

Abstract
Diverging physicochemical properties of HIV drug combinations are challenging to formulate as a single dosage form. We have found that 2-to-4 hydrophilic and hydrophobic HIV drugs in combination can be stabilized with lipid excipients under a controlled solvent removal process to form a novel pharmaceutical powder distinct from typical amorphous material. This discovery has enabled production of a drug combination nanoparticle (DcNP) powder composed of 3 HIV drugs—water-insoluble lopinavir (LogP = 4.7) and ritonavir (LogP = 5.6) and water-soluble tenofovir (LogP = −1.6). DcNP powder, exhibiting repeating units of multi-drug-motifs (referred to as MDM), is made by dissolving all constituents in ethanolic solution, followed by controlled solvent removal. The DcNP powder intersperses chemically diverse drug molecules with lipid excipients to form repeating MDM units. The proposed MDM structure is consistent with data collected with X-ray diffraction, differential calorimetry, and time-of-flight secondary ion mass spectrometry. The successful assembly of chemically diverse drugs in MDM structure is likely due to a novel process of making drug combination powders. The method described here has successfully extended to formulating other clinically prescribed antiviral drug combinations, and thus may serve as a platform technology for developing drug combination nanoparticles for treating a wide range of chronic diseases.
Keywords: HIV/AIDS, Drug-combination particle(s), Spray drying, X-ray powder diffraction (XRD), Nanoparticle(s), Powder technology(s), Physical characterization, Drug-excipient interaction(s), Structure
Introduction
The availability of well-characterized viral protein structures, coupled with success in developing drugs targeted to viral proteins, has significantly improved outcomes in HIV treatments. Chronic oral administration of HIV drug combinations sustains viral suppression and prevents people with HIV from progressing to AIDS. These patients take multiple drug combinations to reduce drug resistance, which occurs in long-term disease.1,2 Multidrug regimens are effective, but are associated with significant pill burden. Patient adherence is necessary to sustain drug levels and mitigate the risk of harboring viruses resistant to oral regimens. A typical dosing regimen can require oral dosing between one to three times a day with multiple tablets, presenting a large burden to patients.3 To improve patient quality of life, decrease multiple-pill fatigue, and risk of missed doses, oral fixed dose combinations (FDCs) have emerged as a “one-pill, once a day” option to treat people with HIV.3,4 This one-pill a day strategy is enabled by the development of advanced manufacturing approaches such as multi-layer tableting, multi-particulate systems, and monolithic systems that allow chemically diverse drugs to be stabilized together in a single oral dosage form.5,6 The challenges in producing physically stable drug combination dosage forms as a single unit are well recognized and require complex, multilayer blending and compression strategies. However, if one can produce multiple chemically diverse HIV drugs as a physically stable and homogenous drug combination powder, then these challenges could be mitigated. Achieving such a product may also simplify the process for producing both oral solid dosage forms and drug-combinations in suspension.
In addition to the “one pill a day” strategy to make solid oral dosage forms for HIV drug combinations, long acting injectable dosage forms of antiretrovirals are an alternative means to treat patients with HIV. Long acting injectables can provide more consistent plasma drug levels over a longer period than daily dosing. Consistent drug levels over time can then, in turn, provide more predictable degrees of sustained viral suppression and improve patient adherence to chronic HIV treatment. The ability to deliver an effective, single injectable dose that potentially replaces 30 pills or more is appealing to patients and clinicians.7 However, parenteral injectable products pose additional physical challenges such as aggregation, chemical degradation, and precipitation that may limit their development. To improve the physical stability of long acting parenteral injections, polymeric excipients such as poloxamer 338, polysorbate 20, or polyethylene glycol are used to stabilize drugs in suspension for use as long acting injectables.8,9 These current polymers are designed to formulate single drug injectables. They are not suitable for the stabilization of multiple combination drugs with diverse physicochemical properties (e.g. compounds that are water soluble and insoluble) in a single parenteral injection. In addition to biopolymers, lipid excipients have been used—typically as liposomes or lipid-membrane vesicles—to formulate nanosuspensions with various physicochemical properties. Thus, certain lipid molecules may serve as excipients for developing multi-drug injectables.10–12 Due to their ability to form various membrane-like structures in water, lipid excipients are typically used to encapsulate APIs within enclosed membrane layers (lipid vesicles or liposomes), or to bind and stabilize APIs within the membrane layers. These properties have allowed lipid excipients to be used extensively for developing parenteral administrations.13 Traditional lipid-based delivery systems (such as liposomes) incorporate hydrophobic drugs within the acyl chains of the bilayer-membrane or encapsulate hydrophilic drugs within the aqueous core of membrane bilayers. However, liposomal drug manufacturing processes are complex and often face challenges in manufacturing scalability, the need to remove of unincorporated (free) drug, and physical instability as a suspension in storage. Ideally, a method to stabilize hydrophobic and hydrophilic HIV drugs together as a single unit, without the need for encapsulation, can aid the development of long acting injectables in suspension.
Recently, our laboratory has reported a process by which drugs with disparate physicochemical properties can be stabilized with lipid excipients in powder form.14–16 In early studies, various lipid-based excipients were tested to incorporate antiviral drugs, beginning with single drug particles and expanding to 3 and 4 drug combination nanoparticles. These drug combination nanoparticles (DcNPs) are suitable for injectable dosage forms and demonstrate long acting effect. One HIV DcNP composed of water-soluble tenofovir (TFV, LogP −1.6) and water-insoluble lopinavir (LPV, LogP = 4.7) and ritonavir (RTV, LogP = 5.2) was reported to be stabilized by the lipid excipients: 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000). In this composition, DSPC is intended to stabilize hydrophobic drugs (such as LPV and RTV) through interactions within the acyl chains. While DSPE-PEG2000 is intended to stabilize hydrophilic drugs (such as TFV) through hydrophilic interactions within the polyethylene polymer. The ratio of the 3 drugs was selected based on the clinically relevant ratios used in oral dosage forms after consideration of their respective pharmacokinetic profiles and therapeutic target drug levels. Total drug-to-lipid ratios (and the ratio of the two lipid excipients) were iteratively tested and described in previous reports. Early studies optimized the composition and process for DcNP formation to yield consistent nano-particle size and association efficiencies for each drug listed. The resulting formulation is suitable for development as an injectable formulation in suspension and was used in multiple pharmacokinetic studies to produce reproducible long acting, in vivo results.17–19 When the 3 drugs—TFV, LPV and RTV in DcNP suspension—were tested in non-human primates (NHPs), only DcNPs and not free drug dosage forms exhibited long-acting pharmacokinetics in both plasma and lymphoid cells.16–20 Due to its reproducibility and in vivo stability, free drug removal—commonly required for alternative liposome encapsulated dosage forms—is not needed in this novel DcNP suspension. The reported results, along with the results from a validated, mechanism-based pharmacokinetic model, indicate that a majority of hydrophilic TFV and hydrophobic LPV and RTV are stabilized together in a DcNP form throughout the 2-week PK study.
In our initial studies, we employed rotary evaporation of drugs and excipients in solution as a controlled solvent removal process to form drug combination powder at a smaller scale. However, rotary evaporation used to produce DcNP powder had limited process control, inconsistent in vitro results and was not readily scalable for clinical evaluation of DcNP product (long acting suspension). With the need to improve process control and monitoring of powder characteristics, a spray drying method was developed. The improved process control is an essential step in forming stable, consistent drug combination powder for use as a suspension to provide long acting, targeted pharmacokinetics in larger scale NHP studies.
Thus, the objective of this study is to define the role of controlled solvent removal in producing drug-combination particles in powder form and to characterize the resultant DcNP powder properties and distinctive molecular properties, which may relate to the stable suspension intended for long-acting pharmacokinetics. After complete solubilization of 3 chemically diverse drugs and 2 lipid excipients together, controlled solvent removal by spray drying is a critical step in locking these constituents together. We found that controlled solvent removal of drugs and lipids in a single solution led to the formation of homogenous repeats of drug-lipid excipients in a unified matrix structure (which we have termed multi-drug-motifs or MDM). This unique structure (and the technology used to achieve that structure) may enable the development of combination powders with chemically diverse drugs to treat a wide range of chronic diseases.
Materials and Methods
Materials
GMP quality lopinavir (LPV), ritonavir (RTV) and tenofovir (TFV)—referred to as active pharmaceutical ingredients or APIs—were a gift from Mylan pharmaceuticals (Morgantown, West Virginia). Dolutegravir (DTG), rilpivirine (RPV) and lamivudine (3TC) were purchased from Carbosynth (Berkshire, United Kingdom). GMP grade 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(- polyethylene glycol)-2000] (DSPE-PEG2000) were purchased from Cordon Pharma (Liestal, Switzerland). Anhydrous ethanol was purchased from Decon Pharmaceuticals (King of Prussia, PA). All other reagents were analytical grade or higher.
Preparation of Drug Combination Nanoparticles in Powder and in Suspension
To produce drug combinations stabilized with lipid excipients in powder form, two lipophilic APIs, LPV and RTV were first dissolved together with two lipid excipients, DSPC and DSPE-PEG2000 at 60 °C in ethanol. To maintain an appropriate ionization state, hydrophilic TFV was dissolved in 200 mM NaHCO3 buffer (pH~7.4) before being added dropwise into the two lipophilic APIs and two lipid excipients in ethanolic solution, and allowed to mix at 60 °C. The bicarbonate buffer keeps TFV in solution and allows for complete mixing with the ethanolic solution containing LPV/RTV and lipid excipients. The final NaHCO3 buffer to ethanol volume ratio is 3:97. Solvent removal from the 3 drugs and lipid excipients in ethanol/buffer solution was achieved with either rotary evaporation (New Castle, Delaware) or an electronically controlled ProCepT 4-M8TriX spray dryer (Zelzate, Belgium) to produce a DcNP powder product containing the 3 drugs and 2 lipids.
To make DcNP powder by rotary evaporation, a typical batch includes 10 g of total lipid and drug dissolved in 200 mL of ethanol/buffer (5% w/v solid). Rotary evaporation is performed at a constant temperature of 60 °C with vacuum pressure increasing from 300 mmHg to 760 mmHg over 1 h. Under these conditions, rotary evaporation can produce approximately 10 g of material (to make ~50 mL of suspension) with 100% recovery.
To make DcNP powder by spray drying, a typical batch includes up to 200 g of total lipid and drug dissolved in 2 L of ethanol/buffer (10% w/v solid). Spray drying is performed at a constant temperature of 70 °C with the feedstock solution introduced via peristaltic pump at a rate of 300 RPM. The spray drying parameters were as follows: inlet air speed of 0.3 m3/min, chamber pressure of 25 mbar, atomization air flow of 25 L/min, cyclone air flow of 0.1 m3/min and a nozzle size of 0.6 mm. With these parameters, typically 65% of the mass in the original feedstock solution is recovered; there is no change in the drug or lipid excipient ratios compared to that of the initial mixture in solution. This spray drying process has been used to generate as much as 200 g of dried product (to make ~ 1 L of suspension). No residual ethanol is detectable in the dried product by GC-MS (with 3000 ppm equal to the lower limit of quantification). Dried product, referred to as DcNP powder, is stored under vacuum desiccation conditions and analyzed for physical characteristics as detailed below. The powder particle size was visually analyzed with transmission electron microscopy (TEM) and ranges from 1 to 5 μm in size.
The methods to prepare DcNPs as injectable suspensions have been described in detail in previous reports.17 Briefly, to produce 1 L of DcNP in aqueous suspension, 220 g of DcNP powder is added to sterile half-normal saline (0.45% NaCl solution) containing 20 mM NaHCO3 (pH = 7.4). This process provides a DcNP in suspension with a nominal drug concentration of 10.7 mg/mL LPV, 3.1 mg/mL RTV, 6.1 mg/mL TFV and total lipid concentration of 180 mM (9:1 DSPC to DSPE-PEG2000 M ratio) with an osmolarity of ~306 mOsm. After suspension, particle size reduction is achieved with homogenization to yield uniform particles with an average diameter of 52.4 ± 9.1 nm as measured by photon correlation spectroscopy (NICOMP 380 ZLS, Particle Sizing Systems, Santa Barbara, CA).17
Powder X-Ray Diffraction
Powder X-ray Diffraction (PXRD) was used to characterize the matrix structure and crystallinity of DcNP powder. PXRD analysis is performed on a Bruker D8 Focus X-ray Diffractor (Madison, WI, USA) with Cu-Kα radiation. Operational voltage and amperage were set to 40.0 kV and 40.0 mA, respectively. Experimental parameters include a step size of 0.035°2θ in an operating range of 5°–50° 2θ. Powder (~100–200 mg) was pressed into a sample container to obtain a flat upper surface. The data collected were plotted as intensity (counts) vs angle (2θ).
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) was performed on the DcNP dry powder to characterize the thermal behavior of individual API, excipient, and the combined product. For this purpose, we used a TA DSC Q20 (New Castle, DE, USA). Baseline calibrations were performed every day prior to collecting DSC data by ramping 10 °C/min up to 200 °C under constant nitrogen (50 mL/min). The samples (1–4 mg each) were placed in a hermetically sealed aluminum pan and subjected to increasing temperature at 10 °C/min from 25 °C to 200 °C. The change in heat flow delta-H (W/g) was collected and plotted against increasing temperature.
Scanning Electron Microscopy (SEM)
Dry DcNP powder composed of LPV, RTV, TFV and two lipid excipients-DSPC and DSPE-PEG2000 was visualized using a FEI Sirion XL30 Scanning Electron Microscope (Hillsboro, Oregon). The powder samples were placed on a conductive and adhesive carbon backplate and placed under a nitrogen stream to remove non-adhered particles. These samples were then sputter coated with Au/Pd for 20 min prior to visualization for an estimated coat depth of 15 nm. The instrument was operated under a working distance of 4.7–5.1 mm and an accelerating voltage of 5–15 kV. The electron micrograph was collected digitally and analyzed based on equivalent amplification for each variant.
Time of Flight Secondary Ion Mass Spectrometry (ToF-SIMS)
Time of Flight Secondary Ion Mass Spectrometry was performed to evaluate molecular distribution within DcNP powders. Distribution profiles for each component in the DcNP (LPV, RTV, TFV and two lipid excipients DSPC and DSPE-PEG2000) were acquired simultaneously with a 25 keV Bi3+ cluster ion source in the pulsed mode. Secondary ions of these components were captured with the IONTOF ToF-SIMS spectrometer (Münster, Germany) to determine the spatial arrangement of the constituents. Depth profiles were acquired in the non-interlaced mode using alternating analysis and sputter cycles. Data was acquired over a mass range of m/z = 0 to 850 using a primary ion current of 0.035 pA in delayed extraction mode over a 100-μm × 100-μm area centered within the sputter crater. Based on these parameters, an estimated depth of 100 nm–200 nm was analyzed for the depth profile. The Secondary ions of a given polarity were extracted and detected using a reflectron time-of-flight mass analyzer. The primary ion dose for each spectrum was 2.3 × 1011 ion/cm2. Sputtering was carried out using a gas cluster ion beam with 10 keV argon 1000 clusters rastered over a 500-μm × 500-μm area for 7 s at a current of 7 nA giving a sputtering dose of 1.22 × 1014 ion/cm2. Positive ion spectra were calibrated using the , , and peaks. The negative ion spectra were calibrated using the CH−, OH−, and C2H− peaks. Calibration errors were kept below 20 ppm. Mass resolution (m/Δm) for a typical spectrum was 3400 for m/z = 27 (pos) and 3600 for m/z = 25 (neg). Data was assembled by overlaying sequential X–Y spectra along the Z-axis of the drug and lipid constituents over a 100 μm by 100-μm grid with each constituent represented by its own color. Further analysis was performed with ImageJ software to estimate the relative abundance of each species across the x-axis of the acquired grid.
Results
Effects of Controlled Solvent Removal by Spray Drying on the Physical Structure of Drug Combination Powder Products Based on X- Ray Diffraction Analysis
We first determined the effects of spray drying as a process to remove solvent under controlled conditions from drugs and lipids mixed in ethanolic solution. To do so, we dissolved LPV, RTV and two lipid excipients—DSPC and DSPE-PEG2000—in ethanol; followed by the addition of TFV in an NaHCO3 buffer such that the final solution contains 3% aqueous buffer. The ethanol-based solvent in the drug-combination mixture was removed under controlled conditions by spray drying. The spray dried drug-combination powder containing 3 drugs and 2 lipids was analyzed with a powder x-ray diffraction (PXRD) instrument. In Fig. 1, the individual constituents within the DcNP powder—LPV, RTV, TFV, DSPC or DSPE-PEG2000—all exhibit crystalline signatures as seen by their distinct X-ray diffraction (peak) patterns. Analysis of each single drug or single excipient control (Fig. 1, Panels a–c, e + f) shows various peaks at the respective 2θ angle positions where Bragg’s law is fulfilled. In Fig. 1 Panel h, the diffraction pattern of the physical mixture of drugs and excipients in the same molar ratio as the DcNP powder is similar to DSPC alone (Panel e) due to the high mass % of this excipient in the DcNP formulation. However, additional peaks attributable to the other constituents are apparent, particularly in lower angle regions (5°—15° 2θ). In contrast to these unique and assignable X-ray diffraction patterns for each component within the DcNP powder, the DcNP powder itself (prepared under controlled solvent removal) did not contain any assignable signature of each constituent. Instead, the 5 components in the DcNP powder are assembled in such a way that they produce two new and distinct diffraction peaks observed at 5.64°2θ and 21.47°2θ (Fig. 1 Panel i).
Fig. 1.
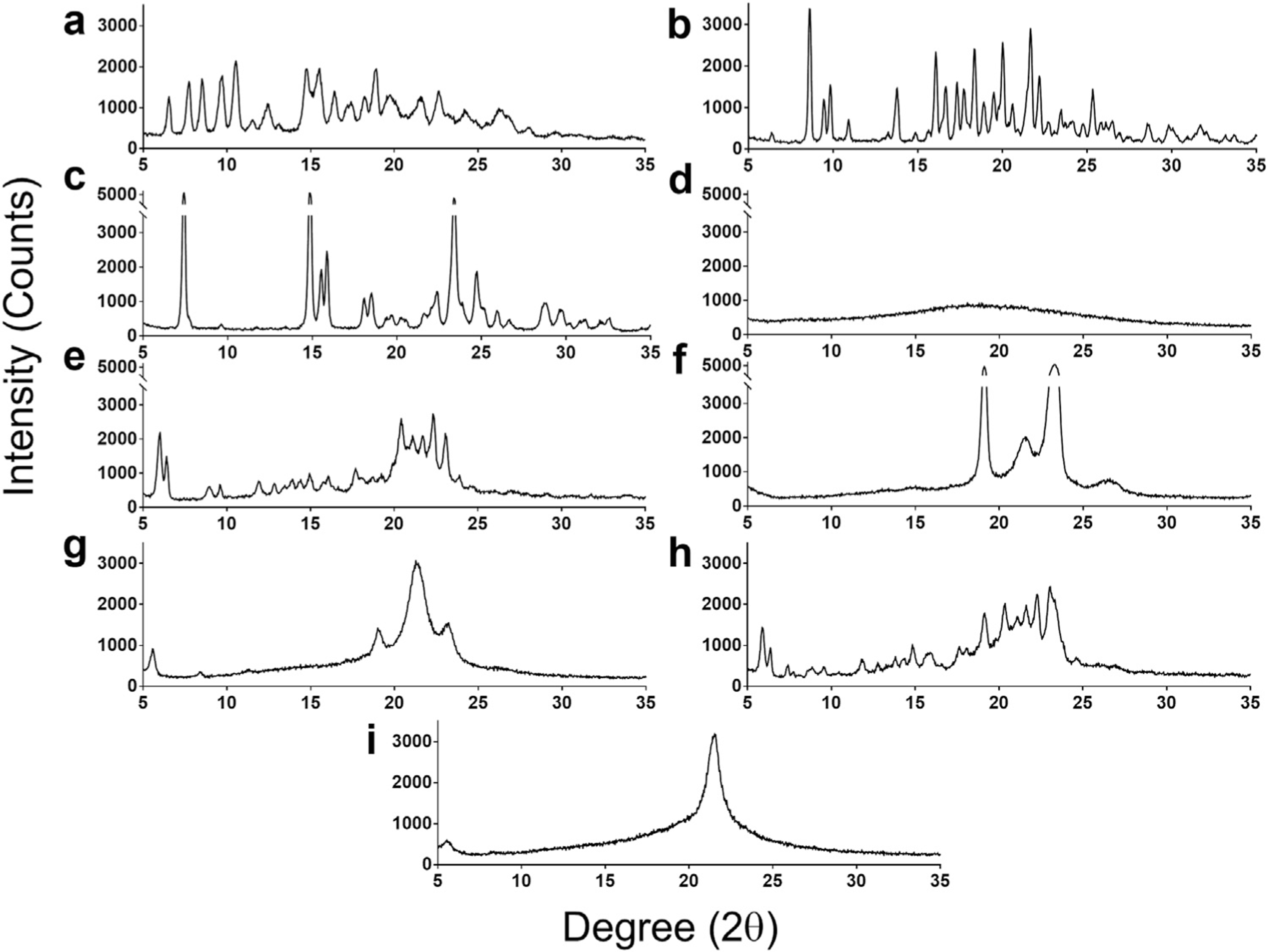
Effects of the controlled solvent removal process on the formation of a unique drug combination powder product containing 3 APIs–lopinavir (LPV), ritonavir (RTV) and tenofovir (TFV) and 2 lipid excipients–DSPC and DSPE-PEG2000. The indicated component or combination of APIs plus lipid excipient products was subjected to powder X-Ray Diffraction (PXRD) analysis (Panels a–i). The PXRD scans are presented as Degrees (in 2θ) vs Intensity (counts). Representative PXRD scans for (a) LPV, (b) RTV, (c) TFV, (d) amorphous mixture of LPV/RTV/TFV after controlled solvent removal without excipients, (e) DSPC, (f) DSPE-PEG2000, (g) DSPC and DSPE-PEG2000 after controlled solvent removal without drugs are presented. (h) A physical mixture of drugs and excipients (LPV/RTV/TFV/DSPC/DSPEPEG2000), and (i) all constituents after controlled solvent removal (LPV/RTV/TFV/DSPC/DSPE-PEG2000) are presented.
To verify this result, a control composed of just the two lipid excipients (9:1 DSPC: DSPE-PEG2000) without the APIs (LPV, RTV, and TFV) was subsequently produced under the identical spray drying process and analyzed with PXRD. By omitting drugs in this composition, this control is used to evaluate the effect of the controlled solvent removal process on lipid excipients. As shown in Fig. 1, Panel g, compared to DcNP products, the excipient product produces two additional peaks at the 19.1°2θ and 23.1°2θ positions, in addition to the main peak observed at 21.47 °2θ for DcNP with all components (Panel i vs g). The additional peaks correspond to the peaks observed in the DSPE-PEG2000 control and may indicate a PEG crystalline structure within the powder product of two lipid excipients (Panel f). Another powder control containing 3 drugs only (LPV, RTV, and TFV) without lipid excipients exhibits a characteristic amorphous halo (Panel d). Collectively, the PXRD data suggests that (1) controlled solvent removal by spray drying produces a new unique structure, revealed in PXRD as one minor and one major peak at 5.64°2θ and 21.47°2θ. (2) None of the original 3 drug signatures are present indicating that there is no unchanged, crystalline API remaining. (3) The structural signature is not achieved with excipient alone undergoing an identical spray drying process, (4) drug alone undergoing complete amorphous transformation, or (5) by a physical admixture of an identical composition. Thus, the controlled solvent removal of this composition leads to a unique repetitive organization of DcNP powder, revealed by PXRD. This repetitive organization is referred to as multi-drug-motif or MDM structure.
Verification of DcNP Powder’s Unique Organizational Structure with Differential Scanning Calorimetry
We next investigated the thermal behavior of the DcNP powder. To do so, we employed differential scanning calorimetry as a complementary technique to characterize this unique structure among 3 drugs and 2 lipid excipients formed from controlled solvent removal. As shown in Fig. 2, line g, the spray dried DcNP product exhibits a single endothermic transition apparent with an onset transition temperature of 70.28 °C and a melting point peak at 74.29 °C. This endotherm occurs at a position independent of individual drug and excipient controls (Fig. 2), indicating that it is not the melting of the crystalline structure for each of the drugs or excipients in the DcNP composition. In agreement with the PXRD data, we do not see any evidence of PEG (from the DSPE-PEG2000 lipid excipient) melting which would be expected around 49 °C–52 °C (Fig. 2, line e). Interestingly, the DcNP thermogram also contains a broad exotherm beginning at temperatures >120 °C and extending until the end of the heating ramp. A possible source of this exotherm is mass loss from heating the drug combination powder formulation, which was observed to be ~3.5% based on thermogravimetric analysis measurements at a ramp rate of 10 °C/min to 200 °C. This weight change may be due to atmospheric water adsorbed to the powder through the spray drying process. It is possible that the exotherm may mask the presence of glass step transitions at higher temperatures. Nevertheless, the thermal characterization of spray dried DcNP powder shows that the individual endothermic transitions of the drugs and excipients are no longer observed in lower temperature ranges and a single endotherm for the spray dried powderoccurs at a unique temperature. This suggests that novel nonbonding interactions are formed through the spray drying process and is likely related to the new MDM or structure in the DcNP powder product.
Fig. 2.

Analysis of test and control products of DcNP constituents by Differential Scanning Calorimetry (DSC). The DSC thermograms of combination antiretroviral drugs and excipients are presented in lines a through h. Panel (a) TFV, (b) LPV, (c) RTV, (d) DSPC, and (e) DSPE-PEG2000. The physical mixture of components a-e is presented in panel f. Panel (g) presents the spray dried LPV/RTV/TFV/DSPC/DSPE-PEG2000 (test powder) and Panel (h) presents the spray dried DSPC/DSPEPEG2000 (lipid excipients control).
Effects of Controlled Spray Dried Process on Distribution of LPV, RTV, TFV, DSPC and DSPE-PEG2000 within DcNP Powder
To further characterize the unique structural organization of drug and excipient molecules within DcNP, we analyzed the powder with a Time of Flight Secondary Ion Mass Spectrometry (ToF SIMS). ToF-SIMS is a surface analysis technique that provides information on the molecular distribution of each of the five components: LPV, RTV, TFV, DSPC, and DSPE-PEG2000 assembled together within the DcNP powder product. We also used scanning electron microscopy (SEM) as a complementary technique to provide the overall morphology of the product compared to that of individual molecules in solid states. If the MDM is organized in a repeated pattern—as suggested by PXRD and consistent with thermal behavior noted in DSC (as described above)—each molecular component in the DcNP powder will be homogenously distributed throughout, and detectable as such, in the time-of flight ion scan under ToF-SIMS analysis. On the other hand, if each constituent in the mixture forms micro domains of each representative molecular species according to their respective physical characteristics and lowest energy state (instead of presenting well-distributed and repeated motifs of 5 distinct molecular species), this technique will identify the formation of such clusters within the DcNP powder.
As shown in Fig. 3, the SEM overall morphological analysis reveals a well-distributed morphology for the DcNP powder prepared by the spray drying process (Fig. 3, b). In contrast, the morphology of drugs and lipids prepared by admixture (Fig. 3, a) retained a heterogeneous polymorphic drug and lipid crystalline morphology. The morphology of the spray dried material does not retain any of the physical characteristics of drugs and lipids prepared by admixture, but rather has a homogeneous, spherical shape (~1–5 μm) associated with the size of atomized droplets from the feedstock solution. Furthermore, ToF-SIMS depth profiles (Fig. 4a–c) of the spherical DcNP particles observed in SEM reveal that the three drugs (LPV, RTV and TFV) as well as the excipients (DSPC and DSPE-PEG2000) are evenly distributed in the X–Y plane of the powder and continue to be well-distributed along the Z-axis through depth profile analysis (100 nm–200 nm). These data suggest that there is no preferential accumulation of API or excipients within a single particle (at the surface or interior) and that each individual particle has a uniform composition and shape. The ToF-SIMS depth analysis provides enough resolution to distinguish individual drug crystals in the physical mixture (Fig. 4, d–f), but no microdomains are observed with the spray dried powder in the X, Y or Z-axis. Taken together with the PXRD and DSC analyses, the molecular and spatial distribution of drugs and excipients revealed by ToF SIMS suggests that controlled solvent removal enables the formation of a stable, non-bonding interactions among drug and excipient molecules (MDM structure). These MDM structures cannot be attained through physical mixture but only when the solubilized drugs and lipids have undergone a controlled solvent removal process.
Fig. 3.

Scanning Electron Microscopy (SEM) analysis of the morphology exhibited by DcNP powder containing 5 constituents. This figure presents the distinct morphology of DcNP prepared by (b) solvent removal process or (a) admixture process. Each bar indicates 10 μm.
Fig. 4.

ToF-SIMs Analysis of 5-component DcNPs prepared by controlled solvent removal (Panels a–c) and by admixture control (Panels d–f). RTV (red, mass fragment of 59 AMU), LPV (green, mass fragment of 101.07 AMU) and TFV (blue, mass fragment of 148.04) are shown in panels a and d. DSPC (red, mass fragment of 58.02) and DSPE-PEG2000 (green, mass fragment of 61.03) are shown in panels b and e. The figures generated are composites of the X, Y, and Z axis, with sequential Z-planes overlaid to present a depth profile of 100–200 nm. The X and Y axis in panels a, b, d and e are a μm scale grid that corresponds to the plane of powder that was analyzed. Panels c and f are a pixel analysis using ImageJ software to show the relative abundance of pixels over the X-coordinate of each image. The X and Y axis in panels c and f represent pixel counts.
Effect of Drug Substitutions on Formation of Multi-Drug-Motif (MDM) Using Controlled Solvent Evaporation
To understand whether the MDM structure observed for LPV, RTV, and TFV with lipid excipients is unique to those three APIs or could be substituted with other HIV drugs, we evaluated additional drug combinations targeted to HIV proteins. These drug combination powders employed the same lipid excipients with similar total drug-to-lipid ratios as the lead LPV, RTV, TFV formulation. The hydrophobic drugs, LPV and RTV, were replaced with an integrase strand inhibitor (dolutegravir, DTG, logP = 2.4), a non-nucleoside reverse transcriptase inhibitor (rilpivirine, RPV, logP = 4.5), or both. In some compositions, the hydrophilic nucleoside reverse transcriptase inhibitor (TFV) was either replaced or added in combination with a drug from the same class (lamivudine, 3TC, logP = −0.9) (Table 1). Drug combinations were chosen based on the current WHO recommended list of anti-HIV drug combination therapies, which typically contains one-to-two hydrophilic nucleoside reverse transcriptase inhibitors plus a hydrophobic protease inhibitor, non-nucleoside reverse transcriptase inhibitor, or integrase strand inhibitor of HIV. The variants to the reference DcNP composition were analyzed with PXRD to assess the structural features of these new DcNP powders and are listed in Table 1. To our surprise, we found that with the listed diverse class of HIV drugs, the controlled solvent removal process for DcNP powder preparation produced the same MDM structure as that observed with LPV, RTV and TFV (Fig. 1, panel i) by PXRD analysis (Table 1). These results highlight the broad range of hydrophobic and hydrophilic drugs that are amenable to produce MDMs by this method and process. Hence, other antiviral combinations could be suitable for this approach. Collectively, these data suggest that the controlled solvent removal of hydrophobic and hydrophilic APIs with lipid excipients in solution may enable the formation of a stable structure with repeating multi-drug-motifs (MDM) with multiple combinations of drugs.
Table 1.
Select Drug Combination Nanoparticle Powder (DcNP) Compositions Evaluated For the Formation of Multi-Drug-Motif Structures.a
| DcNP (Tested) | Hydrophilic Drugs (Log P<1) |
Hydrophobic Drugs (Log P>2) |
Multi-Drug-Motif (MDM) Structure | ||
|---|---|---|---|---|---|
| Lamivudine | Tenofovir | Lopinavir | Ritonavir | ||
| Reference | − | + | + | + | Yesb |
| Variant-1 | + | + | + | + | Yes |
| Variant-2 | − | + | − | − | Yes |
| Variant-3 | − | − | + | + | Yes |
| Variant-4 | − | − | + | − | Noc |
| Lamivudine | Tenofovir | Dolutegravir | Rilpivirine | ||
|
| |||||
| Variant-5 | + | + | + | + | Yes |
| Variant-6 | + | + | + | − | Yes |
A partial list of drug combinations with respective antiretroviral APIs were tested using the controlled solvent removal method as described. The resulting DcNP powder products were evaluated with PXRD for formation of MDM repeating units in powder form.
Yes, indicates formation ofMDM structure as evaluated by PXRD diffraction signature for MDM similar to the lead or “Reference” composition as presented in Fig. 1, panel I.
No, indicates the PXRD diffraction pattern retains significant signature of API or excipient profile.
Discussion
Current HIV treatment requires 2–3 oral antiviral drugs in combination to achieve sustained viral suppression. A typical combination regimen is composed of hydrophilic nucleoside reverse transcriptase inhibitors (NRTIs) with hydrophobic integrase or protease inhibitors to achieve viral suppression in plasma. The varying physicochemical—water-soluble and water-insoluble—properties of these drugs can be challenging to formulate in either oral fixed dose combinations or in long acting injectables. We used a simple controlled solvent removal process to stabilize the physical interactions of chemically distinct drug combinations with lipid excipients in DcNP powder form. The controlled solvent removal process forms multi-drug-motifs (MDM) where drugs and lipids are locked together in repeating units. In this report, we have characterized the molecular organization of that motif in detail. The stabilization of 3 chemically diverse HIV drugs by 2 lipid excipients may address the challenges in co-formulating oral fixed dose combinations or long acting injectables and simplify their development processes.
The poor aqueous solubility of many drugs used in HIV such as protease inhibitors or integrase inhibitors is well described. This insolubility limits the rate and extent of HIV drug absorption in oral dosage forms. One approach to overcome the lower bioavailability of oral dosage forms is to use amorphous solid dispersions (ASDs), instead of crystalline drugs, to enhance drug solubility and bioavailability from oral dosing.21–23 ASDs are meta-stable amorphous API molecules dispersed in polymeric matrices, which can be easily identified as an amorphous halo in PXRD diffraction patterns.21,24 The high energy state of amorphous materials increases solubility at the cost of physical stability; specifically an increased chance of crystallization or phase separation during storage.22 The spontaneous devitrification of amorphous material is a known phenomenon and can limit amorphous dosage forms. In our work, we have stabilized chemically diverse APIs in lipid excipients to form a drug combination nanoparticle (DcNP) powder. This DcNP powder does not appear to follow amorphous characteristics, instead presenting clear PXRD diffraction peaks in the powder (Fig. 1, panel i). The two PXRD peaks of DcNP are not attributable to any of the individual drugs or lipid excipients shown in Fig. 1 a–c, e and f. The solvent removal process generates a new ordering of drugs, lipids, and polymers attached to the DSPE lipid (Fig. 1 panel i). It is likely that this new structure is due to the large overall faction of lipid molecules and their physical interactions in the DcNP powder composition and structure (Fig. 1, h). However, the loss of PEG lipid (DSPE-PEG2000) peaks in PXRD is observed when antiviral drugs are introduced to the lipid excipients through spray drying, suggesting specific PEG polymer-drug interactions within the DcNP powder. Further thermo-analysis of DcNP powder by DSC corroborates the PXRD data. For purely amorphous material, we would expect to observe a glass transition temperature as detailed in literature.25 Instead, when DcNP powder is analyzed with DSC, an endothermic event specific to DcNP powder is observed (Fig. 2, panel g). The endothermic event suggests that new nonbonding physical interactions exist between drug, lipid, and polymer in the DcNP powder that are not present in the physically mixed or excipient only controls (Fig. 2 Panel h & f).
Like ASDs, nanocrystals are an alternative approach to improving the aqueous solubility of poorly aqueous drugs. Small particle size allows for more surface area contact with water and greater achievable concentrations in aqueous environments. This approach has been used to improve the bioavailability of drug products and is well established in the literature.26,27 In a recent study, dual nanocrystals of simvastatin and ezetimibe were assembled together in mannitol microparticles to improve the solubility of both cholesterol lowering drugs, enabling nano-sized combination formulations.28 While DcNP powder achieves a similar outcome (multiple drugs stabilized in powder), there are key distinctions to the DcNP process. Simvastatin (logP = 4.7) and ezetimibe (logP = 4.0) are both hydrophobic drugs that have similar aqueous insolubility. In contrast, TFV (logP = −1.6) and LPV (logP = 5.9) or RTV (logP = 6.0) have very different aqueous solubilities. The ability to incorporate chemically diverse compounds together as a single combination drug powder is particularly important in HIV therapy, where multiple agents with divergent solubility are required in current fixed-dose combination to provide sustained viral suppression. If both hydrophilic and hydrophobic drugs are present in nanocrystal form, it is likely that the hydrophilic drug will rapidly dissolve and separate from the combination powder and lead to asynchronous drug bioavailability and overall exposure.
In addition to stabilizing LPV, RTV, and TFV together, we also discovered that the DcNP process can form a similar structure with other antiviral drug combinations that contains both hydrophilic and hydrophobic drugs. For the hydrophobic drugs (LPV, RTV, DTG, and RPV), the average logP was 4.3 with a range from 6 (RTV) to 2.4 (DTG). For hydrophilic drugs (TFV and 3TC), the average logP was −1.25 with a range of −1.6 to −0.9. When we consider the polar surface across hydrophobic and hydrophilic drugs, we do not observe a large difference (129.65 Å2 vs 124.5 Å2). However, when we consider the % of the molecule that the polar surface area encompasses, we observe a large difference between the hydrophobic and hydrophilic groups (28.6% vs 53.6%, respectively). It is possible that hydrophobic drugs have some portion of their structure exposed to hydrophilic regions (28.6%) while the remainder of the molecule is embedded in the acyl chains of DSPC and DSPE-PEG2000. For hydrophilic drugs, the opposite may be true, where the majority of the molecule is retained in the hydrophilic PEG regions of DSPE-PEG2000, but a fraction of the molecule can interact with the acyl chains. Regardless of the exact mechanism of interaction, multiple pharmacokinetic studies in non-human primates have shown that the MDM structure is a platform technology that can facilitate long acting pharmacokinetics with different drug combination nanoparticles.14–16
While the molecular mechanisms leading to MDM formation in the powder remain elusive, it is possible that after initial dissolution of DSPC, DSPE-PEG2000, LPV, RTV and TFV in 97:3 v/v ethanol/water, the 5 constituents of the DcNP interact freely in ethanolic solution. As ethanolic solvent is gradually removed under controlled conditions, the hydrophobic constituents LPV and RTV align with the hydrophobic fatty acyl chains of DSPC; while hydrophilic TFV aligns or gradually replaces bound water on the polyethylene-glycol (PEG) polymer of the DSPE-PEG2000. With continued solvent removal, it is possible that the PEG moieties of DSPE-PEG2000 and hydrophilic TFV interact with higher affinity and replace bound water on PEG with TFV hydrogen bonding to repeating units of ethylene glycol. As residual bound water is removed, the substitution of hydrogen bonds occupied by water with TFV then stabilizes the overall DcNP powder. Since the controlled solvent removal process is enabled by a spray-dryer, the process of lipid drug interactions within each atomized droplet of drug and lipid excipients in solution produces uniform, repeating units of MDM collected as a spray-dried DcNP powder (Figs. 3 and 4). While the elucidation of these and other mechanisms is of interest, it is beyond the scope of the current report. The interactions between drugs and lipid excipients detected by PXRD and DSC appear to be consistent with a repeating structure in the combination powder. This structure has been referred to as MDM and is graphically represented in Fig. 5.
Fig. 5.

Schematic representation of multi-drug-motifs or MDM repeating units of drugs and excipients in powder state. Based on each of the 5 components in the DcNP, their respective molecular structures and corresponding molar ratios within the composition are shown. A proposed structure with MDM repeating units-representing electron dense and sparse regions-is generated using ChemDraw3D with a top-down field of view (Panel b). The repeating units of MDM are consistent with the PXRD (x-ray diffraction) analysis and scanning electron micrograph (SEM at 2000× magnification) presented in panel a. The mole (molecular) ratio of each type of compound within the repeating unit of MDM is presented in panel c (i.e., LPV/RTV/TFV/DSPC/DSPE-PEG2000 at 4:1:5:36:4 mol ratio). Carbon, oxygen, nitrogen, hydrogen, phosphate and sulfur are shown in grey, red, blue, white, purple and yellow respectively. (Data not drawn to scale).
In this study, spray drying was used to allow for larger batches of powder to be generated with greater consistency compared to previous methods of controlled solvent removal (rotary evaporation). However, spray drying has been shown to produce heterogeneous distribution of drugs and excipients in aerosolized particles.29 For example, a recent study of naproxen and PVPVA spray dried together was characterized as an ASD by PXRD, but had heterogeneous surface distribution of drug and excipients as visualized by ToF-SIMS.29 In contrast, the DcNP powder in our study revealed distinct diffraction peaks with PXRD (unlike the amorphous halo of ASDs) while ToF-SIMS analysis showed much greater homogeneity of not only one, but all three chemically distinct drugs. This homogeneity may prevent the phase separation of individual constituents in DcNP powder and convey greater physical stability compared to other amorphous products.29 Evidence of this physical stability is observed by the storage of DcNP powder for up to 6 months at room temperature (vacuum desiccation) with no observed change in organizational structure as measured by PXRD. After 6 months of storage, DcNP powder does not exhibit changes in suspension behavior such as aggregation or precipitation. Although our data suggest specific interactions between drugs and lipid polymer regions, current techniques and instruments do not provide enough resolution to assign molecular binding domains within the lipid or PEG chains. To further gain mechanistic insights on MDM formation, additional tools and experimental techniques are required. For example, molecular dynamic (MD) simulations have been used to study the effect of pegylated lipid excipients on bilayer assembly in aqueous environments.30 However, for our composition, simulating molecular interactions between 3 chemically diverse drugs molecules, 1 lipid excipient, and 1 pegylated lipid excipient is computationally expensive and requires significant time allocation. Additionally, there is limited information on the molecular structures of drugs and lipid-polymer conjugates. We are in the process of investing in MD simulations, but progress on these studies is limited and have yet to provide clarity. Nevertheless, with additional advancements in knowledge and tools, MD simulation may be a useful tool to test further characterize of the proposed MDM structure.
A key distinction of MDM structures over conventional crystalline or amorphous drug combinations is the ability to stabilize multiple drugs together. Crystalline protease inhibitors, such as LPV, RTV, and darunavir have low aqueous solubility and are strong candidates for formulation as amorphous drugs to increase solubility. Recently, reports have shown that when amorphous protease inhibitors are dissolved in the same aqueous environment, the dissolution of one drug negatively affects the solubility of the other and vice versa.31,32 This observation is important because protease inhibitors are often included in oral fixed dose combination products and, as these multiple drug products dissolve in the stomach, each constituent needs to be able to reach acceptable luminal concentrations for absorption to occur. In contrast, DcNP powder can associate LPV, RTV, and hydrophilic TFV together and—after suspension in buffered saline—no solubility or precipitation issues are observed with hydrophobic LPV or RTV. After particle size reduction, the resultant injectable dosage does not appear to aggregate in suspension. Thus, it may be used as a single injectable dosage form with both hydrophilic TFV and with hydrophobic LPV/RTV together. After injection, this new MDM powder in suspension produces long acting in vivo (plasma drug concentration time-course) behavior in NHPs as previously reported. The suspended DcNP powder continues to be stable in vivo with all three drugs circulating together in plasma and taken up by cells. These DcNPs in suspension were verified in NHPs to transform short-acting oral drug combinations of LPV, RTV, and TFV (terminal t1/2, 5.5, 3.5 and 17 h) to a targeted, long-acting injectable dosage form (t1/2, app 477, 44 and 65 h) with persistent in vivo stability for up to 4 weeks.33–36 The previous in vivo results and the structural results outlined in this report highlight the potential for this DcNP process to develop into a platform technology that can stabilize hydrophilic and hydrophobic drugs together in powder form. We envision that these novel drug combination powders, enabled through controlled solvent removal, can be suspended and homogenized to carry chemically diverse API together to target cells and tissues in vivo.
The presented controlled solvent removal method has the potential to generate novel drug combination powders based on therapeutic compounds with wide ranging and disparate physicochemical properties. However, there are also limitations to this approach. The techniques used to characterize drug combination powders (such as PXRD and DSC) provide evidence that novel interactions are formed through the controlled solvent removal process. Additional studies, such as FTIR or Raman spectroscopy, would further support and identify the specific nonbonding and molecular domain interactions to provide additional insights. Solid state NMR may also be a tool to describe the interactions between drugs and excipients at the molecular and atomic level. These mechanistic insights would be valuable in predicting other drugs that could be included into the DcNP structure and are in consideration for future studies. However, in our experience, the overall diffraction pattern of drug combination powders in the 2 peak orientation (Fig. 1, panel i) has been sufficient in predicting long acting in vivo circulation after SC administration in NHPs. To support future studies with other antiviral or cancer drug combinations, elucidation of the DcNP design space such as drug to drug ratios, drug to lipid ratios, and lipid to lipid ratios will be pursued. It would also be important to further define the drivers of success in the formation of stable MDM structures. Such studies, while of interest, are beyond the scope of this report and are under consideration for future investigation. Given the complexity of the DcNP system, a design of experiment analysis will be considered.
In summary, we have discovered that a number of 2–3 antiviral drug combinations with disparate physical properties (including LPV, RPV, DTG, RTV, TFV or 3TC) can be stabilized together with lipid excipients in a powder form through a controlled solvent removal process. Through physical, morphological, thermal, and X-ray diffraction analysis, we have found that stable DcNP powder exists as a novel MDM structure that is distinct from the crystal polymorphs of individual constituents and from amorphous powder products. DcNP powder itself displays significant molecular homogeneity as seen in ToF-SIMS, which may be related to the observed MDM structural stability. By using a simple spray drying process to unite chemically diverse drugs together as a stable combination powder, this discovery may enable the development of more effective combination APIs in suspension for treatment of wide-ranging chronic diseases such as HIV, cancer, and diabetes.
Acknowledgments
Financial support: This work was supported in part by grants from the National Institute of Health (NIH, US) UM1-AI120176, R61AI149665 and Unitaid (Switzerland) 2020-39-GLAD. J Yu also received training grant support from NIH T32 GM007750.
Abbreviations:
- LPV
lopinavir
- RTV
ritonavir
- TFV
tenofovir
- PXRD
powder x-ray diffraction
- DSC
differential scanning calorimetry
- ToF-SIMS
time of flight secondary ion mass spectrometry
- SEM
scanning electron microscopy
- DcNP
drug combination nanoparticle
- DSPC
1,2-distearoyl-sn-glycero-3-phosphocholine
- DSPE-PEG2000
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]
- RPV
rilpivirine
- 3TC
lamivudine
- DTG
dolutegravir
Footnotes
Declarations of interest: None.
References
- 1.Maartens G, Celum C, Lewin SR. HIV infection: epidemiology, pathogenesis, treatment, and prevention. Lancet. 2014;384:258–271. [DOI] [PubMed] [Google Scholar]
- 2.Autran B, Carcelain G, Li TS, et al. Positive effects of combined antiretroviral therapy on CD4 T cell homeostasis and function in advanced HIV disease. Science. 1997;277:112–116. [DOI] [PubMed] [Google Scholar]
- 3.Nachega JB, Parienti JJ, Uthman O, et al. Lower pill burden and once-daily antiretroviral treatment regimens for HIV infection: a meta-analysis of randomized controlled trials. Clin Infect Dis. 2014;58:1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maitland D, Jackson A, Osorio J, et al. Switching from twice-daily abacavir and lamivudine to the once-daily fixed-dose combination tablet of abacavir and lamivudine improves patient adherence and satisfaction with therapy. HIV Med. 2008;9:667–672. [DOI] [PubMed] [Google Scholar]
- 5.Desai D, Wang J, Wen H, Li X, Timmins P. Formulation design, challenges, and development considerations for fixed dose combination (FDC) of oral solid dosage forms. Pharm Dev Technol. 2013;18:1265–1276. [DOI] [PubMed] [Google Scholar]
- 6.Kelleher JF, Gilvary GC, Madi AM, et al. A comparative study between hot-melt extrusion and spray-drying for the manufacture of anti-hypertension compatible monolithic fixed-dose combination products. Int J Pharm. 2018;545:183–196. [DOI] [PubMed] [Google Scholar]
- 7.Simoni JM, Tapia K, Lee S, et al. A conjoint analysis of the acceptability of targeted long-acting injectable Antiretroviral therapy among persons living with HIV in the U.S. AIDS Behav. 2019;24:1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams PE, Crauwels HM, Basstanie ED. Formulation and pharmacology of long-acting rilpivirine. Curr Opin HIV AIDS. 2015;10:233–238. [DOI] [PubMed] [Google Scholar]
- 9.Trezza C, Ford SL, Spreen W, Pan R, Piscitelli S. Formulation and pharmacology of long-acting cabotegravir. Curr Opin HIV AIDS. 2015;10:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen ML. Lipid excipients and delivery systems for pharmaceutical development: a regulatory perspective. Adv Drug Deliv Rev. 2008;60:768–777. [DOI] [PubMed] [Google Scholar]
- 11.Muller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm. 2002;242:121–128. [DOI] [PubMed] [Google Scholar]
- 12.Muller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev. 2002;54(Suppl 1):S131–S155. [DOI] [PubMed] [Google Scholar]
- 13.Constantinides PP, Chaubal MV, Shorr R. Advances in lipid nanodispersions for parenteral drug delivery and targeting. Adv Drug Deliv Rev. 2008;60:757–767. [DOI] [PubMed] [Google Scholar]
- 14.Perazzolo S, Shireman L, Koehn J, et al. Three HIV drugs, atazanavir, ritonavir, and tenofovir, coformulated in drug-combination nanoparticles exhibit long-acting and lymphocyte-targeting properties in nonhuman primates. J Pharm Sci. 2018;107:3153–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McConnachie LA, Kinman LM, Koehn J, et al. Long-acting profile of 4 drugs in 1 anti-HIV nanosuspension in nonhuman primates for 5 weeks after a single subcutaneous injection. J Pharm Sci. 2018;107:1787–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freeling JP, Koehn J, Shu C, Sun J, Ho RJ. Long-acting three-drug combination anti-HIV nanoparticles enhance drug exposure in primate plasma and cells within lymph nodes and blood. AIDS. 2014;28:2625–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freeling JP, Koehn J, Shu C, Sun J, Ho RJ. Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res Hum Retroviruses. 2015;31:107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinman LM, Brodie SJ, Tsai CC, et al. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: a proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr. 2003;34:387–397. [DOI] [PubMed] [Google Scholar]
- 19.Duan J, Freeling JP, Koehn J, Shu C, Ho RJ. Evaluation of atazanavir and darunavir interactions with lipids for developing pH-responsive anti-HIV drug combination nanoparticles. J Pharm Sci. 2014;103:2520–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraft JC, McConnachie LA, Koehn J, et al. Long-acting combination anti-HIV drug suspension enhances and sustains higher drug levels in lymph node cells than in blood cells and plasma. AIDS. 2017;31:765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arca HC, Mosquera-Giraldo LI, Dahal D, Taylor LS, Edgar KJ. Multidrug, anti-HIV amorphous solid dispersions: nature and mechanisms of impacts of drugs on each other’s solution concentrations. Mol Pharm. 2017;14:3617–3627. [DOI] [PubMed] [Google Scholar]
- 22.Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug DiscovToday. 2007;12:1068–1075. [DOI] [PubMed] [Google Scholar]
- 23.Sharma P, Garg S. Pure drug and polymer based nanotechnologies for the improved solubility, stability, bioavailability and targeting of anti-HIV drugs. Adv Drug Deliv Rev. 2010;62:491–502. [DOI] [PubMed] [Google Scholar]
- 24.Trasi NS, Bhujbal S, Zhou QT, Taylor LS. Amorphous solid dispersion formation via solvent granulation - a case study with ritonavir and lopinavir. Int J Pharm. 2019;1:100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou HH, Rajesh A, Pandya KM, et al. Impact of method of preparation of amorphous solid dispersions on mechanical properties: comparison of coprecipitation and spray drying. J Pharm Sci. 2019;108:870–879. [DOI] [PubMed] [Google Scholar]
- 26.Gao L, Liu G, Ma J, Wang X, Zhou L, Li X. Drug nanocrystals: in vivo performances. J Control Release. 2012;160:418–430. [DOI] [PubMed] [Google Scholar]
- 27.Sverdlov Arzi R, Sosnik A. Electrohydrodynamic atomization and spray-drying for the production of pure drug nanocrystals and co-crystals. Adv Drug Deliv Rev. 2018;131:79–100. [DOI] [PubMed] [Google Scholar]
- 28.Nandi S, Kaur A, Bansal AK. Dual drug nanocrystals loaded microparticles for fixed dose combination of simvastatin and ezetimibe. Pharm Dev Technol. 2020;25:40–53. [DOI] [PubMed] [Google Scholar]
- 29.Bhujbal SV, Zemlyanov DY, Cavallaro A, Mangal S, Taylor LS, Zhou QT. Qualitative and quantitative characterization of composition heterogeneity on the surface of spray dried amorphous solid dispersion particles by an advanced surface analysis platform with high surface sensitivity and superior spatial resolution. Mol Pharm. 2018;15:2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Pastor RW. Coarse-grained model for PEGylated lipids: effect of PEGylation on the size and shape of self-assembled structures. J Phys Chem B. 2011;115:7830–7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen DN, Van den Mooter G. The fate of ritonavir in the presence of darunavir. Int J Pharm. 2014;475:214–226. [DOI] [PubMed] [Google Scholar]
- 32.Trasi NS, Taylor LS. Thermodynamics of highly supersaturated aqueous solutions of poorly water-soluble drugs-impact of a second drug on the solution phase behavior and implications for combination products. J Pharm Sci. 2015;104:2583–2593. [DOI] [PubMed] [Google Scholar]
- 33.Kraft JC, McConnachie LA, Koehn J, et al. Mechanism-based pharmacokinetic (MBPK) models describe the complex plasma kinetics of three antiretrovirals delivered by a long-acting anti-HIV drug combination nanoparticle formulation. J Control Release. 2018;275:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kearney BP, Flaherty JF, Shah J. Tenofovir disoproxil fumarate: clinical pharmacology and pharmacokinetics. Clin Pharm. 2004;43:595–612. [DOI] [PubMed] [Google Scholar]
- 35.Murphy RL, Brun S, Hicks C, et al. ABT-378/ritonavir plus stavudine and lamivudine for the treatment of antiretroviral-naive adults with HIV-1 infection: 48-week results. AIDS. 2001;15:F1–F9. [DOI] [PubMed] [Google Scholar]
- 36.Perazzolo S, Shireman LM, McConnachie LA, et al. Integration of computational and experimental approaches to elucidate mechanisms of first-pass lymphatic drug sequestration and long-acting pharmacokinetics of the injectable triple-HIV drug combination TLC-ART 101. J Pharm Sci. 2020;109: 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]