

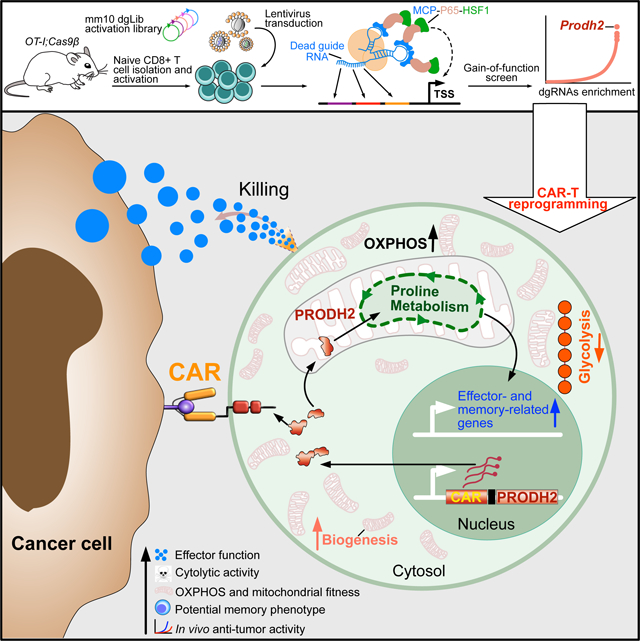
Abstract
Chimeric antigen receptor (CAR)-T cell-based immunotherapy for cancer and immunological diseases has made great strides, but it still faces multiple hurdles. Finding the right molecular targets to engineer T cells toward a desired function has broad implications for the armamentarium of T cell-centered therapies. Here, we developed a dead-guide RNA (dgRNA)-based CRISPR activation screen in primary CD8+ T cells, and identified gain-of-function (GOF) targets for CAR-T engineering. Targeted knock-in or overexpression of a lead target, PRODH2, enhanced CAR-T-based killing and in vivo efficacy in multiple cancer models. Transcriptomics and metabolomics in CAR-T cells revealed that augmenting PRODH2 expression reshaped broad and distinct gene expression and metabolic programs. Mitochondrial, metabolic and immunological analyses showed that PRODH2 engineering enhances the metabolic and immune functions of CAR-T cells against cancer. Together these findings provide a system for identification of GOF immune boosters, and demonstrate PRODH2 as a target to enhance CAR-T efficacy.
eTOC
Ye et al. develop and apply a dead-guide RNA based genome-wide gain-of-function CRISPR activation screen, which identifies PRODH2 as an enzyme that reprograms proline metabolism in CAR-T cells to enhance anti-tumor efficacy.
Graphical Abstract
Introduction
T cells are the cornerstone of adaptive immunity and therefore key players in maintaining human health. Over the past two decades an armamentarium of powerful therapies have been developed centering on T cells or T cell-mediated immunological pathways, several of which have been approved for clinical use or are in active clinical trials (Tang et al., 2018). These include immune checkpoint blockade that targets immunological synapses between cancer cells and T cells (Herbst et al., 2018; Pardoll, 2012; Ribas, 2012), agonist antibodies or small molecules that augment T cell functions (Moran et al., 2013) or enhance production of T cell-secreted or T cell-modulating cytokines and chemokines [AU: correct as edited? - Yes](Silva et al., 2019), neoantigen cancer vaccines based on T cell recognition of peptide – major histocompatibility complexes (MHCs) (Hollingsworth and Jansen, 2019), direct adoptive transfer of tumor infiltrating lymphocytes (TILs) (Rosenberg and Restifo, 2015), and chimeric antigen receptor (CAR)-T cells (June et al., 2018; Xin Yu et al., 2019). Therefore, engineering T cells toward a desired function has a broad range of applications with therapeutic implications.
CAR-T cell-based immunotherapy is a revolutionary approach to treat cancer and is potentially applicable to various other diseases (June et al., 2018; Majzner and Mackall, 2019; Rosenberg and Restifo, 2015). Five CAR-T products have been approved by the US FDA to date for the treatment of B-cell malignancies or multiple myeloma. However, current CAR-T therapies still face several major challenges leading to primary or secondary resistance and relapse of disease in hematological cancers. These challenges are due to a variety of reasons, including loss of antigen, failure of target recognition, cancer immune escape or insufficient persistence (June et al., 2018; Sharma et al., 2017). Therefore, it is important to identify genetic targets that can boost effector function in CD8+ T cells, and to directly harness such factors to engineer more effective T cells for cell-based therapy, including CAR-Ts.
To date, targets for T cell engineering predominantly rely on the immunology literature, leveraging endogenous genes, such as TRAC, TET2, NOTCH/DELTA, that provide significant enhancement of T cell function (reviewed in (Brown and Mackall, 2019; Roybal and Lim, 2017; Sadelain et al., 2017)). Loss-of-function (LOF) screens enable high-throughput identification of essential genes of T cell function using RNA interference (RNAi) (Chen et al., 2014; Zhou et al., 2014) or CRISPR/Cas9-mediated genetic knockout (Henriksson et al., 2019; Shifrut et al., 2018). In contrast, gain-of-function (GOF) screens can directly identify functional boosters that can be harnessed for T cell programming. This makes it possible to identify specific genes, which, regardless of their original physiological function, when overexpressed, knocked in or exogenously supplied can augment the function of immune cells.
GOF screening directly in primary T cells has remained challenging thus far. This is in part due to the difficulty of introducing three separate components (RNA-guided nucleases, and trans-activator and guide RNAs) simultaneously into primary T cells to achieve CRISPR activation (CRISPRa). This is important because unlike previously reported T cell CRISPR knockout screens (Henriksson et al., 2019; LaFleur et al., 2019; Shifrut et al., 2018; Ting et al., 2018) (Dong et al., 2019; Ye et al., 2019), CRISPRa screens, being a GOF approach, can identify a new class of targets that can be harnessed as functional boosters for T cell reprogramming. Here, we designed a genome-scale catalytically dead guide RNA (dgRNA) library as a versatile CRISPRa screening tool for high-throughput identification of GOF targets in any catalytically active Cas9 expressing cells, which is particularly useful for applications in primary immune cells that are less amenable to viral transduction and genetic manipulation. Utilizing this system, we identified and subsequently validated genes that can directly augment the effector function of CD8+ T cells. With target-centered interrogations, we showed that GOF engineering of a top hit, Proline Dehydrogenase 2 (PRODH2/Prodh2), into primary or CAR-T cells, can reshape their metabolic pathways and distinct gene expression programs, significantly improve their functions and enhance their anti-tumor efficacy in vivo.
Results
Genome-scale identification of boosters of effector function for primary CD8+ T cells
Due to the challenges of introducing multiple components including Cas9, co-activators and guide RNAs into primary T cells, we utilized a dgRNA system that is compatible with active Cas9 (Dahlman et al., 2015; Liao et al., 2017), whereby the immune cells can be readily isolated from Cas9 transgenic mice (Platt et al., 2014). We first designed and constructed a lentiviral T cell dgRNA activation (TdgA) vector (Figure 1A). We then designed a mouse genome-scale dgRNA library (mm10dgLib) using the promoter sequences of all annotated protein-coding transcripts from the mm10 genome assembly (Methods) (Figure 1B, Figure S1A). After spacer identification, on-target and off-target mapping and filtering, proximal score ranking, and spacer per gene choices for library balancing, the final mm10dgLib consists of 84,601 dgRNAs that target 22,391 coding transcripts and 1,000 non-targeting controls (NTCs) (Methods) (Figure 1B, Figure S1A, Dataset S1), which was synthesized as a pool and cloned into the TdgA vector. We sequenced the mm10dgLib plasmid library and verified that 82,197/83,601 (98.3%) of gene-targeting spacers (GTSs) and 988/1,000 (98.8%) of NTCs were successfully cloned, and both GTSs and NTCs showed a log-normal distribution (Figure S1B–C). We then packaged the plasmid library into lentiviral delivery system and performed functional titration by flow cytometry to confirm adequate viral titer (Figure S1D–E), enabling genome-scale activation screens for primary cells expressing catalytically active Cas9.
Figure 1. Genome-scale dgRNA activation screen identified genes that boost the effector function of CD8+ T cells.
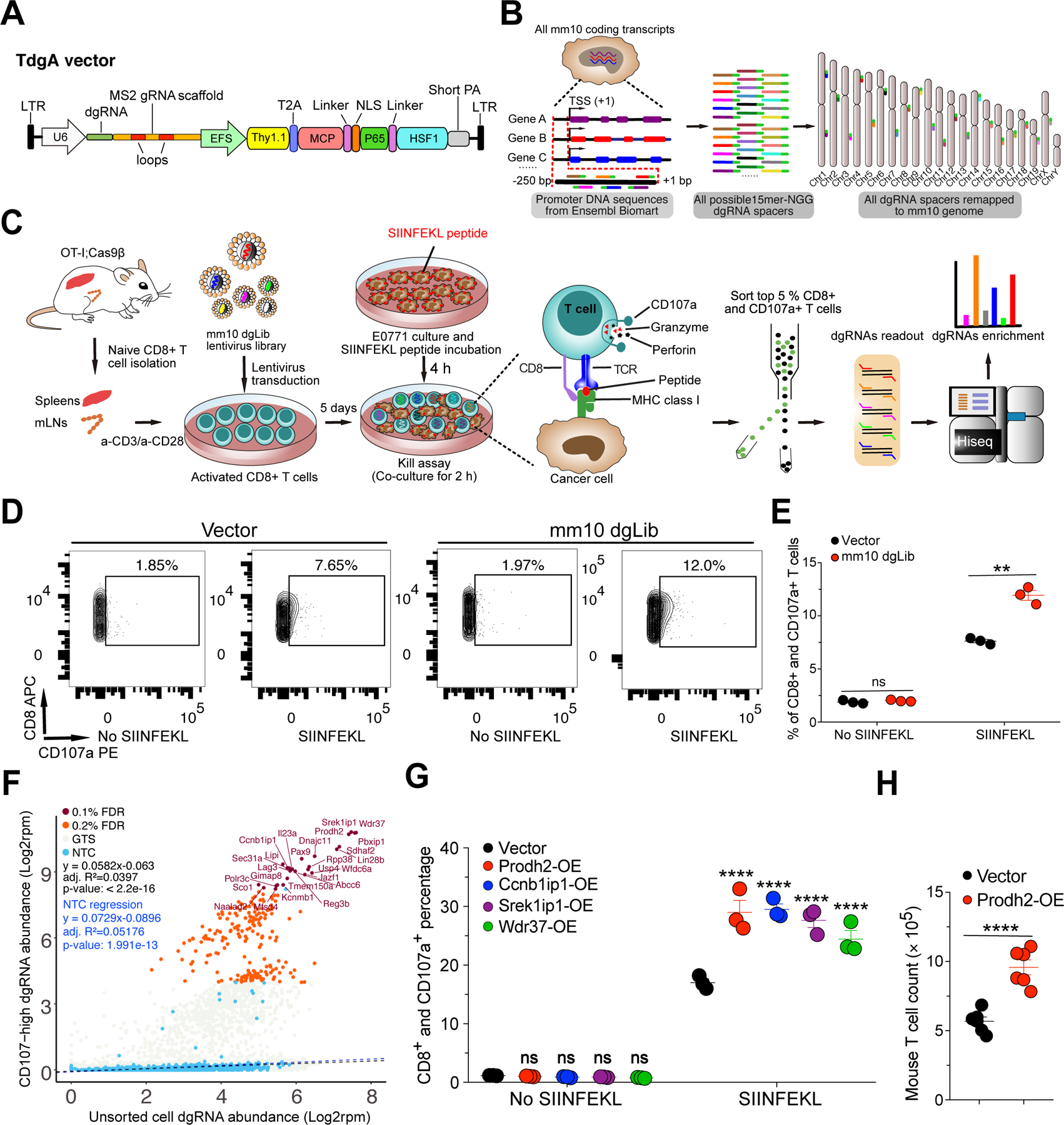
(A) Schematic representation of a T cell dead-guide RNA (dgRNA) activation lentiviral vector (TdgA), which contains a human U6 promoter, a dgRNA scaffold, and Thy1.1-MPH (MCP-p65-HSF1) expression cassette driven by an EFS promoter.
(B) Schematics of experiment: mouse genome-scale dead-guide RNA library (mm10dgLib) design, including 15nt proximal promoter spacer identification, on-target and off-target mapping, scoring, filtering, and prioritization of final spacers (details in STAR Methods).
(C) Schematics of genome-scale dgRNA library-based mouse primary CD8+ T cell kill assay activation screen (dgTKS) to identify genes that boost effector functions of CD8+ T cells. The main procedure includes naïve CD8+ T cell isolation, mm10dgLib transduction, a kill assay (CD8+ T cell degranulation, as measured by CD107a level in CD8 T cells in a T cell : cancer cell co-culture), CD8+;CD107a+ T cell sorting, genomic DNA preparation, dgRNA library readout, and dgRNAs enrichment.
(D) Representative flow cytometry results of the kill assay in the dgTKS experiment. FACS gating plot showing the percentage of CD107a+ cells among all CD8+ cells in vector and mm10dgLib transduced CD8+ T cells co-culture with E0771 cancer cells pulsed with SIINFEKL peptide. (n = 3 biological replicates). Representative data from two independent experiments.
(E) Quantification of CD107a in the mm10dgLib screen (n = 3 biological replicates).
(F) Bulk screen scatterplot of dgTKS screen, showing relative dgRNA abundances in the entire mm10dgLib library, with CD107a+-high FACS sorted CD8+ T cells, as compared to unsorted T cells. Blue dots are NTCs; brown dots are scoring GTSs that passed FDR 0.1% cutoff, with gene name labeled; orange dots are scoring GTSs that passed FDR 0.2% cutoff but did not pass FDR 0.1%; grey dots are remaining GTSs. Black dashed line is a regression line of all data points. Blue dashed line is a regression line of 1,000 NTCs representing a neutral baseline. Regression parameters and p-values were shown. GTSs deviating from the baseline showed enrichment in the CD107a+-high FACS as compared to the behavior of NTCs. The points were shown at the individual gRNA level. Representative top scoring genes targeted by specific sgRNAs were shown.
(G) Quantitative analysis of flow cytometry for kill assay for individual genes overexpressed by lentiviral vectors.
(H) Mouse T cell number quantification at day 4 after IL-2 withdrawal. (n = 6 in total of one independent experiments).
**P<0.01, ****P<0.0001 by multiple t tests (with adjusted P value) (E, G) or unpaired t tests (H).
See also: Figure S1
Degranulation is one of the major mechanisms through which cytotoxic CD8+ T lymphocytes (CTL) mediate the killing of target cells (Trapani and Smyth, 2002). CD107a (also known as LAMP-1) [AU: correct as edited? - Yes]is a marker which can be presented on the cell surface after degranulation (Peters et al., 1991). To identify genes that when activated can enhance the degranulation ability of CD8+ T cells after encountering their cognate antigen presented on the cell surface, we devised and performed a genome-scale dgRNA library-based CD8+ T cell Kill assay activation Screen (dgTKS) (Figure 1C). To do this, we developed a co-culture system (i.e., a kill assay) in which OT-I;Cas9β CD8+ T cells sensitively respond to E0771 breast cancer cells presenting SIINFEKL peptide, the cognate antigen of the CD8+ T cells from OT-I transgenic mice (Figure 1C). In this system, we measured CD107a+ expression among CD8+ T cells co-cultured with E0771 cancer cells with or without SIINFEKL peptide pulsing, and found that mm10dgLib lentivirus transduced CD8+ T cells had significantly higher CD107a+ levels compared to empty vector transduced cells (P < 0.001) (Figure 1D–E). Deep sequencing data showed that the cumulative coverage in each independent experiment was between 93.3 – 98.2% (Figure S1F; Dataset S1). Using fluorescence activated cell sorting (FACS), we sorted the mm10dgLib-transduced CD8+ T cells expressing a high level (top 5%) of CD107a, in three independent biological replicates, for genomic DNA preparation and dgRNA library readout (Methods; Figure 1C). We then used Illumina sequencing to read the dgRNA cassette of both CD107a+-high sorted and unsorted CD8+ T cells, and we quantified the dgRNA abundance in the entire mm10dgLib (Dataset S1). As a neutral baseline, we found that the NTCs were relatively evenly distributed in the unsorted cell population, but we were rarely detected in the CD107a+-high CD8+ T cells (Figure 1F). In contrast, there were two distinct populations of dgRNAs that deviated from the distribution and regression line of NTCs (Figure 1F). With an FDR of 0.1%, we identified significantly enriched dgRNAs in sorted CD107a+-high cells targeting 26 genes, including Prodh2, Srek1ip1, Wdr37, Ccnb1ip1, Pbxip1, and Sdhaf2 (Figure 1F). To determine how a theoretically neutral cell population would behave, we utilized the 1,000 NTCs in the pool, and found that they largely follow a log-linear relation. The regression is statistically significant, showing the baseline dynamics without genetic effect as theoretically random factors alone for NTCs as a population (Figure 1F). A regression using all data points generated a similar curve (Figure 1F). An observed strong shift in a substantial population of dgRNAs from the theoretical neutral regression line suggested that there are a population of dgRNAs as hits under potential selection (Figure 1F). To measure whether and how much each gene-targeting dgRNA deviate from the theoretically neutral line (gene perturbation effect, representative of potential selection), we used an outlier test to calculate the degree of shift and statistical significance, and identified hits such as Prodh2, Srek1ip1, Wdr37, Pbxip1, Sdhaf2, Lin28b, Pax9, Dnajc11, Il23a and Ccnb1ip1 Wdr37 (Figure S1G). Another approach based on direct comparison of the mean difference of dgRNA abundance between sorted and unsorted populations uncovered similar hits including Prodh2, Ccnb1ip1, Srek1ip1, and Wdr37 (Figure S1H). This screen revealed an unbiased picture of GOF effects of endogenous genes on CD8+ T cell degranulation and provided a ranked list of potential targets for T cell engineering.
Hits identified from the GOF screen enhance CD8+ T cell effector function
Based on the enrichment in the GOF screen, we hypothesized that increased expression of high-rank candidate genes from the dgTKS screen might enhance the degranulation ability of CD8+ T effector cells. Using RT-qPCR, we measured the natural expression levels for several high-rank candidate genes, including Defb19, Schip1, Sox5, Il1rap, Srek1ip1, Wdr37, Sdhaf2, Lin28b, Prodh2, Ccnb1ip1 and Bdnf. Results showed that most of these genes have low baseline levels of mRNA expression compared with the house-keeping gene Gapdh in primary CD8+ T cells (Figure S1I). The baseline expression of each of these genes is physiologically low in T cells, leaving sufficient room for gene overexpression in GOF T cell engineering. After confirming overexpression by RT-qPCR (Figure S1J–K), we then performed validation kill assays 4 days after lentiviral transduction of cDNA for overexpressing four top hits (Prodh2, Ccnb1ip1, Srek1ip1, or Wdr37) and found that activation of each significantly enhanced degranulation by CD8+ T cells upon encountering SIINFEKL-pulsed cancer cells (Figure 1G). These data validated that augmented gene expression of Prodh2, Ccnb1ip1, Srek1ip1, or Wdr37 can enhance CD8+ T cell effector function, providing GOF candidates for engineering improved T cell capabilities.
Interestingly, among all targets, we noticed that Prodh2-OE T cells proliferated rapidly in culture as compared to non-transduced or vector-transduced control T cells. Under the IL-2 withdrawal condition, quantification of cell numbers showed that the bulk Prodh2-OE CD8+ T cells proliferate 1.8 times faster than the vector-transduced counterparts (Figure 1H). In addition, Prodh2-OE CD8+ T cells have lower levels of apoptosis (Figure S1L).
GOF PRODH2 engineering by genomic knock-in or lentiviral overexpression boosts CAR-T killing of cancer cells
To harness PRODH2’s function to program T cells for cell therapy, we set out to co-engineer PRODH2 and CAR together in human T cells. We generated an anti-CD22 CAR (CD22-CAR) AAV construct specifically targeted to the TRAC locus, with simultaneous knock-in of the PRODH2 transgene (AAV-CD22-CAR-T2A-PRODH2), to allow expression of CD22-CAR and PRODH2 in the same T cells (Figure 2A). In parallel, for functional studies, because untargeted primary cells or CAR-T cells without any other transgene are imperfect controls, we generated a matched control with a prematurely stopped PRODH2 CDS (AAV-CD22-CAR-T2A-PRODH2(Stop)) (Methods) (Figure 2A). With electroporation of Cas9 ribonucleoprotein (RNP) targeting TRAC exon 1 followed by transduction of the AAV constructs, we introduced these transgenes into the endogenous TRAC locus of primary CD8 T cells to generate CD22-CAR;PRODH2 stable knock-in CAR-T cells, as well as the CD22-CAR;PRODH2(Stop) control knock-in CAR-T cells (Methods) (Figure 2A). Flow cytometry analysis of both AAV knock-in constructs in the CD8+ T cells showed highly efficient TRAC editing by Cas9 RNP, as evident by a population of cells with CAR+ and CD3− expression (as CD3 and TCR form a functional complex) (Figure 2B). We used fluorescence activated cell sorting (FACS) to purify TCR−CAR+ T cells (Figure 2B).
Figure 2. PRODH2 engineering by genomic knock-in or lentiviral overexpression boosts cytotoxic activity of CAR-Ts against cognate cancer cells.
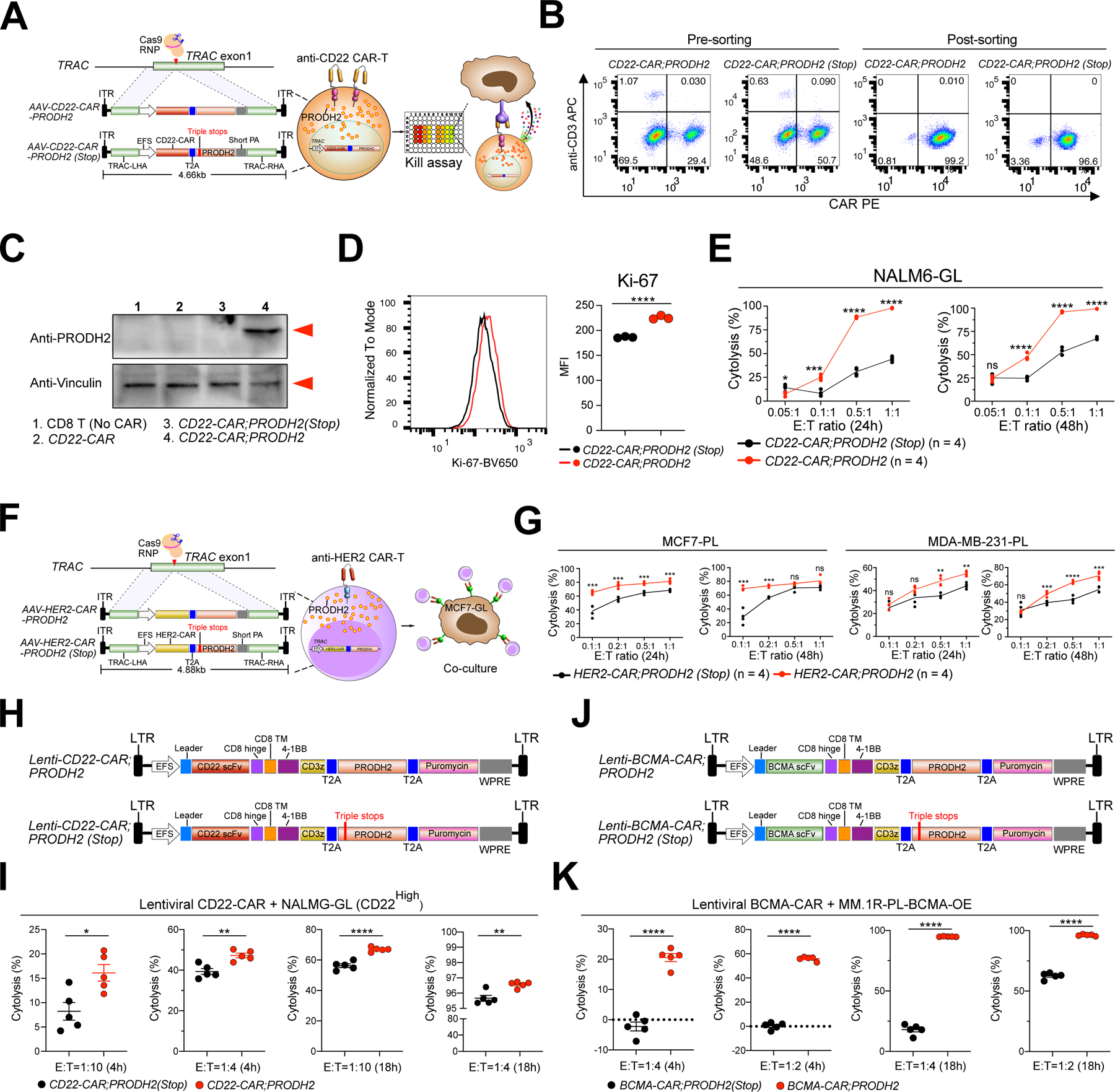
(A) A schematic of human CD22-CAR;PRODH2 (PRODH2 KI CAR-T) and CD22-CAR;PRODH2(Stop) (PRODH2(Stop) KI CAR-T, Control CAR-T) cell generation. In the CD22-CAR;PRODH2(Stop) construct, three artificial pre-mature stop codons were inserted between 318–319bp position of the PRODH2 CDS to generate a truncated mutant version. Knock-in (KI) constructs consist of TRAC locus homology-directed repair (HDR) 5’ and 3’ arms, an EFS promoter, a CD22-CAR expression cassette, a T2A sequence, a PRODH2 or PRODH2(Stop) CDS, and a short polyA. AAV6-packaged KI constructs were introduced into T cells by viral transduction after TRAC first-exon targeting Cas9:crRNA RNP electroporation.
(B) Representative flow cytometry plots of PRODH2 KI and control CAR-T cells before and after Flow Cytometry sorting. Representative data from two independent experiments.
(C) Representative immunoblot for PRODH2 expression in untreated CD8 T cell (no CAR), CD22-CAR, CD22-CAR;PRODH2(Stop), and CD22-CAR;PRODH2 T cells. The red arrows indicated bands of predicted molecular sizes based on the antibody providers. Representative data from three independent experiments.
(D) Flow analysis of PRODH2 KI and control CAR-T cell proliferation by Ki-67 staining.
(E) Kill assay of purified PRODH2 KI and control CAR-T cells with NAML6-GL (NAML6 with GFP and Luciferase reporters) cancer cells, with a titration series of Effector : Target (E:T) ratios, and at two timepoints (24h and 48h). The timepoint of CAR T cells used for co-culture was day 67 after CAR KI. Individual replicate datapoints were shown (n = 4 biological replicates). Representative data from two independent experiments.
(F) A schematic of human HER2-CAR;PRODH2 and HER2-CAR;PRODH2(Stop) CD8 T cell generation. HER2-CAR;PRODH2 and HER2-CAR;PRODH2(Stop) constructs were established by replacing CD22-CAR with HER2-CAR construct in CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) constructs.
(G) Kill assay of HER2-CAR;PRODH2 and HER2-CAR;PRODH2(Stop) KI T cells with MCF7-PL and MDA-MB-231-PL (MCF7 and MDA-MB-231 cells expressed with Puromycin and Luciferase reporters) breast cancer cells, with a titration series of Effector : Target (E:T) ratios, and at two timepoints (24h and 48h). All CAR-T cells were used for co-culture at day 17 after CAR KI. Individual replicate datapoints were shown (n = 4 biological replicates).
(H) A schematic of lentiviral CD22-CAR;PRODH2 (PRODH2-OE CAR-T) and CD22-CAR;PRODH2(Stop) (Control CAR-T) cell generation.
(I) Kill assay of Lenti-CD22-CAR;PRODH2 (Stop) and Lenti-CD22-CAR;PRODH2 T cells with NALM6-GL (CD22high) cancer cells at day3 after lentiviral transduction. (n = 5 biological replicates).
(J) A schematic of lentiviral BCMA-CAR;PRODH2 (PRODH2-OE BCMA CAR-T) and BCMA-CAR;PRODH2(Stop) (Control BCMA CAR-T) cell generation.
(K) Kill assay of Lenti-BCMA-CAR;PRODH2 (Stop) and Lenti-BCMA-CAR;PRODH2 T cells with MM.1R-PL-BCMA-OE cancer cells at day3 after lentiviral transduction. (n = 5 biological replicates).
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns = not significant by multiple t tests (with adjusted P value) (E, G) or unpaired t tests (D, I, K).
See also: Figure S1
We then performed immunoblotting to measure the baseline expression of PRODH2 protein in PRODH2-knock-in CAR-T cells, along with three control T cells (CD8 T cell without CAR, CD22 CAR-T cell without additional transgene, and PRODH2(Stop)-knock-in CD22 CAR-T cell). The results demonstrated that PRODH2 is highly expressed in CD22-CAR;PRODH2 T cells, but undetectable in CD8 T cells (no CAR), CD22-CAR, and CD22-CAR;PRODH2(Stop) T cells (Figure 2C), which showed minimal baseline expression and excluded the possibility of PRODH2(Stop) transgene leakage. We analyzed CAR-T cell proliferation by flow cytometry of Ki-67 and found that PRODH2 KI CD22 CAR-T cells had higher levels of Ki-67 (Figure 2D). We then performed a co-culture assay to test the ability of CAR-T cells to kill cognate leukemic cells, NALM6-GL (GFP and Luciferase positive). As compared to the CD22-CAR;PRODH2(Stop) control, CD22-CAR;PRODH2 CAR-T cells have a significantly stronger ability to kill the NALM6-GL in an antigen-specific manner (Figure 2E). Similarly, we engineered a HER2-specific CAR with PRODH2 overexpression along with matched control (HER2-CAR;PRODH2 and HER2-CAR;PRODH2(Stop)) by knocking the AAV constructs into the TRAC locus (Figure 2F). Co-culture assays also showed that PRODH2-KI HER2 CAR-T cells had stronger cytolytic activity against MCF7-PL (Puromycin and Luciferase positive) and MDA-MB-231-PL breast cancer cells (Figure 2G).
In addition, in order to test if PRODH2 can be engineered in the traditional lentiviral CAR-T system, we also generated lentiviral CD22-CAR and BCMA-CAR with co-cistronic overexpression of PRODH2 or PRODH2(Stop) constructs (Figure 2H, J). The co-culture data again showed PRODH2 overexpressed CD22-CAR and BCMA-CAR T cells significantly enhanced cancer cell killing (Figure 2I, K), where the cytotoxicity enhancement effect of PRODH2 in BCMA CAR is particularly strong (Figure 2K). These data together suggest that PRODH2 GOF engineering via either genomic knock-in or lentiviral overexpression enhanced in vitro killing ability of antigen-specific CAR-T cells, in cancer-specific CARs in three cellular models (leukemia, multiple myeloma and breast cancer).
PRODH2 GOF engineering enhances CAR-T in vivo efficacy against cancer in animal models
We then asked if PRODH2 GOF can enhance CAR-T cells’ therapeutic efficacy in in vivo settings. We first tested a B cell leukemia model with CD22 as the cancer antigen (Figure 3A). The bioluminescence imaging data showed that control CD22-CAR T cells had anti-tumor activity, but the leukemia relapsed quickly (P < 0.0001) (Figure 3B), while PRODH2 knock-in CD22-CAR T cells (CD22-CAR;PRODH2) showed significantly stronger anti-tumor activity compared to control (CD22-CAR;PRODH2(Stop)) T cells, and can more effectively control leukemia progression, in terms of both tumor burden (P < 0.0001) and survival (P < 0.05) (Figure 3B–C; Figure S1M). We also tested PRODH2-OE CAR T in a solid tumor model (anti-HER2-CAR), where breast tumors were induced by an intra-mammary fat pad injection of an established human breast cancer line MCF7-PL-HER2OE (Figure S1N). Tumor growth kinetics showed that control HER2-CAR T cells had a small anti-tumor effect, whereas PRODH2-OE CAR-T cells (HER2-CAR;PRODH2) had significant enhancement of efficacy compared to the control CAR-Ts (P < 0.0001), although all groups had tumor growth potentially due to the challenges in the solid tumor microenvironment (Figure S1O).
Figure 3. PRODH2 engineering by genomic knock-in or lentiviral overexpression enhanced CAR-T in vivo efficacy in mouse models.
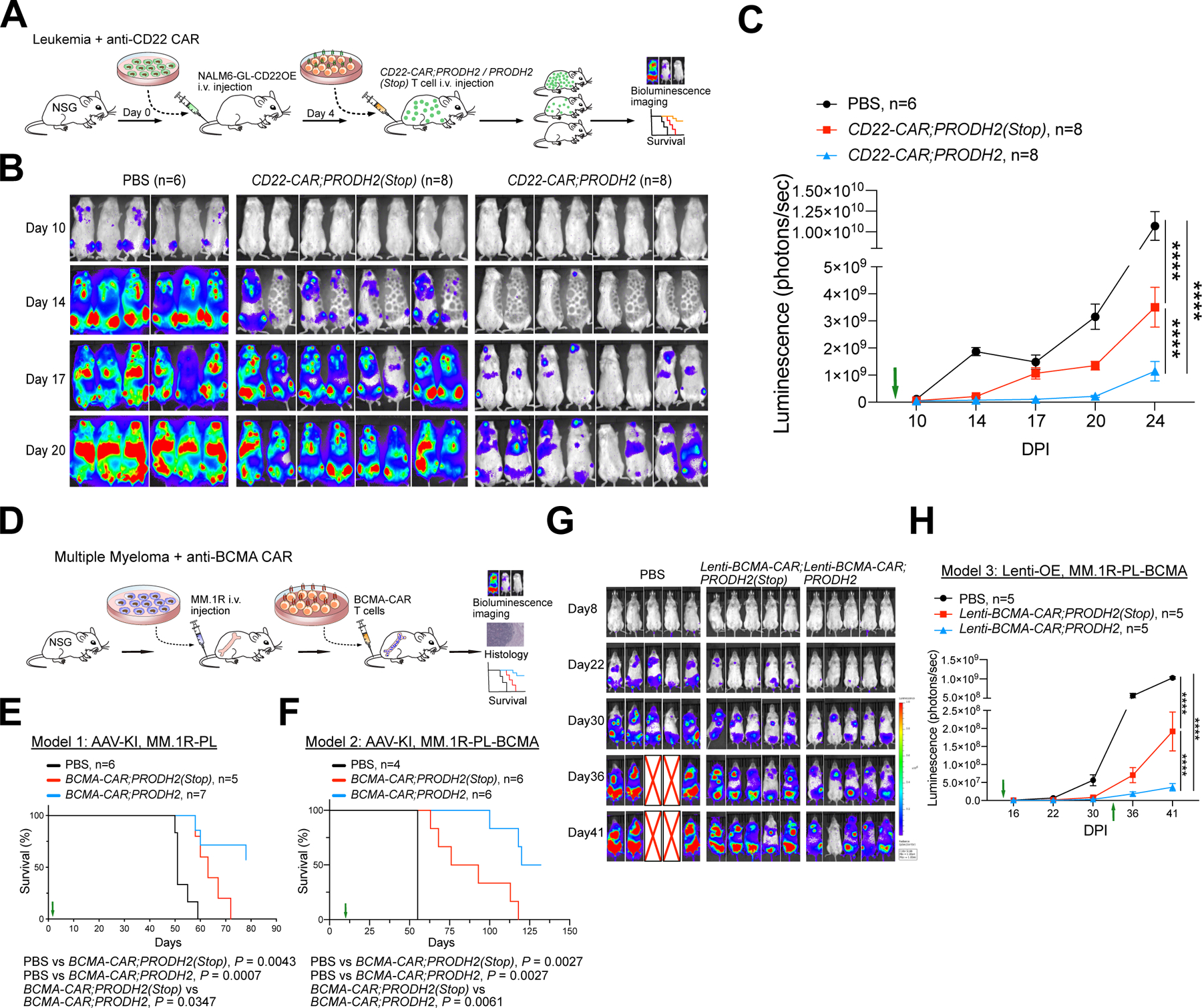
(A) A schematic of the experimental design of leukemia model PRODH2 knock-in CAR-T efficacy testing, showing leukemia induction, CD22-CAR intravenously injection, survival, and bioluminescence imaging. CAR T cells were pre-stimulated with NALM6-GL-CD22OE cancer cells at an E:T ratio = 1:1 at day 25 before injection.
(B) IVIS imaging showing bioluminescence of NSG mice which were injected with NALM6-GL-CD22OE cancer cells and with CD22-CAR therapy. Note: The dark shadow on the mouse at day 14 was induced by the imaging machine but has no influence on bioluminescence quantification.
(C) Quantification of cancer burden by total luminescence. Green arrow indicated CAR-T injection was performed at day 4. (n = 6–8 mice/group).
(D) A schematic of the experimental design of PRODH2 CAR-T efficacy testing in multiple myeloma models, showing induction, BCMA-CAR intravenously injection, and survival.
(E) Survival curve of MM.1R induced myeloma bearing NSG mice after AAV-KI BCMA-CAR;PRODH2 or BCMA-CAR;PRODH2 (Stop) T cell adoptive transfer therapy. (CAR-T adoptive transfer indicated with a green arrow). (n = 5–7 mice/group).
(F) Survival curve of BCMA-OE MM.1R induced myeloma bearing NSG mice after AAV-KI BCMA-CAR;PRODH2 or BCMA-CAR;PRODH2 (Stop) T cell adoptive transfer therapy. (CAR-T adoptive transfer indicated with a green arrow). (n = 4–6 mice/group).
(G) IVIS imaging showing bioluminescence of multiple myeloma bearing NSG mice after lentiviral-based BCMA-CAR;PRODH2 or BCMA-CAR;PRODH2 (Stop) T cell adoptive transfer therapy. “X” represented dead or euthanized animals (Endpoint). (n = 5 mice/group).
(H) Quantification of cancer burden by total luminescence for (G). CAR-T injection indicated with green arrows.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by two-way ANOVA (with multiple comparisons test) (C, H) or Log-rank (Mantel-Cox) tests (E, F).
See also: Figure S1
Because the effect of PRODH2 on BCMA CAR-T is most pronounced in vitro (Figure 2), we focused the subsequent in vivo experiments on PRODH2-engineered BCMA CAR-Ts. Using two independent CAR-T platforms (AAV-KI and lenti-OE), we established, with PRODH2 CAR-T and PRODH2(Stop) control CAR-T, and tested them in parallel against a systemic multiple myeloma model (Figure 3D). We performed three different experiments, one with an AAV PRODH2-KI BCMA CAR against an MM.1R multiple myeloma model; the second also with AAV PRODH2-KI BCMA CAR, against a BCMA-OE MM.1R multiple myeloma model; and the third using a lentiviral PRODH2-OE BCMA CAR (Figure 3D). The MM.1R cells when injected into NSG mice develop into multiple myeloma as confirmed by histology (Figure S1P). Overall survival analysis showed that control AAV-KI BCMA CAR-T cells slightly extended animal overall survival; however, all animals still all succumbed to disease (Figure 3E). AAV-KI PRODH2-OE CAR-T cells (BCMA-CAR;PRODH2) had a significantly stronger in vivo therapeutic effect and significantly enhanced the overall survival (Median survival time: PBS: 51 days; Control CAR: 63 days; PRODH2-OE CAR: not-reached) (P < 0.05), with more than half of the animals surviving longer-term (Figure 3E). We repeated the experiment in a second model with BCMA-OE MM.1R cells. The treatment effect overall is better as the animal survival is longer in the setting of antigen overexpression (Figure 3F). Again, we observed that while control CAR-T slightly extend the overall survival of diseased animals, AAV-KI PRODH2-OE CAR-T cells had a significantly stronger efficacy and significantly enhanced the overall survival (P < 0.01), where >=50% of the animals surviving longer-term (Figure 3F).
Because turnaround time is important for CAR-T production, we also tested the in vivo efficacy of PRODH2 engineered CAR-Ts in a third model, using the traditional lentiviral CAR-T system that requires shorter production time. The in vivo tumor growth data again showed that, while both CAR groups had antitumor effect (Figure 3G–H), PRODH2 overexpressed lentiviral BCMA-CAR T cell adoptive transfer had significantly enhanced efficacy as compared to PRODH2(Stop) control (P < 0.0001) (Figure 3G–H). Together, these in vivo data demonstrated that PRODH2 GOF engineering, either by genomic knock-in or lentiviral overexpression, significantly enhanced CAR-T cells’ efficacy against cancer in several mouse models, although the effect is more pronounced in the BCMA CAR-T as compared to other models.
PRODH2 knock-in restructures CAR-T cell gene expression and immune effector functions
We then asked why PRODH2 GOF engineered CAR T cells showed stronger killing and in vivo efficacy. To provide molecular bases on how PRODH2 GOF engineering programs the human CAR-T cells, we performed a series of mechanistic interrogations by multi-omics profiling (transcriptome profiling, CyTOF, untargeted metabolomics, targeted metabolomics and integrated analyses) as well as in-depth analyses (biochemical immunological and cellular assays) (Methods) (Figure 4A). To unbiasedly understand the PRODH2-mediated global cellular changes, we first performed mRNA-seq to profile the entire transcriptome of CD22-CAR;PRODH2 as well as CD22-CAR;PRODH2(Stop) knock-in CAR-T cells (Datasets S2). DE analysis again uncovered broad transcriptomic changes as a result of PRODH2 knock-in, with 2,810 upregulated and 1,172 downregulated genes (FDR adjusted p-value, q < 0.001) (Figure 4B–C; Figure S2A–B; Datasets S2). The knock-in of PRODH2 co-cistronically with the CAR-T construct again showed strong changes in the transcriptomic programs of cell cycle, metabolism, apoptosis and immune response (Figure 4B–C; Figure S2A–B; Datasets S2). With regard to immune genes and pathways, PRODH2 knock-in CAR-T up-regulated genes and processes including immune cell activation, leukocyte mediated cytotoxicity and largely pro-inflammatory signatures, such as T cell activation, signal transduction and cytokine production (Figure 4B–C; Figure S2A–B; Datasets S2), while the terms are not mutually exclusive because these genes often play important roles in multiple T cell pathways. These data together suggest that PRODH2 knock-in altered the gene expression programs of CD22-CAR-T cells centered on cell cycle, T cell activation and metabolic processes.
Figure 4. Whole transcriptome profiling of PRODH2 knock-in CD22 CAR-T cells and FACS validation of enhanced effector function.
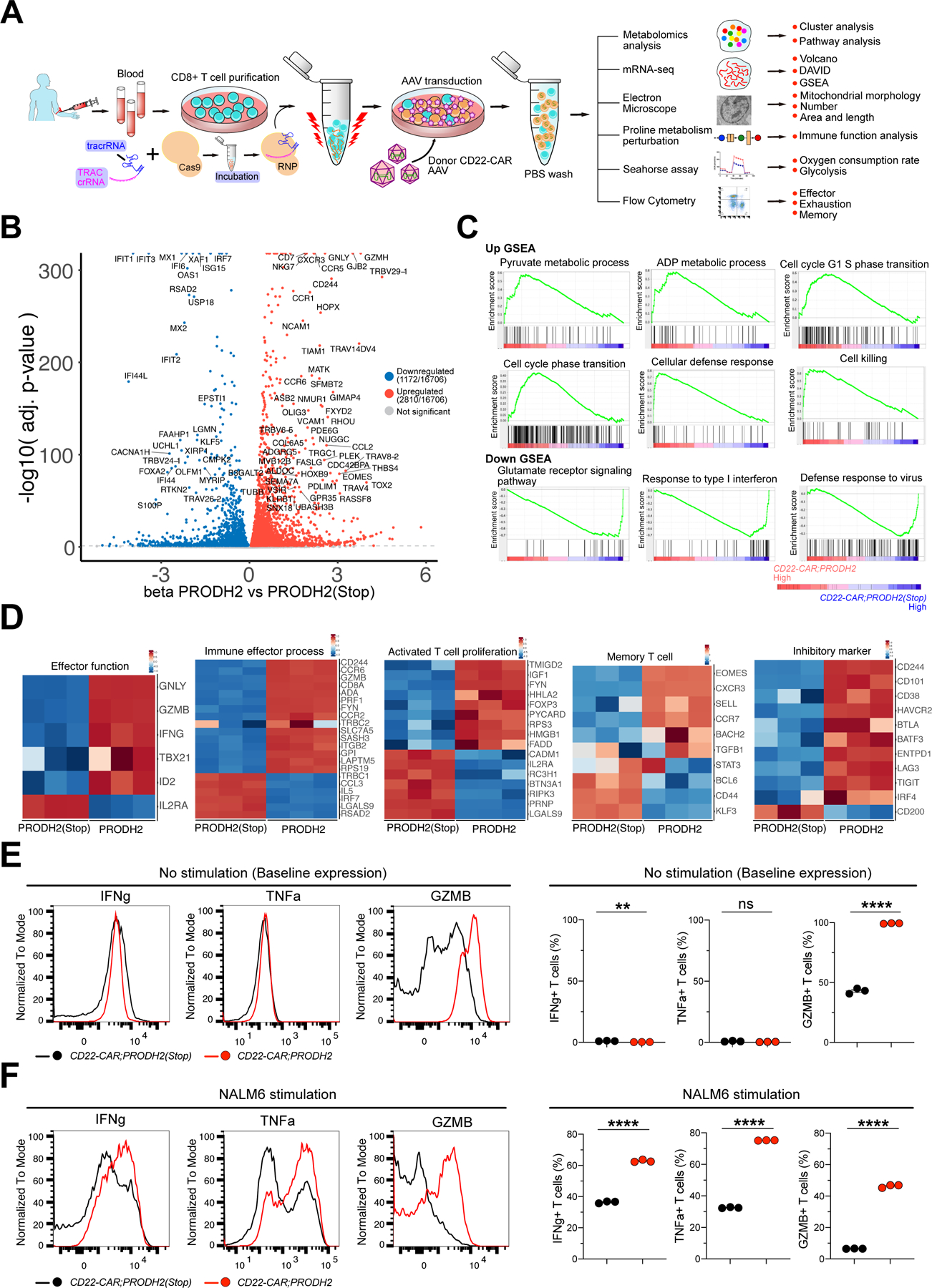
(A) A schematic of the experimental design of PRODH2 CAR-T mechanistic investigation: AAV- or lenti-based CAR-T generation, metabolomics, mRNA-seq, metabolic analysis, immunological analysis, TEM, and other experiments.
(B) Volcano plots of mRNA-seq differential expression between PRODH2 vs. PRODH2(Stop) CD22-CAR-T cells from human donor (n = 3 biological replicates) (FDR adj. q < 0.001). CD22-CAR T cells were collected for RNA extraction and bulk mRNA-seq at day 25 after CAR knock-in.
(C) GSEA individual pathways for mRNA-seq of PRODH2 knock-in CD22-CAR-T cells. GSEA plots of individual pathways from the representative up- and down-regulated gene sets between CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cell groups. (Cut-off criteria: p-value < 0.001)
(D) Heatmaps of differentially expressed genes in representative pathways such as effector function, immune effector process, activated T cell proliferation, memory T cell and inhibitory marker.
(E-F) Intracellular staining of effector function markers, IFNg, TNFa, and GZMB in both PRODH2 overexpressed CD22 CAR and control CD22 CAR T cells, without stimulation (E) or after 12h of NALM6-GL stimulation (F).
See also: Figure S2
**P<0.01, ****P<0.0001, ns = not significant by unpaired t tests (E, F).
A number of highly upregulated genes are directly related to T cell function, such as effector function and immune effector process, activated T cell proliferation, T cell memory and exhaustion (Figure 4D; Figure S2C). Representative highly upregulated genes in these lines include those well-documented in T cell activation (e.g. CCR3, CCR5, CCR9, CXCR3, CXCR4, EOMES, ADA, SIRPG, RHOU), T cell signaling (e.g. PLCG2, ZAP70, FYN, LCK, JAG2, PRKDC, PRKG2, PIK3CG, PIK3R6), and/or effector function (e.g. IFNG, TGFBR2, GZMB, GZMH, GZMK) (Figure 4B–D; Figure S2C). Knocking out a representative gene implicated in T cell activation (Saoudi et al., 2014), RHOU, abolished the effect of PRODH2-mediated enhancement of cytotoxicity (Figure S2D), supporting the validity of the RNA-seq data and differential expression analysis.
To cross-validate the immunological phenotypes of PRODH2 KI CAR-T cells, we first performed an unbiased immune profiling using Mass Cytometry (CyTOF), with a 19-marker panel that include major T cell lineage and functional state markers. We profiled a total number of 216,501 cells across 6 samples, with 3 biological replicates from purified CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) knock-in CAR-T cells, in a baseline state without cancer stimulation (Figure S3A). We randomly sampled 5,000 cells from each sample and quantify the expression of each marker at surface protein level, and we revealed the changes in each marker between control and PRODH2 knock-in CAR-T cells (Figure S3B). We found that PRODH2 knock-in CAR-T cells have significantly higher levels of IFNg and TNFa, major effector cytokines for CD8 T cells (Figure S3B–C). PRODH2 knock-in CAR-T cells also have significantly higher level of CD134/OX40, a co-stimulatory marker, as well as TIM-3, an immune checkpoint but also T cell activation marker (Figure S3B–C). PRODH2 knock-in CAR-T cells have significantly lower level of Cleaved Caspase-3, indicator of apoptosis (Figure S3B–C).
Because effector function stood out as one of the strongest signals, we then performed flow cytometry to validate the findings from mRNA-seq and CyTOF. We measured the effector cytokines, such as IFNg, TNFa, and GZMB, by intracellular staining and flow cytometry analysis of CAR-T cells before and after co-culture with cognate cancer cells. Results showed that PRODH2-GOF significantly increased GZMB in CAR-T cells at base line (Figure 4E), and substantially increase the production of all three cytokines (IFNg, TNFa and GZMB) under cancer stimulation (Figure 4F). Cytokine secretion of IL2, IL15, and IL7 measured by ELISA were not different between PRODH2-GOF and control CAR-T cells (Figure S3D). These data together suggest that PRODH2 GOF enhanced T cell effector function.
PRODH2 GOF alters metabolism of CD8+ T cell and CAR-T cells
PRODH2/Prodh2 encodes an enzyme that catalyze the conversion of 4-Hydroxyproline (IUPAC name (2S,4R)-4-Hydroxypyrrolidine-2-carboxylic acid; Aliases: 4Hyp, Hydroxyproline, L-Hydroxyproline, Trans-4-Hydroxy-L-proline) into 1-Pyrroline-3-hydroxy-5-carboxylate (PHC), a key step of the proline metabolic pathway without known redundancy (Phang et al., 2010). This enzyme is expressed at low levels across most organs or cell types in the human body, including primary CD4+ and CD8+ T cells (Stelzer et al., 2016; Thul et al., 2017; Uhlen et al., 2015). The low baseline expression of PRODH2 makes it a promising candidate to be harnessed for GOF T cell manipulation. We therefore next investigated whether augmenting PRODH2 expression can change the characteristics of human T cells (Figure S4A). We overexpressed human PRODH2 in human primary CD8+ T cells using lentivirus (hPRODH2-OE) and confirmed potent augmentation of gene expression over endogenous level (Figure S4B). Again, we found that the human CD8+ T cells with augmented PRODH2 also proliferate significantly faster than vector control (Figure S4C), similar to the phenotype with mouse T cells reported above. We then performed a targeted metabolomics experiment focused on proline and arginine metabolism (Methods) (Figure S4D; Dataset S4). In hPRODH2-OE CD8+ T cells, we observed that the 4-Hydroxyproline level was significantly decreased, and the PHC level was significantly increased (Figure S4D), suggestive of an increased level of biochemical activity consistent with the augmented gene expression by GOF (on-target metabolic function in T cells). Moreover, hPRODH2-OE CD8+ T cells have significantly lower levels of metabolites including oxoproline, ornithine, pyruvate, aspartate, leucine, malate and oxaloacetate; and higher levels of metabolites including choline, glutamate, cysteine and lactate.
We then performed metabolomics directly in PRODH2 GOF CAR-T cells. Metabolomics data identified a total of 75 metabolites in both CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) knock-in CAR-T cells (Figure S5A; Dataset S3). Among those, 8 metabolites are more abundant in CD22-CAR;PRODH2 knock-in CAR-T cells, whereas 19 metabolites are less abundant (Figure 5A–B). As established in the biochemical literature and KEGG database, these metabolites have clearly defined metabolic pathways with enzymes that catalyze their production and catabolism (Kanehisa and Goto, 2000). By intersecting the genes encoding those enzymes that produce or breakdown these metabolites, we observed multiple DE genes whose upregulation or downregulation are consistent with the predicted metabolic activity, i.e. the directions of metabolite alterations (Figure S5A–B; Datasets S3). We again observed on-target activity of PRODH2 GOF, where the upregulation of PRODH2 is in concert with decreased 4-Hydroxyproline and increased PHC levels, along with alteration of other genes and metabolites (Figure 5A–B; Figure S5A–B). For example, upregulation of GAMT that encodes a guanidinoacetate N-methyltransferase is consistent with the reduction of its substrate arginine and downstream product creatinine; upregulation of glucose-6-phosphatase 3 encoded by the G6PC3 gene is consistent with reduced glucose-6-phosphatase level; and reduced glutamine level is consistent with downregulation of GLUL that produces it from glutamate and upregulation of CAD that converts it into carbamoyl-phosphatase (Figure S5A–B). Furthermore, multi-omics analysis with MetaboAnalyst that integrates mRNA-seq DE analysis and metabolomics data identified enriched metabolic pathways in an unbiased manner, with the note that the significant changes of these metabolic pathways can be in both directions. The second-most enriched metabolic pathway with the DE gene set in CD22-CAR;PRODH2 knock-in CAR-T cells is arginine and proline metabolism, with significantly enriched intersecting DE genes involved in these pathways as well as differentially represented (DR) metabolites (Figure S5C–D; Datasets S3), again highlighting the on-target activity of augmenting PRODH2.
Figure 5. Metabolomic profiling and biochemical-immunological validation of PRODH2 GOF CAR-Ts.
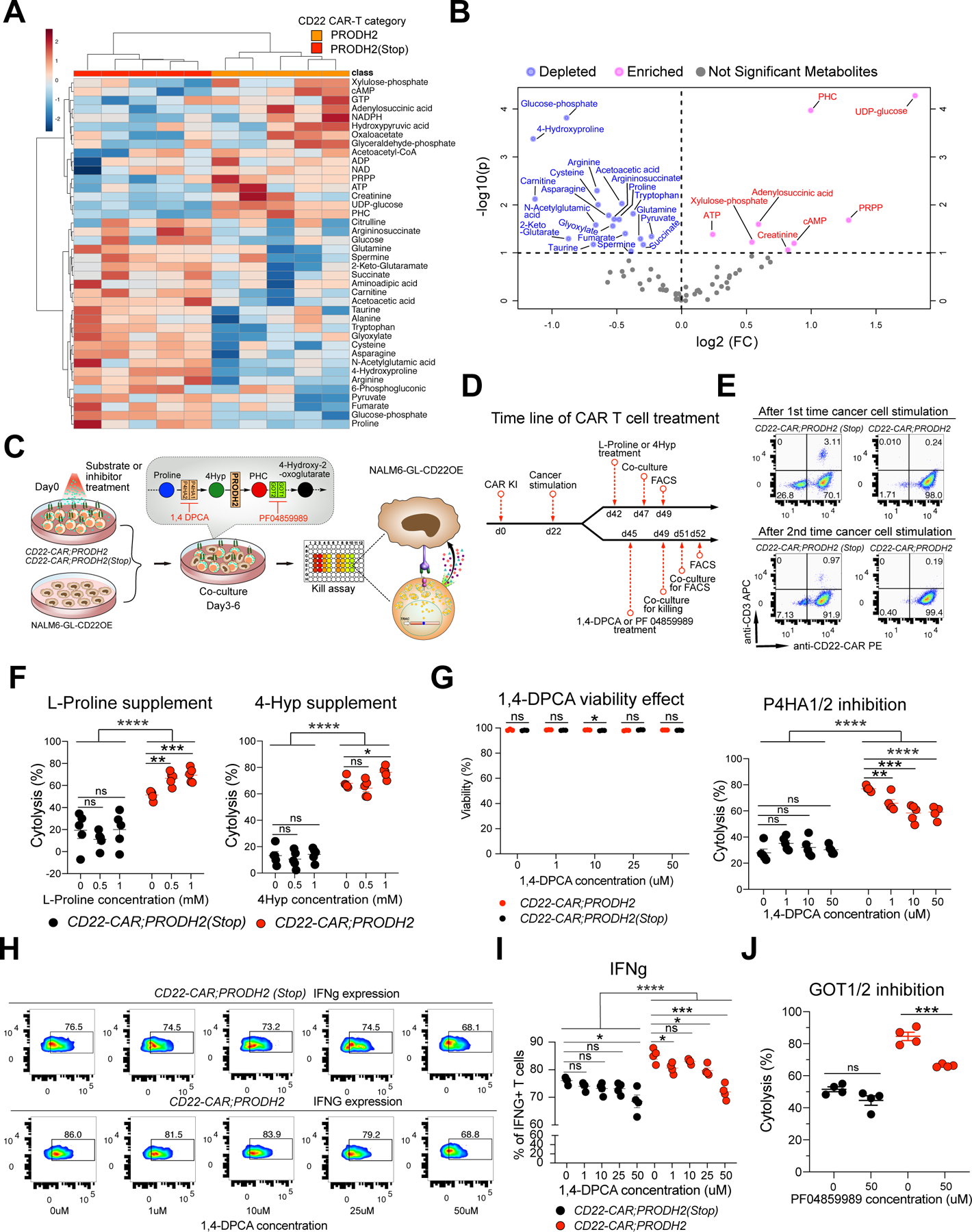
(A) Heatmap of the relative abundance of top 40 QTOF/QQQ detected metabolites of PRODH2 vs. PRODH2(Stop) CD22-CAR-T cells (n = 5 biological replicates). Representative data from two independent experiments.
(B) Volcano plot of differentially represented (DR) metabolites between PRODH2 vs. PRODH2(Stop) CD22-CAR-T cells. Blue dots indicate decreased metabolites, pink dots indicate increased metabolites.
(C) Schematic of biochemical-immunological validation of PRODH2 GOF CAR-Ts, including chemical compound treatment, co-culture and flow cytometry. CAR-T cells were supplied with extra L-proline (substrate for P4HA1 and P4HA2), 4-hydroxyproline (4Hyp, substrates for PRODH2), or 1,4-DPCA (inhibitor of P4HA1 and P4HA2), PF 04859989 (inhibitor of GOT1 and GOT2) before co-culture and flow cytometry analyses.
(D) Timeline of CAR-T cell treated with L-Proline, 4Hyp, 1,4-DPCA, and PF 04859989, co-culture, and FACS.
(E) Representative CAR KI percentages after 1st and 2nd cancer stimulations. Representative data from two independent experiments.
(F) Substrate supplement experiment. Cytolytic activity measurement by co-culture of CD22-CAR;PRODH2 and CD22-CAR;PRODH2 (Stop) T cells with NALM6-GL-CD22OE cancer cells for 6h after T cells pre-treated with different concentration of L-Proline and 4Hyp.
(G) P4HA enzymatic inhibition experiment. (Left) DPCA toxicity analysis. Cell viability measurement of CD22-CAR;PRODH2 and CD22-CAR;PRODH2 (Stop) T cells after treating with different concentration of 1,4-DPCA. (Right) Cytolytic activity measurement by co-culture of CD22-CAR;PRODH2 and CD22-CAR;PRODH2 (Stop) T cells with NALM6-GL-CD22OE cancer cells for 12h after T cells pre-treated with different concentration of 1,4-DPCA.
(H) Representative flow plots of IFNg production of CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells after 1,4-DPCA treatment and co-culture. T cells were pre-stimulated with NALM6-GL-CD22OE cancer cells for 8 days, then treated with the 1,4-DPCA inhibitor for 3 days. The co-culture E:T = 0.5:1. (n = 4 biological replicates)
(I) Quantification of (H).
(J) GOT enzymatic inhibition experiment. Cytolytic activity measurement by co-culture of CD22-CAR;PRODH2 and CD22-CAR;PRODH2 (Stop) T cells with NALM6-GL-CD22OE cancer cells for 24h after T cells pre-treated with PF04859989.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns = not significant by multiple t tests (with adjusted P value) (G), two-way ANOVA and unpaired t tests (F, G, I, J).
Biochemical-immunology investigation demonstrates the proline metabolic pathway activity of PRODH2-enhanced T cell function
To follow on the observations of unbiased profiling by multi-omics, we then investigated the details on how PRODH2 overexpression enhanced CAR-T function. We first tested manipulation of part of the proline metabolic pathway by supplying extra L-proline and 4-hydroxyproline (4Hyp) substrates for P4HA1and P4HA2 and PRODH2, respectively (Cooper et al., 2008; Launay et al., 2019; Summitt et al., 2015). We pre-treated the CAR-T cells with substrates or inhibitor for 3–6 days depending on the specific experiment, then subjected to co-culture or flow-based immunological assays (Figure 5C–D). High efficiency CAR knock-in was confirmed prior to co-culture assays (Figure 5E). As a result, we found that while the baseline (no chemical treatment, PBS only) cytolysis of CD22-CAR;PRODH2 is substantially higher than that of the control, L-Proline significantly increased the cancer killing ability only in CD22-CAR;PRODH2 cells, but not in control CAR-T that normally do not express PRODH2 and thereby lack the enzyme (Figure 5F). At 1mM concentration, 4Hyp also significantly increased the cancer killing ability only in CD22-CAR;PRODH2 cells (Figure 5F). These data together showed that supplementing a direct substrate of PRODH2 significantly improved cancer killing ability only in the presence of PRODH2 overexpressed CAR-T cells, but not in control CAR-T that normally lack the enzyme, which provided direct evidence that the introduced biochemical activity by PRODH2 GOF is responsible for the enhanced cancer cell cytolysis.
Furthermore, to restrict 4Hyp production, we leveraged 1,4-DPCA inhibitor to block P4HA1 and P4HA2 function (Xiong et al., 2014; Xiong et al., 2018). Using a similar biochemical-immunology assay, we found that that blocking P4HA1/2 by its inhibitor 1,4-DPCA reduced cancer killing ability only in CD22-CAR;PRODH2 T cells, but had no influence on control CAR-T (Figure 5G). The live-dead staining data showed 1,4-DPCA treatment itself has no influence on T cell viability (Figure 5G). Consistent with the flow, RNA-seq and CyTOF data above, we again found that the baseline IFNg production level was higher in PRODH2-OE CAR-T cells as compared to controls across all conditions (Figure 5H–I). In concordance with the metabolic function and cytolysis phenotype, at E:T ratio of 0.5:1 where cancer cells can still saturate T cells within the assay period, IFNg level is strongly suppressed by 1,4-DPCA in PRODH2-OE CAR-T cells, to a level around control CAR-T cells without treatment; whereas this effect is moderate in control CAR-T cells (Figure 5H–I). Because PHC, the metabolic product of PRODH2, is transported by GOT1/2 to subsequently enter the TCA cycle, we tested the usage of GOT1 and GOT2 inhibitor PF04859989 was used to block PHC downstream metabolism. The GOT1/2 inhibition – cytolysis experiment showed that PF04859989 reduced cancer killing ability only in the presence of PRODH2 GOF (i.e. only in CD22-CAR;PRODH2 T cells but not in PRODH2(Stop) control group) (Figure 5J). Blocking the one-step-upstream enzyme that produces PRODH2 direct substrate and downstream enzyme of PHC in PRODH2-OE CAR-T had an exactly opposite effect in cancer killing. These results from the proline metabolic pathway perturbation experiments suggested that the mechanism underlying enhanced CAR-T function is mediated by an on-target enhancement of PRODH2 enzymatic function.
Mitochondrial cellular and function analyses revealed PRODH2 GOF-driven energetics in CAR-T cells
We then investigated how PRODH2 engineering drives CAR-T cellular metabolic state. Because PRODH2’s enzymatic activity is primarily in the mitochondria (Mt), we performed a series of cellular and biochemical Mt analyses on PRODH2 CAR-T cells under a longer-term culture. We first used electron microscopy (EM) to examine the morphology of the CAR-T cells. Both purified CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) knock-in control CAR-T cells have similar morphology in cell shape, nucleus and various organelles. Interestingly, as compared to control, the PRODH2 knock-in CAR-T cells have elevated mitochondria representation (Figure 6A–H), which is a signature of memory-like metabolic state (Buck et al., 2016; van der Windt et al., 2012). This is measured in terms of higher mitochondria count per cell (6A, E) as well as larger total mitochondria area per cell (Figure 6G) in CD22-CAR;PRODH2 CAR-T cells, but no difference in mitochondria length between CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) CAR-T cells (Figure 6F). Furthermore, compared to control CAR-T cells, the PRODH2 knock-in CAR-T cells remodeled cristae morphology leading to cristae expansion and widening (Figure 6C), which was also previously implicated in T cell effector phenotypes (Buck et al., 2016). Another interesting phenomenon was that PRODH2 knock-in CAR-T cells have more cytolytic granules (Figure 6D, H), a signature of cytolytic T cell function. To further evaluate the mitochondrial features by independent approaches, we measured mitochondrial mass and depolarization by MitoTracker staining (Bengsch et al., 2016; Scharping et al., 2016), and found that PRODH2 knock-in and lentiviral overexpressed BCMA-CAR-T cells had higher mitochondrial mass (Figure 6I), but no difference of mitochondrial depolarization compared with control CAR-T cells (Figure S6A). We also measured mitochondrial DNA copy number and complex I and IV expression levels by RT-qPCR. The results showed that there was no significant difference between PRODH2 knock-in and control CAR T cells in Mt DNA or Complex I / Complex IV level (Figure S6B). The increased mitochondria load was also cross-validated in an independent CAR-T setting, with a different vehicle (lentiviral vector) and a different CAR transgene (BCMA-CAR) (Figure S6C).
Figure 6. Characterization of long-term mitochondria energetics of PRODH2 knock-in CAR-T cells.
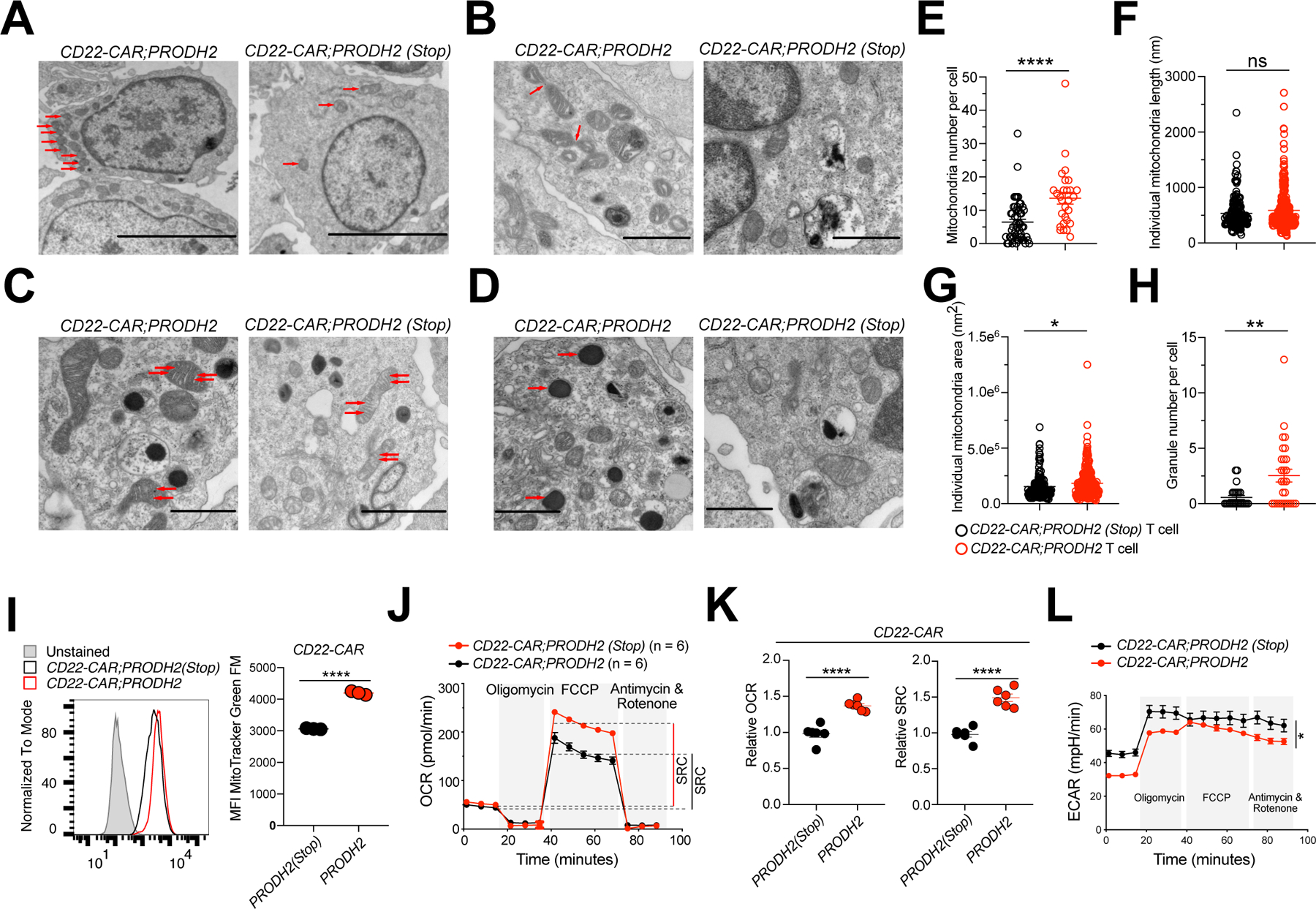
(A-D) TEM analysis of PRODH2 knock-in and Control CAR-T cells. TEM images were examined for mitochondrial numbers (A) (red arrows indicated mitochondria), mitochondrial fission (B) (red arrows indicated mitochondria fission), mitochondrial cristae remodeling (C) (red arrows indicated mitochondrial cristae), and granule numbers (D) (red arrows indicated granules). Scale bars, 5 μm (A), 1 μm (B, C (left image), D), and 2 μm (C, right image).
Data from one experiment with independent replicates.
(E) Quantification of mitochondria number per cells.
(F) Quantification of individual mitochondria length.
(G) Quantification of individual mitochondria area.
(H) Quantification of granule number per cells.
(I) Mitochondrial mass as measured by MitoTracker Green FM staining of indicated CAR-T cells at day 36 after stimulation with NALM6-GL-CD22OE cancer cells.
(J-L) Seahorse experiment of PRODH2 knock-in and Control CAR-T cells, with a density of 2e5 CAR-T cells / well (n = 6). Data are representative of three independent experiments.
(J) Oxygen consumption rate (OCR) was measured at baseline and in response to oligomycin (Oligo), FCCP, and rotenone plus antimycin A.
(K) Relative maximum OCR and relative spare respiratory capacity (SRC) were quantified.
(L) Extracellular acidification rate (ECAR) was measured at baseline and after drug treatment as the OCR measurement (n = 6). Representative data from three independent experiments.
*P<0.05, **P<0.01, ****P<0.0001, ns = not significant by unpaired t tests (E-I, K) and two-way ANOVA (L).
See also: Figure S6
We then performed quantitative Seahorse analysis to measure oxygen consumption rate (OCR), spare respiratory capacity (SRC), and extracellular acidification rate (ECAR) for these CAR-T cells. We found that compared to control CAR-T cells, the PRODH2 knock-in CAR-T cells have higher OCR and SRC (Figure 6J, K). We confirmed this observation with an independent condition in which cell density was doubled (Figure S6D). The ECAR data showed the PRODH2 knock-in CAR-T cells have low ECAR (Figure 6L), an indicator of glycolysis, which further suggested that PRODH2 GOF CAR-T cells were shifted towards the oxidative phosphorylation (OXPHOS) pathway. SRC and OXPHOS are memory-like signatures of T cells (Buck et al., 2016; van der Windt et al., 2012). It has been shown that CAR-T cells with enhanced mitochondria biogenesis and oxidative metabolism have better persistence during cancer immunotherapy (Kawalekar et al., 2016), which are metabolic and mitochondria features similarly observed in PRODH2 knock-in CAR-T cells. The mitochondria EM analysis revealed the cell biology features of PRODH2 GOF CAR-T cells linked to increased memory-like features, as well as effector function.
PRODH2 GOF alters the behaviors of CAR-T cells after antigen-specific cancer stimulation in long-term co-culture
Retaining long-term activity under chronic antigen stimulation is important for effective T cell-based cancer immunotherapies. A recent study showed that reprogramming T cell metabolism and mitochondria fitness can enhance both effector function and long-term memory phenotype simultaneously, for example by targeting the Regnase 1 enzyme (Wei et al., 2019). Along with the mitochondrial function observations, we set out to test PRODH2 overexpression in CAR-T cells over a long course of co-culture with cancer cells, mimicking the chronic antigen stimulation in disease settings (Figure 7A). CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) control T cells manifested drastically different phenotypes at day 27 after CAR knock-in but before encountering cancer antigen (without cancer stimulation), where PRODH2 overexpressed CD22-CAR showed predominant effector phenotype higher than the control (Figure 7B), consistent with the multiple evidence of enhanced effector phenotypes shown above. We then stimulated both groups with antigen-specific cancer cells every 12 days to mimic the chronic cancer antigen stimulation in therapeutic settings (Eyquem et al., 2017) (Figure 7A). We used the well-established canonical CD45RA;CD62L dual marker flow analyses at day 69 post CAR-T knock-in. We found that while PRODH2 CAR-T cells have higher CD45RA-high;CD62L-low population (representing effector cells), which is consistent with the findings above, PRODH2 CAR-T cells interestingly quickly shift away from the CD45RA-high;CD62L-low phenotype during the prolonged culture and cancer stimulation. After two stimulations, they exhibited significantly higher fractions in the CD45RA-low;CD62L-low, CD45RA-low;CD62L-high and CD45RA-high;CD62L-high populations (Figure 7C). Because these three populations resemble the less-differentiated (memory or naïve) phenotypes, this observation is consistent with the EM and Seahorse results. This phenotype was more pronounced after a third stimulation (Figure 7C). In this long-term co-culture, the PRODH2 GOF T cells are associated with a slight increase of PD1, TIM3 and LAG3 (Figure S6E). We also measured several independent surface markers and gene expression regulators in long-term culture (Figure 7D–F). The results showed that CCR7, IL7R, CXCR3, which are indicators of memory-like phenotypes in T cells, were significantly increased after cancer stimulation in long-term culture (Figure 7E). BCL6 and TCF7, which are well-known master regulators of T cell memory differentiation (Chang et al., 2014; Crotty et al., 2010), were also substantially increased in PRODH2 GOF CAR-T cells with cancer stimulation in long term co-culture (Figure 7F).
Figure 7. PRODH2 promotes CAR T cell memory formation after cancer cell engagement in long term co-culture.
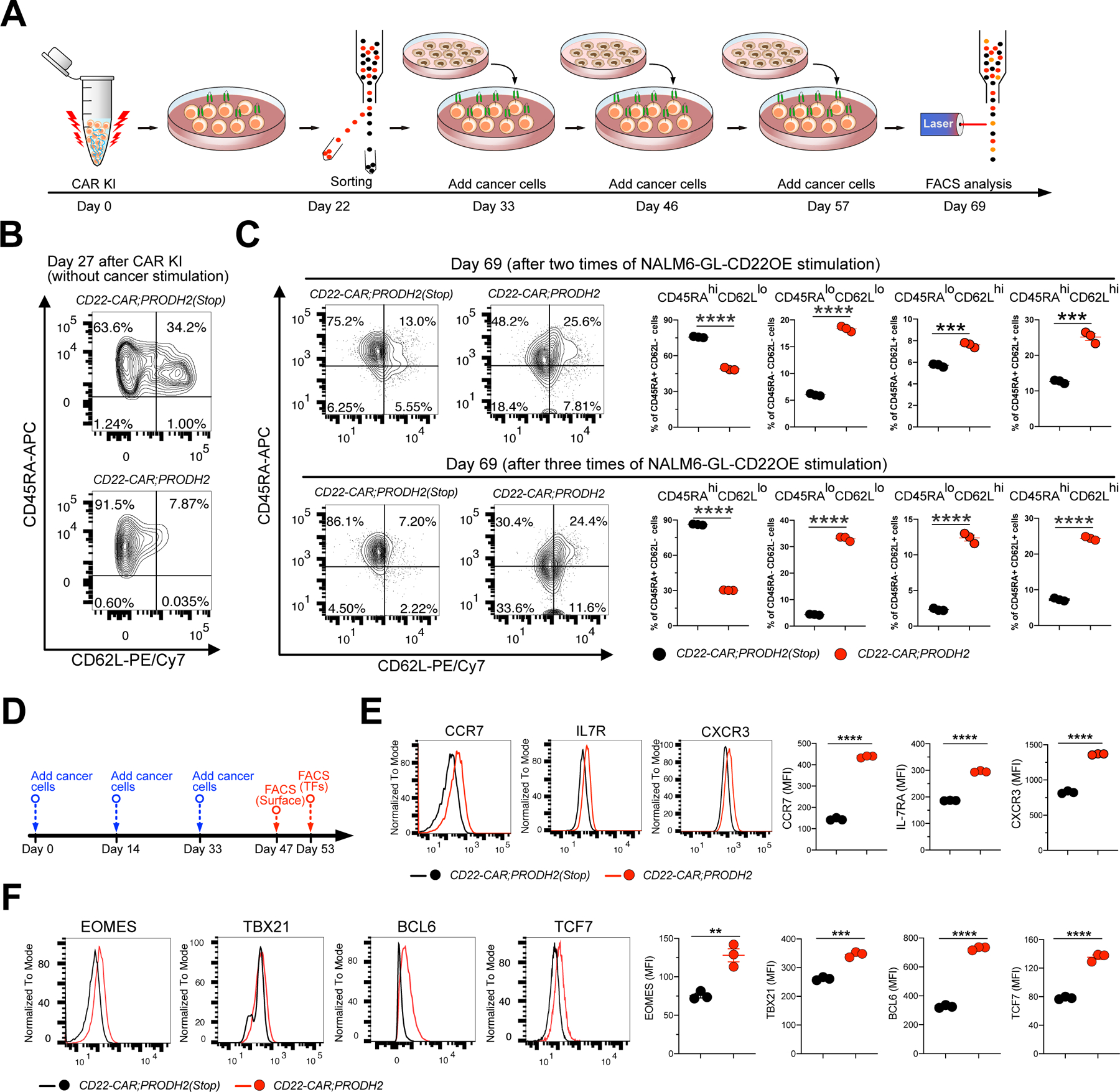
(A) A schematic of the experimental design showing time-line of CAR KI, FACS sorting, cancer stimulation and flow analysis. CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells were purified by FACS sorting at day 22 after CAR KI, then CAR T cells were stimulated with NALM6-GL-CD22OE cancer cells various times, each about every 12 days with a E:T ratio = 1:1. T cells stimulated different times were harvested for analysis at day 69.
(B) Baseline expression of CD45RA and CD62L in CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells analyzed at day 27.
(C) CD45RA and CD62L expression in CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells analyzed at day 69, stimulated either 2 or 3 times with NALM6-GL-CD22OE cancer cells. (hi = high expression, lo = low expression).
(D) Time-line of cancer stimulation and flow cytometry.
(E, F) Analysis of T cell memory in PRODH2 knock-in CAR-T cells after three times of cancer cell stimulation in long term co-culture.
(E) Flow analysis and quantification of human T cell memory surface markers, CCR7, IL7R and CXCR3.
(F) Flow analysis and quantification of human T cell memory transcription regulators, EOMES, TBX21, BCL6 and TCF7.
**P<0.01, ***P<0.0001, ****P<0.0001 by unpaired t tests (C, E, F).
Finally, we investigated the potential risk of malignant transformation of PRODH2 GOF. We first performed an IL-2 withdrawal assay and found that the majority CAR-T cells were dead at day 7 without IL-2 in the medium, suggesting that they were not immortalized or transformed (Figure S6F). Moreover, we analyzed the expression of NOTCH1, GATA3, and RUNX1, malignant transformation markers of T cell acute lymphoblastic leukemias (T-ALL) (Van Vlierberghe and Ferrando, 2012). The flow cytometry data showed that these markers are negative or expressed at a low level across all groups, where PRODH2 GOF CAR-T cells are at levels similar to both PRODH2(Stop) control CAR-T and untransduced T cells (Figure S6G). These data together suggested that PRODH2 GOF is not associated with obvious risk of malignant transformation of the T cells themselves.
Discussion
Catalytically dead guide RNAs (dgRNAs), can be utilized to modulate gene expression with catalytically active Cas9 (Dahlman et al., 2015; Kiani et al., 2015). High-throughput CRISPR screens enabled unbiased discovery of therapeutic targets (Shalem et al., 2015); however, the application of dgRNA technologies has not yet been harnessed at massively-parallel scale. Recently, genetic screens were performed to identify T cell regulators in vitro (Henriksson et al., 2019; LaFleur et al., 2019; Shifrut et al., 2018; Ting et al., 2018). T cell knockout screens have also been recently conducted in vivo (Dong et al., 2019; Ye et al., 2019). While several studies demonstrated genome-scale GOF screen systems for protein-coding genes (Gilbert et al., 2014; Konermann et al., 2015) and long noncoding RNAs (lncRNAs) (Joung et al., 2017) in human melanoma cells, it is noted that in CRISPRa based systems genes having higher fold activation are often lowly expressed. Thus, the basal expression levels of genes affect screening results with GOF screens (Gilbert et al., 2014; Konermann et al., 2015). In Jurkat cells, CRISPRa has been applied to identify stimulation-responsive enhancers (Mumbach et al., 2017; Simeonov et al., 2017), to map T cell signaling pathways (Chi et al., 2016), and to activate master regulator gene expression (Forstnerič et al., 2019). However, Jurkat is an immortalized lymphocyte cell line and not primary T cell, which is not clinically relevant. These previous studies discussed the future application of CRISPRa in primary T cells (Forstnerič et al., 2019). It is challenging to introduce the entire CRISPRa screening machinery (dCas9, transactivator and a large-scale guide RNA library) simultaneously into primary CD8+ T cells.
Here, we leveraged the dgRNA system and the catalytically active Cas9 transgenic mice to achieve the screening capability. We thereby developed an orthogonal system for CD8+ T cell GOF screens and identified endogenous factors for boosting T cell function, by designing a catalytically dead guide RNA library, and leveraging Cas9 transgenic mice where Cas9+ immune cells are readily available to be isolated and cultured at large scale. With the development of orthogonal screening technologies, the dgRNA library system may be used in conjunction with knockout systems to perform complex intersectional screens in individual Cas9+ cells.
Previous studies have demonstrated that overexpression of STAT5 can improve anti-tumor effects (Grange et al., 2012; Grange et al., 2013). However, these studies are not unbiased screens in T cells. Using a systems approach, we unveiled several ranked GOF candidates enriched in T cells with superior effector function. We validated 4 genes (Prodh2, Ccnb1ip1, Srek1ip1, and Wdr37) where augmenting their expression consistently enhanced CD8+ T cell degranulation. These data validated the ability of the dgTKS system for discovery of new GOF targets in primary T cells. This recently developed screening system can potentially be broadly applied to discovery of GOF genes in various other immune and primary cell types.
T cell survival, activation, development, proliferation, differentiation and anti-tumor effector function are virtually all driven by metabolism (Geltink et al., 2018). Naïve T cells are supported by ATP which are predominantly generated from oxidative phosphorylation before activation (MacIver et al., 2013). The T cell proteome and phosphoproteome were mapped in dynamic T cell activation signaling networks during T cell activation (Tan et al., 2017). Activated T cells shifted their metabolic state from oxidative metabolism to glycolysis for cell proliferation and effector functions (Chang et al., 2013; Geltink et al., 2018; MacIver et al., 2013). Glycolysis is a relatively inefficient pathway for generating ATP, which are important for supporting rapid T cell growth and proliferation (O'Neill et al., 2016). The total biomass is dramatically increased after T cell activation for proliferation, which requires substantial nucleotide, lipid, and amino acid synthesis (Fox et al., 2005). Programming T cell metabolism using bioengineering approaches therefore may provide effective strategies for enhancement of T cell function.
Our dgTKS system and genome-scale GOF screen identified Prodh2/PRODH2 as a top hit in cancer cell killing ability. PRODH2 encodes an enzyme in proline metabolism by catalyzing the first step in the catabolism of trans-4-hydroxy-L-proline. Proline and arginine metabolism is important in T cell anti-tumor activity (Geiger et al., 2016), although the exact role of proline metabolism in T cell function is less well-characterized. Because PRODH2 is not expressed in primary T cells, its physiological function is likely minimal and thus LOF perturbation will not interfere with its natural role, making it an ideal GOF target. PRODH2/Prodh2 is a highly tissue-specific enzyme mainly expressed in livers and kidneys and only lowly expressed in most other cell types (Jiang et al., 2012; Knight et al., 2009). To the best of our knowledge to date there is no reported study on PRODH2 functions in T cells. Prodh2 played an important role in proline homeostasis, especially under stress conditions (Funck et al., 2010). PRODH2 is important for primary hyperoxaluria (PH), as it is a disorder of cellular glyoxylate metabolism (Harambat et al., 2011; Hoppe et al., 2009). Public databases including Immgen (Heng et al., 2008) and DICE (Schmiedel et al., 2018) showed that Prodh2 is usually not expressed in immune cells, while it is expressed in the mitochondria of kidney, liver and gallbladder cells. Therefore, in a normal physiological setting, PRODH2 is not expressed or detected in T cells, which explains its absence in prior loss-of-function T cells screens (Chen et al., 2014; Dong et al., 2019; Henriksson et al., 2019; LaFleur et al., 2019; Shifrut et al., 2018; Ye et al., 2019; Zhou et al., 2014). The metabolic effects of PRODH2 makes it an interesting target for T cell metabolic programming in a setting of ectopic engineering of new elements as immune boosters.
The landmark success of CD19 CAR with US FDA approval opens a new era of cell therapy, e.g. (Schuster et al., 2019). There remain many hurdles for CAR-T therapy due to various challenges (Lim and June, 2017). Cancer cells can metabolically restrict T cell anti-tumor functions by nutrient depletion (Chang et al., 2015). Thus, approaches to enhance the T cell metabolism may improve T cell anti-tumor immunity. The second and third generation CARs designed with extra co-stimulatory domains (CD28 or / and 4–1BB) in addition to CD3ζ, had a better anti-tumor efficacy compared with first generation (June et al., 2018). CD28 signaling domain has been found to have an extra function of enhancing aerobic glycolysis, and 4–1BB enhanced mitochondria biogenesis and fatty acid oxidation in the CAR-T cells (Kawalekar et al., 2016). Recently, intracellular L-arginine (involved in arginine and proline metabolism) concentration has been found to impact the metabolic fitness and survival capacity of T cells and anti-tumor activity (Geiger et al., 2016). Programming T cell metabolism is an emerging approach for immunotherapy (Chang and Pearce, 2016). More recently, it was shown that overexpression of canonical AP-1 factor c-Jun in CAR T cells induced exhaustion resistance (Lynn et al., 2019). Engineering CARs with codon optimized CD8 transmembrane domain can induce lower levels of cytokines but retained potent cytolytic activity (Ying et al., 2019). A split, universal, and programmable (SUPRA) CAR system can fine-tune T cell activation strength and sense and logically respond to multiple antigens (Cho et al., 2018). Additional approaches include re-structuring of signaling domains (Sadelain et al., 2017), overexpression of boosting factors (Lynn et al., 2019), co-administration of immunomodulating factors or viral vectors (Ma et al., 2019), or changing costimulatory domains or lowering CAR binding affinity (Ghorashian et al., 2019; Savoldo et al., 2011).
Limitation of study
The overall killing of HER2-CAR T cell was not as striking as CD22-CAR or BCMA-CAR T cells, which was expected as solid tumor therapy using CAR-T cells is still a major challenge. In addition, the memory characterization of CAR-T cells were performed in long-term co-culture in vitro, because the in vivo models also have their own limitations as the study of CAR-T cells in vivo requires the use of immunocompromised animals. While PRODH2’s effect on effector function and cancer killing is strong and the mechanism is clear from this study, PRODH2’s effect on memory establishment is worthy of further investigation in the future.
In conclusion, whole transcriptome analysis, multi-omics analyses, CAR-T cellular and immunological assays, metabolic analyses and mechanistic dissection together showed that GOF engineered PRODH2 via OE or KI can reprogram T cell metabolism; promote T cell proliferation, activation, and effector function; improve effective responses to longer-term cancer stimulation; and enhance anti-tumor efficacy in vivo (Figure S7). More generally, the dgRNA library-based CRISPRa screen technology also provides a versatile and high-throughput activation screening system for identification of new classes of GOF targets directly in T cells or other primary immune cells. Given that the dgRNA library is compatible with catalytically active Cas9, orthogonal activation and knockout screens are possible. These systems might be a broadly useful resource for the field.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sidi Chen (sidi.chen@yale.edu).
Materials Availability
Mouse lines (OT-I;Cas9β mice) and cell lines (NALM6-GL, MM.1R-PL, MCF7-PL, NALM6-GL-CD22OE, MM.1R-PL-BCMAOE, and MCF7-PL-HER2OE) generated in this study are available from the lead contact upon request.
Data and Code Availability
The genomic sequencing raw data, the metabolomics raw data, and CyTOF raw data have been deposited at SRA, Metabolomics Workbench, and Mendeley data, respectively, and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table.
The original codes of data analysis are available from the lead contact upon reasonable request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. The unprocessed data are deposited into public archives as above.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ultra-LEAF™ Purified anti-mouse CD3ε Antibody (clone: 145–2C11) | Biolegend | Cat#100340 |
| Ultra-LEAF(TM) Purified anti-mouse CD28 Antibody (clone: 37.51) | Biolegend | Cat#102116 |
| APC anti-human CD3 Antibody (Clone: UCHT1) | Biolegend | Cat#300412 |
| FITC anti-human IgG Fc | Biolegend | Cat#409310 |
| PE anti-human IgG Fc (HP6017) | Biolegend | Cat#409304 |
| PE anti-DYKDDDDK Tag Antibody (L5) | Biolegend | Cat#637310 |
| APC/Cy7 anti-human CD8a antibody (HIT8a) | Biolegend | Cat#300926 |
| APC anti-human CD3 Antibody (Clone: HIT3a) | Biolegend | Cat#300312 |
| APC anti-mouse CD8a Antibody (53–6.7) | Biolegend | Cat#100712 |
| PE/Cy7 anti-mouse CD3ε Antibody (145–2C11) | Biolegend | Cat#100320 |
| PE anti-rat CD90/mouse CD90.1 (Thy-1.1) Antibody (OX-7) | Biolegend | Cat#202524 |
| PE anti-mouse CD107a (LAMP-1) Antibody (1D4B) | Biolegend | Cat#121612 |
| PE conjungate Cleaved Caspase-3 (Asp175) Rabbit mAb (5A1E) | CST | Cat#9978S |
| Brilliant Violet 510™ anti-human CD8 Antibody (SK1) | Biolegend | Cat#344732 |
| APC/Cy7 anti-human IFN-γ Antibody (4S.B3) | Biolegend | Cat#502530 |
| PE anti-human TNF-α Antibody (MAb11) | Biolegend | Cat#502909 |
| PerCP/Cyanine5.5 anti-human/mouse Granzyme B Recombinant Antibody (QA16A02) | Biolegend | Cat#372212 |
| APC anti-humanCD45RA Antibody (HI100) | Biolegend | Cat#304112 |
| PE/Cy7 anti-human CD62L Antibody (DREG-56) | Biolegend | Cat#304822 |
| PerCP/Cyanine5.5 anti-human CD197 (CCR7) Antibody (Clone: G043H7) | Biolegend | Cat#353219 |
| APC anti-human CD127 (IL-7Rα) Antibody (Clone: A019D5) | Biolegend | Cat#351315 |
| FITC anti-human CD183 (CXCR3) Antibody (Clone: G025H7) | Biolegend | Cat#353703 |
| EOMES Monoclonal Antibody (WD1928), PECyanine7 | eBioscience™ | Cat#25–4877–42 |
| APC anti-T-bet Antibody (Clone: 4B10) | Biolegend | Cat#644813 |
| PE anti-TCF1 (TCF7) Antibody (Clone: 7F11A10 ) | Biolegend | Cat#655207 |
| FITC anti-human/mouse Bcl-6 Antibody (Clone: 7D1) | Biolegend | Cat#358513 |
| APC anti-human Notch 1 Antibody (Clone: MHN1–519 ) | Biolegend | Cat#352107 |
| PerCP/Cyanine5.5 anti-GATA3 Antibody (Clone: 16E10A23) | Biolegend | Cat#653811 |
| RUNX1 Monoclonal Antibody (RXDMC), PE | eBioscience™ | Cat#12–9816–80 |
| Anti-human CTLA4 (CD152)-161Dy | FLUIDIGM | Cat#3161004B |
| Anti-human TIM3–153Eu | Yale CyTOF CORE | |
| Anti-human CD278/ICOS-168Er | FLUIDIGM | Cat#3168024B |
| Anti-human TNFRSF9/CD137–173Yb | FLUIDIGM | Cat#3173015B |
| Anti-human IL2–166Er | FLUIDIGM | Cat#3158007B |
| Anti-human TNF-α-152Sm | FLUIDIGM | Cat#3152001B |
| Anti-human IFN-γ-165Ho | FLUIDIGM | Cat#3165002B |
| Anti-human CXCR3–163Dy | FLUIDIGM | Cat#3163004B |
| Anti-human CD62L-174Yb | Yale CYTOF Core | Cat#V00751 |
| Anti-human Perforin-176Yb | Yale CYTOF Core | |
| Anti-human CD45RA-169Tm | FLUIDIGM | Cat#3143006B |
| Anti-human CD8a-146Nd | FLUIDIGM | Cat#3146001B |
| Anti-human CD45RO-164Dy | FLUIDIGM | Cat#3164007B |
| Anti-human CD3–170Er | FLUIDIGM | Cat#3170001B |
| Anti-human CD45–89Y | FLUIDIGM | Cat#3089003B |
| Anti-Cleaved Caspase3–172Yb | Yale CYTOF Core | |
| Anti-human CD25–149Sm | FLUIDIGM | Cat#3149010B |
| Anti-human CD134(OX40)-150Nd | Yale CYTOF Core | |
| Anti-human LAG3–175Lu | Yale CYTOF Core | |
| Recombinant Human Siglec-2/CD22 Fc Chimera Protein, CF | R&D | Cat#1968-SL-050 |
| Recombinant Human BCMA/TNFRSF17 Fc Chimera Protein, CF | R&D | Cat#193-BC-050 |
| Dynabeads Human T-Activator CD3/CD28 | Invitrogen | Cat#11131D |
| Anti-human PRODH2 | Atlas Antibodies | Cat#HPA051287 |
| Anti-human Vinculin | Abcam | Cat#129002 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemical Competent E. coli | ThermoFisher | Cat#C737303 |
| Endura™ ElectroCompetent Cells | Lucigen | Cat#60242–2 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| PEI MAX -Transfection Grade Linear Polyethylenimine Hydrochloride (MW 40,000) | Polyscience | Cat#24765–1 |
| DPBS, no calcium, no magnesium | Gibco | Cat#14190136 |
| RPMI 1640 Medium | Gibco | Cat#11875–093 |
| Fetal Bovine Serum | Sigma Aldrich | Cat#F4135–500ML |
| DMEM, high glucose, pyruvate | Gibco | Cat#11995065 |
| Opti-MEM™ I Reduced Serum Medium | ThermoFisher | Cat#31985070 |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | Cat#15140122 |
| Lonza™ BioWhittaker™ L-Glutamine (200mM) | Lonza | Cat#BW17605E |
| 2-Mercaptoethanol | Sigma Aldrich | Cat#M6250–10ML |
| X-VIVO 15 Serum-free Hematopoietic Cell Medium | Lonza | Cat#BE02–060F |
| Human AB Serum; Male Donors; type AB | MP Biomedical | Cat#092930949 |
| ACK Lysing Buffer | Lonza | Cat#10–548E |
| Naïve CD8a+ T Cell Isolation Kit, mouse | Miltenyi | Cat#130–096-543 |
| LS Columns | Miltenyi | Cat#130–042-401 |
| Recombinant Mouse IL-2 (carrier-free) | Biolegend | Cat#575404 |
| Recombinant Mouse IL-7 (carrier-free) | Biolegend | Cat#577802 |
| Recombinant Mouse IL-12 (p70) (carrier-free) | Biolegend | Cat#577008 |
| Recombinant Mouse IL-15 (carrier-free) | Biolegend | Cat#566302 |
| Recombinant Human IL-2 (carrier-free) | Biolegend | Cat#589104 |
| Monensin Solution (1,000x) | Biolegend | Cat#420701 |
| Brefeldin A Solution (1,000X) | Biolegend | Cat#420601 |
| QuickExtract DNA Extraction Solution | Epicenter | Cat#QE09050 |
| Proteinase K | Qiagen | Cat#19131 |
| Pierce™ Universal Nuclease for Cell Lysis | ThermoFisher | Cat#88702 |
| RNase A | Qiagen | Cat#19101 |
| PEG 8000, Molecular Biology Grade | Promega | Cat#V3011 |
| Sodium chloride | Sigma | Cat#S3014 |
| Gibson Assembly® Master Mix | NEB | Cat#E2611 |
| Phusion Flash High-Fidelity PCR Master Mix | ThermoFisher | Cat#F548L |
| DreamTaq Green PCR Master Mix (2X) | ThermoFisher | Cat#K1082 |
| E-Gel™ Low Range Quantitative DNA Ladder | ThermoFisher | Cat#12373031 |
| Fixation/Permeabilization Solution Kit | BD | Cat#554714 |
| SuperScript™ IV Reverse Transcriptase | Invitrogen | Cat#18090050 |
| Random Hexamers (50 μM) | Invitrogen | Cat#N8080127 |
| NEBNext® Ultra™ RNA Library Prep Kit for Illumina® | NEB | Cat#E7530S |
| NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1) | NEB | Cat#E7335S |
| TaqMan™ Fast Advanced Master Mix | Invitrogen | Cat#4444557 |
| QIAquick Gel Extraction Kit | Qiagen | Cat#28706 |
| QIAamp Fast DNA Tissue Kit | Qiagen | Cat#51404 |
| EndoFree® Plasmid Maxi Kit | Qiagen | Cat#12362 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Relative Human Mitochondrial DNA Copy Number Quantification qPCR Assay Kit | ScienCell | Catalog#8938 |
| Human IL-2 ELISA Kit | Millipore Sigma | Cat#RAB0286 |
| IL-7 Human ELISA Kit | ThermoFisher | Catalog#EHIL7 |
| IL-15 Human ELISA Kit | ThermoFisher | Catalog#BMS2106 |
| Agilent Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat#103015–100 |
| Agilent Seahorse XF Calibrant solution | Agilent | Cat#100840–000 |
| Agilent Seahorse XF RPMI medium | Agilent | Cat#103576–100 |
| Agilent Seahorse XF sensor cartridges and cell culture microplates | Agilent | Cat#101085–004 |
| Poly-D-lysine | Sigma | Cat#27964–99–4 |
| Seahorse XF 100 mM Pyruvate Solution | Agilent | Cat#103578–100 |
| Seahorse XF 1.0 M Glucose Solution | Agilent | Cat#103577–100 |
| Seahorse XF 200 mM Glutamine Solution | Agilent | Cat#103579–100 |
| Bovine Serum Albumin | Sigma Aldrich | Cat#A9418–100G |
| EDTA | Sigma Aldrich | Cat#E8008–100ML |
| XenoLight D-Luciferin – K+ Salt Bioluminescent Substrate | Perkin Elmer | Cat#122799 |
| Neon™ Transfection System 100 μL Kit | Invitrogen | Cat#MPK10025 |
| HiFi Cas9 protein | IDT | Cat#1081061 |
| Cas9 tracrRNA | IDT | Cat#1072532 |
| crRNA | IDT | Custom, sequence specific |
| Standard metabolites list | Sigma | Table S6 |
| OVA 257–264 | Invivogen | Cat#vac-sin |
| BpiI (BbsI) (10 U/μL) | ThermoFisher | Cat#ER1012 |
| Esp3I (BsmBI) (10 U/μL) | ThermoFisher | Cat#ER0451 |
| 4-Hydroxyproline | Sigma | Cat#H54409 |
| Acetyl CoA | Sigma | Cat#A2056 |
| ADP | Sigma | Cat#A2754 |
| Amino acid standards, physiological | Sigma | Cat#A9906 |
| AMP | Sigma | Cat#1930 |
| Arginine | Sigma | Cat#A5006 |
| ATP | Sigma | Cat#A26209 |
| cAMP | Sigma | Cat#A9501 |
| Choline | Sigma | Cat#C7017 |
| CoA | Sigma | Cat#C4282 |
| CTP | Sigma | Cat#C1506 |
| Fumarate | Sigma | Cat#D95654 |
| Glucose | Sigma | Cat#G8270 |
| Glutathione | Sigma | Cat#G4251 |
| Glyceraldehyde3P | Sigma | Cat#G5251 |
| Glycerate3P | Sigma | Cat#P8877 |
| Glyoxylate | Sigma | Cat#G4502 |
| GTP | Sigma | Cat#G8877 |
| IMP | Sigma | Cat#I4625 |
| Malate | Sigma | Cat#46940 |
| Myo-Inositol | Sigma | Cat#I5125 |
| Orinithine | Sigma | Cat#57197 |
| Oxaloacetate | Sigma | Cat#O4216 |
| Palmitoyl CoA | Sigma | Cat#P9716 |
| Proline | Sigma | Cat#P3350000 |
| PRPP | Sigma | Cat#P8296 |
| Putrescine | Sigma | Cat#51799 |
| Pyruvate | Sigma | Cat#P2256 |
| Ribose5P | Sigma | Cat#83875 |
| Spermidine | Sigma | Cat#S2626 |
| Spermine | Sigma | Cat#S3256 |
| Sphingosine | Sigma | Cat#S7049 |
| Succinate | Sigma | Cat#W327700 |
| Tetrahydrofolic acid | Sigma | Cat#G5251 |
| TTP | Sigma | Cat#T0251 |
| UTP | Sigma | Cat#U6375 |
| Xylose-5-Phosphase | Sigma | Cat#78963 |
| α-Hydroxyglutaric acid | Sigma | Cat#90790 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| human CAR-T cell RNA-seq | This paper | PRJNA806391 |
| human CD8 T cell metabolomics | This paper | ST002085 |
| human CAR-T cell metabolomics | This paper | ST002084 |
| CyTOF data | This paper | DOI: 10.17632/pnbjdtdkfg.1 |
| Experimental Models: Cell Lines | ||
| HEK293FT | ThermoFisher | Catalog Number: R70007 |
| HEK293T | ATCC | Catalog Number: CRL-3216™ |
| NALM6 | ATCC | Catalog Number:3273 |
| MCF-7 | ATCC | Catalog Number: HTB-22 |
| E0771 | CH3 | Catalog Number: 940001 |
| MB-MDA-231 | ATCC | Catalog Number: HTB-26 |
| MM.1R | ATCC | Catalog Number:CRL-2975 |
| Human Peripheral Blood CD8+ T Cells | STEMCELL | Catalog Number: 70027 |
| Human Peripheral Blood Mononuclear Cells | STEMCELL | Catalog Number: 70025.1 |
| Experimental Models: Organisms/Strains | ||
| OT-I | Jackson Laboratory | Stock No: 003831 |
| NSG | Jackson Laboratory | Stock No: 005557 |
| Cas9ß | Platt et al., 2014 | Jackson Lab |
| OT-I;Cas9ß | Dong et al. 2019 Cell | Sidi Chen lab |
| Oligonucleotides | ||
| mm10dg library | This paper | Dataset S1 |
| Taqman probes | This paper | Table S3 |
| Amplicon primers for surveyor or Nextera | This paper | Table S2 |
| gBlocks of U6-dgRNA-MS2 | IDT | Custom, sequence specific |
| EFS-Thy1.1-MCP-p65-HSF1 | IDT | Custom, sequence specific |
| Prodh2 probe | ThermoFisher | Mm00457662-m1 |
| Actb probe | ThermoFisher | Mm00607939-s1 |
| Gapdh probe | ThermoFisher | Mm99999915_g1 |
| Wdr37 probe | ThermoFisher | Mm00552518-m1 |
| Srek1ip1 probe | ThermoFisher | Mm00482686-m1 |
| Ccnb1ip1 probe | ThermoFisher | Rn01414585-m1 |
| PRODH2 probe | ThermoFisher | Hs00560411-m1 |
| ACTB probe | ThermoFisher | Hs99999903-m1 |
| GAPDH probe | ThermoFisher | Hs02786624-g1 |
| Recombinant DNA | ||
| psPAX2 | Addgene | Cat#12260 |
| pMD2.G | Addgene | Cat#12259 |
| pRepCap6 (AAV6 capsid) | Addgene | Cat#110770 |
| pAdDeltaF6 (PDF6) | Addgene | Cat#112867 |
| TdgA Vector | This study | Sidi Chen lab |
| Mm10dgLib library | This study | Sidi Chen lab |
| Software and Algorithms | ||
| FlowJo software 9.9.6 | FlowJo, LLC | https://www.flowjo.com |
| Cutadapt | Martin et al., 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| DAVID | Huang et al., 2009 | https://david.ncifcrf.gov |
| Scran R package | Lun et al., 2016 | https://www.r-project.org |
| Rtsne R package | Maaten, 2014; Maaten and Hinton, 2008 | https://www.r-project.org |
| Edge R package | Robinson et al., 2010 | https://www.r-project.org |
| Kallisto | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| Sleuth | Pimentel et al., 2017 | https://pachterlab.github.io/sleuth/ |
| GSEA | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| PSCAN | Zambelli et al., 2009 | http://159.149.160.88/pscan/ |
| Bowtie 1.1.2 | Langmead et al., 2009 | http://bowtiebio.sourceforge.net |
| Agilent Mass Hunter Qualitative Analysis Software | Agilent | (Version B.07.0.0, build 7.0.7024.0). |
| Agilent Mass Profiler Professional | Agilent | (Version 14.5-Build 2772) |
| Metabolite database | HMDB | http://www.hmdb.ca/ |
| Metabolite database | METLIN | http://metlin.scripps.edu |
| MetaboAnalyst 4.0 | Chong et al., 2018; Xia et al., 2015 | https://www.metaboanalyst.ca |
| Pathvisio v3.3.0 | Pathvisio | https://www.pathvisio.org |
| KEGG | KEGG | http://www.genome.jp/kegg/ |
| Other | ||
Mouse models
All animal work was performed under the guidelines of Yale University Institutional Animal Care and Use Committee (IACUC) with approved protocols (Chen-2015–20068; Chen-2018–20068; Chen-2021–20068). The general health of the mice are in good condition (BAR: bright, alert and responsive) before the cancer-related experiments started. Mice were housed in a free access to water and food, temperature (approximately 22°C) and humidity controlled colony room, maintained on a 12h light/dark cycle (07:00 to 19:00 light on). Mice health checks were performed daily. Mice, both female and male, aged 8–12 weeks were used for experiments. Rosa26-Cas9–2A-EGFP constitutive expressed mice (Cas9β mice) and OT-I TCR transgenic mice (Hogquist et al., 1994) were used in this study. OT-I and Cas9β mice were bred to generate OT-I;Cas9β mice. NOD-scid IL2Rgammanull (NSG) mice were purchased from JAX and bred in-house for in vivo tumor model and T cell based therapeutic efficacy testing experiments.
Cell culture
HEK293FT, HEK293T, E0771, NALM6, MM.1R, MCF7, and MDA-MB-231 cell lines were obtained from commercial sources (ThermoFisher, American Type Culture Collection (ATCC) and CH3) and were tested negative for mycoplasma contamination. HEK293FT, E0771 and MCF7 cells were cultured in DMEM (Gibco) media supplemented with 10 % FBS (Sigma) and 200 U / mL penicillin–streptomycin (Gibco), hereafter referred to as D10. NALM6 and MM.1R cells were cultured in RPMI-1640 (Gibco) media supplemented with 10% FBS and 200 U / mL penicillin–streptomycin. NALM6-GL-CD22OE, MM.1R-PL-BCMAOE, and MCF7-PL-HER2OE cell lines were established by transducing WT cancer cells with GFP-Luciferase (GL) or Puromycin-Luciferase (PL) lentivirus first, then purified by enriching for GFP+ or Puromycin resistant cells by FACS or puromycin selection, respectively. For NALM6-GL, MM.1R-PL, and MCF7-PL cell lines, cells were transduced with CD22-Blasticidin, BCMA-Blasticidin or HER2-Blasticidin lentivirus for overexpression of specific cancer antigen transgenes where appropriate, which were established by Blasticidin selection.
Naïve CD8+ T cell isolation and culture
Mesenteric lymph nodes (mLNs) and spleens were dissected from OT-I;Cas9β mice, then placed into ice-cold PBS supplemented with 2 % FBS. Lymphocytes were released by grinding organs through a 100 μm filter, then re-suspended with 2 % FBS. Red blood cells (RBCs) were lysed with 1 mL ACK lysis buffer (Lonza) for 2 spleens at 1–2 min at room temperature, then neutralized with 2 % FBS PBS at 20 volumes per volume of lysis buffer. RBC-lysed lymphocyte solution was filtered through 40 μm filters to remove cell debris. Naïve CD8a+ T cell purification was performed using Naïve CD8a+ T cell Isolation Kits (Miltenyi Biotec) according to the manufacturer’s protocols. Naïve CD8a+ T cells were cultured at 1 × 10e6 cells / mL density in 2 μg / mL anti-CD3ε (BioLegend) treated plates or dishes, in RPMI-1640 (Gibco) media supplemented with 10 % FBS, 2 mM L-Glutamine, 200 U / mL penicillin–streptomycin (Gibco), and 49 μM β-mercaptoethanol (Sigma), hereafter referred to as cRPMI media. For in vitro experiments, cRPMI media was supplemented with 2 ng / mL IL-2, 1 μg / mL anti-CD28, and 12 ng / mL Il-12p70 cytokines or antibodies. All cytokines and antibodies were purchased from BioLegend.
Pre-clinical efficacy testing using in vivo models
NOD-scid IL2Rgammanull (NSG) mice were purchased from JAX and bred in-house. Eight-to-twelve-week-old both sex mice were used for cancer modeling and efficacy testing.
For the leukemia model, 5e5 NALM6-GL-CD22OE cancer cells were inoculated via intravenous injection. After 4 days of cancer inoculation, 4e6 CD22-CAR;PRODH2 or CD22-CAR;PRODH2 (Stop) T cells were tail vein injected as treatments.
For the myeloma models, model 1, 1.5e6 per mouse of MM.1R-PL cancer cells were injected intravenously. Then 1.5e6 per mouse of BCMA-CAR;PRODH2 and BCMA-CAR;PRODH2 (Stop) T cells were injected intravenously one day after cancer injection as treatments; Model 2, 2e6 per mouse of MM.1R-PL-BCMAOE cancer cells were injected intravenously. Then 9e5 per mouse of BCMA-CAR;PRODH2 and BCMA-CAR;PRODH2 (Stop) T cells were injected intravenously ten day after cancer injection as treatments. For the lenti-BCMA-CAR model, 3e6 MM.1R-PL-BCMAOE cancer cells were injected intravenously. Then 1e6 per mouse of lenti-BCMA-CAR;PRODH2 or lenti-BCMA-CAR;PRODH2 (Stop) T cells were injected intravenously at day 9 after cancer injection as treatments, the second dose of CAR-T cells (1.3e6 per mouse) was injected at day 33. Treatment dose and time-point are indicated in the figure legends.
For the breast cancer model, 1e6 MCF7-PL-HER2 cancer cells were fat-pad inoculated into female NSG mice, 1.5e6 HER2-CAR-PRODH2 or HER2-CAR T cells were injected intravenously one day after cancer inoculation as treatments. Leukemia progression was measured by bioluminescent imaging using the IVIS system. Solid tumor progression was evaluated by tumor volume measurement by caliper, calculated as the formula below:
Mice were euthanized by carbon dioxide asphyxiation. Animal were dissected then the collected tissues were fixed in 4% PFA for 2 days. Samples were embedded in paraffin then sectioned at 4 μm and stained with hematoxylin and eosin (H&E). Slides were scanned using an Aperio digital slide scanner (Leica). All mice were killed once they reached an endpoint according to the IACUC-approved protocols.
METHODS DETAILS
mm10dgLib design
The 250 bp sequences upstream of all mouse protein-coding genes were extracted by Ensembl Biomart. All possible dgRNA spacers (15mer-NGG) were identified, both on the sense and antisense strands. 18mers were then mapped to the mm10 genome by Bowtie with settings bowtie -n 2 -e 120 --best. After discarding any alignments with mismatches in the 17th or 18th position ("GG" in NGG PAM), the number of alignments was tabulated for each spacer. Each alignment was then scored based on the number of mismatches, disregarding any mismatches in the 16th position (the "N" in NGG), using the following formula: (# of 0 mismatch alignments * 1000) + (# of 1 mismatch alignments * 50) + (# of 2 mismatch alignments * 1). Larger mismatch scores indicate decreased genome-wide mapping specificity. Subsequently, all non-uniquely mapping spacers, as well as spacers with mismatch score >= 2000, were discarded. The top 4 dgRNA spacers were then chosen based on shortest distance to the −200 position from the TSS, since transcription factors generally to bind to promoters at around this distance. In total, 83,601 dgRNAs targeting 22,391 coding transcripts and 1,000 non-targeting controls (NTCs) were designed.
Construction of T cell dgRNA activation (TdgA) vector
The gBlocks of U6-dgRNA-MS2 and EFS-Thy1.1-MCP-p65-HSF1 gene fragments were synthesized from Integrated DNA Technologies (IDT), then sub-cloned into lentiviral vectors via Gibson Assembly (NEB). The resulting activation vector, lenti-U6-dgRNA (BsmbI)-MS2-EFS-Thy1.1-MPH, is referred to as TdgA vector.
mm10dgLib cloning
Designed mm10dgLib was synthesized by IDT followed by sub-cloning into TdgA vector via Gibson assembly and electroporation. Approximately > 230x coverage (~2e7 clones were obtained) was achieved after electroporation. The mm10dgLib plasmids were extracted via Maxi preparation (Qiagen), followed by Illumina sequencing confirmation to ensure library representation. A total of 82,197 / 83,601 of GTS and 988 / 1,000 of NTC were cloned.
Lentivirus production
Lentivirus was produced using low-passage HEK239FT cells. One day before transfection, HEK293FT or HEK293T cells were seeded in 15 cm-dish at 50–60 % confluency. 2 h before transfection, D10 media was replaced with 13 mL pre-warmed Opti-MEM medium (Invitrogen). For each plate, 450 μL of Opti-MEM was mixed with 20 μg mm10dgLib plasmid or vector control plasmid, 15 μg psPAX2 (Addgene), 10 μg pMD2.G (Addgene) and 130 μL polyethylenimine (PEI) (1 mg/mL) or 100 μL lipofectamine 2000 (Thermo Fisher). After brief vortex, the mixture was incubated for 15 min at room temperature and then dropwise added to cells. After 6 h of transfection, Opti-MEM media was replaced with 20 mL pre-warmed D10 media. Viral supernatant was collected at 48 h and 72 h post-transfection, then filtered using 0.45 μm filters (Fisher / VWR) to remove cell debris, and then concentrated using AmiconUltra 100 kD ultracentrifugation units (Millipore). All virus was aliquoted and stored in −80 °C.
mm10dgLib lentivirus titration
Naïve CD8+ T cells were infected with lentivirus in different volume ratios between cRPMI and lentivirus after isolation. At day 3 after T cell isolation, T cells were collected and stained with anti-CD8-APC and anti-Thy1.1-PE, the Thy1.1-positive cells represented cells successfully infected by virus and expressing the vector. The ratio of CD8+ and Thy1.1+ cells was determined by flow cytometry. Viral titer was calculated by comparing mm10dgLib-infected cells to uninfected cells, albeit following the same staining procedures. Each group has 3 independent biological replicates.
mm10dgLib lentivirus transduction
Given the lentiviral titer information, for each replicate, 1.5e7 OT-I;Cas9β naïve CD8+ T cells were cultured in a 10-cm plate and transduced with 400 μL mm10dgLib or empty vector lentivirus at a volume ratio of 1:25 (virus : media), which can successfully infect around 75 % T cells (Figure S1).
AAV-HDR based CAR and lentiviral-based CAR construction
TRAC homologous recombination arms (HA) were cloned from the both sides of crRNA binding site located in the first exon of TRAC locus. CD22-CAR sequence contains a CD22 m971 scFv, CD8 hinge, CD8 transmembrane domain, 4–1BB intracellular domain, and CD3ζ intracellular domain. HER2-CAR sequence contains a 4D5 scFv, CD8 hinge, CD8 transmembrane domain, CD28 and 4–1BB intracellular domains, and CD3ζ intracellular domain. BCMA-CAR sequence contains a BCMA scFv, CD8 hinge, CD8 transmembrane domain, 4–1BB intracellular domain, and CD3ζ intracellular domain. These sequences were synthesized as gBlocks from IDT. PRODH2 cDNA was purchased from GenScript, then subcloned into an AAV vector. The final vector was named as LHA-EFS-CD22-CAR-PRODH2-RHA and LHA-EFS-HER2-CAR-PRODH2-RHA. Stop codons were engineered at 5’ coding regions of PRODH2 to generate control CAR-T AAV vectors. For the lenti-CAR constructs, EFS-CD22-CAR-PRODH2 / PRODH2 (Stop) and EFS-BCMA-CAR-PRODH2 / PRODH2 (Stop) DNA sequences were PCR amplified from AAV-CAR plasmids then cloned into a lentiviral backbone.
AAV production
Low-passage HEK293FT cells were used for AAV production. Briefly, 2 h before transfection, D10 media was replaced by pre-warmed DMEM (FBS-free). For each 15 cm-dish, HEK293FT cells were transiently transfected with 5.2 μg transfer, 8.9 μg AAV6 serotype and 10.4 μg pDF6 plasmids using 130 μL PEI (1 mg/mL). After 6–12 h of transfection, DMEM was replaced with 20 mL pre-warmed D10 media. Cells were dislodged and transferred to 50 mL Falcon tubes after 48–72 h post-transfection. For AAV purification, 1 / 10 volume of pure chloroform was added and solution was incubated at 37 °C in the shaker with speed of 200 rpm for 1 h. NaCl was added to a final concentration of 1 M, then pelleted at 20,000 × g at 4 °C for 15 min. The aqueous layer was gently transferred to another clean tube and the chloroform layer was discarded. 10 % (w / v) of PEG8000 (Promega) was added and shaken within the tubes until dissolved. The mixture was incubated on the ice for 1 h followed by centrifuge at 20,000 × g at 4 °C for 15 min. The pellet was resuspended with 5–15 mL PBS including MgCl2 and benzonase (Sigma). After 30–60 min digestion, one volume of chloroform was added, shaken vigorously and spun down at 15,000 × g at 4 °C for 10 min. The aqueous layer was collected carefully and AAV was concentrated using AmiconUltra 100 kD ultracentrifugation units (Millipore). Virus was aliquoted and stored in −80 °C. To measure virus titer, RT-qPCR was performed using Taqman assays (ThermoFisher) targeted to EFS promoter engineered in the AAV vector.
Flow cytometry for surface markers
T cells were collected and washed once using MACS buffer (0.5 % BSA and 2 mM EDTA in PBS) before staining. T cells were stained on ice for 30 min after adding antibodies (1:200 dilution). For the CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) KI T cells were incubated with 0.2 μg CD22-Fc (R&D system) per 106 cells in 100 μL PBS for 30 min on ice, then washed with 1 mL cold PBS and stained with FITC anti-human IgG-Fc or PE anti-human IgG-Fc (Biolegend) for 30 min on ice. Pure CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells were established by sorting CD3 negative and CD22-CAR positive populations. For BCMA-CAR;PRODH2 and BCMA-CAR;PRODH2(Stop) KI T cell detection, same strategy was used as CD22-CAR. For HER2-CAR;PRODH2 and HER2-CAR;PRODH2(Stop) KI human CD8 T cells were stained with PE anti-Flag (Biolegend) antibody for 30 min on ice, then washed with 1mL MACS buffer. All samples were run on a BD FACSAria cytometer, and analysis was performed using FlowJo software 9.9.4 (Threestar, Ashland, OR). Cells were gated by FSC/SSC plot. To distinguish between positive and negative boundaries of the stained cells, non-staining or untreated control samples were analyzed and utilized as background.
Intracellular flow cytometry
Intracellular flow cytometry was performed to detect the expression level of cytokines and immune markers. Purified CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells were co-cultured with NALM6-GL cancer cells at 1:1 ratio for 12h. X-VIVO™ 15 media (Lonza) supplied with 5 % human AB serum, IL-2, and 5 μg/mL Brefeldin A was used as co-culture media. After 12h, at a time point when CAR-T cells killed majority of the cancer cells, CAR-T cells were collected and PBS washed, membrane protein staining was performed prior to intracellular staining. Cells were fixed and permeabilized after membrane protein staining, and then specific antibodies were added.
A list of flow antibodies can be found in Key Resources Table (KRT).
Kill assay and dgLib screen
mm10dgLib lentivirus-infected OT-I;Cas9β CD8+ T cells were cultured on the 10-cm dishes pretreated with anti-CD3ε in the cRPMI-1640 supplemented with 2 ng / mL IL-2, 1 μg / mL anti-CD28, and 2 ng / mL IL-12p70 for 4 days, the media were changed with fresh media every day. About 12 hours before the kill assay, infected OT-I;Cas9β CD8+ T cells were reseeded onto new dishes without treatment with anti-CD3ε antibody, and cultured with cRPMI only supplemented with 2 ng / mL IL-2 and 2 ng / mL IL-12p70 to rest cells. At the same time, 2e6 E0771 cells were seeded in 6-well plates in D10 media. On day 5, E0771 cells were incubated with 1 ng / mL SIINFEKL peptide for 4 hours. Before start kill assay, CD8+ T cells were suspended with fresh cRPMI media supplied with 2 nM monensin and anti-CD107a-PE antibody (BioLegend) (1:400 dilution), the final cell concentration was 2e6 cells / mL. After SIINFEKL peptide incubation, 3e6 CD8+ T cells per replicate (E0771: T cell = 1:1) were added into E0771 cells for 2 h co-culture. At the end of co-culture, T cells were gently washed down with PBS and stained with anti-CD8α-APC, anti-CD3ε-PE/Cy7 for 30 min on ice, cells were analyzed and sorted using BD FACSAria. CD107a-high cells were sorted by FACS for library readout similar to an in vitro T cell cytotoxicity CRISPR screen approach in a previous study (Dong et al., 2019).
Kill assay screen readout and deep sequencing
Sorted cells and cell pellets (2 × 106 cells / replicate, collected before kill assay) were lysed using QuickExtract solution (Epicentre) at 65 °C for 30 min, then 85 °C for 5 min. dgRNA readout was performed using two-step PCR amplification. PCR was performed using Phusion Flash High Fidelity Master Mix (Thermo Fisher). The first round PCR (PCR #1) used primers to amplify dgRNA cassette:
Forward: 5’-aatggactatcatatgcttaccgtaacttgaaagtatttcg-3’,
Reverse: 5’-ctttagtttgtatgtctgttgctattatgtctactattctttccc-3’.
The second round PCR (PCR #2) amplification used uniquely barcoded primers, from SF1 to SF12 and SNR1 to SNR12, to allow multiplexing of samples in a single Hiseq run. For PCR #1, each sample underwent 5 reactions in a 50 μL PCR volume to capture all enriched dgRNAs as deeply as possible. The cycle condition was: 98 °C for 1 min, 25 cycles of (98 °C for 1s, 62 °C for 5 s, 72 °C for 15 s), and 72 °C for 2 min. PCR products of each sample were pooled together for barcoding PCR. For PCR #2, 2 μL of pooled PCR #1 products were used as template in a 50 μL PCR reaction. The PCR cycle conditions were: 98 °C for 1 min, 30 cycles of (98 °C for 1s, 62 °C for 5 s, 72 °C for 15 s), and 72 °C for 2 min. All PCR products were quantified with a gel-based method using the Low-Range Quantitative Ladder (Life Technologies), then gel purified using the QIAQuick gel extraction kit (Qiagen). Purified PCR products were equally pooled in one tube for deep sequencing. Diluted libraries with 5–20 % PhiX were sequenced with HiSeq 2500 or HiSeq 4000 systems (Illumina).
A list of barcoded primers can be found in KRT.
dgRNA screen data analysis
Raw single-end fastq read files were filtered and demultiplexed using Cutadapt (Martin, 2011). To remove extra sequences downstream (i.e. 3’ end) of the sgRNA spacer sequences, the following command was used: cutadapt -a GTTTTAGAGCTAGGC -e 0.2 --discard-untrimmed. As the forward PCR primers used to readout sgRNA representation, they were designed to have a variety of barcodes to facilitate multiplexed sequencing, demultiplexing these filtered reads was performed with the following settings: cutadapt -g file:fbc.fasta --no-trim, where fbc.fasta contained the 12 possible barcode sequences within the forward primers. Finally, to remove extra sequences upstream (i.e. 5’ end) of the sgRNA spacers, the following command was used: cutadapt -g AAAGGACGAAACACCG -e 0.2 -m 14 -M 18. Minimum / maximum read lengths were set at 14 / 18 bp respectively to exclude non-specific reads and enriched for the 15 bp dgRNA spacer sequences. The 15 bp dgRNA spacer sequences from each demulitplexed sample were then mapped the designed dgRNA spacers in the mm10dgLib (Dataset S1). A bowtie index of the dgRNA library was generated using the bowtie-build command in Bowtie 1.1.2 (Langmead et al., 2009). The filtered fastq read files were mapped to the index using the following settings: bowtie -v 1 -m 1 mm10dgLib -q <fastq_file>. Using the resulting mapping output, the number of reads that had mapped to each sgRNA within the library was quantified.
Enrichment analysis of dgRNAs
Enrichment analysis was performed using custom R scripts. The library representation of each sample was quantified by the number of read counts. Raw counts were normalized and log-transformed. Mean values were averaged from biological replicates of the same group (plasmid, unsorted cell, or high-kill). FDR values were calculated from the null distribution of the 1,000 NTCs in each variable. Linear regression for the 1,000 NTCs between samples was performed using the lm() function. The points in waterfall and scatter plots were shown at the individual gRNA level.
Immunoblots
Human CD8+ T cells with CD22-CAR, CD22-CAR;PRODH2 or CD22-CAR;PRODH2 (Stop) KI were collected and washed with PBS to remove media. Cells were lysed with RIPA lysis buffer supplied with protease and phosphatase inhibitor cocktail (Thermo Fisher) and incubated on ice for 30 min, followed by centrifugation at ≥ 15, 000 × g for 15 min at 4 °C. The supernatant was collected for protein quantification. The total protein concentration was quantified by performing a Bradford protein assay (Bio-Rad), a total of 10 μg protein per sample was loaded into SDS-PAGE gel (Bio-Rad), proteins in the gel were transferred into Amersham Protran 0.45 μm NC Nitrocellulose Blotting membrane (GE Healthcare) after electrophoresis. Membranes were blocked with 2 % BSA in TBST buffer for 1 h at room temperature, followed by the primary antibody incubation at 4 °C overnight. Anti-PRODH2 antibody was purchased from Atlas Antibodies (HPA051287) (1:1000 dilution) and anti-Vinculin was purchased from Abcam (ab129002) (1:2000 dilution) which was used as internal control. Antibody binding was detected using horseradish peroxidase-conjugated secondary antibody and ECL substrate (Bio-Rad).
Cytokine secretion assays
CAR-T cells and cancer cells were co-cultured at a E:T = 1:1 ratio for 12h in 200 μL X-VIVO™15 T cell media. The supernatant was collected and analyzed for IL-2, IL-15, and IL-7 secretion using ELISA kits (ThermoFisher), the protocols were provided by the manufacturer.
RT-qPCR
For cDNA overexpression, 48 h and 72 h after lentivirus transduction, OT-I CD8+ T cells were collected for RNA preparation. Human CD8+ T cell PRODH2 overexpression and CD22-CAR;PRODH2 knock-in (KI) qPCR verification were performed as described in the results. All RNA preparations were performed using RNasy Plus Mini Kit (Qiagen). Total mRNA was reverse transcribed into cDNA by using SuperScript™ IV Reverse Transcriptase (Thermo Fisher). Gene expression was quantified using Taqman Fast Universal PCR Master Mix (Thermo Fisher) and Taqman probes (Invitrogen). RNA expression level was normalized to Gapdh / GAPDH or Actb / ACTB (mouse / human). Relative mRNA expression was determined via the ΔΔ Ct method. For the relative human mitochondrial DNA copy number quantification, CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells were collected for DNA isolation (Qiagen). The relative human mitochondrial DNA copy number was quantified by using a qPCR assay kit (ScienCell). The mtDNA primer set provided with the kit was used to recognize and amplify one of the most conserved regions on human mtDNA and will not amplify off-target sequence on nuclear genomic DNA. The reference primer set was used for amplifying a 100 bp-long region on human chromosome 17 and serve as reference for data normalization. Twenty microliter qPCR reactions were set up and a quantification method was used based on the protocol provided by the manufacturer.
A list of probes can be found in KRT.
Human primary CD8+ T cell electroporation and CAR-T knock-in
Human primary CD8+ T cells were isolated from healthy donor PBMCs (StemCell). CD8+ T cells were cultured in X-VIVO™ 15 media (Lonza) supplied with 5 % human AB serum and IL-2. Before electroporation, TRAC crRNA (5’-TCTCTCAGCTGGTACACGGC-3’) and tracrRNA were mixed in 1:1 ratio (final concentration 50 μM), heated at 95 °C for 5 min in the thermal cycler, then cooled to room temperature. 3 μL Cas9 protein (61 μM) was mixed with 2 μL Buffer R for each reaction (Neon Transfection System Kit, Thermo Fisher), then mixed with 5 μL annealed crRNA:tracrRNA duplex, incubated the mixture at room temperature for 10–20 min. Human CD8+ T cells were collected and washed with PBS to completely remove the media. 3 × 106 of T cells per reaction were resuspended in 100 μL Buffer R which included 10 μL RNP complex. 100 μL of cell:RNP mixture was loaded into the Neon Pipette without any bubbles. The electroporation parameter was set at 1600 V, 10 ms for 3 pluses. Cells were immediately transferred to a 24-well plate with pre-warmed media after electroporation. A total of ~ 1.5e9 viral genome copy of AAV6 HDR donor was added into each electroporated T cell reaction within 1 h after electroporation.
Top candidate validation in kill assay
Candidate hits mouse Prodh2, Ccnb1ip1, Srek1ip1, and Wdr37 were selected for further validation. All cDNAs were purchased from the Dharmacon, then cloned into a lentiviral overexpression vector (lenti-EF1a-Flag-WPRE vector). Before lentivirus transduction, OT-I CD8+ T cells were isolated and activated by anti-CD3ε and anti-CD28 antibodies for 2–3 days, then T cells were transduced with concentrated lentivirus. At day 6, infected OT-I CD8+ T cells were reseeded onto new 6-well plates which were untreated with anti-CD3ε antibody and cRPMI only supplemented with 2 ng / mL IL-2 and 2 ng / mL IL-12p70 to rest cells. At the same time, 5e5 / well of E0771 cells were seeded in 24-well plates for kill assay. The next day, E0771 cells were incubated with 1 ng/mL or 10 ng / mL SIINFEKL peptide for 4 hours. The validation kill assay was performed in the same manner as the screen kill assay.
IL-2 withdrawal assay
Mouse OT-I;Cas9β CD8 T cells were activated with anti-CD3ε and anti-CD28 antibodies, T cells were transduced with Prodh2-OE or Vector lentivirus after T cells were completely activated. At day 3 after lentivirus transduction, T cells were collected and washed with PBS, then equal cell numbers were plated in media without IL-2. Cells were stained with PE anti-caspase 3 (Cell signaling technology), FITC anti-CD3, and APC anti-CD8 antibodies at day 1 and 4 after IL-2 withdrawal. To test PRODH2 GOF CAR-T cell potential cancer-like transformation, the same number of CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells were plated in X-VIVO™15 media without IL-2 cytokine, then T cell survival was measured after 7 days by cell counting.
Human CD22-CAR;PRODH2 and HER2-CAR;PRODH2 T cell kill assay
To detect CAR-PRODH2 T cell killing, NALM6-GL, NALM6-GL-CD22OE, MCF7-PL, MCF7-PL-HER2OE or MDA-MB-231-PL cancer cell lines were established as described above, cancer cells were seeded in a 96-well plate first, then different Effector : Target (T cell : cancer cell) ratio (E : T ratio) co-cultures were set up. Cytolysis was measured after co-culture by adding 150 μg / mL D-Luciferin (PerkinElmer) using a multi-channel pipette. Luciferase intensity was measured by a Plate Reader (PerkinElmer).
Lenti-CD22-CAR;PRODH2 and Lenti-BCMA-CAR;PRODH2 T cell transduction and co-culture
Lentivirus was produced by HEK293T cells, lentiviral supernatant was collected and precipitated by Lenti-X Concentrator (Takara). Lentiviral pellets were resuspended with X-VIVO™15 media (LONZA), then aliquoted and stored in −80°C. Human CD8 T cells were transduced with lentivirus onto 1–2e6 T cells plated in a 24-well plate which was pre-coated with Retronectin (Takara) in PBS overnight in 4°C. The spin-infection was performed at 32°C at 900 × g for 90min. The CAR-positive T cells were measured at day3 after transduction, then co-culture assays were performed to determine CAR-T cell killing.
Bulk mRNA sequencing (mRNA-seq) library preparation
The mRNA library preparations were performed using a NEBNext® Ultra™ RNA Library Prep Kit for Illumina and samples were multiplexed using barcoded primers provided by NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1). For the human CD8+ T cell RNA-seq, CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) cassettes were site-specifically KI into TRAC locus, the flow cytometry, qPCR, and western blot were performed to confirm that CD22-CAR-PRODH2 or CD22-CAR-PRODH2(Stop) were successfully KI and expressed before doing the mRNA-seq library preparations. Libraries were sequenced with HiSeq 4000 or Novaseq systems (Illumina).
Bulk mRNA-seq data processing
Raw FASTQ files from mRNA-seq were analyzed for transcript quantification using Kalliso quant algorithm (Bray et al., 2016) with the setting -b 100. Transcriptome references were obtained from Ensembl. Differential gene expression analysis for the effect of PRODH2 overexpression or genomic knock-in was then performed using Sleuth (Pimentel et al., 2017) with gene-level aggregation. Visualization of differentially expressed (DE) genes including volcano plots, bar plots, and Venn diagrams were performed using standard R packages including ggplot2 and VennDiagram.
Gene set level pathway analysis of differentially expressed genes
Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) was performed using the Java application from the Broad Institute. The full gene set from the differential gene expression analysis was ranked by “beta” value and then used as an input for GSEA pre-ranked analysis with database reference C5 Gene Ontology - Biological Process (GO-BP). The Database for Annotation, Visualization and Integrated Discovery (DAVID) (Huang et al., 2009) was used for further annotation enrichment analysis. Upregulated and downregulated genes from the differential gene expression analysis were defined with adjusted p-value cutoff of 0.001 for human CAR-T experiments and 0.05 for mouse experiments. Pathway enrichments for GO-BP annotations were used for visualization.
Metabolite extraction and data collection
For extraction of intracellular metabolites, cell culture media was first aspirated, then harvested and washed twice with PBS. 2e6 alive cells for each sample were used for metabolite extraction. After normalizing cell counts, 800 μL of 80 % (vol / vol) HPLC-grade methanol (Sigma) (precooled to −80 °C on dry ice) was added to fresh cells in a 1.5-mL microcentrifuge tube, then tubes were put on dry ice for 30 minutes (Yuan et al., 2012). The tubes were then incubated on ice for 20 minutes and centrifuged at 15,000 × g for 15 min at 4 °C to pellet the cell debris. The metabolite-containing supernatant was transferred to a new 1.5-mL microcentrifuge tube on dry ice. Metabolite extraction was repeated with 400 μL of 80 % (vol / vol) HPLC-grade methanol. The cell lysate / methanol mixtures were dried by Speedvac at room temperature. The metabolites were dissolved again with 80 % (vol / vol) methanol, then centrifuged at 18,000 × g for 10 min to remove any particulates, and the metabolite mixtures were stored at −80 °C until LC-MS analysis. For the metabolite analysis, the untargeted metabolic profiling was firstly analyzed with an Agilent 6550 Q-TOF LC/MS System first, then targeted metabolites were analyzed with an Agilent 6490 Triple quadrupole (QQQ) LC/MS System. Multiple reaction monitoring (MRM) was employed for the quantitation of purified standard (Sigma). A HILIC liquid chromatograph were optimized with a bioZen™ 2.6 μm Glycan LC Column, 150 × 2.1 mm (Phenomenex) and a Glycan guard column, 4 × 2 mm (Phenomenex). The eluents included buffer A, 0.01 % formic acid in HPLC-grade water, and buffer B, 0.01 % formic acid in acetonitrile. The gradient was set as follows: 0–2 min 94 % B, 2–8 min 94–90 % B, 8–16 min 90–76 % B, 16–36 min 76–50 % B, 36–42 min hold at 50 % B and then back to initial conditions for 2 min for column equilibration. The flow rate was set as 0.3 mL / min. Multiple reaction monitoring (MRM) was employed for the quantitation of purified standard (Sigma).
A list of standard metabolites used in this study can be found in KRT.
Metabolomics data processing
Two metabolomics strategies were adopted, i.e. untargeted metabolomics (aiming to unbiasedly detect all detectable metabolites) and targeted approaches (aiming to detect specifically defined metabolites, such as related metabolites in the proline metabolism and T cell metabolism). For untargeted metabolomics analysis, the optimized workflow consists of automated peak detection and integration, peak alignment, background noise subtraction, and multivariate data analysis. These steps were carried out for comprehensive metabolite phenotyping of the two groups using Agilent Mass Hunter Qualitative Analysis Software (Version B.07.0.0, build 7.0.7024.0) and Agilent Mass Profiler Professional (Version 14.5-Build 2772). The metabolites were first putatively identified based on accurate mass match (accurate mass ± 30 ppm error) and fragmentation pattern match. Putative structural annotation was carried out by searching the metabolite databases HMDB (http://www.hmdb.ca/) and METLIN (http://metlin.scripps.edu) using the mass-to-charge ratio of the metabolic features. For the targeted metabolomics, available metabolites from the significantly changed metabolites of untargeted metabolomics analysis, as well as related proline metabolism and immune system metabolism, were purchased from Sigma. Multiple reaction monitoring (MRM) was employed for the qualitative and quantitative analysis of purified standard (Sigma). The features of spectra were extracted using Agilent Mass Hunter Qualitative Analysis Software (Version B.07.0.0, build 7.0.7024.0). Each peak was manually checked and the abundances of all metabolites were exported. The retention time of the standards was cross-referenced with those detected in the untargeted method for consistency, which also confirmed the accuracy of the untargeted methods. The integration of untargeted and targeted metabolites includes the combination of non-overlapping metabolites and the selection of targeted metabolites of overlapping metabolites. Multivariate data analyses were conducted using Statistical analysis module of MetaboAnalyst 4.0 (Chong et al., 2018; Xia et al., 2015). Briefly, Log transformation and auto scaling (mean-centered and divided by the standard deviation of each variable) were used for data processing. Heatmaps and volcano plots were generated plots with MetaboAnalyst 4.0. Distance Measure was set as Euclidean and the Clustering Algorithm was set as Ward. The functions of the metabolic flowchart were constructed with the software Pathvisio v3.3.0 based on the KEGG database (http://www.genome.jp/kegg/). The integrated analysis of the changed metabolites and genes were done with Joint Pathway analysis module of MetaboAnalyst 4.0.
Intersection of mRNA-seq, metabolomics and KEGG pathways
Differential expressed genes from mRNA-seq analysis were cross-referenced with expected differential expressed genes from metabolomics analysis in order to obtain consensus upregulated and downregulated gene sets due to PRODH2 overexpression or genomic knock-in. For a given metabolite, genes contributing to its production or consumption were defined using the KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathway Database (Kanehisa and Goto, 2000). Genes could be both "producing" and "consuming" when involved in reversible reactions. Expected upregulated genes were defined as those producing metabolites with positive logFC or those consuming metabolites with negative logFC. Expected downregulated genes were defined as those producing metabolites with negative logFC or those consuming metabolites with positive logFC. For consensus "upregulated" genes, first the union of downregulated mRNA-seq genes from human donor 1 and donor 2 from the CAR-T experiments were removed from the union of upregulated mRNA-seq genes (setdiff). Then the resulting restricted gene set was intersected with expected upregulated metabolomics genes. For consensus "downregulated " genes, the union of upregulated mRNA-seq genes was removed from the downregulated mRNA-seq genes (setdiff). Then the resulting restricted gene set was intersected with expected downregulated metabolomics genes.
Multi-Omics analysis
Consensus differential expressed genes from intersection analysis and differentially represented metabolites with fold changes from metabolomics analysis were used as inputs for joint pathway analysis using the MetaboAnalyst Portal (Xia et al., 2009). Default parameters were used, with Hypergeometric Test for enrichment analysis, Degree Centrality for topology analysis, and Gene-metabolite pathways for pathway databases. Pathways were considered statistically significant if the p-values were less than 0.05. For visualization, upregulated gene set and downregulated gene set were separated, each compared against DR metabolites (both increased and decreased, as the gene activity can influence on either direction), using the MetaboAnalyst Portal.
Transmission electron microscopy (TEM) of CAR-T cells
Purified CAR-T cells were collected and washed with PBS, then fixed in 2.5% gluteraldehyde in 0.1M sodium cacodylate buffer pH7.4 for 1 hour. Buffer rinsed cells were spun down in 2% agar and the chilled blocks were trimmed and post fixed in 1% osmium tetroxide for 1 hour then the sample was rinsed in buffer and stained in aqueous 2% uranyl acetate for 1 hour. This was followed by rinsing in distilled water, dehydrating in an ethanol series and infiltrating with Embed 812 (Electron Microscopy Sciences) resin and baked over night at 60 °C in silicone molds. Hardened blocked were sectioned using a Leica UltraCut UC7, 60nm sections were collected on formvar and carbon coated nickel grids and contrast stained using 2% uranyl acetate and lead citrate. The sections were viewed FEI Tencai Biotwin TEM at 80Kv. Images for quantitation were collected randomly and viewed using a Morada CCD and iTEM (Olympus) software. Quantification of TEM data were performed in randomized fields with anonymized images.
Seahorse assay
Oxygen consumption rates (OCR) and extracellular acidification rate (ECAR) were measured in XF media (Agilent) supplied with 25mM glucose, 2mM L-glutamine, 1mM sodium pyruvate, and human IL2 after CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells culture for two months after electroporation, one month after cancer stimulation. 1 μM oligomycin, 1.5 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP) and 50 nM rotenone / 0.5 μM antimycin A were used for testing extra mitochondrial capacity, spare respiratory capacity (SRC), under stress, and measuring extracellular acidification rate (ECAR). Different CD22-CAR T cell densities (2e5 and 4e5 / well) were seeded into XF cell culture microplate (Agilent) that were pre-coated with poly-D-lysine (Sigma) which can facilitate cell adhesion to the plate surface for SRC detection. The microplate was incubated for 30–60 min at 37 °C in a non-CO2 incubator before running in a Seahorse XF96 Analyzer (Agilent). Standard Seahorse program setup was used: Calibration; Equilibration; Base line reading (Loop 3 times), Mix 3 min, Measurement 3 min, End loop; Injection Port A (Loop 3 times), Mix 3 min, Measurement 3 min, End loop; Injection Port B (Loop 5 times), Mix 3 min, Measurement 3 min, End loop; Injection Port C (Loop 3 times), Mix 3 min, Measurement 3 min, End loop; End Program.
Mass cytometry (CyTOF)
Purified CD22-CAR;PRODH2 and CD22-CAR;PRODH2(Stop) T cells (without cancer stimulation) were collected and washed with PBS, resuspended cell to 1e7 / mL in PBS and Cell-ID Cisplatin (Fluidigm) was added to a final concentration of 5 μM. Cells were incubated at room temperature for 5 min, then washed with Maxpar Cell Staining Buffer (Fluidigm). 1.5e6 CAR T cells per replicate were used for staining, each group has three replicates. Cells were stained with surface marker antibody cocktail first, then fixed and permeabilized. Second round staining was performed using cytoplasmic / secreted antibody cocktail. Finally, cells were incubated in intercalation solution (Fluidigm) in a final concentration of 125 nM, then incubated overnight at 4 °C. Before running on a CyTOF machine, cell concentrations were adjusted to 5–7e5/ mL with water. All data were collected on a CyTOF Helios instrument (Fluidigm). All surface and cytoplasmic / secreted antibodies were purchased from Fluidigm or Yale CyTOF core.
A list of CyTOF antibodies used in this study can be found in KRT.
CyTOF data analysis
CyTOF quality prefiltering was performed in FlowJo with CD3 and CD8 gates. Channel values were exported and analyzed with custom scripts in R. Dimensionality reduction by Uniform Manifold Approximation and Projection (UMAP) using the umap package and figures were drawn using ggplot2.
Mitochondria mass and depolarization measurement
CD22-CAR;PRODH2 and CD22-CAR;PRODH2 (Stop) T cells were cultured in X-VIVO™15 media supplied with human IL-2. T cells were stimulated with NALM6-GL-CD22OE cancer cells at a 1:1 ratio. At day 36 after cancer stimulation, T cells were stained with MitoTracker GFP FM and MitoTracker Deep Red FM dyes (ThermoFisher) to measure mitochondrial mass and depolarization.
CAR T cell chemical treatment, co-culture and flow assays
CD22-CAR;PRODH2 and CD22-CAR;PRODH2 (Stop) T cells were pre-treated with P4HA substrate L-Proline (Sigma) or PRODH2 substrate 4-Hydroxyproline (or 4Hyp) (Sigma) for 5 days. T cells were then washed with PBS and used to perform co-culture assay with NALM6-GL-CD22OE cancer cells at a 0.5:1 E:T ratio. T cells also pre-treated with a P4HA1 and P4HA2 inhibitor, 1,4-DPCA (Santa Cruz), or a GOT1 and GOT2 inhibitor, PF04859989 (Axon Medchem) for 3–6 days depending on specific assays, then subjected to the co-culture as described. Flow cytometry was carried out after co-culture to detect T cell immune markers as specified. T cell viability measurement was performed by the live-dead staining after T cells were treated with different concentration 1,4-DPCA as specified for 3 days.
A list of flow antibodies can be found in KRT.
Long-term CAR-T culture with chronic antigen stimulation and memory marker analysis
To measure CD22-CAR;PRODH2 and CD22-CAR;PRODH2 (Stop) T cell memory phenotypes under chronic antigen stimulation in long-term culture, purified CAR T cells were stimulated with NALM6-GL-CD22OE cancer cells every 12 days at an E:T ratio = 1:1, for up to 3 times. CAR T cells were harvested at day 69 after various times of antigen stimulation, and stained with anti-CD45RA and anti-CD62L antibodies.
A list of flow antibodies can be found in KRT.
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample sizes for animal experiments were determined with estimated power to detect significance similar to prior work, cited literature, or similar approaches in the field. Sample sizes for certain experiments, such as in vitro assays, were not predetermined by power calculations, and were predetermined according to the lab's prior work, cited literature, or similar approaches in the field. Most experiments were done with at least two biological replicates. Experimental replications were indicated in detail in the relevant sections above and in each figure panel's legend. In animal experiments, mice were randomized by sex, cage and littermates. For the in vivo validation experiments, tumor-bearing mice were normalized by tumor burden, either caliper-measured tumor size or bioluminescence signal, before being allocated into difference groups. If tumors were unmeasurable (early timepoint) at time of grouping, all mice were randomized by sex, cage, and littermates without considering tumor burden and allocated into control and treatment groups. In vitro cell culture experiments were not randomized. Investigators were not blinded for the in vitro cell culture experiments. Investigators were blinded for the TEM experiments, including sample preparation, image capture, tumor burden measurements, and data quantification. In NGS data analysis, investigators were blinded for initial processing of the original data using key-coded metadata. Various standard statistical analyses were performed. All statistical methods are described in the figure legends and/or supplementary Excel tables. The P values and statistical significance were estimated for all analyses. The unpaired t tests, two sided, was used to compare two groups. Multiple t tests and two-way ANOVA were used to compare multiple groups. Survival curves were compared using the log-rank (Mantel-Cox) tests. Most data showed normal or near-normal distribution and are continuous, in those cases parametric tests were performed. In the cases where data were not normal, transformations such as log were performed to approach normality. In the cases where data were not normal after transformation, or discrete, non-parametric tests were performed. Different levels of statistical significance were accessed based on specific P values and type I error cutoffs (0.05, 0.01, 0.001, 0.0001). Non-NGS standard analysis was performed using GraphPad Prism and RStudio. No data was excluded in this study.
Supplementary Material
Highlights.
Dead-guide RNA based CRISPR activation screen identifies targets for CAR-T engineering
PRODH2 gain-of-function reprograms proline metabolism in CD8+ T cells
PRODH2 re-shapes transcriptome, metabolome, and immune functions in CAR-T cells
PRODH2 reprogramming enhances CAR-T cell therapy both in in vitro and in vivo settings
Acknowledgments
We thank Drs. Krause, Isufi, Seropian, LoRusso, Fuchs and Herbst for discussions. We thank Meizhu Bai and Sung Yeon for critically reading the manuscript. We thank all members in Chen laboratory, as well as various colleagues in Department of Genetics, Systems Biology Institute, Cancer Systems Biology Center, MCGD Program, Immunobiology Program, BBS Program, Cancer Center, Stem Cell Center, Liver Center, RNA Center and Center for Biomedical Data Sciences at Yale for assistance and/or discussion. We thank M Graham, X Liu, T Wu, S Ren, R Kibbey, P Smith, G Wang, C Castaldi and other colleagues in the Center for Genome Analysis, Center for Molecular Discovery, Pathology Tissue Services, Histology Services, Electron Microscopy, CyTOF Facility, Islet, Oxygen consumption, Mass Isotopomer flux Core, High Performance Computing Center, West Campus Analytical Chemistry Core, West Campus Imaging Core, Animal Resource Center, and Keck Biotechnology Resource Laboratory at Yale, for technical support.
S.C. is supported by Yale SBI/Genetics Startup Fund, NIH/NCI/NIDA (DP2CA238295, R01CA231112, U54CA209992-8697, R33CA225498, RF1DA048811, P50CA196530-A10805, P50CA121974-A08306), DoD (W81XWH-17-1-0235, W81XWH-20-1-0072, W81XWH-21-1-0514), Damon Runyon Dale Frey Award (DFS-13-15), Melanoma Research Alliance (412806, 16-003524), St-Baldrick’s Foundation (426685), Breast Cancer Alliance, Cancer Research Institute (CLIP), AACR (499395, 17-20-01-CHEN), The Mary Kay Foundation (017-81), The V Foundation (V2017-022), Alliance for Cancer Gene Therapy, Sontag Foundation (DSA), Pershing Square Sohn Cancer Research Alliance, Dexter Lu, Ludwig Family Foundation, Blavatnik Family Foundation, and Chenevert Family Foundation. R.M. is supported by NIH/NIAID Human Immunology Project Consortium (HIPC) program (U19AI089992). MBD, RDC and JJP are supported by the Yale MSTP training grant from NIH (T32GM007205). RDC is also supported by an NIH/NCI fellowship (F30CA250249). GW is supported by CRI Irvington and RJ Anderson Postdoctoral Fellowships. X.D is supported by Revson Postdoctoral Fellowship.
Footnotes
Declaration of Interests
SC is a co-founder of EvolveImmune Tx. A patent has been filed by Yale University related to this study (SC, LY, JP, LP, RDC and MBD as inventors). Other authors declare no competing interest.
ADDITIONAL RESOURCES
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, et al. (2016). Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 45, 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brown CE, and Mackall CL (2019). CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol 19, 73–74. [DOI] [PubMed] [Google Scholar]
- Buck MD, O'Sullivan D, Geltink RIK, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJW, et al. (2016). Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, and Pearce EL (2016). Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol 17, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen QY, Gindin M, Gubin MM, van der Windt GJW, et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Wherry EJ, and Goldrath AW (2014). Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15, 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Belanger S, Frederick MA, Li B, Johnston RJ, Xiao N, Liu YC, Sharma S, Peters B, Rao A, et al. (2014). In vivo RNA interference screens identify regulators of antiviral CD4(+) and CD8(+) T cell differentiation. Immunity 41, 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi S, Weiss A, and Wang H (2016). A CRISPR-Based Toolbox for Studying T Cell Signal Transduction. Biomed Res Int 2016, 5052369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Collins JJ, and Wong WW (2018). Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 173, 1426–1438 e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li SZ, Bourque G, Wishart DS, and Xia JG (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SK, Pandhare J, Donald SP, and Phang JM (2008). A novel function for hydroxyproline oxidase in apoptosis through generation of reactive oxygen species. J Biol Chem 283, 10485–10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S, Johnston RJ, and Schoenberger SP (2010). Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol 11, 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman JE, Abudayyeh OO, Joung J, Gootenberg JS, Zhang F, and Konermann S (2015). Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nature Biotechnology 33, 1159–U1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, Park JJ, Kim HR, Errami Y, Guzman CD, et al. (2019). Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 178, 1189–1204 e1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gonen M, and Sadelain M (2017). Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstnerič V, Oven I, Ogorevc J, Lainšček D, Praznik A, Lebar T, Jerala R, and Horvat S (2019). CRISPRa-mediated FOXP3 gene upregulation in mammalian cells. Cell & Bioscience 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CJ, Hammerman PS, and Thompson CB (2005). Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol 5, 844–852. [DOI] [PubMed] [Google Scholar]
- Funck D, Eckard S, and Muller G (2010). Non-redundant functions of two proline dehydrogenase isoforms in Arabidopsis. BMC Plant Biol 10, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et al. (2016). L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167, 829–842 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geltink RIK, Kyle RL, and Pearce EL (2018). Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annual Review of Immunology, Vol 36 36, 461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, Albon SJ, Casanovas-Company J, Castro F, and Popova B (2019). Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nature medicine 25, 1408–1414. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange M, Buferne M, Verdeil G, Leserman L, Schmitt-Verhulst AM, and Auphan-Anezin N (2012). Activated STAT5 Promotes Long-Lived Cytotoxic CD8(+) T Cells That Induce Regression of Autochthonous Melanoma. Cancer Res 72, 76–87. [DOI] [PubMed] [Google Scholar]
- Grange M, Verdeil G, Arnoux F, Griffon A, Spicuglia S, Maurizio J, Buferne M, Schmitt-Verhulst AM, and Auphan-Anezin N (2013). Active STAT5 Regulates T-bet and Eomesodermin Expression in CD8 T Cells and Imprints a T-bet-Dependent Tc1 Program with Repressed IL-6/TGF-beta 1 Signaling. J Immunol 191, 3712–3724. [DOI] [PubMed] [Google Scholar]
- Harambat J, Fargue S, Bacchetta J, Acquaviva C, and Cochat P (2011). Primary hyperoxaluria. Int J Nephrol 2011, 864580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TSP, Painter MW, and Project IG (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Henriksson J, Chen X, Gomes T, Ullah U, Meyer KB, Miragaia R, Duddy G, Pramanik J, Yusa K, Lahesmaa R, et al. (2019). Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell 176, 882–896 e818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Morgensztern D, and Boshoff C (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. [DOI] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR (1994). T-Cell Receptor Antagonist Peptides Induce Positive Selection. Cell 76, 17–27. [DOI] [PubMed] [Google Scholar]
- Hollingsworth RE, and Jansen K (2019). Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe B, Beck BB, and Milliner DS (2009). The primary hyperoxalurias. Kidney Int 75, 1264–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jiang J, Johnson LC, Knight J, Callahan MF, Riedel TJ, Holmes RP, and Lowther WT (2012). Metabolism of [13C5]hydroxyproline in vitro and in vivo: implications for primary hyperoxaluria. Am J Physiol Gastrointest Liver Physiol 302, G637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Engreitz JM, Konermann S, Abudayyeh OO, Verdine VK, Aguet F, Gootenberg JS, Anjana NES, Wright JB, Fulco CP, et al. (2017). Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood (vol 548, pg 343, 2017). Nature 549, 418–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, O'Connor RS, Kawalekar OU, Ghassemi S, and Milone MC (2018). CAR T cell immunotherapy for human cancer. Science 359, 1361–1365. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., Patel PR, Guedan S, Scholler J, Keith B, et al. (2016). Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 44, 380–390. [DOI] [PubMed] [Google Scholar]
- Kiani S, Chavez A, Tuttle M, Hal RN, Chari R, Ter-Ovanesyan D, Qian J, Pruitt BW, Beal J, Vora S, et al. (2015). Cas9 gRNA engineering for genome editing, activation and repression. Nature Methods 12, 1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J, Easter LH, Neiberg R, Assimos DG, and Holmes RP (2009). Increased protein intake on controlled oxalate diets does not increase urinary oxalate excretion. Urol Res 37, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–U332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFleur MW, Nguyen TH, Coxe MA, Yates KB, Trombley JD, Weiss SA, Brown FD, Gillis JE, Coxe DJ, Doench JG, et al. (2019). A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nat Commun 10, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay A, Cabassa-Hourton C, Eubel H, Maldiney R, Guivarc'h A, Crilat E, Planchais S, Lacoste J, Bordenave-Jacquemin M, Clement G, et al. (2019). Proline oxidation fuels mitochondrial respiration during dark-induced leaf senescence in Arabidopsis thaliana. J Exp Bot 70, 6203–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao HK, Hatanaka F, Araoka T, Reddy P, Wu MZ, Sui Y, Yamauchi T, Sakurai M, O'Keefe DD, Nunez-Delicado E, et al. (2017). In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell 171, 1495–1507 e1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WA, and June CH (2017). The Principles of Engineering Immune Cells to Treat Cancer. Cell 168, 724–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al. (2019). c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Dichwalkar T, Chang JY, Cossette B, Garafola D, Zhang AQ, Fichter M, Wang C, Liang S, and Silva M (2019). Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 365, 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, and Rathmell JC (2013). Metabolic regulation of T lymphocytes. Annu Rev Immunol 31, 259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzner RG, and Mackall CL (2019). Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 25, 1341–1355. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17, 10–12. [Google Scholar]
- Moran AE, Kovacsovics-Bankowski M, and Weinberg AD (2013). The TNFRs OX40, 4–1BB, and CD40 as targets for cancer immunotherapy. Curr Opin Immunol 25, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, et al. (2017). Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet 49, 1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat Rev Immunol 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters PJ, Borst J, Oorschot V, Fukuda M, Krahenbuhl O, Tschopp J, Slot JW, and Geuze HJ (1991). Cytotoxic Lymphocyte-T Granules Are Secretory Lysosomes, Containing Both Perforin and Granzymes. J Exp Med 173, 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang JM, Liu W, and Zabirnyk O (2010). Proline metabolism and microenvironmental stress. Annu Rev Nutr 30, 441–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 14, 687–690. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen SD, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A (2012). Tumor Immunotherapy Directed at PD-1. New Engl J Med 366, 2517–2519. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, and Restifo NP (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, and Lim WA (2017). Synthetic Immunology: Hacking Immune Cells to Expand Their Therapeutic Capabilities. Annu Rev Immunol 35, 229–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Riviere I, and Riddell S (2017). Therapeutic T cell engineering. Nature 545, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saoudi A, Kassem S, Dejean A, and Gaud G (2014). Rho-GTPases as key regulators of T lymphocyte biology. Small GTPases 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, and Mei Z (2011). CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. The Journal of clinical investigation 121, 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, Ha B, Altay G, Greenbaum JA, McVicker G, et al. (2018). Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 175, 1701–1715 e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jager U, Jaglowski S, Andreadis C, Westin JR, et al. (2019). Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 380, 45–56. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, and Zhang F (2015). High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 16, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971 e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, Labao-Almeida C, Ali LR, Quijano-Rubio A, Ruterbusch M, Leung I, et al. (2019). De novo design of potent and selective mimics of IL-2 and IL-15. Nature 565, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov DR, Gowen BG, Boontanrart M, Roth TL, Gagnon JD, Mumbach MR, Satpathy AT, Lee Y, Bray NL, Chan AY, et al. (2017). Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et al. (2016). The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr Protoc Bioinformatics 54, 1 30 31–31 30 33. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summitt CB, Johnson LC, Jonsson TJ, Parsonage D, Holmes RP, and Lowther WT (2015). Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem J 466, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, Wang X, Cho JH, Wang H, Rankin S, et al. (2017). Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 46, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Pearce L, O'Donnell-Tormey J, and Hubbard-Lucey VM (2018). Trends in the global immuno-oncology landscape. Nat Rev Drug Discov 17, 922. [DOI] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, et al. (2017). A subcellular map of the human proteome. Science 356. [DOI] [PubMed] [Google Scholar]
- Ting PY, Parker AE, Lee JS, Trussell C, Sharif O, Luna F, Federe G, Barnes SW, Walker JR, Vance J, et al. (2018). Guide Swap enables genome-scale pooled CRISPR-Cas9 screening in human primary cells. Nat Methods 15, 941–946. [DOI] [PubMed] [Google Scholar]
- Trapani JA, and Smyth MJ (2002). Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol 2, 735–747. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- van der Windt GJW, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, and Pearce EL (2012). Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8(+) T Cell Memory Development. Immunity 36, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P, and Ferrando A (2012). The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest 122, 3398–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, Wang Y, Wang YD, Qian C, Xu B, et al. (2019). Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Psychogios N, Young N, and Wishart DS (2009). MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res 37, W652–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia JG, Sinelnikov IV, Han B, and Wishart DS (2015). MetaboAnalyst 3.0-making metabolomics more meaningful. Nucleic Acids Res 43, W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Yu J, Hubbard-Lucey VM, and Tang J (2019). The global pipeline of cell therapies for cancer. Nat Rev Drug Discov 18, 821–822. [DOI] [PubMed] [Google Scholar]
- Xiong GF, Deng L, Zhu JQ, Rychahou PG, and Xu R (2014). Prolyl-4-hydroxylase a subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. Bmc Cancer 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong GF, Stewart RL, Chen J, Gao TY, Scott TL, Samayoa LM, O'Connor K, Lane AN, and Xu R (2018). Collagen prolyl 4-hydroxylase 1 is essential for HIF-1 alpha stabilization and TNBC chemoresistance. Nature Communications 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, Du Y, Guo J, Dai X, Wang G, et al. (2019). In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat Biotechnol 37, 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, Guo X, Liu H, Ding N, Zhang T, et al. (2019). A safe and potent anti-CD19 CAR T cell therapy. Nat Med 25, 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, and Asara JM (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, Cremasco V, Dougan SK, Cowley GS, Elpek K, et al. (2014). In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 506, 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomic sequencing raw data, the metabolomics raw data, and CyTOF raw data have been deposited at SRA, Metabolomics Workbench, and Mendeley data, respectively, and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table.
The original codes of data analysis are available from the lead contact upon reasonable request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. The unprocessed data are deposited into public archives as above.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ultra-LEAF™ Purified anti-mouse CD3ε Antibody (clone: 145–2C11) | Biolegend | Cat#100340 |
| Ultra-LEAF(TM) Purified anti-mouse CD28 Antibody (clone: 37.51) | Biolegend | Cat#102116 |
| APC anti-human CD3 Antibody (Clone: UCHT1) | Biolegend | Cat#300412 |
| FITC anti-human IgG Fc | Biolegend | Cat#409310 |
| PE anti-human IgG Fc (HP6017) | Biolegend | Cat#409304 |
| PE anti-DYKDDDDK Tag Antibody (L5) | Biolegend | Cat#637310 |
| APC/Cy7 anti-human CD8a antibody (HIT8a) | Biolegend | Cat#300926 |
| APC anti-human CD3 Antibody (Clone: HIT3a) | Biolegend | Cat#300312 |
| APC anti-mouse CD8a Antibody (53–6.7) | Biolegend | Cat#100712 |
| PE/Cy7 anti-mouse CD3ε Antibody (145–2C11) | Biolegend | Cat#100320 |
| PE anti-rat CD90/mouse CD90.1 (Thy-1.1) Antibody (OX-7) | Biolegend | Cat#202524 |
| PE anti-mouse CD107a (LAMP-1) Antibody (1D4B) | Biolegend | Cat#121612 |
| PE conjungate Cleaved Caspase-3 (Asp175) Rabbit mAb (5A1E) | CST | Cat#9978S |
| Brilliant Violet 510™ anti-human CD8 Antibody (SK1) | Biolegend | Cat#344732 |
| APC/Cy7 anti-human IFN-γ Antibody (4S.B3) | Biolegend | Cat#502530 |
| PE anti-human TNF-α Antibody (MAb11) | Biolegend | Cat#502909 |
| PerCP/Cyanine5.5 anti-human/mouse Granzyme B Recombinant Antibody (QA16A02) | Biolegend | Cat#372212 |
| APC anti-humanCD45RA Antibody (HI100) | Biolegend | Cat#304112 |
| PE/Cy7 anti-human CD62L Antibody (DREG-56) | Biolegend | Cat#304822 |
| PerCP/Cyanine5.5 anti-human CD197 (CCR7) Antibody (Clone: G043H7) | Biolegend | Cat#353219 |
| APC anti-human CD127 (IL-7Rα) Antibody (Clone: A019D5) | Biolegend | Cat#351315 |
| FITC anti-human CD183 (CXCR3) Antibody (Clone: G025H7) | Biolegend | Cat#353703 |
| EOMES Monoclonal Antibody (WD1928), PECyanine7 | eBioscience™ | Cat#25–4877–42 |
| APC anti-T-bet Antibody (Clone: 4B10) | Biolegend | Cat#644813 |
| PE anti-TCF1 (TCF7) Antibody (Clone: 7F11A10 ) | Biolegend | Cat#655207 |
| FITC anti-human/mouse Bcl-6 Antibody (Clone: 7D1) | Biolegend | Cat#358513 |
| APC anti-human Notch 1 Antibody (Clone: MHN1–519 ) | Biolegend | Cat#352107 |
| PerCP/Cyanine5.5 anti-GATA3 Antibody (Clone: 16E10A23) | Biolegend | Cat#653811 |
| RUNX1 Monoclonal Antibody (RXDMC), PE | eBioscience™ | Cat#12–9816–80 |
| Anti-human CTLA4 (CD152)-161Dy | FLUIDIGM | Cat#3161004B |
| Anti-human TIM3–153Eu | Yale CyTOF CORE | |
| Anti-human CD278/ICOS-168Er | FLUIDIGM | Cat#3168024B |
| Anti-human TNFRSF9/CD137–173Yb | FLUIDIGM | Cat#3173015B |
| Anti-human IL2–166Er | FLUIDIGM | Cat#3158007B |
| Anti-human TNF-α-152Sm | FLUIDIGM | Cat#3152001B |
| Anti-human IFN-γ-165Ho | FLUIDIGM | Cat#3165002B |
| Anti-human CXCR3–163Dy | FLUIDIGM | Cat#3163004B |
| Anti-human CD62L-174Yb | Yale CYTOF Core | Cat#V00751 |
| Anti-human Perforin-176Yb | Yale CYTOF Core | |
| Anti-human CD45RA-169Tm | FLUIDIGM | Cat#3143006B |
| Anti-human CD8a-146Nd | FLUIDIGM | Cat#3146001B |
| Anti-human CD45RO-164Dy | FLUIDIGM | Cat#3164007B |
| Anti-human CD3–170Er | FLUIDIGM | Cat#3170001B |
| Anti-human CD45–89Y | FLUIDIGM | Cat#3089003B |
| Anti-Cleaved Caspase3–172Yb | Yale CYTOF Core | |
| Anti-human CD25–149Sm | FLUIDIGM | Cat#3149010B |
| Anti-human CD134(OX40)-150Nd | Yale CYTOF Core | |
| Anti-human LAG3–175Lu | Yale CYTOF Core | |
| Recombinant Human Siglec-2/CD22 Fc Chimera Protein, CF | R&D | Cat#1968-SL-050 |
| Recombinant Human BCMA/TNFRSF17 Fc Chimera Protein, CF | R&D | Cat#193-BC-050 |
| Dynabeads Human T-Activator CD3/CD28 | Invitrogen | Cat#11131D |
| Anti-human PRODH2 | Atlas Antibodies | Cat#HPA051287 |
| Anti-human Vinculin | Abcam | Cat#129002 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemical Competent E. coli | ThermoFisher | Cat#C737303 |
| Endura™ ElectroCompetent Cells | Lucigen | Cat#60242–2 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| PEI MAX -Transfection Grade Linear Polyethylenimine Hydrochloride (MW 40,000) | Polyscience | Cat#24765–1 |
| DPBS, no calcium, no magnesium | Gibco | Cat#14190136 |
| RPMI 1640 Medium | Gibco | Cat#11875–093 |
| Fetal Bovine Serum | Sigma Aldrich | Cat#F4135–500ML |
| DMEM, high glucose, pyruvate | Gibco | Cat#11995065 |
| Opti-MEM™ I Reduced Serum Medium | ThermoFisher | Cat#31985070 |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | Cat#15140122 |
| Lonza™ BioWhittaker™ L-Glutamine (200mM) | Lonza | Cat#BW17605E |
| 2-Mercaptoethanol | Sigma Aldrich | Cat#M6250–10ML |
| X-VIVO 15 Serum-free Hematopoietic Cell Medium | Lonza | Cat#BE02–060F |
| Human AB Serum; Male Donors; type AB | MP Biomedical | Cat#092930949 |
| ACK Lysing Buffer | Lonza | Cat#10–548E |
| Naïve CD8a+ T Cell Isolation Kit, mouse | Miltenyi | Cat#130–096-543 |
| LS Columns | Miltenyi | Cat#130–042-401 |
| Recombinant Mouse IL-2 (carrier-free) | Biolegend | Cat#575404 |
| Recombinant Mouse IL-7 (carrier-free) | Biolegend | Cat#577802 |
| Recombinant Mouse IL-12 (p70) (carrier-free) | Biolegend | Cat#577008 |
| Recombinant Mouse IL-15 (carrier-free) | Biolegend | Cat#566302 |
| Recombinant Human IL-2 (carrier-free) | Biolegend | Cat#589104 |
| Monensin Solution (1,000x) | Biolegend | Cat#420701 |
| Brefeldin A Solution (1,000X) | Biolegend | Cat#420601 |
| QuickExtract DNA Extraction Solution | Epicenter | Cat#QE09050 |
| Proteinase K | Qiagen | Cat#19131 |
| Pierce™ Universal Nuclease for Cell Lysis | ThermoFisher | Cat#88702 |
| RNase A | Qiagen | Cat#19101 |
| PEG 8000, Molecular Biology Grade | Promega | Cat#V3011 |
| Sodium chloride | Sigma | Cat#S3014 |
| Gibson Assembly® Master Mix | NEB | Cat#E2611 |
| Phusion Flash High-Fidelity PCR Master Mix | ThermoFisher | Cat#F548L |
| DreamTaq Green PCR Master Mix (2X) | ThermoFisher | Cat#K1082 |
| E-Gel™ Low Range Quantitative DNA Ladder | ThermoFisher | Cat#12373031 |
| Fixation/Permeabilization Solution Kit | BD | Cat#554714 |
| SuperScript™ IV Reverse Transcriptase | Invitrogen | Cat#18090050 |
| Random Hexamers (50 μM) | Invitrogen | Cat#N8080127 |
| NEBNext® Ultra™ RNA Library Prep Kit for Illumina® | NEB | Cat#E7530S |
| NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1) | NEB | Cat#E7335S |
| TaqMan™ Fast Advanced Master Mix | Invitrogen | Cat#4444557 |
| QIAquick Gel Extraction Kit | Qiagen | Cat#28706 |
| QIAamp Fast DNA Tissue Kit | Qiagen | Cat#51404 |
| EndoFree® Plasmid Maxi Kit | Qiagen | Cat#12362 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Relative Human Mitochondrial DNA Copy Number Quantification qPCR Assay Kit | ScienCell | Catalog#8938 |
| Human IL-2 ELISA Kit | Millipore Sigma | Cat#RAB0286 |
| IL-7 Human ELISA Kit | ThermoFisher | Catalog#EHIL7 |
| IL-15 Human ELISA Kit | ThermoFisher | Catalog#BMS2106 |
| Agilent Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat#103015–100 |
| Agilent Seahorse XF Calibrant solution | Agilent | Cat#100840–000 |
| Agilent Seahorse XF RPMI medium | Agilent | Cat#103576–100 |
| Agilent Seahorse XF sensor cartridges and cell culture microplates | Agilent | Cat#101085–004 |
| Poly-D-lysine | Sigma | Cat#27964–99–4 |
| Seahorse XF 100 mM Pyruvate Solution | Agilent | Cat#103578–100 |
| Seahorse XF 1.0 M Glucose Solution | Agilent | Cat#103577–100 |
| Seahorse XF 200 mM Glutamine Solution | Agilent | Cat#103579–100 |
| Bovine Serum Albumin | Sigma Aldrich | Cat#A9418–100G |
| EDTA | Sigma Aldrich | Cat#E8008–100ML |
| XenoLight D-Luciferin – K+ Salt Bioluminescent Substrate | Perkin Elmer | Cat#122799 |
| Neon™ Transfection System 100 μL Kit | Invitrogen | Cat#MPK10025 |
| HiFi Cas9 protein | IDT | Cat#1081061 |
| Cas9 tracrRNA | IDT | Cat#1072532 |
| crRNA | IDT | Custom, sequence specific |
| Standard metabolites list | Sigma | Table S6 |
| OVA 257–264 | Invivogen | Cat#vac-sin |
| BpiI (BbsI) (10 U/μL) | ThermoFisher | Cat#ER1012 |
| Esp3I (BsmBI) (10 U/μL) | ThermoFisher | Cat#ER0451 |
| 4-Hydroxyproline | Sigma | Cat#H54409 |
| Acetyl CoA | Sigma | Cat#A2056 |
| ADP | Sigma | Cat#A2754 |
| Amino acid standards, physiological | Sigma | Cat#A9906 |
| AMP | Sigma | Cat#1930 |
| Arginine | Sigma | Cat#A5006 |
| ATP | Sigma | Cat#A26209 |
| cAMP | Sigma | Cat#A9501 |
| Choline | Sigma | Cat#C7017 |
| CoA | Sigma | Cat#C4282 |
| CTP | Sigma | Cat#C1506 |
| Fumarate | Sigma | Cat#D95654 |
| Glucose | Sigma | Cat#G8270 |
| Glutathione | Sigma | Cat#G4251 |
| Glyceraldehyde3P | Sigma | Cat#G5251 |
| Glycerate3P | Sigma | Cat#P8877 |
| Glyoxylate | Sigma | Cat#G4502 |
| GTP | Sigma | Cat#G8877 |
| IMP | Sigma | Cat#I4625 |
| Malate | Sigma | Cat#46940 |
| Myo-Inositol | Sigma | Cat#I5125 |
| Orinithine | Sigma | Cat#57197 |
| Oxaloacetate | Sigma | Cat#O4216 |
| Palmitoyl CoA | Sigma | Cat#P9716 |
| Proline | Sigma | Cat#P3350000 |
| PRPP | Sigma | Cat#P8296 |
| Putrescine | Sigma | Cat#51799 |
| Pyruvate | Sigma | Cat#P2256 |
| Ribose5P | Sigma | Cat#83875 |
| Spermidine | Sigma | Cat#S2626 |
| Spermine | Sigma | Cat#S3256 |
| Sphingosine | Sigma | Cat#S7049 |
| Succinate | Sigma | Cat#W327700 |
| Tetrahydrofolic acid | Sigma | Cat#G5251 |
| TTP | Sigma | Cat#T0251 |
| UTP | Sigma | Cat#U6375 |
| Xylose-5-Phosphase | Sigma | Cat#78963 |
| α-Hydroxyglutaric acid | Sigma | Cat#90790 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| human CAR-T cell RNA-seq | This paper | PRJNA806391 |
| human CD8 T cell metabolomics | This paper | ST002085 |
| human CAR-T cell metabolomics | This paper | ST002084 |
| CyTOF data | This paper | DOI: 10.17632/pnbjdtdkfg.1 |
| Experimental Models: Cell Lines | ||
| HEK293FT | ThermoFisher | Catalog Number: R70007 |
| HEK293T | ATCC | Catalog Number: CRL-3216™ |
| NALM6 | ATCC | Catalog Number:3273 |
| MCF-7 | ATCC | Catalog Number: HTB-22 |
| E0771 | CH3 | Catalog Number: 940001 |
| MB-MDA-231 | ATCC | Catalog Number: HTB-26 |
| MM.1R | ATCC | Catalog Number:CRL-2975 |
| Human Peripheral Blood CD8+ T Cells | STEMCELL | Catalog Number: 70027 |
| Human Peripheral Blood Mononuclear Cells | STEMCELL | Catalog Number: 70025.1 |
| Experimental Models: Organisms/Strains | ||
| OT-I | Jackson Laboratory | Stock No: 003831 |
| NSG | Jackson Laboratory | Stock No: 005557 |
| Cas9ß | Platt et al., 2014 | Jackson Lab |
| OT-I;Cas9ß | Dong et al. 2019 Cell | Sidi Chen lab |
| Oligonucleotides | ||
| mm10dg library | This paper | Dataset S1 |
| Taqman probes | This paper | Table S3 |
| Amplicon primers for surveyor or Nextera | This paper | Table S2 |
| gBlocks of U6-dgRNA-MS2 | IDT | Custom, sequence specific |
| EFS-Thy1.1-MCP-p65-HSF1 | IDT | Custom, sequence specific |
| Prodh2 probe | ThermoFisher | Mm00457662-m1 |
| Actb probe | ThermoFisher | Mm00607939-s1 |
| Gapdh probe | ThermoFisher | Mm99999915_g1 |
| Wdr37 probe | ThermoFisher | Mm00552518-m1 |
| Srek1ip1 probe | ThermoFisher | Mm00482686-m1 |
| Ccnb1ip1 probe | ThermoFisher | Rn01414585-m1 |
| PRODH2 probe | ThermoFisher | Hs00560411-m1 |
| ACTB probe | ThermoFisher | Hs99999903-m1 |
| GAPDH probe | ThermoFisher | Hs02786624-g1 |
| Recombinant DNA | ||
| psPAX2 | Addgene | Cat#12260 |
| pMD2.G | Addgene | Cat#12259 |
| pRepCap6 (AAV6 capsid) | Addgene | Cat#110770 |
| pAdDeltaF6 (PDF6) | Addgene | Cat#112867 |
| TdgA Vector | This study | Sidi Chen lab |
| Mm10dgLib library | This study | Sidi Chen lab |
| Software and Algorithms | ||
| FlowJo software 9.9.6 | FlowJo, LLC | https://www.flowjo.com |
| Cutadapt | Martin et al., 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| DAVID | Huang et al., 2009 | https://david.ncifcrf.gov |
| Scran R package | Lun et al., 2016 | https://www.r-project.org |
| Rtsne R package | Maaten, 2014; Maaten and Hinton, 2008 | https://www.r-project.org |
| Edge R package | Robinson et al., 2010 | https://www.r-project.org |
| Kallisto | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| Sleuth | Pimentel et al., 2017 | https://pachterlab.github.io/sleuth/ |
| GSEA | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| PSCAN | Zambelli et al., 2009 | http://159.149.160.88/pscan/ |
| Bowtie 1.1.2 | Langmead et al., 2009 | http://bowtiebio.sourceforge.net |
| Agilent Mass Hunter Qualitative Analysis Software | Agilent | (Version B.07.0.0, build 7.0.7024.0). |
| Agilent Mass Profiler Professional | Agilent | (Version 14.5-Build 2772) |
| Metabolite database | HMDB | http://www.hmdb.ca/ |
| Metabolite database | METLIN | http://metlin.scripps.edu |
| MetaboAnalyst 4.0 | Chong et al., 2018; Xia et al., 2015 | https://www.metaboanalyst.ca |
| Pathvisio v3.3.0 | Pathvisio | https://www.pathvisio.org |
| KEGG | KEGG | http://www.genome.jp/kegg/ |
| Other | ||