

Abstract
The isonitrile moiety is an electron-rich functionality that decorates various bioactive natural products isolated from diverse kingdoms of life. Isonitrile biosynthesis was restricted for over a decade to isonitrile synthases, a family of enzymes catalyzing a condensation reaction between l-Trp/l-Tyr and ribulose-5-phosphate. The discovery of ScoE, a non-heme iron(II) and α-ketoglutarate-dependent dioxygenase, demonstrated an alternative pathway employed by nature for isonitrile installation. Biochemical, crystallographic, and computational investigations of ScoE have previously been reported, yet the isonitrile formation mechanism remains obscure. In this present work, we employed in vitro biochemistry, chemical synthesis, spectroscopy techniques, and computational simulations that enabled us to propose a plausible molecular mechanism for isonitrile formation. Our findings demonstrate that the ScoE reaction initiates with C5 hydroxylation of (R)-3-((carboxymethyl)amino)butanoic acid to generate 1, which undergoes dehydration presumably mediated by Tyr96 to synthesize 2 in a trans configuration. (R)-3-isocyanobutanoic acid is finally generated through radical-based decarboxylation of 2, instead of the common hydroxylation pathway employed by this enzyme superfamily.
Graphical Abstract

Introduction
The isonitrile functional group is an intriguing synthon reported in hundreds of characterized natural products isolated from various organisms (Figure S1).1–10 The isonitrile moiety commonly behaves as a warhead that imparts potent bioactivities associated with virulence, detoxification, metal acquisition, and antimicrobial activities.1–3 There are currently two known biosynthetic pathways for isonitrile formation.11 The first pathway is catalyzed by the isonitrile synthase (IsnA) family that installs isonitrile via condensation of the α-nitrogen atom from l-Tyr/l-Trp and a carbon atom derived from ribulose-5-phosphate (Figure S2A).12 We recently uncovered the second mechanism from studying a conserved gene cluster in some Actinobacteria species involved in isonitrile lipopeptide (INLP) biosynthesis.13 ScoE, a non-heme iron(II) and α-ketoglutarate (α-KG)-dependent dioxygenase (Fe(II)/α-KG-dioxygenase), promoted the conversion of (R)-3-((carboxymethyl)amino)butanoic acid (CABA) to (R)-3-isocyanobutanoic acid (INBA) through an oxidative decarboxylation mechanism (Figure 1, Figure S2B).14
Figure 1.

Overall reaction of ScoE and detection of possible intermediates. (A) Extracted ion chromatograms (EICs) showing glyoxylate and R-3-ABA formation from ScoE assays derivatized with 2-NPH under acidic conditions. Glyoxylate and R-3-ABA are degradation products of possible reaction intermediate 2 under acidic conditions. (B) EICs of 3 generated from the ScoE reaction subject to negative controls. (C) EICs demonstrating production of 4 from assays containing CABA, α-KG, Fe(II), and ScoE. Metabolite 4 shows no evidence of consumption from the time course. A 10-ppm mass error tolerance was used for each trace with the masses listed.
Fe(II)/α-KG-dioxygenases catalyze diverse oxidative reactions including but not limited to hydroxylation, desaturation, epoxidation, and halogenation.15 Fe(II)/α-KG-dioxygenases are thought to possess a conserved activation mechanism involving mononuclear Fe(II) coordinated to a conserved 2-histidine-1-carboxylate facial triad with α-KG bound to the metallocofactor in a bidentate fashion.15 The carboxylate ligand is replaced by an alanine/glycine in the case of Fe(II)/α-KG-halogenases for halide coordination.16,17The final coordination site, usually found in proximity to the substrate binding site, is occupied by molecular oxygen to yield an Fe(III)-superoxo species.15,18 The distal oxygen from the Fe(III)-superoxo intermediate subsequently attacks C2 of α-KG to yield a peroxohemiketal bicyclic species that undergoes oxidative cleavage to yield succinate, CO2, and a high-valent Fe(IV)=O species.19 The Fe(IV)=O species is responsible for the oxidative transformations by abstracting a hydrogen atom from the primary substrate to yield an Fe(III)-OH species and a substrate radical.20 The steps beyond this point in the mechanism depend on the enzymatic reaction in question. For example, with hydroxylation, the Fe(III)-OH bond undergoes homolytic cleavage to promote a “hydroxyl rebound” on the primary substrate.15,19,20 Despite precedence of Fe(II)/α-KG-dioxygenases promoting diverse chemical transformations, isonitrile formation is a newly discovered activity possessed by this enzyme family consisting of a 4-electron oxidation of the primary substrate to yield INBA. This chemical transformation is quite different compared to canonical 2-electron oxidation reactions that are catalyzed by most Fe(II)/α-KG-dioxygenases on the primary substrate, thus warranting mechanistic interrogation to explain the logic behind the two N-C bond desaturation reactions.
In an attempt to reveal the ScoE-catalyzed reaction and mechanism, we previously established the reaction stoichiometry. Two equivalents of oxygen and α-KG were required to produce one equivalent of INBA, with the primary substrate CABA undergoing oxidative decarboxylation, not deformylation nor decarbonylation.21 We obtained several crystal structures of ScoE with and without substrates, and noted an ordered water network extending from the CABA site.14,21 Through site-directed mutagenesis, we confirmed the importance of Tyr96, Tyr97, Tyr101, and Arg195 at maintaining the hydrogen bonding network between three ordered water molecules since alterations of these residues completely abrogated or severely compromised isonitrile formation. Several pathways to isonitrile formation were previously proposed; however, a definitive pathway with evidence for reaction intermediates remains elusive.14,21,22. In this present work, we report putative intermediates and shunt products detected from the ScoE reaction and their apparent formation rates. To supplement our experimental results, we performed DFT cluster calculations to better understand the energetics surrounding the proposed intermediates. We further propose a plausible mechanism that revises previously reported mechanisms for isonitrile formation and is consistent with the recent independent work from Chen et al.23
Results
Detection of putative ScoE reaction intermediates by NMR and LC-HRMS analysis.
To reveal putative reaction intermediates, we attempted to reconstitute the ScoE reaction using 13C-labeled CABA and monitor the reaction at different time points with nuclear magnetic resonance (NMR) and liquid chromatography high-resolution mass spectrometry (LC-HRMS). ScoE from Streptomyces coeruleorubidus was purified from E. coli by removing co-purified metals and reconstituting its activity with fresh Fe(II) before each enzymatic assay (Figure S3, Table S1–S2).14,21 An in vitro biochemical assay incubated for three hours at 4 °C containing 5-13C-CABA, resulted in detection of two new enriched correlations in HSQC and one new enriched correlation in HMBC, respectively, which were not present in assays containing unlabeled CABA (Figures S4–S5). The first HSQC signal at δc~88 ppm was consistent with the C5 signal of C5-OH CABA (1) that was previously reported for an enzymatic assay with SfaA, a homolog of ScoE (85% sequence similarity).22 The second HSQC signal detected at δc~162 ppm was assigned to C5 of imine CABA (2) in comparison to a synthetic standard (Figures S5A–S7). The proton chemical shift from C5 of 2 differed from synthetic 2, likely because the buffered environment favored 2 in the ionized state, thus decreasing the electron cloud density at the sp2 methine.24 Although we were unable to directly detect 2 using LC-HRMS, its presence was supported by detection of (R)-3-aminobutanoic acid (R-3-ABA) and glyoxylate from ScoE assays derivatized with 2-nitrophenylhydrazine (2-NPH) after acidifying reactions with HCl (Figure 1A, Figure S8).25 Furthermore, utilization of 5-13C-CABA and 6-13C-CABA led to the expected mass spectral shift of derivatized glyoxylate, respectively (Figure 1A, Figure S8A–B). It is notable that only a trace amount of R-3-ABA could be detected from enzymatic reactions without acidification, suggesting that the degradation of 2 to R-3-ABA and glyoxylate in the reaction condition was minimal. The signal at δc~154 ppm in HMBC corresponds to C5 of INBA in comparison to previously reported NMR spectra (Figure S5B).22
An overnight assay with 5-13C-CABA revealed the same signals assigned to C5 of 2 and C5 of INBA, while the C5 signal of 1 was no longer detected (Figure S9). We also observed several new enriched NMR signals including correlations at δc~165 ppm in HSQC, and at δc~164 and 168 ppm in HMBC (Figure S9). The HSQC signal was assigned to C5 of (R)-3-formamidobutanoic acid (FABA, 3) based on NMR confirmation using a synthetic standard (Figure 1B, Figure S10). The presence of 3 was further supported with analysis of the reaction with LC-HRMS ([M+H]+ calculated: 132.0656, observed: 132.0658, 1.5 ppm error) (Figure 1B, Figure S10A–B). The two remaining HMBC signals were tentatively assigned to C5 of C5-OH imine CABA (4) and its tautomer 5-oxo CABA (5) due to the following reasons. First, a comparative metabolomics experiment revealed a peak with a mass consistent to both species ([M-H]− calculated: 174.0407, observed: 174.0408, 0.6 ppm error) that was absent from negative controls (Figure 1C, Figure S11A). Second, a synthetic standard of 5 matched the C5 chemical shift with δc~164 ppm but did not match the retention time of the peak detected from LC-HRMS, suggesting the mass peak to be 4 (Figure 1C, Figure S9, Figure S12). Third, the HMBC signal at δc~168 ppm is consistent with C5 of 4 because the carbon atom bonded to the enolic OH group is more deshielded than C5 of 5 and thus yielding a chemical shift further downfield (Figure S9).24 These results suggested that 4 was first generated from the ScoE reaction as detected by LC-HRMS, which could tautomerize to form 5 as supported by NMR analysis (Figure 1C, Figure S9, Figures S11–12).
Identification of on-pathway intermediates and shunt products.
We next sought to assign 1–5 to be reaction intermediates or shunt products. 1 was proposed to be the first intermediate in the pathway based on the previously reported structure of ScoE with 1 and detection of 1 from a 13C-NMR experiment analyzing an enzymatic assay from SfaA.22 Our recent structures of ScoE with bound CABA further demonstrated that one of the C5 hydrogens points toward the iron cofactor, suggesting that C5 was the most likely site of initial C-H activation and hydroxylation.21 Consistently, we observed a transient production of 1 where it was detected from a three-hour assay at 4 °C, but not from an overnight assay based on the NMR analysis, thus supporting the intermediacy of this compound (Figure S4, Figure S9). 2 was proposed to be the next on-pathway intermediate upon dehydration of 1. An in vitro biochemical assay with the chemically synthesized 2, α-KG, Fe(II), and ScoE successfully generated INBA compared to negative controls, confirming that 2 was a reaction intermediate of ScoE (Figure 2A, Figure S13). Notably, only 2 with trans-configuration was chemically synthesized based on the NOESY correlation between the sp2-methine and the nitrogen bearing sp3-methine observed from both the final synthetic compound and its ester synthetic precursor (Figure S6–S7). A previous study reported that cis-imine CABA was not recognized by SfaA to generate INBA22, although this conclusion was questionable based on a recent paper from the same group which confirmed 2 as a reaction intermediate without specifying its configuration.23 We further determined the kinetic parameters of ScoE towards 2 (kcat=21.9 ± 0.6 min−1, KM=369 ± 40 μM), which were comparable to CABA (kcat=21.9 ± 2.1 min−1, KM=286 ± 93 μM) as reported previously (Figure 2B).14
Figure 2.
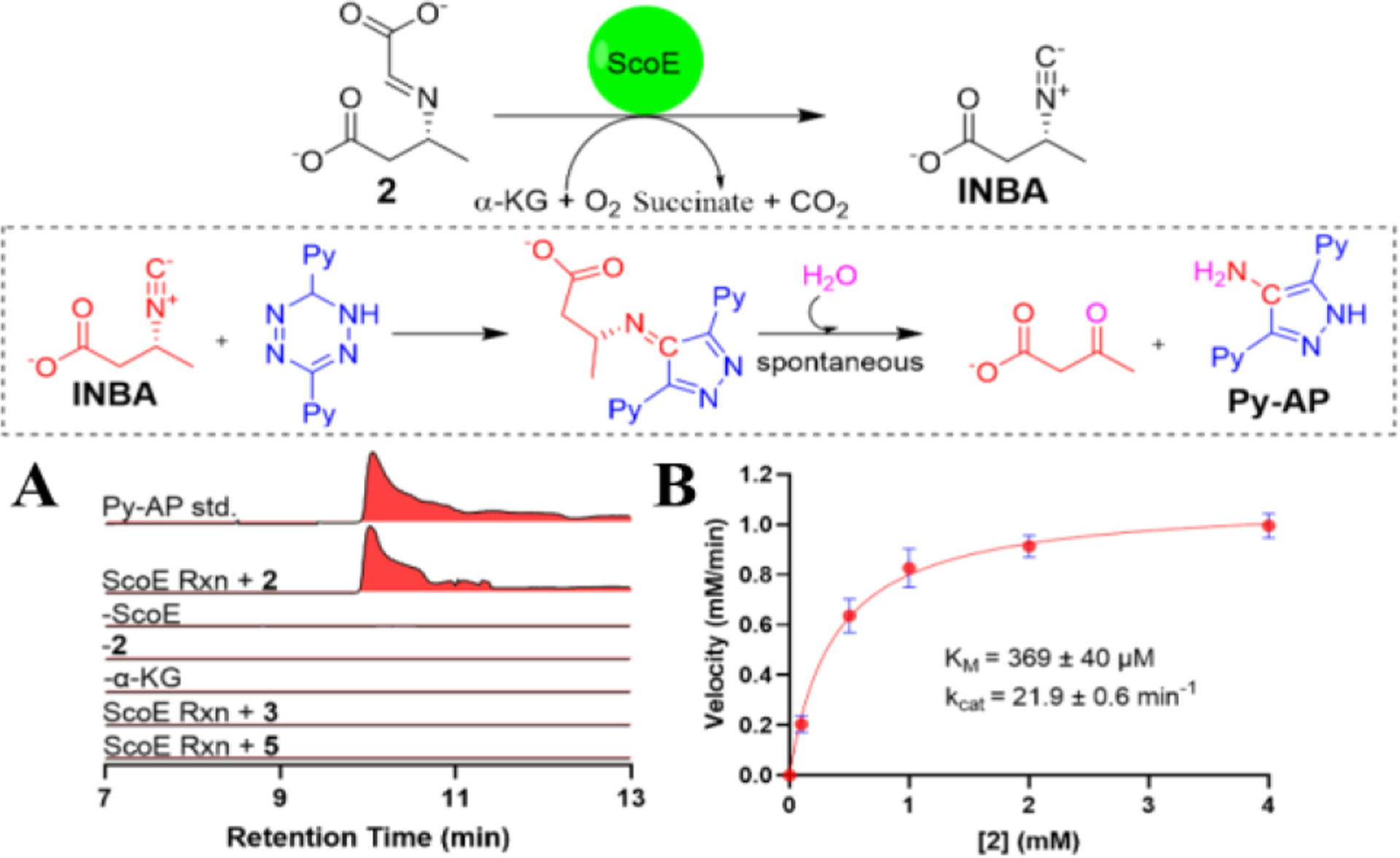
Biochemical analysis of ScoE utilizing various substrates. (A) EICs demonstrating the production of 3,5-di(pyridine-2-yl)-1Hpyrazol-4-amine (Py-AP) as a product of the isonitriletetrazine click reaction when 2 was used as a substrate. Omission of any of the assay components led to abolition of Py-AP. Utilization of 3 and 5 as substrates was not recognized by ScoE. (B) ScoE kinetic parameters for 2 in INBA formation. The data points and error bars represent the average and standard deviation from three independent experiments, respectively. Calculated mass for Py-AP (m/z=238.1088 [M+H]+) was used for each trace with a 10-ppm error tolerance.
In contrast to the proposed intermediacy of 1 and 2, compounds 3–5 were proposed to be shunt products. Specifically, 3 was predicted to be a degradation product of INBA as isonitrile-bearing natural products were reported to undergo de-composition to their corresponding formamides.26–28 Notably, these formamides were shown not to be direct precursors of isonitrile-containing natural products from in vivo feeding experiments of radiolabeled formamide in which radiolabeled isonitrile compounds were not detected from the cultures.26–28 Consistent with in vivo results, our in vitro biochemical assay using 3 in place of CABA failed to generate INBA, arguing against the intermediacy of 3 in ScoE reactions (Figure 2A). In addition, an in vitro ScoE reaction performed in 18O-water resulted in incorporation of an 18O atom from the solvent into 3 based on HRMS analysis, suggesting the formation of 3 via water-based degradation, although the aldehyde may also exchange its oxygen atom with the solvent (Figure S10C). 3 was also detected by LC-HRMS as a degradation product from synthetic INBA (Figure S10D). Similarly, a biochemical assay using synthetic 5 did not generate isonitrile, suggesting that 5 was also not an intermediate (Figure 2A). The failed recognition of 5 by ScoE is in agreement with previous work from Chen et al., but Chen favored 3 as an on-pathway intermediate which is in contrast to our observations.22 We could not synthesize a standard of 4 for ScoE assays due to its inherent propensity to tautomerize to 5 or become hydrolyzed. Nonetheless, we reasoned 4 to be a likely shunt product as the time course experiments of ScoE reactions showed no evidence of 4 consumption after generation, in contrast to 1 and 2 (Figure S5, Figure S8A, Figure S9, Figure 1C).
Quantification and kinetic analysis of ScoE reaction products.
A quantitative analysis of the ScoE reaction was next conducted to obtain insight into the relative amounts of products under different reaction conditions. The amount of INBA, 2, 3, and 4 was quantified through LC-MS analysis under conditions of limited (1:1) and excess α-KG (3:1) with respect to the CABA substrate, which showed a similar trend except that 4 was produced more when α-KG was in excess (Figure 3A). Based on the apparent formation rate of 2 in the overall ScoE reaction (Figure 3B), it could be deduced that the formation of 2 from CABA was much faster than the conversion of 2 to INBA (Figure 2B), suggesting that the second half reaction of 2 to INBA is a rate limiting step in the enzymatic conversion of CABA to INBA by ScoE. In addition, the relatively low formation rates of 3 and 4 compared to INBA demonstrated that these degradation or shunt pathways could not outcompete the on-pathway reaction to form INBA (Figure 3).
Figure 3.

LC-MS quantification and kinetic rate constants for ScoE reaction products. A) Relative amounts of INBA, 2, 3, and 4 quantified by LC-MS from a series of in vitro assays using different ratios of CABA:α-KG (1:1 and 1:3). Error bars correspond to standard deviation of the mean from three replicate experiments. Detailed procedures are found in the Supporting Information. B) Apparent rate constants of formation at excess α-KG conditions (1:3) for compounds 2, 3, and 4. Error bars correspond to standard deviation of the mean from three replicate experiments.
Analysis of DFT QM cluster calculations and transition states for proposed intermediates.
To investigate the energetics of the preferred mechanism, we performed density functional theory (DFT) quantum mechanical (QM) cluster calculations on all proposed intermediates and identified putative transition states (Figure S14, Text S1, Tables S3–S6). The calculations were performed at the UB3LYP/LACVP* level of theory. Single point calculations of geometry optimized intermediates and approximate transition states from the proposed mechanism were also evaluated with the larger def2-TZVP basis set on the metal center, and mechanistic conclusions were qualitatively unchanged (Text S2, Table S6). Although previous computational studies have been performed on ScoE, our calculations incorporate the recently elucidated crystallographic positioning of CABA (Figure S15).21,29,30 We also expanded upon previous ScoE QM models by including backbone atoms of key residues, the second coordination sphere residue Thr145, and the backbone of Tyr135, which forms a hydrogen bond with CABA (Figure 4). For the QM calculations, all sidechain atoms were allowed to move freely while the backbone atoms were restrained to maintain their crystal structure positions. Coordinates for all reaction scans and putative transition states are provided as a zip file with the supplemental information. The charge shift analysis method was used to confirm key residues previously proposed to be involved in charge transfer with the substrate (e.g., Lys193 and Arg310) (Text S3, Figure S16).31,32
Figure 4.

Depiction of the ScoE active site and the amino acids selected for inclusion in the QM cluster model. Key residues, ligands, and interactions are shown with their skeleton structures. Interactions are shown as dotted red lines. All residues and ligands are superimposed on the ScoE backbone with the α-carbon positions denoted with a black dot.
Starting with the first half reaction, we compared the energetics of C5-H activation (TS1A) and the previously proposed N-H hydrogen atom transfer (HAT) reaction (TS1B). Since the N-H hydrogen is oriented a significant distance from the oxo ligand in the crystal structure, 5.6 Å, and forms a strong hydrogen bond with Tyr96, the QM model for the N-H HAT mechanism was generated from the centroid of a 50ns clustered molecular dynamics (MD) simulation (Text S4–S5, Table S7–S11). The simulation sampled conformations with the CABA amine oriented toward the Fe(IV)=O center (Figure S17–S20). From the QM cluster calculations, we found that the crystallographic conformation of CABA favors C5-H activation with a modest energy barrier of 9.3 kcal/mol (TS1A) and a larger barrier of 21.9 kcal/mol for the previously proposed N-H abstraction due to conformational changes and the required breaking of a strong hydrogen bond between Tyr96 and the CABA amine (Figure S21–S22).29 Following the HAT at C5, the relative energy of the iron-hydroxyl intermediate was calculated to be −42.3 kcal/mol. Upon closer analysis, we found no radical character on the substrate in line with previous observations.29 While small model calculations show the radical based mechanism to also be favorable at −9.5 kcal/mol relative to CABA (Figure S23), a carbocation mechanism may also explain the formation of the iron-hydroxyl intermediate with more favorable energetics (Figure S14). The subsequent hydroxyl rebound step (TS2A) was effectively barrierless with a barrier of 0.4 kcal/mol relative to the iron-hydroxyl intermediate (Figure S24). Additionally, we found the dehydration of 1 to form 2 (TS3A), to have a barrier of −33.0 kcal/mol relative to the starting reactant and 17.7 kcal/mol relative to 2 (Figure S25). Conversely, the previously proposed second hydrogen abstraction (TS2B) had an energy barrier of 12.7 kcal/mol relative to IM1B but an energy barrier of 17.4 kcal/mol relative to the starting reactant (Figure 26).29 While we chose to use anionic Tyr96 as the base, due to its ideal positioning and known mechanistic importance, water may also act as the required base.
Applying our QM studies to the second half reaction, we sought to evaluate the proposed shunt product status of 4. One notable difference between our calculations and previous computational studies, is the use of trans-2, which was shown experimentally to generate INBA in this work.22 Starting from 2, we found the second C5-H HAT reaction (TS4AB) to have an energy barrier of 21.2 kcal/mol, in line with the difficulty of abstracting a hydrogen from an sp2 carbon (Figure S27). Following the HAT step, we modelled the hydroxyl rebound that generates 4 (TS5A). In line with previous computational observations, we report an activation barrier higher than is typically observed for the rebound step of non-heme iron(II)-dependent hydroxylases of 18.3 kcal/mol relative to the starting reactant (Figure S28). We also found that 4 was located at an energy significantly lower than INBA, which suggests 4 is not an intermediate consistent with previous reports (Figure S29).29,30 Rather than proceeding via a hydroxylated intermediate, DFT calculations indicate a preference of a potential decarboxylation mechanism triggered by the transfer of the radical to the iron center, which we found to have a low energy barrier (TS5B) of −2.7 kcal/mol relative to the second half-reaction starting complex (Figure S30, Figure S31). Together these results support 1 and 2 as reaction intermediates and suggest that 4 is likely a shunt product (Figure 5).
Figure 5.

Relative energy profiles in kcal/mol for the first and second half reactions with optimized intermediate structures. (A) The first half reaction depicts the energy levels of the first C5-H HAT and the subsequent hydroxyl rebound, as well as the final generation of 2. (B) The second half reaction depicts the energy levels of the second HAT from 2 and the decarboxylation triggered by a radical transfer to the iron center. The intermediates 1 and 2 are labeled in bold. Only the coordinating atoms and substrate are shown for clarity.
Previous studies proposed the possibility of an alternate pathway for the generation of INBA in the second half reaction that proceeds via N-activation of the substrate by the iron-oxo species (Figure S32).21,22 To investigate the feasibility of the N-activation pathway, we performed QM cluster model calculations on this step using the same cluster model as described previously. We identified intermediates and approximate transition states from constrained geometry scans (Figures S33–36). Our calculations reveal several significant energy barriers to the N-activation pathway, which leads us to disfavor it as a mechanism for INBA formation (Figure S37). The largest energy barrier of 74.6 kcal/mol corresponded to the breaking of the N-O bond and the generation of the radical on C5 (Figure S35), which is significantly higher than the 21.2 kcal/mol activation barrier observed for the sp2 C5-H hydrogen atom abstraction.
Discussion
One of the outstanding challenges in synthetic chemistry is the regioselective and stereoselective functionalization of C-H bonds in complex molecules.33–36 Nature cleverly employs a strategy to catalyze C-H bond functionalization by using metal-loenzymes, such as cytochrome P450 monooxygenases (CYPs) and non-heme iron(II) oxygenases, to promote diverse chemical transformations such as dealkylation and epoxidation, among others.15,18,37–41 In this study, we probed the isonitrile-forming mechanism of ScoE, a Fe(II)/α-KG-dioxygenase, by monitoring the reaction with NMR, LC-HRMS, and performing computational studies to aid our mechanistic understanding and ultimately propose a plausible molecular mechanism.
After identifying putative reaction intermediates and shunt products, we proposed the most likely pathway for ScoE-catalyzed synthesis of INBA from CABA (Scheme 1). Our results indicate that ScoE catalyzes two distinct and sequential C-H bond functionalization steps involving hydroxylation and oxidative decarboxylation, respectively (Scheme 1). The pathway commences with the first half-reaction of α-KG generating the Fe(IV)=O species, followed by C5-H abstraction, and hydroxylation to yield 1 (Scheme 1). A catalytic base near the active site was proposed to aid in the subsequent dehydration of 1 to form 2. One candidate is Tyr96, which forms a hydrogen bond with the CABA amine.21 The importance of Tyr96 in the mechanism is further highlighted by the inability of Y96F ScoE to synthesize INBA from CABA.21 Furthermore, a biochemical assay with Y96F ScoE was found to generate succinate from α-KG together with a trace amount of 2, which was likely generated from dehydration of 1 halted at the active site and supports our proposed role for Tyr96 (Figure S38). Alternatively, one of the water molecules near the CABA binding site can potentially act as a base because the architecture of the water network was previously shown to be important for isonitrile formation.21 The dehydration of 1 was presumably also assisted by the ferric species to yield 2 (Scheme 1). In vitro biochemical assays utilizing trans-2 in place of CABA demonstrated direct synthesis of INBA, consistent with a recent report.23 Our kinetic analysis of the ScoE reaction showed a relatively high apparent rate of 2 formation (Figure 3B), indicating that the conversion of CABA to 2 is not a rate-limiting step in INBA formation.
Scheme 1.
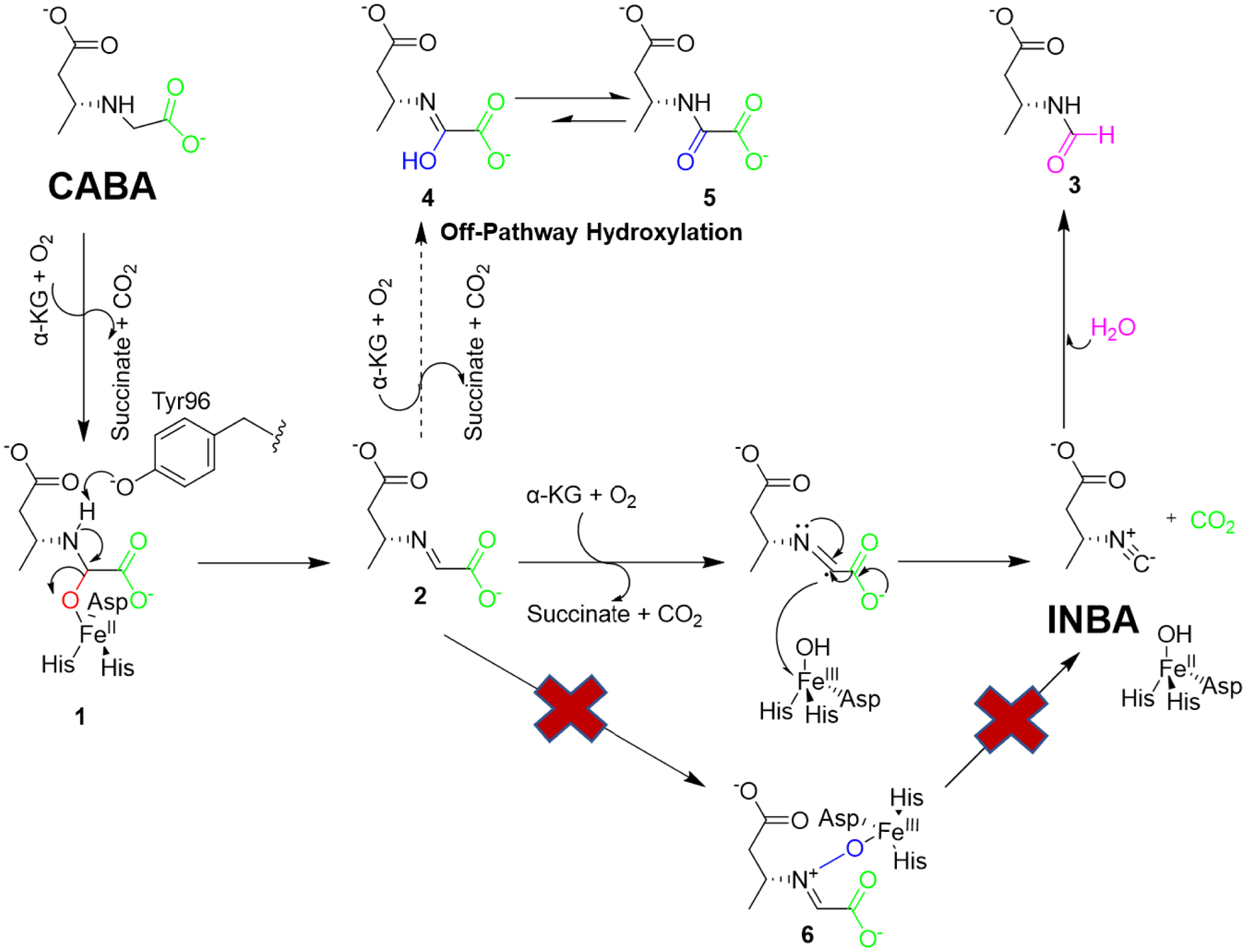
Proposed mechanism for isonitrile formation by ScoE.
The pathway continues with an additional α-KG half-reaction generating another Fe(IV)=O species to mediate C5-H abstraction of 2. The second site of hydroxylation was previously proposed to occur on the amine of 2 to yield metabolite 6 (Scheme 1)22, which is unfavored from our NMR analysis because we did not detect a typical sp2-methine signal around 130 ppm from HSQC analysis.42 Our computational studies also demonstrated that the nitrogen activation pathway was disfavored due to the high energy barriers (Figure S37). Our findings instead support a radical-based oxidative decarboxylation mechanism of 2. Specifically, we propose C5-H abstraction of 2 to generate a Cα radical that is transferred to the Fe(III)-OH species to drive decarboxylation and generate INBA (Scheme 1, Figure S30). Although we cannot rule out the possibility of 4 as a reaction intermediate, we disfavor this pathway because our time course experiments did not yield any visible consumption of 4, unlike the on-pathway intermediates 1 and 2 (Figure S5, Figure S8A, Figure S9, Figure 1C). Our computational analysis also showed that 4 was located at a lower energy level than INBA, further indicating that 4 is a shunt product in the pathway (Figure S29).29,30 Furthermore, we confirmed that 3 was a degradation product of INBA with a low apparent formation rate, in contrast to a previous report which proposed 3 to be an on-pathway reaction intermediate.22
Our proposed mechanism for ScoE is composed of two distinct C-H functionalization steps that both occur on C5 of CABA to yield a Cα radical. Cα-H abstraction has precedence in several enzymes such as the bicopper enzyme PHM, lysyl oxidases, enolase superfamily, and flavocytochrome b2.43–46 The first half-reaction of ScoE is proposed to generate 2 via hydroxylation and dehydration, in a similar manner to the Fe(II)/α-KG-dioxygenase AsqJ involved in cyclopeptin biosynthesis that generates a desaturated product via a hydroxylated intermediate.47 The second half reaction of ScoE proceeds likely through a Cα radical-based decarboxylation of 2 to yield INBA, similar to olefin-forming enzymes such as the CYP OleTJE, non-heme Fe(II) oxidase UndA, and the Fe(II)/α-KG-dioxygenase IsnB, which also catalyze oxidative decarboxylation but the initial hydrogen abstraction of these enzymes takes place at a Cβ position.29,30,42,48–52 The analogous position for ScoE would involve activation of the nitrogen, a pathway that is disfavored from our computational work and NMR experiments.
Conclusion
In conclusion, through analysis of enzymatic reactions with labelled substrates, we were able to identify and characterize two putative intermediates en route to isonitrile biosynthesis. The first α-KG half-reaction yields 1, which undergoes dehydration to yield 2. 2 then undergoes oxidative decarboxylation through a radical-based mechanism to yield INBA. We expect this mechanism to be conserved amongst other homologs of ScoE, including Rv0097 that is related to virulence of Mycobacterium tuberculosis.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Dr. Jeff Pelton for helping with NMR spectroscopic analysis. This research was financially supported by grants to W.Z. from the NIH (R01GM136758 and DP2AT009148) and the Chan Zuckerberg Biohub Investigator Program. This work was supported in part by the National Science Foundation (CBET-1704266 and CBET-1846426 for H.J.K., V.V., and D.W.K.). H.J.K. holds a Career Award at the Scientific Interface from the Burroughs-Wellcome Fund which supported this work (H.J.K., V.V., and D.W.K.).
Footnotes
Supporting Information
The supporting information is available free of charge on the ACS publications website.
Experimental details, chemical synthesis schemes, analytical data, primers utilized for plasmid construction, NMR spectra, supplemental figures containing LC-MS data, QM model details and calculations, and TS scans. (Figures S1–S33, Tables S1–S10, and Text S1–S5).
The authors declare no competing financial interests.
REFERENCES
- (1).Clarke-Pearson MF; Brady SF Paerucumarin, a New Metabolite Produced by the Pvc Gene Cluster from Pseudomonas Aeruginosa. J. Bacteriol 2008, 190 (20), 6927–6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Crawford JM; Portmann C; Zhang X; Roeffaers MBJ; Clardy J Small Molecule Perimeter Defense in Entomopathogenic Bacteria. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (27), 10821–10826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang L; Zhu M; Zhang Q; Zhang X; Yang P; Liu Z; Deng Y; Zhu Y; Huang X; Han L; Li S; He J Diisonitrile Natural Product SF2768 Functions As a Chalkophore That Mediates Copper Acquisition in Streptomyces Thioluteus. ACS Chem. Biol 2017, 12 (12), 3067–3075. [DOI] [PubMed] [Google Scholar]
- (4).Gloer JB; Rinderknecht BL Nominine : A New Insecticidal Indole Diterpene from the Sclerotia of Aspergillus Nomius. J. Org. Chem 1989, No. 6, 2530–2532. [Google Scholar]
- (5).Lim Fang Yun, Won Tae Hyung, Raffa Nicholas, Baccile Joshua A., Wisecaver Jen, Rokas Antonis, Schroeder Frank C., Keller NP Fungal Isocyanide Synthases and Xanthocillin Biosynthesis in Aspergillus Fumigatus. Am. Soc. Microbiol 2018, 9 (3), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hillwig ML; Zhu Q; Liu X Biosynthesis of Ambiguine Indole Alkaloids in Cyanobacterium Fischerella Ambigua. ACS Chem. Biol 2014, 9 (2), 372–377. [DOI] [PubMed] [Google Scholar]
- (7).M Soledade C; Yaya EE The First Isocyanide of Plant Origin Expands Functional Group Diversity in Cruciferous Phytoalexins : Synthesis, Structure and Bioactivity Of. Org. Biomol. Chem 2012, 10, 3613–3616. [DOI] [PubMed] [Google Scholar]
- (8).Emsermann J; Kauhl U; Opatz T Marine Isonitriles and Their Related Compounds. Mar. Drugs 2016, 14 (16), 1–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schnermann MJ; Shenvi RA Syntheses and Biological Studies of Marine Terpenoids Derived from Inorganic Cyanide. Nat. Prod. Rep 2016, 32 (4), 543–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Manzo E; Ciavatta ML; Gavagnin M; Mollo E; Guo Y; Cimino G Isocyanide Terpene Metabolites of Phyllidiella Pustulosa, a Nudibranch from the South China Sea. J. Nat. Prod 2004, 1701–1704. [DOI] [PubMed] [Google Scholar]
- (11).Zhang X; Evanno L; Poupon E Biosynthetic Routes to Natural Isocyanides. European J. Org. Chem 2020, 2020 (13), 1919–1929. [Google Scholar]
- (12).Brady SF; Clardy J Cloning and Heterologous Expression of Isocyanide Biosynthetic Genes from Environmental DNA. Angew. Chemie - Int. Ed 2005, 44 (43), 7063–7065. [DOI] [PubMed] [Google Scholar]
- (13).Harris NC; Sato M; Herman NA; Twigg F; Cai W; Liu J; Zhu X; Downey J; Khalaf R; Martin J; Koshino H; Zhang W Biosynthesis of Isonitrile Lipopeptides by Conserved Nonribosomal Peptide Synthetase Gene Clusters in Actinobacteria. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (27), 7025–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Harris NC; Born DA; Cai W; Huang Y; Martin J; Khalaf R; Drennan CL; Zhang W Isonitrile Formation by a Non-Heme Iron(II)-Dependent Oxidase/Decarboxylase. Angew. Chemie - Int. Ed 2018, 57 (31), 9707–9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Martinez S; Hausinger RP Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-Dependent Oxygenases. J. Biol. Chem 2015, 290 (34), 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Blasiak LC; Vaillancourt FH; Walsh CT; Drennan CL Crystal Structure of the Non-Haem Iron Halogenase SyrB2 in Syringomycin Biosynthesis. Nature 2006, 440 (7082), 368–371. [DOI] [PubMed] [Google Scholar]
- (17).Neugebauer ME; Kissman EN; Marchand JA; Pelton JG; Sambold NA; Millar DC; Chang MCY Reaction Pathway Engineering Converts a Radical Hydroxylase into a Halogenase. Nat. Chem. Biol 2021, 1–26. [DOI] [PubMed] [Google Scholar]
- (18).Grzyska PK; Appelman EH; Hausinger RP; Proshlyakov DA Insight into the Mechanism of an Iron Dioxygenase by Resolution of Steps Following the FeIV=O Species. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (9), 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bollinger JM; Price JC; Hoffart LM; Barr EW; Krebs C Mechanism of Taurine: α-Ketoglutarate Dioxygenase (TauD) from Escherichia Coli. Eur. J. Inorg. Chem 2005, No. 21, 4245–4254. [DOI] [PubMed] [Google Scholar]
- (20).Price JC; Barr EW; Glass TE; Krebs C; Bollinger JM Evidence for Hydrogen Abstraction from C1 of Taurine by the High-Spin Fe(IV) Intermediate Detected during Oxygen Activation by Taurine:α -Ketoglutarate Dioxygenase (TauD). J. Am. Chem. Soc 2003, 125 (43), 13008–13009. [DOI] [PubMed] [Google Scholar]
- (21).Jonnalagadda R; Del Rio Flores A; Cai W; Mehmood R; Narayanamoorthy M; Ren C; Zaragoza JPT; Kulik HJ; Zhang W; Drennan CL Biochemical and Crystallographic Investigations into Isonitrile Formation by a Non-Heme Iron-Dependent Oxidase/Decarboxylase. J. Biol. Chem 2021, 296 (Ii), 100231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen T; Chen J; Tang Y; Zhou J; Guo Y; Chang W Pathway from N-Alkylglycine to Alkylisonitrile Catalyzed by Iron(II) and 2-Oxoglutarate-Dependent Oxygenases. Angew. Chemie 2020, 132 (19), 7437–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chen T-Y; Zheng Z; Zhang X; Chen J; Cha L; Tang Y; Guo Y; Zhou J; Wang B; Liu H; Chang W Deciphering the Reaction Pathway of Mononuclear Iron Enzyme-Catalyzed N≡C Triple Bond Formation in Isocyanide Lipopeptide and Polyketide Biosynthesis. ACS Catal. 2022, 2270–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pretsch E, Buhlmann P Badertscher M Structure Determination of Organic Compounds, Springer Berlin Heidelberg, 2000. [Google Scholar]
- (25).He HY; Ryan KS Glycine-Derived Nitronates Bifurcate to O-Methylation or Denitrification in Bacteria. Nat. Chem 2021, 13 (6), 599–606. [DOI] [PubMed] [Google Scholar]
- (26).Iengo A; Santacroce C; Sodano G Metabolism in Porifera. X. On the Intermediary of a Formamide Moiety in the Biosynthesis of Isonitrile Terpenoids in Sponges. Experientia 1979, 35 (1), 10–11. [Google Scholar]
- (27).Hagadone MR; Scheuer PJ; Holm A On the Origin of the Isocyano Function in Marine Sponges. J. Am. Chem. Soc 1984, 106 (8), 2447–2448. [Google Scholar]
- (28).Massarotti A; Brunelli F; Aprile S; Giustiniano M; Tron GC Medicinal Chemistry of Isocyanides. Chem. Rev 2021, 10742–10788. [DOI] [PubMed] [Google Scholar]
- (29).Ali HS; Ghafoor S; de Visser SP Density Functional Theory Study into the Reaction Mechanism of Isonitrile Biosynthesis by the Nonheme Iron Enzyme ScoE. Top. Catal 2021, No. Iv, 1–16. [Google Scholar]
- (30).Li H; Liu Y Mechanistic Investigation of Isonitrile Formation Catalyzed by the Nonheme Iron/α-KG-Dependent Decarboxylase (ScoE). ACS Catal. 2020, 10 (5), 2942–2957. [Google Scholar]
- (31).Kulik HJ; Zhang J; Klinman JP; Martínez TJ How Large Should the QM Region Be in QM/MM Calculations? The Case of Catechol O-Methyltransferase. J. Phys. Chem. B 2016, 120 (44), 11381–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Karelina M; Kulik HJ Systematic Quantum Mechanical Region Determination in QM/MM Simulation. J. Chem. Theory Comput 2017, 13 (2), 563–576. [DOI] [PubMed] [Google Scholar]
- (33).Wu LF; Meng S; Tang GL Ferrous Iron and α-Ketoglutarate-Dependent Dioxygenases in the Biosynthesis of Microbial Natural Products. Biochim. Biophys. Acta - Proteins Proteomics 2016, 1864 (5), 453–470. [DOI] [PubMed] [Google Scholar]
- (34).Brückl T; Baxter RD; Ishihara Y; Baran PS Innate and Guided C-H Functionalization Logic. Acc. Chem. Res 2012, 45 (6), 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wencel-Delord J; Dröge T; Liu F; Glorius F Towards Mild Metal-Catalyzed C–H Bond Activation. Chem. Soc. Rev 2011, 40 (9), 4740–4761. [DOI] [PubMed] [Google Scholar]
- (36).Newhouse T; Baran PS If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chemie - Int. Ed 2011, 50 (15), 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Li X; Awakawa T; Mori T; Ling M; Hu D; Wu B; Abe I Heterodimeric Non-Heme Iron Enzymes in Fungal Meroterpenoid Biosynthesis. J. Am. Chem. Soc 2021, 143 (50), 21425–21432. [DOI] [PubMed] [Google Scholar]
- (38).Shimo S; Ushimaru R; Engelbrecht A; Harada M; Miyamoto K; Kulik A; Uchiyama M; Kaysser L; Abe I Stereodivergent Nitrocyclopropane Formation during Biosynthesis of Belactosins and Hormaomycins. J. Am. Chem. Soc 2021, 143 (44), 18413–18418. [DOI] [PubMed] [Google Scholar]
- (39).McQuarters AB; Wolf MW; Hunt AP; Lehnert N 1958 – 2014: After 56 Years of Research, Cytochrome P450 Reactivity Is Finally Explained. Angew. Chemie - Int. Ed 2014, 53 (19), 4750–4752. [DOI] [PubMed] [Google Scholar]
- (40).Guengerich FP; Munro AW Unusual Cytochrome P450 Enzymes and Reactions. J. Biol. Chem 2013, 288 (24), 17065–17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Del Rio Flores A; Twigg FF; Du Y; Cai W; Aguirre DQ Biosynthesis of Triacsin Featuring an N-Hydroxytriazene Pharmacophore. Nat. Chem. Biol 2021, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Cordero FM; Bonollo S; Machetti F; Brandi A The Synthesis of 4-Hydroxypipecolic Acids by Stereoselective Cycloaddition of Configurationally Stable Nitrones 2006, 3235–3241. [Google Scholar]
- (43).Abad E; Rommel JB; Kästner J Reaction Mechanism of the Bicopper Enzyme Peptidylglycine α-Hydroxylating Monooxygenase. J. Biol. Chem 2014, 289 (20), 13726–13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Williamson PR; Kagan HM Alpha-Proton Abstraction and Carbanion Formation in the Mechanism of Action of Lysyl Oxidase. J. Biol. Chem 1987, 262 (17), 8196–8201. [PubMed] [Google Scholar]
- (45).Babbitt PC; Hasson MS; Wedekind JE; Palmer DRJ; Barrett WC; Reed GH; Rayment I; Ringe D; Kenyon GL; Gerlt JA The Enolase Superfamily: A General Strategy for Enzyme-Catalyzed Abstraction of the α-Protons of Carboxylic Acids. Biochemistry 1996, 35 (51), 16489–16501. [DOI] [PubMed] [Google Scholar]
- (46).Sobrado P; Daubner SC; Fitzpatrick PF Probing the Relative Timing of Hydrogen Abstraction Steps in the Flavocytochrome B2 Reaction with Primary and Solvent Deuterium Isotope Effects and Mutant Enzymes. Biochemistry 2001, 40 (4), 994–1001. [DOI] [PubMed] [Google Scholar]
- (47).Liao HJ; Li J; Huang JL; Davidson M; Kurnikov I; Lin TS; Lee JL; Kurnikova M; Guo Y; Chan NL; Chang WC Insights into the Desaturation of Cyclopeptin and Its C3 Epimer Catalyzed by a Non-Heme Iron Enzyme: Structural Characterization and Mechanism Elucidation. Angew. Chemie - Int. Ed 2018, 57 (7), 1831–1835. [DOI] [PubMed] [Google Scholar]
- (48).Pickl M; Kurakin S; Cantú Reinhard FG; Schmid P; Pöcheim A; Winkler CK; Kroutil W; De Visser SP; Faber K Mechanistic Studies of Fatty Acid Activation by CYP152 Peroxygenases Reveal Unexpected Desaturase Activity. ACS Catal. 2019, 9 (1), 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Faponle AS; Quesne MG; De Visser SP Origin of the Regioselective Fatty-Acid Hydroxylation versus Decarboxylation by a Cytochrome P450 Peroxygenase: What Drives the Reaction to Biofuel Production? Chem. - A Eur. J 2016, 22 (16), 5478–5483. [DOI] [PubMed] [Google Scholar]
- (50).Yu CP; Tang Y; Cha L; Milikisiyants S; Smirnova TI; Smirnov AI; Guo Y; Chang WC Elucidating the Reaction Pathway of Decarboxylation-Assisted Olefination Catalyzed by a Mononuclear Non-Heme Iron Enzyme. J. Am. Chem. Soc 2018, 140 (45), 15190–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Rui Z; Li X; Zhu X; Liu J; Domigan B; Barr I; Cate JHD; Zhang W Microbial Biosynthesis of Medium-Chain 1-Alkenes by a Nonheme Iron Oxidase. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (51), 18237–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Manley OM; Fan R; Guo Y; Makris TM Oxidative Decarboxylase UndA Utilizes a Dinuclear Iron Cofactor. J. Am. Chem. Soc 2019, 141 (22), 8684–8688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.