

Abstract
Herein we report a novel chlorooxime mediated modification of native peptides and proteins under physiologic conditions. This method features fast reaction kinetics (apparent k2 = 306 ± 4 M−1s−1 for GSH) and exquisite selectivity for cysteine residues. This cysteine conjugation reaction can be carried out with just single-digit micromolar concentrations of the labeling reagent. The conjugates show high stability towards acid, base, and external thiol nucleophiles. A nitrile oxide species generated in situ is likely involved as the key intermediate. Furthermore, a bis-chlorooxime reagent is synthesized to enable facile Cys-Cys stapling in native peptides and proteins. This highly efficient cysteine conjugation and stapling was further implemented on bacteriophage to construct chemically modified phage libraries.
Keywords: Cysteine modification, Protein modification, Peptide stapling, Chlorooxime, Phage modification
Graphical Abstract
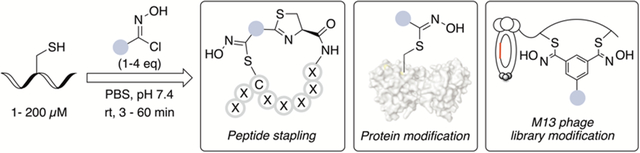
Ultrafast and cysteine-specific modifications are reported using chlorooximes. This cysteine conjugation reaction can be carried out with single-digit micromolar concentrations of the labeling reagent under physiological conditions. The conjugates show high stability towards acid, base, and external thiol nucleophiles. A nitrile oxide species generated in situ is likely involved as the key intermediate. These methods are applied to native peptide modification, proteins modification, and chemically modified phage libraries construction.
Introduction
Biocompatible and chemo-selective reactions for protein modification[1] have been heavily sought after as they enable a myriad of applications, including labeling a protein of interest in situ and constructing antibody-drug conjugates as therapeutics. Among the 20 canonical amino acids, cysteine has emerged as the residue of choice for site-specific protein modification due to its low abundance and strong nucleophilicity.[2] Well-developed reagents for cysteine conjugation include activated disulfides,[3] alkyl halides,[3–4] Michael acceptors,[5] and maleimide derivatives[6] (Figure 1a). Despite the long history of these cysteine-reactive reagents, they do have their limitations. The majority of these cysteine-reactive reagents exhibit modest to slow kinetics, which demands for their use at high concentrations. In contrast to other commonly used reagents, maleimide derivatives are known to elicit fast cysteine conjugation (k2: ~102 M−1s−1).[7] However, they are also known to yield conjugates prone to hydrolysis.[6d] There remains substantial interest in developing highly efficient and biocompatible cysteine conjugation chemistries,[8] as exemplified by the recent reports of hypervalent iodine reagent,[9] strain-release reagent[10] and organometallic reagent.[11]
Figure 1.

Reagents for cysteine-selective modifications in peptides and proteins.
Nitrile oxide is a reactive organic compound and it reacts with a wide range of alkenes, alkynes,[12] as well as various nucleophiles including those seen in proteins such as cysteine thiols. Nucleophile-nitrile oxide addition reactions have been known for decades in organic chemistry, however, their applications to biological systems are rarely reported as the nitrile oxide formation had been only documented in organic solvent with an organic base. The nitrile oxides’ vulnerability to hydrolysis and the potential challenge of selectivity control towards different nucleophilic amino acid residues limit their development in biological sciences. Recently, water-assisted nitrile oxide formation has been reported using chlorooxime as the precursor, which enabled facile conjugation with alkynes and isonitriles in aqueous media.[13] On a related front, Schreiber and co-workers successfully applied masked nitrile-oxide electrophiles for selective covalent targeting of glutathione peroxidase 4 (GPX4).[14] Inspired by these publications, we examined the potential of nitrile oxide chemistry for chemo-selective modification of proteins. Our results show that chlorooxime functions as a highly efficient reagent for cysteine-specific modification of peptides and proteins under mild conditions (neutral pH, room temperature) (Fig. 1b).
Results and Discussion
Reaction profiles
Inspired the report of the Kittakoop group[13b] and more recently the Wennemers group,[13a] which describe water-assisted nitrile oxide generation from chlorooximes, we set out to examine the chemoselectivity of nitrile oxides by reacting (Z)-N-hydroxybenzimidoyl chloride (FC1) with glutathione (GSH). Remarkably, when mixed at 50 μM under physiological conditions (10 mM phosphate-buffered saline (PBS), pH 7.4, room temperature), the conjugation went to completion in less than 3 min with quantitative conversion to a thiohydroximate product 2 (Figure 2a). Importantly, the use of organic co-solvent is not required for the efficient bioconjugation despite the fact that the chlorooxime FC1 has a low solubility in water. As a control experiment, oxidized glutathione (GSSG) was subjected to the chlorooxime FC1 and it remained intact after incubation for 20 mins (Figure 2b), indicating that the FC1-GSH conjugation occurs exclusively at the thiol group. We then systematically examine the amino acids with potential reactive functional groups, including lysine (primary amino group), serine and tyrosine (hydroxyl group), histidine (imidazole group), and tryptophan (indole group) (Figure 2c). None of these amino acids gave any detectable conjugation product with chlorooxime FC1 except cysteine (entry 1, Figure 2c). Interestingly, the conjugation of a free or N-terminal cysteine (NCys) with chlorooxime FC1 gave 2-thiazoline 3 as the product, which presumably results from the intramolecular cyclization of the thiohydroximate followed by elimination of hydroxylamine (Figure 2d).[15] These results demonstrate the exquisite chemoselectivity of FC1 towards the cysteine thiol over other proteinogenic nucleophiles. It is worth noting that FC1 can be easily prepared from corresponding aldehyde in 2 steps on gram scale and they can remain good quality for months if stored properly (inert atmosphere, −4 to −20 °C).
Figure 2.

Identification of chlorooxime-cysteine bioconjugation. a) LC-MS of GSH and the crude reaction mixture of GSH with FC1. Reaction conditions: GSH (50 μM), FC1 (50 μM) in PBS(10 mM, pH 7.4) at room temperature for 3 min. b) from top to bottom: 1H NMR of FC1, a mixture of FC1 (2 mM) with GSH (4 mM) for 3 min, isolated conjugate 2, and a mixture of FC1 (2 mM) with GSSG (4 mM) for 20 min. c) Conjugation of amino acids with FC1. Reaction conditions: amino acid (50 μM), chlorooxime FC1 (100 μM), in PBS (10 mM, pH 7.4) at rt for 1 hour. 5% of co-solvent acetone was used for Fmoc-Cys-OH to increase the solubility. aWith 200 μM of FC1. d) Conjugation of L-cysteine with chlorooxime and proposed mechanism.
Next, we studied the stability of conjugated product thiohydroximate 2 towards acid, base and thio-exchange reagent see Supporting Information, Figure S19). The conjugate 2 is proved to be stable in aqueous solution with pH range from 2 to 8.8 and no hydrolysis happened over one week (entry 1 to 4, Figure S19). When conjugate 2 was incubated with free cysteine in basic or acidic aqueous solution, the conjugate stayed intact and the signals C–S bond cleavage was not detected as assessed by 1H NMR and mass spectrometry (entry 5 to 8, Figure S19). The addition of 5 eq of iodoacetamide didn’t give any thiol alkylated product, indicating that the formation of the C–S bond is irreversible (entry 9, Figure S19). These results demonstrate the high stability of conjugate product to acid, base as well as thio-exchange conditions.
Kinetic and mechanism studies
Kinetic experiments were carried out by studying the conjugation of chlorooxime FC1 with glutathione (GSH) (Figure 3). The reaction progression was recorded by monitoring the UV-Vis absorption (247 nm) changes of the reaction mixture. As stated above, the reaction of FC1 with GSH exhibit extremely fast kinetics with a half-life of 41 seconds (Figure 3a). As shown in Figure 3b, the linear correlation of Ln[FC1] versus time indicates a first-order decay of the chlorooxime.[16] Interestingly, increasing the initial concentration of GSH did lead to the proportional increase of the reaction rate, even though the chlorooxime decay followed first order kinetics in all cases. This (pseudo)first order behavior can be potentially rationalized by a postulated low concentration of the reactive nitrile oxide species at any given time of the reaction. Plotting the observed first-order rate constant k1 versus the initial concentration of GSH gives a linear relationship, from which an apparent rate constant k2, app was extracted to be 306 ± 4 M−1s−1 (Figure 3c). Both GSH and chlorooxime are likely involved in the rate-determining step. A similar kinetic profile was observed for FC1 conjugation with β-mercaptoethanol (BME): the reaction proceeds rapidly (t1/2 = 147 s) under the same experimental conditions, albeit being a bit slower than that of GSH (see Supporting Information, Figure S12). The slower reaction rate observed for BME can be rationalized by its higher pKa value (9.61 for BME[17] vs. 8.66 for GSH[18]). The kinetics of FC1-GSH conjugation compares favorably even to the fastest cysteine conjugation reactions such as those using iodoacetamide[4b] or maleimide[6c, 7b, 7c]. In the competition experiment performed, a 1:1 equivalent mixture of iodoacetamide and chlorooxime was treated with glutathione and it was found that the conjugation with chlorooxime is overwhelming, giving thiohydroximate as the only product (see Supporting Information, Figure S26). In the absence of a reactive partner, FC1 in aqueous solution (PBS, pH 7.4) does undergo slow degradation with a half-life of 9.8 hours (see Supporting Information, Figure S18). Considering the ultrafast conjugation with thiols, this small degradation of FC1 does not appear to present any complication for cysteine modification, as seen in the peptide and protein labeling studies described below.
Figure 3.

Kinetic and mechanism studies of chlorooxime-thiol conjugations. a) Real-time monitor of conjugate reaction using UV-Visible absorbance at 247 nm (max absorbance wavelength for chlorooxime FC1). Reaction conditions: GSH (50 μM), chlorooxime (50 μM) in PBS pH 7.4 at room temperature. b) Plotting the natural logarithm of the concentration of FC1 (M) versus time (s) for the reactions of FC1 (50 μM) with BME (50 μM), GSH (50 μM), GSH (100 μM), GSH (250 μM), respectively. c) Plotting observed first-order rate constant k1 versus the initial concentration of GSH. Based on the observation k1 = k2, app.×[GSH]0, the slope of the curve gives the overall rate constant k2, app. of 306 ± 4 M−1s−1. d) Proposed mechanisms for cysteine-chlorooxime conjugation.
A plausible mechanism for this novel bioconjugation was proposed (Figure 3d). The formation of a reactive nitrile oxide intermediate int I is likely involved through the water-assisted α-elimination of hydrochloric acid from chlorooxime.[13] Then the nitrile oxide int I reacts predominately with the more nucleophilic sulfhydryl group from cysteine residue to form the C–S bond over other potential nucleophiles, achieving the high selectivity. An alternative mechanism involves the nucleophilic addition of sulfhydryl group to chlorooxime followed by β-elimination of a molecule of hydrochloric acid to form the bioconjugation product. A control experiment using 3-chlorobenzo[d]isoxazole didn’t afford any conjugate with GSH, implying the chlorooxime-cysteine bioconjugation is more likely to proceed through a nitrile oxide intermediate (see Supporting information for detail).
Cysteine-specific peptide modification and stapling.
We further examined the cysteine specificity of the chlorooxime conjugation using model peptides that incorporate various residues. The peptides were synthesized to carry a fluorophore (FAM or TAMRA) on their C-terminus, which facilitates monitoring the reaction progression using LC-MS. As shown in Figure 4, the labeling of peptide P1 with chlorooxime FC1 went smoothly at physiological conditions, affording a double thiohydroximate modified peptide 4 at 94% conversion. Double labeling of peptide P3, which harbors an N-terminal cysteine (NCys) and an internal cysteine, was readily achieved using FC1 as well, yielding a thiazoline- and thiohydroximate-conjugated peptide 5 in near complete conversion (Figure 4b). Excited by the efficiency and thiol-specificity of FC1, we synthesized a bis-chlorooxime FC2, which we envisioned would allow facile Cys-Cys crosslinking to give peptide macrocycles. As expected, the macrocyclized peptide P1 was obtained with near complete conversion by treating the peptide with FC2 (entry 1, Figure 4c). Excitingly, the Cys-Cys stapling efficiency remained intact when decreasing the reaction concentration down to 1 micromolar (entry 2, Figure 4c). The Cys-Cys stapling of peptide P2 using FC2 was completed in less than 15 min with 94% conversion (entry 3, Figure 4c). Under identical conjugation conditions, commonly used crosslinking reagents (DCA[19], DBB[20], DBM[21]) showed much lower efficiency (entry 4–6, Figure 4c). Interestingly, cyclization of N-terminal cysteine-bearing peptide P3 can be readily accomplished with FC2 as well, yielding a novel macrocycle with an embedded thiazoline ring (Figure 4d).
Figure 4.
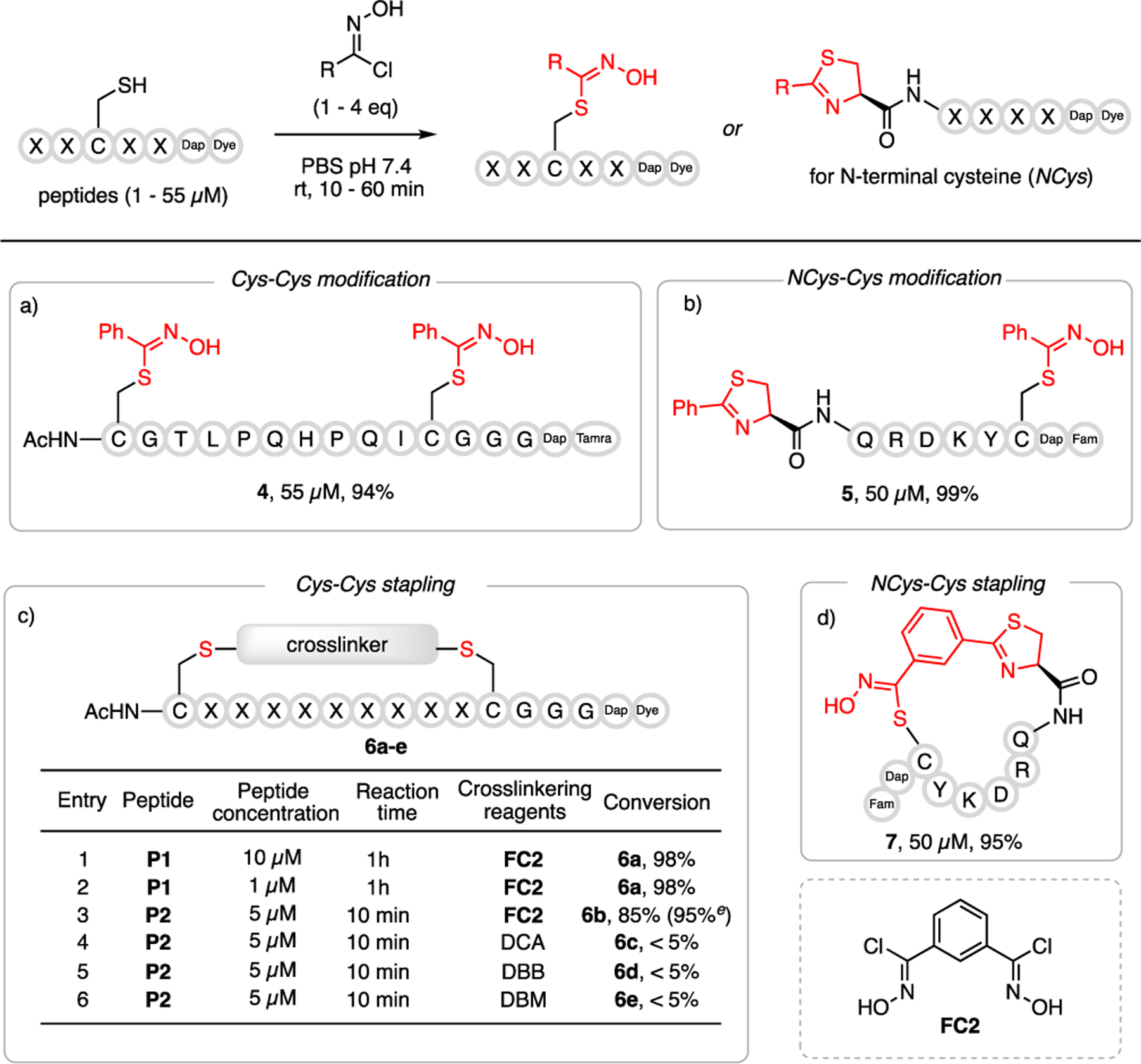
Cysteine-specific peptide modification and stapling. a-d) Reaction conditions: peptide (1 to 55 μM), chlorooxime FC1 (4 eq) or crosslinking reagents (2 eq) with TCEP (2 eq) in PBS (10 mM, pH 7.4) at room temperature for 10–60 min. Incubation overnight for N-terminal cysteine. e) incubation for 15 min. P1: (Ac)CGTLPQHPQICGGG(Dap)(TAMRA). P2: (Ac)CEFHYK NTAQCGGGDap(FAM). P3: CQRDKYC(Dap)(FAM). DCA: 1,3-dichloropropan-2-one. DBB: 1-(azidomethyl)-3,5-bis(bromomethyl)benzene. DBM: 2,3-dibromomaleimide. TCEP: tris(2-carboxyethyl)phosphine.
Site-specific protein modification.
We further assessed the chlorooxime-cysteine conjugation for site-specific labeling of large proteins. We first used Tobacco Etch Virus (TEV) protease as a model protein for this line of studies. The engineered TEV protease in this study has four cysteine residues in total, two of which are surface exposed while the other two are buried in the protein interior (Figure 5a). The TEV protease (24 μM) was incubated with chlorooxime FC1 at physiological conditions (Tris pH 8.0, aqueous solution, room temperature) and the reaction was monitored by LC-MS. Deconvoluted mass showed only two cysteine residues modification occurred affording conjugate 8, and over modification was not observed after 1-hour incubation. This result implied both of the two surface-exposed cysteines were converted to the corresponding thiohydroximate, and the other two buried cysteine residues remained intact. Another protein we tested against bis-chlorooxime FC2 is the pIII coat protein of the M13 phage, which was engineered to carry a disulfide-cyclized peptide on its N-terminus. Selective reduction of this disulfide was accomplished by treating this protein with TCEP. The reduced protein was then subjected to crosslinking and recyclization with bis-chlorooxime FC2. According to the results of LC-MS analysis, the bis-chlorooxime FC2 afforded complete pIII protein cyclization product 9 with 10 μM reagent over just 10 minutes.
Figure 5.

Cys-specific proteins modification. a) TEV protease modification using FC1. Reaction conditions: TEV protease (24 μM), chlorooxime FC1 (100 μM) in Tri pH 8.0 at room temperature for 60 min. b) Deconvoluted mass spectra of protein for the starting TEV protease (left) and reactions with FC1 (right). c) pIII protein cyclization using FC2. Reaction conditions: reduced pIII protein (5 μM), chlorooxime FC2 (10 μM) in PBS pH 7.4 at room temperature for 10 min. d) Deconvoluted mass spectra of protein for the starting reduced pIII protein (left) and reactions with FC2 (right).
Novel chemically modified phage libraries.
An appealing application of site-specific protein modification is to create chemically modified phage libraries. Phage display is a powerful technique for constructing and screening peptide libraries,[22] which however had been limited to natural peptide libraries. Through site-specific modification of phage coat proteins, several designer peptide libraries have been recently reported.[20, 23] To realize the full potential of phage display of non-natural peptide libraries, diverse chemistries are needed for efficient and site-specific modification of phage-displayed peptides.[24] We envisioned the bis-chlorooxime can be used to construct novel macrocyclic peptide libraries on phage.
Encouraged by the pIII modification result, we went on to test the potential of using chlorooxime and bis-chlorooxime for phage modification. Towards this end, we attempted to synthesize a biotin conjugate of the chlorooximes with no success as biotin was found prone to oxidation in the final NCS oxidation step in chlorooxime synthesis.[25] Alternatively, we decided to use biotin sulfone (BSO) as an affinity tag. Similar to biotin, BSO is known to bind streptavidin, albeit with somewhat reduced affinity.[26] The details of synthesis for the BSO-chlorooxime conjugates FC4 and FC5 (Figure 6) are reported in the Supporting Information. We tested these compounds for phage modification using a C6C peptide library displayed on M13 phage. The phage library was treated with TCEP to convert the C6C disulfide into free cysteines and then mixed with the chlorooxime compounds (10 μM) for labeling. After 30–60 min of incubation, the phage library was subjected to PEG precipitation to remove excess labeling reagents. The resuspended phage was subjected to pulldown by the streptavidin beads (Figure 6a). The phage suspension was titered before and after the pulldown to assess the degree of BSO modification of the phage. The results (Figure 6b) show that both FC4 and FC5 modified phage libraries allowed efficient pulldown by streptavidin beads, yielding a similar pulldown percentage as the Biotin-IA treated phage used as a positive control. These results showcase the application of chlorooxime-cysteine conjugation chemistry in the construction of chemically modified phage libraries.
Figure 6.

Phage modification via chlorooxime-cysteine conjugation. a) Illustration of phage modification with FC4 and FC5, and the streptavidin pulldown assay to quantify labeling efficiency. b) Results of streptavidin pull-down assay showing FC4/5 elicited efficient phage modification at 10 μM within 30–60 min. The positive control molecule, Biotin-IA, was used at 1 mM for 2 hr to give a similar percentage of labeling.
Conclusion
This contribution reports a systematic investigation of the nitrile oxide chemistry for modifying native peptides and proteins. Specifically, we find that chlorooximes can elicit ultrafast and cysteine-specific conjugation to native peptides and proteins, via a nitrile oxide species autonomously generated in situ. We note that a nice paper by Wang and coworkers appeared during the preparation of our manuscript,[27] which also describes the efficient and cysteine-specific reactivity of the chlorooximes. While sharing the main conclusion, our work herein provides quantitative description of the chlorooxime-cysteine conjugation kinetics, which gives an apparent k2 of 306 ± 4 M−1 s−1, comparable to the fastest cysteine conjugations known to date. Owing to its fast kinetics, we further show the chlorooxime-mediated cysteine modification can be carried out with just single digit micromolar concentrations of the reagent. The resulting conjugates show robust stability towards acid, base and thio-exchange reagent. Importantly, we report bis-chlorooxime derivatives that installs an affinity handle to elicit Cys-Cys and NCys-Cys stapling of model proteins as well as bacteriophage, which allows facile construction of phage displayed novel macrocyclic peptide libraries. Although cysteine modification has been extensively investigated, few known reactions come close to the fast kinetics and exquisite cysteine selectivity seen in this study as well as in the paper of the Wang group.[27] We believe this highly efficient chemistry will find wide applications in chemical biology research as well as in protein engineering towards novel therapeutics.
Supplementary Material
Acknowledgements
Financial support of this work is provided by the National Science Foundation (CHE-1904874), the National Institutes of Health (GM102735), and the Ono Pharma Foundation.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Boutureira O, Bernardes GJL, Chem. Rev 2015, 115, 2174–2195; [DOI] [PubMed] [Google Scholar]; b) Tamura T, Hamachi I, J. Am. Chem. Soc 2019, 141, 2782–2799; [DOI] [PubMed] [Google Scholar]; c) Baslé E, Joubert N, Pucheault M, Chem. Biol 2010, 17, 213–227; [DOI] [PubMed] [Google Scholar]; d) Chalker JM, Bernardes GJL, Davis BG, Acc. Chem. Res 2011, 44, 730–741; [DOI] [PubMed] [Google Scholar]; e) Tang KC, Cao J, Boatner LM, Li L, Farhi J, Houk KN, Spangle J, Backus KM, Raj M, Angew. Chem. Int. Ed 2022, 61, e202112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Ochtrop P, Hackenberger CPR, Curr. Opin. Chem. Biol 2020, 58, 28–36; [DOI] [PubMed] [Google Scholar]; b) Gunnoo SB, Madder A, ChemBioChem 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- [3].Hemantha HP, Bavikar SN, Herman-Bachinsky Y, Haj-Yahya N, Bondalapati S, Ciechanover A, Brik A, J. Am. Chem. Soc 2014, 136, 2665–2673. [DOI] [PubMed] [Google Scholar]
- [4].a) Lindley H, Biochem. J 1962, 82, 418–425; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nelson KJ, Day AE, Zeng B-B, King SB, Poole LB, Anal. Biochem 2008, 375, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Ariyasu S, Hayashi H, Xing B, Chiba S, Bioconjugate Chem. 2017, 28, 897–902; [DOI] [PubMed] [Google Scholar]; b) Silva M, Faustino H, Coelho JAS, Pinto MV, Fernandes A, Companon I, Corzana F, Gasser G, Gois PMP, Angew. Chem. Int. Ed 2021, 60, 10850–10857; [DOI] [PubMed] [Google Scholar]; c) Bernardim B, Cal PMSD, Matos MJ, Oliveira BL, Martínez-Sáez N, Albuquerque IS, Perkins E, Corzana F, Burtoloso ACB, Jiménez-Osés G, Bernardes GJL, Nat. Commun 2016, 7, 13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Kim Y, Ho SO, Gassman NR, Korlann Y, Landorf EV, Collart FR, Weiss S, Bioconjugate Chem. 2008, 19, 786–791; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moore JE, Ward WH, J. Am. Chem. Soc 1956, 78, 2414–2418; [Google Scholar]; c) Wu C-W, Yarbrough LR, Wu FYH, Biochemistry 1976, 15, 2863–2868; [DOI] [PubMed] [Google Scholar]; d) Ravasco JMJM, Faustino H, Trindade A, Gois PMP, Chem. Eur. J 2019, 25, 43–59. [DOI] [PubMed] [Google Scholar]
- [7].a) Saito F, Noda H, Bode JW, ACS Chem. Biol 2015, 10, 1026–1033; [DOI] [PubMed] [Google Scholar]; b) Chen Y, Tsao K, De Francesco É, Keillor JW, J. Org. Chem 2015, 80, 12182–12192; [DOI] [PubMed] [Google Scholar]; c) Bednar RA, Biochemistry 1990, 29, 3684–3690. [DOI] [PubMed] [Google Scholar]
- [8].Cal PMSD, Bernardes GJL, Gois PMP, Angew. Chem. Int. Ed 2014, 53, 10585–10587. [DOI] [PubMed] [Google Scholar]
- [9].Ceballos J, Grinhagena E, Sangouard G, Heinis C, Waser J, Angew. Chem. Int. Ed 2021, 60, 9022–9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Lopchuk JM, Fjelbye K, Kawamata Y, Malins LR, Pan C-M, Gianatassio R, Wang J, Prieto L, Bradow J, Brandt TA, Collins MR, Elleraas J, Ewanicki J, Farrell W, Fadeyi OO, Gallego GM, Mousseau JJ, Oliver R, Sach NW, Smith JK, Spangler JE, Zhu H, Zhu J, Baran PS, J. Am. Chem. Soc 2017, 139, 3209–3226; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tokunaga K, Sato M, Kuwata K, Miura C, Fuchida H, Matsunaga N, Koyanagi S, Ohdo S, Shindo N, Ojida A, J. Am. Chem. Soc 2020, 142, 18522–18531. [DOI] [PubMed] [Google Scholar]
- [11].a) Vinogradova EV, Zhang C, Spokoyny AM, Pentelute BL, Buchwald SL, Nature 2015, 526, 687–691; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Waddington MA, Zheng X, Stauber JM, Hakim Moully E, Montgomery HR, Saleh LMA, Král P, Spokoyny AM, J. Am. Chem. Soc 2021, 143, 8661–8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Heaney F, Eur. J. Org. Chem 2012, 2012, 3043–3058. [Google Scholar]
- [13].a) Schäfer RJB, Monaco MR, Li M, Tirla A, Rivera-Fuentes P, Wennemers H, J. Am. Chem. Soc 2019, 141, 18644–18648; [DOI] [PubMed] [Google Scholar]; b) Kesornpun C, Aree T, Mahidol C, Ruchirawat S, Kittakoop P, Angew. Chem. Int. Ed 2016, 55, 3997–4001. [DOI] [PubMed] [Google Scholar]
- [14].a) Eaton JK, Ruberto RA, Kramm A, Viswanathan VS, Schreiber SL, J. Am. Chem. Soc 2019, 141, 20407–20415; [DOI] [PubMed] [Google Scholar]; b) Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, Zimmermann K, Cai LL, Niehues M, Badock V, Kramm A, Chen S, Hillig RC, Clemons PA, Gradl S, Montagnon C, Lazarski KE, Christian S, Bajrami B, Neuhaus R, Eheim AL, Viswanathan VS, Schreiber SL, Nat. Chem. Biol 2020, 16, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang W, Gao J, J. Org. Chem 2020, 85, 1756–1763. [DOI] [PubMed] [Google Scholar]
- [16].Blackmond DG, Angew. Chem. Int. Ed 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]
- [17].Jencks WP, Salvesen K, J. Am. Chem. Soc 1971, 93, 4433–4436. [Google Scholar]
- [18].Pirie NW, Pinhey KG, J. Biol. Chem 1929, 84, 321–333. [Google Scholar]
- [19].Chen S, Lovell S, Lee S, Fellner M, Mace PD, Bogyo M, Nat. Biotechnol 2020, 39, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Heinis C, Rutherford T, Freund S, Winter G, Nat. Chem. Biol 2009, 5, 502–507. [DOI] [PubMed] [Google Scholar]
- [21].Smith MEB, Schumacher FF, Ryan CP, Tedaldi LM, Papaioannou D, Waksman G, Caddick S, Baker JR, J. Am. Chem. Soc 2010, 132, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Smith GP, Petrenko VA, Chem. Rev 1997, 97, 391–410. [DOI] [PubMed] [Google Scholar]
- [23].a) Ng S, Lin E, Kitov PI, Tjhung KF, Gerlits OO, Deng L, Kasper B, Sood A, Paschal BM, Zhang P, Ling CC, Klassen JS, Noren CJ, Mahal LK, Woods RJ, Coates L, Derda R, J. Am. Chem. Soc 2015, 137, 5248–5251; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tharp JM, Hampton JT, Reed CA, Ehnbom A, Chen P-HC, Morse JS, Kurra Y, Pérez LM, Xu S, Liu WR, Nat. Commun 2020, 11, 1392; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Owens AE, Iannuzzelli JA, Gu Y, Fasan R, ACS Cent. Sci 2020, 6, 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zheng X, Li Z, Gao W, Meng X, Li X, Luk LYP, Zhao Y, Tsai Y-H, Wu C, J. Am. Chem. Soc 2020, 142, 5097–5103. [DOI] [PubMed] [Google Scholar]
- [25].a) Bates HA, Rosenblum SB, J. Org. Chem 1986, 51, 3447–3451; [Google Scholar]; b) Kruse CG, Poels EK, Jonkers FL, Van der Gen A, J. Org. Chem 1978, 43, 3548–3553. [Google Scholar]
- [26].a) GREEN N, Biochem. J 1963, 89, 599–609; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Green NM, in Adv. Protein Chem , Vol. 29 (Eds.: Anfinsen CB, Edsall JT, Richards FM), Academic Press, 1975, pp. 85–133; [DOI] [PubMed] [Google Scholar]; c) Zempleni J, Mock DM, J. Nutr 1999, 129, 494S–497S; [DOI] [PubMed] [Google Scholar]; d) Luong JHT, Vashist SK, ACS Omega 2020, 5, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen Q, Long T, Zheng J, Sheng W, Sun S, Wei W, Zhao J, Wang H, CCS Chemistry 2022, 1–9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.