

Summary
At least 20% of B cells in the periphery express an antigen receptor with a degree of self-reactivity. If activated, these autoreactive B cells pose a risk as they can contribute to the development of autoimmune diseases. To prevent their activation, both B cell-intrinsic and extrinsic tolerance mechanisms are in place in healthy individuals. In this review article, I will focus on B cell-intrinsic mechanisms that prevent the activation of autoreactive B cells in the periphery. I will discuss how inhibitory signaling circuits are established in autoreactive B cells, focusing on the Lyn-SHIP-1-SHP-1 axis, how they contribute to peripheral immune tolerance, and how disruptions of these circuits can contribute to the development of autoimmunity.
Keywords: B cells, Tolerance/Suppression/Anergy, Signal Transduction, Protein Kinases/Phosphatases, Autoimmunity
Introduction
Autoreactive B cells play a significant role in autoimmune disease. They can produce autoantibodies that can wreak havoc and act as antigen-presenting cells and cytokine-producing cells that initiate and influence autoimmune responses (1, 2). To prevent unwanted activation, autoreactive B cells must be controlled by tolerance mechanisms. Autoreactive B cells are generated during B cell development. Up to 75% of newly formed B cells have detectable autoreactivity (3). They can also arise during immune responses due to somatic hypermutation (4). The B cell tolerance mechanisms in place to control these autoreactive B cells fall into two categories: mechanisms that eliminate autoreactive B cells and mechanisms that prevent their activation and subsequent immune response.
Tolerance mechanisms that eliminate autoreactive B cells include central tolerance mechanisms such as receptor editing and clonal deletion (5, 6). Immature autoreactive B cells that receive antigen receptor signals above a certain threshold are halted in their development, due to high avidity interactions with self-antigen. Such self-antigens will often be expressed on the cell surface of cells in the body, increasing the avidity of the interaction. Halted B cells will rearrange and express new light chains to change the specificity of their B cell receptor (BCR) away from self-reactivity. If successful, the B cell will resume B cell development towards maturity. If unsuccessful, the B cells will remain self-reactive and undergo apoptosis if bound strongly to self-antigen (clonal deletion). These processes purge the repertoire of a significant portion of autoreactive B cells, causing a drop in the frequency of autoreactivity from ~ 75% to ~40% of B cells (3). Maturing B cells that leave the bone marrow (transitional B cells/new emigrants) have an increased sensitivity to antigen receptor engagement and resultant activation-induced death (7, 8). This leads to the deletion of newly emigrated autoreactive B cells that have high-avidity interactions with self-antigens in the periphery (9, 10). This peripheral deletion checkpoint is likely a major contributor to the drop from 40% to 20% self-reactivity observed within the peripheral B cell repertoire, when comparing newly emigrated B cells with mature B cells (3). Another contributing factor to this decline of autoreactive B cell frequency in the mature population is the decreased lifespan of autoreactive B cells silenced by B cell anergy (11). As discussed below, anergic autoreactive B cells persist in the periphery in a state of functional unresponsiveness. Anergic B cells have an approximate 80% reduction in half-life due to a competitive disadvantage to receive sufficient B cell-activating factor (BAFF) receptor signals needed for survival (12). Combined, these mechanisms reduce the likelihood of developing an autoimmune response by limiting the availability of autoreactive B cells. Disruptions in central tolerance mechanisms or elevated BAFF levels increase the size of the peripheral autoreactive B cell pool, which is correlated with an increased risk of developing autoimmunity (13, 14).
Under homeostatic conditions, autoreactive B cells present in the periphery are either ignorant or actively suppressed. If autoreactive B cells do not encounter their self-antigen, or if the avidity of the interaction of the self-antigen with their BCR is not high enough to cause effective antigen receptor crosslinking (low concentration and not high enough receptor affinity), they will be in a naïve/ignorant state (15–18). Autoreactive B cells that do interact with their self-antigen are actively suppressed. Both B cell-intrinsic and extrinsic mechanisms can contribute to this suppressed state. B cell-extrinsic factors that prevent B cell activation include the absence of T cell help, due to T cell tolerance mechanisms, or active suppression by regulatory T cells (19–21). B cell-intrinsic mechanisms act by limiting the signals required for B cell activation below a threshold that supports immune responses. These include physical changes in antigen receptor availability and active inhibitory signaling circuits. In this review, I will discuss how inhibitory signaling circuits are established in autoreactive B cells, focusing on the Lyn-SHIP-1-SHP-1 axis, how they contribute to peripheral immune tolerance, and how disruptions of these circuits contribute to the development of autoimmunity.
Anergic B cells: a case study of autoreactive B cells in the periphery.
Estimates of the percentage of autoreactive B cells present within the normal mature B cell repertoire vary between 15–20% (3, 22). However, the actual percentages of B cells with a degree of self-reactivity in the periphery could be higher (23, 24). Various degrees of autoreactivity can be found in different populations of B cells within healthy individuals. Both the more innate-like B-1 B cells and marginal zone B cells have a more polyreactive B cell receptor repertoire that often has a degree of self-reactivity. This self-reactivity provides a positive selection signal that is important for their development (25, 26). The remaining autoreactive B cells can be found both in the transitional B cell compartment and among mature follicular B cells. In these populations, reduced surface IgM expression is the best indicator of autoreactivity, due to interactions with self-antigen. As discussed below, reduced surface antigen receptor expression may be sufficient to reduce self antigen-induced BCR signaling below the threshold required for B cell activation. However, this reduced surface IgM is often accompanied by increased expression or activation of inhibitory signaling proteins. A textbook example of this can be found among autoreactive B cells that are present in the periphery in a state of functional unresponsiveness or B cell anergy.
Within the two-signal model of lymphocyte activation (27), B cell anergy is thought to occur if B cells receive an initial signal through their antigen receptor (signal 1), but fail to receive a second signal (signal 2) from cognate T cells, required for full activation. In the absence of that second signal, the B cell becomes tolerant. Nossal and colleagues coined the term B cell anergy to explain experimental observations they made following a low antigen dose in vivo tolerization protocol (28). The observed tolerance was not because of deletion of antigen-specific B cells, because antigen-specific B cells were still detected in the periphery; instead, these B cells were in a state of functional unresponsiveness, since they failed mount an antibody response upon antigen challenge. Subsequent pioneering work by Chris Goodnow and colleagues using the double transgenic MD4 x ML5 mouse model first identified many of the characteristics associated with B cell anergy (29). In this model, a BCR transgenic mouse (MD4) with a virtually monoclonal repertoire of B cells reactive with the non-self antigen hen egg lysozyme (HEL) are crossed with transgenic mice (ML5) that express soluble HEL as a neo-self antigen. This system allowed them to compare B cells with the same BCR under conditions where they are either naïve (antigen inexperienced) or exposed to their antigen as a self-antigen.
While the affinity of the MD4 BCR for HEL is very high, the largely monovalent nature of soluble HEL makes the overall avidity of the self-antigen-BCR interaction sufficiently low that the signal strength during B cell development is below the threshold that induces clonal deletion. Since the construct encoding the transgenic BCR is inserted outside of the immunoglobulin locus, receptor editing is not possible in these mice. Increasing the avidity of the BCR-self antigen interaction, by exposing developing MD4 B cells to HEL in membrane-bound form, results in clonal deletion of these MD4 B cells (30).
As observed by Nossal, anergic MD4xML5 B cells persist in peripheral lymphoid organs, but fail to mount an autoantibody response when challenged with exogenous HEL (29). They have a shortened half-life (11) and low IgM surface expression yet maintain a high expression of surface IgD. MD4xML5 B cells have elevated intracellular calcium and phospho-Erk levels, due to continuous interaction with self-antigen (31). Upon additional antigen receptor stimulation, both early and later biochemical signaling events are significantly reduced (32), and biological responses (upregulation of activation markers, proliferation) are absent. The BCR signals derived from interactions with self-antigen are not completely unproductive. They are sufficient to induce nuclear translocation of some transcription factors such as NFAT but not others such as NFκB (31, 33, 34). These signals induce a transcriptional program found in anergic B cell populations with different antigen-specificities (34, 35) (Isaac Harley, manuscript in preparation).
It should be noted that MD4xML5 B cells are not representative of all autoreactive B cells silenced by B cell anergy. Additional transgenic models have been developed in which B cells are anergic in the sense that they are present in the periphery, are exposed to self-antigen, display different levels of unresponsiveness to external stimulation, and do not mount an autoantibody response (36). This suggests that anergy exists on a spectrum, where the signal strength induced by self-antigen interactions determines the depth of functional impairment required for tolerance. Anergic B cell populations have been identified in the normal peripheral B cell repertoire in mice and humans. In mice, the T3/An1 population (B220+ CD93+ CD23+ IgMlo) (37–39) and a B220+ CD93- CD23+ IgMlo population (39) are enriched for autoreactive B cells with an anergic phenotype. In humans, the BND population (CD27- IgD+ IgM-) (40) and the more encompassing CD27- IgD+ IgMlo (41) population are enriched for autoreactive B cells with an anergic phenotype.
Despite a level of transcriptional reprogramming, B cell anergy is not an irreversible state. Instead, it requires continuous interaction with self-antigen to be maintained (42, 43). Ars/A1 mice express a transgenic BCR that binds the hapten arsonate (Ars) but cross-reacts with a lower affinity with self-antigen (DNA/chromatin), rendering Ars/A1 B cells anergic (44). A useful feature of this model is that self-antigen can be dissociated from antigen receptors with a monovalent antigen (Ars-Tyr) that binds with a higher affinity but cannot crosslink receptors. Upon dissociation of self-antigen, Ars/A1 B cells regain antigen receptor responsiveness within minutes (42). Notably, this reversal in responsiveness happens before antigen receptor levels change. This suggests that continuous antigen receptor engagement by self-antigen induces active inhibitory signaling circuits that can be rapidly reversed upon self-antigen dissociation.
The reversible nature of B cell anergy has implications for both the development of autoimmune responses and normal immunity. Anergic B cells are a likely source of pathogenic B cells during autoimmune disease. Indeed, autoreactive B cells with specificities relevant to autoimmune diseases, such as anti-nuclear antibodies (ANA) (45) and anti-insulin (46), can be found with an anergic phenotype in healthy individuals. Importantly, loss of B cell anergy precedes the development of systemic lupus erythematosus (SLE) (45), rheumatoid arthritis (47), autoimmune thyroid disease (48), and type 1 diabetes (46).
With their potential to contribute to autoimmune disease, why keep anergic B cells around? Some data suggests that they may play a role in maintaining immune tolerance to associated antigens (49) and support peripheral T cell tolerance (50, 51). Intriguing data from the Goodnow lab suggests that anergic B cells can participate in germinal center responses under certain conditions. If the BCR mutates away its self-reactivity during somatic hypermutation, these B cells can contribute to a protective response, a process called clonal redemption (52, 53). How this process exactly works is still unclear, but it suggests that while being a liability, anergic B cells may be present to avoid gaps in the B cell repertoire that otherwise could be detrimental when exposed to pathogens.
Regulation of BCR signaling in B cells.
Biochemical signaling pathways are tightly regulated to ensure that the duration of signaling and signal amplitude are appropriate for a functional response upon signal initiation. B cell receptor signaling is no exception (reviewed in (54, 55) (Figure 1). Upon B cell receptor cross-linking, Src-family kinases phosphorylate the tyrosines in the Immunoreceptor Tyrosine-based Activation Motifs (ITAM) of the CD79A/B (Igα/Igβ) heterodimer that is associated with the B cell receptor. ITAMs contain two conserved tyrosines; once both tyrosines are phosphorylated, the Syk family kinase Syk can bind with its two Src Homology 2 (SH2) domains resulting in the activation of Syk (56). When recruited to the BCR, Syk can become phosphorylated by Src-family kinases, further promoting its enzymatic activity. Once activated, Syk can amplify signaling by phosphorylating ITAM tyrosines (57). Src-family kinases can interact with unphosphorylated ITAMs using a sequence within their unique domain. Once CD79A and B ITAMs become phosphorylated Src-family kinases can bind to these phospho-tyrosines with a higher affinity via their SH2-domain (58). In contrast to Syk, which requires that both ITAM tyrosines be phosphorylated to enable binding, Src family kinases can bind mono-phosphorylated ITAMs (59).
Figure 1. Regulation of BCR signaling.
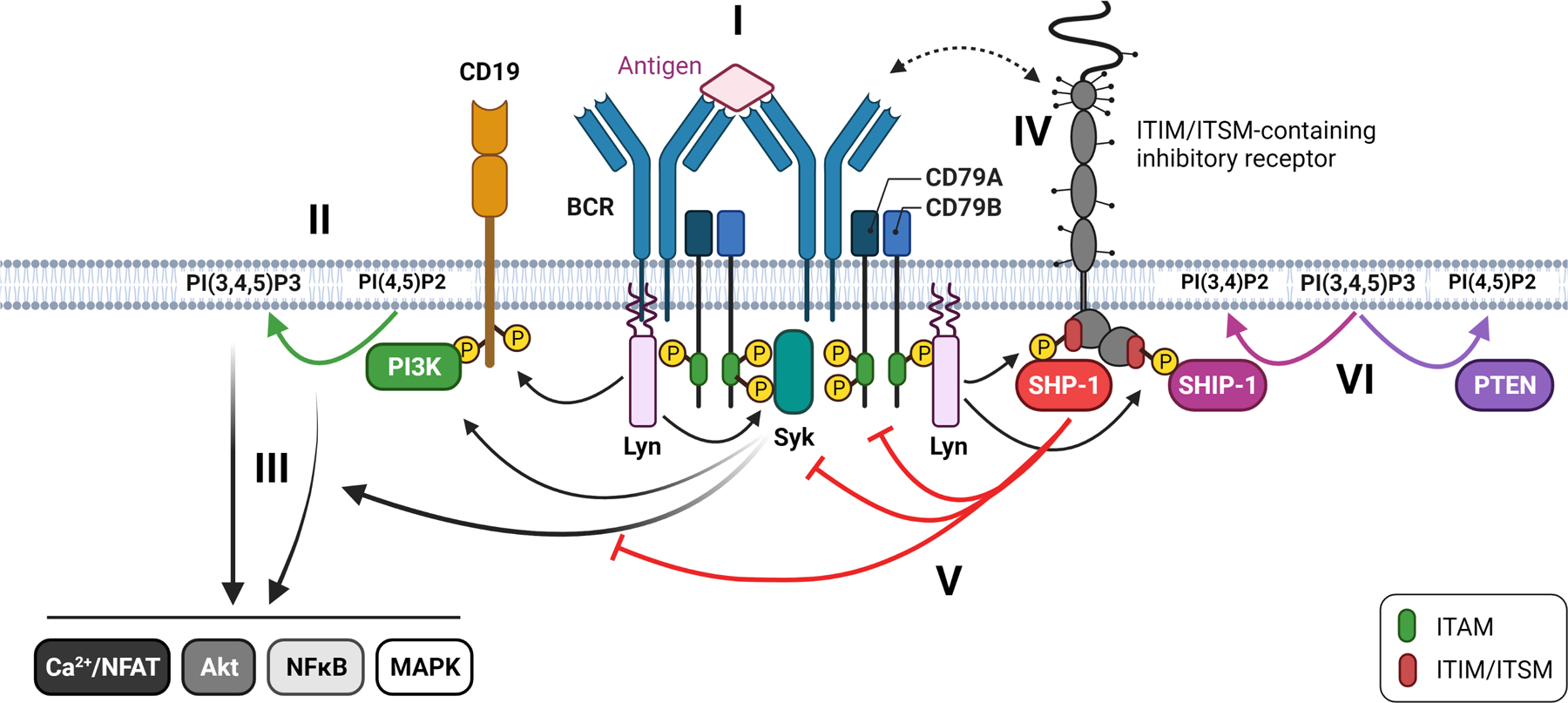
I) Upon BCR crosslinking Lyn phosphorylates the ITAMs of CD79A/CD79B resulting in the recruitment and activation of Syk. II) Lyn and Syk activate PI3K which phosphorylates PI(4,5)P2 to generate PI(3,4,5)P3. III) A signaling cascade activates multiple pathways important for B cell activation. PI(3,4,5)P3 plays an important role in facilitating activating signaling via the recruitment of key proteins that contain PH domains. IV) ITIM/ITSM-containing receptors come in proximity to actively signaling BCRs. Lyn phosphorylates the ITIM/ITSM tyrosine leading to the recruitment and activation of SHP-1 and SHIP-1. V) The tyrosine phosphatase SHP-1 inhibits signaling by dephosphorylating key tyrosines. VI) The inositol phosphatase SHIP-1 inhibits PI3K-dependent signaling by dephosphorylating PI(3,4,5)P3 to PI(3,4)P2. PTEN also counteracts PI3K signaling by dephosphorylating PI(3,4,5)P3 to PI(4,5)P2.
B cells express multiple Src family kinases, but the main Src family kinases involved in initiation of BCR signaling are Lyn and Fyn. The activity of Src family kinases is regulated by two conserved tyrosines, an inhibitory tyrosine in the C-terminus and an activating tyrosine in the kinase domain (reviewed in (60)). When phosphorylated, the C-terminal tyrosine interacts with an SH2 domain within the Src-family kinase, folding it into a closed conformation. Dephosphorylation of the inhibitory C-terminal tyrosine by CD45 and CD148 allows the protein to assume an open conformation that can be activated. Other Src family kinases, associated with BCRs and brought into proximity due to the antigen-mediated crosslinking, trans-autophosphorylate the activating tyrosine, fully activating the kinase. Several phosphatases have been implicated in dephosphorylating the activating tyrosine to control Src family kinase activity, including CD45, ptpn22, ptpn2, and SHP-1. The kinase Csk phosphorylates the C-terminal tyrosine to deactivate the kinase entirely. This is a dynamic process. Acute inhibition of Csk results in complete dephosphorylation of the inhibitory tyrosine and phosphorylation of the activating tyrosine within two minutes without additional stimulation (61). This suggests that Src family kinases are in an equilibrium between activated and inactivated states and that upon antigen receptor crosslinking, this equilibrium shifts towards a more activated state.
Once Lyn/Fyn and Syk are activated, several activating signaling events follow. Phosphoinositide 3-kinase (PI3K) is activated in a Lyn/CD19-dependent manner (62), with contributions by Syk and BCAP (63). PI3K phosphorylates PtdIns(4,5)P2, leading to a local increase in PtdIns(3,4,5)P3 in the cell membrane. Certain PH-domain-containing proteins can bind PtdIns(3,4,5)P3, localizing them on the cell membrane at a site of active signaling. This can bring them into the proximity of their substrates or kinases required for further activation. An example of the latter is Akt, which plays an important role in regulating B cell metabolism and survival. An example of the former is PLCγ2. Upon BCR stimulation, the scaffold protein BLNK is recruited to the BCR by a phosphorylated non-ITAM tyrosine on CD79A. BLNK becomes phosphorylated by Syk and serves as an organizer by recruiting several proteins, including Btk and PLCγ2 (64–66). Upon phosphorylation by Btk, PLCγ2 becomes activated, and when recruited to the plasma cell membrane via its PH domain, it hydrolyzes PtdIns(4,5)P3 into diacylglycerol (DAG) and Ins(1,4,5)P3 (IP3). IP3 will bind to the IP3 receptor in the ER membrane resulting in calcium release into the cytosol. This initiates a cascade that results in the dephosphorylation of members of the NFAT family of transcription factors, resulting in their translocation into the nucleus. DAG activates Protein Kinase C (PKC), which initiates a signaling cascade that leads to the activation of NFκB and contributes to the activation of the MAPK pathway.
Almost immediately after the initiation of activating signaling, inhibitory feedback mechanisms kick in to limit and eventually terminate signaling. Both tyrosine and inositol phosphatases are recruited to actively signaling antigen receptors and become activated within minutes after BCR crosslinking. Typically, a receptor containing one or more Immunoreceptor Tyrosine-based Inhibitory Motif (ITIM) or Immunoreceptor Tyrosine-based Switch Motif (ITSM) will be brought into proximity to an actively signaling receptor, resulting in the phosphorylation of the conserved tyrosine within the ITIM/ITSM by Src-family kinases. Lyn is the primary Src family kinase responsible for this, thus having a dual role in B cell signaling by initiating activating signaling and inhibitory feedback loops. Lyn-deficient B cells have an almost complete loss of phosphorylation of the ITIM motifs of canonical inhibitory receptors such as CD22 and FcγRIIB, resulting in a failed recruitment of their associated phosphatases SHP-1 and SHIP-1, respectively (67–69).
SHP-1 is a tyrosine phosphatase reported to dephosphorylate key tyrosines in ITAMs, Syk, BLNK, and other early signaling mediators (70, 71). Accordingly, SHP-1 deficient B cells have elevated and prolonged signaling upon BCR stimulation (72). B cells also express the related tyrosine phosphatase SHP-2, although its role in regulating B cell responses is less understood. SHP-1 and SHP-2 have two SH2 domains, and upon tandem SH2 binding to phosphorylated ITIMs, these proteins undergo a conformational change leading to their activation (73–76). SHP-1 becomes phosphorylated, and this has been suggested to regulate its activity and facilitate the ability to sustain activation independent of SH2 domain engagement (77, 78).
SHIP-1 is an inositol phosphatase that dephosphorylates the PI3K product PtdIns(3,4,5)P3 into PtdIns(3,4)P2, counteracting the PI3K pathway (reviewed in (79)). Besides a phosphatase domain, SHIP-1 contains multiple domains that facilitate protein-protein interactions. These include 1) an SH2 domain that can interact with phosphorylated tyrosines in ITIMs and ITAMs, 2) C terminal proline-rich domains that can interact with SH3 domains of the adaptor protein Grb2 and possibly other signaling proteins, and 3) two PNxY motifs that are phosphorylated by Src-family kinases and mediate interactions with the adaptor proteins Dok-1, Dok-3 and Shc1. These protein-protein interactions play an important role in stabilizing phosphatase-receptor interactions and localizing SHIP-1 to the cell membrane. While SHIP-1 has a PH-related domain that may contribute to membrane localization (80), when complexed with the PH-domain containing Dok-1/Dok-3, these proteins can facilitate the recruitment of SHIP-1 to the membrane to sites enriched for SHIP-1’s substrate PtdIns(3,4,5)P3 (81). In addition, at least two mechanisms have been described by which SHIP-1 can negatively regulate the MAPK pathway via Shc (82) or Dok-1 (83, 84). Accordingly, SHIP-1 deficient B cells have enhanced PI3K-dependent signaling (e.g., calcium responses) and Erk signaling (85).
While this Lyn-SHP-1-SHIP-1 axis is vital to regulate the magnitude and duration of antigen receptor signaling, it also plays a critical role in peripheral tolerance by preventing the activation of autoreactive B cells.
The role of Lyn in peripheral B cell tolerance.
Lyn’s role in immune tolerance was unveiled when Lyn-deficient mice were generated (86–88). These mice develop a multi-organ autoinflammatory disease preceded by the development of autoantibodies. Lyn is expressed in both B cells and myeloid cells. Consequences of lineage-specific deletion of Lyn have demonstarted that Lyn expression in both lineages is imperative for immune tolerance (89, 90). B cell-restricted deletion of Lyn is sufficient to lead to autoantibody development, with titers comparable to those observed in whole-body Lyn deficient mice, demonstrating a B cell-intrinsic requirement for Lyn for appropriate B cell tolerance (89). Since Lyn plays a role both in BCR signal initiation and in establishing inhibitory feedback mechanisms that restrict signaling, theoretically, Lyn could alter B cell tolerance in two ways. Loss of Lyn could result in decreased BCR signal strength during B cell development, allowing autoreactive clones that would normally be deleted during B cell development to mature instead. Alternatively, loss of inhibitory signaling in Lyn deficient cells enhances BCR signal strength, thereby decreasing the activation threshold of autoreactive B cells in the periphery. As discussed below most evidence points towards the latter possibility, although there is some indirect evidence that the former may also occur (91).
Besides autoantibody production, Lyn deficiency leads to a reduction in peripheral B cell numbers and an increase in plasma cells and B1a cells (86–89). While B cell development appears to be unaffected in the bone marrow of Lyn deficient mice, there is a stark reduction in B cells during maturation in the periphery when transitional stage 1 B cells mature into transitional stage 2 B cells and beyond. This is accompanied by an increase in BCR responsiveness, starting at the transitional stage of B cell development. This indicates that Lyn’s suppressive signaling function becomes dominant from this stage onward (92–95). Combined, these data suggest that increased BCR signal strength at the transitional stage leads to increased peripheral deletion of (mildly) autoreactive B cells. Etopic expression of survival factors can rescue these cells from activation-induced death (93, 95). This idea is further supported by our observations in the Ars/A1 model, where B cell restricted loss of Lyn phenocopies the effects of B cell restricted loss of the downstream phosphatases SHIP-1 or SHP-1. In all cases, the loss of the respective protein leads to enhanced signaling and a stark reduction in peripheral autoreactive B cells ((96) and Fiske and Getahun, manuscript in preparation).
Interestingly, the Lyn deficient Ars/A1 B cells that can be found in the periphery are responsive to antigen receptor stimulation and differentiate into antibody-forming cells, indicating that B cell anergy is lost in this model upon Lyn deletion (Fiske and Getahun, manuscript in preparation). The requirement for Lyn to establish B cell anergy in this model is noteworthy, because earlier studies using two other models of B cell anergy suggested that Lyn does not play a major role in B cell anergy. The results in the MD4xML5 model of B cell anergy were hard to interpret since very few HEL specific B cells could be found in the periphery and those that were present had an immature phenotype (69). This suggests that due to increased BCR signal strength, Lyn-deficient MD4xML5 B cells were deleted before they could be anergized. In VH3H9 heavy chain transgenic mice on the Balb/c background, λ1-expressing B cells are DNA-reactive and anergic. Lyn deficiency did not result in their deletion, nor did these B cells develop into antibody-forming cells, suggesting that B cell anergy was intact in this model (97). Of note, the responsiveness to LPS was restored in λ1+ VH3H9 B cells. Lyn also plays a role in controling TLR signaling. TLR signaling is required to developing pathogenic IgG antibodies in Lyn deficient mice, suggesting that Lyn deficiency may promote these responses by both derepressing BCR and TLR signaling (89, 90, 98, 99). The difference in dependence on Lyn for establishing or maintaining a state of unresponsiveness between different models of B cell anergy strengthens the idea that B cell anergy exists as a continuum (23, 100). The dependence on suppressive signaling mediated by the Lyn-SHP-1-SHIP-1 axis in anergic B cells may be contingent on the signal strength caused by the antigen-specificity and the antigen receptor affinity and avidity of the self antigen-BCR interaction.
Besides weakening tolerance mechanisms that prevent the activation of autoreactive B cells, Lyn also controls plasma cell differentiation and survival, thereby contributing to overall autoantibody production. The transcription factor Ets1 suppresses plasma cell differentiation and is downregulated following BCR or TLR signaling. Enhanced signaling, due to loss of Lyn, promotes reduced Ets expression and thus plasma cell differentiation (101). Loss of Lyn also enhances plasma cell survival, as Lyn normally plays a role in restricting plasma cell responsiveness to cytokines important for their survival (102).
Combined, these studies fit the notion that, while Lyn plays a role in both activating and inhibitory signaling, Lyn’s inhibitory function prevails in peripheral B cells. In agreement with this, acute activation of Lyn, by chemical inhibition of Csk, results in a hyporesponsive state of B cells (61). This was due to a Src-family kinase-dependent suppression of PI3K signaling by SHIP-1. Recent work from the Freedman and Rivera groups suggests an intriguing mechanism by which Lyn function may be skewed towards inhibition (60, 103). Lyn is expressed in two forms: the 56 kDa Lyn A form and the 53kDa Lyn B form, due to alternative splicing. Lyn A has a 21 amino acid insertion in the N terminal unique region that makes it susceptible to activation-induced degradation (61, 104). By making mice that only express either the Lyn A or Lyn B form, the Freedman group found that the Lyn A form is more associated with activating signaling, while the Lyn B form is predominantly responsible for inhibitory signaling and associated B cell tolerance (105). While it is unclear how the Lyn B form is biased towards activating inhibitory signaling, it suggests a mechanism by which initial activating signaling by Lyn is followed by more inhibitory signaling, due to a relative skewing towards the more inhibitory form.
While all these data corroborate the dominance of Lyn’s inhibitory function, there is a paradoxical observation in mice that express a constitutively active form of Lyn (106). In these mice, inhibitory pathways (SHP-1/SHIP-1) are constitutively activated and become more activated upon further BCR stimulation, yet upon stimulation, the B cells from these mice are hyperactive. These mice also develop autoantibodies and autoimmune disease. While these results support a role for Lyn in activating SHIP-1 and SHP-1, it is unclear why the net result in signaling is enhanced, instead of suppressed. The authors suggest that the loss of B cell tolerance is due to enhanced signaling in autoreactive B cells, because of the activating effects of Lyn during BCR signaling. While this could explain the observed autoantibody production, another possibility may be that this is due to Lyn’s other functions during B cell activation; Lyn is activated following CD40 ligation (107, 108). Ongoing work in our lab suggests that Lyn plays a pivotal role in PI3K activation following CD40 stimulation, providing a survival signal in activated autoreactive B cells (unpublished results).
In light of the predominant suppressive nature of Lyn, it is noteworthy that reduced expression of Lyn has been observed in B cells from SLE patients (109–111). Similarly, increased Csk expression and resultant augmented Lyn inactivation has been observed in SLE patients (112).
The importance of suppression of the PI3K pathway for peripheral tolerance.
Multiple signaling pathways are suppressed in anergic B cells, including the PI3K pathway, the MAPK pathway, and the NFkB pathway (31, 33, 113). It has become apparent that because of its upstream location, regulation of the PI3K signaling is of particular importance for B cell anergy and peripheral B cell tolerance in general.
Rickert and colleagues found that, unlike naïve MD4 B cells, anergic MD4xML5 B cells do not accumulate the PI3K product PtdIns(3,4,5)P3 in the plasma membrane following BCR stimulation (113). They found that MD4xML5 B cells have elevated levels of the inositol phosphatase PTEN, a negative regulator of the PI3K pathway that dephosphorylates PtdIns(3,4,5)P3 to generate PtdIns(4,5)P2. Upon PTEN deletion, MD4xML5 B cells regained B cell responsiveness, suggesting a critical role for suppressing PI3K signaling to maintain B cell anergy. To determine the contribution of PTEN overexpression on the anergic phenotype of MD4xML5 mice, we generated MD4 mice in which we can induce overexpression of PTEN in MD4 B cells in the absence of self-antigen. We found that induction of PTEN levels in MD4 B cells comparable to those observed in MD4xML5 B cells was sufficient to recapitulate some but not all the characteristics of anergic B cells (114). These B cells had dampened PI3K-dependent signaling and made reduced antibody responses upon antigen challenge, but were not entirely suppressed in their ability to mount antibody responses as MD4xML5 B cells are. Unlike in MD4xML5 mice, their B cells express normal surface IgM levels and show normal early signaling events such as Syk activation. This alludes that IgM downregulation per se and additional inhibitory signaling pathways play an important role in getting to the level of unresponsiveness observed in MD4xML5 B cells.
PTEN is upregulated following stimulation through multiple receptors, including the BCR (115). Studies in mice and humans have revealed an inverse relationship between surface IgM levels and intracellular PTEN expression ((24) and unpublished observations). In humans, B cells with elevated PTEN expression include the IgMlo IgD+ CD27- anergic BND population (40), which accounts for 2.5% of the lowest IgM expressing B cells, but also includes up to 40% of naïve B cells that have reduced IgM expression. This speaks to the notion that autoreactive B cells down-regulate IgM and upregulate a negative regulator such as PTEN in proportion to the strength of the signal they perceive following self-antigen engagement, to reach a state of unresponsiveness where the self-antigen engagement does not induce a signal that surpasses the threshold needed for activation (23, 100). The importance of PTEN-mediated suppression for B cell tolerance is further suggested by the reduction of PTEN expression in B cells from subjects that have developed SLE (115), Type 1 Diabetes and autoimmune thyroid disease (24), as well as B cells from the non-obese diabetic (NOD) mouse (116). These changes in PTEN expression in autoreactive B cells appear to be due to differences in miRNA regulation (24, 115), highlighting a role for miRNA regulation in peripheral B cell tolerance (117, 118)
PTEN is not the only negative regulator of the PI3K pathway important for controlling the activation of autoreactive B cells in the periphery. Deletion of SHIP-1 in B cells causes high titers of autoantibodies and lupus-like autoimmune disease (119, 120). Anergic B cells display elevated phosphorylation of SHIP-1 and the associated adaptor protein Dok-1, indicating increased activity. B cell restricted deletion of SHIP-1 leads to a loss of B cell anergy in two different models (119, 121). Anergic B cells need continuous interaction with self-antigen to remain unresponsive (42). To determine if continuous suppression of the PI3K pathway is required for the maintenance of anergic B cell unresponsiveness, we used a system in which we could induce acute deletion of either SHIP-1 or PTEN, or induce expression of a constitutively active PI3K in anergic B cells (96). All changes lead to enhanced PI3K dependent signaling, loss of B cell anergy, and a resultant autoantibody response.
Like the changes in PTEN expression observed in B cells of autoimmune patients, other forms of dysregulation of the PI3K pathway have also been associated with a loss of peripheral tolerance. B cells from SLE patients have been reported to have reduced levels of SHIP-1 activation (109). Individuals with gain-of-function mutations in the P110δ catalytic subunit of PI3K are immunocompromised and have defects in both the B and T cell compartment (122). About 40% of patients present with a loss of B cell tolerance, as evident by the production of IgM autoantibodies (123, 124). Analysis of patients and murine models with equivalent mutations suggests that this loss of tolerance is B cell-intrinsic and affects both central and peripheral tolerance (125, 126).
Given its central role in peripheral tolerance, correction of PI3K signal strength could be an attractive strategy to silence autoreactive B cells, thus preventing or treating autoimmunity. Indeed, in preclinical models, both genetic (127) and pharmaceutical (128) attenuation of PI3Kδ activation was shown to silence autoreactive B cells and ameliorate autoimmune disease while leaving the ability of naïve B and T cells to respond to antigen challenge intact.
How SHIP-1 becomes activated in anergic autoreactive B cells is not completely clear. Lyn plays a critical role in activating both SHIP-1 and SHP-1 following B cell activation (61, 67, 106). All three proteins are crucial for B cell tolerance (72, 89, 119, 120) and genetic complementation experiments support a role for the Lyn-SHIP-1 and Lyn-SHP-1 pathways in maintaining B cell tolerance (129). In the Ars/A1 model, inducible deletion of Lyn in Ars/A1 B cells leads to a selective loss of suppression of PI3K dependent signaling events and MAPK (Erk) activation (Fiske and Getahun, manuscript in preparation), both pathways that are suppressed by SHIP-1 (85). This suggests that Lyn plays a key role in suppressing PI3K activation, as observed by others (130).
How does Lyn promote the activation of SHIP-1 and SHP-1 in autoreactive B cells? ITIM/ITSM-bearing receptors are activated by being brought into the proximity of sites that contain activated Lyn. For example, in B cells, the ITIM of FcγRIIB only becomes phosphorylated, leading to SHIP-1 recruitment, if it is co-aggregated with the BCR (67). If inhibitory receptors are involved, how are they recruited to the BCR (Figure 2)? If the receptor’s ligand is part of the self-antigen, this will imply that some self-antigens may recruit additional negative regulators to silence the B cells that are specific for that self-antigen. Or are there mechanisms in place that recruit phosphatases directly to activated BCRs, independent of self-antigen specificity, either because the BCR itself recruits phosphatases or because the inhibitory receptor is constitutively associated with the BCR? It is likely a combination of these possibilities.
Figure 2. Initiation of inhibitory signaling in autoreactive B cells.
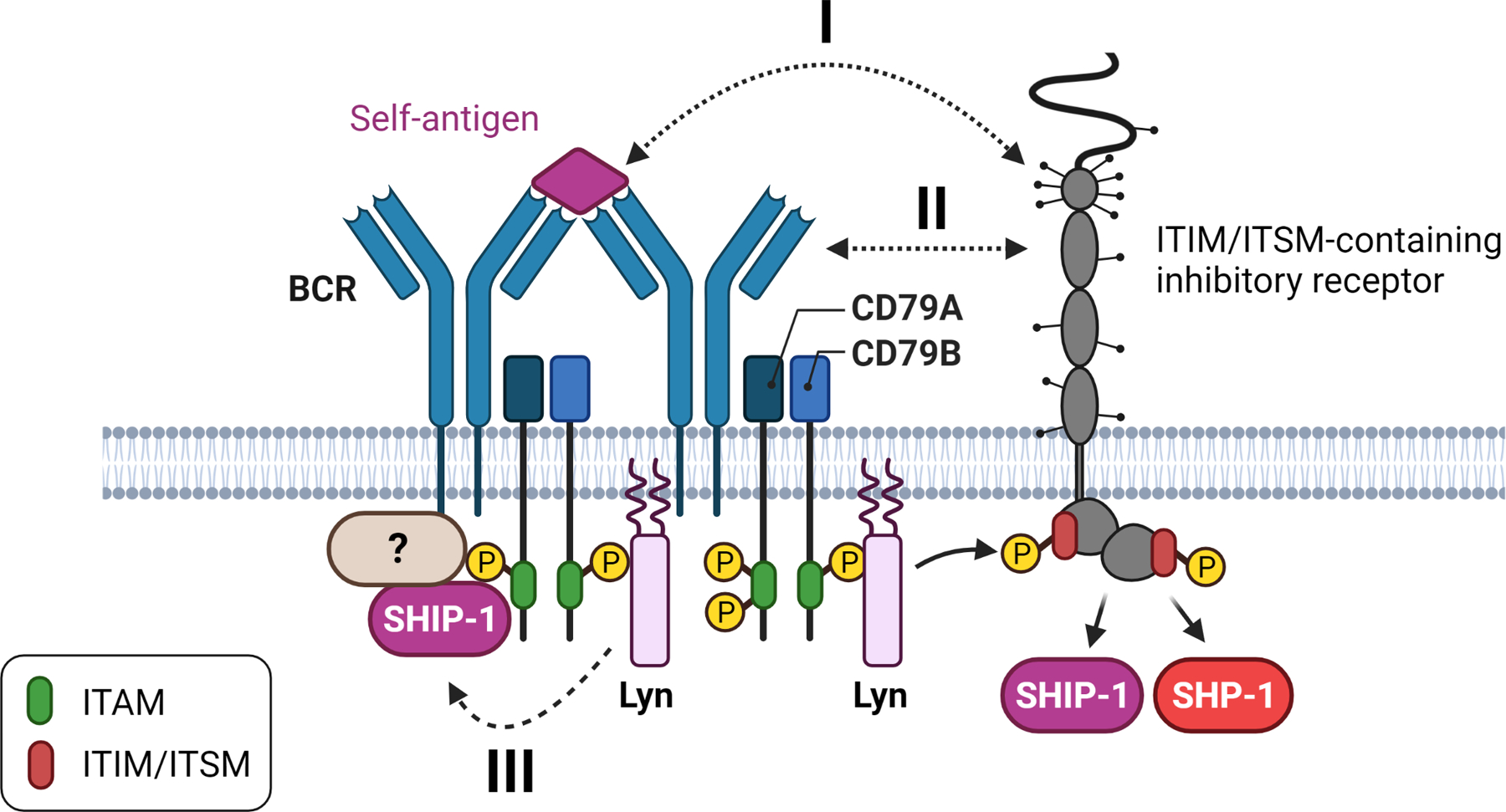
I) Self-antigens that contain a ligand for an inhibitory receptor can directly recruit this inhibitory receptor to the BCR. II) Ligands for an inhibitory receptor can be part of the BCR (e.g. glycans) or recruited to the BCR, resulting in recruitment of the inhibitory receptor to the BCR. III) The BCR may act as an inhibitory receptor via ITAMi signaling.
Activation of SHIP-1 in autoreactive B cells
The inhibitory low-affinity IgG Fc receptor, FcγRIIB, is the best characterized activator of SHIP-1 in B cells (131). IgG-antigen immune complexes crosslink FcγRIIB with the BCR, leading to Lyn mediated phosphorylation of the ITIM tyrosine in FcγRIIB and subsequent recruitment and activation of SHIP-1 (67, 88, 132). FcγRIIB deficient mice have enhanced IgG antibody responses, indicating that FcγRIIB is part of an important B cell-intrinsic feedback mechanism that controls the magnitude of humoral responses (133, 134). Depending on genetic background, FcγRIIB deficient animals develop autoantibodies and lupus-like disease (135). While FcγRIIB can be specifically involved in silencing IgG (rheumatoid factor)-specific B cells (136), given the ligand-specificity of FcγRIIB, it is more likely to play a role in peripheral tolerance when self-reactive IgG is being produced. Indeed, the effects of FcγRIIB deficiency on central tolerance mechanisms and B cell anergy are minimal (137). FcγRIIB does play an significant role in maintaining more distal peripheral tolerance during germinal center responses (137, 138).
SHIP-1 also becomes activated and is recruited to the cell membrane following BCR crosslinking without the involvement of FcγRIIB (81, 85, 139, 140). The exact mechanism by which this occurs is not completely clear, but CD79A and Lyn are instrumental. This is part of normal feedback regulation of antigen receptor signaling. In the context of B cell tolerance, the involvement of structural components of the BCR complex (CD79A&B) is intriguing. It suggests a mechanism by which inhibitory signaling is initiated in a manner independent of antigen-receptor specificity and thus could silence autoreactive B cells without the need for additional inhibitory receptors.
We and others have shown that the SH2 domain of SHIP-1 can directly bind to phosphorylated ITAMs of CD79A and B (81, 141, 142). It preferentially binds to the membrane-proximal ITAM tyrosine of CD79A (142). Recruitment of SHIP-1 to phosphorylated ITAMs is not unique to CD79A; it has been observed during signaling by other ITAM-bearing receptors as well (143, 144).
CD79A has previously been suggested to play a dual role in BCR signaling, inducing regulatory signaling and mediating B cell activation. Deletion of the intracellular tail of CD79A, including the ITAM, results in a block in development at the immature B cell stage. Upon BCR crosslinking, these immature B cells display delayed but enhanced signaling, suggesting a positive and negative role for CD79A during BCR signaling (145, 146). The inhibitory effect of CD79A on signaling is dependent on the ITAM tyrosines (147). The Nussenzweig lab made mice that effectively had a CD79A-CD79A dimer associated with the BCR instead of a CD79A-CD79B heterodimer, due to the replacement of the intracellular domain of CD79B with that of CD79A. In these mice B cells developed into mature B cells. Still, these B cells had features of anergic B cells, including a decreased ability to mount antibody responses that was likely due to both decreased surface BCR expression and suppressed signaling (148). CD79B can also regulate BCR signal strength/duration, but this appears to be due to its role in BCR internalization (149, 150).
The Wienands lab used the DT-40 chicken B cell model in an effort to define the mechanism by which SHIP-1 is recruited and activated following BCR stimulation (81). Using a proteomics approach, they identified Dok-3 and Grb2 as key adaptor proteins that interact with SHIP-1 following BCR stimulation (81). Dok-3 interacts with phosphorylated C-terminal NPXY motifs in SHIP-1, while Grb2 interacts with the SH3 domain of SHIP-1. Using mutated versions of SHIP-1, they found that following BCR stimulation, all three motifs in SHIP-1, the SH2 domain, the SH3 domain, and NPXY motif, were needed to recruit SHIP-1 to the membrane and cause its subsequent suppressive activity. Mutation of the SH2 domain had the most considerable effect on both parameters, indicating an essential role for CD79A. CD79A/Lyn are necessary for the phosphorylation of the NPXY motifs of SHIP-1 (81, 142), which, in turn, is vital for Dok-3-SHIP-1 complex formation. Whether SHIP-1 is directly recruited to CD79A, or if other proteins like Grb2 (151) or Dok-3 (152) aid or stabilize this interaction is not fully clear. In another cell system, the adaptor protein Shc1 has been implicated in SHIP-1 recruitment to the BCR instead (139). The reality is that CD79A can interact with many different proteins, both via their ITAM tyrosines and via residues outside of the ITAM (64–66, 153–155). These interactions could either promote the binding of SHIP-1 or sterically hinder this interaction. Understanding how these interactomes are established and how they may differ in autoreactive B cells is an active field of research. Of note, in anergic B cells, Dok-1 appears to interact with SHIP-1 instead of Dok-3 (119), suggesting that these interactomes may differ between naïve and autoreactive B cells.
Limiting antigen receptor availability vs. active suppression of signaling
Most autoreactive B cells in the periphery express reduced surface IgM due to interactions with self-antigen. Surface IgD levels often remain high on these autoreactive B cells, even though IgD has the same antigen-binding domains as the downregulated IgM (23, 29, 40, 44). However, exceptions have been observed where IgD is downregulated as well (156, 157). The reason for this difference in the behavior of IgM and IgD is not completely clear. There are structural differences between surface IgM and IgD; IgD is shorter due to one less immunoglobulin domain (2 less in mice), and IgM has a more rigid structure. In contrast, IgD has a long and flexible hinge region resulting in a more T-like shape vs. the classical Y-like shape of IgM. These structural differences have been proposed to make IgD less sensitive to low-valency antigen (158), although this has been challenged (35). Regardless, in systems where immunoglobin surface expression was restricted to one class, indirect readouts suggest that IgD is less sensitive to self-antigen (159, 160).
The availability of normal surface levels of IgD is often used to determine if an autoreactive B cell with reduced IgM expression is actively suppressed. If crosslinking of equivalent amounts of surface IgD results in dampened calcium mobilization in the autoreactive B cells, when compared to a naïve B cell, this suggests that active inhibitory signaling suppresses activation of the autoreactive B cell in concert with reduced availability of surface IgM. Indeed, autoreactive B cells from BCR transgenic models like MD4xML5 (32) or Ars/A1 (unpublished data) as well as CD27- IgMlo/- IgD+ BND cells found in peripheral blood (40, 46) have dampened calcium mobilization following IgD crosslinking compared to naïve B cells, despite equal levels of surface IgD expression.
To gain insight into the degree to which active suppression plays a role in silencing autoreactive B cells within the wild-type repertoire, the Zikherman lab studied their Nur77-eGFP model, in which GFP expression is controlled by the regulatory region of the immediate early gene Nur77 (NR4A1). Thus, GFP fluorescence is a reporter of antigen receptor signal strength (161). Previously they provided evidence that the range of GFP expression observed within the B cell repertoire of a wild-type mouse can be used as a proxy for the degree of self-antigen interaction (self-reactivity) by individual B cells. In these mice, GFP expression is inversely correlated with surface IgM expression, fitting with the idea that the downregulation of surface IgM is correlated with the signal strength induced by self-antigen binding. Importantly they find no correlation between GFP expression and surface IgD expression levels. By comparing GFP high (presumed autoreactive) versus GFP low (presumed not autoreactive) populations and restricting their analysis to B cells with equal surface IgM expression within these populations, they observed no difference in calcium mobilization following IgM crosslinking (23). This suggests there were no additional suppressive forces at play in the GFP+ (presumed autoreactive) B cells and that IgM downregulation is the main B cell-intrinsic mechanism that prevents their activation, as suggested by others (162). When they simply compared the ability of B cells with high or low IgM expression to respond to IgD crosslinking, they found that only the lowest IgM expressing cells (~1% of the total population) had reduced calcium responses following IgD crosslinking (23). This would suggest that only the B cells with the lowest IgM, presumable due to strong interactions with self-antigen, are anergic.
While these data make a compelling case for the importance of down-regulation of IgM as a B cell-intrinsic tolerance mechanism, there are some potential caveats. The assumption is that GFP is directly correlated to signal strength received, and the highest 10% and lowest 10% of GFP expressing B cells are compared as a representative of self-reactive and not self-reactive, respectively. If you compare the B cells that overlap in surface IgM expression within these populations, then either IgM downregulation does not always reflect self-reactivity or the GFP signal strength does not capture all self-reactivity. Using IgD responsiveness as a readout for active suppression also has caveats that may underestimate active inhibitory signaling. First, IgM and IgD have been proposed to reside in different protein islands on B cells (163), and if so, they may be differentially regulated due to proximity to inhibitory receptors (164). Secondly, if predominantly IgM productively interacts with self-antigen, then maybe self-antigen-BCR (IgM) interactions are locally suppressed by inhibitory signaling, without affecting distal sites assayed in these experiments. For example, CD40 signaling is intact in anergic B cells (165). In Ars/A1 B cells, where early events in BCR mediated PI3K signaling (pAkt/pS6) are actively suppressed, these same PI3K signaling events are not suppressed upon CD40 stimulation (unpublished observations), supporting the idea of localized suppressive signaling.
To determine the relative contribution of active inhibitory signaling versus IgM downregulation in anergic B cells, we analyzed early signaling events in Ars/A1 B cells following IgM cross-linking. When we correct for surface IgM levels and thus the degree of IgM crosslinking, the magnitude of most signaling events (e.g., Syk, BLNK, PLCγ2 phosphorylation) are indistinguishable from those observed in B cells from C57BL/6 mice with a wild-type repertoire. There were two exceptions: PI3K-dependent signaling events are suppressed in Ars/A1 B cells, and phosphorylation of the 182 tyrosine of CD79A, the membrane-proximal ITAM tyrosine of CD79A is more robust in Ars/A1 B cells (Fiske and Getahun, manuscript in preparation). A potential caveat in these observations is that the subpopulation of B cells in the wild-type repertoire with comparable IgM levels to Ars/A1 B cells, which was used for comparison, is likely enriched for autoreactive B cells. Not all B cells with low IgM expression need to be autoreactive per se. Other biological processes may also influence IgM expression causing variation in expression levels. Even mice that express a monoclonal BCR not thought to be autoreactive have a gaussian distribution of surface IgM expression levels on their B cells (95). Nonetheless, the autoreactive B cells present in the control group may also have active inhibitory signaling that suppresses early signaling events, which could confound interpretation. Still, actively suppressed PI3K signaling is consistent with active Lyn-SHIP-1-mediated suppression in Ars/A1 B cells. The absence of reduced phosphorylation of proteins that are targets of the SHP-1 was surprising, as discussed in the next section. The elevated basal and post-stimulation phosphorylation of CD79AY182 likely represent monophosphorylated ITAMs, as observed previously (119). This study observed that additional crosslinking of IgM on Ars/A1 B cells led to more ITAM monophosphorylation with little dual phosphorylation of ITAMs. This suggests that the continuous interaction with self-antigen predisposed these cells to induce ITAM monophosphorylation.
Could ITAM monophosphorylation and Lyn-SHIP-1 mediated suppression be related? ITAM monophosphorylation has also been observed during TCR signaling (166, 167), often under conditions of suboptimal stimulation, and results in inhibitory signaling (so-called inhibitory ITAM (ITAMi) activity). Suboptimal stimulation of Fc receptors can also lead to ITAMi signaling (78, 168). Crosslinking of chimeric receptors that contain a CD79A ITAM that can only be monophosphorylated cannot activate Syk, but still efficiently can activate Lyn, SHIP-1, and Dok-1 (119). This is likely due to the ability of Lyn to efficiently bind monophosphorylated ITAMs, while Syk requires dually phosphorylated ITAMs to bind (59), tipping the balance towards inhibitory signaling. If ITAM monophosphorylation is associated with suboptimal stimulation, and interactions with self-antigen result in downregulation of IgM, it is tempting to speculate that in an autoreactive B cell, the continuous interaction of self-antigen with reduced surface IgM results in suboptimal crosslinking, that promotes ITAMi signaling of the IgM-associated CD79A and possibly CD79B. Within the signal 1 without signal 2 model of tolerance induction (27), this would suggest that an autoreactive B cell that encounters its self-antigen will internalize antigen-bound IgM and initiate the early steps of B cell activation. However, when the second signal is not received due to T cell tolerance mechanisms, and surface levels of IgM are low due to continuous self-antigen stimulation, the continuous suboptimal crosslinking of IgM present on the cell surface by self-antigen could lead to ITAMi signaling by CD79A/B and subsequent activation of the Lyn-SHIP-1 axis.
Activation of SHP-1 in autoreactive B cells
Most of the known ITIM/ITSM-containing inhibitory receptors expressed in B cells use SHP-1 to suppress B cell activation. B cell-targeted deletion of SHP-1 leads to defects in B cell development and an increase in B-1a B cells at the expense of follicular B cells. These mice have partially impaired antibody responses, and their B cells display increased antigen receptor signaling (72, 96). At five months of age, mice accumulate autoantibodies and develop lupus-like disease, demonstrating a loss of B cell tolerance.
To study the effects of SHP-1 deficiency on B cell tolerance, Cyster and Goodnow crossed MD4 mice onto the viable motheaten background, that expresses a splice variant of SHP-1 that has ~ 20% enzymatic activity. They used a bone marrow transfer approach to assay the effect of SHP-1 deficiency on B cell tolerance induction under conditions that normally induce B cell deletion (membrane-bound HEL) or B cell anergy (soluble HEL) (169). They found that enhanced BCR signaling in immature B cells led to a lowered threshold for clonal deletion. As a result, SHP-1 deficient MD4 B cells were not only efficiently deleted when exposed to membrane bound HEL, but also when exposed to soluble HEL. The few MD4 B cells found in the periphery in mice that expressed soluble HEL had an immature phenotype, making it hard to determine the effects of SHP-1 deficiency on B cell anergy. To address this issue, we used mb1-cre driven deletion of SHP-1 in the Ars/A1 model (96). As observed in the MD4xML5 model, deletion of SHP-1 resulted in a drastic reduction of Ars/A1 B cells in the periphery. However, the Ars/A1 B cells present in the periphery made an autoantibody response. Inducible deletion of SHP-1 caused Ars/A1 B cells to regain antigen responsiveness and resulted in proliferation and differentiation into autoantibody forming plasmablasts. These results show that in the Ars/A1 model of B cell anergy, SHP-1 is required to both establish and maintain B cell anergy.
How SHP-1 is activated in Ars/A1 B cells is still unclear. At the onset of the experiment described in the previous section, we had expected that the reduced phosphorylation of known SHP-1 targets observed in Ars/A1 B cells after IgM crosslinking, when compared to naïve B cells, was due to SHP-1 activity. Instead, it appears that reduced surface IgM can account for those observations. While SHIP-1-mediated suppression of PI3K signaling may be directly linked to the BCR via CD79A, SHP-1-mediated suppression of Ars/A1 B cells may depend more on its self-antigen to recruit the appropriate inhibitory receptor. This may be missed when B cells are stimulated with a polyclonal anti-IgM antibody. A candidate inhibitory receptor could be CD72, which Ars/A1 B cells express at elevated levels (Isaac Harley, manuscript in preparation). CD72 is a type II membrane protein that contains an extracellular C-type lectin-like domain and an ITIM in its cytoplasmic domain. It binds to Sm/ribonucleoprotein (RNP) antigen (170) and plays a role in suppressing anti-nuclear antigen reactive B cells, like Ars/A1, by recruitment of SHP-1 (171). CD72 deficient animals develop anti-nuclear antibodies and lupus-like disease (171, 172), and decreased expression of CD72 has been observed in B cells from SLE patients and correlates with disease activity (173, 174).
B cells express multiple SHP-1-activating receptors such as CD22/Siglec-2, Siglec-G, CD72, PIR-B, BTLA, CD5, and members of the FcRL family. Best studied are CD22/Siglec-2 and the related Siglec-G (reviewed in (175)). These receptors bind sialic acid groups on glycans. CD22 binds α2,6-linked sialic acid, and Siglec-G binds both α2,6 and α2,3-linked sialic acid. They can interact with ligands expressed on the same cell (cis), including glycans present on the BCR, CD22, and CD45, or on other cells (trans). Interactions with the BCR restrict signaling in a Lyn/SHP-1 dependent manner. Consequently, CD22 deficient B cells (176–179) have a higher basal activation level and respond more strongly to BCR stimulation. The ligand for Siglec-G is more prevalent on B-1 B cells (180). Accordingly, Siglec-G deficient mice have an expanded B1-population (181), resembling SHP-1 deficient animals (72), and their B-1 cells are hyperresponsive to BCR stimulation. Depending on the generic background, (aged) CD22 deficient mice (176, 182) and Siglec-G deficient mice (183, 184) develop autoantibodies, which can synergize with other drivers of loss of tolerance to cause more severe autoimmune disease (185, 186). CD22 and Siglec-G play a role in B cell tolerance by controlling the threshold and magnitude of BCR signaling and by aiding in self - non-self discrimination (187). While their ligands have not always been identified, other SHP-1 activating receptors such as PIR-B (188–190), BTLA (191, 192), CD5 (193), and FcRL5 (194) can play similar roles to contribute to B cell tolerance by controlling BCR signal strength. Changes in their expression on B cells correlate with autoimmune disease, further emphasizing the importance of this axis in tolerance (195, 196).
Inhibitory circuits beyond Lyn-SHP-1-SHIP-1 in peripheral tolerance
This review focused primarily on the Lyn-SHP-1-SHIP-1 axis of inhibitory signaling and its role in peripheral B cell tolerance. Particular attention was given to the regulation of BCR signaling in autoreactive B cells, that prevent “signal 1” required for B cell activation. However, (autoreactive) B cells require multiple temporally spaced signals to become fully activated, survive, and differentiate into antibody-secreting cells. Inhibitory signaling circuits also regulate the receptors that process these additional signals. When these circuits become dysregulated, they can also contribute to a loss of B cell tolerance and autoimmunity. For example, the tyrosine phosphatase ptpn1 negatively regulates CD40 and BAFF-R signaling in B cells (197), receptors important to provide “signal 2” during B-T interactions and promote B cell survival, respectively. Deletion of ptpn1 in B cells results in autoantibody production and lupus-like disease manifestations. Rheumatoid arthritis patients are reported to have reduced expression of this phosphatase in their B cells which correlates with increased responsiveness to CD40 stimulation (197). The tyrosine phosphatase ptpn2 negatively regulates cytokine receptor signaling, and ptpn2 deficient B cells have increased responsiveness to IL-21 (198). IL-21 promotes plasmablast differentiation and is important for autoimmune responses (199). Multiple non-coding PTPN2 SNPs have been linked with an increased risk to develop autoimmune disease (200) and are associated with a decrease in ptpn2 expression (201). These are merely examples of how changes in inhibitory signaling can lower the threshold to develop an autoimmune response at different steps of B cell activation.
Concluding remarks
Inhibitory signaling plays an important role in maintaining peripheral tolerance of autoreactive B cells. Much remains to be learned about the exact pathways by which individual autoreactive B cells establish the inhibitory circuits needed to maintain unresponsiveness. Clinical data show that changes in expression levels or activation status of inhibitory effector proteins can be associated with a loss of B cell tolerance (24, 109–112, 115, 173, 174, 195–197, 202). Reductionistic models are essential to delineate which proteins play an important role in B cell tolerance and how their interplay creates an inhibitory circuit. A challenge remains in identifying the genetic and environmental factors that disrupt regulatory signaling and determining how they contribute to autoimmunity, as they may affect multiple interdependent cell lineages and B cell development/central tolerance. While it will depend on the magnitude of the change in expression or function of individual proteins as well as the timing of these changes, multiple synergistic hits within a pathway or related pathways are likely needed to significantly impact escape from tolerance. But as our understanding increases, so does our ability to intervene to prevent or treat autoimmune disease. Understanding inhibitory signaling is crucial in this regard because of the potential to therapeutic use pharmacological agents (128) or biologics (203, 204) to correct dysregulation that leads to a loss of tolerance.
Acknowledgements
I wish to thank John Cambier, Isaac Harley, Brigita Fiske and Bergren Crute for helpful suggestions while preparing this review. Figures were created with BioRender.com. This work was supported by the National Institute of Health grant AI149019, the Arthritis National Research Foundation and the University of Colorado Human Immunology and Immunotherapy Initiative. I have no conflicts of interest to declare.
References
- 1.Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, et al. Mechanisms of Autoantibody-Induced Pathology. Front Immunol 2017;8:603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Getahun A, Cambier JC. Non-Antibody-Secreting Functions of B Cells and Their Contribution to Autoimmune Disease. Annu Rev Cell Dev Biol 2019;35:337–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science 2003;301(5638):1374–7. [DOI] [PubMed] [Google Scholar]
- 4.Brink R, Phan TG. Self-Reactive B Cells in the Germinal Center Reaction. Annu Rev Immunol 2018;36:339–57. [DOI] [PubMed] [Google Scholar]
- 5.Nemazee D Mechanisms of central tolerance for B cells. Nat Rev Immunol 2017;17(5):281–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pelanda R, Greaves SA, Alves da Costa T, Cedrone LM, Campbell ML, Torres RM. B-cell intrinsic and extrinsic signals that regulate central tolerance of mouse and human B cells. Immunol Rev 2022. [DOI] [PMC free article] [PubMed]
- 7.Norvell A, Monroe JG. Acquisition of surface IgD fails to protect from tolerance-induction. Both surface IgM- and surface IgD-mediated signals induce apoptosis of immature murine B lymphocytes. J Immunol 1996;156(4):1328–32. [PubMed] [Google Scholar]
- 8.Su TT, Rawlings DJ. Transitional B lymphocyte subsets operate as distinct checkpoints in murine splenic B cell development. J Immunol 2002;168(5):2101–10. [DOI] [PubMed] [Google Scholar]
- 9.Yau IW, Cato MH, Jellusova J, Hurtado de Mendoza T, Brink R, Rickert RC. Censoring of self-reactive B cells by follicular dendritic cell-displayed self-antigen. J Immunol 2013;191(3):1082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrews SF, Zhang Q, Lim S, Li L, Lee JH, Zheng NY, et al. Global analysis of B cell selection using an immunoglobulin light chain-mediated model of autoreactivity. J Exp Med 2013;210(1):125–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fulcher DA, Basten A. Reduced life span of anergic self-reactive B cells in a double-transgenic model. J Exp Med 1994;179(1):125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 2004;20(4):441–53. [DOI] [PubMed] [Google Scholar]
- 13.Meffre E, O’Connor KC. Impaired B-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol Rev 2019;292(1):90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol 2002;2(7):465–75. [DOI] [PubMed] [Google Scholar]
- 15.Aplin BD, Keech CL, de Kauwe AL, Gordon TP, Cavill D, McCluskey J. Tolerance through indifference: autoreactive B cells to the nuclear antigen La show no evidence of tolerance in a transgenic model. J Immunol 2003;171(11):5890–900. [DOI] [PubMed] [Google Scholar]
- 16.Adelstein S, Pritchard-Briscoe H, Anderson TA, Crosbie J, Gammon G, Loblay RH, et al. Induction of self-tolerance in T cells but not B cells of transgenic mice expressing little self antigen. Science 1991;251(4998):1223–5. [DOI] [PubMed] [Google Scholar]
- 17.Akkaraju S, Canaan K, Goodnow CC. Self-reactive B cells are not eliminated or inactivated by autoantigen expressed on thyroid epithelial cells. J Exp Med 1997;186(12):2005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang H, Kearney JF, Grusby MJ, Benoist C, Mathis D. Induction of tolerance in arthritogenic B cells with receptors of differing affinity for self-antigen. Proc Natl Acad Sci U S A 2006;103(10):3734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell IK, Kinkel SA, Drake SF, van Nieuwenhuijze A, Hubert FX, Tarlinton DM, et al. Autoimmune regulator controls T cell help for pathogenetic autoantibody production in collagen-induced arthritis. Arthritis Rheum 2009;60(6):1683–93. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Liu A, Iikuni N, Xu H, Shi FD, La Cava A. Regulatory CD4+ T cells promote B cell anergy in murine lupus. J Immunol 2014;192(9):4069–73. [DOI] [PubMed] [Google Scholar]
- 21.Aschermann S, Lehmann CH, Mihai S, Schett G, Dudziak D, Nimmerjahn F. B cells are critical for autoimmune pathology in Scurfy mice. Proc Natl Acad Sci U S A 2013;110(47):19042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe A, Su KY, Kuraoka M, Yang G, Reynolds AE, Schmidt AG, et al. Self-tolerance curtails the B cell repertoire to microbial epitopes. JCI Insight 2019;4(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan C, Noviski M, Huizar J, Zikherman J. Self-reactivity on a spectrum: A sliding scale of peripheral B cell tolerance. Immunol Rev 2019;292(1):37–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith MJ, Ford BR, Rihanek M, Coleman BM, Getahun A, Sarapura VD, et al. Elevated PTEN expression maintains anergy in human B cells and reveals unexpectedly high repertoire autoreactivity. JCI Insight 2019;4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayakawa K, Asano M, Shinton SA, Gui M, Allman D, Stewart CL, et al. Positive selection of natural autoreactive B cells. Science 1999;285(5424):113–6. [DOI] [PubMed] [Google Scholar]
- 26.Meyer-Bahlburg A, Andrews SF, Yu KO, Porcelli SA, Rawlings DJ. Characterization of a late transitional B cell population highly sensitive to BAFF-mediated homeostatic proliferation. J Exp Med 2008;205(1):155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bretscher P, Cohn M. A theory of self-nonself discrimination. Science 1970;169(3950):1042–9. [DOI] [PubMed] [Google Scholar]
- 28.Nossal GJ, Pike BL. Clonal anergy: persistence in tolerant mice of antigen-binding B lymphocytes incapable of responding to antigen or mitogen. Proc Natl Acad Sci U S A 1980;77(3):1602–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988;334(6184):676–82. [DOI] [PubMed] [Google Scholar]
- 30.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature 1991;353(6346):765–9. [DOI] [PubMed] [Google Scholar]
- 31.Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, et al. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity 1997;6(4):419–28. [DOI] [PubMed] [Google Scholar]
- 32.Cooke MP, Heath AW, Shokat KM, Zeng Y, Finkelman FD, Linsley PS, et al. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med 1994;179(2):425–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 1997;386(6627):855–8. [DOI] [PubMed] [Google Scholar]
- 34.Glynne R, Akkaraju S, Healy JI, Rayner J, Goodnow CC, Mack DH. How self-tolerance and the immunosuppressive drug FK506 prevent B-cell mitogenesis. Nature 2000;403(6770):672–6. [DOI] [PubMed] [Google Scholar]
- 35.Sabouri Z, Perotti S, Spierings E, Humburg P, Yabas M, Bergmann H, et al. IgD attenuates the IgM-induced anergy response in transitional and mature B cells. Nat Commun 2016;7:13381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol 2007;7(8):633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ, et al. Identification of anergic B cells within a wild-type repertoire. Immunity 2006;25(6):953–62. [DOI] [PubMed] [Google Scholar]
- 38.Teague BN, Pan Y, Mudd PA, Nakken B, Zhang Q, Szodoray P, et al. Cutting edge: Transitional T3 B cells do not give rise to mature B cells, have undergone selection, and are reduced in murine lupus. J Immunol 2007;178(12):7511–5. [DOI] [PubMed] [Google Scholar]
- 39.Nojima T, Reynolds AE, Kitamura D, Kelsoe G, Kuraoka M. Tracing Self-Reactive B Cells in Normal Mice. J Immunol 2020;205(1):90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duty JA, Szodoray P, Zheng NY, Koelsch KA, Zhang Q, Swiatkowski M, et al. Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J Exp Med 2009;206(1):139–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quach TD, Manjarrez-Orduno N, Adlowitz DG, Silver L, Yang H, Wei C, et al. Anergic responses characterize a large fraction of human autoreactive naive B cells expressing low levels of surface IgM. J Immunol 2011;186(8):4640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol 2005;6(11):1160–7. [DOI] [PubMed] [Google Scholar]
- 43.Goodnow CC, Brink R, Adams E. Breakdown of self-tolerance in anergic B lymphocytes. Nature 1991;352(6335):532–6. [DOI] [PubMed] [Google Scholar]
- 44.Benschop RJ, Aviszus K, Zhang X, Manser T, Cambier JC, Wysocki LJ. Activation and anergy in bone marrow B cells of a novel immunoglobulin transgenic mouse that is both hapten specific and autoreactive. Immunity 2001;14(1):33–43. [DOI] [PubMed] [Google Scholar]
- 45.Malkiel S, Jeganathan V, Wolfson S, Manjarrez Orduno N, Marasco E, Aranow C, et al. Checkpoints for Autoreactive B Cells in the Peripheral Blood of Lupus Patients Assessed by Flow Cytometry. Arthritis Rheumatol 2016;68(9):2210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith MJ, Packard TA, O’Neill SK, Henry Dunand CJ, Huang M, Fitzgerald-Miller L, et al. Loss of anergic B cells in prediabetic and new-onset type 1 diabetic patients. Diabetes 2015;64(5):1703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liubchenko GA, Appleberry HC, Striebich CC, Franklin KE, Derber LA, Holers VM, et al. Rheumatoid arthritis is associated with signaling alterations in naturally occurring autoreactive B-lymphocytes. J Autoimmun 2013;40:111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith MJ, Rihanek M, Coleman BM, Gottlieb PA, Sarapura VD, Cambier JC. Activation of thyroid antigen-reactive B cells in recent onset autoimmune thyroid disease patients. J Autoimmun 2018;89:82–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aviszus K, Macleod MK, Kirchenbaum GA, Detanico TO, Heiser RA, St Clair JB, et al. Antigen-specific suppression of humoral immunity by anergic Ars/A1 B cells. J Immunol 2012;189(9):4275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morlacchi S, Soldani C, Viola A, Sarukhan A. Self-antigen presentation by mouse B cells results in regulatory T-cell induction rather than anergy or clonal deletion. Blood 2011;118(4):984–91. [DOI] [PubMed] [Google Scholar]
- 51.Murray SE, Toren KG, Parker DC. Peripheral CD4(+) T-cell tolerance is induced in vivo by rare antigen-bearing B cells in follicular, marginal zone, and B-1 subsets. Eur J Immunol 2013;43(7):1818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reed JH, Jackson J, Christ D, Goodnow CC. Clonal redemption of autoantibodies by somatic hypermutation away from self-reactivity during human immunization. J Exp Med 2016;213(7):1255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sabouri Z, Schofield P, Horikawa K, Spierings E, Kipling D, Randall KL, et al. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc Natl Acad Sci U S A 2014;111(25):E2567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol 2004;41(6–7):599–613. [DOI] [PubMed] [Google Scholar]
- 55.Kurosaki T, Shinohara H, Baba Y. B cell signaling and fate decision. Annu Rev Immunol 2010;28:21–55. [DOI] [PubMed] [Google Scholar]
- 56.Kurosaki T, Johnson SA, Pao L, Sada K, Yamamura H, Cambier JC. Role of the Syk autophosphorylation site and SH2 domains in B cell antigen receptor signaling. J Exp Med 1995;182(6):1815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WW, Zurn C, et al. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol Cell 2002;10(5):1057–69. [DOI] [PubMed] [Google Scholar]
- 58.Pleiman CM, Abrams C, Gauen LT, Bedzyk W, Jongstra J, Shaw AS, et al. Distinct p53/56lyn and p59fyn domains associate with nonphosphorylated and phosphorylated Ig-alpha. Proc Natl Acad Sci U S A 1994;91(10):4268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pao LI, Famiglietti SJ, Cambier JC. Asymmetrical phosphorylation and function of immunoreceptor tyrosine-based activation motif tyrosines in B cell antigen receptor signal transduction. J Immunol 1998;160(7):3305–14. [PubMed] [Google Scholar]
- 60.Brian BF, Freedman TS. The Src-family Kinase Lyn in Immunoreceptor Signaling. Endocrinology 2021;162(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu W, Skrzypczynska KM, Weiss A. Acute Csk inhibition hinders B cell activation by constraining the PI3 kinase pathway. Proc Natl Acad Sci U S A 2021;118(43). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buhl AM, Pleiman CM, Rickert RC, Cambier JC. Qualitative regulation of B cell antigen receptor signaling by CD19: selective requirement for PI3-kinase activation, inositol-1,4,5-trisphosphate production and Ca2+ mobilization. J Exp Med 1997;186(11):1897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okada T, Maeda A, Iwamatsu A, Gotoh K, Kurosaki T. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity 2000;13(6):817–27. [DOI] [PubMed] [Google Scholar]
- 64.Engels N, Wollscheid B, Wienands J. Association of SLP-65/BLNK with the B cell antigen receptor through a non-ITAM tyrosine of Ig-alpha. Eur J Immunol 2001;31(7):2126–34. [DOI] [PubMed] [Google Scholar]
- 65.Kabak S, Skaggs BJ, Gold MR, Affolter M, West KL, Foster MS, et al. The direct recruitment of BLNK to immunoglobulin alpha couples the B-cell antigen receptor to distal signaling pathways. Mol Cell Biol 2002;22(8):2524–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patterson HC, Kraus M, Kim YM, Ploegh H, Rajewsky K. The B cell receptor promotes B cell activation and proliferation through a non-ITAM tyrosine in the Igalpha cytoplasmic domain. Immunity 2006;25(1):55–65. [DOI] [PubMed] [Google Scholar]
- 67.Nishizumi H, Horikawa K, Mlinaric-Rascan I, Yamamoto T. A double-edged kinase Lyn: a positive and negative regulator for antigen receptor-mediated signals. J Exp Med 1998;187(8):1343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith KG, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by CD22: a requirement for Lyn. J Exp Med 1998;187(5):807–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, et al. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity 1998;8(4):497–508. [DOI] [PubMed] [Google Scholar]
- 70.Mizuno K, Tagawa Y, Mitomo K, Arimura Y, Hatano N, Katagiri T, et al. Src homology region 2 (SH2) domain-containing phosphatase-1 dephosphorylates B cell linker protein/SH2 domain leukocyte protein of 65 kDa and selectively regulates c-Jun NH2-terminal kinase activation in B cells. J Immunol 2000;165(3):1344–51. [DOI] [PubMed] [Google Scholar]
- 71.Adachi T, Wienands J, Wakabayashi C, Yakura H, Reth M, Tsubata T. SHP-1 requires inhibitory co-receptors to down-modulate B cell antigen receptor-mediated phosphorylation of cellular substrates. J Biol Chem 2001;276(28):26648–55. [DOI] [PubMed] [Google Scholar]
- 72.Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity 2007;27(1):35–48. [DOI] [PubMed] [Google Scholar]
- 73.Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci Adv 2020;6(5):eaay4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang J, Liu L, He D, Song X, Liang X, Zhao ZJ, et al. Crystal structure of human protein-tyrosine phosphatase SHP-1. J Biol Chem 2003;278(8):6516–20. [DOI] [PubMed] [Google Scholar]
- 75.Wang W, Liu L, Song X, Mo Y, Komma C, Bellamy HD, et al. Crystal structure of human protein tyrosine phosphatase SHP-1 in the open conformation. J Cell Biochem 2011;112(8):2062–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Famiglietti SJ, Nakamura K, Cambier JC. Unique features of SHIP, SHP-1 and SHP-2 binding to FcgammaRIIb revealed by surface plasmon resonance analysis. Immunol Lett 1999;68(1):35–40. [DOI] [PubMed] [Google Scholar]
- 77.Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol Cell 2001;8(4):759–69. [DOI] [PubMed] [Google Scholar]
- 78.Mkaddem SB, Murua A, Flament H, Titeca-Beauport D, Bounaix C, Danelli L, et al. Lyn and Fyn function as molecular switches that control immunoreceptors to direct homeostasis or inflammation. Nat Commun 2017;8(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pauls SD, Marshall AJ. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur J Immunol 2017;47(6):932–45. [DOI] [PubMed] [Google Scholar]
- 80.Ming-Lum A, Shojania S, So E, McCarrell E, Shaw E, Vu D, et al. A pleckstrin homology-related domain in SHIP1 mediates membrane localization during Fcgamma receptor-induced phagocytosis. FASEB J 2012;26(8):3163–77. [DOI] [PubMed] [Google Scholar]
- 81.Manno B, Oellerich T, Schnyder T, Corso J, Losing M, Neumann K, et al. The Dok-3/Grb2 adaptor module promotes inducible association of the lipid phosphatase SHIP with the BCR in a coreceptor-independent manner. Eur J Immunol 2016;46(11):2520–30. [DOI] [PubMed] [Google Scholar]
- 82.Tridandapani S, Phee H, Shivakumar L, Kelley TW, Coggeshall KM. Role of SHIP in FcgammaRIIb-mediated inhibition of Ras activation in B cells. Mol Immunol 1998;35(17):1135–46. [DOI] [PubMed] [Google Scholar]
- 83.Yamanashi Y, Tamura T, Kanamori T, Yamane H, Nariuchi H, Yamamoto T, et al. Role of the rasGAP-associated docking protein p62(dok) in negative regulation of B cell receptor-mediated signaling. Genes Dev 2000;14(1):11–6. [PMC free article] [PubMed] [Google Scholar]
- 84.Tamir I, Stolpa JC, Helgason CD, Nakamura K, Bruhns P, Daeron M, et al. The RasGAP-binding protein p62dok is a mediator of inhibitory FcgammaRIIB signals in B cells. Immunity 2000;12(3):347–58. [DOI] [PubMed] [Google Scholar]
- 85.Helgason CD, Kalberer CP, Damen JE, Chappel SM, Pineault N, Krystal G, et al. A dual role for Src homology 2 domain-containing inositol-5-phosphatase (SHIP) in immunity: aberrant development and enhanced function of b lymphocytes in ship −/− mice. J Exp Med 2000;191(5):781–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell 1995;83(2):301–11. [DOI] [PubMed] [Google Scholar]
- 87.Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, et al. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity 1995;3(5):549–60. [DOI] [PubMed] [Google Scholar]
- 88.Chan VW, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 1997;7(1):69–81. [DOI] [PubMed] [Google Scholar]
- 89.Lamagna C, Hu Y, DeFranco AL, Lowell CA. B cell-specific loss of Lyn kinase leads to autoimmunity. J Immunol 2014;192(3):919–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA. Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U S A 2013;110(35):E3311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest 2011;121(9):3635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shahaf G, Gross AJ, Sternberg-Simon M, Kaplan D, DeFranco AL, Mehr R. Lyn deficiency affects B-cell maturation as well as survival. Eur J Immunol 2012;42(2):511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gross AJ, Proekt I, DeFranco AL. Elevated BCR signaling and decreased survival of Lyn-deficient transitional and follicular B cells. Eur J Immunol 2011;41(12):3645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gross AJ, Lyandres JR, Panigrahi AK, Prak ET, DeFranco AL. Developmental acquisition of the Lyn-CD22-SHP-1 inhibitory pathway promotes B cell tolerance. J Immunol 2009;182(9):5382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meade J, Fernandez C, Turner M. The tyrosine kinase Lyn is required for B cell development beyond the T1 stage in the spleen: rescue by over-expression of Bcl-2. Eur J Immunol 2002;32(4):1029–34. [DOI] [PubMed] [Google Scholar]
- 96.Getahun A, Beavers NA, Larson SR, Shlomchik MJ, Cambier JC. Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic B cells. J Exp Med 2016;213(5):751–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seo S, Buckler J, Erikson J. Novel roles for Lyn in B cell migration and lipopolysaccharide responsiveness revealed using anti-double-stranded DNA Ig transgenic mice. J Immunol 2001;166(6):3710–6. [DOI] [PubMed] [Google Scholar]
- 98.Hua Z, Gross AJ, Lamagna C, Ramos-Hernandez N, Scapini P, Ji M, et al. Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Immunol 2014;192(3):875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Silver KL, Crockford TL, Bouriez-Jones T, Milling S, Lambe T, Cornall RJ. MyD88-dependent autoimmune disease in Lyn-deficient mice. Eur J Immunol 2007;37(10):2734–43. [DOI] [PubMed] [Google Scholar]
- 100.DeFranco AL. Multilayer Control of B Cell Activation by the B Cell Antigen Receptor: Following Themes Initiated With Bill Paul. Front Immunol 2018;9:739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luo W, Mayeux J, Gutierrez T, Russell L, Getahun A, Muller J, et al. A balance between B cell receptor and inhibitory receptor signaling controls plasma cell differentiation by maintaining optimal Ets1 levels. J Immunol 2014;193(2):909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Infantino S, Jones SA, Walker JA, Maxwell MJ, Light A, O’Donnell K, et al. The tyrosine kinase Lyn limits the cytokine responsiveness of plasma cells to restrict their accumulation in mice. Sci Signal 2014;7(338):ra77. [DOI] [PubMed] [Google Scholar]
- 103.Alvarez-Errico D, Yamashita Y, Suzuki R, Odom S, Furumoto Y, Yamashita T, et al. Functional analysis of Lyn kinase A and B isoforms reveals redundant and distinct roles in Fc epsilon RI-dependent mast cell activation. J Immunol 2010;184(9):5000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brian BFt, Jolicoeur AS, Guerrero CR, Nunez MG, Sychev ZE, Hegre SA, et al. Unique-region phosphorylation targets LynA for rapid degradation, tuning its expression and signaling in myeloid cells. Elife 2019;8. [DOI] [PMC free article] [PubMed]
- 105.Brian BF, Sauer ML, Ruis B, Auger JL, Senevirathne SE, Swanson WL, Nunez MG, Moriarity BS, Lowell CA, Binstadt BA, Freedman TS Splice-specific lyn knockout mice reveal a dominant function of LynB in preventing autoimmunity. BioRxiv 2021.
- 106.Hibbs ML, Harder KW, Armes J, Kountouri N, Quilici C, Casagranda F, et al. Sustained activation of Lyn tyrosine kinase in vivo leads to autoimmunity. J Exp Med 2002;196(12):1593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Faris M, Gaskin F, Parsons JT, Fu SM. CD40 signaling pathway: anti-CD40 monoclonal antibody induces rapid dephosphorylation and phosphorylation of tyrosine-phosphorylated proteins including protein tyrosine kinase Lyn, Fyn, and Syk and the appearance of a 28-kD tyrosine phosphorylated protein. J Exp Med 1994;179(6):1923–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ren CL, Morio T, Fu SM, Geha RS. Signal transduction via CD40 involves activation of lyn kinase and phosphatidylinositol-3-kinase, and phosphorylation of phospholipase C gamma 2. J Exp Med 1994;179(2):673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Taher TE, Parikh K, Flores-Borja F, Mletzko S, Isenberg DA, Peppelenbosch MP, et al. Protein phosphorylation and kinome profiling reveal altered regulation of multiple signaling pathways in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum 2010;62(8):2412–23. [DOI] [PubMed] [Google Scholar]
- 110.Flores-Borja F, Kabouridis PS, Jury EC, Isenberg DA, Mageed RA. Decreased Lyn expression and translocation to lipid raft signaling domains in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum 2005;52(12):3955–65. [DOI] [PubMed] [Google Scholar]
- 111.Liossis SN, Solomou EE, Dimopoulos MA, Panayiotidis P, Mavrikakis MM, Sfikakis PP. B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J Investig Med 2001;49(2):157–65. [DOI] [PubMed] [Google Scholar]
- 112.Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet 2012;44(11):1227–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Browne CD, Del Nagro CJ, Cato MH, Dengler HS, Rickert RC. Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of B cell anergy. Immunity 2009;31(5):749–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Getahun A, Wemlinger SM, Rudra P, Santiago ML, van Dyk LF, Cambier JC. Impaired B cell function during viral infections due to PTEN-mediated inhibition of the PI3K pathway. J Exp Med 2017;214(4):931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu XN, Ye YX, Niu JW, Li Y, Li X, You X, et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med 2014;6(246):246ra99. [DOI] [PubMed] [Google Scholar]
- 116.Smith MJ, Hinman RM, Getahun A, Kim S, Packard TA, Cambier JC. Silencing of high-affinity insulin-reactive B lymphocytes by anergy and impact of the NOD genetic background in mice. Diabetologia 2018;61(12):2621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thai TH, Patterson HC, Pham DH, Kis-Toth K, Kaminski DA, Tsokos GC. Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Fas(lpr) mouse. Proc Natl Acad Sci U S A 2013;110(50):20194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gonzalez-Martin A, Adams BD, Lai M, Shepherd J, Salvador-Bernaldez M, Salvador JM, et al. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat Immunol 2016;17(4):433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.O’Neill SK, Getahun A, Gauld SB, Merrell KT, Tamir I, Smith MJ, et al. Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP-1 phosphatase-mediated inhibitory signaling cascade required for B cell anergy. Immunity 2011;35(5):746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maxwell MJ, Duan M, Armes JE, Anderson GP, Tarlinton DM, Hibbs ML. Genetic segregation of inflammatory lung disease and autoimmune disease severity in SHIP-1−/− mice. J Immunol 2011;186(12):7164–75. [DOI] [PubMed] [Google Scholar]
- 121.Akerlund J, Getahun A, Cambier JC. B cell expression of the SH2-containing inositol 5-phosphatase (SHIP-1) is required to establish anergy to high affinity, proteinacious autoantigens. J Autoimmun 2015;62:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Preite S, Gomez-Rodriguez J, Cannons JL, Schwartzberg PL. T and B-cell signaling in activated PI3K delta syndrome: From immunodeficiency to autoimmunity. Immunol Rev 2019;291(1):154–73. [DOI] [PubMed] [Google Scholar]
- 123.Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: A large patient cohort study. J Allergy Clin Immunol 2017;139(2):597–606 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dulau Florea AE, Braylan RC, Schafernak KT, Williams KW, Daub J, Goyal RK, et al. Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J Allergy Clin Immunol 2017;139(3):1032–5 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lau A, Avery DT, Jackson K, Lenthall H, Volpi S, Brigden H, et al. Activated PI3Kdelta breaches multiple B cell tolerance checkpoints and causes autoantibody production. J Exp Med 2020;217(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Preite S, Cannons JL, Radtke AJ, Vujkovic-Cvijin I, Gomez-Rodriguez J, Volpi S, et al. Hyperactivated PI3Kdelta promotes self and commensal reactivity at the expense of optimal humoral immunity. Nat Immunol 2018;19(9):986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maxwell MJ, Tsantikos E, Kong AM, Vanhaesebroeck B, Tarlinton DM, Hibbs ML. Attenuation of phosphoinositide 3-kinase delta signaling restrains autoimmune disease. J Autoimmun 2012;38(4):381–91. [DOI] [PubMed] [Google Scholar]
- 128.Franks SE, Getahun A, Cambier JC. A Precision B Cell-Targeted Therapeutic Approach to Autoimmunity Caused by Phosphatidylinositol 3-Kinase Pathway Dysregulation. J Immunol 2019;202(12):3381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tsantikos E, Maxwell MJ, Kountouri N, Harder KW, Tarlinton DM, Hibbs ML. Genetic interdependence of Lyn and negative regulators of B cell receptor signaling in autoimmune disease development. J Immunol 2012;189(4):1726–36. [DOI] [PubMed] [Google Scholar]
- 130.Xu Y, Huntington ND, Harder KW, Nandurkar H, Hibbs ML, Tarlinton DM. Phosphatidylinositol-3 kinase activity in B cells is negatively regulated by Lyn tyrosine kinase. Immunol Cell Biol 2012;90(9):903–11. [DOI] [PubMed] [Google Scholar]
- 131.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature 1996;383(6597):263–6. [DOI] [PubMed] [Google Scholar]
- 132.Bewarder N, Weinrich V, Budde P, Hartmann D, Flaswinkel H, Reth M, et al. In vivo and in vitro specificity of protein tyrosine kinases for immunoglobulin G receptor (FcgammaRII) phosphorylation. Mol Cell Biol 1996;16(9):4735–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 1996;379(6563):346–9. [DOI] [PubMed] [Google Scholar]
- 134.Li F, Smith P, Ravetch JV. Inhibitory Fcgamma receptor is required for the maintenance of tolerance through distinct mechanisms. J Immunol 2014;192(7):3021–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 2000;13(2):277–85. [DOI] [PubMed] [Google Scholar]
- 136.Avalos AM, Uccellini MB, Lenert P, Viglianti GA, Marshak-Rothstein A. FcgammaRIIB regulation of BCR/TLR-dependent autoreactive B-cell responses. Eur J Immunol 2010;40(10):2692–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Espeli M, Bashford-Rogers R, Sowerby JM, Alouche N, Wong L, Denton AE, et al. FcgammaRIIb differentially regulates pre-immune and germinal center B cell tolerance in mouse and human. Nat Commun 2019;10(1):1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fukuyama H, Nimmerjahn F, Ravetch JV. The inhibitory Fcgamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nat Immunol 2005;6(1):99–106. [DOI] [PubMed] [Google Scholar]
- 139.Pauls SD, Ray A, Hou S, Vaughan AT, Cragg MS, Marshall AJ. FcgammaRIIB-Independent Mechanisms Controlling Membrane Localization of the Inhibitory Phosphatase SHIP in Human B Cells. J Immunol 2016;197(5):1587–96. [DOI] [PubMed] [Google Scholar]
- 140.Okada H, Bolland S, Hashimoto A, Kurosaki M, Kabuyama Y, Iino M, et al. Role of the inositol phosphatase SHIP in B cell receptor-induced Ca2+ oscillatory response. J Immunol 1998;161(10):5129–32. [PubMed] [Google Scholar]
- 141.Crute BW, Sheraden R, Ott VL, Harley ITW, Getahun A, Cambier JC. Inhibitory Receptor Trap: A Platform for Discovery of Inhibitory Receptors That Utilize Inositol Lipid and Phosphotyrosine Phosphatase Effectors. Front Immunol 2020;11:592329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mukherjee O, Weingarten L, Padberg I, Pracht C, Sinha R, Hochdorfer T, et al. The SH2-domain of SHIP1 interacts with the SHIP1 C-terminus: impact on SHIP1/Ig-alpha interaction. Biochim Biophys Acta 2012;1823(2):206–14. [DOI] [PubMed] [Google Scholar]
- 143.Osborne MA, Zenner G, Lubinus M, Zhang X, Songyang Z, Cantley LC, et al. The inositol 5’-phosphatase SHIP binds to immunoreceptor signaling motifs and responds to high affinity IgE receptor aggregation. J Biol Chem 1996;271(46):29271–8. [DOI] [PubMed] [Google Scholar]
- 144.Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal 2010;3(122):ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Torres RM, Hafen K. A negative regulatory role for Ig-alpha during B cell development. Immunity 1999;11(5):527–36. [DOI] [PubMed] [Google Scholar]
- 146.Kraus M, Saijo K, Torres RM, Rajewsky K. Ig-alpha cytoplasmic truncation renders immature B cells more sensitive to antigen contact. Immunity 1999;11(5):537–45. [DOI] [PubMed] [Google Scholar]
- 147.Kraus M, Pao LI, Reichlin A, Hu Y, Canono B, Cambier JC, et al. Interference with immunoglobulin (Ig)alpha immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation modulates or blocks B cell development, depending on the availability of an Igbeta cytoplasmic tail. J Exp Med 2001;194(4):455–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Reichlin A, Gazumyan A, Nagaoka H, Kirsch KH, Kraus M, Rajewsky K, et al. A B cell receptor with two Igalpha cytoplasmic domains supports development of mature but anergic B cells. J Exp Med 2004;199(6):855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gazumyan A, Reichlin A, Nussenzweig MC. Ig beta tyrosine residues contribute to the control of B cell receptor signaling by regulating receptor internalization. J Exp Med 2006;203(7):1785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Reichlin A, Hu Y, Meffre E, Nagaoka H, Gong S, Kraus M, et al. B cell development is arrested at the immature B cell stage in mice carrying a mutation in the cytoplasmic domain of immunoglobulin beta. J Exp Med 2001;193(1):13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Losing M, Goldbeck I, Manno B, Oellerich T, Schnyder T, Bohnenberger H, et al. The Dok-3/Grb2 protein signal module attenuates Lyn kinase-dependent activation of Syk kinase in B cell antigen receptor microclusters. J Biol Chem 2013;288(4):2303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Reth M, Gold MR. What goes up must come down: A tripartite Dok-3/Grb2/SHIP1 inhibitory module limits BCR signaling. Eur J Immunol 2016;46(11):2507–11. [DOI] [PubMed] [Google Scholar]
- 153.Patterson HC, Kraus M, Wang D, Shahsafaei A, Henderson JM, Seagal J, et al. Cytoplasmic Ig alpha serine/threonines fine-tune Ig alpha tyrosine phosphorylation and limit bone marrow plasma cell formation. J Immunol 2011;187(6):2853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Clark MR, Campbell KS, Kazlauskas A, Johnson SA, Hertz M, Potter TA, et al. The B cell antigen receptor complex: association of Ig-alpha and Ig-beta with distinct cytoplasmic effectors. Science 1992;258(5079):123–6. [DOI] [PubMed] [Google Scholar]
- 155.Castello A, Gaya M, Tucholski J, Oellerich T, Lu KH, Tafuri A, et al. Nck-mediated recruitment of BCAP to the BCR regulates the PI(3)K-Akt pathway in B cells. Nat Immunol 2013;14(9):966–75. [DOI] [PubMed] [Google Scholar]
- 156.Liu X, Manser T. Antinuclear antigen B cells that down-regulate surface B cell receptor during development to mature, follicular phenotype do not display features of anergy in vitro. J Immunol 2005;174(8):4505–15. [DOI] [PubMed] [Google Scholar]
- 157.Liu X, Wysocki LJ, Manser T. Autoantigen-B cell antigen receptor interactions that regulate expression of B cell antigen receptor Loci. J Immunol 2007;178(8):5035–47. [DOI] [PubMed] [Google Scholar]
- 158.Ubelhart R, Hug E, Bach MP, Wossning T, Duhren-von Minden M, Horn AH, et al. Responsiveness of B cells is regulated by the hinge region of IgD. Nat Immunol 2015;16(5):534–43. [DOI] [PubMed] [Google Scholar]
- 159.Noviski M, Mueller JL, Satterthwaite A, Garrett-Sinha LA, Brombacher F, Zikherman J. IgM and IgD B cell receptors differentially respond to endogenous antigens and control B cell fate. Elife 2018;7. [DOI] [PMC free article] [PubMed]
- 160.Renna V, Surova E, Khadour A, Datta M, Amendt T, Hobeika E, et al. Defective Allelic Exclusion by IgD in the Absence of Autoantigen. J Immunol 2021. [DOI] [PubMed]
- 161.Zikherman J, Parameswaran R, Weiss A. Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature 2012;489(7414):160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kirchenbaum GA, St Clair JB, Detanico T, Aviszus K, Wysocki LJ. Functionally responsive self-reactive B cells of low affinity express reduced levels of surface IgM. Eur J Immunol 2014;44(4):970–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Maity PC, Blount A, Jumaa H, Ronneberger O, Lillemeier BF, Reth M. B cell antigen receptors of the IgM and IgD classes are clustered in different protein islands that are altered during B cell activation. Sci Signal 2015;8(394):ra93. [DOI] [PubMed] [Google Scholar]
- 164.Gasparrini F, Feest C, Bruckbauer A, Mattila PK, Muller J, Nitschke L, et al. Nanoscale organization and dynamics of the siglec CD22 cooperate with the cytoskeleton in restraining BCR signalling. EMBO J 2016;35(3):258–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Eris JM, Basten A, Brink R, Doherty K, Kehry MR, Hodgkin PD. Anergic self-reactive B cells present self antigen and respond normally to CD40-dependent T-cell signals but are defective in antigen-receptor-mediated functions. Proc Natl Acad Sci U S A 1994;91(10):4392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wu W, Zhou Q, Masubuchi T, Shi X, Li H, Xu X, et al. Multiple Signaling Roles of CD3epsilon and Its Application in CAR-T Cell Therapy. Cell 2020;182(4):855–71 e23. [DOI] [PubMed] [Google Scholar]
- 167.Kersh EN, Kersh GJ, Allen PM. Partially phosphorylated T cell receptor zeta molecules can inhibit T cell activation. J Exp Med 1999;190(11):1627–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ben Mkaddem S, Hayem G, Jonsson F, Rossato E, Boedec E, Boussetta T, et al. Shifting FcgammaRIIA-ITAM from activation to inhibitory configuration ameliorates arthritis. J Clin Invest 2014;124(9):3945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cyster JG, Goodnow CC. Protein tyrosine phosphatase 1C negatively regulates antigen receptor signaling in B lymphocytes and determines thresholds for negative selection. Immunity 1995;2(1):13–24. [DOI] [PubMed] [Google Scholar]
- 170.Akatsu C, Shinagawa K, Numoto N, Liu Z, Ucar AK, Aslam M, et al. CD72 negatively regulates B lymphocyte responses to the lupus-related endogenous toll-like receptor 7 ligand Sm/RNP. J Exp Med 2016;213(12):2691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li DH, Winslow MM, Cao TM, Chen AH, Davis CR, Mellins ED, et al. Modulation of peripheral B cell tolerance by CD72 in a murine model. Arthritis Rheum 2008;58(10):3192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xu M, Hou R, Sato-Hayashizaki A, Man R, Zhu C, Wakabayashi C, et al. Cd72(c) is a modifier gene that regulates Fas(lpr)-induced autoimmune disease. J Immunol 2013;190(11):5436–45. [DOI] [PubMed] [Google Scholar]
- 173.Nakano S, Morimoto S, Suzuki J, Mitsuo A, Nakiri Y, Katagiri A, et al. Down-regulation of CD72 and increased surface IgG on B cells in patients with lupus nephritis. Autoimmunity 2007;40(1):9–15. [DOI] [PubMed] [Google Scholar]
- 174.Asmiyou A, Bakr AM, Shahin DA, Wahba Y. CD40 and CD72 expression and prognostic values among children with systemic lupus erythematosus: a case-control study. Lupus 2020;29(10):1270–6. [DOI] [PubMed] [Google Scholar]
- 175.Meyer SJ, Linder AT, Brandl C, Nitschke L. B Cell Siglecs-News on Signaling and Its Interplay With Ligand Binding. Front Immunol 2018;9:2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nitschke L, Carsetti R, Ocker B, Kohler G, Lamers MC. CD22 is a negative regulator of B-cell receptor signalling. Curr Biol 1997;7(2):133–43. [DOI] [PubMed] [Google Scholar]
- 177.O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Hyperresponsive B cells in CD22-deficient mice. Science 1996;274(5288):798–801. [DOI] [PubMed] [Google Scholar]
- 178.Otipoby KL, Andersson KB, Draves KE, Klaus SJ, Farr AG, Kerner JD, et al. CD22 regulates thymus-independent responses and the lifespan of B cells. Nature 1996;384(6610):634–7. [DOI] [PubMed] [Google Scholar]
- 179.Sato S, Miller AS, Inaoki M, Bock CB, Jansen PJ, Tang ML, et al. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: altered signaling in CD22-deficient mice. Immunity 1996;5(6):551–62. [DOI] [PubMed] [Google Scholar]
- 180.Hutzler S, Ozgor L, Naito-Matsui Y, Klasener K, Winkler TH, Reth M, et al. The ligand-binding domain of Siglec-G is crucial for its selective inhibitory function on B1 cells. J Immunol 2014;192(11):5406–14. [DOI] [PubMed] [Google Scholar]
- 181.Hoffmann A, Kerr S, Jellusova J, Zhang J, Weisel F, Wellmann U, et al. Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat Immunol 2007;8(7):695–704. [DOI] [PubMed] [Google Scholar]
- 182.O’Keefe TL, Williams GT, Batista FD, Neuberger MS. Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med 1999;189(8):1307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Muller J, Lunz B, Schwab I, Acs A, Nimmerjahn F, Daniel C, et al. Siglec-G Deficiency Leads to Autoimmunity in Aging C57BL/6 Mice. J Immunol 2015;195(1):51–60. [DOI] [PubMed] [Google Scholar]
- 184.Jellusova J, Wellmann U, Amann K, Winkler TH, Nitschke L. CD22 x Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J Immunol 2010;184(7):3618–27. [DOI] [PubMed] [Google Scholar]
- 185.Mary C, Laporte C, Parzy D, Santiago ML, Stefani F, Lajaunias F, et al. Dysregulated expression of the Cd22 gene as a result of a short interspersed nucleotide element insertion in Cd22a lupus-prone mice. J Immunol 2000;165(6):2987–96. [DOI] [PubMed] [Google Scholar]
- 186.Bokers S, Urbat A, Daniel C, Amann K, Smith KG, Espeli M, et al. Siglec-G deficiency leads to more severe collagen-induced arthritis and earlier onset of lupus-like symptoms in MRL/lpr mice. J Immunol 2014;192(7):2994–3002. [DOI] [PubMed] [Google Scholar]
- 187.Duong BH, Tian H, Ota T, Completo G, Han S, Vela JL, et al. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J Exp Med 2010;207(1):173–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kubo T, Uchida Y, Watanabe Y, Abe M, Nakamura A, Ono M, et al. Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. J Exp Med 2009;206(9):1971–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Maeda A, Scharenberg AM, Tsukada S, Bolen JB, Kinet JP, Kurosaki T. Paired immunoglobulin-like receptor B (PIR-B) inhibits BCR-induced activation of Syk and Btk by SHP-1. Oncogene 1999;18(14):2291–7. [DOI] [PubMed] [Google Scholar]
- 190.Blery M, Kubagawa H, Chen CC, Vely F, Cooper MD, Vivier E. The paired Ig-like receptor PIR-B is an inhibitory receptor that recruits the protein-tyrosine phosphatase SHP-1. Proc Natl Acad Sci U S A 1998;95(5):2446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Vendel AC, Calemine-Fenaux J, Izrael-Tomasevic A, Chauhan V, Arnott D, Eaton DL. B and T lymphocyte attenuator regulates B cell receptor signaling by targeting Syk and BLNK. J Immunol 2009;182(3):1509–17. [DOI] [PubMed] [Google Scholar]
- 192.Oya Y, Watanabe N, Owada T, Oki M, Hirose K, Suto A, et al. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum 2008;58(8):2498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hippen KL, Tze LE, Behrens TW. CD5 maintains tolerance in anergic B cells. J Exp Med 2000;191(5):883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Haga CL, Ehrhardt GR, Boohaker RJ, Davis RS, Cooper MD. Fc receptor-like 5 inhibits B cell activation via SHP-1 tyrosine phosphatase recruitment. Proc Natl Acad Sci U S A 2007;104(23):9770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wiedemann A, Lettau M, Weissenberg SY, Stefanski AL, Schrezenmeier EV, Rincon-Arevalo H, et al. BTLA Expression and Function Are Impaired on SLE B Cells. Front Immunol 2021;12:667991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Melissaropoulos K, Liossis SN. Decreased CD22 expression and intracellular signaling aberrations in B cells of patients with systemic sclerosis. Rheumatol Int 2018;38(7):1225–34. [DOI] [PubMed] [Google Scholar]
- 197.Medgyesi D, Hobeika E, Biesen R, Kollert F, Taddeo A, Voll RE, et al. The protein tyrosine phosphatase PTP1B is a negative regulator of CD40 and BAFF-R signaling and controls B cell autoimmunity. J Exp Med 2014;211(3):427–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wiede F, Sacirbegovic F, Leong YA, Yu D, Tiganis T. PTPN2-deficiency exacerbates T follicular helper cell and B cell responses and promotes the development of autoimmunity. J Autoimmun 2017;76:85–100. [DOI] [PubMed] [Google Scholar]
- 199.Long D, Chen Y, Wu H, Zhao M, Lu Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J Autoimmun 2019;99:1–14. [DOI] [PubMed] [Google Scholar]
- 200.Sharp RC, Abdulrahim M, Naser ES, Naser SA. Genetic Variations of PTPN2 and PTPN22: Role in the Pathogenesis of Type 1 Diabetes and Crohn’s Disease. Front Cell Infect Microbiol 2015;5:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Long SA, Cerosaletti K, Wan JY, Ho JC, Tatum M, Wei S, et al. An autoimmune-associated variant in PTPN2 reveals an impairment of IL-2R signaling in CD4(+) T cells. Genes Immun 2011;12(2):116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Huck S, Le Corre R, Youinou P, Zouali M. Expression of B cell receptor-associated signaling molecules in human lupus. Autoimmunity 2001;33(3):213–24. [DOI] [PubMed] [Google Scholar]
- 203.Ozgor L, Brandl C, Shock A, Nitschke L. Epratuzumab modulates B-cell signaling without affecting B-cell numbers or B-cell functions in a mouse model with humanized CD22. Eur J Immunol 2016;46(9):2260–72. [DOI] [PubMed] [Google Scholar]
- 204.Sieger N, Fleischer SJ, Mei HE, Reiter K, Shock A, Burmester GR, et al. CD22 ligation inhibits downstream B cell receptor signaling and Ca(2+) flux upon activation. Arthritis Rheum 2013;65(3):770–9. [DOI] [PubMed] [Google Scholar]