

Abstract
The ubiquitous and cancer-associated Epstein-Barr virus (EBV) is associated with nearly all cases of nasopharyngeal carcinoma (NPC). Nasopharyngeal tissue is comprised of both pseudostratified and stratified epithelium, which are modeled in three-dimensional (3-D) cell culture. The cellular origin of EBV-associated NPC is as yet unknown, but both latent and lytic infections are likely important for preneoplastic mechanisms and replenishing the compartmentalized viral reservoir. Conventional 2-D cultures of nasopharyngeal epithelial cells (as primary cells or immortalized cell lines) are difficult to infect with EBV and cannot mimic the tissue-specific biology of the airway epithelium, which can only be captured in 3-D models. We have shown that EBV can infect the pseudostratified epithelium in air-liquid interface (ALI) culture using primary conditionally reprogrammed cells (CRCs) derived from the nasopharynx. In this protocol, we provide a step-by-step guide for the (i) conditional reprogramming of primary nasopharyngeal cells, (ii) differentiation of CRCs into pseudostratified epithelium in ALI culture (known as pseudo-ALI), and (iii) EBV infection of pseudo-ALI cultures. Additionally, we show that nasopharyngeal CRCs can be grown as organotypic rafts and subjected to EBV infection. These nasopharyngeal-derived 3-D cell cultures can be used to study EBV latent and lytic infection in relation to cell type and donor variation, by immunostaining and single-cell RNA-sequencing methods ( Ziegler et al., 2021 ). These methods are useful for studies of EBV molecular pathogenesis, and can overcome many of the limitations associated with conventional 2-D cell cultures.
Graphic abstract:
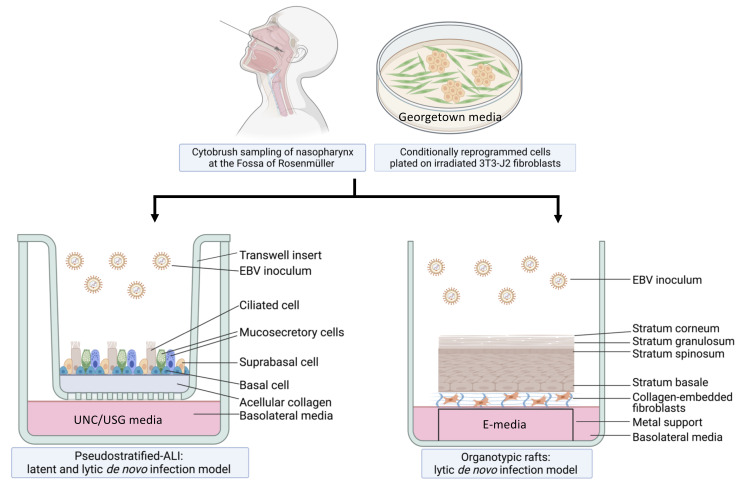
Workflow of nasopharyngeal-derived conditionally reprogrammed cells grown into pseudostratified-ALI and organotypic rafts in 3-D cell culture. Created with Biorender.com.
Keywords: 3-D model, Pseudostratified, Air-liquid interface (ALI), Organotypic rafts, Epstein-Barr virus (EBV), Nasopharynx, Nasopharyngeal epithelium
Background
EBV is a gammaherpes virus with both latent and lytic infection programs ( Young et al., 2016 ). Latent infection is characteristic of nasopharyngeal carcinoma (NPC), but lytic infection and the production of progeny virus is important for replenishing the local viral reservoir ( Tsao et al., 2012 ; Raab- Traub et al., 2015 ; Shair et al., 2018 ). More than 90% of people acquire EBV by adulthood and these chronic carriers continue to shed and transmit EBV in the nasal and oral cavities (Sitki- Green et al., 2003 ; Hadinoto et al., 2009 ). However, only a subset of individuals develops EBV-associated NPC, which occurs at high incidence in specific populations, such as South East Asians and Alaskan Innuits (Yu and Yuan, 2002). Both latent and lytic infection are thought to be important in the development of NPC, but this process has been difficult to study because of the rarity of EBV-infected nasopharyngeal cells in asymptomatic carriers, and the lack of physiologically relevant in vitro infection models ( Caves et al., 2018 ; Shair, 2020). To address the question of which epithelial cell types in the nasopharynx are susceptible to EBV infection, and which cell types result in latent vs. lytic infection, we have built two distinct experimental models of the nasopharynx. The EBV infection program depends on epithelial differentiation, which is more accurately modeled in three-dimensional (3-D) culture. Thus, 3-D cell culture models overcome the shortcomings of previous EBV infection studies conducted in 2-D culture, which are limited to studies of EBV latency in outgrowing cell lines.
For the pseudostratified air-liquid interface model (pseudo-ALI model), a pseudostratified epithelium derived from nasopharyngeal conditionally reprogrammed cells (CRC) is exposed to de novo EBV infection by co-culture with anti-human IgG-reactivated EBV-positive B-cell inoculum ( Maruo et al., 2001 ), or by cell-free virus infection. In the presence of Rho-associated protein kinase inhibitors (ROCKi), primary epithelial cells grown with feeder irradiated 3T3-J2 mouse fibroblasts are induced into a highly proliferative state by conditional reprogramming, which allows for propagation of primary cells while maintaining their differentiation potential ( Chapman et al., 2010 ; Liu et al., 2012 ; Suprynowicz et al., 2012 ). Subsequently, UNC/USG (University of North Carolina/Ultroser G) specialized media is used to differentiate CRCs into mature pseudostratified airway epithelia in air-liquid interface cultures. Originally developed at the University of North Carolina (Fulcher and Randell, 2013), the UNC component has additives that induce the differentiation of human bronchial epithelial (HBE) and nasopharyngeal epithelial cells, which do not grow well in serum-containing media; thus, it’s necessary to use bovine pituitary extract (BPE) as a serum substitute. Other key constituents of the UNC media include a multitude of growth factors and trace elements that are essential for airway epithelial differentiation (Fulcher and Randell, 2013). These include retinoic acid, which promotes mucocilliary differentiation, and hydrocortisone, which promotes growth and epithelial sodium channel (ENaC) activity. The USG component is needed to promote robust electrophysiology ( Gentzsch et al., 2017 ). The protocol we describe here can be used to differentiate both nasopharyngeal and bronchial epithelial cells (Fulcher and Randell, 2013; Serrano Castillo et al., 2019 ), but the EBV infection experiments we describe have only been attempted with nasopharyngeal cells.
Akin to EBV infection in organotypic rafts generated from primary oral keratinocytes ( Temple et al., 2014 , 2017), we generated nasopharyngeal-derived organotypic rafts to study EBV infection in the nasopharyngeal stratified epithelium, using CRCs which were exposed to reactivated EBV by the co-culture method. The principle of organotypic rafts is to generate multilayered stratified epithelia by differentiating keratinocytes at the air-liquid interface ( Kopan et al., 1987 ) in defined E-media, which was originally described for the differentiation of oral ( Temple et al., 2017 ) and cervical epithelial cells (Meyers, 1996). Here, we generated 3-D cell cultures using nasopharyngeal cells from donors undergoing skull base surgery (without any upper airway pathology), and from patients undergoing sinus surgery. CRCs can also be derived from the inferior turbinate from healthy persons without any co-pathology, provided such donors with no reason for surgery can be recruited. In sufficient quantity, the CRCs can be cryopreserved and thawed to generate both types of 3-D cell culture. A major advantage of using cryobanked cells is reproducibility and assessment of donor variability. These 3-D culture methods, when combined with EBV infection, are expected to reveal susceptible cell types in the upper airway, and uncover host determinants of EBV (latent and lytic) infection. A schematic of the workflow and measurable experimental outcomes are described in the Diagram below.
Part I: Differentiation into Pseudostratified-ALI Culture
Materials and Reagents
-
3T3-J2 Feeder Cells
Low passage 3T3-J2 cells (Kerafast, catalog number: EF3003)
-
3T3-J2 Growth Media
DMEM (Corning, catalog number: MT10013CV)
Bovine calf serum, iron-supplemented (GE Healthcare, catalog number: SH3007202)
0.05% Trypsin-EDTA (Corning, catalog number: MT25052CI)
DMSO (Fisher, catalog number: BP231-100)
100 mm tissue culture plates (Greiner, catalog number: 664160)
150 mm tissue culture plates (Greiner, catalog number: 639160)
-
Primary Nasopharyngeal Cells
Georgetown Media (see Recipes)
DMEM high glucose (Gibco, catalog number: 11965092)
Ham’s F-12 nutrient mix (Gibco, catalog number: 11765054)
100× Glutamine (Gibco, catalog number: 25030-081)
Fetal bovine serum (FBS) (Gibco, catalog number: 16140071)
Penicillin-streptomycin (Gibco, catalog number: 15140122)
Hydrocortisone (Sigma, catalog number: H0888)
EGF (Invitrogen, catalog number: PHG0311)
Insulin (Sigma, catalog number: I5500)
Amphotericin B, 250 µg/mL (Fisher, catalog number: BP264550)
Gentamicin, 10 mg/mL (Gibco, catalog number: 15710064)
Cholera toxin (Sigma, catalog number: C8052)
-
Y-27632 (Tocris, catalog number: 1254)
UNC/USG Media (see Recipes)
DMEM/F12 (Gibco, catalog number: 11320-033)
Penicillin-streptomycin (Gibco, catalog number: 15140122)
FeSO4·7H2O (Sigma, catalog number: F8633-250G)
MgCl2·6H2O (Sigma, catalog number: M2393-100G)
CaCl2·2H2O (Sigma, catalog number: C7902-500G)
ZnSO4·7H2O (Sigma, catalog number: Z0251-100G)
Selenium from Sodium selenite (Sigma, catalog number: S5261-100G)
Manganese from Manganese Chloride tetrahydrate (Sigma, catalog number: M8054-100G)
Silicon from Sodium Silicate nonahydrate (Sigma, catalog number: S4392-250G)
Molybdemun from Ammonium Molybdate tetrahydrate (Sigma, catalog number: M1019-100G)
Vanadium from Ammonium Metavanadate (Sigma, catalog number: 398128-50G)
Tin from Tin Chloride dihydrate (Sigma, catalog number: 243523-5G)
Transferrin (Sigma, catalog number: T8158-1G)
Hydrocortisone (Sigma, catalog number: H0888)
Insulin (Sigma, catalog number: I9278-5ML)
Epinephrine (Sigma, catalog number: E4250-10G)
T3 (Sigma, catalog number: T6397-100MG)
EGF (Corning, catalog number: 354052)
BSA (Sigma, catalog number: A9647-100G)
Retinoic Acid (Sigma, catalog number: R2625-50MG)
BPE (Pel-Freez, catalog number: 57136-3)
Phosphorylethanolamine (Sigma, catalog number: P-0503)
Ethanolamine (Sigma, catalog number: E9508-500ML)
Ultroser G (Pall, catalog number: 15950-017)
Amphotericin B, 250 µg/mL (Fisher, catalog number: BP264550)
0.25% Trypsin (HyClone, catalog number: SH3004201)
0.05% Trypsin/EDTA (Corning, catalog number: MT25052CI)
Hanks’ Balanced Salt Solution (HBSS) 1× with calcium and magnesium (Gibco, catalog number: 14025076)
Dulbecco’s Phosphate Buffered Saline (PBS) 1× without calcium and magnesium (Lonza, catalog number: 17512F)
Collagen IV (Sigma, catalog number: C7521)
Glacial Acetic Acid (Fisher, catalog number: BP2401-212)
Transwell Inserts, 24 well plate, polyester membrane, 0.4 µm pore size (Corning, catalog number: 3470)
Sterile Cytology Brush (Medical Packaging Corp, catalog number: CYB-1)
Equipment
Gamma-irradiator. We used the Gammacell 1000D model (Atomic Energy of Canada Ltd)
Humidified incubator at 37°C and 5% CO2, for cell culture
Biosafety cabinet Class II Type A2, for sterile cell culture work and biocontainment
Procedure
-
Preparing Irradiated 3T3-J2 Feeder Cells
-
Defrost 1 vial of 3T3-J2 cells (frozen at ~1 × 106 cells per vial) into a 100 mm dish in 3T3-J2 growth media. Do not centrifuge the cells prior to plating, as this will greatly decrease the recovery of viable cells. Change growth media the next day.
TIP: Do not heat inactivate bovine calf serum. It is best to use low-passage cells (≤ 20 passages).
Expand 3T3-J2 cells by seeding at a density of ~3.3 × 103 cells/cm2. Do not allow 3T3-J2 cells to grow beyond 80% confluence, or they will begin to senesce and will no longer expand. Cells should reach no more than ~5 × 106–6 × 106 cells per 150 cm2 of culture.
Passage cells by trypsinizing and seeding at a density of 1:5–1:4.
Cells can be irradiated at each passage. For irradiation, prepare ~ 2.5 × 107–3 × 107 cells in 20–25 mL of 3T3-J2 growth media, and irradiate with 30 Gy.
Plate approximately 1 × 106 cells in a 100 mm dish and monitor for growth, to ensure successful irradiation. Successfully irradiated cells will not expand and will lift off within 5–6 days.
Freeze stocks of irradiated cells at 1.5 × 106/mL, in 3T3-J2 growth media plus 10% DMSO.
-
-
Conditional Reprogramming of Primary Nasal Epithelial Cells
Coat 100 mm tissue culture plates with collagen IV stock solution diluted 1:10 in 0.1 M Na2CO3 at 37°C for 2 h, or at 4°C overnight. Prepare 2× 100 mm plates per donor.
-
The day prior to receiving donor cells, seed 5 × 105 irradiated 3T3-J2 cells from frozen stock in DMEM plus 10% FBS to each 100 mm dish (if using freshly irradiated 3T3-J2 cells, seed 2 × 105 per 100 mm or 5 × 105 per 150 mm dish); leave overnight. The irradiated fibroblasts can be seeded as soon as 3 h before seeding donor cells, and will still attach. To maximize recovery of viable cells, plate directly into growth media without centrifuging cells, and change media the next day.
Tip: Aim for 1/3rd confluence, but no more than 50%. If the fibroblasts are too light, you can top up with more irradiated fibroblasts, as long as you add them 3 h before seeding the donor cells.
-
Before beginning the procedure, appropriate institutional approval for research with human subjects and consent forms must be in place. Collect nasopharyngeal cells by scraping the nasopharynx with a cytobrush—ten times to the left and ten times to the right. We scrape cells from the Fossa of Rosenmüller, where NPC most often arises. Transport cells on cytobrushes to the lab immediately, in a tube with 10 mL of PBS on ice (DO NOT FREEZE). At the lab, vortex the cytobrush briefly (15–20 s), and scrape it on the edge of the tube to dislodge cells. Remove the cytobrush with autoclaved sterile forceps and discard it.
Note: Cells from the nasopharynx are collected from patients under general anesthesia undergoing head and neck surgery. We have collected cells from patients undergoing sinus surgery or skull base surgery. Inclusion and exclusion criteria should be in place to exclude any donors that have a history of head and neck cancer. Potential donors should be pre-screened for COVID-19 before surgery. Cells can also be collected from the inferior turbinate of healthy donors not undergoing surgery. If inferior turbinate cells are collected from donors without anesthesia, the yield is usually sufficient to seed two plates of 24 well-sized transwells, but not enough for bulk cryopreservation. We have not attempted to collect nasopharyngeal cells without general anesthesia, but it is likely to yield low quantities of cells insufficient for cryobanking.
Centrifuge the dislodged cells at 500 × g and room temperature for 10 min.
While cells are centrifuging, aspirate DMEM media from plates with irradiated fibroblasts. Rinse with PBS. Add 5.5 mL of Georgetown media per 100 mm dish.
Aspire supernatant from centrifuged cells. Do not pour off the supernatant. If there is a large upper white/pink layer (consisting of the lymphocytes and epithelial cells) in the pellet, it may dislodge from the lower portion of the pellet (which are the red blood cells) very easily. It is not necessary to lyse the red blood cells.
Resuspend cells in fresh Georgetown media prewarmed to 37°C. Volume should be 2.5 mL per 100 mm dish. Seed the entire pellet. Lymphocytes and red blood cells will die and wash off over the next several days. Label the plate passage 0.
Each day, aspirate media. Add 5 mL (100 mm dish) of 1× PBS and gently rock the plate to wash off lymphocytes and blood cells. Aspirate PBS, removing as many lymphocytes and blood cells as possible, and add 8 mL of Georgetown media. It is important to refresh media every day to prevent contamination.
Monitor cell growth and proceed to passaging when the cells appear confluent. The fibroblasts will lift off approximately 5–6 days after seeding, so it is important to have passaged the cells by day 6. Ideally, you should see scattered colonies of epithelial cells growing out on 30% or more of the plate by this point (Figure 1). The outgrowing islands of primary cells are known as conditionally reprogrammed cells (CRCs).
-
Passaging Primary CRCs
One day before passaging, collagen coat 1× 100 mm dish and 1× 150 mm dish per donor, and seed irradiated 3T3-J2 fibroblasts as before (seed 1.5 × 106 cells/150 mm dish).
Aspirate media from plates.
Gently rinse with PBS.
Dilute stock of 0.25% trypsin to 0.1% in PBS.
Add 2.5 mL of 0.1% trypsin per plate and place in the 37°C incubator for 1–2 min, to trypsinize fibroblasts from plate. Observe the cells under a microscope at least every minute—the fibroblasts should dissociate from the plate easily; waiting too long will cause the primary epithelial cells to detach. If the fibroblasts are not detached after 2 min, gently tap on the sides of the plate. Do not leave trypsin on for more than 3 min.
Remove trypsin and fibroblasts by aspirating and discarding. Add 2.5 mL of 0.05% trypsin to each dish, and incubate at 37°C until all cells detach.
Centrifuge at 500 × g for 10 min, to pellet the cells. While pelleting cells, aspirate media from plates with new irradiated 3T3-J2 feeder cells, rinse once with PBS, and add Georgetown media (5.5 mL per 100 mm dish, and 12.5 mL per 150 mm dish).
Resuspend the cell pellet in 10 mL of Georgetown media. Seed 2.5 mL to the 100 mm dish and 7.5 mL to the 150 mm dish (split ratio of 1:4). It is not necessary to count cell density because the main purpose is to recover all cells.
Next day, replace media. At passage 1 and beyond, replace media every 2–3 days.
Monitor cell growth, and split to transwells or freeze when cells are confluent, or when fibroblasts start to lift off (5–6 days). Cryopreserve at 1.5 million cells/mL in Georgetown media plus 10% DMSO. The number of vials obtained varies depending on the donor, and ranges from 2 to 10 vials. It is best to freeze at passage 1, but we have successfully differentiated cells that were frozen at passage 2.
-
Lifting and Differentiating CRCs into Pseudostratified-ALI Cultures
Differentiation into pseudo-ALI cultures can be generated from freshly expanded or cryopreserved CRCs.
Collagen coat Corning 3470 transwells as described above, using 100 µL of collagen IV solution per insert. Rinse with PBS before seeding cells.
When cells are ready to passage, repeat harvesting process as before. Use 5 mL of trypsin for 150 mm dishes. Combine cells from both plates (150 mm and 100 mm) in one tube.
Resuspend cells in 10 mL of Georgetown media and count using a hemocytometer.
Add 400 µL of Georgetown media to the basolateral side of each transwell.
Seed 2 × 105 cells in 200 µL of Georgetown media to the apical surface of each transwell.
Any remaining cells can be frozen at 1.5 × 106 cells/mL in Georgetown media plus 10% DMSO.
Once a confluent monolayer has formed (this should take 1–2 days), aspirate the media in the apical and basolateral sides.
Rinse wells once with 200 µL of HBSS in the apical and 400 µL of HBSS in the basolateral sides, and aspirate. HBSS supplied with calcium and magnesium helps to prevent cell loss.
Add 400 µL of UNC/USG media to the basolateral side.
-
Continue monitoring cultures, and change basolateral media three times a week for 4 weeks (28 days), to allow cells to differentiate. Rotating cilia are visible beginning at 3 weeks ( Video 1 ).
Tip: Debris and mucus may build up on the apical surface. If this happens, gently rinse the culture with HBSS, taking care not to damage the culture.
-
At 28 days, mature pseudo-ALI cultures can be fixed in 4% paraformaldehyde (PFA) and subjected to whole-mount immunofluorescence staining or formalin-fixed paraffin-embedded (FFPE) histology stains ( Figure 2 , Ziegler et al., 2021 ), to control for differentiation into ciliated, basal, and mucosecretory cells.
Note: Differentiated pseudo-ALI cultures are mature by 28 days post-lifting. Although UNC/USG media can maintain bronchial-derived pseudo-ALI cultures for several months, we have found that nasopharyngeal-derived pseudo-ALI cultures will begin leaking one week after reaching maturation. This can be monitored by watching for liquid media leaking into the apical side of the pseudo-ALI culture. It is common to observe thick mucus on the apical side, but this is visibly distinct from leaking liquid media. Therefore, it is best to conduct experiments as soon as the nasopharyngeal-derived pseudo-ALIs are mature. It is possible that some commercial airway differentiation media (e.g., Stemcell Technologies PneumaCult-Ex) can extend the lifespan of the mature pseudo-ALI cultures, but we have not tested different types of commercial media.
Figure 1. Images of growing CRCs and CRCs seeded into pseudo-ALI culture.

Left, image of nasopharyngeal CRCs grown on a collagen-coated tissue culture plate co-cultured with irradiated 3T3-J2 feeder fibroblasts (captured with a phase-contrast microscope). Right, images of pseudo-ALI cultures with CRCs seeded into the apical side of a 24 well plate-sized transwell.
Video 1. Cilia beating in pseudo-ALI culture.
Video was captured on an inverted phase-contrast microscope at day 15 post-lifting. The primary nasopharyngeal cells were harvested from a donor at the time of skull base surgery.
Figure 2. Representative histology stains and fluorescent imaging of pseudo-ALI cultures.

A. Histology stains of a representative nasopharyngeal pseudo-ALI culture. Shown is an uninfected pseudo-ALI culture grown from a donor collected at the time of skull base surgery. H&E, hematoxylin and eosin stain shows a pseudostratified epithelium with columnar ciliated cells. Ki67, nuclear protein Ki67 is a proliferation marker that marks cycling cells located in the basal layer. AB/PAS, Alcian blue/periodic acid-Schiff stains acidic and neutral mucins, marking different types of mucosecretory cells. B. Fluorescent Z-stack images of EBV-infected cells in pseudo-ALI culture showing GFP-positivity (green), counterstained with DAPI (blue). The pseudo-ALI culture was infected with an epithelial-tropic recombinant EBV strain (M81, Tsai et al., 2013 ) that co-expresses GFP, fixed in 4% paraformaldehyde at 4 days post-infection (p.i.), and imaged by confocal microscopy. 3-D renderings were assembled in Nikon NIS Elements. EGF treatment (10 ng/mL) enhances cell-free virus infection ( Wang et al., 2015 ). If live cell imaging is preferred, the images would have to be captured directly in the transwell (without mounting of the excised membrane), and a suitable objective with an extended focal length would have to be determined by the end user. Note that the B-cell co-culture infection method would be clouded by GFP-positivity from input B-cells, and is, therefore, not recommended as a method for assessing infection.
Part II: EBV Infection of Pseudostratified-ALI Cultures
Materials and Reagents
-
EBV infection
Producer B-cell line. We use Akata B-cells infected with recombinant EBV, herein referred to “rAkata” ( Maruo et al., 2001 ), gift of Dr. George Tsao (Hong Kong University) with permission from Dr. Kenzo Takada (Nihon University School of Medicine, Japan).
RPMI (HyClone, catalog number: SH30027FS) plus 10% fetal bovine serum (Seradigm, VWR, catalog number: 97068-085)
α-human IgG goat antiserum at 10 mg/mL (Fisher, catalog number: ICN55087) for preparation of B-cell derived virus
Human EGF, recombinant (Sigma, catalog number: E9644), for cell-free virus infection
T-25 tissue culture flask (Greiner, catalog number: 690175)
T-75 tissue culture flask (Greiner, catalog number: 658175)
-
Histology
Paraformaldehyde (Sigma, catalog number: P6148)
Fine tipped forceps (Electron Microscopy Sciences, catalog number: 78149-SS)
#11 scalpel (Aspen Surgical, Thermo Scientific, catalog number: 02-688-79)
-
Quantitative PCR
Turbo DNase 2 units/µL (Fisher, catalog number: AM2238)
Proteinase K (Thermo, catalog number: EO0491)
Plasmid containing segment of EBV genome and primers. We use a pUC57 plasmid with a BALF5 fragment (Akata strain, NCBI Accession, catalog number: KC207813.1)
Primers (Eurofins MWG Operon): qBALF5-F (5′ GAGCGATCTTGGCAATCTCT 3′), qBALF5-R (5′ TGGTCATGGATCTGCTAAACC 3′)
PowerUp SYBR-green 2× master mix (Thermo, catalog number: A25780), or preferred qPCR reagent
Equipment
QuantStudio 3 Real-Time PCR System (Applied Biosystems), or preferred real-time PCR instrument
Procedure
-
Preparing/Harvesting Virus Stocks from EBV-Infected Cells
-
For B-cell co-culture infection:
-
Expand EBV-infected producer B-cells. Use 2.5 × 106 cells for each well to be infected. Split B-cell stock cultures when cells are at an approximate density of 1 × 106–1.5 × 106/mL; do not allow them to exceed this density, otherwise they will begin to reactivate and die.
Tip: The recombinant EBV in rAkata cells has a neomycin resistance gene and an EGFP expression cassette inserted in the non-essential BXLF1 (viral thymidine kinase) locus. Keep cells with neomycin selection (0.8 mg/mL) to maintain recombinant EBV during serial passage.
Note: All work with EBV-infected cell cultures should be performed under BSL-2 conditions.
Seed T-75 flasks with 1 × 106 cells/mL.
Add 5 µL (50 µg) of α-human IgG antibody per milliliter of cells, to reactivate EBV.
Incubate at 37°C for 2 days.
-
-
For cell-free virus infection:
Reactivate virus from producer cell lines (e.g., 293-M81 EBV, Tsai et al., 2013 ), by transfecting cells with plasmids expressing EBV Zebra (aka Zta) and gp110 (homolog of herpes simplex virus gB) proteins. Harvest supernatant 5–6 days after transfection for optimal virus yield. Spin cells down at 500 × g and collect supernatant as inoculum.
Alternatively, reactivate virus from EBV-infected B-cells as above, but incubate cells for 5–6 days after adding α-human IgG. Spin cells down at 500 × g and collect supernatant as inoculum.
-
-
Infection of Pseudostratified-ALI Cultures by the B-cell Co-culture Method
The B-cell co-culture method uses reactivated B-cells as the inoculum because cell-to-cell contact encourages infection in epithelial cells ( Imai et al., 1998 ). Cell free virus infection of epithelial cells is possible, but much less efficient ( Imai et al., 1998 ).
Spin cultures of reactivated B-cells down (from step A.1.d) after 48 h of reactivation (500 × g for 5 min).
-
Resuspend B-cells at 1.25 × 107/mL in PBS. Each well is co-cultured with 2.5 × 106 B-cells.
Note: A reactivation control can be included by preparing a parallel reactivation B-cell culture similarly resuspended in PBS (at 1.25 × 107 cells/mL) and grown in a 24-well plate for an additional 2 days, to match the timepoint for removal of inoculum. Perform qPCR on clarified supernatant as described in Part II Procedure F .
Aspirate basolateral media from ALI cultures.
-
Add 200 µL of PBS to the apical side and 400 µL of PBS to the basolateral side of each well. Incubate at 37°C for 5 min.
Tip: Do NOT exceed 5 min, or the cell junctions will be too disrupted and cells will fall off.
-
Aspirate PBS. Rinse basolateral and apical sides twice with PBS, aspirating carefully to remove. For every wash, use 200 µL of PBS in the apical and 400 µL of PBS in the basolateral sides.
Add 400 µL of PBS to the basolateral side, and add 200 µL of the reactivated B cell suspension (in PBS) to the apical surface of each culture. The multiplicity of infection (MOI) is irrelevant because, in the B-cell co-culture infection protocol, reactivated B-cells are continually producing virus. However, for the cell-free virus infection protocol, MOI can be calculated at the time of inoculation, as determined by qPCR for EBV genome equivalents (refer to Part II.F).
Note: We have only attempted apical infection, but basolateral infection of the pseudo-ALI culture may be possible.
Tips:
If a mock infection is desired, use EBV-negative Akata cells similarly treated with anti-human IgG.
-
As input control for qPCR use either:
Fixed EBV-positive B-cells. Fix reactivated B-cells with 4% PFA at room temperature for 10 min, wash twice with PBS by spinning cells at 500 × g for 5 min, and resuspend in 200 µL of PBS for each well.
Or, apply live reactivated EBV-positive B-cell inoculum to a fixed ALI culture. Fix an ALI culture with 4% PFA at room temperature for 10 min, wash twice with 200 µL of PBS on the apical side and 400 µL of PBS on the basolateral side, then proceed with inoculation with live reactivated B-cell inoculum, as described above in step 6.
Incubate at 37°C for 2 h. Then, remove PBS from the basolateral portion of the culture and replace with 400 µL of UNC/USG media.
Return cultures to the 37°C incubator for 2 days.
Remove B-cells and basolateral media from ALI cultures by aspirating gently.
Rinse wells (apical and basolateral sides) gently three times with 1× HBSS at room temperature. Aspirate to remove the HBSS very carefully. The cultures will be somewhat fragile at this point, so be careful not to touch the membrane with the tip. The HBSS should be added to a different side of the transwell with each wash, to ensure minimal disruption of the cell culture.
-
Replace basolateral media with 400 µL of fresh UNC/USG media and incubate at 37°C until fixation/harvest. Replenish with fresh UNC/USG media every 2 days.
Tip: Harvest times are usually between 2–5 days post-infection, beginning with the day of B cell co-culture. As infection proceeds, transwells may begin to leak into the apical side. Do not remove any leaked media until the harvest time point.
At this point, you can visually monitor infection each day on an inverted fluorescent microscope, if using a recombinant virus expressing GFP.
-
Infection of Pseudostratified-ALI Cultures by the Cell-Free Virus Infection Method
Treat ALI cultures with 10 ng/mL EGF diluted in UNC/USG media for 24 h prior to infection, 200 µL of media in the apical and 400 µL of medial in the basolateral sides. The addition of EGF encourages the expression of the EBV epithelial cell receptor Ephrin receptor A2 ( Zhang et al., 2018 ).
Remove media with EGF from cultures by aspiration.
Rinse once with PBS and aspirate.
Add 1 mM EDTA in PBS (a volume of 200 µL to the apical and 400 µL to the basolateral sides), to weaken cell-to-cell junctions. This facilitates the diffusion of cell-free EBV inoculum throughout the culture. Return cells to the incubator at 37°C for 5 min.
Wash off EDTA with three rinses of PBS.
Add 200 µL of virus stock to the apical surface and 400 µL of UNC/USG media to the basolateral side of each well. A target MOI of 100–1000 EBV genome copies (as determined by qPCR) is applied.
Incubate at 37°C for 24 h.
Remove virus and basolateral media by aspiration.
Rinse cultures three times with HBSS.
Replace basolateral media and incubate at 37°C until fixation/harvest.
-
Fixation of Pseudostratified-ALI Cultures for Whole Mount Immunofluorescence Staining or Histology Sectioning (FFPE)
It is important to note that some infected cells do not have detectable GFP fluorescence. Depending on the recombinant EBV molecular clone used, the GFP introduced is expressed from various exogenous promoters, such as pCMV and pSV40 ( Maruo et al., 2001 ; Tsai et al., 2013 ). Thus, while monitoring GFP fluorescence can be done ( Figure 2B ), it is not the preferred method to assess infectivity. Whole mount immunofluorescence staining and immunostaining of FFPE sections is the preferred method.
Before starting, transfer wells to be fixed to a new 24 well tissue culture plate.
Rinse cultures three times with HBSS.
-
(Optional) For whole-mount staining, if WGA-488 stain is desired (to show cilia and cell membrane contour), after the first wash incubate with a WGA-488 Antibody diluted to 1 µg/mL in HBSS at room temperature in the dark for 10 min.
Note: If PBS is used after adding WGA-488 (before PFA fixation), the WGA will unbind and there will be no staining. Wash three times with HBSS before fixation.
Tip: For FFPE sections, WGA staining is not preserved due to delipidation of the membranes by solvents used in the deparaffinization process.
-
For wells that will be used for whole mount staining:
Add 4% PFA (a volume of 200 µL to the apical and 400 µL to the basolateral sides). Fix at 37°C for 10 min.
Remove PFA (by tipping not aspirating) and transfer wells to a new Corning 24 well transwell culture plate.
Wash cells three times with PBS. Tip, but do not aspirate liquid from the apical surface.
-
Store fixed cells in 70% ethanol, 0.1% PFA, or 0.02% sodium azide in PBS at 4°C. This step depends on the intended use for the culture. Wells that will be used for RNA-in situ hybridization should be stored in 70% ethanol for up to 1 week. Wells that will be used for whole mount immunofluorescent stains should be stored in 0.1% PFA or 0.02% sodium azide in PBS.
Note: Cryosectioning preserves the fluorescence of fluorescent proteins (e.g., GFP) and does not often require further optimization of the immunofluorescent staining procedure. However, we find that, unlike FFPE sectioning which is reliable, the success of cryosectioning on pseudo-ALI cultures is variable. There are times when the pseudostratified cells are torn apart by cryosectioning and no remaining cells are visible. Therefore, we do not recommend cryosectioning.
Proceed with immunostaining. Examples of whole-mount immunostains are presented in Figure 3 .
-
For FFPE processing and sectioning:
Add 4% PFA (a volume of 200 µL to the apical and 400 µL to the basolateral sides).Fix overnight at 4°C.
Store fixed cells in 0.1% PFA or 0.02% sodium azide in PBS at 4°C, until proceeding with FFPE processing (Figure 4) .
Proceed with staining for EBV or cell type-specific marker ( Ziegler et al., 2021 ).
Note: 3-D cell cultures can also be analyzed by single cell RNA-sequencing for host and viral transcripts (Ziegler et al., 2021). Single cells can be dissociated from the transwells by treatment with 0.05% trypsin-EDTA. We have found that single cell RNA-sequencing is more sensitive than immunostaining in identifying infected cells. Beware that the presence of a few EBV transcripts does not confidently assign a latent or lytic infection. Instead, a global transcriptome analysis is needed to distinguish latent and lytic infection from abortive infection. We showed that latent and lytic transcriptional signatures can be identified in EBV-infected pseudo-ALI cultures, using 10× Genomics single cell RNA-sequencing. The success of single cell RNA-sequencing using 10× Genomics depends on the abundance of transcript per cell (EBV transcripts can be as low as 0.01–0.1% of the total transcriptome). It also depends on accurate exon annotation of the reference genome. Many EBV (and herpesvirus) transcripts overlap. Thus, the reads aligning to these overlapping regions are indistinguishable, and will only be mapped if these overlapping transcripts are annotated as “fused” exons. The merged EBV+hg38 (human) genome annotation file is available in NCBI Gene Expression Omnibus (GEO) GSE157243. Filtering criteria and data processing steps are provided in the GEO submission. The Akata EBV genome (NCBI KC207813.1) with updated annotations is available in Github ( https://github.com/TangLabGOT/Reference-Genomes ).
-
Collecting Viral DNA from Pseudostratified-ALI Cultures
-
At the time of removing the inoculum, collect a background wash. This will inform you on how many EBV genomes were not successfully washed off of your cultures. Note that this MUST be done for EVERY WELL that will be used for a harvest, as it is essential to show an increase in viral titer.
Aspirate virus from apical surface.
Wash three times with HBSS, carefully aspirating liquid after each wash.
After the third wash, pipette 100 µL of HBSS onto the apical surface of the culture. Pipette up and down gently several times to collect any remaining viral genomes, and store the collected HBSS at 4°C, until ready to harvest extracellular and cell-associated virus.
Replace basolateral media and incubate until desired timepoint to harvest genomes. Leave the apical side dry.
At the desired timepoint, pipette 50 µL of HBSS onto the apical surface of the culture.
Using a 200 µL-sized pipette tip, scrape back and forth across the membrane to dislodge cells. Pipette up and down continuously to detach cells from the membrane.
When the cells are in suspension in the HBSS, transfer to a microcentrifuge tube for storage.
Add another 50 µL of HBSS to the apical surface and repeat steps 4–5, being careful to collect all remaining cells in this second wash. The final volume of the cell suspension should be 100 µL.
Pellet the cells by centrifuging at 500 × g for 5 min.
Transfer the supernatant to a new tube marked as apical virus and store at 4°C—this supernatant contains any unbound, extracellular virus produced by the culture. DO NOT DISCARD!
Prepare an ethanol-dry ice slurry in a covered ice bucket, by filling with dry ice and spraying with ethanol, and ensure the water bath is set to 37°C.
Resuspend the cell pellet by adding 100 µL of fresh HBSS and vortexing.
Place tubes containing the cell suspension in dry ice-ethanol slurry, and cover the ice bucket for 3 min, to freeze.
Transfer tubes to water bath for 3 min, to thaw.
Repeat steps 11–12 twice, for a total of three freeze-thaw cycles.
Spin tubes down at 1,000 × g for 5 min, to pellet cell debris.
-
Transfer supernatant, which now contains viral genomes released by lysing cells, to a new tube marked as cell-associated virus, and store at 4°C for up to 1 week or -20°C for longer, until quantification.
Tip: Under sterile conditions the collected GFP+ virus can be used to titer infectious units by the Green Raji Unit (GRU) assay (Caves et al., 2018). In this case, the harvested virus should be stored at 4°C for up to 1 week.
-
-
Quantifying EBV DNA by qPCR using SYBR-green Detection
-
Add 2 units of DNase I directly to 50 µL of viral supernatant and incubate at 37°C for 30 min.
Tip: As little as 25 µL of viral supernatant can be used, if a repeat is desired.
Heat inactivate at 70°C for 10 min.
Add Proteinase K (stock: 20 mg/mL) to a final concentration of 0.1 mg/mL at 50°C for 30 min.
Heat inactivate at 75°C for 20 min.
Dilute samples 1:10 in dH2O before performing qPCR, if desired.
-
Create standards by preparing 10-fold serial dilutions of BALF5 plasmid (109 → 100 copies per reaction). Use a DNA Copy Number and Dilution Calculator. An example is provided below:
Base pair mass: 650 (g/mol)/bp
Fragment (plasmid) length: 2786 bp
Molar mass (specific to plasmid size): 1810900 g/mol
332541830 copies/ng
-
Prepare master mix for 2 µL of template DNA (either the known BALF5 plasmid or the DNase-treated supernatant), in a total volume of 10 µL per reaction, according to the amounts in Table 1.
Table 1. qPCR reaction composition.
1× Reaction Volume (µL) Primer 1: qBALF5-F (10 µM), [final] = 0.3 µM 0.3 Primer 2: qBALF5-R (10 µM), [final] = 0.3 µM 0.3 PowerUp SYBR Green (2×)* 5 Template DNA (undiluted or 1:10 dilution) 2 dH2O 2.4 *If desired, substitute with preferred brand of qPCR reagent.
-
Perform absolute quantification by qPCR.
Part III: Generation of Nasopharyngeal Organotypic Rafts and EBV Infection
-
Figure 3. Whole-mount immunostains of nasopharyngeal pseudo-ALIs recombinantly-infected with EBV.

Maximum intensity projections of confocal images. Shown are representative results from two donors (NP-H10-A and NP-H15-A, Ziegler et al., 2021 ) grown from cryobanked CRCs. Immunostain for the EBV lytic protein (Zebra). EBV-encoded RNAs detected by fluorescent in-situ hybridization (EBER-ISH) are abundantly expressed during latency. EBER-positivity in the absence of Zebra staining denotes latent infection. Zebra and EBERs are localized to the nucleus. Histogram shows pixel intensity for the Zebra or EBER signal in the mock sample (blue), or in the EBV-infected sample at day 5 p.i. (red). See Part IV: Image Data Analysis, section A, for how to create an overlaid histogram.
Figure 4. Paraffin-embedding and sectioning of pseudostratified-ALI cultures and organotypic rafts.

A. FFPE processing of pseudostratified-ALI. (1) With the transwell inverted, excise the transwell membrane using a #11 scalpel without disturbing the cell side. Handle the membrane at the edge using fine-tipped forceps. (2) Once removed, the membrane is inverted (cell side up) on a dissection board. Using the scalpel, bisect the membrane. (3) Place both halves of the cut membrane onto a tissue biopsy pad and carefully cover with another pad, taking caution not to move the pads after they make contact with the membrane. Place the pads into a histology tissue cassette and load onto a tissue processor. (4) Following a shortened (40 min) routine processing schedule, open the cassette and gently remove the top pad. The membrane halves are lifted off the pad and embedded on an angle in a paraffin mold with the cut edge facing down. (5) Once cooled, cut the paraffin blocks in 4-µm-thick slices with a microtome, and place the cut sections onto a 44°C waterbath. Pick the sections up from the waterbath using positive charged slides. The slides are baked in an oven at 60°C for 60 min. Following drying/melting, the slides are ready for staining. B. FFPE processing of organotypic rafts. (1) Handle organotypic rafts by the outer edge, using fine-tipped forceps. Lay the raft flat on a biopsy pad in a tissue cassette. Gently place another pad on top of the raft taking caution to not move the pad around. (2) Following routine tissue processing, the raft is removed from the cassette and cut into strips using a #11 scalpel. (3) Using a warm dry embedding mold, each strip is put on edge into the mold as it chills to hold the strips in place. The mold is then filled with paraffin and chilled. When the paraffin block is removed from the mold, the surface is lightly rubbed on the warming plate to remove surface air bubbles. Once cooled, follow the sectioning guidelines described in Panel A, step 5 above.
Materials and Reagents
-
3T3-J2 Feeder Cells (non-irradiated)
Low passage 3T3-J2 cells (Kerafast, catalog number: EF3003)
-
3T3-J2 Growth Media
DMEM (Corning, catalog number: MT10013CV)
Bovine calf serum, iron-supplemented (GE Healthcare, catalog number: SH3007202)
0.05% Trypsin-EDTA (Corning, catalog number: MT25052CI)
-
Collagen Plug
Type I Collagen, high concentration from rat tail (Corning, catalog number: 354249)
Type I Collagen, low concentration from rat tail (Corning, catalog number: 354236)
10× reconstitution buffer (62 mM NaOH, 260 mM NaHCO3, 200 mM HEPES, pH 8.2), filter sterilized
10 M NaOH
10× DMEM (made from powdered DMEM, Fisher, catalog number: 12-00-061), made fresh, filter sterilized
-
Raft Culture (source: Temple et al., 2017 )
E-media, filter sterilized (store in the dark at 4°C for up to 1 month) (see Recipes)
Powdered DMEM (Fisher, catalog numer: 12-00-061)
Powdered Ham’s media (Fisher, catalog number: 21-700-075)
Nystatin (Fisher, catalog number: BP29495)
Penicillin-streptomycin 100× solution (Cytiva, catalog number: SV30010)
Cholera toxin (Sigma, catalog number: C8052)
Adenine stock solution, 180 mM (Sigma, catalog number: A2786) (see Recipes)
Transferrin (R&D, Fisher, catalog number: 2914HT100MG)
T3 solution (Fisher, catalog number: 66-665-0) (see Recipes)
Hydrocortisone solution (Sigma, catalog number: H0888-1G) (see Recipes)
Insulin from bovine pancreas (Sigma, catalog number: I5500)
-
Fetal bovine serum (Seradigm, catalog number: VWR 97068-085)
EGF solution (see Recipes)
Human EGF, recombinant (Sigma, catalog number: E9644-.2MG)
-
EBV infection
EBV-infected Akata cells (these can be recombinantly-infected rAkata cells, or wild-type Akata cells)
RPMI (HyClone, SH30027FS) plus 10% fetal bovine serum (Seradigm, VWR, catalog number: 97068-085)
α-human IgG goat antiserum at 10 mg/mL (Fisher, catalog number: ICN55087), for preparation of B-cell derived virus
-
Metal Grid and Reusable Materials:
Chromic sulfuric acid (Fisher, catalog number: SC88-50)
Metal grid (McMaster-Carr, catalog number: 2930T39), stainless steel wire cloth discs, 3" diameter, 40 × 40 mesh size, packs of 10, fits a 100 mm dish. Bend the edges at three spots to create a “tripod”, taking care to create an even surface. Before each use, wash the metal grid by soaking in a glass beaker of chromic sulfuric acid for 1 h, then remove acid and rinse continuously with tap water for a minimum of 1 h to overnight, followed by a 1 h rinse in distilled water. Autoclave before use.
9" spoon spatulas, stainless steel, 3/4" wide end, 5/16" narrow end (Amazon ASIN: B00ARJOX16). Autoclave before use.
Procedure
The procedure for organotypic raft culture and EBV infection for primary oral keratinocytes has been described in detail ( Temple et al., 2017 ). Here, we show that organotypic rafts can also be generated from nasopharyngeal CRCs. For nasopharyngeal-derived organotypic rafts, CRCs are similarly seeded on top of a collagen plug embedded with live 3T3-J2 cells and lifted to ALI on a metal grid. The protocol by Temple et al. (2017) is provided below with modifications (underlined) and additional tips:
-
Generation of Raft Culture
The following procedure is for collagen plugs prepared in 6-well plate sized wells.
To prepare the 3T3-J2 collagen plugs, use low-passage 3T3-J2 cells grown in 3T3-J2 growth medium (10% bovine calf serum, iron-supplemented) as described for pseudostratified-ALI in Section A above. Note, for organotypic rafts do not irradiate 3T3-J2 cells.
-
Prepare Type 1 collagen from rat tail to exactly 4 mg/mL, by mixing low and high concentration Type I collagen on ice. Prepare extra, as some collagen will be lost in pipetting.
Tip: Mixing high and low concentration is required because diluting high concentration collagen with acetic acid, as recommended by the manufacturer, results in a very low pH and is also difficult to achieve accurately due to high viscosity.
Trypsinize subconfluent 3T3-J2 cells with 0.05% Trypsin-EDTA. Neutralize with 3T3-J2 growth media and pellet cells. For each raft, resuspend 6.25 × 105 3T3-J2 cells in 0.25 mL of 10× reconstitution buffer. All work moving forward should be performed on ice, to prevent cell clumping and premature collagen solidification.
Add 0.25 mL of 10× DMEM and mix well by pipetting.
Add 2 mL of 4 mg/mL Type I collagen. Adjust pH with 6 µL of 10 N NaOH per 2.5 mL collagen plug. Mix thoroughly by inverting the tube.
Aliquot 2.5 mL of collagen-3T3-J2 cell mixture into one well of a 6-well plate, to form the collagen plug. Tilt to distribute evenly, and remove air bubbles with a sterile pipette tip.
Incubate the collagen plugs at 37°C and 5% CO2 for 1–2 h, to allow the collagen plug to solidify.
Once the collagen plug has solidified, add 2–3 mL of E-media without EGF, and equilibrate at room temperature for at least 10 min. The presence of EGF can damage the 3T3-J2 fibroblasts. E-media should completely cover the collagen plug. The collagen plug in E-media can be prepared up to 2 days in advance, and left in a 37°C tissue culture incubator until ready to seed nasopharyngeal CRCs.
-
When ready to seed nasopharyngeal CRCs, aspirate the E-media covering the collagen plug. Seed 3 × 106 nasopharyngeal CRCs onto the collagen plug in 1 mL of E-media supplemented with 5 ng/mL EGF. Incubate at 37°C in a tissue culture incubator for 2–4 h, to allow the CRCs to adhere. Do not rock the culture, as this may result in uneven distribution of the CRCs. After 2 h, check that the CRCs have adhered using an inverted phase/contrast microscope. The adhered CRCs should fill the entire surface of the collagen plug. This is critical, or the resulting organotypic rafts will be thin. Do not leave the CRCs on the collagen plug with EGF-supplemented media for extended periods beyond 4 h, as this will damage the 3T3-J2 fibroblasts.
Note: It is possible to transduce nasopharyngeal CRCs with lentivirus expression vectors at the time of expansion before seeding into organotypic rafts. Stable selection of transduced cells is not recommended, as increased passage number will affect the quality of the organotypic raft. A transient selection is possible if the transduction is efficient.
Once the CRCs have adhered, gently aspirate residual media, and carefully transfer the collagen plug with adhered CRCs from the 6-well plate to an autoclave-sterilized metal grid in a 100 mm plate. Loosen the plug with a sterile spatula around the perimeter and spread the collagen plug onto the metal grid, avoiding folds or air bubbles underneath. Up to two collagen plugs can be placed onto each metal grid. Apply E-media without EGF to the bottom of the plate until it reaches the bottom surface of the raft. Aspirate any air bubbles. In order to generate an air-liquid interface, do not allow any portion of the raft to be submerged (see Video 2 ). Incubate the raft cultures in a tissue culture incubator at 37°C for 9–12 days, to allow differentiation. Extending the differentiating period will result in thinning of the rafts. Change the underlying E-media every other day (Figure 5A) .
-
Infecting Raft Cultures with EBV ( Video 3 )
Prepare anti-human IgG-reactivated Akata cells one day prior to infection (day 3 post air-lifting). Resuspend Akata cells to 5 × 105 cells/mL in RPMI media with 10% FBS. Add 100 µg/mL α-human IgG goat antiserum. Incubate at 5% CO2 and 37°C for 24 h.
-
At day 4 post air-lifting, aspirate E-media from the raft, and score the raft surface in a grid format with a sterile scalpel, ten times horizontally and ten times vertically. Scoring is a form of wounding of the tissue that enables the inoculum to directly contact all epithelial cell layers in the raft.
Note: EBV infection without scoring the raft is possible with oral keratinocyte-derived organotypic rafts (Temple et al., 2017), although we have not attempted this with nasopharyngeal organotypic rafts.
-
Inoculate the scored raft surface with reactivated Akata cells from step B1, by spinning down the reactivated Akata cells and resuspending 2.5 × 106 cells in 200 µL of PBS. Add the inoculum dropwise and evenly distribute across the surface of the raft, using a 200 µL pipette.
Note: The inoculum size was optimized for 2.5 × 106 Akata cells per raft. Because this is a co-culture method of infection, the size of the inoculum is simply calculated by the number of co-cultured EBV producing B-cells. The reactivated Akata inoculum is continually producing infectious EBV while co-cultured with the raft.
Allow the inoculum to absorb, and then replace the basolateral E-media. Place in a 5% CO2 tissue culture incubator at 37°C for the desired length of time (4–7 days post-infection). Do not wash off the EBV inoculum. Replenish with fresh E-media every other day.
-
Harvesting Raft Cultures by FFPE for Histology and Immunostaining ( Video 4 )
Aspirate media from the raft.
Rinse the raft twice with PBS, by gently pipetting into the 100 mm plate and allowing the raft to submerge.
Fix the raft directly on the metal grid, by submerging in 4% PFA at room temperature for 1 h.
-
Using a spatula, transfer the fixed raft to a biopsy sponge soaked in PBS. Close the tissue cassette lid and store in 70% ethanol for FFPE processing (Figure 4) .
Note: Unlike ethanol, tert-Butanol preserves the fluorescence of fluorescent proteins (Zhanmu et al., 2019). If visualization of a reporter fluorescent protein is desired after fixation, store the PFA-fixed raft in 50% tert-Butanol and dehydrate in graded (50%, 75%, 95%, and 100%) tert-Butanol for 30 mins each, before FFPE processing. Rehydration should also be carried out in graded (100%, 95%, 75%, and 50%) tert-Butanol.
Proceed to histology and immunostaining. Representative immunostains for cellular and EBV proteins denoting lytic infection (EBV Zebra+) in the suprabasal layers (involucrin+) are shown in Figure 6 .
Video 2. Lifting of organotypic rafts onto a metal grid.
Figure 5. Example of an organotypic raft.

A. An organotypic raft growing at the air-liquid interface on a metal grid, and fed with basolateral E-media. B. Zoomed image of a mature organotypic raft at the time of harvest. A white layer of differentiated cells is visible on top of the pink collagen plug which contains the embedded 3T3-J2 fibroblasts.
Video 3. Scoring and EBV infection of organotypic rafts.
Video 4. Harvesting of fixed organotypic raft for FFPE processing.
Figure 6. Immunostains of FFPE sections from nasopharyngeal organotypic rafts mock-treated or recombinantly-infected with EBV. EBV-infected rafts were infected by the co-culture method with Akata B-cells.

Shown are representative FFPE sections stained for cellular or EBV marker proteins and counterstained with DAPI or wheat germ agglutinin (WGA). A. Representative FFPE sections stained for cellular marker proteins (keratin 5 [K5], involucrin [Inv], and human IgG [hIgG]). Basal cells are strongly positive for K5 but weakly positive for Inv. Suprabasal cells are strongly positive for Inv and positive for K5. The green dotted line denotes the border between the basal and suprabasal layers. B. Representative FFPE sections stained for EBV (Zebra) protein and hIgG. Zebra+ cells can be quantified, and the percent of Zebra+ nuclei per image plotted (see Part IV: Image Data Analysis). In the images shown, no contaminating B-cells (Akata inoculum) were observed with the anti-hIgG stain. H&E, hematoxylin and eosin. Arrow, keratin pearl. Scale bars = 50 µm.
Part IV: Image Data Analysis
-
Generating Overlaid Histograms with CellSens Dimension Software
(CellSens Dimension software available from Olympus)
The purpose of generating an overlaid histogram is to show signal-to-noise ratio. A typical example would compare the Zebra-stained signal in a pseudo-ALI culture infected with EBV compared to the mock control, as shown in Figure 3 .
Open the multichannel Z-stack infected and mock images in CellSens Dimension.
Generate a maximum intensity projection of the z-stacks by selecting Process -> Maximum Intensity, and save as new images.
Select Image -> Separate -> Channels, so that each color channel is now displayed as an individual image.
Create a new image containing the channel displaying the stain by selecting Image -> Combine -> Channels, and select the images to compare. There are options to change the pseudocolor of each channel, so ensure that each is displayed as a different color. This color will appear in the histogram.
Ensure that both channels are visible. The histograms of each channel are now overlaid in the histogram window. Select the button labeled with an arrow in Figure 7 , to switch from logarithmic to linear view. Screenshot and crop the histogram window to display as desired. Additional examples of overlaid histograms are shown in Figure 3 .
-
Quantification of Fluorescent Particles with ImageJ Software and Fiji Plugin
(ImageJ software and Fiji plugin available at https://imagej.net/software/)
The purpose of Particle Analysis is to score for Zebra+ nuclei. A typical example may show the percent DAPI+ nuclei that are Zebra+ in an EBV-infected organotypic raft compared to a mock control, as shown in Figure 3 .
Set LUTs and adjust images as necessary in the microscope software. Export/save separate channels as TIFF images.
Open the DAPI and TRITC images in Fiji.
-
First rotate and crop to the region of interest (the membrane/cells).
Select the area to crop in the DAPI image. Switch to the red image and select Edit -> Selection -> Restore selection (or press Ctrl – Shift – E). Then select crop (or push Ctrl – Shift – X in each) for each image. This will crop the same area of each channel.
In the DAPI image, Select Image -> Adjust -> Color Threshold, and adjust the brightness threshold, so that the background is removed while nuclei are still observed.
Analyze -> Analyze Particles. Adjust settings to include all nuclei. This number is the number of DAPI positive cells.
When analyzing, ensure “Add to Manager” is selected.
Click in the ROI manager, then select More -> Fill, and it should fill particles in green color.
Select Image -> Color -> Split, to create a binary image containing only the DAPI nuclei.
Select Edit -> Selection -> Create Selection to select the nuclei in the binary image.
In the red channel image (Zebra staining), press Ctrl – Shift – E or select restore selection, to select only the nuclei area from the blue image.
Threshold again (set the value as high as needed, so that each nuclei area is counted as one cell) and click apply, to generate a black and white image containing only the Zebra staining inside the DAPI nuclei area. Analyze particles again to determine the number of Zebra positive cells. An example of the commands and graphed result are shown in Figure 8 .
Figure 7. Generating an overlaid histogram with CellSens Dimension Software to illustrate signal to noise.

The cropped images illustrate steps 2–5 described in Part IV: Image Data Analysis (A). This example compares the signal of a Zebra-stained image, from a nasopharyngeal pseudo-ALI culture (donor NPH10A) infected with EBV harvested at 5 days p.i., with a mock-infected control ( Ziegler et al., 2021 ). A. Maximum intensity projection of a Zebra-stained z-stack image (red) with DAPI (blue) in the EBV-infected sample. B. The red channel displaying the signal of the Zebra stain is separated for further analysis. C. The two red channel images corresponding to the Zebra stain in the EBV-infected and mock control are selected, and the color for the mock image is pseudocolored blue. D. The final overlaid histogram for the Zebra-stained channel is shown. Red denotes the EBV-infected sample and blue denotes the mock-infected sample. The arrow indicates the button to toggle between the logarithmic and linear display scale, whichever best shows the signal-to-noise ratio between the EBV-infected vs mock sample.
Figure 8. Particle Analysis with ImageJ software and Fiji plugin to quantify positively stained nuclei.

The cropped images illustrate steps 4–11 described in Part IV: Image Data Analysis (B). This example compares the signal of a Zebra-stained image from nasopharyngeal organotypic rafts infected with EBV or mock control. Shown are results from donor NPH17A ( Ziegler et al., 2021 ). A. A threshold is applied to the DAPI channel in ImageJ. B. Results from Particle Analysis on the thresholded DAPI images. Each number on the left column denotes a particle. C. Zebra-stained images are evaluated for percent DAPI+ nuclei that are Zebra+. Zebra staining is displayed as a binary image within the DAPI nuclei, and the results from Particle Analysis are shown. Note that the settings detect no Zebra+ particles in the mock, but many Zebra+ particles in the EBV-infected sample. D. An example of percent Zebra+ nuclei in organotypic rafts at 7 days p.i., calculated with ImageJ software. This example is calculated from the images in Figure 6B.
Recipes
-
Georgetown Media Preparation
Preparing Stock Solutions
Dissolve hydrocortisone in 100% ethanol to a concentration of 0.5 mg/mL. Combine 1 mL of the hydrocortisone with 19 mL of DMEM and 2.5 µg EGF. Store 1.1-mL aliquots of hydrocortisone/EGF at -20°C.
Dissolve 100 mg insulin in 20 mL of distilled water containing 200 µL of glacial acetic acid. Filter sterilize and store in 1.1 mL-aliquots at -20°C.
Dissolve 25 mg of Y-27632 (ROCKi) with sterile water to a concentration of 5 mM. Make 1-mL aliquots and store at -20°C.
Dissolve 1 mg vial of cholera toxin in 2 mL of distilled water and filter sterilize. Stock solution is stable at 4°C for 1 year.
-
Prepare Complete DMEM media, by adding 5.5 mL of glutamine, 5.5 mL of penicillin-streptomycin, and 50 mL of heat inactivated fetal bovine serum to 500 mL of DMEM (H).
To make 500 mL of Georgetown media:
In a sterile tissue culture hood combine 373 mL of complete DMEM media, 125 mL of F-12 nutrient mix, 0.5 mL of hydrocortisone/EGF mix, 0.5 mL of insulin, 0.5 mL of amphotericin B, 0.5 mL of gentamicin, and 4.3 µL of cholera toxin.
Add 0.5 mL of Y-27632 to the Georgetown media. This component MUST be added last.
Store Georgetown media at 4°C for up to one month.
-
UNC/USG Media
Storage Condition: Must be made fresh every 1–2 weeks, and stored at 4°C. Filter sterilize.
Preparing Stock Solutions
-
Stock 4: FeSO4·7H2O (0.042 g/L)
MgCl2·6H2O (12.20 g/L)
CaCl2·2H2O (0.441 g/L)
Stock 11: ZnSO4·7H2O (0.863 g/L)
Trace elements:
Selenium from Sodium selenite (3 µM)
Manganese from Manganese Chloride tetrahydrate (0.1 µM)
Silicon from Sodium Silicate nonahydrate (50 µM)
Molybdemun from Ammonium Molybdate tetrahydrate (0.1 µM)
Vanadium from Ammonium Metavanadate (0.5 µM)
Tin from Tin Chloride dihydrate (0.05 µM)
To make 500 mL of UNC/USG media, mix the following components:
DMEM/F12 (500 mL)
Penicillin/streptomycin (1×, 1:100)
Stock 4 (1:100)
Stock 11 (1:1,000)
Trace elements (1:100)
Transferrin (10 mg/mL, 1:1,000)
Hydrocortisone (200 µM, 1:1,000)
Insulin (10 mg/mL, 1:2,000)
Epinephrine (3.3 mM, 1:1,000)
T3 (10 µM, 1:1000)
EGF (5 µg/mL, 1:5,000)
BSA (0.25 g/mL, 1:500)
Retinoic Acid (5 mM, 1:1 × 105)
BPE (0.8% v/v)
Phosphorylethanolamine (500 µM, 1:1,000)
Ethanolamine (500 µM, 1:1,000)
Ultroser G (0.5% v/v)
Amphotericin B (250 µg/mL, 1:1,000)
-
-
E-Media
Preparing Stock Solutions
Adenine stock solution: Add 486 mg adenine to 15 mL of dH2O. Add approximately 10 drops of concentrated HCl until dissolved, and then adjust volume to 20 mL with dH2O.
T3 solution: Dissolve 13.6 mg T3 in 100 mL of 0.02 N NaOH. Make two 100-fold serial dilutions in sterile PBS, to obtain the final stock solution.
Hydrocortisone solution: Dissolve 25 mg hydrocortisone in 5 mL of 100 % ethanol. To generate the stock solution, add 4.8 mL of the hydrocortisone solution to 55.2 mL of 1 M HEPES, pH 7.
To make 500 mL of E-medium:
Dissolve 5.015 g powdered DMEM (DMEM with 4.5 g/L D-glucose and L-glutamine without sodium pyruvate and sodium bicarbonate) and 1.33 g powdered Ham’s medium (F12 nutrient mixture with L-glutamine, without sodium bicarbonate) in 300 mL of dH2O.
Add 1.534 g sodium bicarbonate, 5 mL of penicillin-streptomycin solution (10,000 U/mL penicillin and 10,000 μg/mL streptomycin stock solution), 5 μL of cholera toxin (1 mg/mL stock dissolved in dH2O), 0.5 mL of adenine stock solution, 0.5 mL of transferrin (5 mg/mL stock dissolved in PBS), 0.5 mL of T3 stock solution, 0.5 mL of hydrocortisone stock solution, and 0.5 mL of insulin (5 mg/mL dissolved in 0.1 N HCl).
Adjust volume to 470 mL with dH2O, and adjust pH to 7.1–7.15.
Filter sterilize with a 0.2 μm pore-size low-protein-binding filter, and store at 4°C in the dark. Prior to use, add 5 mL of nystatin (10,000 U/mL suspension), and 25 mL of FBS.
-
EGF solution
Dissolve at 1 µg/mL in dH2O and supplement with 1 mg/mL BSA.
Acknowledgments
We thank Mary Ferguson and Ian Hayman for help with optimizing the organotypic rafts. K.S. received funding from the Hillman Foundation. M. M. received funding from the Cystic Fibrosis Foundation Research Development Program (to the University of Pittsburgh) through the Cystic Fibrosis Research Center Cell Core. This work was supported in part by the University of Pittsburgh School of Medicine Dean’s Faculty Advancement Award and used the Hillman Tissue and Research Pathology Services shared resource that is supported in part by the National Institutes of Health award P30CA047904. This protocol is based on the manuscript “A primary nasopharyngeal three-dimensional air-liquid interface cell culture model of the pseudostratified epithelium reveals differential donor- and cell type-specific susceptibility to Epstein-Barr virus infection” ( Ziegler et al., 2021 ). The protocol for EBV infection in organotypic rafts was adapted from the manuscript “Generation and Infection of Organotypic Cultures with Epstein-Barr Virus” ( Temple et al., 2017 ).
Competing interests
The authors have declared no competing interests.
Ethics
The methods were performed in accordance with relevant guidelines and regulations approved by the University of Pittsburgh Institutional Review Board (IRB). The study received approval under IRB: STUDY19030014 and were conducted in compliance with guidelines approved for the University of Pittsburgh Sinus Fluid and Tissue Bank. All individuals involved have given written formal consent.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1. Caves E. A., Cook S. A., Lee N., Stoltz D., Watkins S. and Shair K. H. Y.(2018). Air-Liquid Interface Method To Study Epstein-Barr Virus Pathogenesis in Nasopharyngeal Epithelial Cells. mSphere 3(4): e00152-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chapman S., Liu X., Meyers C., Schlegel R. and McBride A. A.(2010). Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest 120(7): 2619-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fulcher M. L. and Randell S. H.(2013). Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol Biol 945: 109-121. [DOI] [PubMed] [Google Scholar]
- 4. Gentzsch M., Boyles S. E., Cheluvaraju C., Chaudhry I. G., Quinney N. L., Cho C., Dang H., Liu X., Schlegel R. and Randell S. H.(2017). Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells. Am J Respir Cell Mol Biol 56(5): 568-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hadinoto V., Shapiro M., Sun C. C. and Thorley-Lawson D. A.(2009). The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog 5(7): e1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Imai S., Nishikawa J. and Takada K.(1998). Cell-to-cell contact as an efficient mode of Epstein-Barr virus infection of diverse human epithelial cells. J Virol 72(5):4371-4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kopan R., Traska G. and Fuchs E.(1987). Retinoids as important regulators of terminal differentiation: examining keratin expression in individual epidermal cells at various stages of keratinization. J Cell Biol 105(1): 427-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu X., Ory V., Chapman S., Yuan H., Albanese C., Kallakury B., Timofeeva O. A., Nealon C., Dakic A., Simic V., et al.(2012). ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 180(2): 599-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maruo S., Yang L. and Takada K.(2001). Roles of Epstein-Barr virus glycoproteins gp350 and gp25 in the infection of human epithelial cells. J Gen Virol 10): 2373-2383. [DOI] [PubMed] [Google Scholar]
- 10. Meyers C.(1996). Organotypic(raft) epithelial tissue culture system for the differentiation-dependent replication of papillomavirus. Methods Cell Sci 18: 201-210. [Google Scholar]
- 11. Raab-Traub N.(2015). Nasopharyngeal Carcinoma: An Evolving Role for the Epstein-Barr Virus. Curr Top Microbiol Immunol 1): 339-363. [DOI] [PubMed] [Google Scholar]
- 12. Serrano Castillo F., Bertrand C. A., Myerburg M. M., Shapiro M. E., Corcoran T. E. and Parker R. S.(2019). A physiologically-motivated model of cystic fibrosis liquid and solute transport dynamics across primary human nasal epithelia. J Pharmacokinet Pharmacodyn 46(5): 457-472. [DOI] [PubMed] [Google Scholar]
- 13. Shair K. H. Y.(2020). mSphere of Influence: 3-D Culture Models Influence Studies on Epstein-Barr Virus Molecular Pathogenesis in the Epithelium. mSphere 5(5): e00954-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shair K. H. Y., Reddy A. and Cooper V. S.(2018). New Insights from Elucidating the Role of LMP1 in Nasopharyngeal Carcinoma. Cancers(Basel) 10(4): 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sitki-Green D., Covington M. and Raab-Traub N.(2003). Compartmentalization and transmission of multiple epstein-barr virus strains in asymptomatic carriers. J Virol 77(3): 1840-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suprynowicz F. A., Upadhyay G., Krawczyk E., Kramer S. C., Hebert J. D., Liu X., Yuan H., Cheluvaraju C., Clapp P. W., Boucher R. C. Jr. et al.(2012). Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci U S A 109(49): 20035-20040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Temple R. M., Meyers C. and Sample C. E.(2017). Generation and Infection of Organotypic Cultures with Epstein-Barr Virus. Methods Mol Biol 1532: 65-78. [DOI] [PubMed] [Google Scholar]
- 18. Temple R. M., Zhu J., Budgeon L., Christensen N. D., Meyers C. and Sample C. E.(2014). Efficient replication of Epstein-Barr virus in stratified epithelium in vitro . Proc Natl Acad Sci U S A 111(46): 16544-16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsai M. H., Raykova A., Klinke O., Bernhardt K., Gartner K., Leung C. S., Geletneky K., Sertel S., Munz C., Feederle R., et al.(2013). Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep 5(2): 458-470. [DOI] [PubMed] [Google Scholar]
- 20. Tsao S. W., Tsang C. M., Pang P. S., Zhang G., Chen H. and Lo K. W.(2012). The biology of EBV infection in human epithelial cells. Semin Cancer Biol 22(2): 137-143. [DOI] [PubMed] [Google Scholar]
- 21. Wang H. B., Zhang H., Zhang J. P., Li Y., Zhao B., Feng G. K., Du Y., Xiong D., Zhong Q., Liu W. L., et al.(2015). Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat Commun 6: 6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Young L. S., Yap L. F. and Murray P. G.(2016). Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer 16(12): 789-802. [DOI] [PubMed] [Google Scholar]
- 23. Yu M. C. and Yuan J. M.(2002). Epidemiology of nasopharyngeal carcinoma. Semin Cancer Biol 12(6): 421-429. [DOI] [PubMed] [Google Scholar]
- 24. Zhang H., Li Y., Wang H. B., Zhang A., Chen M. L., Fang Z. X., Dong X. D., Li S. B., Du Y., Xiong D., et al.(2018). Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat Microbiol 3(2):164-171. [DOI] [PubMed] [Google Scholar]
- 25. Zhanmu O., Zhao P., Yang Y., Yang X., Gong H. and Li X.(2019). Maintenance of Fluorescence During Paraffin Embedding of Fluorescent Protein-Labeled Specimens. Front Neurosci 13: 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ziegler P., Tian Y., Bai Y., Abrahamsson S., Backerholm A., Reznik A. S., Green A., Moore J. A., Lee S. E., Myerburg M. M., et al.(2021). A primary nasopharyngeal three-dimensional air-liquid interface cell culture model of the pseudostratified epithelium reveals differential donor- and cell type-specific susceptibility to Epstein-Barr virus infection. PLoS Pathog 17(4): e1009041. [DOI] [PMC free article] [PubMed] [Google Scholar]