

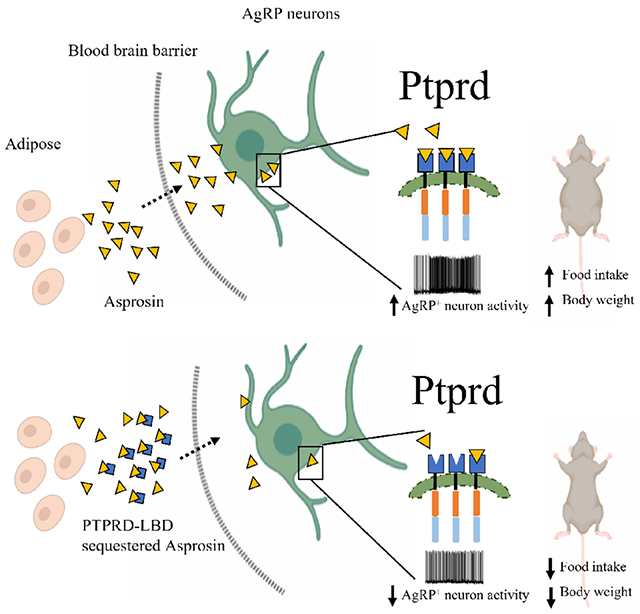
Abstract
Asprosin is a fasting-induced glucogenic and centrally-acting orexigenic hormone. The olfactory receptor Olfr734 is known to be the hepatic receptor for asprosin that mediates its effects on glucose production, but the receptor for asprosin’s orexigenic function has been unclear. Here, we have identified Protein Tyrosine Phosphatase Receptor δ (Ptprd) as the orexigenic receptor for asprosin. Asprosin functions as a high-affinity Ptprd ligand in hypothalamic AgRP neurons, regulating the activity of this circuit in a cell autonomous manner. Genetic ablation of Ptprd results in a strong loss of appetite, leanness and an inability to respond to the orexigenic effects of asprosin. Ablation of Ptprd specifically in AgRP neurons causes resistance to diet-induced obesity. Introduction of the soluble Ptprd ligand binding domain in the circulation of mice suppresses appetite and blood glucose levels by sequestering plasma asprosin. Identification of Ptprd as the orexigenic asprosin-receptor creates a new avenue for development of anti-obesity therapeutics.
Keywords: Asprosin, receptor, Ptprd, AgRP, metabolism, obesity
Graphical Abstract
eTOC blurb:
We have identified Protein Tyrosine Phosphatase Receptor δ (Ptprd) as the receptor for the orexigenic activity of the hormone asprosin. Ptprd genetic ablation leads to loss of appetite, leanness and unresponsiveness to both endogenous and exogenous asprosin. And strikingly, treatment of mice with the soluble ligand binding domain of Ptprd suppresses appetite and blood glucose by sequestering plasma pools of asprosin.
Introduction
In 2016 our laboratory identified asprosin; a novel, fasting-induced, glucogenic and orexigenic adipokine (Romere et al., 2016). In cell-autonomous actions at the level of hepatocytes and hypothalamic AgRP neurons, asprosin induces hepatic glucose release into the circulation and appetite stimulation, respectively (Duerrschmid et al., 2017; Romere et al., 2016). White adipose tissue generates and secretes asprosin, although other tissues such as salivary glands and cartilage have also been implicated in this process (Morcos et al., 2022; Romere et al., 2016; Ugur and Aydin, 2019). Plasma asprosin crosses the blood-brain-barrier, and its levels in the cerebrospinal fluid (CSF) and in the plasma increase during fasting. Identification of asprosin was made possible through study of a human genetic disease called Neonatal Progeroid syndrome (NPS), also known as Marfan Lipodystrophy syndrome. Individuals with NPS display plasma asprosin deficiency, marked leanness, hypophagia and robust insulin sensitivity compared with age- and sex-matched controls (Duerrschmid et al., 2017). Mice (and rabbits) engineered to harbor NPS mutations (Fbn1NPS/+) mimic the plasma asprosin deficiency, hypophagia, leanness and insulin sensitivity displayed by individuals with NPS (Chen et al., 2018; Duerrschmid et al., 2017; Romere et al., 2016). Moreover, Fbn1NPS/+ mice display remarkable resistance to metabolic syndrome despite being subjected to severe obesogenic and diabetogenic stress (Duerrschmid et al., 2017). Depressed AgRP neuron activity and low appetite displayed by NPS mice can be completely corrected upon replenishing plasma asprosin, thereby demonstrating a causal link (Duerrschmid et al., 2017). Importantly, asprosin was found elevated in individuals (and in rodent models) with metabolic syndrome in multiple studies (Alan et al., 2018; Baykus et al., 2019; Duerrschmid et al., 2017; Janoschek et al., 2020; Ugur and Aydin, 2019; Wang et al., 2020; Zhang et al., 2019). This observation combined with the fact that genetic asprosin deficiency completely protects Fbn1NPS/+ mice from diet-induced metabolic syndrome suggested that pharmacologic asprosin inhibition could be a valuable new avenue for metabolic syndrome treatment. In an extensive proof-of-concept study, we showed that monoclonal antibody (mAb)-mediated asprosin neutralization potently improved metabolic syndrome in multiple preclinical models of the disease (Mishra et al., 2021).
Since the discovery of asprosin, one central question has remained. What is the identity of the asprosin receptor? Li et al. identified OR4M1 (Olfr734 is the mouse ortholog) as the hepatic asprosin receptor (Li et al., 2019). This study provided evidence that OR4M1 functions as the liver receptor of asprosin, demonstrating high affinity receptor-ligand interaction and prevention of asprosin-mediated hepatic glucose release with knockout of Olfr734 in mice. This study also served as the first independent replication of the original study in 2016 (Romere et al., 2016) identifying asprosin as a glucogenic hormone. Interestingly, genetic knockout of Olfr734, while phenocopying the glucogenic deficit associated with asprosin deficiency, did not phenocopy the feeding or body weight deficits associated with genetic asprosin deficiency or pharmacologic asprosin inhibition. Specifically, Olfr734−/− mice do not display a reduction in appetite under ad libitum fed conditions, nor do they display a reduction in body weight (Liu et al., 2020). A marked reduction in ad libitum appetite and body weight are the hallmarks of genetic and pharmacologic inhibition of asprosin (Duerrschmid et al., 2017; Mishra et al., 2021). Given the absence of key orexigenic phenotypes with complete loss of Olfr734, it seemed unlikely that OR4M1/Olfr734 could serve as the orexigenic receptor for asprosin. Additionally, a study that mapped the entire mouse AgRP neuron transcriptome, the cell type directly activated by asprosin for appetite stimulation, including during fasted and fed conditions, reported zero copies of Olfr734 mRNA in this neuronal population (Henry et al., 2015). In contrast, OR4M1/Olfr734 is highly expressed in the olfactory epithelium and bulb and its loss results in impairment of olfaction-aided food seeking without impairing body weight (Liu et al., 2020). These results are consistent with a role for OR4M1/Olfr734 in asprosin-mediated olfactory behavior that aids food-seeking in the wild, but not with a role in asprosin-mediated core orexigenic drive and maintenance of body weight. Therefore, in the present study we embarked on a search for the cell-surface receptor that enables asprosin-mediated orexigenesis and maintenance of optimal body weight.
Results
Identification of an asprosin-interacting receptor in the mouse brain
We confirmed the reported absence (Henry et al., 2015) of the Olfr734 mRNA in mouse AgRP neurons via three independent methodologies. To that effect, we performed fluorescence RNAscope hybridization labeling for AgRP and Olfr734 transcripts in the hypothalamus of wild-type (WT) mice and found no evidence of coexpression of Olfr734 and AgRP transcripts (Figure 1a). Further, Olfr734 mRNA was undetectable by qPCR in visually enriched tdTOMATO expressing AgRP neurons (Figure 1b), and in ribo-tag pulled-down active transcripts from AgRP-Cre mice (Figure 1b). Given our inability to detect Olfr734 expression in AgRP neurons using multiple techniques, and the lack of phenotypic concordance between the genetic loss of Olfr734 and asprosin, or pharmacologic asprosin inhibition, when it comes to body weight and ad libitum appetite deficits (Liu et al., 2020; Duerrschmid et al., 2017; Mishra et al., 2021), we ruled out Olfr734 as a potential orexigenic receptor for asprosin.
Figure 1: Identification of an asprosin-interacting receptor in the mouse brain.
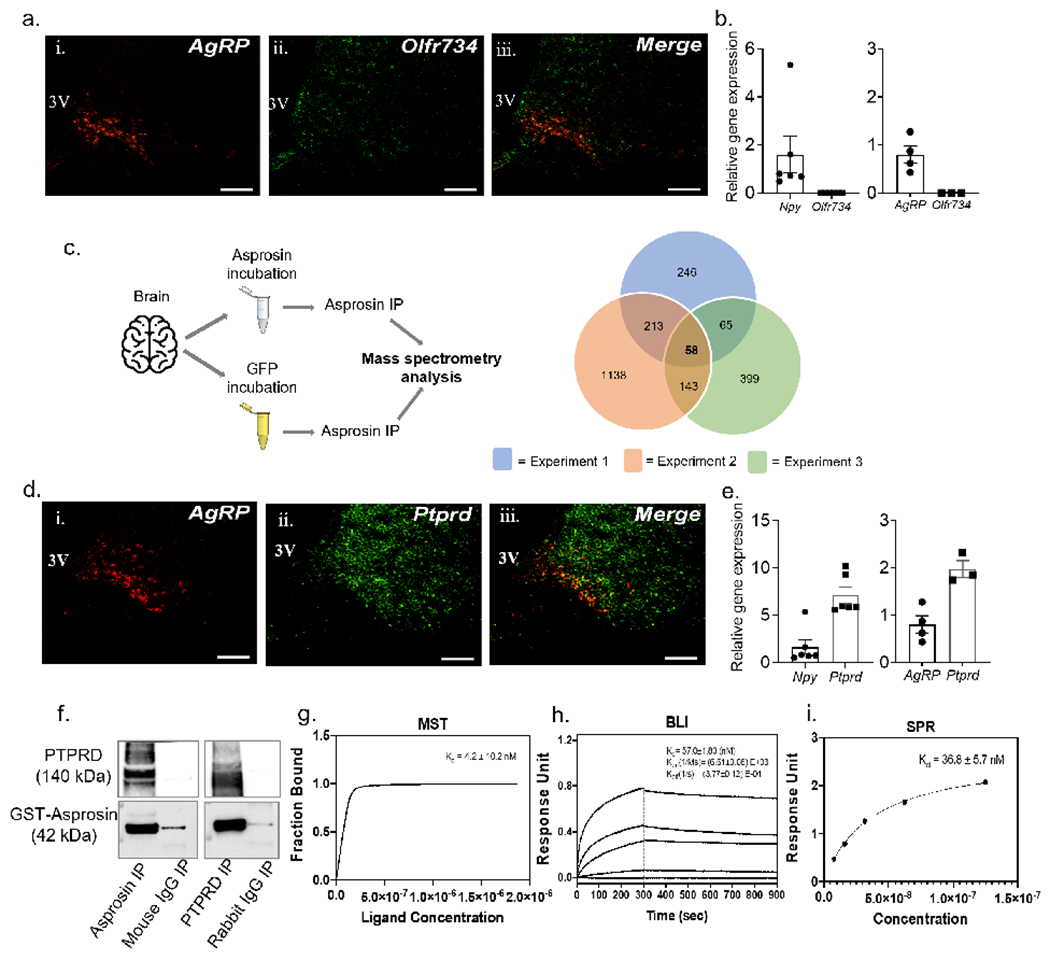
(a) Dual-label immunofluorenscence RNAscope hybridization showing relative overlap between AgRP (red) and Olfr734 (green) expressing cells in the arcuate (ARC) nucleus of wild-type (WT) mice. Scale bars, 100 μm. 3V, 3rd ventricle.
(b) qPCR results showing relative mRNA levels of Olfr734 (left) from visually enriched tdTOMATO expressing neurons of AgRP-cre mice and (right) cre-responsive ribotag enriched transcripts from AgRP neurons of AgRP-cre mice.
(c) Schematic overview of asprosin immunoprecitation (IP; left) of brain tissue incubated with recombinant asprosin or recombinant GFP before mass spectrometry (MS) analysis, (right) overlap of candidate asprosin-interacting proteins from 3 repeats of IP/MS. 58 candidate proteins, including Ptprd appeared in all 3 repeats.
(d) Dual-label immunofluorenscence RNAscope hybridization showing relative overlap between AgRP (red) and Ptprd (green) expressing cells in the ARC nucleus of WT mice. Scale bars, 100 μm.
(e) qPCR results showing relative mRNA levels of Ptprd (left) from visually enriched tdTOMATO expressing neurons of AgRP-cre mice and (right) cre-responsive ribotag enriched transcripts from AgRP neurons of AgRP-cre mice.
(f) Reciprocal in vitro immunoprecipitation of recombinant GST-asprosin and extracellular domain of his-tagged PTPRD (PTPRD-6his), with mouse IgG and rabbit IgG as negative controls.
(g-i) Quantification of the binding affinity between Asprosin and PTPRD by Microscale thermophoresis (k; MST; Kd of 4.2 ±10.2 nM), Bio-layer interferometry (l; BLI; Kd of 57.0 ±1.83 nM), and by Surface plasmon resonance analysis (m; SPR; Kd of 36.8 ±5.7 nM). Data are represented as mean ± SEM.
To identify the legitimate orexigenic receptor for asprosin, we incubated recombinant asprosin or GFP with mouse brain homogenate followed by immunoprecipitation with a high-affinity anti-asprosin mAb and subjected the immunoprecipitates to mass spectrometry to screen for potential asprosin-interacting proteins (Figure 1c). Of the 58 proteins that were identified as potential asprosin-interactors in three separate experiments, we found only one membrane-bound receptor - protein tyrosine phosphatase receptor type δ (Ptprd) - that is reported to be strongly expressed in AgRP neurons (Henry et al., 2015). Olfr734 was not detected as an asprosin-interacting protein in any of the three experiments. Therefore, we focused on the plausibility of Ptprd to function as an asprosin-receptor and modulate asprosin-mediated AgRP neuron activation and orexigenic and body weight modulating functions.
Ptprd, encoded by the Ptprd gene, is a single-pass membrane receptor belonging to the leukocyte common antigen-related protein (LAR)/type IIa class of receptor protein tyrosine phosphatases (Chagnon et al., 2004). Similar to other important appetite modulating receptors such as LEPR, GLP-1R and MC-4R (Jensen et al., 2018; Kishi et al., 2003; Scott et al., 2009), Ptprd is extensively expressed in the brain (Shishikura et al., 2016). The association of PTPRD variants with several neuronal disorders, including addiction, restless leg syndrome, neurofibrillary pathology in Alzheimer’s disease, obsessive-compulsive disorder and cognitive impairment, has stimulated an interest in its neurobiology and genomics (Bienvenu et al., 2020; Drgonova et al., 2015; Maciukiewicz et al., 2019; Ortiz et al., 2014; Tomita et al., 2020; Uhl and Martinez, 2019). While the precise neural function of Ptprd remains unknown, its intracellular phosphatase domain is reported to function as a regulator of phosphorylation-based signaling (Ortiz et al., 2014; Pevser et al., 2015; Tomita et al., 2020). Its extracellular domain has been implicated as a synaptic specifier and neural cell adhesion molecule; however, with no known ligands identified that bind this protein, Ptprd is characterized as an orphan receptor (Uhl and Martinez, 2019).
To establish the relevance of Ptprd in asprosin’s orexigenic signaling, we first verified the expression of Ptprd in AgRP neurons employing three distinct methodologies. Fluorescence RNAscope hybridization analysis indicated a strong overlap between AgRP and Ptprd transcripts in the hypothalamus of WT mice (Figure 1d). Further, high expression of Ptprd was seen in visually enriched tdTOMATO-expressing AgRP neurons, and in ribo-tag pulled down active transcripts from AgRP-Cre mice (Figure 1e). These results confirmed that Ptprd is strongly expressed in AgRP neurons. Finally, reciprocal co-immunoprecipitation assays biochemically confirmed asprosin binding with the PTPRD extracellular domain (Figure 1f).
Asprosin interacts with the Ptprd extracellular domain with nanomolar affinity
Next, three different techniques, namely Microscale thermophoresis (MST), Biolayer interferometry (BLI) and Surface plasmon resonance (SPR), were employed to assess the affinity (i.e., the disassociation constant; KD) of asprosin for the PTPRD extracellular domain (Figure 1g–i). The three assays found single- to double-digit nanomolar affinity between the two proteins (MST: 4.2 ± 10.2 nM; BLI: 57 ± 1.83 nM; SPR: 36.8 ± 5.7 nM; Figure 1g–i), well within the range typically displayed by hormone-receptor interactions (Li et al., 2019; Mistrík et al., 2004; Whittaker et al., 2008).
Ptprd ablation phenocopies the body weight and feeding deficits associated with asprosin dysfunction
Ptprd−/− mice were found to be markedly lean and hypophagic, when compared to their WT littermates, with significantly lower subcutaneous and intraperitoneal adipose mass (Figure 2a–g). The hypophagia was accompanied by a likely compensatory reduction in energy expenditure (Figure 2h–j; S1). Similar to males, Ptprd−/− female mice were also lean, hypophagic and expended significantly lower energy per day (Figure 2k–r). Importantly, energy expenditure and food intake were both found to be significantly lower in male and female Ptprd−/− mice compared with WT mice upon analysis of covariance (ANCOVA; Figure 2g,j,o,r). ANCOVA statistically adjusts for the influence of total body mass disparity from group comparisons of energy balance (Mina et al., 2018; Müller et al., 2021). Thus, the reduced food intake and energy expenditure observed in Ptprd−/− mice is independent of lower body weight (Figure 2g,j,o,r). Further, the lower energy expenditure displayed by Ptprd−/− mice during the active (dark) phase normalized during the inactive (light) phase (Figure 2i,q; S1), suggesting that loss of Ptprd has little impact on core basal metabolic rate, and that the reduction in active phase energy expenditure is likely a compensatory mechanism to withstand reduced food intake. Therefore, in a comprehensive assessment of energy balance, Ptprd−/− mice phenocopy Fbn1NPS/+ mice (Duerrschmid et al., 2017), a model of genetic asprosin deficiency, as well as mice treated with anti-asprosin mAbs (Mishra et al., 2021), a model of pharmacologic asprosin inhibition.
Figure 2. Ptprd ablation phenocopies the body weight and orexigenic deficits associated with genetic asprosin deficiency and pharmacologic asprosin inhibition.
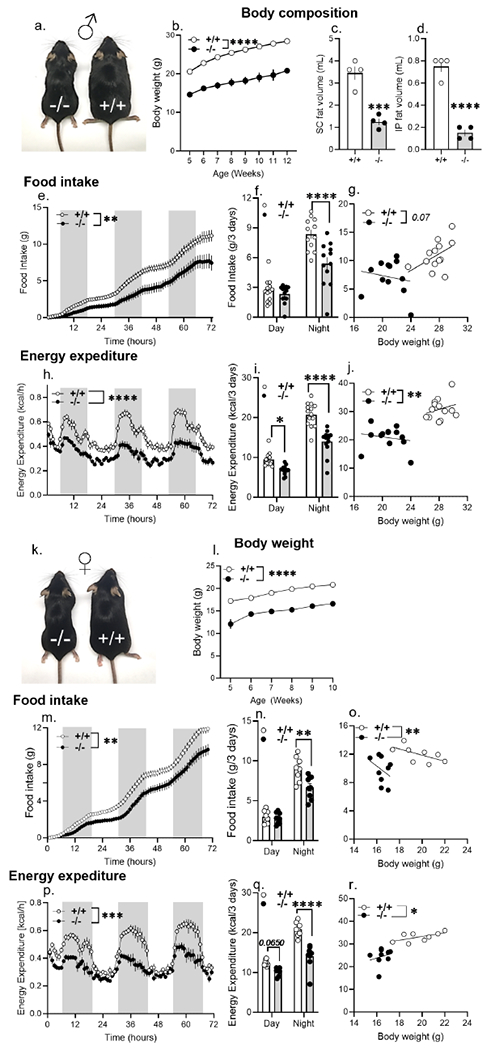
(a) A representative photograph of 5-month-old male Ptprd+/+ and Ptprd−/− littermates (n=1 photograph from n = 3 mice per genotype).
(b) Weekly body weights of wildtype (Ptprd+/+, n = 25) and Ptprd null (Ptprd−/−, n = 11) male mice from 5- to 12-weeks of age.
(c-d) Body composition data using Magnetic Resonance Imaging (MRI) on 5-month-old Ptprd+/+ and Ptprd−/− male mice on normal chow (n = 4/group).
(e-g) Cumulative food intake (e), total food intake in the dark and light phase (f), and ANCOVA analysis (g) of total food intake of Ptprd+/+ and Ptprd−/− male mice measured over 3 days using the Promethion metabolic system (n=12 or 13/group).
(h-j) Hourly energy expenditure (h) total energy expenditure in the light and dark phase (i), and ANCOVA analysis (j) of energy expenditure of Ptprd+/+ and Ptprd−/− male mice measured over 3 days using the Promethion metabolic system (n = 13/group).
(k) A representative photograph of 6-month-old female Ptprd+/+ and Ptprd−/− littermates (n=1 photograph from n = 3 mice per genotype).
(l) Weekly body weights of wildtype (Ptprd+/+, n = 16) and Ptprd null (Ptprd−/−, n = 11) female mice from 5- to 10-weeks of age.
(m-o) Cumulative food intake (m), total food intake in the dark and light phase (n), and ANCOVA analysis (o) of total food intake of Ptprd+/+ (n = 8) and Ptprd−/− (n = 9) female mice measured over 3 days using Promethion metabolic system.
(p-r) Hourly energy expenditure (p) and total energy expenditure in the light and dark phase (q), and ANCOVA analysis (r) of energy expenditure of Ptprd+/+ (n = 8) and Ptprd−/− (n = 9) female mice measured over 3 days using the Promethion metabolic system.
*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; ‘effect of genotype’ determined by two-tailed Student’s t-test (c,d,f,i,n,q), Repeated measures two-way ANOVA (b,e,h,l,m,p) or ANCOVA (g,j,o,r). Data are represented as mean ± SEM. See also Figure S1, S2, S3 and S4.
Increased plasma glycerol implicated enhanced mobilization of lipids to meet the energy demands of hypophagic Ptprd−/− mice (Figure S2). Plasma total cholesterol, high density lipoprotein (HDL), free fatty acids, triglycerides and circulating asprosin, adiponectin, ghrelin, and leptin levels were not statistically different between the Ptprd−/− and WT littermates (Figure S2), although plasma leptin showed a strong downward trend. Ptprd−/− mice did not display lethargy or signs of sickness in general, with no deficits seen in general locomotor activity (Figure S3). No difference in nutrient preference via respiratory exchange ratio analysis was detected (Figure S3) between Ptprd−/− and WT littermates.
Ptprd mRNA was found to be undetectable in hepatic tissue (Figure S4a) from WT mice. Therefore, Ptprd seemed unlikely to modulate the peripheral effect of asprosin on hepatic glucose release. Nonetheless, plasma glucose levels were monitored under fed and fasted conditions, along with a glucose tolerance test, in both male and female mice. Unlike Olfr734−/− mice (Li et al., 2019), Ptprd−/− male (Figure S4b–d) and female (Figure S4e–g) mice maintained euglycemia, similar to age- and sex-matched WT littermates. Together these results suggested that Ptprd potentially mediates the orexigenic but not the glucogenic effects of asprosin.
Whole-body loss of a single allele and AgRP neuron-specific loss of both alleles of Ptprd is protective against diet-induced obesity in females
To test whether global loss of a single Ptprd allele is sufficient to perturb metabolism in mice, we tracked the body weight of Ptprd+/+ and Ptprd+/− weanlings to adulthood on a normal chow (NC) diet. 10-week-old Ptprd+/− females weighed slightly less than their WT counterparts (Figure 3a). This marginal reduction in body weight was accompanied by a statistically significant reduction in food intake (Figure 3b). Energy expenditure was unaltered in Ptprd+/− females on NC (Figure 3c). However, these metabolic differences between Ptprd+/− and Ptprd+/+ females were exacerbated upon placing mice on a high-fat diet (HFD). Ptprd+/− females consumed nearly 40% less food and weighed nearly 4g less than WT littermates after 10 weeks of HFD (Figure 3d–e). Thus, loss of a single Ptprd allele protected female mice from HFD-induced obesity. Interestingly, the metabolic consequences of loss of one allele of Ptprd were sexually dimorphic, with no differences seen in body weight, food intake or energy expenditure of Ptprd+/ − and Ptprd+/+ males on NC or HFD (Figure S5).
Figure 3. Whole-body loss of a single allele and AgRP neuron-specific loss of both alleles of Ptprd is protective against diet-induced obesity in females.
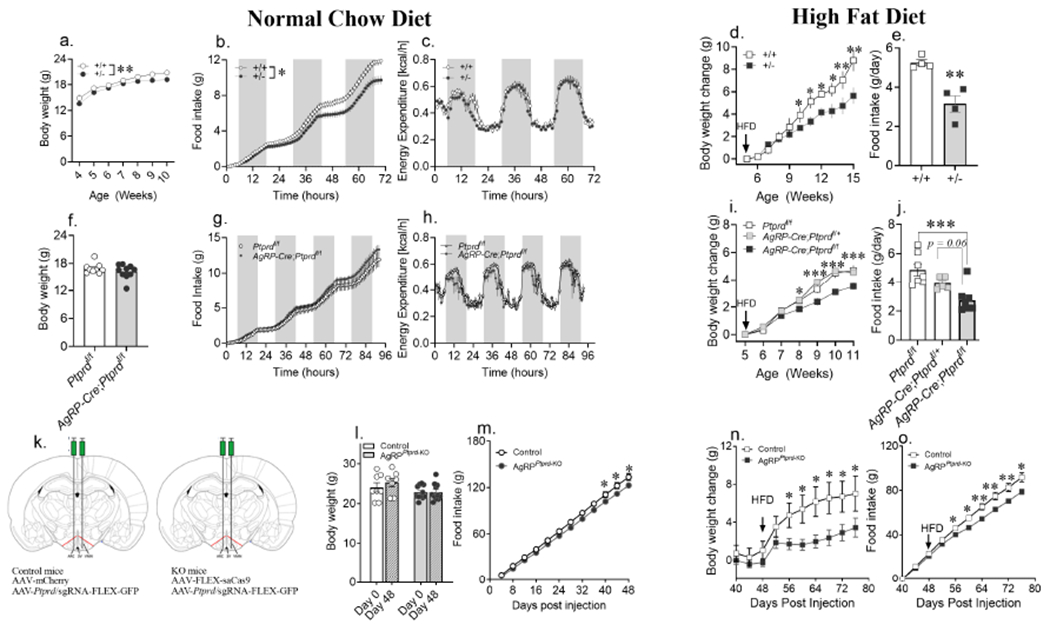
(a) Weekly body weights of wildtype (Ptprd+/+; n = 16) and heterozygous Ptprd knockout Ptprd+/−; n = 24) female mice maintained on ad libitum normal chow (NC) diet.
(b,c) Cumulative food intake and hourly energy expenditure of 10-week-old Ptprd+/+ and Ptprd+/− female mice on NC over 3 days using the Promethion metabolic system (food intake: Ptprd+/+ n = 8; Ptprd+/− n = 10; Energy expenditure: Ptprd+/− n = 8; Ptprd+/− n = 14).
(d,e) Weekly body weight change of Ptprd+/+ and Ptprd+/− females maintained on high fat diet (HFD) from 5-weeks of age, with food intake measured in a randomly selected cohort of 16-week-old female mice from each group (body weight change: Ptprd+/+ n = 9, Ptprd+/− n = 12; food intake: Ptprd+/+ n = 4; Ptprd+/− n = 4).
(f) Body weight of 6-week-old Ptprdflox/flox and AgRP-cre; Ptprdflox/flox maintained on ad libitum NC diet (Ptprdflox/flox n = 9, AgRP-cre; Ptprdflox/flox n = 10)
(g,h) Cumulative food intake and hourly energy expenditure of 8-week-old Ptprdflox/flox and AgRP-cre; Ptprdflox/flox mice maintained on ad libitum NC over 4 days using the Promethion system (n = 4/group).
(i,j) Weekly body weight change of Ptprdflox/flox (n = 9), AgRP-cre; Ptprdflox/+ (n = 4) and AgRP-cre; Ptprdflox/flox (n = 10) females maintained on high fat diet (HFD) from 5 weeks of age, with food intake measured in 14-week-old females from each group (Ptprdflox/flox :n = 8, AgRP-cre; Ptprdflox/+ : n = 5 and AgRP-cre; Ptprdflox/flox: n = 8).
(k) Schematic figure showing bilateral stereotactic injection of virus (AAV) containing Ptprd sgRNA with AAV expressing mCherry (control) or Cas9 (AgRPPtprd-KO) in the arcuate (ARC) nucleus of adult AgRP-cre female mice.
(l) Body weight of control and AgRPPtprd-KO female mice maintained on NC diet from day 0 to day 48 post stereotactic injection (control: n = 8; AgRPPtprd-KO: n = 11).
(m) Cumulative food intake measured every fourth day of control and AgRPPtprd-KO female mice maintained on NC diet (control: n = 6; AgRPPtprd-KO: n = 11).
(n,o) Body weight change and cumulative food intake of control and AgRPPtprd-KO female mice subjected to HFD on day 48 post stereotactic injection of AAVs coding cas9 and Ptprd sgRNA (control: n = 6-7; AgRPPtprd-KO: n = 11).
*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; by Student’s t-test (e,f,j,l), ‘effect of genotype’ determined by repeated measures two-way ANOVA (a,b,c,g,h) with point-comparison by multiple unpaired t-test (d,i,m,n,o). Circle and square symbol represent mice maintained on NC diet and HFD, respectively. Data are represented as mean ± SEM. See also Figure S5–S7.
To understand the spatial relevance of Ptprd in regulation of food intake, conditional knockout of Ptprd in AgRP neurons was accomplished by crossing Ptprdflox/flox mice with AgRP-Cre mice. On NC diet, AgRP-Cre+;Ptprdf/f females were found to be metabolically similar to Ptprdf/f control mice, with no difference noted in body weight, food intake or energy expenditure (Figure 3f–h). This is in contrast to the marked reductions in body weight, appetite and energy expenditure seen with whole-body loss of Ptprd and suggests that AgRP neuron-specific Ptprd loss on a NC diet can be compensated for by cell types with intact asprosin-Ptprd signaling. However, on a HFD, AgRP neuron-specific ablation of Ptprd protected female mice from diet-induced obesity with AgRP-Cre+;Ptprdf/f female mice weighing significantly less than age- and sex-matched control Ptprdf/f and AgRP-Cre+; Ptprdf/+ mice (Figure 3i; Figure S6a) after 6 weeks of high fat diet. Compared to Ptprdf/f mice, protection from HFD-induced obesity in AgRP-Cre+;Ptprdf/f mice was accompanied by a significant reduction in food intake (Figure 3j), with no changes seen in energy expenditure, activity and respiratory exchange ratio (Figure S6b–d). Interestingly, male mice with AgRP neuron-specific loss of Ptprd gained weight and consumed food in quantities similar to the control Ptprdf/f and AgRP-Cre+ Ptprdf/+ mice on NC and HFD (Figure S7a–e), again displaying a sexually dimorphic phenotype in contrast with whole-body Ptprd−/− mice where both sexes are equally affected.
As a third strategy, to avoid any developmental impacts of Ptprd loss, AgRP neuron-specific Ptprd ablation was accomplished in adult mice (Figure 3k–o). For this, Agrp-Cre mice received a bilateral stereotaxic injection of two Adeno-Associated Virus, serotype 8 (AAV) vectors in the arcuate nuclei (Arc) of the hypothalamus (Figure 3k). The first AAV-expressed a Ptprd-specific small guide RNA (sgRNA), while the second AAV expressed a cre-inducible cas9, leading to in-dels in the Ptprd gene only in AgRP neurons. Control mice received the same sgRNA as experimental mice, however the cre-inducible cas9 was replaced with mCherry. Mice were monitored for food intake and body weight changes for 80 days after stereotaxic injection. On a NC diet, day 40-48 post treatment, AgRPPtprd-KO females showed a mild reduction in food intake, which was not accompanied by a body weight reduction (Figure 3l,m). Once again, sexually dimorphic protection from HFD-induced obesity was seen with AgRP neuron-specific loss of Ptprd. Over a 40-day period on HFD, AgRPPtprd-KO female mice consumed 14g less food and gained half-as-much weight compared with control female mice (Figure 3n,o). AgRPPtprd-KO males consumed as much food and gained as much weight as control mice on NC and HFD (Fig. S7f–j). These results contrast with whole-body Ptprd−/− mice where both sexes are equally affected, showing that sexual dimorphism only appears with less-than-total loss of Ptprd. Further, mice with AgRP neuron-specific loss of Ptprd are relatively unaffected at baseline (NC) and need to be put on a HFD for the feeding and body weight reductions to manifest. This indicates that the effects of Ptprd loss on food intake and body weight are not confounded by general sickness, learning/memory deficits or strength deficits as those would also be evident at baseline.
Deletion of Ptprd renders AgRP neurons unresponsive to asprosin
We accomplished unilateral knockout of Ptprd in AgRP neurons in adult mice to determine the impact of Ptprd loss on asprosin-mediated AgRP neuron activation (Figure 4). For this, Agrp-Cre mice received a stereotaxic injection of two AAV vectors in the arcuate nucleus of the hypothalamus on the experimental side (Figure 4a). The first AAV expressed a Ptprd-specific small guide RNA (sgRNA), while the second AAV expressed a cre-inducible cas9, leading to in-dels in the Ptprd gene only in AgRP neurons. On the control side, mice received the same sgRNA as the experimental side, however the AgRP-cre inducible cas9 was replaced with mCherry. 40 days after stereotaxic injections, an endogenous ‘high-asprosin’ condition was induced in mice with overnight fasting. Increased activity of AgRP neurons via an increase in firing rate and resting membrane potential upon fasting was noted in neurons from the control side (Figure 4b–d). The fasting-induced increase in AgRP neuron activity was abolished in AgRPPtprd-KO neurons from the experimental side (Figure 4b–d). Similarly, increase in AgRP neuron activity in response to recombinant asprosin displayed by neurons from the control side was completely abolished in AgRPPtprd-KO neurons from the experimental side (Figure 4e–g). These results suggest absolute Ptprd necessity for asprosin-mediated AgRP neuron activation.
Figure 4. Deletion of Ptprd renders AgRP neurons unresponsive to asprosin.
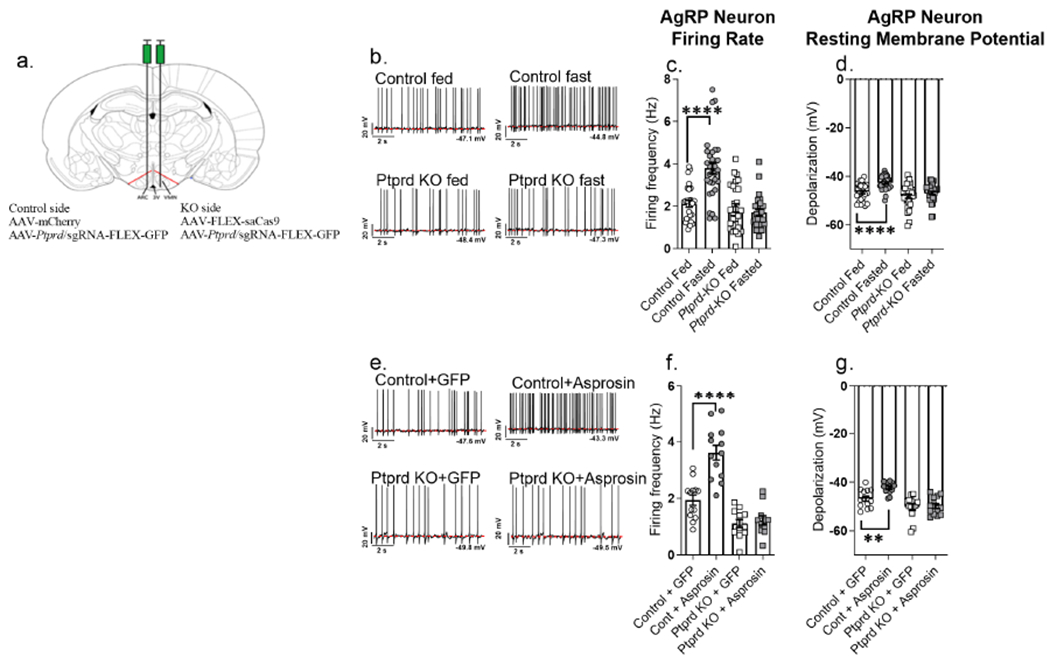
(a) Schematic figure showing unilateral stereotactic injection of virus (AAV) containing Ptprd sgRNA with AAV expressing mCherry (control) or Cas9 (AgRPPtprd-KO) in the arcuate (ARC) nucleus of adult AgRP-cre male mice
(b-d) Representative action potential firing traces, baseline neuronal firing rate and membrane potential of AgRP neurons from AgRP-cre mice subjected to overnight fasting or ad libitum feeding (control + fed: n = 28; control + fasted: n = 31; Ptprd KO + fed: n = 31; Ptprd KO + fasted: n = 28) after unilateral stereotactic injection of Ptprd sgRNA + mCherry expressing AAVs on one side (control side), and Ptprd sgRNA + Cas9 expressing AAVs in the other side (AgRPPtprd-KO; KO side) ARC of hypothalamus
(e-g) Representative action potential firing traces, neuronal firing rate and membrane potential of AgRP neurons from AgRP-Cre mice, incubated with recombinant GFP or asprosin for 2 hours (Control+GFP: n = 13, Control+Asprosin: n = 13, Ptprd KO+GFP: n = 13, Ptprd KO+Asprosin: n = 11) after unilateral stereotactic injection of Ptprd sgRNA + mCherry expressing AAVs on one side (control side), and Ptprd sgRNA + Cas9 expressing AAVs in the other side (AgRPPtprd-KO; KO side) of ARC of the hypothalamus.
*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; by two-tailed Student’s t-test. Data are represented as mean ± SEM.
Ptprd−/− mice are unresponsive to the orexigenic effects of asprosin while responding normally to its glucogenic effects
To produce sustained plasma asprosin elevation in mice without relying on recombinant preparations of variable activity, we used an adenovirus-mediated gain-of-function (GOF) strategy that we have successfully used in the past (Mishra et al., 2021;Figure S8 Figure S7). We tail-vein injected WT and Ptprd−/− mice with adenoviruses (Ad5) encoding human FBN1 (with native signal peptide) or human cleaved asprosin (with an IL2 signal peptide to promote secretion, Figure S8), and then tested whether asprosin-GOF-induced metabolic phenotypes were observed in the setting of complete Ptprd loss (Figure 5). Like our previous reports (Romere et al., 2016; Duerrschmid et al., 2017; Mishra et al., 2021), WT mice transduced with Ad5-FBN1 (Figure 5a–c) or Ad5-Asprosin (Figure 5d–f) exhibited hyperphagia, higher baseline blood-glucose and lower glucose tolerance when compared with Ad5-empty control mice. Interestingly, asprosin-GOF had no impact at all on appetite in Ptprd−/− mice compared with the hyperphagia seen in WT mice (Figure 5a,d), while the impact on baseline blood-glucose and glucose tolerance was essentially identical between WT and Ptprd−/− mice (Figure 5b,c,e,f).
Figure 5. Ptprd−/− mice are unresponsive to the orexigenic effects of asprosin while responding normally to its glucogenic effects.
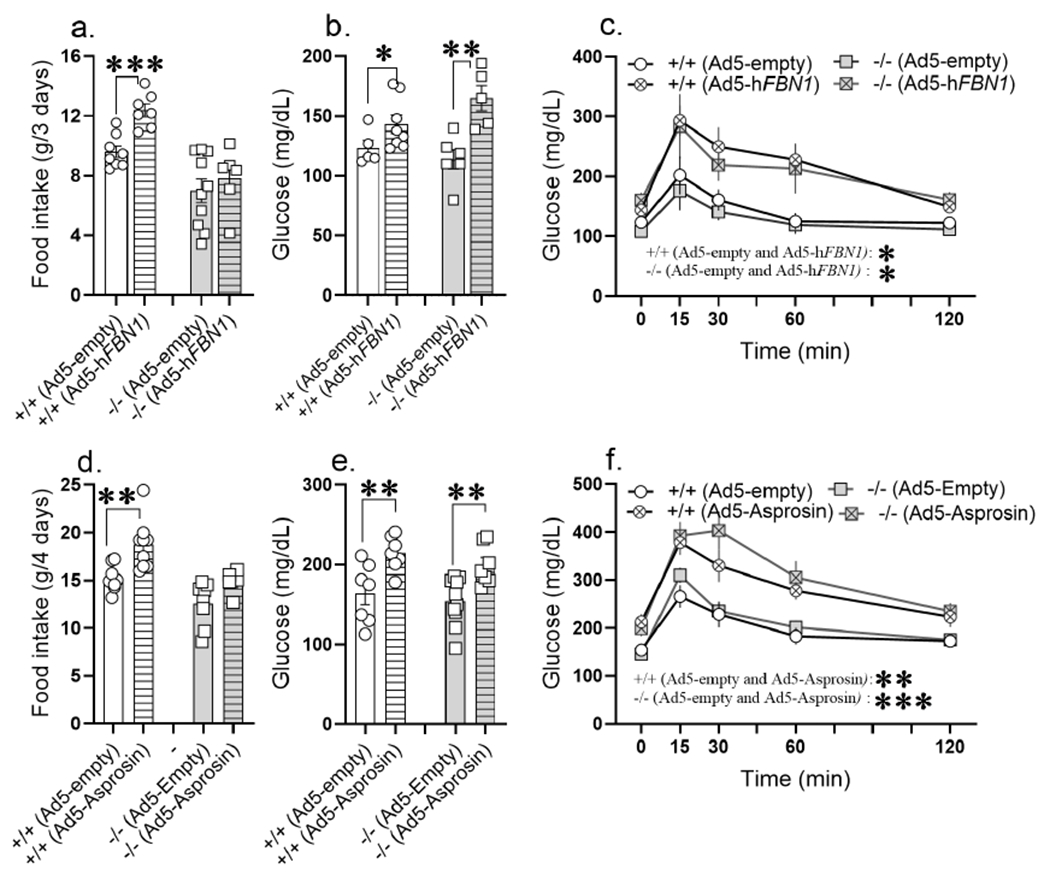
(a-c) 14-week-old normal chow fed Ptprd+/+ and Ptprd−/− male mice tail-vein transduced with Ad-empty or Ad-hFBN1 (3.6 x 109 pfu/mouse) viruses. (a) Cumulative food intake measured in Promethion metabolic chamber over 3 days; day 8 to day 11 post adenoviral vector transduction (Ad-empty: n = 8 Ptprd+/+ and n = 10 Ptprd−/−; Ad-hFBN1: n = 7 Ptprd+/+ and n = 6 Ptprd−/−. (b,c) ad libitum fed baseline blood glucose levels and glucose tolerance assay in response to intraperitoneal glucose injection (2mg/kg) on day 14 post adenoviral transduction (baseline glucose: Ad-empty: n = 5 Ptprd+/+ and n = 6 Ptprd−/−; Ad-hFBN1: n = 8 Ptprd+/+ and n = 6 Ptprd−/−, GTT: Ad-empty: n = 5 Ptprd+/+ and n = 4 Ptprd−/−; Ad-hFBN1: n = 7 Ptprd+/+ and n = 4 Ptprd−/−).
(d-f) 12-week-old normal chow fed Ptprd+/+ and Ptprd−/− male mice tail-vein transduced with Ad-empty or Ad-hAsprosin (5 x 1010 pfu/mouse) viruses. (d) Cumulative food intake measured in promethion metabolic chamber over 4 days; day 8 to day 12 post adenoviral vector transduction (Ad-empty: n = 8 Ptprd+/+ and n = 8 Ptprd−/−; Ad-hAsprosin: n = 9 Ptprd+/+ and n = 6 Ptprd−/−. (e,f) ad libitum fed baseline blood glucose levels and glucose tolerance assay (GTT) in response to intraperitoneal glucose injection (2mg/kg) on day 15 post adenoviral transduction (Ad-empty: n = 7 Ptprd+/+ and n = 10 Ptprd−/−; Ad-hAsprosin1: n = 7 Ptprd+/+ and n = 7 Ptprd−/−).
*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; by two-tailed Student’s t-test (a,b,d,e), and repeated measures two-way ANOVA (c,f). Data are represented as mean ± SEM. See also Figure S8.
Asprosin activates Ptprd in a cell autonomous manner
Ptprd phosphatase domain 1 (PD1) directly interacts with the transcription factor Stat3, leading to Stat3 dephosphorylation at tyrosine residue 705 (p-Stat3) and inhibition of its transcriptional activity (Böhmer and Friedrich, 2014). This suggests that Stat3 phosphorylation and transcriptional activity can serve as a measure of asprosin-mediated Ptprd signaling. In support of this, we found p-Stat3 levels to be significantly lower in the hypothalamus of WT mice exposed to higher circulating asprosin via tail-vein injection with Ad5-FBN1 compared with mice that received Ad5-empty (Figure 6a). Conversely, we observed significantly higher hypothalamic p-Stat3 levels in Ptprd−/− mice when compared with WT (Figure 6b–d). We also noted a trend of higher hypothalamic p-Stat3 levels in diet-induced obese (DIO) mice subjected to asprosin neutralization (Figure S9a).
Figure 6. Asprosin activates Ptprd in a cell autonomous manner.
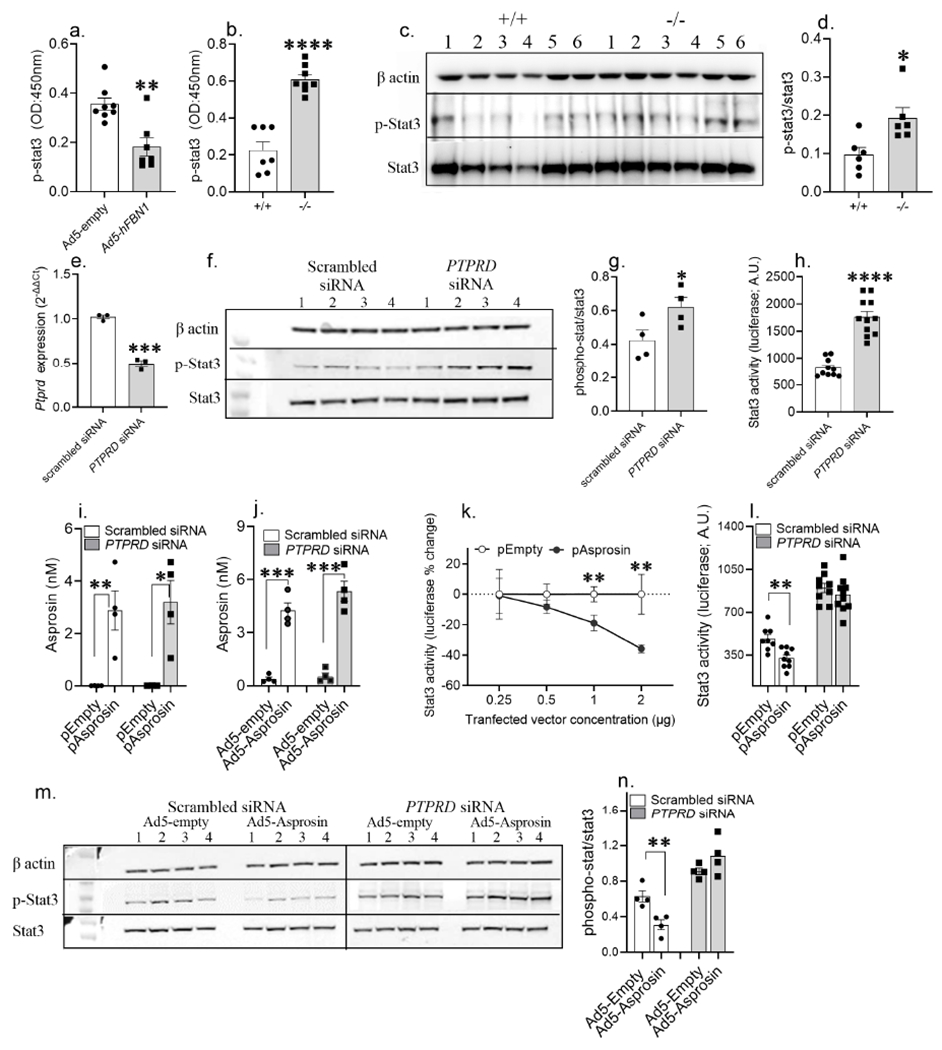
(a) phosphorylated-Stat3 (p-Stat3) levels measured using ELISA in hypothalamic neural lysate of WT normal chow fed lean mice, 15 days post tail-vein transduction with Ad-empty or Ad-hFBN1 (3.6 x 109 pfu/mouse; three technical replicates of n= 7 or 8 biological replicates per group) viruses.
(b) p-Stat levels measured using ELISA in hypothalamic neural lysate of normal chow fed lean Ptprd+/+ and Ptprd−/− male mice (three technical replicates of n= 7 or 8 biological replicates per).
(c-d) A representative western blot and relative quantification of β-actin, p-Stat3 and Stat3 in hypothalamic neural lysate of normal chow fed Ptprd+/+ and Ptprd−/− male mice (three technical replicates of n = 6 biological replicates per group).
(e) PTPRD mRNA levels measured in HEK293T cells 24h post transfection with scrambled (control) and Ptprd siRNA (25nM; three technical replicates of n = 3 biological replicates per treatment).
(f-g) A representative western blot and relative quantification of beta actin, p-Stat3 and Stat3 in HEK293T cell lysate 48h post transfection with control and PTPRD siRNA (25nM; three technical replicates of n = 4 biological replicates per treatment).
(h) Stat3-response element driven luciferase activity measured in HEK293T cells transduced with 2μg 4xM67 pTATA-TK-Luc plasmid with control or PTPRD siRNA treatment (25nM; three technical replicates of n = 11 biological replicates per treatment).
(i,j) Validation of asprosin overexpression in HEK293T cells using IL2-his-asprosin expressing mammalian expression plasmid (empty plasmid as control;i) and Ad5-IL2-his-Asprosin (Ad5-empty as control; j) under Ptprd knockdown condition (non-targeted scrambled siRNA as control; four technical replicates of n =4 biological replicates per group).
(k) Percent change in Stat3-response element driven luciferase activity measured in HEK293T cells 72 hours post co-transfection of 2μg 4xM67 pTATA-TK-Luc plasmid with serial dilution of empty or IL2-his-asprosin expressing mammalian expression plasmid (0.25, 0.5, 1 and 2 μg plasmid; two technical replicates of n = 5 or 6 biological replicates per treatment).
(l) Stat3-response element driven luciferase activity measured in HEK293T cells 72 hours post co-transfection of 2μg 4xM67 pTATA-TK-Luc plasmid with 1 pg empty or asprosin expressing plasmid under conditions of control or PTPRD knockdown (25nM; three technical replicates of n = 9 or 10 biological replicates pertreatment).
(m,n) A representative western blot and relative quantification of β-actin, p-Stat3 and Stat3 of HEK293T cell lysate, 72 h post co-treatment of control and PTPRD siRNA (25 nM) with control or asprosin expressing Ad5 vectors (100vp/cell; three technical replicates of n = 4 biological replicates per treatment).
*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; by two-tailed Student’s t-test. Data are represented as mean ± SEM. See also Figure S9.
To interrogate whether asprosin could activate endogenous Ptprd in a cell autonomous manner, we used HEK293T cells that naturally express PTPRD and STAT3. PTPRD knockdown using siRNA significantly increased p-Stat3 levels and Stat3 transcriptional activity (Figure 6e–h). Recombinant asprosin has been shown to display variable activity (Mishra et al., 2021). Therefore, we transduced or transfected HEK293T cells with adenovirus or mammalian-expression plasmid encoding human cleaved asprosin (with an IL2 signal peptide to induce secretion) to enhance media asprosin levels (Figure 6i,j). This resulted in a dose-dependent decline in Stat3 transcriptional activity (Figure 6k). Additionally, we found that introduction of the anti-asprosin mAb in the media enhances Stat3 activity in cells subjected to asprosin overexpression or exposed to asprosin conditioned media compared with the control IgG (Figure S9b), showing that extracellular asprosin neutralization completely prevents asprosin signaling independent of whether asprosin is overexpressed and secreted or delivered via conditioned media. Importantly, Ptprd knockdown rendered the cells unresponsive to asprosin-mediated suppression of p-Stat3 levels and Stat3 transcriptional activity (Figure 6l–n). Collectively, these results demonstrate cell-autonomous activation of Ptprd by asprosin.
The PTPRD ligand-binding-domain (PTPRD-LBD) sequesters asprosin in vivo to decrease appetite, body weight and blood glucose in obese mice
We have previously validated the use of asprosin-neutralizing monoclonal antibodies as a therapeutic approach in the treatment of metabolic syndrome in mice (Mishra et al., 2021). Similar to the anti-asprosin mAb approach, here we tested whether asprosin can be sequestered, and metabolic syndrome ameliorated, with introduction of the PTPRD ligand-binding-domain (PTPRD-LBD) in the circulation of DIO mice. The level of free asprosin detected by ELISA was significantly reduced when recombinant asprosin was incubated with recombinant PTPRD-LBD (Figure 7a), indicating asprosin sequestration by the PTPRD-LBD. DIO mice were exposed to elevated plasma PTPRD-LBD by transducing mice with adenovirus (Ad5) encoding PTPRD-LBD (tagged with an IL2 signal peptide to induce secretion). We again chose the adenoviral strategy to avoid the unreliability of the recombinant protein approach in vivo. We developed a sandwich ELISA to detect his-tagged PTPRD-LBD in mouse plasma (Figure S10) and demonstrated an elevation in the PTPRD-LBD levels in the plasma of DIO mice injected with Ad5-IL2-PTPRD-LBD concurrent with increased PTPRD-LBD expression in the liver (Figure 7b,c). Of note, DIO mice serve as a model of pathologically high plasma asprosin (Romere et al., 2016; Duerrschmid et al., 2017; Li et al., 2019). We found significantly reduced levels of free asprosin in the plasma of DIO mice with elevated plasma PTPRD-LBD (Figure 7d) indicating that plasma asprosin can be successfully sequestered by the circulating PTPRD-LBD. Introduction of the PTPRD-LBD in the plasma was associated with a significant reduction in daily food intake, body weight, blood glucose levels and glucose tolerance in DIO mice (Figure 7e–h). These results are virtually identical to the effects of anti-asprosin mAbs in DIO mice (Mishra et al., 2021).
Figure 7. Ectopic introduction of the PTPRD ligand-binding-domain (PTPRD-LBD) into the circulation functions as an “asprosin-trap” to ameliorate appetite, body weight and blood glucose in obese mice.
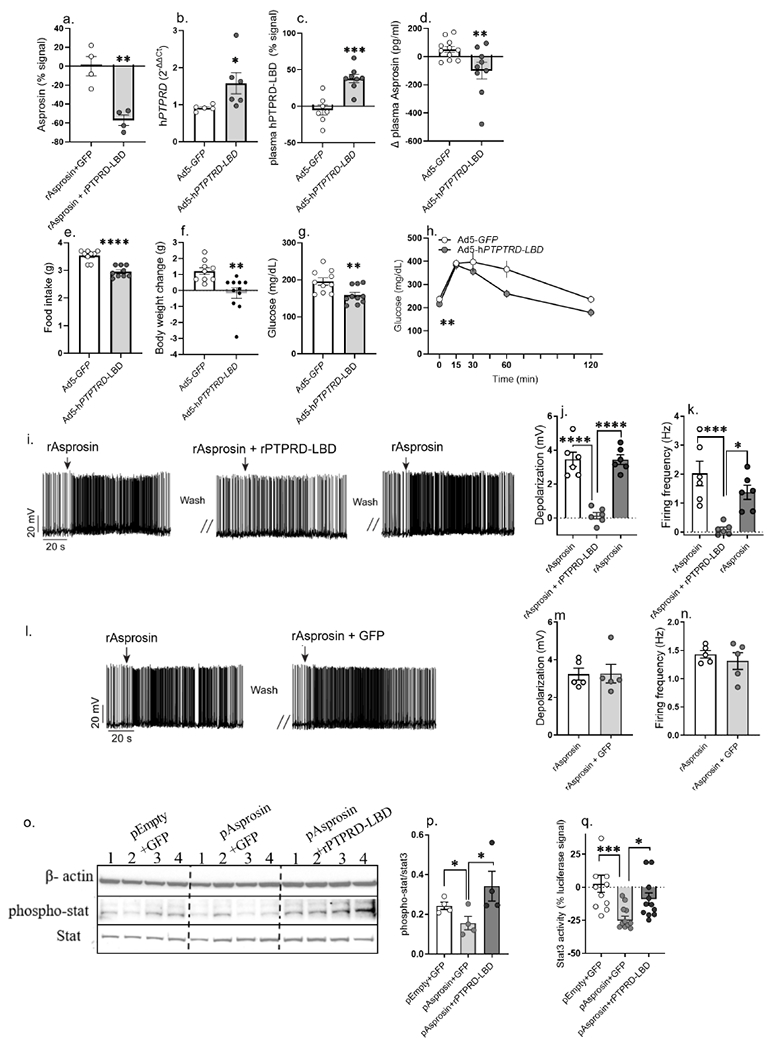
(a) Asprosin detected by sandwich ELISA in 5nM recombinant asprosin (rAsprosin) preincubated with 500ng GFP or recombinant PTPRD-ligand binding domain (rPTPRD-LBD) for one hour in phosphate buffer saline (two technical replicates of n = 4 biological replicates per treatment). Asprosin levels plotted relative to signal detected in rAsprosin+GFP.
(b-c) Adenoviral vector (Ad5) mediated overexpression of the IL2-tagged PTPRD ligand-binding-domain (Ptprd-LBD) detected at mRNA level in hepatic tissue (b) and as protein in plasma (c) of 18-week-old DIO mice, 15 days post tail vein injection of Ad5-GFP or Ad5-hPTPRD-LBD (1 x 1011 vp/mouse) viruses.
(d) Plasma asprosin levels of 18-week-old DIO mice, 4 days post tail vein injection of Ad5-GFP or Ad5-hPTPRD-LBD (2 x 1011 vp/mouse) viruses. Plasma asprosin plotted relative to day 0 asprosin levels for each mouse (one technical replicate of n = 11 Ad5-GFP and 10 Ad5-hPTPRD-LBD biological replicates).
(e-h) Food intake, body weight change, baseline glucose and glucose tolerance test (GTT) measured in 18-week-old male DIO mice 12-14 days post tail-vein-injection of Ad5-GFP or Ad5-hPTPRD-LBD (1 x 1011 vp/mouse) viruses (body weight change, food intake: Ad5-GFP: n = 9; Ad5-hPTPRD-LBD: n = 10; baseline glucose: n = 10/treatment; GTT: Ad5-GFP: n = 8; Ad5h-PTPRD-LBD: n = 7).
(i-k) Representative action potential firing traces, depolarization and firing frequency of AgRP neurons after recombinant asprosin (rAsprosin), rAsprosin + recombinant PTPRD-LBD (rPTPRD-LBD), and rAsprosin treatment (three technical replicates of n = 6 biological replicates per treatment).
(l-n) Representative action potential firing traces, depolarization and firing frequency of AgRP neurons after rAsprosin and rAsprosin + GFP treatment, to test the effect of irrelevant protein (GFP) treatment (three technical replicates of n = 5 biological replicates per treatment).
(o,p) A representative western blot and relative quantification of β-actin, p-Stat3 and Stat3 in 10μg cell lysate from HEK293T cells transfected with empty-backbone (pEmpty) or asprosin coding (pAsprosin) mammalian expression plasmid (2 μg) for 48 hours, followed by 8 hours of treatment with rGFP or rPTPRD-LBD (1 μg; three technical replicates of n = 4 biological replicates per treatment).
(q) Percent change in Stat3-response element driven luciferase activity measured in HEK293T cells treated with GFP or rPTPRD-LBD (1 μg) for 12 hours, 72 hours post co-transfection of 2μg 4xM67 pTATA-TK-Luc plasmid with 1 μg pEmpty or pAsprosin (pEmpty+ GFP: n = 11; pAsprosin+ GFP: n = 12; pAsprosin+ rPTPRD-LBD: n = 11).
*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; by two-tailed Student’s t-test and 2-way ANOVA (h). Data are represented as mean ± SEM.See also Figure S10.
As a second line of evidence, we found that recombinant PTPRD-LBD, but not recombinant GFP, was able to completely prevent asprosin-mediated AgRP neuron activation in ex vivo hypothalamic slice electrophysiology (Figure 7i–n). Again, these results are virtually identical to the effects of anti-asprosin mAbs (Mishra et al., 2021). We extended these studies to an in vitro setup to interrogate whether introducing the PTPRD-LBD into the media could interfere with asprosin-mediated PTPRD signaling in a cell autonomous manner, as measured by p-Stat3 levels and Stat3 transcriptional activity. We transfected HEK293T cells with a plasmid encoding human cleaved asprosin (tagged with an IL2 signal peptide to induce secretion) and found that asprosin-mediated reduction in p-Stat3 levels and Stat3 transcriptional activity was abrogated upon introduction of recombinant PTPRD-LBD into the media (Figure 7o–q).
Discussion
To achieve the goal of identifying the asprosin receptor that modulates its orexigenic and body weight maintaining functions, we performed unbiased screening of asprosin-interacting proteins in the mouse brain. Three independent screens identified protein tyrosine phosphatase receptor type δ (Ptprd) as an asprosin-interactor. This observation was confirmed biochemically, with single- to double-digit nanomolar affinity with multiple assays, well within the range traditionally displayed by hormones and their receptors (Li et al., 2019; Mistrík et al., 2004; Whittaker et al., 2008).
Published AgRP neuron transcriptome analysis showed that Ptprd is highly expressed in AgRP neurons (Henry et al., 2015), the key cell type we have previously shown to respond to asprosin in a cell autonomous manner (Duerrschmid et al., 2017). Based on these results, we embarked on a multi-pronged experimental strategy to test the necessity of Ptprd for asprosin-mediated AgRP neuron activation and orexigenic function. Whether Ptprd is genetically ablated, or its ligand-binding domain (Ptprd-LBD) is ectopically introduced in the circulation to sequester plasma asprosin, the result is a corresponding decrease in appetite and body weight. Marked leanness, reduced adiposity, hypophagia and a compensatory reduction in energy expenditure of Ptprd−/− mice completely recapitulates the energy balance and body weight perturbations associated with genetic asprosin deficiency in humans (NPS patients) and mice (Fbn1NPS/+; Duerrschmid et al., 2017; Romere et al., 2016).
Further, like Fbn1NPS/+ mice, mice with Ptprd ablated specifically in AgRP neurons, constitutively or in the adult setting, were protected from diet-induced-obesity. Importantly, deletion of Ptprd from AgRP neurons rendered them unresponsive to exogenous asprosin. At the whole organism level, asprosin gain-of-function (via increase in plasma asprosin through an adenoviral-mediated approach) failed to increase appetite in Ptprd−/− mice compared with WT, validating the absolute necessity of Ptprd for asprosin’s orexigenic function. Interestingly, Ptprd loss is not associated with alterations in blood glucose or glucose tolerance, and asprosin gain-of-function in Ptprd−/− mice results in changes in blood glucose and glucose tolerance comparable to WT mice, suggesting that Ptprd is necessary for only the orexigenic function of asprosin, and that the glucogenic effects of asprosin are mediated by a different receptor. This is consistent with Olfr734/OR4M1 serving as the hepatic (glucogenic) receptor for asprosin (Li et al., 2019), and supported by the observed absence of the Ptprd transcript in the liver and that of the Olfr734 transcript in AgRP neurons.
While total loss of Ptprd (whole-body loss of both alleles) presents as hypophagia and leanness in both sexes, a less-than-total loss (whole-body loss of just one allele, or loss of both alleles in just one cell type; namely, AgRP neurons) only manifests in females. Such sexual dimorphism, with males being relatively insensitive to lower magnitude insults, is well known to exist in the regulation of feeding behavior and energy homeostasis. For example, AgRP neuron-specific knockout of the leptin receptor leads to a significant increase in adiposity and body weight in female, but not male mice (Egan et al., 2017). Leptin treatment sufficiently restores neonatal AgRP neuronal innervation in leptin deficient (Lepob/ob) females, but not in males (Kamitakahara et al., 2018) and central injection of leptin is more effective at causing anorexia in female rats as opposed to males (Clegg et al., 2003). Key transcription factors known to regulate feeding such as FoxO1 (Forkhead box protein O1) and Stat3 (Signal transducer and activator of transcription 3) also display similar phenomena. Female mice with hypothalamic ablation of FoxO1 weigh significantly less than their wild type counterparts, but males do not (Heinrich et al., 2014). Inactivation of Stat3 in POMC neurons causes mild obesity in female, but not male mice (Xu et al., 2007). These examples of sexual dimorphism, with penetrance of the genotype limited to females, encompass multiple signaling pathways and cell types within the realm of energy homeostasis (Liu et al, 2021; Wang and Xu, 2019). Therefore, the sexual dimorphism seen with less-than-total loss of Ptprd is likely a general phenomenon, rather than a direct consequence of altered Ptprd signaling.
Several clinical and pre-clinical studies have implicated PTPRD variants and SNPs as associative factors for metabolic disturbances, including type 2 diabetes, anorexia nervosa and anti-psychotic medication-induced weight gain (Bienvenu et al., 2020; Drgonova et al., 2015; Maciukiewicz et al., 2019; Peyser et al., 2015; Tomita et al., 2020; Uhl and Martinez, 2019). We previously demonstrated the potent efficacy of anti-asprosin mAbs in multiple mouse models of metabolic syndrome (Mishra et al., 2021). Given the human and rodent association of asprosin deficiency with marked hypophagia, leanness and protection from diet-induced metabolic syndrome, and its elevation in individuals with metabolic syndrome, neutralization of asprosin or its downstream signaling seems to be an exceptionally promising therapeutic strategy for the treatment of metabolic syndrome. The identification of Ptprd as an asprosin receptor provides another therapeutic strategy for the pharmacological inhibition of the asprosin pathway for the treatment of metabolic syndrome. As proof-of-concept, we demonstrate that the PTPRD ligand-binding domain (PTPRD-LBD), when ectopically introduced in the circulation of DIO mice, leads to a reduction in appetite, body weight and blood glucose - similar to that seen with use of anti-asprosin mAbs (Mishra et al., 2021). Concordant with reduced appetite, asprosin-mediated AgRP neuron activation and Stat3-dephosphorylation, and thus its deactivation, were both suppressed by the PTPRD-LBD. Interestingly, in contrast to Ptprd−/− mice, which display no alterations in glucose homeostasis, this “receptor-trap” strategy also improved hyperglycemia in DIO mice, indicating sequestration of asprosin from both its receptors (i.e., Ptprd and Olfr734). Thus, like anti-asprosin mAbs, use of the soluble PTPRD-LBD is a dual-effect therapy that targets the two spatio-temporally distinct functions of asprosin - orexigenic and glucogenic. This pharmacologic approach could serve as stand-alone or combination therapy with anti-asprosin mAbs and is reminiscent of the TNFɑ targeting approach against inflammatory diseases (Humira® – anti-TNFɑ mAb, Enbrel® – TNFɑ receptor trap). We envision further studies assessing the potential for off-target or toxic effects in the future as we contemplate a drug development path to the clinic. However, the discovery of Ptprd as an orexigenic asprosin-receptor, and demonstration of the PTPRD-LBD as a pharmacologic inhibitor of the asprosin pathway, provide a new approach toward anti-metabolic syndrome pharmacotherapy.
Limitations of the study
The current study identified Ptprd as the orexigenic asprosin receptor and focused only on its role in AgRP neuron-mediated physiology. However, Ptprd is extensively expressed in numerous brain regions (Shishikura et al., 2016), similar to other important appetite modulating receptors such as LEPR, GLP-1R and MC-4R (Jensen et al., 2018; Kishi et al., 2003; Scott et al., 2009). Additionally, emerging evidence indicates that many more brain regions, such as the hippocampus (Sweeney et al., 2015), septum (Xu et al., 2019), and cortex (Azevedo et al., 2021), play previously underappreciated roles in feeding control. Thus, while not investigated in this study, non-AgRP neuron mediated roles of the asprosin-Ptprd axis represent a rich avenue for future studies. We found single- to double-digit nanomolar affinity between asprosin and PTPRD with multiple assays. However, the absence of possible binding potentiators (Sarrazin et al., 2011) and/or the absence of the correct in vivo context from the purely in vitro MST/BLI/SPR techniques, could have potentially underestimated the binding affinity in the present study. Further, it is imperative to point out that sex differences in pharmacology of anti-obesity drugs (Cataldi et al., 2019) is well documented. Thus, a sex-dependent variation in response to asprosin sequestration or neutralization cannot be ruled out, and this issue needs further investigation. Lastly, given our previous finding that cAMP signaling is indispensable for asprosin-mediated AgRP neuron activation (Duerrschmid et al., 2017), the identification of a putative brain receptor that is not a GPCR is somewhat surprising. However, there is precedence for non-GPCRs influencing cellular cAMP levels (Eriksson et al., 1995; Rahn et al., 1994) and precedence for a hormone to function via two vastly different receptor families in different tissues (Martinot et al., 2017; Prossnitz et al., 2007) as asprosin appears to do (Olfr734 and Ptprd). The focus on Stat3 in the present study is not meant to imply that other signaling pathways (e.g., cAMP) are unimportant for asprosin action. Rather it simply serves as the first of many such future efforts to define the asprosin mechanism in relevant cells.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Atul R. Chopra (atul.chopra@case.edu)
Materials Availability
Reagents and viral vectors used in the study are available from the lead contact upon request.
Data and Code Availability
The study did not generate any standardized data that needed to be deposited in a public repository.
This paper does not report original code.
Data S1 (Related to Figures 1–7; Supplemental Figures 1–10) represents an Excel file containing the values that were used to create all the graphs in the paper, as well as a PDF file containing the full-length, unprocessed Western blots. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
Wild-type C57BL/6 mice (WT mice; Jackson Laboratory, JAX:000664), diet induced obese mice (DIO mice; JAX:380050), Rosa26-Flipase+ mice (Jackson Laboratory, JAX: 003946), AgRP-RES-Cre (C57BL/6-Agrptm1(cre)Lowl; Jackson Laboratory, JAX: 012899), Rosa26-LSL-tdTOMATO (JAX: 007905), Rosa26-Ribotag (Jackson Laboratory, JAX: 029977) were purchased from The Jackson Laboratory. Two different strains of Ptprd mice were used in this work and were maintained as heterozygous. B6;129-Ptprd< tm1YiW > mice were purchased from the RIKEN BioResource Center. Mice with Ptprd cKO potential (C57BL/6N-A<tm1Brd>Ptprd<tm2a(KOMP)Wtsi>/WtsiOrl; MEXY mice) was purchased from Wellcome Trust Sanger Institute and crossed to Flpase+ mice to remove the neomycin selection cassette and LacZ reporter, thereby making a conditional allele. Thereafter, homozygous conditionally ready floxed mice (Ptprd tm2c(KOMP)Wtsi) were mated with AgRP-IRES-Cre (C57BL/6-Agrptm1(cre)Lowl) to create AgRP neuron specific knock-out (KO) of Ptprd. For cKO of Ptprd in adult mice, CRISPR-cas9 mediated unilateral and bilateral KO was done in AgRP-IRES-Cre (C57BL/6-Agrptm1(cre)Lowl) mice. Littermates from in-house matings served as controls in all experiments, except for WT lean and DIO mice that were bought from The Jackson Laboratory and used for experimentation after acclimation to the mouse housing facility.
Mice were housed in microventilators on a 12-hour light cycle in animal facility maintained at 20-25°C and 40-60% humidity. Mice had ad libitum access to water and normal chow (5V5R, Lab Supply), dustless pellet diet (F0173, Bio-Serv) or high fat diet (60% calories from fat, TD.06414, Envigo Teklad). Animal housing, husbandry, and euthanasia were conducted under animal protocols approved by the Case Western Reserve University Institutional Animal Care and Use Committee (protocol# 2018-0042). General health of mice was monitored by the CWRU animal resource center.
Postnatal knockdown of Ptprd in AgRP neurons
CRISPR/Cas9 approach was used for unilateral and bilateral disruption of Ptprd selectively in AgRP neurons. Briefly, targeting efficiency of sgRNA GTCAGCAACCAGAGATTTGA against Ptprd was tested and confirmed with TIDE analysis (Brinkman et al., 2014) before being cloned into AAV-ITR-U6-sgRNA plasmid.
To fully ablate the function of Ptprd selectively in AgRP neurons, a bilateral knockout of Ptprd was done in AgRP-IRES-Cre male and female mice (12 weeks old). AgRP-IRES-Cre mice received bilateral stereotaxic injections of AAV-FLEX-saCas9 (Vector Biolabs, #7122) and AAV-Ptprd/sgRNA-FLEX-mCherry in the arcuate nucleus of the hypothalamus (ARC). Wild type mice received the same viruses and AgRP-IRES-Cre mice received bilateral AAV-Ptprd/sgRNA-FLEX-mCherry and the AAV-mCherry (no Cas9) were served as two groups of control mice. Seven weeks post injection, mice were subjected to high fat diet for the next 5 weeks. Mice were monitored for changes in body weight and food intake throughout the experiment.
For unilateral knockdown of Ptprd, AgRP-IRES-Cre male mice (10-12 weeks old) received stereotaxic injections of AAV-FLEX-saCas9 (Vector Biolabs, #7122) with AAV-Ptprd/sgRNA-FLEX-GFP on one side and AAV-Ptprd/sgRNA-FLEX-GFP with AAV-mCherry (no Cas9) virus on the other side in the ARC. Six weeks after the stereotaxic injections, electrophysiology recordings in GFP-labeled AgRP neurons from each side of ARC (control vs. Ptprd-KO side) was performed.
Cell culture
HEK293T cells were maintained at 37°C and 5% CO2 in DMEM, containing 10% FBS (HyClone) and 100 mg ml-1 penicillin-streptomycin. In general, siRNA was transfected using transfection reagent (Dharmacon, T-2001-02) following manufacturer’s protocol, and asprosin was overexpressed using Ad5-Asprosin or Asprosin coding mammalian expression plasmid with Ad5-empty or CMV6-Entry-empty as controls. For luciferase readout, HEK293T cells were transduced with 4xM67 pTATA-TK-Luc (Addgene; 8688) plasmid. HEK293T cell transfection, lysis and luciferase readouts were done with Fugene HD transfection reagent (Promega; E2312), Reporter Lysis 5X Buffer (Promega, E3971) and Luciferase Assay Reagent (Promega, E1483), respectively, using standard manufacturer’s protocol.
METHOD DETAILS
Immunoprecipitation and mass spectrometry
Cerebella were dissected, chopped into small pieces, and pooled from 5 adult mice and placed in 5 ml of DMEM media at room temperature. 300 μg of recombinant asprosin or GFP was added to each sample and incubated at room temperature for 10 min with rotation. Formaldehyde was added to a final concentration of 0.5% and samples were incubated at room temperature for 10 minutes with rotation. Crosslinking was quenched with addition of 125 mM glycine. Samples were incubated for another 5 minutes at room temperature with rotation. Cerebellar tissue was centrifuged at 2000 rpm for 5 minutes to collect tissue and then washed 3X with cold PBS. Crosslinked cerebellar tissue was then lysed with 3 times packed tissue volumes of NETN (170mM NaCl, 1mM EDTA, 50mM Tris, and 0.5% NP40) by sonication (Sonics $ Materials, Inc., CT) with 25amp for total 3 min operation with 60s rest every 30 sec. After ultracentrifugation (100kg, 20min, 4°c), supernatant was pre-cleared with protein A beads. Pre-cleared supernatant was incubated with anti-asprosin antibody and antibody-protein complex were pulled down by protein A bead. Immunoprecipitated proteins complex were extracted from bead by boiling in laemmli buffer and separated by SDS-PAGE. After in-gel digestion by trypsin, peptides recovered from gel was analyzed with LC-MS/MS (Ultimate 3000 LC coupled with Orbitrap Fusion™ Tribrid™) based on a previous study (Jung et al., 2017). Search of the obtained mass spectrum was done in Proteome Discoverer 1.4 interface with the Mascot algorithm 2.4. In order to quantify asprosin and Ptprd, searched data and raw data was imported into Skyline proteomics application (MacCoss lab, Seattle, WA) and the AUC of unique peptides of each gene were calculated. Four unique peptides were used for each gene including 2 miscleavages without any static or dynamic modification (Asprosin - DYLSGELGDNLK, KKPVAGTYSLQISSTPLYK, KPVAGTYSLQISSTPLYK, KPVAGTYSLQISSTPLYKK, YDKDYLSGELGDNLK, YLIESGNEDGFFK, YLIESGNEDGFFKINQK; Ptprd – NVLELNDVR, SPQGLGASTAEISAR, SYSFVLTNR, VVAVNNIGR).
For assessment of recombinant receptor-ligand interaction, 1 μg of recombinant (r) GST-Asprosin and rPTPRD-his protein (Creative BioMart, PTPRD-36H) were mixed and incubated at room temperature with rotation in IP Lysis Buffer (ThermoFisher Scientific, 87787) supplemented with 1X Halt protease inhibitor cocktail (ThermoFisher Scientific, 87786). Aliquots of this suspension were incubated with rabbit anti-PTPRD (ABclonal, A15713, 1:1000), mouse anti-asprosin, mouse IgG (Southern Biotech, 0107-01, 1:1000) or Rabbit IgG (Southern Biotech, 0111-01, 1:1000) for two hours. Thereafter, protein-antibody samples were loaded onto Protein G magnetic beads (ThermoFisher Scientific, 88847) and incubated for 1 hour with rotation. Elute from magnetic beads was subjected to western blot analysis of asprosin and PTPRD.
Asprosin-PTPRD affinity assessment
Surface plasmon resonance:
Surface plasmon resonance (SPR) studies were performed using Biacore T200 with PTPRD (Acro Biosystems, PTD-H52H9) covalently immobilized on an S series CM5 sensor chip via amine coupling. To avoid protein aggregation, caspase-1 used in SPR experiments were purified by gel filtration in a buffer containing 25 mM Tris-HCl (pH8.0), 100 mM NaCl and 5 mM DTT, stored at 4 °C, and used within 48 hr with minimum concentration process. Asprosin was injected in a series of concentrations from 1 μM to 7.8125 nM was injected at 30 μl/min over the sensor chip at room temperature. Sensorgram traces subtracted with the reference and zero-concentration traces were analyzed using BIA Evaluation software (GE Healthcare Bio-Sciences). Nonlinear steady-state analysis was performed to compute Kd.
Microscale thermophoresis:
The microscale thermophoresis (MST) assay was performed with the Monolith NT.115 from NanoTemper Technologies. PTPRD (Acro Biosystems, PTD-H52H9) was fluorescently labeled according to the manufacturer’s protocol (His-Tag Labeling Kit RED-tris-NTA, NanoTemper). A solution of unlabeled asprosin was serially diluted according to the manufacturer’s protocol. After labeled PTPRD and unlabeled asprosin were mixed and incubated at room temperature for 30 minutes, samples were loaded into glass capillaries (Monolith NT.115 Capillaries, NanoTemper). Measurements were carried out according to manufacturer’s protocol: 40% power and 40% MST power. Assays were repeated three times and KD’s were calculated using the NanoTemper analysis software.
Biometric layer interference:
Binding affinity of asprosin to PTPRD was measured on Pall ForteBio’s Octet RED96 system. The PTPRD protein (Creative BioMart, PTPRD-36H) was labeled with Biotin using EZ-Link-Sulfo-NHS-Biotin (ThermoFisher Scientific, 21217) and desalted by Zeba™ Spin Desalting Columns. The biotin-labeled PTPRD (20μg/ml) was loaded on the super streptavidin (SSA) biosensors for 300 seconds. Following 20 seconds of baseline in kinetics buffer, the loaded biosensors were dipped into a series of 3-fold diluted human asprosin (1.23–900 nM) for 300 seconds to record association kinetics and then dipped into a kinetic buffer for 600 seconds to record dissociation kinetics. Kinetic buffer without asprosin was set to correct the background. For fitting of KD value, ForteBio’s data analysis software was used to fit the curve by a 1:1 binding model and the global fitting method was applied.
RNAScope analysis of Ptprd and Olfr734
Mice were anesthetized and perfused transcardially with 0.9% saline followed by 10% formalin. Brains were removed and post fixed in 10% formalin for 16 h at 4°C and cryoprotected in 30% sucrose for 48 hours. Brains were frozen, sectioned at 14 μm using the cryostat, washed in DEPC-treated phosphate buffered saline for 10 min. Sections were mounted on DEPC-treated charged slides, dried for 0.5 hour at room temperature and stored at −80°C. On the day of the RNAScope assay, the slides were thawed and slides were rinsed 2 times in PBS 1X and placed in an oven for 30 min at 60°C. After that, slides were post fixed in 10% formalin for 15 minutes at 4°C. Slides were then gradually dehydrated in ethanol (50, 70 and 100%, 5 min each) and underwent target retrieval for 5 minutes at 100°C. Slides were incubated in protease III (#322337, ACDBio) for 30 minutes at 40°C. Slides were then rinsed in distilled water and incubated in RNAScope probes for AgRP (Mm-AgRP; #400711, ACDBio) and Olfr734 (Mm-Olfr734-C3; #878653-C3, ACDBio) or Prprd (Mm-Ptprd-C2; #474651-C2, ACDBio,) for 2 hours at 40°C. Sections were then processed using the RNAScope Fluorescent Multiplex Detection Reagents (#320851, ACDBio) according to the manufacturer instructions. Slides were cover-slipped and analyzed using a fluorescence microscope.
AgRP neuron labelling and Electrophysiology
AgRP neuron labeling, and electrophysiology experiments were performed as previously described (Duerrschmid et al., 2017; He et al., 2016). Briefly, to identify AgRP neurons, we crossed the Rosa26-LSL–tdTOMATO mice with AgRP-IRES-Cre mice to generate AgRP-IRES–Cre/Rosa26-LSL–tdTOMATO mice, which express tdTOMATO selectively in AgRP neurons. The entire brains of AgRP-IRES-Cre/Rosa26-LSL-tdTOMATO mice were removed and immediately submerged in ice-cold sucrose-based cutting solution (adjusted to pH 7.3) containing (in mM) 10 NaCl, 25 NaHCO3, 195 Sucrose, 5 Glucose, 2.5 KCl, 1.25 NaH2PO4, 2 Na pyruvate, 0.5 CaCl2, 7 MgCl2 bubbled continuously with 95% O2 and 5% CO2. The slices (250 μm) were cut with a Microm HM 650V vibratome (Thermo Scientific and recovered for 1 h at 34°C and then maintained at room temperature in artificial cerebrospinal fluid (aCSF, pH 7.3) containing 126 mM NaCl, 2.5 mM KCl, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 11.1 mM glucose, and 21.4 mM NaHCO3 saturated with 95% O2 and 5% CO2. tdTOMATO(+) neurons were visualized using epifluorescence and IR-DIC imaging on an upright microscope equipped with a moveable stage (MP-285, Sutter Instrument). Individual neurons were manually picked up by the pipette and 20 neuron cells were combined as a sample for qPCR analysis of Ptprd and Olfr734.
In mice with unilateral knock out of Ptprd using CRISPR-Cas9, six weeks after the stereotaxic injections of AAV-FLEX-saCas9+AAV-Ptprd/sgRNA-FLEX-GFP on one side and AAV-Ptprd/sgRNA-FLEX-GFP+AAV-mCherry (no Cas9) on the other side, electrophysiology recording of GFP-labeled or mCherry-labeled AgRP neurons was performed in AgPR-IRES-Cre mice. Mice maintained under ad libitum feeding or after overnight fasting were deeply anesthetized with isoflurane and were transcardially perfused, brain slices containing the ARH prepared and maintained in artificial CSF as described above. GFP or mCherry-labeled neurons in the ARH were visualized using epifluorescence and infrared–differential interference contrast (IR-DIC) imaging on an upright microscope (Eclipse FN-1, Nikon).
For electrophysiological recording, brain slices were superfused at 34°C in oxygenated artificial CSF at a flow rate of 1.8-2 ml/min. Patch pipettes with resistances of 3-5 MΩ were filled with intracellular solution (pH 7.3) containing 128 mM K-Gluconate, 10 mM KCl, 10 mM HEPES, 0.1 mM EGTA, 2 mM MgCl2, 0.05 mM Na-GTP and 0.05 mM Mg-ATP. Recordings were made using a MultiClamp 700B amplifier (Axon Instrument), sampled using Digidata 1440A, and analyzed offline with pClamp 10.3 software (Axon Instruments). Series resistance was monitored during the recording, and the values were generally <10 MΩ and were not compensated. Data were excluded if the series resistance increased dramatically during the experiment or without overshoot for the action potential. Currents were amplified, filtered at 1 kHz, and digitized at 20 kHz. The current clamp was engaged to measure neural firing frequency and resting membrane potential (RM) in control and Ptprd KO AgRP neurons from fed and fasted mice. Some experiments were performed in brain slices from fed mice after one-hour incubation with GFP or 34nM asprosin prior to recording.
In another experiment, brain slices containing the ARC were prepared from AgRP-IRES-Cre/Rosa26-LSL-tdTOMATO mice (10-12 weeks age) using same method described above. tdTOMATO(+) AgRP neuron depolarization and firing rate were recorded in response to asprosin alone and asprosin preincubated with rPTPRD-LBD or a physiologically irrelevant protein (GFP). For this, rAsprosin was preincubated with either PTPRD-LBD or a physiologically irrelevant protein (GPF) at a 1:10 ratio and then kept on ice for 1 h before treating the brain. tdTOMATO(+) AgRP neurons were first exposed to a puff of rAsprosin (34nM), followed by perfusion with a mixture of rAsprosin+ PTPRD-LBD (or rAsprosin+GFP), each for 4 minutes. After a wash, AgRP neuron were treated with another puff of asprosin (34nM).
Adenovirus injection
Twelve-fourteen-week-old normal chow-fed lean Ptprd−/− and Ptprd+/+(WT; wildtype) littermate male mice were injected intravenously via the tail-vein with adenovirus (Ad5) as previously described (Mishra et al., 2021). Mice injected with Ad5-empty (3.6 x 109 pfu/ mouse) served as controls for experimental mice that received Ad5-FBN1 virus (3.6 x 109pfu/ mouse) containing the human FBN1 coding region under control of a CMV promoter. Mice injected with Ad5-empty (5 x 1010 pfu/mouse) served as controls for experimental mice that received Ad5-IL2-Asprosin (5 x 1010 pfu/mouse) containing an N-terminal his-tagged human asprosin coding region preceded by an IL2 signal peptide, under control of an EF1 promoter.
Eighteen-week-old DIO (diet induced obese; C57BL/6J) male mice were intravenously injected via the tail vein with Ad5-IL2-hPTPRD-LBD or Ad5-eGFP (1 x 1011 vp/mouse or 2 x 1012 vp/mouse). Mice transduced with Ad5-eGFP served as controls for experimental mice that received Ad5-IL2-hPTPRD-LBD containing an N-terminal his-tagged ligand-binding-domain (LBD) of PTPRD coding region preceded by an IL2 signal peptide, under control of an EF1 promoter.
Western blot analysis
HEK293T cell protein lysates were prepared in Pierce immunoprecipitation lysis buffer (ThermoFisher Scientific, 87787) supplemented with 1X Halt protease inhibitor cocktail (ThermoFisher Scientific, 87786). 5 or 10 μg cell lysate samples were mixed with Bolt LDS Sample Buffer (Fisher Scientific, B00008) and run on a Bolt 4–12% Bis-Tris Plus gradient gel (Thermo Fisher Scientific). Protein lysate from Snapfrozen hypothalamic tissue of Ptprd −/− and WT mice was prepared using N-PER Neuronal protein extraction reagent (Thermo Fisher, 87792) supplemented with protease inhibitor and phosphatase inhibitor. 60 μg protein lysate samples were mixed with NuPAGE™ sample buffer (Fisher Scientific, NP0007) and run on a NuPAGE™ 3 to 8%, Tris-Acetate gel (Thermo Fisher Scientific). Proteins were transferred to nitrocellulose membranes using the Invitrogen Power Blotter for 7-10 minutes at room temperature. The membranes were blocked with Clear Milk Blocking Buffer or 5% BSA blocking buffer (in 1X TBST). Membranes were incubated with primary antibody at 4° C overnight followed by incubation with HR-labelled secondary antibody for 2 hr at room temperature. HRP was detected using chemiluminiscent substrates (Thermo Fisher Scientific, 34577, 34094). Membranes blotted for p-Stat3 were stripped, washed and reblotted for stat3 detection.
Antibodies used were rabbit polyclonal anti-PTPRD (1:500; ABclonal, A15713), mouse monoclonal anti-asprosin (1:2000), mouse monoclonal anti-stat3 (1:2000; Cell signaling technology, 9139), rabbit monoclonal anti-phospho Stat3-Tyr705 (1:500; Cell signaling technology, catalog#9145); Mouse βActin (1:2000; Cell signaling; 8H10D10) mAb; HRP-conjugated anti-rabbit (1:10,000; GE Healthcare), and HRP-conjugated anti-mouse (1:10,000; KPL Scientific).
Quantitative PCR
Ptprd expression was measured in hypothalamus, cerebellum, heart, liver, white adipose tissue, brown adipose tissue, and skeletal muscle of wild type mice. Expression of PTPRD was measured in liver of mice transduced with Ad5-GFP and Ad5-PTPRD-LBD, and in HEK293T cells transfected with scrambled or PTPRD siRNA. Aliquots of these tissues and cells were used for RNA extraction using the RNeasy kit (Qiagen) following manufacturer’s instructions. First-strand cDNA was synthesized with Iscript™ cDNA synthesis kit (Bio-Rad) and subjected to qPCR analysis. For assessment of Ptprd and Olfr734 in AgRP neurons, individual neurons were manually picked up by the pipette and 20 neuron cells were combined as a sample RNA extraction and reverse transcription using the Ambion Single-Cell-to-CT Kit (Ambion, Life Technologies) according to the manufacturer’s instruction. Briefly, 10 μl Single Cell Lysis solution with DNase I was added to each sample, and then the entire cell contents were used for cDNA synthesis (25°C for 10 min, 42°C for 60 min, and 85°C for 5 min).
For Ribotag pulldown of transcripts from AgRP neurons, RNA was isolated using Ribo-Tag strategy as described before (Wang et al., 2018). Briefly, 350 μl lysate from hypothalamic punches containing the entire ARC region of AgRP-IRES-Cre/ Rosa26-LSL-RiboTag mice was incubated with 1 μl anti-HA antibody (MMS-101P, Covance) for 4 h at 4 °C, and then 50 μl of protein A/G agarose beads (Santa Cruz) overnight at 4 °C, followed by high salt buffer wash. The RNA from inputs (before incubation) and immunoprecipitates were extracted using Qiagen RNeasy Micro Plus kit (Qiagen) and used for RT-PCR.
Real-time qPCR was performed using iTaq™ Universal Probes Supermix (Bio-Rad) and the Bio-Rad CFX96 Real-Time system (Bio-Rad). Target gene primer sets were designed to be compatible with the Universal ProbeLibrary (Roche). The following primers were used: Ptprd-Fwd: 5’tctgaggccaggaactgttt3’, Ptprd-Rev: 5’tggaacccttttagagcttgc3’, Gapdh-Fwd: 5’gggttcctataaatacggactgc3’, Gapdh-Rev: 5’ccattttgtctacgggacga3’; Olfr734-Fwd: 5’gcagggctatatccacttgttatt3’; Olfr734-Rev: 5’gatggatggtccaaacattagc3’; Npy-Fwd: 5’ccgctctgcgacactacat3’; Npy-Rev: 5’tgtctcagggctggatctct3’, AgRP-Fwd: 5’ttccaggctatataacaaaatctgtg3’, AgRP-Rev: 5’tgtagccagggcatgagg3’; GAPDH-Fwd: 5’gagtccactggcgtcttcac3’, GAPDH-Rev: 5’gttcacacccatgacgaaca3’; PTPRD-Fwd: 5’ctgtgacagcccatacagatg3’, PTPRD-Rev: 5’gcgaggaggaccactagga3’; PTPRD (for detection of PTPRD-ligand binding domain in mouse liver)-Fwd: 5’aatatgagtgtgttgccaccaa3’; PTPRD (for detection of PTPRD-ligand binding domain in mouse liver)-Rev: 5’gacacggcgaacttctcg3’. The following Universal ProbeLibrary probes (Roche) were used with Ptprd: 7, Gapdh: 52, Npy: 9; AgRP: 95; Olfr734: 9; GAPDH: 45; PTPRD: 81; PTPRD-LBD: 51.
Metabolic caging and manual food intake assessment
Metabolic caging experiments were performed at the Cardiovascular Research Institute Mouse Metabolic and Phenotyping Core at CWRU (IACUC# 2019-0029). Mice were housed with a 12-h light/dark cycle (7am/7pm) at 22°C with controlled humidity. Metascreen software (V2.3.15.11) controlled system data acquisition. Respirometry (VO2, VCO2, H2O vapor), activity and ad libitum food and water intake measures were collected individually using a Promethion metabolic cage system (Sable Systems, Las Vegas, NV, USA). Gas analyzers were calibrated before each run. Gas measurements were multiplexed over 8 cages and baselined to a cage-equivalent volume of room air twice per 5-minute cycle, while maintaining a 2L/min/cage, negative pressure-derived flow rate. Acquired data was processed using Macro Interpreter (V2.34) running Macro V2.33.3-slice1hr. Energy expenditure was calculated using the Weir equation (Weir, 1949).
In some experiments, for manual measurement of food intake, DIO mice were acclimated to crushed high fat diet (60% calories from fat, TD.06414, Envigo Teklad) in single housing for three days. The diet was replenished, weighed, and re-weighed after 24 hours to establish food intake.
Blood parameters
For glucose tolerance test (GTT), mice were intraperitoneally injected with glucose solution (2g/kg body mass) and blood glucose levels were measured at 0, 15-, 30-, 60- and 120-min post treatment. Mouse glucose was determined using a hand-held glucometer (OneTouch Ultra2, LifeScan) from a droplet of tail blood. Plasma Asprosin and Ghrelin levels were measured using mouse asprosin (Amsbio, AMS.ELK6516 and Abbexa, abx585287) and mouse/rat total Ghrelin (EMD Millipore, EZRGRT-91K) ELISA kits. Mouse plasma total cholesterol, HDL (high-density lipoprotein cholesterol), LDL (low-density lipoprotein cholesterol), triglyceride (TG), free fatty acids (FFA), glycerol, leptin, and adiponectin levels were measured by the mouse metabolism and phenotyping core at Baylor College of Medicine, Houston, Texas (MMPC at BCM & NIH fund RO1DK114356 & UM1HG006348).
Phospho-Stat3 assays and luciferase assays
In vivo assessment of phospho-Stat3:
16-week-old lean, WT mice were fasted overnight followed by 2 hours of refeeding, two weeks post transduction with ad5-hFBN1 or ad5-empty (3.6 x 109pfu/mouse). Mice were decapitated under deep isoflorane anesthesia and hypothalami excised, snapfrozen, and later subjected to phosphor-Stat3 quantification by ELISA (Abcam; ab126458), and by western blot analysis of β-actin, Stat3 and phospho-Stat3. Similarly, 16-week-old Ptprd−/− and wildtype (WT) normal chow fed male littermates were subjected to overnight fast followed by 2h refeeding, for assessment of hypothalamic phosphor-Stat3 quantification by western blot and ELISA. To compare pSTAT3 in animals matched for adiposity, phosphorylated-Stat3 (p-Stat3) levels were measured using ELISA in hypothalamic neural lysate of overnight fasted 20-week-old diet-induced obese (DIO) male mice, 16h post intraperitoneal treatment with anti-asprosin monoclonal antibody (mAb) or IgG control antibody (500μg in 500 ml saline/mouse; n = 6/group).
In vitro phospho-Stat3 studies:
Phospho-stat3 levels were quantified by western blot and luciferase readout in HEK293T cells under conditions of PTPRD knockdown, with or without asprosin overexpression.
To evaluate the effect of PTPRD loss on Stat3 activity, HEK293T cells were co-transfected with 2μg pTATA-TK-Luc (2μg) and PTPRD siRNA (25nM) or scrambled siRNA (25nM). Cells were lysed for assessment of siRNA efficiency, and phospho-Stat3 quantification by western blot and luciferase readout, 72 hours post transfection. In another experiment, Stat3-response element driven luciferase activity was assessed in HEK293T co-transfected with pTATA-TK-Luc (lμg/well) and serial dilution of asprosin-coding mammalian expression plasmid, with empty plasmid as control treatment (2μg, 1 μg, 0.5 μg and 0.25 μg/well). Luciferase assay readout was performed 72h post transfection. To ascertain the effect of PTPRD knockdown on asprosin mediated signaling, 24 hours after PTPRD or scrambled siRNA (25nM) treatment, asprosin was overexpressed using Ad5-asprosin or Ad5-empty (100vp/cell) in HEK293T cells. 48 hours post asprosin overexpression, media was collected for measuring secreted asprosin levels and cells were collected for western blot of β-actin, stat3 and p-Stat3. Similarly, asprosin was overexpressed using asprosin coding mammalian expression plasmid (1 μg/well) under conditions of PTPRD knockdown (with scrambled siRNA as control) and pTATA-TK-Luc (1 μg/well) transfection, and luciferase assay was done 72h post treatment for assessment of asprosin induced changes in cellular stat3 activity under receptor knockdown conditions.
For in vitro neutralization of asprosin, HEK293T cells were transfected with empty or asprosin coding mammalian expression plasmid (1 μg/well). 48h post transfection, cells were treated with 1μg recombinant eGFP or rPTPRD protein (Acro Biosystems, PTD-H52H9) for 8 hours. Cells were lysed for western blot of βactin, stat3 and p-Stat3. Similar experiment was done under conditions of pTATA-TK-Luc (1 μg/well) transfection, and luciferase assay was performed 8 hours post ectopic protein treatment. Additionally, effect of asprosin neutralization was tested in HEK293T cells expressing asprosin or cells exposed to asprosin conditioned media. For this, HEK293T cells were co-transfected with pTATA-TK-Luc (2 ug/well) and asprosin coding mammalian expression plasmid (1 μg/well) for 48h, followed by ectopic treatment of anti-asprosin mAb or IgG control. Stat3-response element driven luciferase activity was measured 18h post antibody treatment. Additionally, above experiment was repeated using asprosin-conditioned media. Conditioned media was collected from HEK293T cells transfected with asprosin expressing mammalian plasmid, and incubated with IgG control or anti-asprosin mAb (500 ng/ml) for 2h. Stat3-response element driven luciferase activity was measured in pTATA-TK-Luc (2 ug/well) transfected cells, 18 h post treatment with asprosin conditioned media+IgG and asprosin conditioned media+anti-asprosin mAb.
Mouse body composition, magnetic resonance imaging
Mouse MRI imaging experiments were performed at the Case Center for Imaging Research. Each animal was anesthetized in isoflurane and placed in the prone position at isocenter in a Bruker Biospec 7T MRI scanner. A 72-mm inner diameter volume coil was used to acquire the images. Following initial localizer scans, a fat-water technique was used to obtain three separate high resolution coronal image sets with multiple echo shifts in order to generate separate fat and water images using the Relaxation-Compensated Fat Fraction (RCFF) MRI as described previously (Johnson et al., 2012). These quantitative fat fraction maps were then used to automatically segment out peritoneal and subcutaneous adipose tissue in each imaging slice. The total volume of the peritoneal and subcutaneous adipose tissue was then calculated by summing over all imaging slices.
Asprosin and PTPRD ligand binding domain ELISA detection procedures
Media collected from HEK293T cells transduced with Ad5-asprosin or Ad5-empty was subjected to asprosin ELISA, as previously described (Duerrschmid et al., 2017; Mishra et al., 2021). For validation of asprosin sequestration with PTPRD-ligand binding domain, 5nM solution of recombinant human asprosin preincubated with recombinant PTPRD or GFP protein was subjected to asprosin ELISA as previously described (Duerrschmid et al., 2017; Mishra et al., 2021). For detection of human asprosin in plasma of mice treated with human asprosin and FBN1 expressing Ad5 vectors, plasma samples were first processed for IgG and albumin removal using Proteome purify2 columns (R&D systems, Catalog # IDR002) and concentrated using vivaspin 500 (VS0131) PES filters before running the ELISA (Mishra et al., 2021).
Briefly, a sandwich ELISA custom built using mouse monoclonal anti-asprosin antibody against human asprosin amino acids 106–134 (human profibrillin amino acids 2838–2865) as the capture antibody, and a rabbit anti-asprosin monoclonal antibody as the detection antibody was used for detection of human asprosin (Duerrschmid et al., 2017; Mishra et al., 2021). An anti-rabbit secondary antibody linked to HRP was used to generate a signal, and mammalian-cell produced recombinant human asprosin was used to generate a standard curve.
For detection of human PTPRD ligand binding domain (LBD) in plasma of mice treated with PTPRD-LBD expressing adenoviruses, a sandwich ELISA was custom built using rabbit anti-PTPRD (100ng/well; ABclonal; A15713) as the capture antibody, and mouse anti-his (100ng/well; Genscript; A00186) as the detection antibody. An anti-mouse secondary antibody linked to HRP was used to generate a signal, and recombinant PTPRD (Acro Biosystems, PTD-H52H9) was used to generate a standard curve.
QUANTIFICATION AND STATISTICAL ANALYSES
All results are presented as mean ± standard error of the mean (s.e.m.). Statistical significance of continuous data was tested using unpaired two-tailed Student’s t-tests or Analysis of Variance (one-way and two-way ANOVA, when appropriate) followed by the Bonferroni multiple test corrections post-hoc analysis using GraphPad Prism 6 & 7. For metabolic caging experiments, data was additionally analyzed using analysis of covariance (ANCOVA) to account for covariates on R software (4.0.3). Repeated measures analysis was used in experiments that involved multiple measures of the same variable. Age- and sex-matched mice were assigned to groups randomly for all experiments. Promethion data collection and analysis, qPCR studies and RNAscope studies were done in a blinded fashion. Power analysis was not done for predetermining the sample size, instead an n ≥ 6 mice for in vivo studies and an n ≧ 4 biological replicates for all in vitro studies was considered. Data outliers were identified with Grubb’s outlier analysis with Q = 1% in GraphPad Prism 7. Grubb’s outlier analysis identified an outlier in plasma leptin and ghrelin levels of Ptprd−/− mice. Food intake data of one mouse with malocclusion was excluded from Ad5-hFBN1 experiment. Promethion metabolic caging data (body weight, energy expenditure, food intake, respiratory exchange ratio, and locomotor activity) of two Ptprd−/− mice was excluded from analysis due to malocclusion. Alpha (α) for statistical significance was set at 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-PTPRD | ABclonal | A15713 |
| Mouse anti-asprosin | This paper | N/A |
| mouse IgG | Southern Biotech | 0107-01 |
| Rabbit IgG | Southern Biotech | 0111-01 |
| mouse monoclonal anti-stat3 | Cell signaling technology | 9139 |
| rabbit monoclonal anti-phospho Stat3-Tyr705 | Cell signaling technology | 9145 |
| Mouse βActin | Cell signaling technology | 8H10D10 |
| HRP-conjugated anti-rabbit | GE Healthcare | NA934-1ML |
| HRP-conjugated anti-mouse | KPL Scientific | 474-1806 |
| anti-HA antibody | Covance | MMS-101P |
| mouse anti-his | Genscript | A00186 |
| Bacterial and virus strains | ||
| AAV-FLEX-saCas9 | Vector Biolabs | 7122 |
| AAV-Ptprd/sgRNA-FLEX-mCherry | This paper | N/A |
| Ad5-empty Adenovirus | Mishra et al., 2021 | N/A |
| Ad5-FBN1 virus Adenovirus | Mishra et al., 2021 | N/A |
| Ad5-IL2-Asprosin Adenovirus | Mishra et al., 2021 | N/A |
| Ad5-IL2-hPTPRD-LBD Adenovirus | This paper | N/A |
| Ad5-eGFP Adenovirus | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| GST-Asprosin | This paper | N/A |
| His-Asprosin | Biolegend | 761902 |
| PTPRD protein | Creative BioMart | PTPRD-36H |
| PTPRD protein | Acro Biosystems | PTD-H52H9 |
| Green Fluorescent Protein | United States Biological | G8965-10E |
| His-Tag Labeling Kit RED-tris-NTA | NanoTemper | SKU: MO-L018 |
| Protease III | ACDBio | 322337 |
| RNAScope probes for AgRP | ACDBio | 400711 |
| RNAScope probes for Olfr734 | ACDBio | 878653-C3 |
| RNAScope probes for Prprd | ACDBio | 474651-C2 |
| IP Lysis Buffer | ThermoFisher Scientific | 87787 |
| 1X Halt protease inhibitor cocktail | ThermoFisher Scientific | 87786 |
| Protein G magnetic beads | ThermoFisher Scientific | 88847 |
| Bolt LDS Sample Buffer | Fisher Scientific | B00008 |
| Bolt 4–12% Bis-Tris Plus gradient gel | Thermo Scientific | NW04122BOX |
| N-PER Neuronal protein extraction reagent | Thermo Fisher | 87792 |
| NuPAGE™ sample buffer | Fisher Scientific | NP0007 |
| NuPAGE™ 3 to 8%, Tris-Acetate gel | Thermo Scientific | EA03755BOX |
| chemiluminiscent substrates | Thermo Fisher Scientific | 34577 |
| chemiluminiscent substrates | Thermo Fisher Scientific | 34094 |
| Critical commercial assays | ||
| Mouse Asprosin ELISA kit | Amsbio | AMS.ELK6516 |
| Mouse Asprosin ELISA kit | Abbexa | Abx585287 |
| mouse/rat total Ghrelin ELISA kit | EMD Millipore | EZRGRT-91K |
| Phospho-Stat3 ELISA kit | Abcam | ab126458 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| C57BL/6J Mus musculus | Jackson Laboratory | JAX:000664 |
| ROSA26::FLPe knock in | Jackson Laboratory | JAX:003946 |
| C57BL/6-Agrptm1(cre)Lowl | Jackson Laboratory | JAX:012899 |
| B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | Jackson Laboratory | JAX:007905 |
| B6J.129(Cg)-Rpl22tm1.1Psam/SjJ Mus musculus | Jackson Laboratory | JAX: 029977 |
| B6;129-Ptprdtm1Yiw/YiwRbrc Mus musculus | RIKEN BioResource | RBRC04925 |
| C57BL/6N-Atm1Brd Ptprdtm2a(KOMP)Wtsi/Mmucd | Wellcome Trust Sanger Institute | MMRRC_065397-UCD |
| Oligoneucleotides | ||
| Ptprd-Fwd: 5’tctgaggccaggaactgttt3’ | This paper | N/A |
| Ptprd-Rev: 5’tggaacccttttagagcttgc3’ | This paper | N/A |
| Gapdh-Fwd: 5’gggttcctataaatacggactgc3’ | This paper | N/A |
| Gapdh-Rev: 5’ccattttgtctacgggacga3’ | This paper | N/A |
| Olfr734-Fwd: 5’gcagggctatatccacttgttatt3’ | This paper | N/A |
| Olfr734-Rev: 5’gatggatggtccaaacattagc3’ | This paper | N/A |
| Npy-Fwd: 5’ccgctctgcgacactacat3’ | This paper | N/A |
| Npy-Rev: 5’tgtctcagggctggatctct3’ | This paper | N/A |
| AgRP-Fwd: 5’ttccaggctatataacaaaatctgtg3’ | This paper | N/A |
| AgRP-Rev: 5’tgtagccagggcatgagg3’ | This paper | N/A |
| GAPDH-Fwd: 5’gagtccactggcgtcttcac3’ | This paper | N/A |
| GAPDH-Rev: 5’gttcacacccatgacgaaca3’ | This paper | N/A |
| PTPRD-Fwd: 5’ctgtgacagcccatacagatg3’ | This paper | N/A |
| PTPRD-Rev: 5’gcgaggaggaccactagga3’ | This paper | N/A |
| PTPRD (for detection of PTPRD-LBD)-Fwd: 5’aatatgagtgtgttgccaccaa3’ | This paper | N/A |
| PTPRD (for detection of PTPRD-LBD)-Rev: 5’gacacggcgaacttctcg3’ | This paper | N/A |
| Ptprd sgRNA (gtcagcaaccagagatttga) | This paper | N/A |
| siGENOME Non-Targeting siRNA Pool #1, 5 nmol | Dharmacon | D-001206-13-20 |
| siGENOME Human PTPRD siRNA - SMARTpool | Dharmacon | M-008527-01-0010 |
| Recombinant DNA | ||
| 4xM67 pTATA-TK-Luc | Addgene | 8688 |
| pCMV-Asprosin | This Paper | N/A |
| pCMV-Empty | This Paper | N/A |
| Software and algorithms | ||
| Graphpad Prism version 6 & 7 | GraphPad | https://www.graphpad.com |
| R version 4.0.3 | R software | https://www.R-project.org/ |
| Other | ||
| DharmaFECT 1 Transfection Reagent | Dharmacon | T-2001-02 |
| RNeasy Mini Kit (250) | Qiagen | 74106 |
| Iscript™ cDNA Synthesis Kit | Bio-Rad | 1708891 |
| Itaq™ Universal Probes Supermix | Bio-Rad | 172-5131 |
| Ambion Single-Cell-to-CT Kit | Thermo Fisher Scientific | 44-582-37 |
| Reporter Lysis 5X Buffer | Promega | E3971 |
| Luciferase Assay Reagent | Promega | E1483 |
| FuGENE HD Transfection Reagent | Promega | E2311 |
| Teklad High Fat Diet | Envigo | TD.06414 |
Highlights:
Protein Tyrosine Phosphatase Receptor δ is the orexigenic asprosin receptor.
Ptprd loss results in appetite reduction and unresponsiveness to exogenous asprosin.
AgRP neuron-specific Ptprd ablation leads to protection from diet-induced-obesity.
The Ptprd Ligand-binding-domain functions as an appetite suppressing therapeutic.
Acknowledgements
We thank Andrew Pieper, Seth Field, and members of the Chopra lab for helpful suggestions and critical reading of the manuscript. This work was supported by the NIDDK (DK102529, DK118290).
Declaration of Interests
Atul Chopra has been awarded asprosin-related patents, and is a co-founder, director and officer of Vizigen, Inc., and Aceragen, Inc., and holds equity in both companies. The other authors declare that no competing interests exist.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alan M, Gurlek B, Yilmaz A, Aksit M, Aslanipour B, Gulhan I, Mehmet C, and Taner CE (2018). Asprosin: a novel peptide hormone related to insulin resistance in women with polycystic ovary syndrome. Gynecol Endocrinol 35, 1–4. [DOI] [PubMed] [Google Scholar]
- Azevedo EP, Ivan VJ, Friedman JM, and Stem SA (2021). Higher-Order Inputs Involved in Appetite Control. Biol Psychiatry S0006-3223, 01468–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baykus Y, Yavuzkir S, Ustebay S, Ugur K, Deniz R, and Aydin S (2019). Asprosin in umbilical cord of newborns and maternal blood of gestational diabetes, preeclampsia, severe preeclampsia, intrauterine growth retardation and macrosemic fetus. Peptides 120, 170132. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Lebrun N, Clarke J, Duriez P, Gorwood P, and Ramoz N (2020). De novo deleterious variants that may alter the dopaminergic reward pathway are associated with anorexia nervosa. Eat Weight Disord - Stud Anorexia Bulimia Obes 25, 1643–1650. [DOI] [PubMed] [Google Scholar]
- Böhmer F-D, and Friedrich K (2014). Protein tyrosine phosphatases as wardens of STAT signaling. Jak-Stat 3, e28087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168–e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldi M, Muscogiuri G, Savastano S, Barrea L, Guida B, Taglialatela M, and Colao A (2019). Gender-related issues in the pharmacology of new anti-obesity drugs. Obes Rev 20, 375–384. [DOI] [PubMed] [Google Scholar]
- Chagnon MJ, Uetani N, and Tremblay ML (2004). Functional significance of the LAR receptor protein tyrosine phosphatase family in development and diseases. Biochem Cell Biol 82, 664–675. [DOI] [PubMed] [Google Scholar]
- Chen M, Yao B, Yang Q, Deng J, Song Y, Sui T, Zhou L, Yao H, Xu Y, Ouyang H, et al. (2018). Truncated C-terminus of fibrillin-1 induces Marfanoid-progeroid-lipodystrophy (MPL) syndrome in rabbit. Dis Model Mech 11, dmm031542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg DJ, Riedy CA, Smith KAB, Benoit SC, and Woods SC (2003). Differential Sensitivity to Central Leptin and Insulin in Male and Female Rats. Diabetes 52, 682–687. [DOI] [PubMed] [Google Scholar]
- Drgonova J, Walther D, Wang KJ, Hartstein GL, Lochte B, Troncoso J, Uetani N, Iwakura Y, and Uhl GR (2015). Mouse Model for Protein Tyrosine Phosphatase D (PTPRD) Associations with Restless Leg Syndrome or Willis-Ekbom Disease and Addiction: Reduced Expression Alters Locomotion, Sleep Behaviors and Cocaine-Conditioned Place Preference. Mol Med 21, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerrschmid C, He Y, Wang C, Li C, Bournat JC, Romere C, Saha PK, Lee ME, Phillips KJ, Jain M, et al. (2017). Asprosin is a centrally acting orexigenic hormone. Nat Med 23, 1444–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan OK, Inglis MA, and Anderson GM (2017). Leptin Signaling in AgRP Neurons Modulates Puberty Onset and Adult Fertility in Mice. J Neurosci 37, 3875–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson H, Ridderstråle M, Degerman E, Ekholm D, Smith CJ, Manganiello VC, et al. (1995). Evidence for the key role of the adipocyte cGMP-inhibited cAMP phosphodiesterase in the antilipolytic action of insulin. Biochimica et biophysica acta 1266, 101–7. [DOI] [PubMed] [Google Scholar]
- He Y, Shu G, Yang Y, Xu P, Xia Y, Wang C, Saito K, Hinton A, Yan X, Liu C, et al. (2016). A Small Potassium Current in AgRP/NPY Neurons Regulates Feeding Behavior and Energy Metabolism. Cell Reports 17, 1807–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich G, Meece K, Wardlaw SL, and Accili D (2014). Preserved Energy Balance in Mice Lacking FoxO1 in Neurons of Nkx2.1 Lineage Reveals Functional Heterogeneity of FoxO1 Signaling Within the Hypothalamus. Diabetes 63, 1572–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry FE, Sugino K, Tozer A, Branco T, and Sternson SM (2015). Cell type-specific transcriptomics of hypothalamic energy-sensing neuron responses to weight-loss. Elife 4, e09800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoschek R, Hoffmann T, Morcos YAT, Sengle G, Dötsch J, and Hucklenbruch-Rother E (2020). Asprosin in pregnancy and childhood. Mol Cell Pediatrics 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CB, Pyke C, Rasch MG, Dahl AB, Knudsen LB, and Secher A (2018). Characterization of the Glucagonlike Peptide-1 Receptor in Male Mouse Brain Using a Novel Antibody and In Situ Hybridization. Endocrinology 159, 665–675. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Fox NC, Sperling RA, and Klunk WE (2012). Brain Imaging in Alzheimer Disease. Csh Perspect Med 2, a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SY, Choi JM, Rousseaux MWC, Malovannaya A, Kim JJ, Kutzera J, Wang Y, Huang Y, Zhu W, Maity S, et al. (2017). An Anatomically Resolved Mouse Brain Proteome Reveals Parkinson Disease-relevant Pathways*. Mol Cell Proteomics 16, 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitakahara A, Bouyer K, Wang C, and Simerly R (2018). A critical period for the trophic actions of leptin on AgRP neurons in the arcuate nucleus of the hypothalamus. J Comp Neurol 526, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, and Elmquist JK (2003). Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol 457, 213–235. [DOI] [PubMed] [Google Scholar]
- Li E, Shan H, Chen L, Long A, Zhang Y, Liu Y, Jia L, Wei F, Han J, Li T, et al. (2019). OLFR734 Mediates Glucose Metabolism as a Receptor of Asprosin. Cell Metab 30, 319–328.e8. [DOI] [PubMed] [Google Scholar]
- Liu T, Xu Y, Yi C-X, Tong Q, and Cai D (2021). The hypothalamus for whole-body physiology: from metabolism to aging. Protein Cell 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Long A, Chen L, Jia L, and Wang Y (2020). The Asprosin–OLFR734 module regulates appetitive behaviors. Cell Discov 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciukiewicz M, Gorbovskaya I, Tiwari AK, Zai CC, Freeman N, Meltzer HY, Kennedy JL, and Müller DJ (2019). Genetic validation study of protein tyrosine phosphatase receptor type D (PTPRD) gene variants and risk for antipsychotic-induced weight gain. J Neural Transm 126, 27–33. [DOI] [PubMed] [Google Scholar]
- Martinot E, Sèdes L, Baptissart M, Lobaccaro JM, Caira F, Beaudoin C, et al. (2017). Bile acids and their receptors. Mol Aspects Med 56, 2–9. [DOI] [PubMed] [Google Scholar]
- Mina AI, LeClair RA, LeClair KB, Cohen DE, Lantier L, and Banks AS (2018). CalR: A Web-Based Analysis Tool for Indirect Calorimetry Experiments. Cell Metab 28, 656–666.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra I, Duerrschmid C, Ku Z, He Y, Xie W, Silva ES, Hoffman J, Xin W, Zhang N, Xu Y, et al. (2021). Asprosin neutralizing antibodies as a treatment for metabolic syndrome. Elife 10, e63784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistrík P, Moreau F, and Allen JM (2004). BiaCore analysis of leptin–leptin receptor interaction: evidence for 1:1 stoichiometry. Anal Biochem 327, 271–277. [DOI] [PubMed] [Google Scholar]
- Morcos YAT, Lütke S, Tenbieg A, Hanisch FG, Pryymachuk G, Piekarek N, Hoffmann T, Keller T, Janoschek R, Niehoff A, Zaucke F, Dötsch J, Hucklenbruch-Rother E, and Sengle G (2022). Sensitive asprosin detection in clinical samples reveals serum/saliva correlation and indicates cartilage as source for serum asprosin. Scientific reports, 12(1), 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller TD, Klingenspor M, and Tschöp MH (2021). Revisiting energy expenditure: how to correct mouse metabolic rate for body mass. Nat Metab 3, 1134–1136. [DOI] [PubMed] [Google Scholar]
- Ortiz B, Fabius AWM, Wu WH, Pedraza A, Brennan CW, Schultz N, Pitter KL, Bromberg JF, Huse JT, Holland EC, et al. (2014). Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis. Proc National Acad Sci 111, 8149–8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyser ND, Du Y, Li H, Lui V, Xiao X, Chan TA, and Grandis JR (2015). Loss-of-Function PTPRD Mutations Lead to Increased STAT3 Activation and Sensitivity to STAT3 Inhibition in Head and Neck Cancer. Plos One 10, e0135750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, Sklar LA (2007). GPR30: A G protein-coupled receptor for estrogen. Mol Cell Endocrinol 265-266, 138–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn T, Ridderstråle M, Tornqvist H, Manganiello V, Fredrikson G, Belfrage P, et al. (1994). Essential role of phosphatidylinositol 3-kinase in insulin-induced activation and phosphorylation of the cGMP-inhibited cAMP phosphodiesterase in rat adipocytes. Studies using the selective inhibitor wortmannin. FEBS Lett 350, 314–8. [DOI] [PubMed] [Google Scholar]
- Romere C, Duerrschmid C, Bournat J, Constable P, Jain M, Xia F, Saha PK, Del Solar M, Zhu B, York B, et al. (2016). Asprosin, a Fasting-Induced Glucogenic Protein Hormone. Cell 165, 566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin S, Lamanna WC, and Esko JD (2011). Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 3, a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM and Elmquist JK (2009). Leptin targets in the mouse brain. J Comp Neurol 514, 518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishikura M, Nakamura F, Yamashita N, Uetani N, Iwakura Y, and Goshima Y (2016). Expression of receptor protein tyrosine phosphatase δ, PTPδ, in mouse central nervous system. Brain Res 1642, 244–254. [DOI] [PubMed] [Google Scholar]
- Sweeney P, and Yang Y (2015). An excitatory ventral hippocampus to lateral septum circuit that suppresses feeding. Nat Commun 6, 10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita H, Cornejo F, Aranda-Pino B, Woodard CL, Rioseco CC, Neel BG, Alvarez AR, Kaplan DR, Miller FD, and Cancino GI (2020). The Protein Tyrosine Phosphatase Receptor Delta Regulates Developmental Neurogenesis. Cell Reports 30, 215–228.e5. [DOI] [PubMed] [Google Scholar]
- Ugur K, and Aydin S (2019). Saliva and Blood Asprosin Hormone Concentration Associated with Obesity. Int J Endocrinol 2019, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl GR, and Martinez MJ (2019). PTPRD: neurobiology, genetics, and initial pharmacology of a pleiotropic contributor to brain phenotypes. Ann Ny Acad Sci 1451, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, and Xu Y (2019). Mechanisms for sex differences in energy homeostasis. J Mol Endocrinol 62, R129–R143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, He Y, Xu P, Yang Y, Saito K, Xia Y, Yan X Jr, A. H, Yan C, Ding H, et al. (2018). TAp63 contributes to sexual dimorphism in POMC neuron functions and energy homeostasis. Nat Commun 9, 1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yin C, Wang L, Liu Y, Li H, Li M, Yi X, and Xiao Y (2020). Serum Asprosin Concentrations Are Increased and Associated with Insulin Resistance in Children with Obesity. Ann Nutr Metab 75, 205–212. [DOI] [PubMed] [Google Scholar]
- Weir J.B. de V. (1949). New methods for calculating metabolic rate with special reference to protein metabolism. J Physiology 109, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker L, Hao C, Fu W, and Whittaker J (2008). High-Affinity Insulin Binding: Insulin Interacts with Two Receptor Ligand Binding Sites. Biochemistry-Us 47, 12900–12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lu Y, Cassidy RM, Mangieri LR, Zhu C, Huang X, Jiang Z, Justice NJ, Xu Y, Arenkiel BR, and Tong Q (2019). Identification of a neurocircuit underlying regulation of feeding by stress-related emotional responses. Nat Commun 10, 3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Ste-Marie L, Kaelin CB, and Barsh GS (2007). Inactivation of Signal Transducer and Activator of Transcription 3 in Proopiomelanocortin (Pomc) Neurons Causes Decreased Pome Expression, Mild Obesity, and Defects in Compensatory Refeeding. Endocrinology 148, 72–80. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chen C, Zhou N, Fu Y, and Cheng X (2019). Circulating asprosin concentrations are increased in type 2 diabetes mellitus and independently associated with fasting glucose and triglyceride. Clin Chim Acta 489, 183–188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study did not generate any standardized data that needed to be deposited in a public repository.
This paper does not report original code.
Data S1 (Related to Figures 1–7; Supplemental Figures 1–10) represents an Excel file containing the values that were used to create all the graphs in the paper, as well as a PDF file containing the full-length, unprocessed Western blots. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.