

Abstract
Hepatic steatosis is the main characteristic of some liver metabolism diseases. However, unclear molecular mechanism of hepatic steatosis impedes the therapy of this hepatic steatosis. Glutathione-S-transferase mu 2 (GSTM2), as a member of phase II drug metabolizing enzymes (DMEs), regulates cellular antioxidant and detoxificant. GSTM2 was highly up-regulated in hepatic steatosis tissues and high-fat diet (HFD) fed mice. Loss-of-function GSTM2 mouse model demonstrated that GSTM2 protected mice from excess fat accumulation. Mechanistically, GSTM2 interacted with ASK1 and suppressed its phosphorylation and the activation of subsequent downstream p38-JNK signalling. Moreover, GSTM2 overexpression in the liver effectively ameliorated hepatic lipid accumulation. Therefore, we identified GSTM2 as an important negative regulator in progression of hepatic steatosis via both its detoxification/antioxidant and inhibition of ASK1-p38/JNK signalling. This study showed potential therapeutic function of the DME in progression of hepatic steatosis.
Subject terms: Cell biology, Lipids, Metabolic disorders
Jin et al. investigate the underlying mechanisms of hepatic steatosis and show that Glutathione-S-transferase mu 2 (GSTM2), which is a drug metabolizing enzyme (DME), is upregulated in hepatic steatosis tissue from mice fed with a high fat diet. They show that GSTM2 is a negative regulator of hepatic steatosis via both detoxification/antioxidant and inhibition of ASK1-p38/JNK signalling, which sheds light on its potential as a therapeutic target.
Introduction
Hepatic steatosis, characterized by excessive accumulation of triglycerides (TGs) in hepatocytes, is considered the cause of non‐alcoholic fatty liver disease (NAFLD)1–3. Disordered lipid metabolism leads to the hepatic steatosis causing lots of excess fat accumulation in hepatocytes. High hepatic fat content is the important inducement of the impairment of redox imbalance and insulin resistance. However, the molecular mechanisms of hepatic steatosis occurrence and progression are poorly understood.
Drug-metabolising enzymes (DMEs) play an important role in scavenging the waste products of lipid metabolism and oxidative metabolism and maintaining homeostasis of liver4,5, consisted of groups of enzymes, such as glutathione S-transferases (GSTs). Several studies reported DMEs could respond to the hepatic steatosis by protecting hepatocytes from free radicals6,7, additionally, GSTs are always considered as the scavenger of reactive oxygen species. Among other GSTs, glutathione S-transferase M2 (GSTM2) seems to be different. Previous researches from Huenchuguala et al. reported that they found cells with GSTM2 knockdown accumulated more lipid droplets8–11. Moreover, GSTM2 was highly up-regulated in high-fat diet mouse liver6,7. These studies suggested GSTM2 regulated lipid metabolism specially beyond detoxification/antioxidant, the basic functions of GSTs.
In the present study, the close association between GSTM2 knockout and hepatic steatosis was examined by using GSTM2-null mice. We demonstrated that GSTM2 protects against hepatic steatosis by inhibiting excess fat accumulation by interacting with and suppressing activation of apoptosis signal-regulating kinase1 (ASK1) and subsequent p38-JNK signalling, besides its antioxidant capacity. Moreover, GSTM2 overexpression reversed methionine choline-deficient diet (MCDD)-induced steatosis. Thus, we identified GSTM2 as an important negative regulator in hepatic steatosis progress.
Results
GSTM2 was upregulated in hepatic tissues of mice fed HFD and MCDD
The high-fat diet and methionine choline deficient diet were used to make hepatic steatosis. We validated the expression pattern of GSTM2 in the HFD and MCDD mouse models. The level of GSTM2 mRNA and protein were detected. GSTM2 was significantly up-regulated in mice fed HFD (Fig. 1a–c) and MCDD (Fig. 1d–f).
Fig. 1. Loss-of-function of GSTM2 promoted hepatic fat storage.

a–c GSTM2 mRNA and protein expression in liver samples of 8-week HFD fed mice (n = 3) and 8-week chow diet fed mice (n = 3); *p < 0.05. d–f GSTM2 mRNA and protein expression in liver samples of MCDD fed mice and corresponding control mice; *p < 0.05. GAPDH served as the reference gene. g Haematoxylin and eosin (HE) and Oil red O staining analysis of liver samples of GSTM2 knockout (KO) and control mice; bar = 20 μm. h Hepatic TG concentrations of WT (n = 7) and GSTM2 KO (n = 10) mice were detected; ***p < 0.001. i Oil red O analysis of liver samples of GSTM2 KO and control mice challenged with HFD at 0 d, 5 d, 10 d, 20 d, and 30 d. j Hepatic TG concentrations of WT and GSTM2 KO mice with high-fat diet treatment at 0 d, 5 d, 10 d, 20 d, and 30 d; *p < 0.05. bar, 50 μm.
GSTM2 deletion strongly promotes hepatic fat accumulation
To investigate the function of GSTM2 in the progression of hepatic steatosis, we generated a GSTM2 knockout (GSTM2 KO) mouse (Supplementary Fig. 1a, b). Hepatic histomorphology and TG content detection showed that GSTM2 KO mice had higher hepatic fat content (Fig. 1g, h). Moreover, there was no significant difference in the ratio of liver weight to body weight between GSTM2 KO and WT mice (Supplementary Fig. 1c, d). We also investigated the fat content in white adipose and skeletal muscle tissues via Oil Red O staining. More intramuscular fat was observed in GSTM2 KO mice, whereas no significant difference was found in white adipose tissue between KO and WT mice (Supplementary Fig. 1e, f). To further investigate the function of GSTM2 in hepatic lipid metabolism, we challenged the mice with HFD. KO mice accumulated more fat at a faster rate than WT mice (Fig. 1i, j, Supplementary Fig. 1g). Moreover, the hepatic alanine aminotransferase (ALT) level was higher in KO mice, while the γ-glutamyltranspetidase (GGT) level was not changed (Supplementary Fig. 1h, i). We next challenged the mice with 0.2 mM oleic acid medium (dissolved by BSA in 0.9% NaCl solution) to investigate the effect of GSTM2 on the rapid formation of LDs. Hepatic morphology analysis showed that GSTM2-KO-livers formed more LDs than WT livers (Supplementary Fig. 1j, k). These results suggest that GSTM2 plays an important role in excess hepatic fat accumulation.
GSTM2 overexpression suppresses hepatic steatosis
Although GSTM2 was up-regulated in mice with hepatic steatosis, we also overexpressed this protein in mouse model fed with MCDD (Supplementary Fig. 2a). Western blot analysis showed that GSTM2 was highly expressed in liver tissues (Supplementary Fig. 2b). As the feeding time increased, control mice accumulated much more hepatic fat, while GSTM2 overexpression suppressed hepatic fat accumulation (Fig. 2a). Control mice showed significant hepatic steatosis after 1 week of MCDD feeding, whereas mice with GSTM2 overexpression showed this effect at 4 weeks. Additionally, GSTM2-overexpressing mice showed mild LD accumulation in the first 3 weeks (Fig. 2b). Because fibrosis is the key signal of steatosis aggravation, we further detected the fibrosis level in liver tissues. Masson staining analysis indicated that control mice had significant hepatic fibrosis compared to mice with GSTM2 overexpression (Supplementary Fig. 2c, d). A significant degree of fibrosis was observed in liver tissues of control mice at 3 weeks, whereas GSTM2-overexpressing mice showed mild fibrosis at 8 weeks (Supplementary Fig. 2c, d). Moreover, we detected the marker genes of fibrosis α-SMA and CoL1A1. The results showed the expression of α-SMA and CoL1A1 was up-regulated at 8 weeks significantly (Supplementary Fig. 2e). These results suggest that GSTM2 overexpression inhibits hepatic steatosis development. We subsequently performed a rescue test. A GSTM2 overexpression vector was transfected into mice fed MCDD for 2 weeks, which was then changed to a chow diet (Supplementary Fig. 2f). Overexpression efficiency was detected by Western blotting (Supplementary Fig. 2g). As expected, GSTM2 overexpression promoted the recovery of hepatic steatosis compared to control (Fig. 2c). Although both GSTM2-overexpressing mice and control mice showed good recovery of hepatic steatosis at 2 weeks, GSTM2-overexpressing mice showed lower hepatic fat content at 1 week (Fig. 2d). The rescue experiment suggests that GSTM2 may be a useful therapeutic target for hepatic steatosis.
Fig. 2. GSTM2 overexpression protected against hepatic fat storage.

a HE staining analysis of liver samples of GSTM2 overexpression (OE) and control mice challenged with MCDD at 1 w, 2 w, 3 w, 4 w, 6 w, and 8 w. b Hepatic TG concentrations of GSTM2 OE and control mice with MCD diet treatment at 0 w, 1 w, 2 w, 3 w, 4 w, 6 w, and 8 w; *p < 0.05. c Oil red O staining analysis of liver samples of GSTM2 OE and control mice fed MCDD for 2 w and then chow diet. d Hepatic TG concentrations of corresponding mice in (c); *p < 0.05. bar, 50 μm.
GSTM2 modulates size and number of LDs in HepG2 cells
To further investigate the potential regulatory mechanism, we generated a GSTM2-knockdown stable cell line (shGSTM2-HepG2) that expressed less GSTM2 mRNA and protein than control cells (shNC-HepG2) (Fig. 3a, b). Interestingly, shGSTM2-HepG2 cells contained more LDs (~5 to 6 fold) in normal culture than shNC-HepG2 cells (Fig. 3c, d) and formed more and larger LDs (>2 μm) under fatty acid (FA)-rich (200 μM oleic acid medium) culture conditions (Fig. 3e, f).
Fig. 3. GSTM2 modulated cellular lipid droplets accumulation via regulating cellular ROS level.

a, b Expression level of GSTM2 was detected in shGSTM2-HepG2 cells and corresponding control cells. c More LDs were observed in shGSTM2-HepG2 cells. d Statistics of LD number in shGSTM2-HepG2 cells and shNC-HepG2 cells. e More larger LDs were found in shGSTM2-HepG2 cells treated with 1 mM oleic acid medium for 12 h compared to control cells. f Statistics of LD diameter in shGSTM2-HepG2 cells and shNC-HepG2 cells treated with 1 mM oleic acid medium for 12 h compared to control cells. g Cellular ROS level was detected in shGSTM2-HepG2 and shNC-HepG2 cells in the presence or absence of oleic acid medium; *p < 0.05, **p < 0.01. h More LDs were observed in cells treated with ROSup compared to control cells. i Statistics of LD number in cells treated with ROSup and control cells. Bar, 10 μm.
Effects of GSTM2 on LD content do not absolutely depend on regulating ROS level
Because GSTM2 has antioxidant activity (as described in the Introduction), the potential effect of ROS on cellular lipid formation should be examined. We determined ROS levels in shGSTM2-HepG2 and shNC-HepG2 cells in normal and FA-rich culture medium, respectively. As expected, the ROS level was significantly increased (~15–20%) by GSTM2 knockdown (p < 0.01) (Fig. 3g). We further investigated whether the increased cellular ROS levels affected the LD content by treating cells with an ROS activator (ROSup). As predicted, more cellular LDs (~2 to 3 fold compared to control) were observed in ROSup-treated cells (Fig. 3h, i). Indeed, the higher ROS level increased the LD number, but only approximately 30 LDs were observed in ROSup-treated cells (Fig. 3i), which was much lower than the number detected in shGSTM2-HepG2 cells. Therefore, the increase in ROS did not greatly alter the cellular LD content.
We next investigated the changes in LD formation and expression of breakdown-related genes at different ROS levels. The expression of PLIN2, PLIN5, and ATGL was significantly increased by ROSup treatment (p < 0.05) (Supplementary Fig. 3a). PLIN2, PLIN3, and SREBF1 increased and PCYT1A decreased significantly following GSTM2 knockdown (p < 0.05) (Supplementary Fig. 3b). As expected, GSTM2 overexpression significantly reduced the cellular ROS level (Supplementary Fig. 3c); therefore, we examined gene expression changes after GSTM2 overexpression. In contrast to the results of GSTM2 knockdown, PLIN2 and SREBF1 were down-regulated and PCYT1A was up-regulated, showing significant differences (p < 0.05) (Supplementary Fig. 3d). Because N-acetylcysteine (NAC) is an effective ROS scavenger (Supplementary Fig. 3e), we investigated whether a reduced ROS level would alter the LD content by treating cells with 1 mM NAC. Interestingly, NAC did not significantly change the number of LDs in either normal or FA-rich cultural medium (Supplementary Fig. 3f, g). To evaluate the contribution of the GSTM2 oxidative ability in promoting LD formation, we added ROS scavenger (NAC) in GSTM2 knockdown cells and detected the LD number. The result showed that NAC did suppress the LD number induced by GSTM2 knockdown (Supplementary Fig. 3h). However, the LD number in NAC treatment group was still more than NC group (Supplementary Fig. 3h). These results suggest that modulating ROS levels by GSTM2 is not the only approach for regulating the cellular lipid content.
GSTM2 modulates p38-JNK signalling by inhibiting ASK1 activity
ASK1-p38/JNK signalling plays an important role in NAFLD development, thus we investigated whether this signalling was affected by GSTM2 expression. The phosphorylation level of ASK1 was regulated by GSTM2. The p-ASK1 level was increased by GSTM2 knockdown and decreased by GSTM2 overexpression (Fig. 4a, b). Moreover, GSTM2 knockdown and overexpression had different effects on p38-JNK signalling. The phosphorylation levels of p38 and JNK were increased by GSTM2 knockdown, but decreased by GSTM2 overexpression (Fig. 4d). We further examined the effect of GSTM2 on ASK1-p38/JNK signalling in the presence of palmitic acid (PA), an ASK1 activator. As expected, GSTM2 overexpression reduced ASK1 signalling activation (Fig. 4e). AKS1 signalling was activated and enhanced in liver tissues with the development of NAFLD12, which leads to severe inflammation and fibrosis. This result suggests that GSTM2 has a potential therapeutic function in NAFLD.
Fig. 4. GSTM2 regulated lipid droplet accumulation via ASK1-p38/JNK signalling.

a, b Phosphorylation of ASK1 was up-regulated by GSTM2 KD and down-regulated by GSTM2 OE. c, d p-p38 and p-JNK levels were up-regulated by GSTM2 knockdown (KD) and down-regulated by GSTM2 OE. e GSTM2 OE down-regulated ASK1-p38/JNK signalling in the presence of palmitic acid treatment. f GSTM2 interacted with ASK1 by co-IP detection. g GSTM2 C-terminal, not N-terminal, down-regulated ASK1 activity. h SREBF1 and PLIN2 expression level was up-regulated by GSTM2 KD. i SREBF1 and PLIN2 expression level was down-regulated by GSTM2 OE. j More LDs were observed in cells with SREBF1 or PLIN2 overexpression. k Statistical analysis of number of LDs in cells with SREBF1 or PLIN2 overexpression. l Expression levels of SREBF1 and PLIN2 were down-regulated by GS-4997 treatment (ASK1 activity inhibitor). m Statistical analysis of number of LDs in shGSTM2 cells and shGSTM2 cells treated with GS-4997. n WB detection of SREBF1 and PLIN2 in shGSTM2 cells treated with PA. o WB detection of SREBF1 and PLIN2 in shGSTM2 cells treated with GS-4997. Bar, 10 μm.
C-terminus of GSTM2 inhibits ASK1 phosphorylation by binding to ASK1
We then investigated the mechanism of GSTM2 regulation of ASK1 activity. Previous studies reported that the C-terminus of GSTM1 binds to ASK1 and suppresses the phosphorylation of ASK113,14. Thus, we compared the amino acid sequences and 3D structures of these two proteins. GSTM1 and GSTM2 shared highly similar sequences and structures (Supplementary Fig. 4a, b), with >90% similarity. Moreover, another study reported that GSTM2 binds to ASK115. We next examined whether GSTM2 interacts with ASK1 by conducting a co-immunoprecipitation assay. The result indicated that GSTM2 binds to ASK1 (Fig. 4f). We then investigated the structural details of this binding interaction. GSTM2 contains two main functional domains, GST-binding (N-terminal) and C-terminal domains (Supplementary Fig. 4c). Overexpression of the C-terminus of GSTM2 decreased the p-ASK1 level, whereas N-terminal overexpression did not (Fig. 4g).
GSTM2 regulates expression levels of SREBF1 and PLIN2
SREBF1 and PLIN2 play important roles in cellular lipid metabolism. A previous study reported that SREBF1 is regulated by p38-JNK signalling16,17 and PLIN2 expression is regulated by SREBF1. We detected the effects of GSTM2 on the expression of these two genes. As expected, the expression levels of SREBF1 and PLIN2 were increased by GSTM2 knockdown and decreased by GSTM2 overexpression (Fig. 4h, i). We confirmed the effects of these two genes on cellular LD content by overexpressing SREBF1 and PLIN2. More LDs were observed in cells with both SREBF1 and PLIN2 overexpression (Fig. 4j, k). Next, GS-4997, a specific ASK1 inhibitor, was used to confirm that the expression of SREBF1 and PLIN2 is regulated by ASK1-p38/JNK signalling. SREBF1 and PLIN2 expression was decreased in cells treated with GS-4997 (Fig. 4l).
To further validate the role of ASK1 in GSTM2 knockdown cells, we detected LDs number in GSTM2 knockdown cells treated with GS-4997. The result showed that GS-4997 greatly decreased LDs number compared to GSTM2 knockdown cells (Fig. 4m). We further detected the protein level of SREBF1 and PLIN2 in GSTM2 knockdown cells treated with PA or GS-4997. The results indicated that PA increased the expression level of SREBF1 and PLIN2 (Fig. 4n), whereas GS-4997 suppressed the expression level of SREBF1 and PLIN2 (Fig. 4o). The results indicated that GSTM2 regulated SREBF1 and PLIN2 expression by affecting ASK1 activity.
GSTM2 modulates growth of LDs via ASK1-p38/JNK signalling
The growth of LDs is important in increasing the cellular capacity for lipid storage. Previous studies demonstrated that PCYT1A, GPAT4 and DGAT2 are responsible for increasing the size of LDs18–20. The expression level of PCYT1A was positively correlated with GSTM2 expression, which was confirmed by GSTM2 knockdown and overexpression assays (Fig. 5a–c). Furthermore, PCYT1A knockdown induced the formation of larger LDs in FA-rich cultural medium (Fig. 5d). In addition to regulating LD growth, PCYT1A is related to LD fusion or coalescence. We examined the events involved in LD contact using a living cell station. The supplementary movie shows that GSTM2 knockdown induced more LD–LD contact events (Supplementary movie S1) than in the control (Supplementary movie S2).
Fig. 5. GSTM2 modulated lipid droplet growth via ASK1 signalling.

a–c PCYT1A expression level was down-regulated by GSTM2 KD and up-regulated by GSTM2 OE; *p < 0.05, **p < 0.01. d PCYT1A KD induced LD expansion. e, f GSTM2 affected recruitment of GPAT4 and DGAT2 on LD surface (in the presence of oleic acid medium), bar, 10 μm, 1 μm (inlay). g Expression level of GPAT4 and DGAT2 was regulated by treatment of palmitic acid (ASK1 activator) and GS-4997 (ASK1 activity inhibitor). h Recruitment of GPAT4 on LD surface was regulated by palmitic acid or GS-4997 treatment, bar, 10 μm, 1 μm (inlay). i, j qPCR detection of PCYTA1 expression with PA or GS-4997 treatment respectively.
DGAT2 and GPAT4 targeting the surface of LDs is important for LD expansion. As described above, GSTM2 modulated the size of LDs. We investigated whether this regulation was dependent on modulation of DGAT2 and GPAT4 targeting the LD surface. We found that more DGAT2 and GPAT4 were recruited to the LD surface by GSTM2 knockdown (Fig. 5e, f). Next, we investigated the effect of ASK1-p38/JNK signalling on this progression. The expression level of DGAT2 and GPAT4 was detected in the presence of PA (ASK1 activator) or GS-4997 (ASK1 inhibitor) in normal or FA-rich cultural medium. The expression level of these protein was up-regulated by ASK1 signalling activation and down-regulated by ASK1 signalling suppression (Fig. 5g). Furthermore, recruitment of GPAT4 to the LD surface was enhanced by PA treatment and reduced by GS-4997 treatment (Fig. 5h). We further detected the effect of ASK1 activity on PCYT1A expression. The expression of PCYT1A was decreased by PA treatment, whereas was up-regulated by GS-4997 treatment (Fig. 5i, j, p < 0.05).
GSTM2 KO induced hepatic fat accumulation was suppressed by GS-4997 in vivo
We then investigated whether GS-4997 treatment could rescue the hepatic steatosis induced by GSTM2 knockout. GSTM2 KO mice were treated with 80 μM GS-4997 by intraperitoneal injection, and control mice were treated with an equal amount of DMSO solution (Fig. 6a). Then live tissues were collected and examined. We found that the GS-4997 treatment group showed less hepatic fat accumulation compared to the control group (Fig. 6b), and the TG content detection also supported this result (Fig. 6c). The results indicated that GS-4997 greatly suppressed the increase of TG accumulation induced by GSTM2 KO. Moreover, we detected the p-ASK1 level in GSTM2 KO mice, which showed the ASK1 was activated by GSTM2 deletion (Fig. 6d). Previous study suggested that autophagy affected the TG utilization in liver21,22. We detected the expression level of LC3 in liver of GSTM2 KO mice. The result showed the expression level of LC3-I and LC3-II was not changed in GSTM2 KO mice (Fig. 6d). We then investigated the protein level of ASK1-p38/JNK signalling pathway. As expected, the levels of p-ASK1, p-p38 and p-JNK in the GS-4997 treatment group were decreased significantly (p < 0.05) (Fig. 6e, f).
Fig. 6. GSTM2 KO induced hepatic fat accumulation was suppressed by GS-4997 in vivo.

a The GSTM2 KO mice were fed with chow diet and treated with GS-4997 or DMSO at day 1 and 3, then the liver tissues were collected for further analysis. b GSTM2 KO mice were treated with GS-4997 (ASK-1 inhibitor) by intraperitoneal injection. Hepatic fat was detected by HE and ORO staining. c TG content examination of WT mice fed MCDD with GS-4997 or DMSO treatment. d WB detection of expression level of p-ASK1 and LC3 in GSTM2 KO mice liver. e Investigation of protein levels of ASK1 signalling pathway in GSTM2 KO mice with GS-4997 or control, DMSO treatment by Western blot. f Fray value analysis of (e) by ImageJ software. g The WT and GSTM2 KO mice were fed with MCD diet for one month. The mice were treated with GS-4997 or DMSO at day 14, 21 and 28, and then the liver tissues were collected for further Analysis. h GSTM2 KO mice were fed MCDD and treated with GS-4997 or DMSO by intraperitoneal injection. Then hepatic fat and fibrosis level were detected by HE, ORO and Masson staining. i TG content examination of GSTM2 KO mice fed MCDD with GS-4997 or DMSO treatment. bar, 50 μm.
Subsequently, we detected whether GS-4997 treatment could block the progress of hepatic steatosis in KO and WT mice with MCD diet. The mice were fed with MCD for one month. During that time, GS-4997 was injected at 14 d, 21 d and 28d (Fig. 6g). Then live tissues were collected for further investigation. Histological examination showed that GS-4997 treatment decreased lipid accumulation in WT mice with MCDD (Supplementary Fig. 5a); additionally, the TG content in live tissues was also decreased compared to the DMSO treatment group (Supplementary Fig. 5b). The results of Western blot showed that p-ASK1, p-p38 and p-JNK protein levels were decreased significantly by GS-4997 injection in WT mice with MCDD (p < 0.05) (Supplementary Fig. 5c). Furthermore, GS-4997 also inhibited fat accumulation in liver tissues of KO mice with MCDD (Fig. 6h). The TG content of the GS-4997 treatment group was decreased (p < 0.05) (Fig. 6i). Moreover, the protein level of p-ASK1, p-p38 and p-JNK was decreased in KO mice with MCDD by GS-4997 treatment (p < 0.05) (Supplementary Fig. 5d). Besides detecting the effect of GS-4997 treatment on hepatic fat accumulation, we also investigated the degree of hepatic fibrosis of the samples. As expected, GS-4997 suppressed hepatic fibrosis in both WT and KO mice with MCDD (Fig. 6g, Supplementary Fig. 5a), as detected by Masson staining. These results support that GSTM2 knockout induced hepatic steatosis resulting from ASK1 signalling activation and can be rescued by treatment with GS-4997, an ASK1 inhibitor, in vivo.
Discussion
The risk of hepatic steatosis is greatly increased by an unhealthy lifestyle and excess energy intake. The regulatory mechanism of steatosis is unclear. Here, we showed that GSTM2 negatively regulated hepatic steatosis. GSTM2 inhibited p38 and JNK signalling by suppressing activation of the upstream kinase ASK1. More importantly, we showed that overexpression GSTM2 had a therapeutic effect on hepatic steatosis. Mechanistically, GSTM2 modulated the formation of hepatic fat mainly by regulating the number and size of cellular LDs.
Few studies have evaluated the relationship of DME and hepatic steatosis. Our study demonstrated that the hepatic phase II DME, GSTM2, was involved in the development of hepatic steatosis. A previous study showed that hepatic steatosis increased the toxicity of hepatic drugs by down-regulating the expression of some DMEs23. Moreover, many studies showed that hepatic steatosis increased the risk of drug-induced liver disease24–27. hepatic steatosis may impair hepatic self-protection. Previous studies reported that GSTM2 is highly up-regulated in NAFLD or in an HFD-induced fatty liver mice model (by more than 4-fold)6,7, but the mechanism was not evaluated. As a hepatic functional gene, up-regulation of GSTM2 may function in hepatic self-protection. Knockout and overexpression of GSTM2 in a mouse model revealed its regulatory function in the development of hepatic steatosis. GSTM2 KO enhanced the development of hepatic steatosis. Although GSTM2 overexpression did not block the development of hepatic steatosis, it delayed the progression and reduced lipid accumulation and fibrosis progression. These results suggest that a liver with steatosis protects itself against disease injury by up-regulating GSTM2.
In addition to drug-induced toxicity, ROS is another metabolic by-product that can induce potential cytotoxicity. Many studies have demonstrated a relationship between ROS and lipid accumulation, particularly in fatty liver disease and hepatoma carcinoma28–30. Superoxide dismutase 1 (SOD1)-knockout mice showed higher hepatic fat storage, and more LDs were observed in SOD1 KO hepatocytes17,31. We also found that ROSup treatment induced greater LD formation in HepG2 cells in this study. As an important cellular antioxidant, glutathione is responsible for eliminating ROS. Therefore, GSTM2 can promote ROS degradation by enhancing the activity of glutathione, as demonstrated by our results. We predicted that GSTM2 could alleviate ROS-induced neutral lipid accumulation and increase the fat content in NAFLD. Interestingly, in a previous study glomerulonephritis was treated by injecting GSTM2-transduced mesenchymal stem cells. The researchers found that GSTM2 expression significantly reduced oxidation and inflammation in the disease tissues and greatly ameliorated glomerulonephritis32. In our study, we observed a lower occurrence of fibrosis in GSTM2-overexpressing mice than in control mice. This phenotype is attributed to the function of GSTM2 which ameliorates cellular stress and inflammation. We detected the antioxidant expression level in the GSTM2 KO mice model and found that most antioxidants including SOD1, CAT, and GPX1 were down-regulated. The higher level of ROS and inflammation may impair the function and expression of these antioxidants. Notably, GSTM1 shows high similarity in sequence and structure (>90%) to GSTM2, and Bhattacharjee et al. reported that GSTM2 can play a compensatory role in the absence of GSTM133. Indeed, GSTM1 has a similar function as GSTM2, but several single-nucleotide polymorphisms may lead to gene inactivation. Moreover, no inactivating mutation of GSTM2 has been found in human or house mouse. We considered that GSTM2 was likely be more stable than GSTM1 from a genetic perspective, and therefore, up-regulating GSTM2 may be a better choice for inducing protective effects.
We demonstrated that GSTM2 suppressed the progression of hepatic steatosis by inhibiting ASK1-p38/JNK signalling. This signalling has been reported to play an important role in the development of NAFLD and NASH. ASK1, also known as MAP3K5, activates the downstream p38-JNK1/2 signalling pathway, thereby promoting inhibition of lipid and glucose metabolism34–36 and driving a strong inflammatory response37. ASK1 is currently considered as a target site for NASH therapy38,39, and ASK1 signalling is abnormally active in the liver of NAFLD and obese individuals. Inhibition of ASK1 activity significantly inhibits the development of NASH. For example, selonsertib (GS-4997) is a highly selective and potent ASK1 inhibitor with potential anti-inflammatory, anti-tumour, and anti-fibrotic activities40. Clinical trial results of the patient showed that GS-4997 has a good therapeutic effect on NASH. However, GS-4997 completely inhibits the activity and normal function of ASK1 with potential side effects41,42. In our study, hepatic GSTM2 was found to suppress steatosis by inhibiting ASK1-p38/JNK signalling. Moreover, GSTM2 inhibits hepatic steatosis by affecting the capability to detoxify and function as an antioxidant. Some previous studies supported our results. Han et al. found that GSTM2 expression was much higher in ovarian teratoma mesenchymal stem cell-like cells than in normal cells and that p38 was highly inhibited in the teratomas43. Some early references also described the function of GSTs in regulating the activity of kinases such as ASK1 via protein-protein interactions14,44–46. Currently, the effect of ASK1-p38/JNK signalling on NAFLD and NASH has attracted the attention of researchers. It is important to note that there are currently some negative results of ASK1-related small molecule inhibitors (such as CFLAR-mimicking peptide) in clinical trials. Some studies have observed that these small molecule inhibitors may promote inflammation and amplify NASH liver injury47. Our findings may provide new insights into the role of ASK1 in NAFLD and contribute to the development of related molecular agents.
We also investigated the effect of GSTM2 on LD growth. Inhibition of GSTM2 resulted in a significant increase in the diameter of LDs formed by cells in a fatty acid-rich environment, and thus GSTM2 appears to affect the LD growth process. According to previous studies, LD growth depends on lipid synthesis genes such as DGAT2 and GPAT4, and the key to the growth of LDs is the recruitment of DGAT2 and GPAT4 to the LD surface18. We inhibited the expression of GSTM2 and found that recruitment of DGAT2 and GPAT4 to the surface of LDs was increased. Because hydrogen peroxide treatment did not significantly affect the LD diameter, intracellular ROS may have minimal effects on LD growth. We further examined whether ASK1 signalling affects the LD growth process. When cells were treated with 400 mM palmitic acid or 5 μM GS-4997, the recruitment of DGAT2 and GPAT4 on the surface of LDs showed significant differences. When ASK1 was activated, DGAT2 and GPAT4 were recruited to the LD surface, whereas when ASK1 activity was inhibited, recruitment of GPAT4 to the surface of LDs was reduced. Therefore, ASK1 may affect the growth of LDs by affecting the recruitment of DGAT2 and GPAT4 to the LD surface. No previous studies reported the effect of GSTM2 or ASK1-p38/JNK signalling on LD growth. This is the first study to demonstrate that ASK1-p38/JNK regulated LD growth by promoting DGAT2 and GPAT4 targeting to the surface of LDs. We did not investigate the mechanism of this regulatory role; therefore, further studies are needed.
In summary, we identified GSTM2 as a protective factor against hepatic steatosis. GSTM2 interacted with ASK1 and suppressed its phosphorylation and the activation of downstream p38-JNK signalling. Moreover, GSTM2 plays an important role in detoxification and antioxidant activities, which protect tissues against injury induced by drugs or oxidative stress (Fig. 7). GSTM2 showed a therapeutic effect on hepatic steatosis, providing a potential strategy for the clinical treatment of NAFLD.
Fig. 7. GSTM2 protects against the development of hepatic steatosis via its antioxidant and suppressing ASK1-p38/JNK signaling.
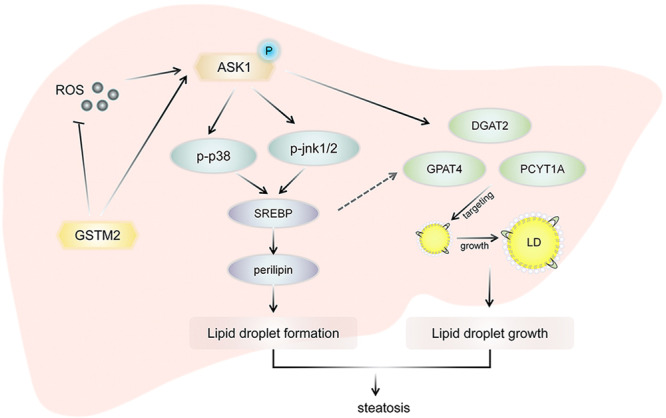
GSTM2 was highly up-regulated in hepatic steatosis tissues. As the member of phase II drug metabolizing enzymes, GSTM2 could detoxify the toxicity of metabolic products such as ROS. ROS could enhance the inflammation and fibrosis progression. Furthermore, GSTM2 interacted with ASK1 directly and suppressed ASK1 phosphorylation. Therefore, GSTM2 down-regulated expression of SREBP and perilipins to inhibit the LD accumulation. ASK1 signaling also regulated the expression level of DGAT2, GPAT4 and PCYT1A and the recruitment of DGAT2 and GPAT4 on LD surface to modulate the LD growth. In conclusion, GSTM2 protects against the hepatic steatosis of NAFLD via its antioxidant and suppressing ASK1-p38/JNK signaling.
Materials and methods
Animals and cell lines
GSTM2 knockout mice were prepared by Cyagen Bioscience, Inc. (Santa Clara, CA, USA). The C57/BL-6 mice were used for the knockout construction. One single-base deletion was induced in exon3 of the GSTM2 (NM_008183.3) gene using the TALEN method. All mice were housed in a normal environment and provided with food and water. The 6–8 week ages male mice were used in this study. All experimental protocols were approved by the Ethics Committee of Huazhong Agricultural University. The HepG2 cell line was gifted by Xianghua Yan’s lab (Huazhong Agricultural University).
Ethics approval and consent to participate
All mouse were housed in a normal environment provided with food and water. The methods were performed in accordance with the approved guidelines from Huazhong Agricultural University, and scientific, ethical and legal principles of the Hubei Regulations for the Administration of Affairs Concerning Experimental Animals. All experimental protocols were approved by the Ethics Committee of Huazhong Agricultural University.
Antibodies
The following rabbit polyclonal antibodies were used: anti-GSTM2 (#ab125,102, Abcam, Cambridge, UK, 1:1000), anti-GSTM2 (#A13,496, ABclonal, Wuhan, China, 1:2000), anti-GAPDH (#CSB-PA00025A0Rb Flarebio Biotech LLC, College Park, MD, USA, 1:1000), anti-CIDEC (#D222114, Sangon Biotech, Shanghai, China, 1:1000), anti-CIDEC (#12287-1-AP, Proteintech, Rocky Hill, NJ, USA, 1:1000), anti-ADRP/perilipin2 (#15294-1-AP, Proteintech), anti-DGAT2 (#bs-12,998R, BIOSS, Beijing, China), anti-DGAT2 (#A13,891, ABclonal, Wuhan, China, 1:2000), anti-GPAT4 (#bs-15,587R, BIOSS), anti-SREBP1 (#14,088-1-AP, Proteintech), and anti-PCYT1A (#bs-11306R, BIOSS), anti-PCYT1A (#A5935, ABclonal, Wuhan, China, 1:1000). The mouse monoclonal antibody used was anti-tubulin (#GB13017-2, Servicebio, Wuhan, China, 1:1000). The following secondary antibodies were used: Alexa Fluor 555-labelled donkey anti-rabbit IgG (H+L) (#A0453, Beyotime, Shanghai, China, 1:200), horseradish peroxidase (HRP)-labelled goat anti-rabbit IgG (H+L) (#GB23303-1, Servicebio, 1:8000), HRP-labelled goat anti-mouse IgG (H+L) (#GB23301, Servicebio, 1:8000), and HRP-conjugated goat anti-rabbit IgG (#D110,058, Sangon Biotech, 1:8000).
Plasmid DNA construction
Full-length coding sequences encoding GSTM2 (NM_001142368.1) and PCYT1A (NM_005017.3) were amplified using a cDNA library of HepG2 cells and then subcloned into the pcDNA3.1 vector (gifted from Prof. Dequan Xu’s lab, Huazhong Agricultural University) and pEGFP-C1 and pmCherry-C1 vectors (gifted from Prof. Xianghua Yan’s lab, Huazhong Agricultural University). Sequences of the primers used for the GSTM2 RNAi experiment were referred to study of Huenchuguala48. The sequences were subcloned into the pSuperior.puro vector for the RNAi experiment (gifted from Prof. Xianghua Yan’s lab).
Cell culture and transfection
WT HepG2 or stably transfected cells (pSuperior.puro-GSTM2) were cultured in DMEM (HyClone, Logan, UT, USA) containing 10% foetal bovine serum (Clark Bioscience, Richmond, VA, USA), 100 U/mL penicillin, and 100 g/mL streptomycin in dishes at 37 °C and transfected with Lipo6000™ Transfection Reagent (#C0528, Beyotime). HepG2 cells were seeded onto a cell slide in a 24-well plate and transfected with plasmid vectors according to the transfection reagent instructions.
HE staining and Oil Red O staining of histological sections
For HE staining, the liver tissues of three GSTM2 knockout mice and three wild-type mice were divided and fixed in 4% paraformaldehyde for 12 h. Then the samples were performed the HE and Oil Red O staining by Servicebio (Wuhan, China) (http://www.servicebio.com/).
ROS level detection
Cells were seeded into a 96-well black plate. ROS levels in the cells were detected using an ROS Assay Kit (#S0033, Beyotime) based on the DCFH-DA method. The ROS level was detected using a microplate spectrophotometer (PerkinElmer EnSpire, Waltham, MA, USA). The excitation and emission wavelengths were 488 and 525 nm, respectively. ROSup was used as an activator to increase the ROS level in cells, which was added to the wells at 1:1000 (v:v) with DMEM and incubated at 37 °C.
Western blot and real-time PCR
Real-time PCR was performed using a Roche LightCycler 480 detection system (Roche, Basel, Switzerland). One negative control reaction in which the cDNA template was replaced with water was performed as a control. Each sample was amplified in triplicate, and the comparative Ct (ΔΔCt) value method was used for relative quantification. GAPDH was used as a reference gene. The Western blot method referred to our previous study49.
Immunofluorescence assay
The immunofluorescence method referred to our previous study49. The confocal laser scanning microscope (Carle Zeiss, German) was used to observe the slide of cells. The images were analysed by ZEN pro software (Carle Zeiss, German), ImageJ and Photoshop CS6 (Adobe).
Living cells workstation
The cells were seeded into a 35 mm confocal dish for 24 h culture and OA treatment, and then were washed 3 times with PBS and then added to the DMEM culture containing BODIPY (1:1000, v/v), followed by incubation for 20 min at 37 °C. The dish was placed in a living cell workstation, and intracellular LDs were observed using a 63 × oil immersion objective. The exposure time was set to 200 ms, the definitely focused 2.0 strategy was used, the interval of image capture was 15 s, and the duration of capture was 1 h. The data were analysed using ZEN pro and ZEN 2.3 (blue edition) software (ZEISS, Oberkochen, Germany).
HFD and MCDD feeding experiment
Fifteen 4-week-old healthy male GSTM2 knockout mice and 15 wild-type mice were selected for high-fat diet (HFD) feeding. The mice were divided into five groups of 3 individuals each. The mice were sacrificed on days 0, 5, 10, 20, and 30 after the start of HFD feeding and then liver tissues were collected. The formula of the HFD was ordinary mixed diet (88.5%), lard (10%), cholesterol (1%), and pig bile salt (0.5%). Six-week-old male mice were divided into two groups and fed an MCDD. The tissue collection was conducted as described above.
Triglyceride content detection
The TG content of the liver tissues was detected by fully automatic chemistry analyser (#Chemray 240, Rayto Life and Analytical Sciences Co.,Ltd., Shenzhen, China). The total protein concentration was detected by Enzyme Label Detector (#Epoch, BioTek Instruments, Inc., headquartered in Winooski, VT, USA). The TG content data was normalized by protein concentrations, mmol/g.
Plasmid and inhibitor injection
The GSTM2 overexpression or control plasmid (200 μg) was mixed with HiGene I (14.4 μL) (#C1507, Applygen Technologies Inc., Beijing, China). Then the mixture was incubated at room temperature (RT) for 30 min. The mixture was injected intraperitoneally. By detection of GSTM2 expression in liver, skeletal muscle, heart and brain, no significant change was observed except in liver tissues. In addition, 5 mg of GS-4997 (also called selonsertib; #S8292, Selleck, Shanghai, China), an ASK1 inhibitor, was mixed with 1 mL of dimethyl sulfoxide (DMSO) to prepare the 10 mM mother liquor. Then the mother liquor was diluted by normal saline for 80 μM working fluid, and the inhibitor was injected intraperitoneally.
Statistics and reproducibility
All experiments were repeated three times. Data are shown as the mean ± SD. Student’s t-test was used for statistical comparisons. For experiments comparing more than two groups, one-way ANOVA and two-way ANOVA were used. P value < 0.05 was considered as statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFF1000601), the National Natural Science Foundation of China (32172700) and the Joint Funds of the National Natural Science Foundation of China (U20A2052), and the Fundamental Research Funds for the Central Universities (grant number 2662018PY043).
Author contributions
Y.J. and Z.-Q.R. participated in research design; Y.J., Y.-J.T., P.-X.Z., Y.G. and S.-L.C. performed experiments; Y.J. drafted the paper; Y-J.T., J.W. and Z.-Q.R. contributed to the writing of the manuscript and supervised the study. All authors read and approved the final manuscript.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Gabriela da Silva Xavier and Karli Montague-Cardoso.
Data availability
The source data for all the graphs in main figures in Supplementary Data 1. The data used to support the findings of this study are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jian Wu, Email: wujian@mail.hzau.edu.cn.
Zhuqing Ren, Email: renzq@mail.hzau.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-022-03251-w.
References
- 1.Thompson MD, et al. Enhanced steatosis and fibrosis in liver of adult offspring exposed to maternal high-fat diet. Gene Expr. 2016;17:47–59. doi: 10.3727/105221616X692135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherif ZA, et al. Global epidemiology of nonalcoholic fatty liver disease and perspectives on US minority populations. Dig. Dis. Sci. 2016;61:1214–1225. doi: 10.1007/s10620-016-4143-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat. Rev. Gastro Hepat. 2013;10:686–690. doi: 10.1038/nrgastro.2013.171. [DOI] [PubMed] [Google Scholar]
- 4.Beuckmann CT, Fujimori K, Urade Y, Hayaishi O. Identification of Mu-class glutathione transferases M2-2 and M3-3 as cytosolic prostaglandin E synthases in the human brain. Neurochem. Res. 2000;25:733–738. doi: 10.1023/a:1007579507804. [DOI] [PubMed] [Google Scholar]
- 5.Chanas SA, et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 2002;365:405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Si X, Strappe P, Blanchard C, Zhou Z. Enhanced anti-obesity effects of complex of resistant starch and chitosan in high fat diet fed rats. Carbohydr. Polym. 2017;157:834–841. doi: 10.1016/j.carbpol.2016.10.042. [DOI] [PubMed] [Google Scholar]
- 7.Wang C, Tao Q, Wang X, Wang X, Zhang X. Impact of high-fat diet on liver genes expression profiles in mice model of nonalcoholic fatty liver disease. Environ. Toxicol. Pharmacol. 2016;45:52–62. doi: 10.1016/j.etap.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Saify K, Saadat I, Saadat M. Down-regulation of antioxidant genes in human SH-SY5Y cells after treatment with morphine. Life Sci. 2016;144:26–29. doi: 10.1016/j.lfs.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 9.Huenchuguala S, et al. Glutathione transferase mu 2 protects glioblastoma cells against aminochrome toxicity by preventing autophagy and lysosome dysfunction. Autophagy. 2014;10:618–630. doi: 10.4161/auto.27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuevas C, et al. Glutathione transferase-M2-2 secreted from glioblastoma cell protects SH-SY5Y cells from aminochrome neurotoxicity. Neurotox. Res. 2015;27:217–228. doi: 10.1007/s12640-014-9500-1. [DOI] [PubMed] [Google Scholar]
- 11.Huenchuguala, S., Muñoz, P. & Seguraaguilar, J. The importance of mitophagy in maintaining mitochondrial function in U373MG cells. bafilomycin a1 restores aminochrome-induced mitochondrial damage. Acs Chem. Neurosci.8, 2247–2253 (2017). [DOI] [PubMed]
- 12.Li DJ, et al. Melatonin safeguards against fatty liver by antagonizing TRAFs-mediated ASK1 deubiquitination and stabilization in a beta-arrestin-1 dependent manner. J. pineal Res. 2019;67:e12611. doi: 10.1111/jpi.12611. [DOI] [PubMed] [Google Scholar]
- 13.Dorion S, Lambert H, Landry J. Activation of the p38 signaling pathway by heat shock involves the dissociation of glutathione S-transferase Mu from Ask1. J. Biol. Chem. 2002;277:30792. doi: 10.1074/jbc.M203642200. [DOI] [PubMed] [Google Scholar]
- 14.Cho SG, et al. Glutathione S-transferase Mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J. Biol. Chem. 2001;276:12749–12755. doi: 10.1074/jbc.M005561200. [DOI] [PubMed] [Google Scholar]
- 15.Dolado I, et al. p38 alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–190. doi: 10.1016/j.cell.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurahashi T, et al. An SOD1 deficiency enhances lipid droplet accumulation in the fasted mouse liver by aborting lipophagy. Biochem. Biophys. Res. Commun. 2015;467:866–871. doi: 10.1016/j.bbrc.2015.10.052. [DOI] [PubMed] [Google Scholar]
- 18.Wilfling F, et al. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. ELife. 2013;3:e01607. doi: 10.7554/eLife.01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Y, et al. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krahmer N, et al. Phosphatidylcholine Synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 2011;14:504–515. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong XW, Tao RY, DePinho RA, Dong XC. The autophagy-related gene 14 (Atg14) is regulated by forkhead box o transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J. Biol. Chem. 2012;287:39107–39114. doi: 10.1074/jbc.M112.412569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L, Li P, Fu SN, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes er stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, et al. Hepatic pharmacokinetics of cationic drugs in a high-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Drug Metab. Dispos. 2011;39:571–579. doi: 10.1124/dmd.110.036806. [DOI] [PubMed] [Google Scholar]
- 24.Tarantino G, Di Minno MND, Capone D. Drug-induced liver injury: is it somehow foreseeable? World J. Gastro. 2009;15:2817–2833. doi: 10.3748/wjg.15.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fisher CD, et al. Experimental non-alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur. J. Pharm. 2009;613:119–127. doi: 10.1016/j.ejphar.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang WV, Ramzan I, Murray M. Impaired microsomal oxidation of the atypical antipsychotic agent clozapine in hepatic steatosis. J. Pharm. Exp. Ther. 2007;322:770–777. doi: 10.1124/jpet.107.124024. [DOI] [PubMed] [Google Scholar]
- 27.Tarantino G, et al. A prospective study of acute drug-induced liver injury in patients suffering from non-alcoholic fatty liver disease. Hepatol. Res. 2007;37:410–415. doi: 10.1111/j.1872-034X.2007.00072.x. [DOI] [PubMed] [Google Scholar]
- 28.Videla LA, Rodrigo R, Araya J, Poniachik J. Insulin resistance and oxidative stress interdependency in non-alcoholic fatty liver disease. Trends Mol. Med. 2006;12:555–558. doi: 10.1016/j.molmed.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Gawrieh S, Opara EC, Koch TR. Oxidative stress in nonalcoholic fatty liver disease: Pathogenesis and antioxidant therapies. J. Invest Med. 2004;52:506–514. doi: 10.1136/jim-52-08-22. [DOI] [PubMed] [Google Scholar]
- 30.Videla LA, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004;106:261–268. doi: 10.1042/CS20030285. [DOI] [PubMed] [Google Scholar]
- 31.Lee J, Homma T, Kurahashi T, Kang ES, Fujii J. Oxidative stress triggers lipid droplet accumulation in primary cultured hepatocytes by activating fatty acid synthesis. Biochem. Biophys. Res. Commun. 2015;464:229–235. doi: 10.1016/j.bbrc.2015.06.121. [DOI] [PubMed] [Google Scholar]
- 32.Li, Y. J. et al. Glutathione S-transferase Mu 2-transduced mesenchymal stem cells ameliorated anti-glomerular basement membrane antibody-induced glomerulonephritis by inhibiting oxidation and inflammation. Stem Cell Res. Ther.5, 19 (2014). [DOI] [PMC free article] [PubMed]
- 33.Bhattacharjee P, et al. Functional compensation of glutathione S-transferase M1 (GSTM1) null by another GST superfamily member, GSTM2. Sci. Rep. 2013;3:2704. doi: 10.1038/srep02704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun, P. et al. Caspase recruitment domain protein 6 protects against hepatic steatosis and insulin resistance by suppressing Ask1. Hepatology. 68, 2212–2229 (2018). [DOI] [PubMed]
- 35.Ye, P. et al. Dual-specificity Phosphatase 9 Protects Against Non-alcoholic Fatty Liver Disease in Mice via ASK1 Suppression. Hepatology. 69, 76–93 (2018). [DOI] [PMC free article] [PubMed]
- 36.Lawan A, et al. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulate glucose metabolism and energy homeostasis. Mol. Cell. Biol. 2015;35:26. doi: 10.1128/MCB.00503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cingolani, F. & Czaja, M. J. Oxidized albumin—a Trojan Horse for p38 MAPK-mediated Inflammation in decompensated Cirrhosis. Hepatology.68,1678–1680 (2018). [DOI] [PMC free article] [PubMed]
- 38.Nakagawa H, et al. Apoptosis signal-regulating kinase 1 inhibits hepatocarcinogenesis by controlling the tumor-suppressing function of stress-activated mitogen-activated protein kinase. Hepatology. 2011;54:185–195. doi: 10.1002/hep.24357. [DOI] [PubMed] [Google Scholar]
- 39.Win S, et al. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology. 2017;67:2013–2024. doi: 10.1002/hep.29689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loomba R, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology. 2018;67:549–559. doi: 10.1002/hep.29514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villanueva MT. Liver disease: conscious uncoupling in NASH. Nat. Rev. Drug Discov. 2017;16:238. doi: 10.1038/nrd.2017.60. [DOI] [PubMed] [Google Scholar]
- 42.Nishida, T., Hattori, K. & Watanabe, K. The regulatory and signaling mechanisms of the ASK family. Adv Biol. Regul. 66, 2–22 (2017). [DOI] [PubMed]
- 43.Han I, et al. Proteomic analysis of mesenchymal stem-like cells derived from ovarian teratoma: potential role of glutathione S-transferase M2 in ovarian teratoma. Proteomics. 2011;11:352–360. doi: 10.1002/pmic.201000475. [DOI] [PubMed] [Google Scholar]
- 44.McIlwain CC, Townsend DM, Tew KD. Glutathione S-transferase polymorphisms: cancer incidence and therapy. Oncogene. 2006;25:1639–1648. doi: 10.1038/sj.onc.1209373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ichijo H, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 46.Hsieh CC, Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. Faseb J. 2006;20:259–268. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537–2564. doi: 10.1016/j.cell.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 48.Huenchuguala S, et al. Glutathione transferase mu 2 protects glioblastoma cells against aminochrome toxicity by preventing autophagy and lysosome dysfunction. Autophagy. 2014;10:618–630. doi: 10.4161/auto.27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin, Y., Tan, Y. J., Chen, L. P., Liu, Y. & Ren, Z. Q. Reactive oxygen species induces lipid droplet accumulation in HepG2 cells by increasing perilipin 2 expression. Int. J . Mol. Sci.19, 3445 (2018). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The source data for all the graphs in main figures in Supplementary Data 1. The data used to support the findings of this study are available from the corresponding author upon request.