RCB and platform fit. The start of late-stage GMP manufacturing was accelerated using MCB and the platform process before the final process lock. Then formal PC studies and pivotal GMP campaign were started right after the process lock. Overall accelerated timeline from DNA to BLA would be shortened from a standard of 10-15 years to 2-4 years.
Alt Text: Monthly process development activities for mAb1 and mAb2 from a vector construction to IND submission for early-stage and from platform fit lab runs to the PPQ campaign for late stage. The early-stage timeline from DNA to IND was shortened from 12–16 to 6 months for mAb1 and mAb2 using the accelerated timeline, while overall accelerated timeline from DNA to BLA would be shortened from 10–15 to 2–4 years.
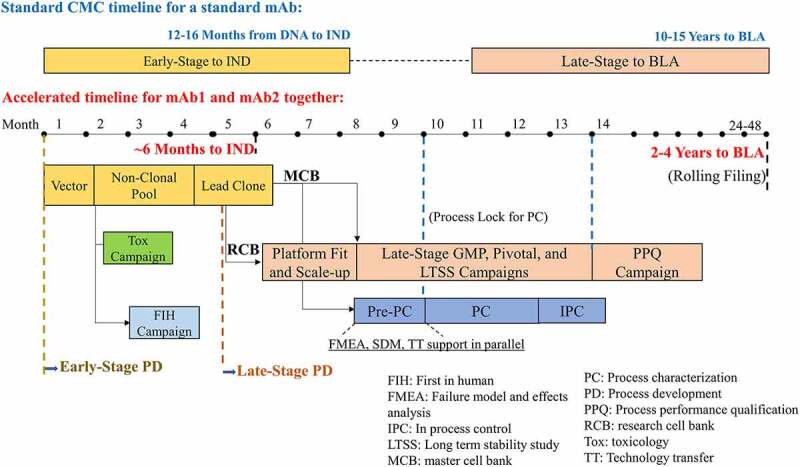
Although late-stage process development may not be on the critical path of product development for a standard mAb due to much longer time requirements for clinical trials, late-stage process development may be on the critical path for a highly accelerated mAb product, such as neutralizing mAb cocktails for COVID-19 treatment.15 To shorten the timeline for late-stage process development of mAb1 and mAb2, we started commercial process development using our CHO cell platform media and process16,32,33 using a RCB right after lead clone selection (Figure 1). The platform fit in lab-scale bioreactors followed by scale-up runs using the RCB vials were performed in parallel with other activities, including technology transfer to a GMP manufacturing facility, MCB manufacturing and conditional release, and pre-PC activities (e.g., SDM screening and development, process risk assessment by FMEA) (Figure 1). It took about 2–3 months to manufacture MCB and conditionally release MCB. Thus, using RCB could save 2–3 months to start late-stage process development in comparison to using MCB. Instead of a WCB, to shorten the time, the MCB was used for PC studies and the PPQ campaign. In addition, we leveraged the N-1 seed cultures and media prepared during the large-scale GMP campaign as much as possible for PC studies, which saved resources and time for lab operations.
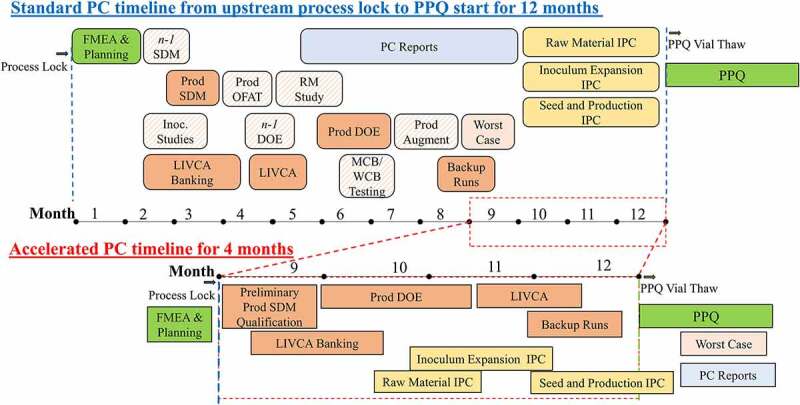
However, the major focus of this report is how to shorten the timeline from 12 to 4 months for mAb1 and mAb2 from the upstream process lock for the initiation of PC studies to the start of the PPQ campaign (Figure 2), which are described in the following sections in detail.
Figure 2.
Standard mAb PC timeline versus the accelerated PC timeline of mAb1 and mAb2 togetherTiming of FMEA, SDM qualification, PC lab work, and IPC and PC report writing was shuffled for a more compacted schedule. Lab bioreactor run number for SDM development and PC studies was reduced leveraging platform knowledge. Based on different PPQ steps, IPC reports were approved in a rolling order when the data was ready. The accelerated PC timeline from upstream lock to PPQ start was shortened from a standard of 12 months to 4 months.
Alt Text: Detailed PC activities over 12-month duration for a standard PC timeline versus detailed PC activities over 4-month duration for the accelerated PC timeline of mAb1 and mAb2 together. The accelerated PC timeline from upstream lock to PPQ start was shortened from a standard of 12 months to 4 months.
Failure mode and effects analysis
Pre-PC risk assessment of different process parameters (PPs) and performance attributes (PAs) by FMEA is required as the first step for all PC studies. The description nomenclature of PAs and PPs at different criticalities may be different for IPC strategies used in different companies.29–31 In this study, the IPC nomenclature is shown in Table 1. Process outputs are measurements that cannot be directly manipulated or controlled, such as in-process measurements, and are indicators of process performance and product quality. Process outputs are defined as three levels (Table 1). Critical performance attributes (CPAs) correspond to a property linked to critical quality attributes (CQAs), and excursions could lead to lot rejection. PAs are monitored to confirm process consistency, and excursions will lead to investigation. Monitored attributes (MAs) are not significantly correlated to product quality and are used to monitor the process. Process inputs are measurements that can be directly manipulated or controlled and are classified based on their impact on process performance and product quality. Process inputs are also defined as three similar levels (Table 1). Critical process parameters (CPPs) may affect CPAs or CQAs, and excursions could lead to lot rejection. PPs may affect PAs, and excursions will lead to investigation. Monitored parameters (MPs) are unrelated to product quality and are used to monitor the process.
Table 1.
Definition and criticalities of performance attributes and process parameters
| Process Performance/ Consistency | Control of CQA | ||
|---|---|---|---|
| Process Output | Monitored Attribute | Performance Attribute | Critical Performance Attribute |
| (MA) | (PA) | (CPA) | |
| Process Input | Monitored Parameter | Process Parameter | Critical Process Parameter |
| (MP) | (PP) | (CPP) | |
Identification of all potential significant inputs from media, seed train, and production culture via FMEA are critical to the selection of parameters for PC studies and IPC strategy development. The important FMEA steps are shown in Supplemental Figure 1. The impact of each process parameter on CQAs and PAs was first quantified through cause-and-effect analysis with different scores (Supplemental Figure 1A). Based on the scores, risk ranking (Supplemental Figure 1B) and PC strategies (Supplemental Figure 1C) were determined for all different PPs.
Due to an urgent timeline for development of this combination therapy and highly prioritized clinical trials, late-stage CMC development activities, including PC studies, were on the critical path. To save time, we leveraged cell culture platform knowledge, other late-stage project experience using the same parental cell line and media, and a limited set of ongoing GMP manufacturing data for mAb1 and mAb2 to de-risk factors. The outcomes of FMEA exercise are visualized as an Ishikawa diagram (Figure 3a). For inoculum expansion and N-1 and N-2 seed bioreactor steps, the incubation temperature, culture duration, final viable cell density (VCD), and final cell viability were identified as potential PPs or PAs with medium risk. For media preparation step, final pH and osmolality were identified as potential PAs with medium risk. No potential CPP or CPA was identified. Based on platform knowledge and other project experience, the risk of identified factors was decreased, and PC studies were not needed for the inoculum expansion, seed bioreactors, and medium preparation steps for mAb1 and mAb2. Instead, we used the existing operation ranges, already successfully implemented in the ongoing GMP campaigns for mAb1 and mAb2, to write IPC reports for inoculum expansion, media preparation, N-1 and N-2 seed bioreactor steps.
Figure 3.
Failure mode and effects analysis (FMEA) results for mAb1 and mAb2. A) risk assessment on all upstream performance attributes (PAs) and process parameters. B) key PAs and quality attributes identified at the production bioreactor step. Parameters and attributes are color coded as high (red), medium (blue), and low (black) risk.
Alt Text: A) Fishbone diagram shows performance attributes (PAs) and process parameters at different cell culture steps, e.g., inoculum expansion, N-1 and N-2 seed bioreactor, and media preparation steps, and production bioreactor step. B) Key PAs and quality attributes identified at production bioreactor step to be evaluated in PC studies.
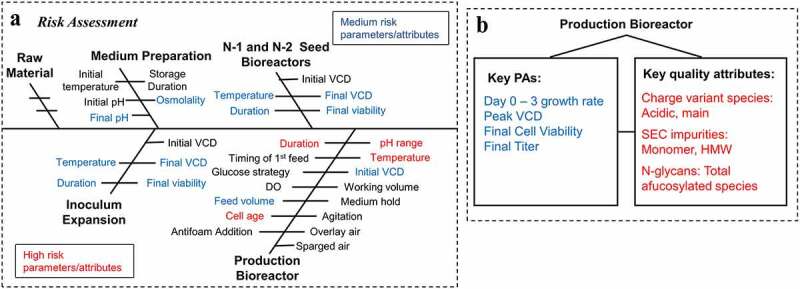
For the production bioreactor step, cell age was identified as a high-risk parameter and potential CPP due to lack of existing clone-specific cell-line stability data (Figure 3a), which was evaluated in a limit of in vitro cell age (LIVCA) PC study using 5-L SDM. Based on ICH guidance, the LIVCA for production use should be based on data derived from production cells expanded under pilot plant scale or commercial scale conditions to the proposed limit of in vitro cell age for production use or beyond,34 while a LIVCA study at lab scale may not be required for regulatory filing. Because we did not have LIVCA results at manufacturing scale before PPQ campaign, to ensure that the cell line would meet the LIVCA requirement at large scale, we performed the LIVCA study using lab 5-L SDM. The major goal of this lab PC LIVCA is to reduce the cell age risk before PPQ campaign.
The culture duration, pH, and temperature were identified as high-risk parameters, while initial VCD and feed volume were identified as medium risk parameters (Figure 3a). These 5 parameters were included in a production design of experiments (DOE) PC study. In summary of performance and quality attributes at the production bioreactor step (Figure 3b), Day 0–3 growth rate, peak VCD, final viability, and final titer were considered as key PAs with medium risk, while charge variant species including main peak and acidic species, size-exclusion chromatography (SEC) impurities including monomer and high molecular weight (HMW) species, and N-glycans including total afucosylated species (i.e., sum of G0, G0-GN, G1, G2, Man3, Man5 and other high mannose species) were the key quality attributes with high risk to be evaluated in lab PC studies. However, host cell proteins (HCP) and low molecular weight species (LMW), two typical CQAs for biologics, were not listed as the key quality attributes for this upstream PC study because we do not have any HCP and LMW issues from upstream and both can be easily removed by the downstream process for this project. In addition, HCP is included as a key quality attribute in downstream PC studies to ensure that the final product quality is under control. LMW was only monitored for information only without a numerical specification limit when the PC studies were performed. SEC monomer and HMW were in specification. Thus, LMW, equal to 100 minus monomer and HMW, is also indirectly controlled.
Scale-down model development and qualification
Since performing PC studies is not practically feasible at manufacturing scale, development of a SDM that represents the manufacturing process is essential to achieve reliable PC results. Normally, all scale-dependent parameters are carefully examined in lab-scale bioreactors. Manufacturing data from multiple runs are then used to determine acceptable limits and demonstrate scale equivalency through two one-sided t-test (TOST) analysis.29,31 A qualified SDM must be used for all PC studies. In this report, to minimize time to start PC studies, a single screening run with different scale-dependent parameters in 5-L bioreactors was performed to generate a preliminary SDM at-risk for formal PC studies, leveraging limited process development, scale-up 500-L, and 2000-L data. Near completion of PC studies, we retrospectively qualified the SDM using multiple PC control runs at 5-L scale and additional GMP campaign data at 2000-L scale. Based on our platform knowledge and other project experience, the risk of this retrospective approach was acceptable with significant time saving. We applied a tiered strategy for the SDM qualification with statistical TOST analysis first. If the TOST analysis failed for a small number of parameters, then practical equivalence evaluation using secondary criteria, such as product specification limit or assay equipment variation range.
SDM screening experiment
For the first 5-L SDM experiment, different scale-dependent parameters were screened, e.g., agitation rates (220, 260, and 300 rpm), overlay air flow rates (0.02, 0.04, and 0.06 liter per minute (LPM)), and sparged air flow rates (0.005, 0.013, and 0.021 LPM), while keeping scale-independent parameters constant (e.g., temperature, pH, and dissolved oxygen (DO)). Both overlay and sparged air flow rates were constant for the same bioreactor during the entire run, while sparged oxygen flow rates were variable to maintain 40% DO via cascade control mode. The 220 rpm agitation conditions were unable to maintain 40% DO due to oxygen flow limitation, so the agitation was increased by 40 rpm when the sparged oxygen flow rate reached 90% of maximum for those 5-L bioreactors with an initial agitation of 220 rpm (Supplemental Table 1). Minimum differences were observed in all cell culture PAs and CQAs under all screening conditions for mAb1 (Supplemental Table 1), which indicated that the cell culture process was consistent and robust with respect to scale-dependent parameters. Based on platform knowledge and other project experience, 290 rpm, 0.04 LPM top air and 0.013 LPM bottom air were selected as SDM conditions for mAb1.
A similar SDM screening run was performed for mAb2 except agitation rates shifted to higher levels (e.g., 260, 290, and 320 rpm) because 220 rpm was too low to maintain DO at 40%. All cell culture performance and quality attributes also had minimum differences across the 5-L screening conditions for mAb2 (Supplemental Table 2). It should be noted that it was acceptable that the in-process SEC monomer values at harvest were slightly lower than the specification limit for mAb2 (Supplemental Table 2), because downstream purification could remove HMW and LMW species, and thus the monomer results for mAb2 met the specification limit in final drug substance after downstream processing. In summary, the same preliminary SDM condition as mAb1 was selected for mAb2 (e.g., 290 rpm, 0.04 LPM overlay air and 0.013 LPM sparged air).
Retrospective SDM qualification
The cell culture performance and metabolite profiles between scales are shown in Supplemental Figure 2. Most trends between scales were similar or exhibited small differences for both mAbs, except that NH4 for mAb1 and VCD for mAb2 in the second half of cell culture duration showed a relatively large difference. Overall results were satisfactory for a SDM, in which only one of 10 PAs showed a relatively large difference.
To demonstrate equivalence between SDM and manufacturing-scale, a TOST analysis for equivalence was performed with SAS JMP 13.1 software for each attribute using θ as the maximum allowable difference. For evaluation of quality and PAs, ±θ was set to ±3 standard deviations of the manufacturing scale data to account for process variability. θ is used for TOST analysis as the first approach for equivalence testing. A conclusion of equivalence is achieved if the lower confidence limit and upper confidence limit differences are contained in the -θ to +θ interval.29,31 If an attribute failed in TOST analysis, practical equivalence was evaluated based on secondary criteria, e.g., 20% of the product specification limit or normal variation ranges of the assay method and instrument. The arbitrary number of 20% above is based on our platform knowledge and previous project experience, which is agreed within different function groups in our company. TOST analysis is just a pure statistical method. Some of the key quality attributes even with a small difference between SDM and manufacturing data may fail in TOST analysis, if the standard deviations for those attributes are very small. Thus, a second criterion, such as 20% specification limit, is necessary to evaluate the practical significance.
To fully qualify the SDM, 5-L SDM or control data (n = 9) during PC studies and 2000-L GMP campaign data (n = 7) were compared before the PPQ campaign. Nine of the important attributes were selected for TOST analysis, including 4 key cell culture PAs and 5 key quality attributes (Figure 3b). Main peak, total afucosylated species and titer failed, while the other 6 attributes passed TOST equivalence analysis for mAb1 (Figure 4). Those three failed attributes were further evaluated for practical equivalence. The normalized titer difference between scales was only 0.12, which was less than 0.2, 20% of 1 (normalized weight/L), the normalized titer limit as described in the process description for mAb1. Therefore, the final titer difference between scales was not considered to be practically different (Supplemental Table 3). For the practical evaluation of charge variant species, 20% of the specification limit for acidic species was used for all three groups, i.e., acidic, main, and basic species, because the acidic species has the greatest impact on product quality among the three groups. Since the main peak difference between scales was less than 20% of the acidic species limit, the main peak attribute passed the practical equivalence evaluation for mAb1 (Supplemental Table 3). Using the same strategy, the total afucosylated species passed the practical equivalence evaluation for mAb1 (Supplemental Table 3).
Figure 4.
TOST analysis for key quality and performance attributes of mAb1 and mAb2 between 5-L SDM and 2000-L manufacturing.Maximum allowable difference (MAD) was set to ±3 standard deviations of the manufacturing scale data (blue dash line). The difference between 5-L and 2000-L (■) was normalized by setting 2000-L average as zero (red dash line). Titer and main peak of mAb1, and main peak and afucosylation of mAb2 were equivalent in mean only because one side of the error bas was out of MAD interval. Afucosylation of mAb1 was inequivalent. Final cell viability of mAb2 failed to be equivalent. As these attributes did not pass TOST analysis, a practical equivalence was evaluated based on secondary criteria in Supplemental Table 3.
Alt Text: Five key quality attributes and four key performance attributes at the production bioreactor step are compared between 5-L SDM and 2000-L manufacturing for mAb1 and mAb2 via this TOST analysis graph. A conclusion of equivalence is achieved for most of the attributes as the 90% confident interval of 5-L are contained in the MAD.
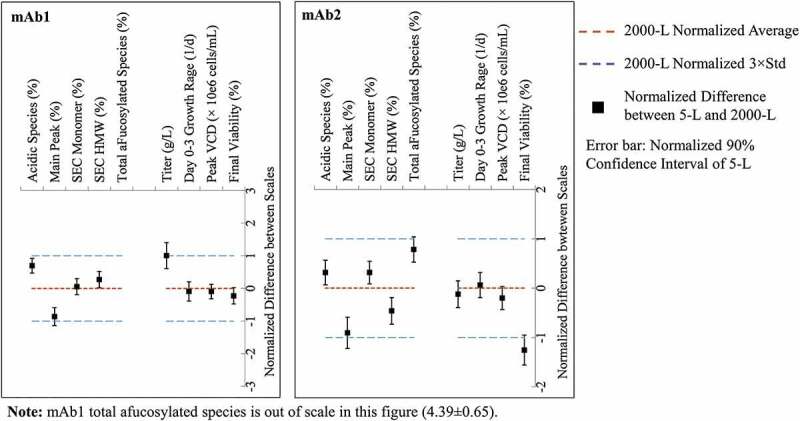
For mAb2, main peak, total afucosylated species and final cell viability failed, while the other 6 attributes passed TOST equivalence analysis (Figure 4). Using the same strategy as mAb1, the main peak and total afucosylated species passed the practical equivalence evaluation for mAb2 (Supplemental Table 3). For cell counting attributes for mAb2, a large variation range of 16.8% for final cell viability and 6.3 × 106 cells/mL for final VCD were observed across 7 different Vi-Cell instruments in the same 5-L bioreactor laboratory (unpublished data), of which the Vi-Cell with the lowest counting numbers was randomly chosen for this PC study. It should be noted that all 7 Vi-Cell instruments tested in the development lab and the Vi-Cell used in GMP facility were maintained by the vendor and passed the quality test using the vendor’s protocol. The difference of 14.9% in final cell viability between SDM data measured using the lab Vi-Cell and GMP campaign data measured by the Vi-Cell in the GMP facility (Supplemental Table 3) was smaller than 16.8%, the difference observed between different Vi-Cell instruments. In addition, most other quality attributes and cell culture attributes were comparable between scales for mAb2 (Figure 4 and Supplemental Figure 2). Therefore, it was considered that the difference in final cell viability between scales was most likely caused by different Vi-Cell instruments rather than true cell culture difference between scales for mAb2.
In summary, most key attributes for both mAb1 and mAb2 SDM passed TOST analysis (Figure 4), while those that failed in TOST analysis passed the practical equivalency evaluation using secondary criteria (Supplemental Table 3). Thus, the SDM used in PC studies was fully qualified for both mAb1 and mAb2.
Fed-batch production PC studies using 5-L SDM bioreactors
The purpose of the PC studies was to demonstrate manufacturing process robustness by understanding the relationship between operational parameters and final cell culture performance and quality attributes using the SDM described above. Two PC studies each for mAb1 and mAb2 were performed for this highly accelerated program, including a production DOE experiment using 23 × 5-L bioreactors and a LIVCA experiment using 8 × 5-L bioreactors, respectively. For the worst-case experiments, key PPs are combined to create a worst-case outcome to a particular parameter for that step. This worst-case material is passed on to subsequent steps to determine if the proposed key parameter ranges are acceptable.30 To save time, the worst-case linkage PC study was moved out after PPQ because it is required for BLA submission and not for PPQ specifically. The seed expansion and raw material PC experiments were not required for mAb1 and mAb2, based on the pre-PC risk assessment using our established platform knowledge from other projects, as described previously.
The overall workflow of the production DOE study is presented in Supplemental Figure 3A. As described previously (Figure 3), 5 production bioreactor process inputs ranked as medium or high risk were included in a multivariate DOE study (Supplemental Table 4). The effects of these process inputs on cell culture process PAs and product quality attributes were examined in the DOE study. This DOE study was designed to probe some two-factor interactions and quadratic terms among the factors of interest. The design included 20 experimental conditions (n = 1) and 3 center-point (control) conditions using 23 × 5-L bioreactors. Main effects, two-way interactions, and quadratic effects were evaluated for all 5 factors. The prediction variance was below 1 for up to 99% of the design space, with a relative prediction variance of 0.35. The power of all main effects was ≥96.5%, interactions ≥ 92.6%, and quadratics ≥ 62%, assuming a signal-to-noise ratio of 3:1. There were no significant effect correlations in the final design. These 4 design diagnostics suggested that the overall experimental design was acceptable. Statistical models were generated to determine if each parameter significantly influenced each attribute of interest.
Production DOE experiment
The results of this DOE study were assessed by statistical evaluations for the operation of the production bioreactor step. Model scaled estimate and the associated impact factor following the equation in Supplemental Figure 3B were used to evaluate whether parameters and interactions significantly affected key process attributes. For calculation of the impact factor, the 2000-L attribute mean values were used for the cell culture process PAs, e.g., peak VCD, final viability and final titer, in the impact factor calculation, while the specification tolerance limits were used for the key quality attributes, e.g., acidic species, main peak, SEC monomer, SEC HMW, and total afucosylated species (Supplemental Figure 3B). The impact factors for different PPs on cell culture performance and quality attributes from the DOE study were summarized for both mAb1 and mAb2 in Supplemental Table 5. Generally, only attributes for which the range was >10% of mean were used for model generation. A parameter is considered to have a statistically significant impact on an attribute if the impact factor is ≥0.2 for a PA and ≥0.1 for a key quality attribute (Supplemental Figure 3B). In this study, the impact factor, measured attribute data, and platform cell culture knowledge were all used to determine if each parameter had a practically significant impact on each attribute of interest. As summarized in Supplemental Table 5, attributes of mAb1 and mAb2 were impacted differently by the tested parameters.
The DOE study outcomes with recommended ranges and criticality of tested parameters for IPC strategies are presented in Table 2. The impact factors for pH in the tested range were <0.1 on all process and quality attributes (Supplemental Table 5), so pH for both mAbs did not have practically significant effects in the range tested and was recommended as a potential MP (Table 2). For the same reason, both daily feed amount and culture duration for mAb1 did not have practically significant effects on all attributes and were recommended as MPs (Table 2). Although the impact factors for temperature and initial VCD in the tested ranges were <0.2 on performance attributes and <0.1 on presumptive CQAs (Supplemental Table 5), one 5-L bioreactor using the combination of lower temperature and higher initial VCD in the production DOE study resulted in <50% final cell viability at harvest on day 14, which missed the harvest specification on cell viability > 50% in the process description. Therefore, both temperature and initial VCD had practically significant effects and was recommended as a potential PP (Table 2). Based on the impact factors in Supplemental Table 5 and the criteria in Supplemental Figure 3B, temperature and culture duration for mAb2 had practically significant effects on one or more presumptive CQAs and were recommended as PPs, while the initial VCD for mAb2 did not have practically significant effects and was recommended as MP (Table 2). Although the impact factor on SEC HMW for the mAb2 feed amount was >0.1, all SEC HMW data for mAb2 were generated from in-process samples at harvest and all were within the HMW specification limit for final drug substance after downstream processing (data not shown). Additionally, SEC HMW could be removed by downstream purification, so the feed amount for mAb2 was considered not to have a practically significant effect and thus recommended as a MA (Table 2).
Table 2.
DOE output summary with recommended ranges and criticality of tested parameters for IPC strategies
| Studied Parameter | Studied/ Recommended Range* | Attributes with Practically Significant Effects |
|
|---|---|---|---|
| mAb1 | mAb2 | ||
| pH Upper/Lower Deadband | 6.7/7.3 to 6.9/7.5 | No (MP) | No (MP) |
| Temperature Setpoint (°C) | 35.5 to 37.5 | Final Viability (PP) | UPLC Titer, Main Peak, Acidic Species, Total afucosylated Species (PP) |
| Daily Feed Amount (% initial volume) | 3.28 to 4.00 | No (MP) | No (MP) |
| Initial VCD (×106 cells/mL) | 4.0 to 6.5 | Final Viability (PP) | No (MP) |
| Culture Duration (days) | 13 to 15 | No (MP) | Total afucosylated Species (PP) |
*Recommended ranges are the same as studied ranges, combining in one column
Limit of in vitro cell age experiment
The LIVCA study was the second PC experiment performed in this report, used to define an acceptable range for allowable number of cell generations. The strategy for the LIVCA study is shown in Supplemental Figure 4. For each mAb, two trains, starting from the respective MCB, were sub-cultured and DCBs were made at passages 6, 12, and 18. Each vial of the MCB and three DCBs was thawed and expanded through a series of 6 shake flask passages and a last scale-up passage in 5-L N-1 seed bioreactors. The 5-L production bioreactors (n = 2) were inoculated at 25 generations for MCB, and at 43, 63, and 83 generations beyond the MCB for the DCBs (Supplemental Figure 4). There were no significant differences in cell culture performance attributes for all 5-L bioreactors with different cell generations, except that NH4 was significantly higher for mAb1 at 63 and 83 generations during the second half of production (Supplemental Figure 5). NH4 has been reported as a well-known inhibitor on CHO cell culture performance.35 However, we do not understand why the NH4 difference was observed for mAb1 between different seed passages (Supplemental Figure 5) and between SDM and manufacturing (Supplemental Figure 2), which will be further studied in future. Nonetheless, all the quality attributes and genetic stability data were similar from all 5-L samples for both mAb1 and mAb2 (data not shown). These results indicate that both cell lines are stable up to 83 generations from MCB.
In-process control strategy development
The IPC strategy is a planned set of controls derived from current product and process understanding that ensures consistent process performance and product quality.36 The IPC strategy (e.g., parameter criticality, setpoints and ranges) obtained from the PC studies is important to define allowable operating ranges for the subsequent PPQ campaign, as required by regulatory agencies before BLA submission. An IPC strategy is essential to ensure controlled and reproducible production of drug substance. The process inputs (e.g., MP, PP, CPP) and outputs (e.g., MA, PA, CPA) were previously discussed for IPC strategy development (Table 1). For this highly accelerated program, to initiate the PPQ campaign as soon as possible, we wrote IPC reports using PC study memorandums as appendices rather than final approved PC reports. Although PC study appendices contained the same PC data as final approved PC reports, writing technical memorandums as appendices to support IPC reports took much less time than writing and approving full PC reports, allowing PC reports to be completed later.
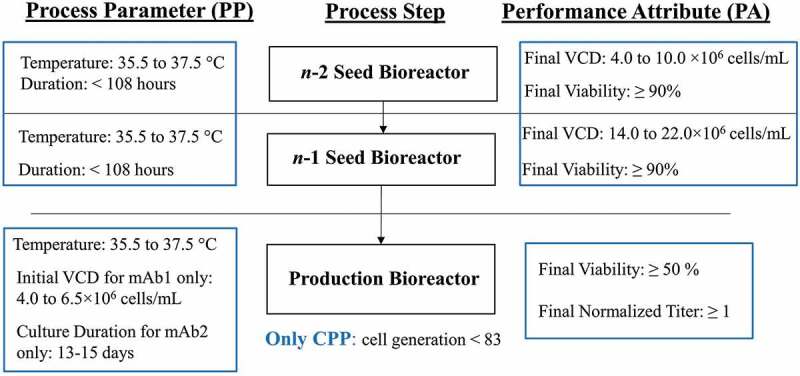
In this work, we wrote three IPC strategy development documents for both mAbs, which constitute the typical number of IPC documents as required for a standard mAb within our company. As presented in the FMEA section, we wrote two IPC reports, e.g., Inoculum Expansion IPC report and Raw Material and Media IPC report (data not shown), fully based on pre-PC risk assessments using platform knowledge, other project experience and limited at-scale GMP campaign data. The Seed and Production Bioreactor IPC report was the third document that required analysis from the production DOE and LIVCA studies. The IPC control ranges for PPs and PAs in seed and production bioreactor steps for mAb1 are shown in Figure 5, in which the only CPP identified was cell generation, while all MAs and MPs are not listed. The only two differences between mAb1 and mAb2 IPC strategy were that the initial VCD was a PP for mAb1, while culture duration was a PP for mAb2 (Figure 5).
Figure 5.
IPC strategies in seed and production bioreactor steps for mAb1 and mAb2.Initial VCD was a PP for mAb1, while culture duration was a PP for mAb2. The only CPP identified was cell generation. The low limit for the final titer at harvest, as described in the process description, was normalized as 1 normalized weight/L. PA had no difference between two mAbs.
Alt Text: PP ranges for all PPs are listed on the left, PA ranges for all PAs are listed on the right, MAs and MPs are not listed, and only CPP identified is cell generation for mAb1 and mAb2 IPC strategies for n-2 seed bioreactor, n-1 seed bioreactor, and n production bioreactor steps.
Additionally, the IPC reports were written on a rolling basis in the order of Raw Material and Media IPC, Inoculum Expansion IPC, and Seed and Production Bioreactor IPC. Thus, the manufacturing batch records (MBRs) for medium preparation were written and approved prior to GMP medium preparation on the floor, while the MBRs for the vial thaw and inoculum expansion steps were approved afterward. Therefore, the seed and production bioreactor MBRs had more flexibility to be written and approved after the initiation of PPQ. In summary, all strategies described here were applied for the PPQ campaign support of mAb1 and mAb2, which was successfully completed at pandemic speed.
Discussion
To address a highly unmet patient need, such as treatments for COVID-19 patients,37 fast product development timelines for vaccines and neutralizing mAbs at pandemic speed have been required. Such a pace put CMC development, including both early- and late-stage cell culture process development, on the critical paths.2,15,38 In this report, we presented a case study for the comprehensive late-stage process development at pandemic speeds for two mAbs as a combination therapy. To the best of our knowledge, this represents the first such case study that the timeline for PC studies and IPC writing and approval can be shortened to 4 months from the upstream process lock to initiation of first vial thaw for the PPQ campaign. For comparison, a standard timeline for mAb PC studies and IPC report writing is approximately one year.
The 8 key strategies used for shortening PC studies and IPC strategy development timeline under the pandemic pace are as follows (Table 3 and Figure 2): 1) Use RCB vial and platform process fit, including cell line, media, and operating parameters for late-stage commercial process development and scale-up runs; 2) Use MCB instead of WCB for pivotal and PPQ GMP campaigns, while subsequent bridging studies between MCB and WCB will be done later; 3) Use platform knowledge, other related project experience, and limited GMP campaign data for pre-PC FEMA to de-risk factors, and in particular, focus PC studies on the most important parameters, e.g., production DOE and LIVCA studies, while not performing formal PC studies for other steps, i.e., vial thaw, inoculum expansion, and N-2 and N-1 bioreactors; 4) Retrospectively fully qualify SDM, when sufficient data from lab PC studies and GMP campaign batches are available, and in particular, run only one lab SDM screen with focus on scale-dependent parameters, while keeping scale-independent parameters constant and preliminarily qualify SDM for formal PC studies at-risk based on limited process development, scale-up, and GMP campaign data; 5) Use satellite run strategy for lab PC studies, including media and seed culture manufactured in GMP facility, which can save resources and time for lab PC studies; 6) Expedite IPC report writing by using technical memorandums including all PC study data analysis and operating ranges already implemented in the GMP campaigns before PPQ as appendices to support IPC report writing, which saved approximately two months in comparison to the standard approach of writing and approving the formal PC reports prior to writing the IPC reports; 7) Move the worst-case linkage PC study after the PPQ campaign, because this PC study is nice to have but not required for PPQ; 8) Write and approve the IPC reports on a rolling basis in the order of Raw Material and Media IPC, Inoculum Expansion IPC, and Seed and Production Bioreactor IPC. Regarding this final point, the MBR for medium preparation was approved first before GMP medium preparation on the floor. Then, the MBRs for the vial thaw and inoculum expansion steps were approved before the initiation of the PPQ campaign. Finally, Seed and Production Bioreactor IPC and MBRs were approved after the PPQ initiation, but before seed and production bioreactor runs.
Table 3.
Summary of strategies used for a standard and accelerated mAbs
| Category | Standard strategy | Accelerated strategy |
|---|---|---|
| Late-stage process development (PD): | ||
| Cell bank used | MCB or WCB | RCB |
| Medium and process conditions | Optimization for 3-6 months | Platform fit within 1-2 months |
| Scale-up runs | MCB or WCB | RCB |
| Late-stage, pivotal, and PPQ GMP campaigns: | ||
| PD, tech transfer, and MBR preparation | Staged one by one | Simultaneously |
| Cell bank | WCB | MCB |
| Process characterization (PC): | ||
| FMEA | After completion of PD and pivotal campaign with all data generated | In parallel with PD and pivotal campaign, when partial PD and pivotal campaign data generated, while leveraging platform knowledge and other project experiences |
| Media and seed culture used in PC | Generated in lab only | Generated in either lab or GMP facility |
| SDM development | Multiple screening runs for all parameters | One run for scale-dependent parameters only |
| SDM qualification before PC studies | With sufficient SDM lab and large-scale data | Preliminary qualification with limited PD and large-scale data |
| SDM qualification before PPQ campaign | NA | Retrospectively qualify SDM using sufficient PC lab and pivotal campaign data |
| PC studies before PPQ | Raw materials and medium preparation, inoculum expansion, seed and production bioreactors, LIVCA, and worst-case linkage studies | Focus on the most important PC studies: production bioreactor DOE and LIVCA |
| PC studies after PPQ | NA | Worst-case linkage PC study |
| PC report writing and approval | To support IPC reports Before PPQ initiation | After PPQ initiation |
| PC memo writing and approval | NA | To support IPC reports |
| In-process control (IPC): | ||
| PC data format to support IPC | Final PC reports | PC memos as appendices |
| Raw Materials, Inoculum, and Seed Bioreactors IPC reports | Written before PPQ using PC data | Written before PPQ using PD and pivotal campaign data as memos, platform knowledge, and other project experiences |
| Production Bioreactor IPC report | Written before PPQ using PC data | Written after PPQ vial thaw, but before first production bioreactor run using PC data |
It is worth noting that all these strategies may well set a standard for our future PC studies to shorten timelines. If a project is urgent, we should be able to use RCB for process development and MCB for late-stage, pivotal, and PPQ campaigns. During FEMA assessment, platform knowledge is always a good justification to reduce PC study run numbers. After preliminary qualification of SDM with a limited number of replicates, the retrospective SDM qualification is a good strategy. Satellite 5-L runs can be used for lab PC studies, as long as the SDM protocol is followed. PC and IPC report writing strategies and moving the worst-case linkage PC study after the PPQ campaign are also applicable to our future PC studies and PPQ campaign for other biologics.
In conclusion, cell culture upstream PC studies and IPC strategy development were shortened from 1 year to approximately 4 months from the upstream process lock to the initiation of PPQ campaign. In the end, the PPQ campaign for mAb1 and mAb2 were successfully completed within a tight timeline, but without sacrificing the quality of the subsequent PPQ campaign. The strategies presented in this report may be applied for other biologics in a risk-based approach to accelerate late-stage process development.
Materials and methods
Cell line and media
The recombinant CHO K1 cell line with GS knockout (GS−/−) was used for IgG1 mAb1 and mAb2 production. The same proprietary chemically defined platform media, i.e., seed medium, basal medium and feed medium, were used for both mAb1 and mAb2, except that methionine sulfoximine was added into the seed medium as a selection agent for mAb1, but not mAb2 for the seed expansion steps.
Cell culture operation
The same platform cell culture process was used in GMP campaigns at 2000-L bioreactor scale for mAb1 and mAb2. Both GMP campaigns for mAb1 and mAb2 were run at 2000-L bioreactor scale as follows. One MCB vial was thawed and passaged in shake flasks, wave bioreactors, and then 200-L seed bioreactor at N-2 step for cell expansion in a batch mode every 3–4 days. N-1 seed bioreactor at 500-L scale was run at fed-batch mode to ensure a sufficient final VCD for the inoculation of 2000-L production bioreactor with a target initial VCD of 5 × 106 cells/mL.33 The fed-batch production bioreactor was controlled at 36.5°C with daily feeding starting on day 2 using proprietary pH and DO control strategies for a full duration of 14 days.
The 5-L bioreactors for mAb1 and mAb2 SDMs were inoculated with either N-1 500-L seed bioreactor dropout or N-1 lab 5-L bioreactor seed culture using the same vial thaw protocol and similar passaging schedules as the N-1 500-L bioreactor seeds. All scale-dependent parameters at 5-L scale were controlled the same as 2000-L scale. Scale-independent parameters at 5-L scale were described in the Results section.
In-process assays for cell culture performance and quality attributes
In-process cell culture performance assays were performed as follows. Cell culture broth was sampled from the bioreactor daily and was directly analyzed for gases, cell count, nutrients, and metabolites. Offline pH, pCO2 and pO2 were measured using a BioProfile pHOx analyzer (Nova Biomedical). VCD and cell viability were quantified off-line using a Vi-Cell XR automatic cell counter (Beckman Coulter). Glucose, glutamine, glutamate, lactate, and ammonia were quantified using a BioProfile FLEX analyzer (Nova Biomedical) for GMP campaigns, while using a CEDEX Bio HT analyzer (Roche) for 5-L bioreactors. Titer was analyzed using a Protein A UPLC method. The normalized titer, expressed as normalized weight/L, is equal to the true titer (g/L) at each time point divided by the day 14 titer specification limit in the process description.
Cell culture supernatant samples were purified by Protein A chromatography prior to all product quality measurements. These samples were then run for several in-process tests as follows. Charge variant species (reported as percent area for acidic, main, and basic peaks) were measured by imaged capillary isoelectric focusing. N-glycan profiles (e.g., G0, G0F, G1F, G2F, Man5) were measured using a commercially available RapiFluor-MS N-Glycan kit from Waters. SEC was used to measure percent areas for monomer, HMW and LMW species
Supplementary Material
Acknowledgments
Thanks to our colleagues from Process Development Cell Line, Upstream, Downstream, Analytical, Scale-Up Lab, and Clinical Manufacturing Facility for supporting this study at Bristol-Myers Squibb.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Authors’ contributions
JX and JO wrote the original manuscript. JX, JO, and KM conceived the original research idea, designed the study, and performed lab work and data analysis. MB and AK contributed to the project administration and funding acquisition. All authors reviewed, edited, and approved the manuscript.
Abbreviations
BLA: Biologics License ApplicationCHO: Chinese hamster ovaryCMC: chemistry, manufacturing, and controlsCOVID-19: coronavirus disease 2019CPA: critical performance attributeCPP: critical process parameterCQA: critical quality attributeDCB: development cell bankDO: dissolved oxygenDOE: design of experimentsEUA: Emergency Use AuthorizationmAb: monoclonal antibodyFDA: Food and Drug AdministrationFIH: first in humanFMEA: failure mode and effects analysisGMP: good manufacturing practiceHCP: host cell proteinsHMW: high molecular weight speciesIND: Investigational New DrugIPC: in-process control strategyLIVCA: limit of in vitro cell ageLPM: liter per minuteLMW: low molecular weight speciesLTSS: long-term stability studyMA: monitored attributeMCB: master cell bankMP: monitored parameterPA: performance attributePC: process characterizationPP: process parameterPPQ: process performance qualificationQbD: quality by designRCB: research cell bankSDM: scale-down modelSEC: size-exclusion chromatographyTOST: two one-sided testsTox: toxicologyVCD: viable cell densityWCB: working cell bank
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.WHO Coronavirus Dashboard . World Health Organization, 2022. https://covid19.who.int/. Accessed February 27, 2022.
- 2.Pecetta S, Finco O, Seubert A.. Quantum leap of monoclonal antibody (mAb) discovery and development in the COVID-19 era. Semin Immunol. 2020;50:101427. doi: 10.1016/j.smim.2020.101427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Development of Monoclonal Antibody Products Targeting SARSCoV-2, Including Addressing the Impact of Emerging Variants, During the COVID-19 Public Health Emergency. Guidance Industry US Food Drug Administration. 2021. [Google Scholar]
- 4.Cao Y, Su B, Guo X, Sun W, Deng Y, Bao L, et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients. B Cells Cell. 2020;182:73–84.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zost SJ, Gilchuk P, Case JB, Binshtein E, Chen RE, Nkolola JP, Schäfer A, Reidy JX, Trivette A, Nargi RS, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. 2020;584(7821):443–11. doi: 10.1038/s41586-020-2548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robbiani DF, Gaebler C, Muecksch F, Lorenzi JCC, Wang Z, Cho A, Agudelo M, Barnes CO, Gazumyan A, Finkin S, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584(7821):437–42. doi: 10.1038/s41586-020-2456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, Musser BJ, Soo Y, Rofail D, Im J, et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N Engl J Med. 2021;384(3):238–51. doi: 10.1056/NEJMoa2035002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, Huhn G, Cardona J, Mocherla B, Stosor V, et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N Engl J Med. 2021;384(3):229–37. doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta A, Gonzalez-Rojas Y, Juarez E, Crespo Casal M, Moya J, Falci DR, Sarkis E, Solis J, Zheng H, Scott N, et al. Early Treatment for Covid-19 with SARS-CoV-2 Neutralizing Antibody Sotrovimab. N Engl J Med. 2021;385(21):1941–50. doi: 10.1056/NEJMoa2107934. [DOI] [PubMed] [Google Scholar]
- 10.Wu L, Zhou L, Mo M, Liu T, Wu C, Gong C, Lu K, Gong L, Zhu W, Xu Z, et al. SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduction Targeted Therapy. 2022;7(1):1–3. doi: 10.1038/s41392-021-00863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplon H, Chenoweth A, Crescioli S, Reichert JM. Antibodies to watch in 2022. mAbs. 2022;14(1):2014296. doi: 10.1080/19420862.2021.2014296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.COVID-19 Biologics Tracker . The Antibody Society, 2022. www.antibodysociety.org/covid-19-biologics-tracker/. Accessed February 27, 2022.
- 13.Torjesen I. Drug development: the journey of a medicine from lab to shelf. Pharmaceutical Journal. 2015. [Google Scholar]
- 14.How long a new drug takes to go through clinical trials. Cancer Research UK; 2019. [Google Scholar]
- 15.Kelley B. Developing therapeutic monoclonal antibodies at pandemic pace. Nat Biotechnol. 2020;38(5):540–45. doi: 10.1038/s41587-020-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J, Rehmann MS, Xu M, Zheng S, Hill C, He Q, Borys MC, Li ZJ. Development of an intensified fed-batch production platform with doubled titers using N-1 perfusion seed for cell culture manufacturing. Bioresources Bioprocessing. 2020;7(1). doi: 10.1186/s40643-020-00304-y. [DOI] [Google Scholar]
- 17.Rehberger B, Wodarczyk C, Reichenbächer B, Köhler J, Weber R, Müller D. Accelerating stable recombinant cell line development by targeted integration. BMC Proceedings 2013; 7:P111. [Google Scholar]
- 18.Stuible M, Gervais C, Lord-Dufour S, Perret S, L’Abbé D, Schrag J, St-Laurent G, Durocher Y. Rapid, high-yield production of full-length SARS-CoV-2 spike ectodomain by transient gene expression in CHO cells. J Biotechnol. 2021;326:21–27. doi: 10.1016/j.jbiotec.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan L, Rizzi G, Bierilo K, Tian J, Yee JC, Russell R, Das TK. Comparative study of therapeutic antibody candidates derived from mini-pool and clonal cell lines. Biotechnol Prog. 2017;33(6):1456–62. doi: 10.1002/btpr.2477. [DOI] [PubMed] [Google Scholar]
- 20.Bolisetty P, Tremml G, Xu S, Khetan A. Enabling speed to clinic for monoclonal antibody programs using a pool of clones for IND-enabling toxicity studies. mAbs. 2020;12(1):8. doi: 10.1080/19420862.2020.1763727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Chen J, Wang J, Gao Q, Ma Z, Xu S, et al. Reshaping cell line development and CMC strategy for fast responses to pandemic outbreak. Biotechnol Prog. 2021;n/a:e3186. [DOI] [PubMed] [Google Scholar]
- 22.Xu G, Yu C, Wang W, Fu C, Liu H, Zhu Y, Li Y, Liu C, Fu Z, Wu G, et al. Quality comparability assessment of a SARS-CoV-2-neutralizing antibody across transient, mini-pool-derived and single-clone CHO cells. mAbs. 2022;14(1):2005507. doi: 10.1080/19420862.2021.2005507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, Rehmann MS, Xu X, Huang C, Tian J, Qian NX, Li ZJ. Improving titer while maintaining quality of final formulated drug substance via optimization of CHO cell culture conditions in low-iron chemically defined media. mAbs. 2018;10(3):488–99. doi: 10.1080/19420862.2018.1433978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li F, Vijayasankaran N, Shen AY, Kiss R, Amanullah A. Cell culture processes for monoclonal antibody production. mAbs. 2010;2(5):466–77. doi: 10.4161/mabs.2.5.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Böhl OJ, Schellenberg J, Bahnemann J, Hitzmann B, Scheper T, Solle D. Implementation of QbD strategies in the inoculum expansion of a mAb production process. Eng Life Sci. 2021;21(3–4):196–207. doi: 10.1002/elsc.202000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, Woodcock J. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–83. doi: 10.1208/s12248-014-9598-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finkler C, Krummen L. Introduction to the application of QbD principles for the development of monoclonal antibodies. Biologicals. 2016;44(5):282–90. doi: 10.1016/j.biologicals.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Q8(R2) Pharmaceutical Development. US Food Drug Administration. 2009. [Google Scholar]
- 29.Hakemeyer C, McKnight N, John RS, Meier S, Trexler-Schmidt M, Kelley B, Zettl F, Puskeiler R, Kleinjans A, Lim F, et al. Process characterization and design space definition. Biologicals. 2016;44(5):306–18. doi: 10.1016/j.biologicals.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Abu‐Absi SF, Yang L, Thompson P, Jiang C, Kandula S, Schilling B, Shukla AA. Defining process design space for monoclonal antibody cell culture. Biotechnol Bioeng. 2010;106(6):894–905. doi: 10.1002/bit.22764. [DOI] [PubMed] [Google Scholar]
- 31.Li F, Hashimura Y, Pendleton R, Harms J, Collins E, Lee B. A systematic approach for scale‐down model development and characterization of commercial cell culture processes. Biotechnol Prog. 2006;22(3):696–703. doi: 10.1021/bp0504041. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Xu XK, Huang C, Angelo J, Oliveira CL, Xu MM, Xu X, Temel D, Ding J, Ghose S, et al. Biomanufacturing evolution from conventional to intensified processes for productivity improvement: a case study. mAbs. 2020;12(1):13. doi: 10.1080/19420862.2020.1770669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yongky A, Xu J, Tian J, Oliveira C, Zhao J, McFarland K, Borys MC, Li ZJ. Process intensification in fed-batch production bioreactors using non-perfusion seed cultures. mAbs. 2019;11(8):1502–14. doi: 10.1080/19420862.2019.1652075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geigert J. The challenge of CMC regulatory compliance for biopharmaceuticals and other biologics. Springer; 2013. [Google Scholar]
- 35.Pereira S, Kildegaard HF, Andersen MR. Impact of CHO metabolism on cell growth and protein production: an overview of toxic and inhibiting metabolites and nutrients. Biotechnol J. 2018;13(3):1700499. doi: 10.1002/biot.201700499. [DOI] [PubMed] [Google Scholar]
- 36.Kepert JF, Cromwell M, Engler N, Finkler C, Gellermann G, Gennaro L, Harris R, Iverson R, Kelley B, Krummen L, et al. Establishing a control system using QbD principles. Biologicals. 2016;44(5):319–31. doi: 10.1016/j.biologicals.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 37.Siemieniuk RAC, Bartoszko JJ, Ge L, Zeraatkar D, Izcovich A, Kum E, Pardo-Hernandez H, Qasim A, Martinez JPD, Rochwerg B, et al. Drug treatments for covid-19: living systematic review and network meta-analysis. BMJ. 2020;370:m2980. doi: 10.1136/bmj.m2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelley B, Renshaw T, Kamarck M. Process and operations strategies to enable global access to antibody therapies. Biotechnol Prog. 2021;37(3):e3139. doi: 10.1002/btpr.3139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.