

Summary of the article:
The recognition of pathogen-associated nucleic acid (NA) promotes effective immunity against invading pathogens. However, endosomal Toll-like receptor (TLR) activation by self-NA also underlies the pathogenesis of systemic autoimmune diseases, such as systemic lupus erythematosus (SLE). For this reason, the activation thresholds of NA-sensing TLRs must be tightly regulated to balance protective and pathogenic immune responses. In this review, we will provide an overview of the evolutionary mechanisms designed to limit the aberrant activation of endosomal TLRs by self-ligands, focusing on four broad strategies. These include: 1) the production of nucleases able to degrade self-DNA and RNA; 2) the cell-specific regulation of endosomal TLR expression; 3) the spatial and temporal control of TLR positioning at a sub-cellular level; and 4) the modulation of downstream TLR signaling cascades. Given the critical role for B cells in lupus pathogenesis, where possible, we will describe evidence for B cell-specific induction of these regulatory mechanisms. We will also highlight our own work showing how modulation of B cell endolysosomal flux tunes NA-sensing TLR activation signals. In the face of inevitable generation of self-NA during normal cellular turnover, these parallel mechanisms are vital to protect against pathogenic inflammation.
Keywords: Systemic Lupus Erythematosus, B cells, Toll-like receptors, Endolysosomal trafficking, Non-canonical autophagy
Introduction
The COVID19 pandemic continues to exert a devastating toll on human health. As of November 2021, a total of >240 million confirmed cases and almost 5 million deaths have been attributed to SARS-CoV-2 (severe acute respiratory syndrome coronavirus) infection1, with estimates for excess global mortality reaching 17 million2. This potential for transmissible pathogens to cause widespread morbidity and mortality emphasizes the importance of robust immune responses to infection. One strategy adopted by the humoral arm of the immune system is to integrate B cell receptor (BCR) and endosomal TLR signals to drive the rapid activation of virus-specific B cells. Specifically, it has long been appreciated that TLR ligands serves as effective vaccine adjuvants. However, whereas soluble protein antigens chemically linked to TLR agonist drive dendritic cell (DC)-specific activation, intact virus particles (in which viral NA is incorporated within viral capsids) promote robust B cell activation via B cell-intrinsic activation of the signaling adaptor Myd883. As such, B cells are “hard-wired” to respond vigorously to viral particles as an immune defense via engagement of antigen-specific BCR and endosomal TLR recognition of pathogen-associated nucleic acid (NA).
Unfortunately, this ability of B cells to recognize nucleic acids via engagement of endosomal TLRs acts as a double-edged sword. Despite the wide scope of potential autoantigens, it was recognized in the 1950’s that systemic lupus erythematosus (SLE) is characterized by relatively restricted autoantibodies targeting nuclear antigens4. The biology underlying this observation remained enigmatic until the seminal discovery that nuclear antigens can induce similar dual BCR and TLR activation of autoreactive B cells5. Specifically, autoreactive B cells recognizing apoptotic particles traffic nucleic acid-containing antigens to endosomal compartments resulting in TLR engagement. The Myd88-dependent receptors TLR7 and TLR9 are critical in this context, with TLR7 required for the generation of Abs targeting RNA and RNA-associated proteins, while TLR9 activation promotes production of Abs targeting double-stranded DNA (dsDNA) and chromatin5,6. These data provided a unifying model for the generation of anti-nuclear antibodies (ANA), but also highlighted the risks inherent in this arrangement. On the one hand, B cell-intrinsic activation by viral DNA/RNA markedly increases titers, affinity, and durability of specific antibody, facilitating both viral clearance and long-term protection from pathogen rechallenge. On the other hand, B cell sensing of self-nucleic acids initiates the development of several humoral autoimmune diseases, including SLE.
Endosomal TLR signals exert B cell-intrinsic contributions to lupus pathogenesis
As detailed below, limiting the expression of NA-sensing TLRs to specific cell lineages serves as a strategy to prevent aberrant activation by self. Of specific relevance to lupus pathogenesis, the endosomal receptors TLR7 and TLR9 are expressed by both B cells and myeloid lineages, in particular plasmacytoid dendritic cells (pDC). For this reason, each lineage might conceivably promote the development of SLE via cell-intrinsic mechanisms. The prevailing model holds that dual BCR and TLR engagement promotes anti-nuclear antibody production by B cells, while Fc receptor-dependent uptake of circulating immune complexes promotes Myd88-dependent pro-inflammatory cytokine and type 1 interferon production by pDC7,8. The logical conclusion from this model is that autoreactive B cell activation is the initiating event driving breaks in tolerance, since this is required for autoantibody:autoantigen immune complex formation. In keeping with this idea, B cell-intrinsic deletion of the TLR signaling adaptor Myd88 abrogated autoimmunity in independent murine lupus strains9–12. In contrast, dendritic cell (DC)-specific Myd88 deletion exerted a more limited impact on dermatitis in MRL.Faslpr mice, without affecting ANA production11. These animal studies are consistent with human data derived from pre-clinical lupus cohorts, in which ANA positivity develops years prior to clinical symptoms, but the type 1 IFN signature develops shortly before disease onset13,14. Together, these studies highlight a critical role for B cell TLR signals in initiating the inflammatory cascade leading to clinical SLE.
In retrospect, the role for TLR7 and TLR9 in facilitating the production of RNA- and DNA-associated autoantibodies was consistent with each receptor’s ligand specificity. However, despite data linking anti-dsDNA autoantibodies titers with disease activity in human SLE15,16, Tlr9 deletion unexpectedly worsened systemic inflammation in murine lupus17–20. In contrast, TLR7 is required for the development of lupus-like disease in multiple independent mouse models18,19,21, including for accelerated disease in Tlr9−/−.MRLlpr and Tlr9−/−.B6.Nba2 mice20,22. While the mechanism underlying these opposing impacts of TLR7 and TLR9 on lupus pathogenesis have yet to be adequately explained, an important additional question is whether myeloid- or B cell-driven TLR signals explain these effects.
The original description of accelerated autoimmunity in TLR9-deficient lupus implicated a myeloid-specific mechanism. Since Tlr9−/−.MRL.Mplpr/lpr lupus-prone mice exhibited increased plasmacytoid dendritic cell (pDC) activation and elevated type 1 interferon levels, these data suggested that loss of TLR9 promotes myeloid dysregulation18. It was against this backdrop that our group sought to directly compare the impact of B cell specific TLR7 vs. TLR9 deletion during lupus pathogenesis. Using a chimeric model of murine SLE, we showed that B cell-intrinsic loss of TLR7 both prevented RNA-associated autoantibody production and abated systemic autoimmunity. In contrast, B cell Tlr9 deletion exerted an isolated impact on anti-dsDNA/chromatin autoantibodies and exacerbated disease23. Thus, B cell-specific TLR7 vs. TLR9 deletion was sufficient to recapitulate the phenotype of global knockout models, despite intact expression of endosomal TLRs in the myeloid compartment.
Importantly, these B cell-intrinsic effects for endosomal TLRs in regulating disease severity have been confirmed in multiple independent murine models. For example, transgenic Tlr7 over-expression exerts a B cell-specific impact on lupus risk24,25, while spontaneous germinal centers in C57BL/6 mice depend on TLR7 engagement by B cells19. In contrast, B cell-intrinsic Tlr9 deletion in MRL/lpr mice exacerbates lupus nephritis despite loss of anti-nucleosome antibodies26. In keeping with predominantly B cell focused effects during lupus pathogenesis, loss of myeloid TLR9 expression exerted no detectable impact on systemic autoimmunity. Most notably, the converse experiment highlighted a critical protective role for TLR9 in SLE, since B cell-specific Tlr9 over-expression was able to protect against progressive nephritis26. In summary, while not seeking to downplay the important role for myeloid TLR signals in driving lupus disease, these combined studies emphasize how tight regulation of endosomal TLR signaling in B cells is required to maintain immune tolerance.
Regulation of endosomal TLR signals
Since endosomal TLR signals promote robust immune responses to viral pathogens and also drive immune tolerance breaks in humoral immunity, activation thresholds must be finely tuned. To balance protective and pathogenic responses, the nucleic acid (NA)-sensing TLRs, like other arms of the immune system, have been shaped by evolutionary forces. Specifically, endosomal TLRs employ various mechanisms to reduce the likelihood that self-ligand activation promotes pathologic inflammation, in the face of the continuous production of billions of apoptotic cells per day. These mechanisms can be categorized into two broad classes: those that reduce the likelihood that NA-sensing TLRs will encounter self-nucleic acids and those that dampen responses to them. Given the importance of these regulatory processes to the pathophysiology of human autoimmunity, significant effort has been directed to delineating these cellular mechanisms. However, the bulk of these studies have focused on the regulation of myeloid TLR signaling, with less emphasis placed on B cell focused studies. For this reason, in addition to providing a general overview of endosomal TLR regulation, we will highlight research, including from our own groups, that aims to uncover B cell-intrinsic regulatory mechanisms.
As a broad framework, we propose that four major mechanisms limit pathogenic activation of NA-sensing TLRs (Figure 1). First, specific nucleases and membrane channels degrade or remove self-NA to prevent receptor binding. Second, the expression of NA-sensing TLRs is limited to a subset of immune lineages required for effective pathogen response, while TLR signals are dampened in tissue macrophages optimized for silent clearance of apoptotic material. Third, NA-sensing TLRs are directed to endosomal compartments to prevent pathogenic activation by extra-cellular self-NA. Moreover, following ligand binding, the induction of endolysosomal trafficking regulates both the amplitude and duration of TLR signaling. Finally, genetic variation modulates signaling cascades downstream of endosomal TLR ligation, as evidenced by the enrichment of TLR-associated gene polymorphisms in subjects with SLE.
Figure 1: Regulatory mechanisms controlling NA-sensing TLR activation.
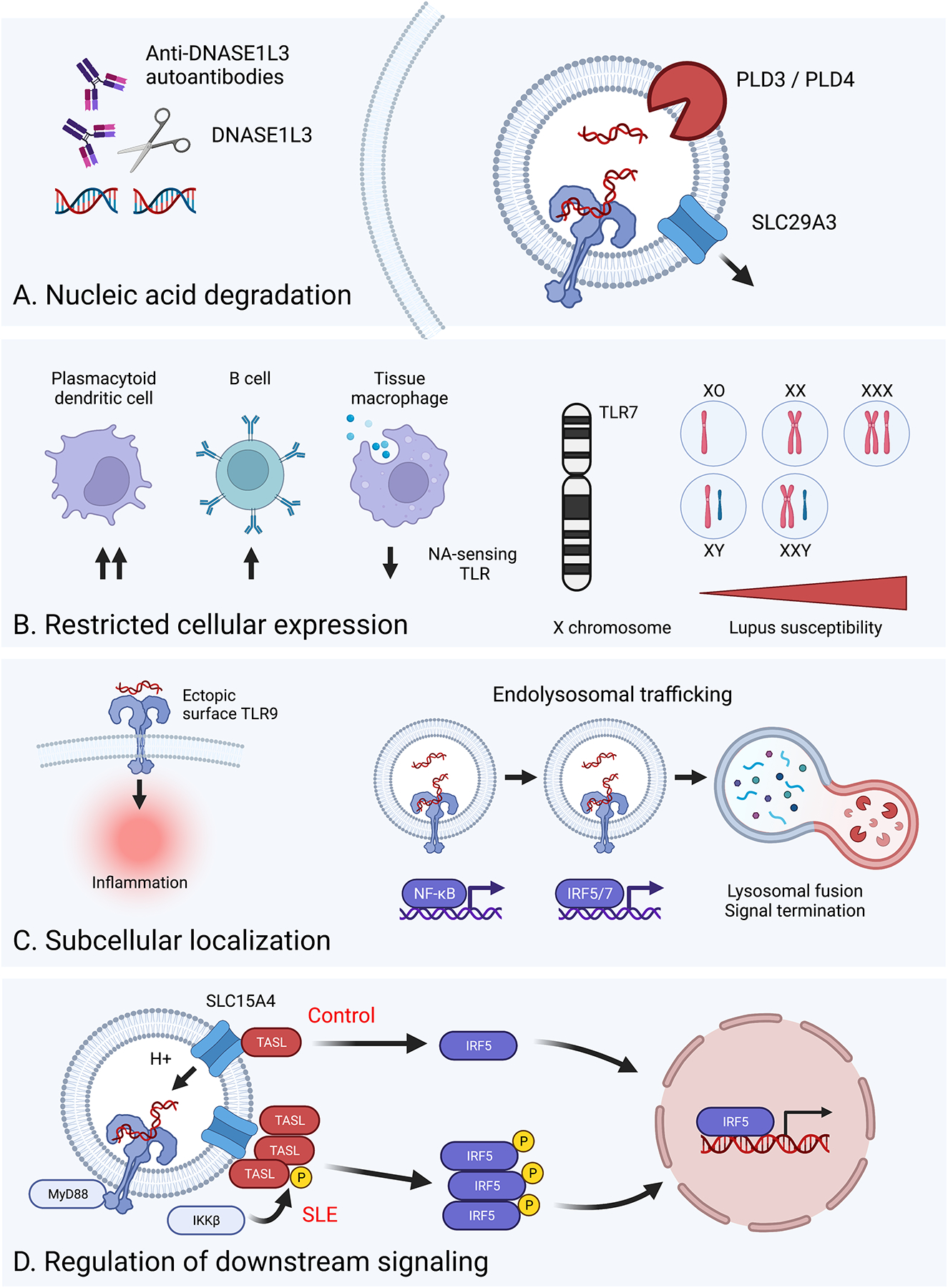
Schematic detailing the various strategies employed by the immune system to limit aberrant activation of NA-sensing TLRs. A. Nucleic acid degradation: Expression of specific nucleases, such as DNASE1L3, PLD3, and PLD4, or the lysosomal transporter SLC29A3, degrade or remove self-NA prior to recognition by NA-sensing TLRs. This regulatory mechanism can be subverted by either genetic defects in specific genes or via the development of anti-DNASE1L3 autoantibodies. B. Restricted cellular expression: The expression of NA-sensing TLRs is limited to certain immune lineages, such as pDCs and B cells, while specifically reduced on tissue macrophages designed for apoptotic cell clearance (left). Aberrant activation of NA-sensing TLRs is further controlled via regulated mRNA expression, as evidenced by X chromosome gene dosage driving TLR7-dependent lupus pathogenesis (right). C. Subcellular localization: NA-sensing TLRs traffic to endosomal compartments to prevent pathogenic engagement by circulating NA (left). Following endosomal TLR ligation, induction of endolysosomal trafficking serves to both control the amplitude of and terminate TLR signaling (right). D. Regulation of downstream signaling: Amongst many genetic variants impacting lupus risk, CXorf21 polymorphisms increase TASL levels resulting in increased TLR-dependent nuclear translocation of IRF5.
1. Degradation of self-nucleic acids by specific nucleases
Given the potential for exposure to ubiquitous self-nucleic acids, various nucleases have evolved to remove NA prior to recognition by endosomal TLRs. Most notably, a secreted DNA nuclease, DNase I-like 3 (DNASE1L3), acts extracellularly to degrade both cell-free DNA and DNA within circulating apoptotic microparticles. In keeping with this enzyme acting to prevent autoimmunity, homozygous loss-of-function DNASE1L3 mutations result in familial, early-onset SLE27,28, while a hypomorphic variant with reduced enzymatic activity has been linked with increased SLE and scleroderma risk29,30. In addition, Dnase1l3−/− mice exhibit lupus-like disease characterized by high-titer anti-DNA antibodies confirming the degradation of extracellular self-nucleic acid is required to maintain immune tolerance31–33. Surprisingly, both TLR7 and TLR9 signals facilitated NA responses in Dnase1l3−/− mice, with each receptor partially redundant for autoantibody production. Although the relative importance of B cell vs. myeloid TLRs in Dnase1l3−/− murine autoimmunity has not been addressed, genetic ablation of type 1 IFN signaling was dispensable for initial tolerance breaks but required for feed-forward amplification of anti-DNA reactivity33; data consistent with the murine and human studies described above.
While these data indicate that loss of DNASE1L3 activity can drive a familial form of early onset SLE, the contribution of this nuclease to the pathogenesis of sporadic SLE was less clear. However, a recent study described anti-DNASE1L3 autoantibodies in ~50% of lupus nephritis patients, which inhibited serum nuclease activity resulting in increased poly-nucleosomal cell-free DNA (cfDNA) within circulating microparticles. Accumulation of these DNASE1L3-sensitive antigens correlated with higher autoantibody titers and lupus disease activity, highlighting a novel non-genetic mechanism by which alterations in DNASE1L3 function impact lupus pathogenesis34.
Predictably, additional nuclease enzymes evolved to limit pathogenic responses to endogenous nucleic acids. For example, lack of the secreted nuclease DNAse I (DNASE1) promotes lupus-like disease in both murine models and human subjects35,36. In addition, deletion of the endolysosome localized nucleases phospholipase D3 (PLD3), phospholipase D4 (PLD4), or DNAse II each result in severe murine autoinflammatory disease characterized by lethality in utero or early in life. Disease development in Pld3−/− and Pld4−/− animals is predictably driven by TLR9 signals, although autoinflammation in Dnase2−/− mice is dependent on activation of the cGAS–STING pathway37–39.
In summary, multiple independent exo- and endonucleases have evolved to prevent the otherwise inevitable activation of DNA sensors following normal cellular turnover. Surprisingly, similar null mutations in RNAse enzymes have yet to be linked to murine or human lupus pathogenesis. Although transgenic RNAse A overexpression protects against the development of TLR7-driven murine SLE40, this limited phenotype of RNAse enzyme knock-out strains suggests either functional redundancy across RNAse enzymes or the existence of alternate mechanisms to limit RNA accumulation. In keeping with this latter hypothesis, mutations in Slc29a3, a lysosomal transporter which traffics nucleoside from lysosomes to the cytoplasm, promotes TLR7-dependent histiocytosis in mice41. Although the predominant clinical phenotypes of human SLC29A3 deficiency include monogenic histiocytic disorders such as H syndrome and familial Rosai-Dorfman disease42,43, rather than SLE, an intronic polymorphism in SLC29A3 that limited monocyte expression was recently identified in an Asian lupus cohort44. Thus, rather than RNA degradation, nucleoside removal from the endolysosomal compartment may be the dominant mechanism limiting inadvertent TLR7 engagement.
2. Regulation of NA-sensing TLR expression across cell lineages
As an additional strategy to limit inadvertent autoimmune activation, the expression of NA-sensing TLRs is restricted to specific cell types. In contrast to broad expression of intracellular NA sensors designed to recognize direct cell infection (e.g. MDA5, MAVS, STING, RIG-I), high-level expression of NA-sensing TLRs is relatively restricted to plasmacytoid dendritic cells (pDCs) and B cells. Low level receptor expression is also observed on specific immune (including macrophages and myeloid DCs) and non-immune (keratinocytes, epithelial cells, hepatocytes) populations, with the potential for induction of TLR7 mRNA in response to inflammatory cytokines45. The evolutionary rationale underlying this arrangement is that TLR7 and TLR9 are designed to recognize NA from extracellular pathogens. Thus, expression is restricted to cell lineages designed for antigen uptake, either by phagocytosis, by Fc-receptor binding in pDCs, or by B cell receptor (BCR)-mediated recognition of specific antigen determinants by B cells.
An additional example of how regulated expression of NA-sensing TLRs prevents autoimmune activation includes the development of specific macrophages designed for silent clearance of apoptotic material. In a wide range of organs, the phenotype of tissue resident macrophages includes expression of receptors specific for apoptotic cells, together with low TLR9 expression and limited NA responsiveness46. The importance of this arrangement in maintaining homeostasis is supported by the observation that defects in apoptotic cell clearance are linked to lupus pathogenesis47.
Finally, even among TLR7/TLR9-expressing immune cells, expression levels are restrained as a mechanism to limit aberrant activation. In murine models, increased Tlr7 gene dose promotes lupus-like disease48–50. Indeed, Tlr7 over-expression can act in a B cell-intrinsic manner to drive SLE, as evidenced by competitive recruitment of TLR7-transgenic B cells into autoimmune germinal centers24, and reduced anti-RNA autoantibodies following Cre-mediated normalization of B cell TLR7 expression in a low-copy Tlr7 transgenic lupus-prone strain25. Moreover, an important contributor to female sex predominance of human SLE is likely X chromosome dose, based on the observation that disease risk is increased in males with Klinefelter syndrome (47,XXY) and Trisomy X (47,XXX) females51,52. Of the multiple X-linked genes potentially contributing to this observation, TLR7 evades X inactivation in immune cells, including both B cells and pDCs53. Strikingly, naïve B cells with biallelic TLR7 expression exhibit a selective advantage during TLR-driven activation, providing additional support for a B cell-intrinsic impact of TLR7 during lupus pathogenesis.
3. Spatial and temporal regulation of NA-sensing TLR expression
Another strategy limiting aberrant activation of NA-sensing TLRs by self-NA is their restricted expression within endosomal compartments. Following internalization and degradation of microbial antigens, pathogen-derived NA activate endosomal TLRs to promote robust immune responses. In addition to allowing pathogen recognition without direct cellular infection, this subcellular localization also sequesters NA-sensing TLRs away from extracellular apoptotic or necrotic material. However, engineering signaling-competent TLR9 on the cell surface results in lethal systemic inflammation54. These findings are consistent with a marked inflammatory potential of mislocalized NA sensors and suggest that specific mechanisms must have evolved to deliver NA-sensing TLRs to the appropriate subcellular compartments.
Targeting of NA-sensing TLRs to endosomal compartments by UNC93B1
Individual TLRs differ in their requirements for signaling initiation depending on their localization and ligand availability. The endosomal NA-sensing TLRs have a specific requirement of translocation to endolysosomal acidic compartment where they are cleaved and recognize ligands55,56. The TLR trafficking protein UNC93B1 binds several TLR families (including TLR3, TLR7, TLR9, TLR11, TLR12, TLR13) and controls their movement from the endoplasmic reticulum (ER) to the endolysosomal compartment where they are proteolytically processed to generate signaling-competent receptors. How UNC93B1 regulates this trafficking process has not been completely defined, although important differences exist between TLR family members. For example, UNC93B1 recruitment of adaptor protein complex 2 (AP-2) is required for TLR9 delivery to the endosome, whereas other TLRs use different UNC93B1-dependent trafficking pathways57. While UNC93B1 is not required for ligand binding or signaling, deficiencies in UNC93B1 binding leads to TLR trafficking defects and subsequent defective signaling. Consistent with these data, loss-of-function Unc93b1 mutations abrogate murine lupus and recessive mutations in human UNC93B1 have been identified in children with Herpes simplex virus-1 (HSV-1) encephalitis (HSE)58,59.
Beyond facilitating endosomal TLR trafficking, UNC93B1 also regulates endosomal TLR signaling and likely contributes to differential functional responses to TLR7 vs. TLR9 activation. For example, the missense mutation Unc93b1D34A prevents TLR9 binding, resulting in the preferential export and function of TLR7. In this setting, increased TLR7 signaling drives lethal systemic inflammation. Although the relative contribution of B cell and myeloid signals to this phenotype has not been addressed, it is notable that B cell depletion prevents pathogenic CD4+ T cell activation in Unc93b1D34A mice60. More recent data have further characterized differential regulation of TLR7 and TLR9 by UNC93B1. Whereas UNC93B1 targets both TLR7 and TLR9 to the endosome, TLR9 is released within the endosomal compartment and this dissociation is required for normal signaling61. In contrast, TLR7 remains bound to UNC93B1 within the endosome which allows an additional layer of regulation. Specifically, UNC93B1 promotes the interaction of TLR7 with Syntenin-1, which facilitates termination of signaling by trafficking the TLR7-UNC93B1 complex into multivesicular bodies. In keeping with this mechanism limiting dysregulated TLR7 activation, mice expressing mutant UNC93B1 unable to bind Syntenin-1 develop spontaneous, TLR7-dependent autoimmunity62. As with other Unc93b1-mutant murine models, the relative contribution of B cell vs. myeloid lineages to the autoimmune phenotype has not been addressed. However, B cells from Unc93b1PKP/PKP mice (unable to interact with Syntenin-1) exhibit increased responses to TLR7 ligands ex vivo, implicating dysregulated B cell activation in disease pathogenesis.
Regulation of TLR signaling by endolysosomal trafficking
In addition to correctly localizing NA-sensing TLRs within the appropriate endosomal compartment for signaling, additional trafficking events regulate the type of signals generated upon ligand binding. These events are mediated by a series of adaptor proteins, as well as engagement of a non-canonical autophagy pathway (Figure 2). Ligand binding to endosomal TLR7 and TLR9 leads to activation of transcription factors NFκB and IRF7, required for production of proinflammatory cytokines and Type I IFN, respectively. While the adaptor protein Myd88 coordinates both signaling pathways, studies in macrophages, plasmacytoid dendritic cells (pDCs), DCs, and B cells have indicated that NFκB and IRF7 activation occurs in distinct endosomal compartments. For example, stimulating pDCs with CpG DNA designed to localize to specific compartments uncovered distinct spatiotemporal regulation of TLR9-dependent IRF7 and NFκB activation63. Iwasaki and colleagues showed that the adaptor protein AP3 is a key regulator controlling this switch from NFκB to IRF7 activation in response to TLR9 engagement64. Mechanistically, AP-3 promotes the ordered transition of TLR9 though distinct endolysosomal stages, from early endosomes (marked by EEA1 or Vamp3 expression), to late endosomes, and finally endosomal fusion with LAMP2+ lysosomes. In this context, NFκB activation occurs within early endosomes (termed NFκB endosomes), while IRF7 is activated in late endosomal compartments (termed IRF7 endosomes). Whereas initial studies suggested that the order of NF-κB vs. IRF7 activation is reversed in plasmacytoid dendritic cells (pDC)63,65, subsequent evidence confirmed that TLR-dependent IRF7 signaling requires endolysosomal maturation and occurs after initial NF-κB activation64,66. Consistent with this model, AP3 deletion in mice or cell lines limits IRF7-dependent type 1 IFN production but promotes a parallel increase in NFκB-driven pro-inflammatory cytokines. This spatial regulation of IRF7 is maintained by a requirement for the adaptor TRAF3, such that modified TRAF3 able to localize to early endosomal compartment was sufficient to induce IRF7 signaling within early endosomes64. This location-specific signaling requirement is not specific for the endosomal NA-sensors TLR7 and TLR9, since even cell surface TLRs, such as TLR4, which activate NF-κB at the plasma membrane must relocate to endosomes for IRF3-dependent type 1 IFN production67,68. In keeping with this regulatory framework, AP-3 activity is also required for TLR4 trafficking to endosomes and induction of type 1 IFN64.
Figure 2: A B cell-intrinsic non-canonical autophagy pathway promotes endolysosomal flux to regulate TLR signaling.
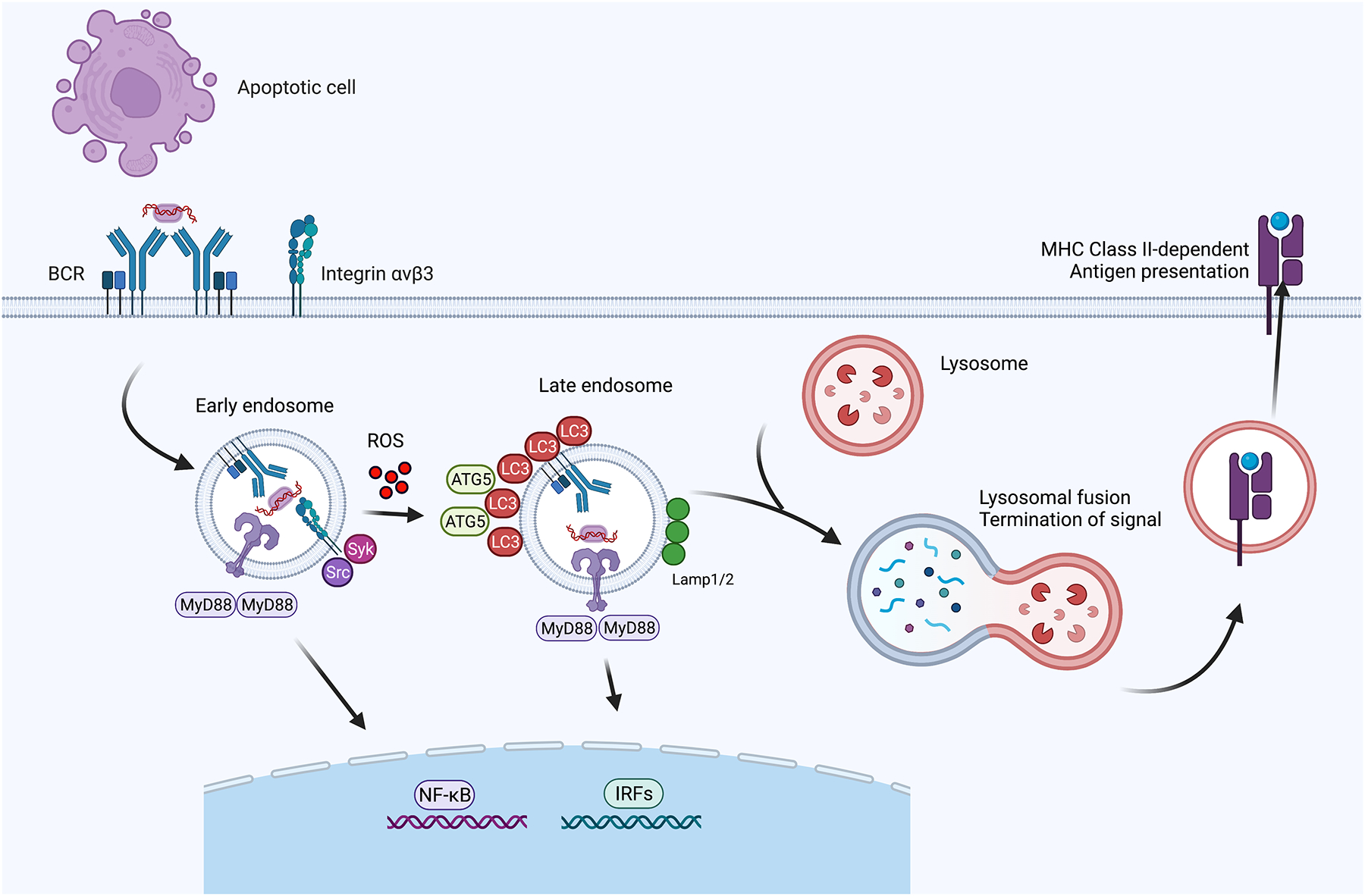
Following recognition of apoptotic cells, self-reactive BCRs traffic NA-containing particles to early endosomes, initiating TLR7/TLR9-induced NF-κB activation. TLR signaling also triggers integrin activation and internalization leading to phosphorylation of Src and Syk kinases. The activation of these kinases causes ATG5-dependent lipidation of LC3 through a mechanism requiring ROS production. Subsequently, LC3-II recruitment causes transition of the TLR containing compartment into a late endosomal compartment permissive for IRF7 activation and abolishes NF-κB signals. Ultimately, lysosomal fusion terminates TLR signals, in addition to facilitating antigen degradation and MHC Class II-dependent antigen presentation. Studies in myeloid cells have revealed additional details in this process such as adaptor proteins involved in transition of signaling.
Non-canonical autophagy regulates TLR-dependent endolysosomal trafficking
In addition to AP-3, more recent studies have identified an important role for autophagy proteins in regulating endolysosomal trafficking. Classical autophagy (also termed macroautophagy) is the physiologic process whereby cellular components are degraded during stress and nutrient starvation. In this “canonical” autophagy pathway, ATG proteins orchestrate the formation of a double-membrane “autophagosome” containing cellular debris and lipidation of the ubiquitin-like protein LC3 which is recruited to autophagosomes. Subsequently, lipidated LC3-I (termed LC3-II) recruitment promotes lysosomal fusion and the degradation of cellular constituents69,70. Importantly, key components of the autophagy machinery also impact other intracellular processes such as endosomal TLR signaling without requiring autophagosome formation. In pDCs, IRF7 activation occurs in compartments positive for autophagy proteins such as LC3, such that type 1 IFN production depends on the recruitment of autophagy proteins71–73.
This non-canonical form of autophagy, which is termed as LC3 associated phagocytosis (LAP) in phagocytic cells, has specific relevance to the pathogenesis of SLE71,74. The ingestion of extracellular pathogens by phagocytic cells and the engagement of pathogen-recognition receptors (e.g. TLRs) results in the recruitment of lipidated LC3-II to phagosomes, resulting in lysosomal fusion and the degradation of ingested pathogens75. Importantly, in addition to pathogen defense, LAP is also critical for efferocytosis, the immunologically silent clearance of dead/dying cells by phagocytes47,76,77. In keeping with this model, Martinez et al. demonstrated that mice deficient in non-canonical LAP autophagy components, but not canonical autophagy-specific genes, develop spontaneous lupus characterized by class-switched antinuclear antibodies and lupus nephritis78. Mechanistically, apoptotic cells are appropriately taken up by myeloid cells from LAP component-deficient mice, but efficient degradation of engulfed material is perturbed, resulting in the production of pro-inflammatory cytokines. Repeated injections of dying cells accelerated lupus-like disease in these animals, supporting dysregulated efferocytosis as the driver of disease development. These combined studies highlight how distinct TLR signaling programs are induced from separate subcellular compartments, with the regulation of endosomal trafficking by adaptor proteins and autophagy components regulating this temporal switch. As we will describe in detail below, disruption of non-canonical autophagy can also result in B cell-intrinsic dysregulation of endosomal TLRs resulting in breaks in immune tolerance.
B cell-intrinsic regulation of TLR signaling thresholds by endolysosomal flux
Germline deletion of several autophagy components results in embryonic or peri-natal lethality. For this reason, Martinez et al. used LysM-Cre mice to conditionally ablate relevant non-canonical autophagy genes in macrophages, monocytes, and DC subsets78. Thus, by definition, the observed lupus-like features in LAP-deficient animals are attributed to myeloid-specific dysregulation of TLR signaling. However, since regulation of B cell TLR signaling is critical to maintain immune tolerance, we hypothesized that endolysosomal trafficking exerts a parallel B cell-specific impact on autoimmune risk.
We recently identified an important role for a family of integrins and autophagy proteins in processing of B cell TLR signals. Integrins are heterodimeric membrane proteins that regulate multiple immune functions, including cell adhesion and migration, by linking the cytoskeleton with extracellular cues. Less well appreciated, is the fact that integrins can also modulate intracellular trafficking events, which prompted us to test whether a specific integrin heterodimer αvβ3 from the αv integrin family regulates B cell activation. Notably, B cells deficient in either αv or β3 subunits exhibit increased TLR responses in vitro and in vivo (Figure 3). Dissecting the underlying mechanisms, we observed that in response to CpG stimulation, αvβ3 traffics to early endosomes together with TLR9, where it promotes Src/Syk kinase activation and production of reactive oxygen species (ROS). This promotes Atg5 activation and the delivery of lipidated LC3 to TLR-containing endosomes; events which ultimately facilitate endolysosomal trafficking, a switch from NFκB to IRF7 activation, and subsequently endosome-lysosome fusion and the termination of TLR signaling79. As predicted by earlier studies, B cells lacking αv or the autophagy components LC3 and Atg5 manifested dysregulated endosomal trafficking, with αv-null B cells exhibiting increased NFκB and delayed IRF7 signals, and LC3- or Atg5-deficient B cells expressing increased TLR-driven NFκB and absent IRF7 induction. Thus, in addition to temporally controlling NF-κB vs. IRF7 activation, the rate of endolysosomal flux also regulates the duration and intensity of TLR signaling. These findings were strikingly reminiscent of earlier experiments using myeloid cells, with the important insight that this ordered trafficking of TLRs through endolysosomal compartments via the autophagy proteins serves to both regulate TLR signaling thresholds and facilitates termination of TLR signaling. After trafficking to late endosomes, lysosomal fusion promotes the degradation of internalized cargo and ultimately terminates TLR signaling.
Figure 3: Impact of non-canonical autophagy gene variants on endosomal TLR signals.
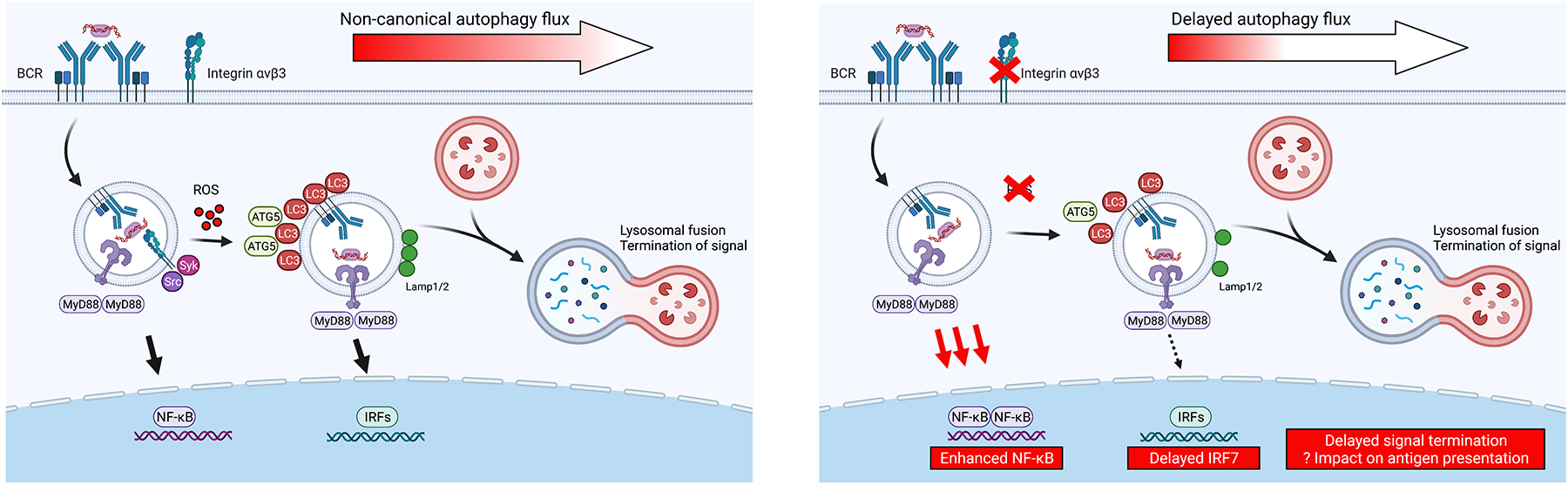
We propose a model in which variants in specific genes, such as αvβ3 integrin or non-canonical autophagy components (ATG5, ATG7, NCF1 and NCF2), result in delayed endolysosomal trafficking and consequently enhanced TLR-dependent NF-κB activation and delayed termination of TLR signaling. Left panel shows normal physiological context in which response to TLR ligands and associated antigens is limited through the integrin autophagy pathway. Right panel shows impact of lack of αvβ3 integrin or reduced NADPH oxidase-dependent ROS production which may drive enhanced NF-κB/MAPK activation and prolong TLR signals, ultimately impacting B cell proliferation, affinity maturation, and plasma cell differentiation. It remains to be determined how these changes in TLR signaling and lysosomal fusion affect processing and presentation of antigens by B cells.
To determine the functional significance of these observations, we quantified TLR-enhanced humoral responses in B cell αv-deficient mice. While TLR signals activate both myeloid and B cell lineages, the relative contribution of each cell type to TLR-enhanced antibody (Ab) titers depends on the physical context of antigen and adjuvant. Specifically, when soluble protein antigen is chemically linked to TLR agonist, DC-specific TLR signals facilitate increased Ab responses. In contrast, immunization with virus-like particles (VLP) or inactivated influenza virus (where TLR activating nucleic acid is incorporated within viral capsid structures) promotes robust B cell activation and GC responses that is dependent on B cell-intrinsic Myd88 expression3. For this reason, we immunized B cell-intrinsic αv-deficient mice with ssRNA-containing Qβ-VLP. Notably, lack of B cell αv integrin resulted in a prominent increase in anti-VLP Ab titers, GC B cells, GC B cell affinity maturation and somatic hypermutation, and the expansion of memory B cells and long-lived plasma cells80. Despite the potential for integrin-mediated extracellular matrix interactions altering B cell migration and trafficking within the GC, we attribute the bulk of these phenotypes to enhanced endosomal TLR signaling. Evidence in support of this hypothesis includes unaffected GC responses after TLR-independent protein immunization in B cell αv-null mice (indicating no major impact of αv integrin on humoral immunity) and similar delayed endosomal trafficking, enhanced TLR signals, and increased GCs in mice deficient in other autophagy genes80. Strikingly, enhanced GC responses in B cell αv-deficient mice were sufficient to protect mice from live influenza challenge, raising the possibility that genetic polymorphisms in autophagy genes might have been selected during evolution.
Previous studies have demonstrated that the autophagy machinery traffics BCR to the endosome following IgM stimulation81, and a switch from canonical to non-canonical autophagy in activated GC B cells regulates B cell differentiation and cell fate82. However, our studies highlight a new role for integrins and autophagy genes in limiting B cell responses following TLR engagement. We predict that this pathway evolved as a tolerance mechanism to prevent excessive B cell responses to NA-containing self-antigens. Testing this hypothesis in vivo is complicated by the requirement for macroautophagy in plasma cell differentiation83,84, confounding assessment of autoantibody titers in lupus-prone mice. However, in support of a direct role for non-canonical autophagy in regulating B cell tolerance, we recently reported accelerated systemic autoimmunity in lupus-prone mice following B cell-specific αv deletion85.
The impact of adaptor proteins and autophagy components on endolysosomal flux is not limited to the strength and duration of TLR signaling. Rather, these specialized intracellular compartments are also intimately linked with antigen presentation via endolysosomal acidification, antigen degradation, and the recruitment of antigen processing machinery. The overall outcome for B cell uptake of NA-containing autoantigens thus depends on the connection between these two processes. For example, elevated TLR signals following B cell-intrinsic αv deletion promotes B cell activation but it is unclear how loss of αv-mediated intracellular trafficking affects antigen presentation to T cells86 . Similarly, AP-3 deficiency, which results in defective Type I IFN production and enhanced pro-inflammatory cytokine secretion, also affects antigen presentation to cognate T cells making it difficult to delineate its exact role in context of autoimmunity87. Further understanding of these intracellular trafficking events and their connection with antigen processing machinery will be important to understand how aberrant B cell activation promotes the development of autoimmunity.
Genetic defects in several autophagy components, including ATG5, ATG7, NCF1 and NCF2, have been linked with the development of human SLE88–92. While studies by our group and others implicate non-canonical autophagy regulation of endolysosomal flux as the mechanism underlying lupus development, whether these variants act in a B cell-intrinsic manner has not been addressed. We speculate that genetic modulation of B cell endolysosomal trafficking is an underappreciated contributor to lupus risk. As one specific example, gene variants in the phagocytic NADPH oxidase (NOX2) complex are linked to the pathogenesis of SLE and other humoral autoimmune diseases. Specifically, missense variants in NCF1 (Arg90His; encoding p47phox NOX2) and NCF2 (His389Gln; encoding NOX2 p67phox), NCF1 copy number variation, and CYBB haploinsufficiency in mothers of boys with X-linked chronic granulomatous disease (CGD), all result in reduced ROS production and are associated with an increased risk of SLE91,93–98. While many core autophagy components are shared between canonical and non-canonical autophagy, activation of the phagocytic NADPH oxidase (NOX2) complex specifically enhances LC3-associated phagocytosis (LAP)74,99. A likely mechanism underlying this observation is NOX2 dependent activation of LC3-associated phagocytosis (LAP) and the resulting attenuation of endosomal TLR signaling. Indeed NOX2-deficient cells are unable to undergo LC3-associated phagocytosis (LAP) and NOX2 family gene deletion accelerates autoimmunity in independent lupus-prone strains and aged C57BL/6 mice74,78,99–101. In addition, a recent study demonstrated that a murine knock-in model expressing the human NCF1 Arg90His lupus risk allele exhibits increased pristane-induced lupus102. Bone marrow-derived macrophages (BMDM) from Ncf1Arg90His knock-in mice exhibit reduced phagosomal acidification and maturation following apoptotic cell ingestion, linking the NCF1 Arg90His variant with defects in the immunologically silent clearance of dead/dying cells by phagocytes47.
Although defects in macrophage efferocytosis likely increases exposure to NA-containing autoantigens, we predict that reduced NOX2 activity also promotes lupus pathogenesis in a B cell-intrinsic manner. In support of this hypothesis, Nox2−/−.MRL.Faslpr mice exhibit a shift towards a speckled ANA pattern and increased anti-ribonucleoprotein (RNP) autoAb titers that parallels exacerbated autoimmunity100. Given the critical role for B cell TLRs signals in regulating the ANA repertoire, these data suggest that parallel myeloid- and B cell-specific mechanisms might underlie the striking increase in lupus risk among NCF1 and NCF2 variant carriers; an important topic for future research.
4. Modulation of signals downstream of NA-sensing TLRs
Genome wide association studies (GWAS) have identified >100 polymorphisms impacting the risk of developing SLE or other humoral autoimmune diseases. Although our understanding of how individual genetic variants contribute to disease risk remains poor, risk polymorphisms frequently cluster within specific immune pathways. In this context, an increasing number of lupus-associated genetic variants have been identified within endosomal TLR signaling pathways. These include TLR7 itself103,104; IRAK1 (within the Myddosome complex)90; TNFAIP3, TNIP1 and UBE2L3 (downstream of NF-κB activation)90,105; and IRF5 and IRF790,106. A detailed description of each of these variants and their contribution to lupus risk is beyond the scope of this review. However, recent mechanistic insights into the biology of lupus risk genes SLC15A4 and CXorf21 are illustrative of how the functional regulation of endolysosomal trafficking and endosomal TLR signaling contributes to lupus pathogenesis107,108. Using interaction proteomics, Heinz et al. demonstrated that the lysosomal proton channel SLC15A4 interacts with the CXorf21-encoded protein they name “TLR adaptor interacting with SLC15A4 on the lysosome” (TASL)109. Genetic disruption of either SLC15A4 or TASL expression exerted no major impact on TLR-dependent NF-κB activation but disrupted IRF-driven transcripts, indicating that the SLC15A4/TASL complex functions downstream of initial endosomal TLR activation. Additional biochemical analyses demonstrated that TASL selectively promotes IRF5 signaling via a functional pLxIS motif in a manner analogous to IRF3 binding the adaptors STING, MAVS and TRIF110.
The structural interaction with SLC15A4 maintains TASL protein levels and the CXorf21 lupus risk haplotype results in increased expression levels107,109. Moreover, CXorf21 is an X-linked gene that escapes X inactivation suggesting that regulation of TASL protein expression might be an additional driver of the female sex bias in SLE107. However, in addition to regulating IRF5 phosphorylation, TASL likely imparts biologically significant impacts on SLC15A4 function. As a lysosomal protein transporter, SLC15A4 promotes the endolysosomal acidification which is required for endosomal TLR7 signaling111. Interestingly, TASL promotes SLC15A4-dependent acidification based on the observation that B cells from healthy females exhibit lower endosomal pH relatively to healthy males, with this pH reduction being dependent on CXorf21 expression112. The mechanism by which TASL modulates endolysosomal pH and how changes in endolysosomal acidification impacts TLR signals and lupus pathogenesis remains to be addressed. Moreover, whether genetic regulation of the SLC15A4/TASL complex facilitates breaks in tolerance via modulation of B cell or myeloid endosomal TLR signaling has not been studied. Ultimately, these recent data indicate that the lupus risk genes SLC15A4, CXorf21, and IRF5 functionally converge within the endolysosomal TLR pathway; findings which highlight how genetic modulation of downstream signaling cascades serves to modulate TLR thresholds and drive lupus risk.
Conclusions and remaining questions
Toll-like receptors are a family of evolutionarily related receptors that induce innate and adaptive immunity via the recognition of pathogen-associated molecular patterns (PAMPs). In contrast with pathogen-specific molecules (such as the TLR5 agonist flagellin), NA-sensing TLRs are not specific for foreign NA and carry the risk of autoimmune activation. For this reason, the immune system has evolved overlapping strategies to prevent pathologic activation of NA-sensing TLRs. In this review, we have highlighted how these protective mechanisms can be divided into two broad categories. First, strategies aimed at reducing the likelihood that self-NA will engage NA-sensing TLRs, including production of nucleases and the lineage-specific regulation of TLR expression. Second, mechanisms to downregulate TLR signaling, such has the induction of endolysosomal trafficking to extinguish TLR activation and the modulation of downstream signaling cascades. While the majority of studies delineating these processes have focused on myeloid cells, we have sought to highlight emerging data implicating B cell-intrinsic contributions to the risk of humoral autoimmunity.
Despite these new insights, several open questions remain which we have noted throughout this review. First, whether each regulatory strategy is similarly adopted by different immune lineages remains unclear. Our work has focused predominantly on B cell-specific regulatory mechanisms, but it is likely that myeloid cells and B cells use distinct strategies to limit aberrant activation by self-NA. Second, the contradictory effect of TLR7 vs. TLR9 deletion on lupus pathogenesis remains enigmatic. Although TLR7 and TLR9 share a requirement for UNC93B1 binding to facilitate appropriate trafficking to the endosomal compartment, recent data from the Barton group highlights that these receptors differ with respect to structural interactions with UNC93B1, need for UNC93B1 release to allow signaling, and the role for UNC93B1 in extinguishing activation signals61,62. These findings suggest a more fundamental difference in responses to TLR7 vs. TLR9 engagement than had previously been assumed. Importantly, work from our lab and others using B cell-intrinsic deletion or over-expression of TLR7/TLR9 suggests that B cells should be the primary focus of future investigations in this arena23–26.
Finally, it will be important to address how NA-sensing TLR signaling and endolysosomal trafficking impacts antigen presentation. The ability of B cells to acquire, process and present antigens is essential not only for receiving T cell help, but also for the expansion and maintenance of antigen-specific T cells. Indeed, studies using murine lupus models have identified a critical role for B cell antigen presentation and costimulatory signals in driving breaks CD4+ T cell tolerance113–115. Studies using DCs have shown that the rate and route of endosomal trafficking of phagocytic products are critical steps in determining both the type of signaling response as well as the efficiency of antigen presentation. However, little is known about these processes in B cells. For example, do B cells differ in their ability to extract and degrade specific (auto)antigens and does the rate of antigen processing impact the capacity for presentation to T cells? Moreover, does induction of endosomal TLR signals impact this process such that modulation of endolysosomal flux either enhances or perturbs the presentation of self-ligands? Ultimately, a more detailed understanding of these molecular events promises to both inform lupus immunopathogenesis and uncover new therapeutic targets for the treatment of human SLE.
Acknowledgements:
This work was supported by the National Institutes of Health under award numbers: 5R01AI151167 (MA), R03AI139716 (SWJ), R01AR073938 (SWJ), R01AR075813 (SWJ)). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support provided by the the Arthritis National Research Foundation (ANRF) Eng Tan Scholar Award (SWJ); and by Lupus Research Alliance, Novel Research Grants (MA and SWJ). Figures were created with BioRender.com. The authors declare that no conflict of interest exists.
References:
- 1.World Health Organization Weekly epidemiological update on COVID-19 – 2 November 2021. In. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---2-november-2021.
- 2.The pandemic’s true death toll. In. The Economist 2021. https://www.economist.com/graphic-detail/coronavirus-excess-deaths-estimates. [Google Scholar]
- 3.Hou B, Saudan P, Ott G, et al. Selective utilization of Toll-like receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity. 2011;34(3):375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holborow EJ, Weir DM, Johnson GD. A serum factor in lupus erythematosus with affinity for tissue nuclei. Br Med J. 1957;2(5047):732–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416(6881):603–607. [DOI] [PubMed] [Google Scholar]
- 6.Shlomchik MJ. Activating systemic autoimmunity: B’s, T’s, and tolls. Current opinion in immunology. 2009;21(6):626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–1533. [DOI] [PubMed] [Google Scholar]
- 8.Gregersen PK, Behrens TW. Genetics of autoimmune diseases--disorders of immune homeostasis. Nat Rev Genet. 2006;7(12):917–928. [DOI] [PubMed] [Google Scholar]
- 9.Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, et al. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med. 2011;208(10):2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groom JR, Fletcher CA, Walters SN, et al. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. The Journal of experimental medicine. 2007;204(8):1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38(3):528–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua Z, Gross AJ, Lamagna C, et al. Requirement for MyD88 Signaling in B Cells and Dendritic Cells for Germinal Center Anti-Nuclear Antibody Production in Lyn-Deficient Mice. J Immunol. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu R, Munroe ME, Guthridge JM, et al. Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies. J Autoimmun. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munroe ME, Lu R, Zhao YD, et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis. 2016;75(11):2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isenberg DA, Manson JJ, Ehrenstein MR, Rahman A. Fifty years of anti-ds DNA antibodies: are we approaching journey’s end? Rheumatology (Oxford). 2007;46(7):1052–1056. [DOI] [PubMed] [Google Scholar]
- 16.ter Borg EJ, Horst G, Hummel EJ, Limburg PC, Kallenberg CG. Measurement of increases in anti-double-stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. A long-term, prospective study. Arthritis and rheumatism. 1990;33(5):634–643. [DOI] [PubMed] [Google Scholar]
- 17.Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol. 2017;17(7):421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–428. [DOI] [PubMed] [Google Scholar]
- 19.Soni C, Wong EB, Domeier PP, et al. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol. 2014;193(9):4400–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santiago-Raber ML, Dunand-Sauthier I, Wu T, et al. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2010;34(4):339–348. [DOI] [PubMed] [Google Scholar]
- 21.Fairhurst AM, Hwang SH, Wang A, et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol. 2008;38(7):1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nickerson KM, Christensen SR, Shupe J, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184(4):1840–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson SW, Scharping NE, Kolhatkar NS, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol. 2014;192(10):4525–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh ER, Pisitkun P, Voynova E, et al. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell-mediated autoimmunity. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(40):16276–16281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang SH, Lee H, Yamamoto M, et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. Journal of immunology. 2012;189(12):5786–5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tilstra JS, John S, Gordon RA, et al. B cell-intrinsic TLR9 expression is protective in murine lupus. J Clin Invest. 2020;130(6):3172–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Mayouf SM, Sunker A, Abdwani R, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43(12):1186–1188. [DOI] [PubMed] [Google Scholar]
- 28.Ozcakar ZB, Foster J 2nd, Diaz-Horta O, et al. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum. 2013;65(8):2183–2189. [DOI] [PubMed] [Google Scholar]
- 29.Zochling J, Newell F, Charlesworth JC, et al. An Immunochip-based interrogation of scleroderma susceptibility variants identifies a novel association at DNASE1L3. Arthritis Res Ther. 2014;16(5):438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acosta-Herrera M, Kerick M, Gonzalez-Serna D, et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann Rheum Dis. 2019;78(3):311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sisirak V, Sally B, D’Agati V, et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell. 2016;166(1):88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weisenburger T, von Neubeck B, Schneider A, et al. Epistatic Interactions Between Mutations of Deoxyribonuclease 1-Like 3 and the Inhibitory Fc Gamma Receptor IIB Result in Very Early and Massive Autoantibodies Against Double-Stranded DNA. Front Immunol. 2018;9:1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soni C, Perez OA, Voss WN, et al. Plasmacytoid Dendritic Cells and Type I Interferon Promote Extrafollicular B Cell Responses to Extracellular Self-DNA. Immunity. 2020;52(6):1022–1038 e1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartl J, Serpas L, Wang Y, et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J Exp Med. 2021;218(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25(2):177–181. [DOI] [PubMed] [Google Scholar]
- 36.Yasutomo K, Horiuchi T, Kagami S, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. 2001;28(4):313–314. [DOI] [PubMed] [Google Scholar]
- 37.Gavin AL, Huang D, Huber C, et al. PLD3 and PLD4 are single-stranded acid exonucleases that regulate endosomal nucleic-acid sensing. Nat Immunol. 2018;19(9):942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109(47):19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6(1):49–56. [DOI] [PubMed] [Google Scholar]
- 40.Sun X, Wiedeman A, Agrawal N, et al. Increased ribonuclease expression reduces inflammation and prolongs survival in TLR7 transgenic mice. J Immunol. 2013;190(6):2536–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibata T, Taoka M, Saitoh S-I, et al. Nucleosides drive histiocytosis in SLC29A3 disorders by activating TLR7. bioRxiv. 2019:2019.2012.2016.877357. [Google Scholar]
- 42.Morgan NV, Morris MR, Cangul H, et al. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 2010;6(2):e1000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Priya TP, Philip N, Molho-Pessach V, Busa T, Dalal A, Zlotogorski A. H syndrome: novel and recurrent mutations in SLC29A3. Br J Dermatol. 2010;162(5):1132–1134. [DOI] [PubMed] [Google Scholar]
- 44.Wang YF, Wei W, Tangtanatakul P, et al. Multi-ancestral GWAS identifies shared and Asian-specific loci for SLE and links type III interferon signaling and lysosomal function to the disease. Arthritis Rheumatol. 2021. [DOI] [PubMed] [Google Scholar]
- 45.Petes C, Odoardi N, Gee K. The Toll for Trafficking: Toll-Like Receptor 7 Delivery to the Endosome. Front Immunol. 2017;8:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity. 2017;47(5):913–927 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng Y, Martin DA, Kenkel J, Zhang K, Ogden CA, Elkon KB. Innate and adaptive immune response to apoptotic cells. J Autoimmun. 2007;29(4):303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deane JA, Pisitkun P, Barrett RS, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27(5):801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312(5780):1669–1672. [DOI] [PubMed] [Google Scholar]
- 50.Subramanian S, Tus K, Li QZ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103(26):9970–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu K, Kurien BT, Zimmerman SL, et al. X Chromosome Dose and Sex Bias in Autoimmune Diseases: Increased Prevalence of 47,XXX in Systemic Lupus Erythematosus and Sjogren’s Syndrome. Arthritis Rheumatol. 2016;68(5):1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scofield RH, Bruner GR, Namjou B, et al. Klinefelter’s syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008;58(8):2511–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Souyris M, Cenac C, Azar P, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol. 2018;3(19). [DOI] [PubMed] [Google Scholar]
- 54.Mouchess ML, Arpaia N, Souza G, et al. Transmembrane mutations in Toll-like receptor 9 bypass the requirement for ectodomain proteolysis and induce fatal inflammation. Immunity. 2011;35(5):721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ewald SE, Lee BL, Lau L, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456(7222):658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Latz E, Schoenemeyer A, Visintin A, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5(2):190–198. [DOI] [PubMed] [Google Scholar]
- 57.Lee BL, Moon JE, Shu JH, et al. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife. 2013;2:e00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casrouge A, Zhang SY, Eidenschenk C, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314(5797):308–312. [DOI] [PubMed] [Google Scholar]
- 59.Kono DH, Haraldsson MK, Lawson BR, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A. 2009;106(29):12061–12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fukui R, Saitoh S, Kanno A, et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity. 2011;35(1):69–81. [DOI] [PubMed] [Google Scholar]
- 61.Majer O, Liu B, Woo BJ, Kreuk LSM, Van Dis E, Barton GM. Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature. 2019;575(7782):371–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Majer O, Liu B, Kreuk LSM, Krogan N, Barton GM. UNC93B1 recruits syntenin-1 to dampen TLR7 signalling and prevent autoimmunity. Nature. 2019;575(7782):366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Honda K, Ohba Y, Yanai H, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434(7036):1035–1040. [DOI] [PubMed] [Google Scholar]
- 64.Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science. 2010;329(5998):1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guiducci C, Ott G, Chan JH, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. The Journal of experimental medicine. 2006;203(8):1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gotoh K, Tanaka Y, Nishikimi A, et al. Selective control of type I IFN induction by the Rac activator DOCK2 during TLR-mediated plasmacytoid dendritic cell activation. The Journal of experimental medicine. 2010;207(4):721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Husebye H, Halaas O, Stenmark H, et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25(4):683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9(4):361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuballa P, Nolte WM, Castoreno AB, Xavier RJ. Autophagy and the immune system. Annu Rev Immunol. 2012;30:611–646. [DOI] [PubMed] [Google Scholar]
- 70.Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell. 2019;176(1–2):11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Henault J, Martinez J, Riggs JM, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315(5817):1398–1401. [DOI] [PubMed] [Google Scholar]
- 73.Hayashi K, Taura M, Iwasaki A. The interaction between IKKalpha and LC3 promotes type I interferon production through the TLR9-containing LAPosome. Sci Signal. 2018;11(528). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martinez J, Malireddi RK, Lu Q, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17(7):893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 75.Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. LC3-Associated Phagocytosis and Inflammation. J Mol Biol. 2017;429(23):3561–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101(4):890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21(5):643–653. [DOI] [PubMed] [Google Scholar]
- 78.Martinez J, Cunha LD, Park S, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 79.Acharya M, Sokolovska A, Tam JM, et al. alphav Integrins combine with LC3 and atg5 to regulate Toll-like receptor signalling in B cells. Nat Commun. 2016;7:10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raso F, Sagadiev S, Du S, et al. alphav Integrins regulate germinal center B cell responses through noncanonical autophagy. J Clin Invest. 2018;128(9):4163–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28(6):799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martinez-Martin N, Maldonado P, Gasparrini F, et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science. 2017;355(6325):641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Conway KL, Kuballa P, Khor B, et al. ATG5 regulates plasma cell differentiation. Autophagy. 2013;9(4):528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pengo N, Scolari M, Oliva L, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. 2013;14(3):298–305. [DOI] [PubMed] [Google Scholar]
- 85.Acharya M, Raso F, Sagadiev S, et al. B Cell alphav Integrins Regulate TLR-Driven Autoimmunity. J Immunol. 2020;205(7):1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Munz C Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol Rev. 2016;272(1):17–27. [DOI] [PubMed] [Google Scholar]
- 87.Mantegazza AR, Guttentag SH, El-Benna J, et al. Adaptor protein-3 in dendritic cells facilitates phagosomal toll-like receptor signaling and antigen presentation to CD4(+) T cells. Immunity. 2012;36(5):782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Salloum R, Franek BS, Kariuki SN, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum. 2010;62(2):553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sigurdsson S, Nordmark G, Goring HH, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76(3):528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun C, Molineros JE, Looger LL, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. 2016;48(3):323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao J, Ma J, Deng Y, et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet. 2017;49(3):433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou XJ, Lu XL, Lv JC, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70(7):1330–1337. [DOI] [PubMed] [Google Scholar]
- 93.Yu B, Chen Y, Wu Q, et al. The association between single-nucleotide polymorphisms of NCF2 and systemic lupus erythematosus in Chinese mainland population. Clin Rheumatol. 2011;30(4):521–527. [DOI] [PubMed] [Google Scholar]
- 94.Alarcon-Riquelme ME, Ziegler JT, Molineros J, et al. Genome-Wide Association Study in an Amerindian Ancestry Population Reveals Novel Systemic Lupus Erythematosus Risk Loci and the Role of European Admixture. Arthritis Rheumatol. 2016;68(4):932–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim-Howard X, Sun C, Molineros JE, et al. Allelic heterogeneity in NCF2 associated with systemic lupus erythematosus (SLE) susceptibility across four ethnic populations. Hum Mol Genet. 2014;23(6):1656–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jacob CO, Eisenstein M, Dinauer MC, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A. 2012;109(2):E59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Olsson LM, Johansson AC, Gullstrand B, et al. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann Rheum Dis. 2017;76(9):1607–1613. [DOI] [PubMed] [Google Scholar]
- 98.Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148(1):79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang J, Canadien V, Lam GY, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106(15):6226–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. 2012;4(157):157ra141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jacob CO, Yu N, Yoo DG, et al. Haploinsufficiency of NADPH Oxidase Subunit Neutrophil Cytosolic Factor 2 Is Sufficient to Accelerate Full-Blown Lupus in NZM 2328 Mice. Arthritis Rheumatol. 2017;69(8):1647–1660. [DOI] [PubMed] [Google Scholar]
- 102.Geng L, Zhao J, Deng Y, et al. Human SLE variant NCF1-R90H promotes kidney damage and murine lupus through enhanced Tfh2 responses induced by defective efferocytosis of macrophages. Ann Rheum Dis. 2021. [DOI] [PubMed] [Google Scholar]
- 103.Shen N, Fu Q, Deng Y, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15838–15843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deng Y, Zhao J, Sakurai D, et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS genetics. 2013;9(2):e1003336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Adrianto I, Wen F, Templeton A, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. 2011;43(3):253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Deng Y, Tsao BP. Updates in Lupus Genetics. Curr Rheumatol Rep. 2017;19(11):68. [DOI] [PubMed] [Google Scholar]
- 107.Odhams CA, Roberts AL, Vester SK, et al. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in Systemic Lupus Erythematosus. Nat Commun. 2019;10(1):2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bentham J, Morris DL, Graham DSC, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47(12):1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Heinz LX, Lee J, Kapoor U, et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7–9. Nature. 2020;581(7808):316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347(6227):aaa2630. [DOI] [PubMed] [Google Scholar]
- 111.Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity. 2014;41(3):375–388. [DOI] [PubMed] [Google Scholar]
- 112.Harris VM, Harley ITW, Kurien BT, Koelsch KA, Scofield RH. Lysosomal pH Is Regulated in a Sex Dependent Manner in Immune Cells Expressing CXorf21. Front Immunol. 2019;10:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chiang K, Largent AD, Arkatkar T, et al. Cutting Edge: A Threshold of B Cell Costimulatory Signals Is Required for Spontaneous Germinal Center Formation in Autoimmunity. J Immunol. 2021;207(9):2217–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jackson SW, Jacobs HM, Arkatkar T, et al. B cell IFN-gamma receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med. 2016;213(5):733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Giles JR, Kashgarian M, Koni PA, Shlomchik MJ. B Cell-Specific MHC Class II Deletion Reveals Multiple Nonredundant Roles for B Cell Antigen Presentation in Murine Lupus. J Immunol. 2015;195(6):2571–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]